 Barak Blonder1
Barak Blonder1 Valeria Boyko1
Valeria Boyko1 Alexandra V. Turchyn2
Alexandra V. Turchyn2 Gilad Antler2
Gilad Antler2 Uriel Sinichkin1
Uriel Sinichkin1 Nadav Knossow1
Nadav Knossow1 Rotem Klein1
Rotem Klein1 Alexey Kamyshny Jr.1*
Alexey Kamyshny Jr.1*- 1Department of Geological and Environmental Sciences, Faculty of Natural Sciences, Ben-Gurion University of the Negev, Beer Sheva, Israel
- 2Department of Earth Sciences, University of Cambridge, Cambridge, United Kingdom
The Gulf of Aqaba is an oligotrophic marine system with oxygen-rich water column and organic carbon-poor sediments (≤0.6% at sites that are not influenced by anthropogenic impact). Aeolian dust deposition from the Arabian, Sinai, and Sahara Deserts is an important source of sediment, especially at the deep-water sites of the Gulf, which are less affected by sediment transport from the Arava Desert during seasonal flash floods. Microbial sulfate reduction in sediments is inferred from the presence of pyrite (although at relatively low concentrations), the presence of sulfide oxidation intermediates, and by the sulfur isotopic composition of sulfate and solid-phase sulfides. Saharan dust is characterized by high amounts of iron minerals such as hematite and goethite. We demonstrated, that the resulting high sedimentary content of reactive iron(III) (hydr)oxides, originating from this aeolian dry deposition of desert dust, leads to fast re-oxidation of hydrogen sulfide produced during microbial sulfate reduction and limits preservation of reduced sulfur in the form of pyrite. We conclude that at these sites the sedimentary sulfur cycle may be defined as cryptic.
Introduction
Reduction–oxidation (redox) reactions in marine sediments are the only source of metabolic energy for microorganisms in the absence of light. The dominant electron donor in marine sediments is organic carbon due to its high metabolic energy yield and general availability in marine environments (Boudreau, 1992; Orcutt et al., 2011). Microbially mediated oxidation of this organic carbon utilizes electron acceptors according to the metabolic energy yield of the respective redox reaction. Electron acceptors are generally consumed according to the Gibbs free energy yield of respiratory pathways associated with organic matter oxidation (Froelich et al., 1979). Depending on the availability and lability of organic carbon in the sediment, and the concentration and speciation of electron acceptors, zones characterized by the reduction of various electron acceptors may overlap (Canfield et al., 1993; Thamdrup, 2000; Hansel et al., 2015). In marine and marginal marine sediments, the oxidation of organic carbon coupled to iron reduction and microbial sulfate reduction are of key importance. Recent work has shown that the sedimentary iron and sulfur cycles may be coupled in interesting and complicated ways (Holmkvist et al., 2011; Mills et al., 2016).
There are three possible fates for hydrogen sulfide produced during microbial sulfate reduction. The first fate is removal through precipitation as iron monosulfide followed by conversion to pyrite (Burdige and Nealson, 1986). The second fate is possible in iron-limited environments: hydrogen sulfide can be incorporated into sedimentary organic matter through polysulfide formation (Werne et al., 2008; Amrani, 2014). The third fate is re-oxidation by chemical or microbially mediated processes to sulfide oxidation intermediates (Sn2-, S0, S2O32-, SO32-, and S4O62-) and/or to the terminal oxidation product, sulfate (Chen and Morris, 1972; Yao and Millero, 1996; Lichtschlag et al., 2013; Holmkvist et al., 2014). Thus, if microbial sulfate reduction takes place in sediments rich in electron acceptors such as reactive Fe(III) (hydr)oxides and manganese oxides (MnO2), hydrogen sulfide may be either totally reoxidized to sulfate or only partially preserved as trace amounts of pyrite in the sediment. If re-oxidation of hydrogen sulfide is fast enough, aqueous sulfide concentrations may remain low and no products of microbial sulfate reduction (e.g., iron sulfide, pyrite, organic sulfur) are preserved. In this case the sulfur cycle has been called “cryptic” (Turchyn et al., 2006; Holmkvist et al., 2011; Hansel et al., 2015; Mills et al., 2016).
Transformation of sulfate to hydrogen sulfide is associated with a large sulfur isotopic fractionation (Harrison and Thode, 1958); up to ε≈70‰ at ambient temperature has been suggested using models of microbial metabolism (Farquhar et al., 2003; Brunner and Bernasconi, 2005), sulfur isotopic fractionation up to ε = 66‰ has been measured in microbial pure cultures (Sim et al., 2011a), and sulfur isotope fractionation up to 77‰ has been measured in deep ocean sediments at elevated temperatures (Rudnicki et al., 2001). The extent of sulfur isotope fractionation depends on sulfate concentrations, microbial sulfate reduction rate, substrate type, and temperature (Habicht et al., 2002; Sim et al., 2011b). In the case of a cryptic sulfur cycle, where most hydrogen sulfide is re-oxidized to sulfate and neither free hydrogen sulfide nor pyrite are present, sulfur isotopes are less useful for study of the biogeochemical sulfur cycle because they are quantitatively conserved and do not necessarily exhibit a large sulfur isotope fractionation (Mills et al., 2016). In this case study, the oxygen isotope composition of sulfate has been shown to be a very powerful tool for studying the redox cycling of sulfur. Similar to sulfur isotopes, oxygen isotopes in dissolved sulfate (δ18OSO4) also increase during microbial sulfate reduction as the light 16O isotope is preferentially reduced, but at some stage stop increasing and reach a value that is in oxygen isotopic equilibrium with water (Fritz et al., 1989; Böttcher et al., 1999). As sulfate and water do not exchange oxygen isotopes easily, this observed oxygen isotope exchange is attributed to the intracellular exchange of oxygen atoms between sulfur intermediate species (such as sulfite) and water (Mizutani and Rafter, 1973; Fritz et al., 1989). The observed 18O isotope enrichment over the isotopic composition of water (magnitude 22 - 30‰) reflects intracellular oxygen isotope exchange (Böttcher et al., 1998, 1999; Brunner et al., 2005, 2012; Turchyn et al., 2006, 2010; Antler et al., 2013). Therefore, if a significant fraction of sulfate is reduced and reoxidized to sulfate, exchange of oxygen isotopes between sulfate and water can be detected, and thus the oxygen isotopic composition of sulfate may be a proxy for any quantitative cycling of sulfur such as during a cryptic sulfur cycle (Bishop et al., 2013; Johnston et al., 2014).
Airborn dust is an important source of nutrients and essential metals to wide areas of the ocean (Duce et al., 1991). The input of mineral dust may affect seawater biogeochemistry and sedimentation processes (Jickells, 1995; Chen et al., 2008). Aeolian dust deposition is especially significant in marine systems that are situated in arid environments where fluvial input is limited. Wind-transported dust with high iron and manganese content, which mainly consists of minerals derived from arid and semi-arid mid-latitude regions in the Northern Hemisphere, has been suggested as a crucial factor that may control primary productivity in high nutrients - low chlorophyll marine ecosystems (Duce and Tindale, 1991; Street and Paytan, 2005). After deposition of dust at the ocean surface, only a small fraction of the aeolian iron flux becomes bioavailable due to the low solubility of the colloidal and particulate Fe(III) (mostly between 1 and 12%) that comprises the bulk of the aerosol iron pool (Mahowald et al., 2005). In the Red Sea, ≤2% of dry-deposited aerosol iron is dissolved during the transport of dust particles to the sediment (Chen et al., 2006).
High fluxes of aeolian dry deposition of dust from adjacent deserts makes the Gulf of Aqaba an ideal playground for the study of the impact of high concentrations of reactive iron and manganese on sulfur cycling in sediments in an oligotrophic marine system. In this work, we report on the concentrations of iron, manganese and sulfur (in various valence states) as well as the isotopic composition of sulfur in the sediments, across a range of water depths, including a site influenced by anthropogenic activity. We evaluate the importance of the reoxidative part of the sulfur cycle, and how this varies with changes in water depth, and identify a cryptic sulfur cycle at the deep-water sites.
Study Site
The Gulf of Aqaba is 180 km long, 6 to 25 km wide and up to 1850 m deep (Ben-Avraham et al., 1979) (Figure 1). A transform fault located at the center of the Gulf of Aqaba causes steep-slope topography. The limited water influx from the Red Sea through the narrow (800 m wide) and shallow (80–250 m depth) Straits of Tiran results in high water column temperature (21–27°C) throughout the year, as warm surface waters flow from the Red Sea into the Gulf and deep waters flow out from the Gulf into the Red Sea (Ben-Avraham et al., 1979; Biton and Gildor, 2011). During the summer months, the water column is stratified and water mixing occurs only to a depth of 20 m. During winter, the water column typically mixes to a depth of 300-400 m but can be mixed down to 860 m depth during exceptionally cold winters (Genin et al., 1995). Nevertheless, the bottom waters of the Gulf of Aqaba are oxic throughout the year with a reported oxygen minimum of 168 μM at 800 m (Reiss and Hottinger, 1984; Badran, 2001). The Gulf of Aqaba is an oligotrophic marine system. Primary production in photic zone (upper 170 m) of the Gulf of Aqaba varies in the range of 0.05–3.38 mg C m-3 h-1 (Levanon-Spanier et al., 1979; Stambler, 2006). The productivity rates are limited due to the seasonal nutrient (N, P) depletion during summer stratification (Reiss and Hottinger, 1984; Li et al., 1998; Labiosa et al., 2003).
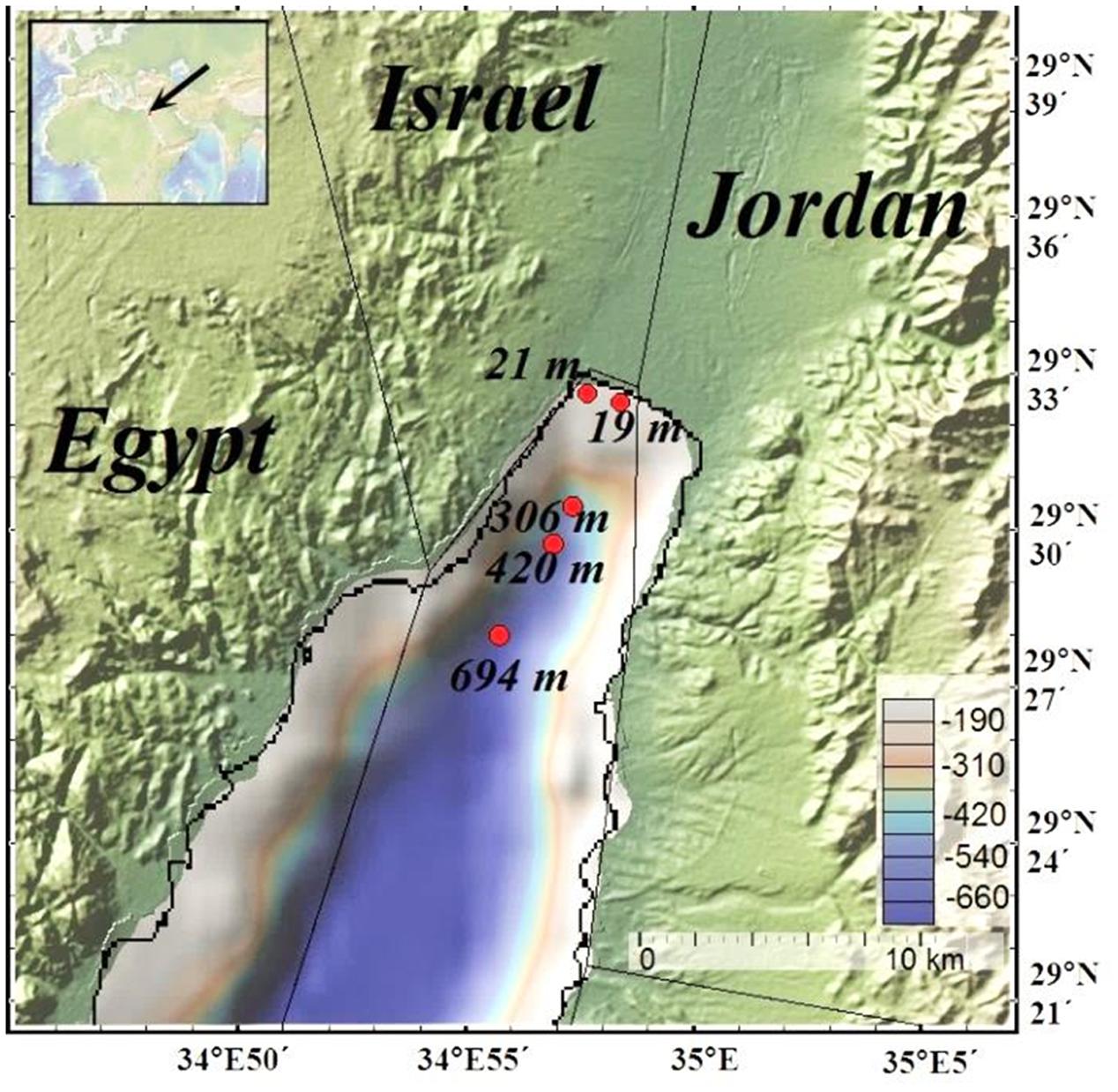
FIGURE 1. Map of the Gulf of Aqaba. Red dots represent sampling sites (map was generated using GeoMapApp, http://www.geomapapp.org).
Particulate input is mostly of aeolian origin and other inputs are relatively low due to scarse local rainfall (22 mm y-1). Recently, it was reported that possible sources of fluvial material to the Gulf are desert flash floods from the Arava desert after rare rainfall events (Katz et al., 2015), but deposition of this fluvial material would impact only sediments in the immediate vicinity of the shoreline. However, in the vicinity of coral reefs the coarse sediments are nearly all biogenic, as depositional rates of terrigenous material from wadies are equal to coral growth (Reiss and Hottinger, 1984). Reported iron content in suspended flashflood sediments is 41 mg g-1 with total inflow into the Gulf of 10 g Fe m-2 (179 mmol m-2) (Katz et al., 2015). Sediments at the north shore of the Gulf, which is adjacent to Arava Valley, are fluvial silts and fine sands with a mean grain size of 257 μm and c.a. 20–23% silt (Katz et al., 2002). Sediments overlain by deep waters (825 m) are characterized by smaller grain size, with a mean grain size of 45 μm and total organic carbon of 0.4% (Rasheed et al., 2003).
Sedimentation rates in the deep waters (up to 826 m) near the Jordanian coast are 42–72 cm ky-1 (Al-Rousan et al., 2004). Lower rates, 4–10 cm ky-1 were reporetd by Reiss et al. (1980) at <900 m depth. Much higher rates, 220–330 cm ky-1, were reported for shallow (10 m water depth, coral reef) sites by Larsen (1978). The strong variation in sedimentation rates in the Gulf of Aqaba may be caused by different degrees of resuspension, which depends on the textural properties of the sediments (e.g., finer sediments with low density are resuspended to larger areas) as well as on storm events (Schumacher et al., 1995; Al-Rousan, 1998).
The Gulf of Aqaba is strongly affected by dust storm events, which supply dust particles with iron, aluminum and manganese from adjacent (e.g., the Negev Desert, the Sinai, and Arabian Peninsulas) and distant (e.g., the Sahara) arid environments (Chase et al., 2006). The total deposition rate of aeolian mineral dust to the Gulf of Aqaba is 34.7 g m-2 year-1 (Al-Taani et al., 2015), whereas the aerosol iron deposition flux varies in range of 1.5–116 μmol m-2 d-1 with a mean of 10 μmol m-2 d-1 (Chase et al., 2006). Inputs of terrigenous sedimentary material are restricted to seasonal hyperpycnal flows, which may reach reef sediments during rare rain events.
Along the northern shore of the Gulf of Aqaba near the Israel-Jordan border, a fish farm (‘Ardag’) operated from 1989 to 2008. The fish farm contributed large amounts of organic carbon to the previously organic matter-poor sediments. In 2002, total organic carbon content as high as 6.85% was detected in the sediments at the fish farm site (Black et al., 2012).
Materials and Methods
Sampling
Five sampling campaigns were carried out between May 2012 and February 2013 at water depths of 19 m (site RS-II-19, ex-fish farm location), 21 m (site RS-V-21), 306 m (site RS-IV-306), 420 m (site RS-III-420), and 694 m (site RS-I-694) (Figure 1 and Table 1). Sampling sites map was generated using GeoMapApp1 (Ryan et al., 2009). At the deep-water sites, cores were retrieved by a multicorer from the IUI-Eilat research vessel. Near-shore sampling was performed manually by divers. The core liners were 50 cm long Perspex tubes with 9.5 cm internal diameter. Three cores were retrieved during each sampling. These cores were used for high resolution dissolved oxygen concentration measurements (Core 1), total sediment composition analysis (Core 2), pore-water extraction and chemical analyses (Core 3).
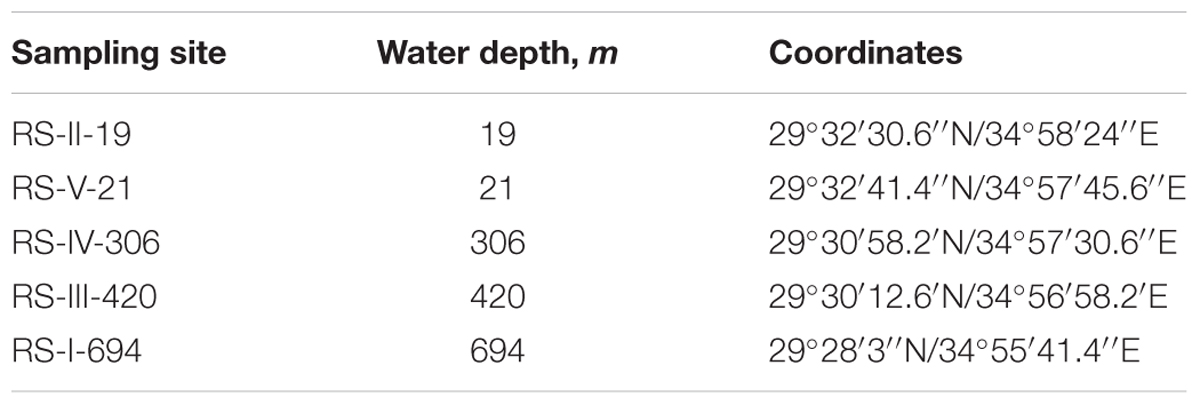
TABLE 1. Water depths and coordinates of sampling sites.
Pore-Water Extraction
The pore-waters were extracted at 4 cm intervals by Rhizon samplers (MacroRhizon, 9 cm long, 4.5 mm diameter, Rhizosphere Research Products, Netherlands) with a filter pore size of 150 ± 30 nm, under a nitrogen atmosphere in a glove box (Seeberg-Elverfeldt et al., 2005).
The first 1 mL of every sample was discarded to avoid sample contamination. The volume of pore-water extracted at each core depth was c.a. 50–80 ml. The extraction of large pore-water volumes was crucial in order to perform numerous geochemical analyses for a variety of dissolved species. The vertical overlap between pore fluid samples retrieved with the Rhizons was calculated according to Seeberg-Elverfeldt et al. (2005) and was found to be in the range of 0–14%.
Pore-Water Analysis
Ferrous (Fe2+) iron was quantified spectrophotometrically by the ferrozine method according to Stookey (1970). The minimum detection limit (MDL) of this method is 1 μmol L-1. Manganese in pore-waters was also measured spectrophotometerically (LaMotte, SMART Spectro) using the 1-(2-Pyridylazo)-2-naphthol (PAN) method (LaMotte test kit 3660-sc) according to Goto et al. (1977). Ascorbic acid was used to reduce total manganese to dissolved Mn2+.
Hydrogen sulfide was measured by two methods. For samples with hydrogen sulfide concentration >1.5 μmol L-1 samples were pre-treated with 50 g L-1 zinc acetate at a ratio of at least 3:20 (V/V) immediately after sampling and the concentration was quantified by the methylene blue method according to Cline (1969). This method accounts for the following species: H2S, HS-, S2-, S(II) in polysulfides, as well as nanoparticles of acid-soluble metal sulfides (e.g., MnS, FeS), which can pass through the pores of the Rhizon sampler. The MDL was 1.5 μmol L-1. For samples with hydrogen sulfide concentration <1.5 μmol L-1, quantification was performed by high-precision liquid chromatography (HPLC, Agilent Technologies 1260 Infinity) with fluorescence detection after derivatization with monobromobimane (Kosower et al., 1979; Fahey and Newton, 1987; Zopfi et al., 2004). The sulfur species measured by this method are: H2S, HS-, S2- and, possibly, the S(II) of polysulfides. The MDL for the sum of these species was 2 nmol L-1. Dissolved zero-valent sulfur (ZVS, includes dissolved and colloidal elemental sulfur and well as polysulfide zero-valent sulfur) was quanified by cyanolysis according to Kamyshny (2009a). The MDL was 60 nmol L-1. The concentrations of polysulfides were below the detection limit of chromatographic quantification after derivatization to dimethylpolysulfanes with methyl trifluoromethanesulfonate (MDL = hundreds of nmol L-1 for individual polysulfides, Kamyshny et al. (2006)) in all samples. The analyses of thiosulfate and sulfite were carried out simultaneously by the same HPLC analysis procedure as for low hydrogen sulfide concentrations. The MDL was 2 nmol L-1. Free thiocyanate (SCN-) and tetrathionate (S4O62-) concentrations were below the detection limit (60 nmol L-1 and 500 nmol L-1, respectively) of chromatographic analyses (Rong et al., 2005; Kamyshny, 2009a). Sulfate (SO42-) was quantified by ion chromatography (DIONEX, DX500) with ASRS-300 suppressor. The eluent, which was composed of 1.8 mmol L-1 sodium carbonate and 1.7 mmol L-1 sodium bicarbonate, was pumped at a flow rate of 2 mL min-1 through a guard column (AG4A-SC), an anion exchange column (AS4A-SC). The MDL of this method was 10 μmol L-1.
Solid Phase Analysis
Sediments were sliced in the field at 2 cm intervals using a core extruder and a spatula. For acid-volatile sulfur (hereafter AVS), non-S0 chromium reducible sulfur (CRS) and elemental sulfur measurements, c.a. 15 mL of sediment were placed in a 50 mL volume falcon tube containing 25 mL zinc acetate in the field (50 g L-1). Additionally, for metal speciation measurements, c.a. 25 mL of sediments were placed in a 50 mL falcon tube and frozen the same day on arrival to the laboratory. For porosity measurements, 10 mL of sediment were placed in a 15 mL volume falcon tube and stored at 4°C in the laboratory.
Elemental sulfur was extracted from the sediment that had been pre-treated with zinc acetate by shaking with methanol for 16 h on a rotary shaker (Zopfi et al., 2004). The sample-to-methanol ratio was c.a. 1/20 [w/v]. After separation of the sediment by centrifugation, elemental sulfur (S0) in the methanol extract was quantified by HPLC. An Agilent Quaternary pump – G1311B, UV-VIS Detector (Agilent G1365D), and Prevail C18 Grace reverse phase column (250 mm × 4.6 mm × 5 μm) were used. Pure methanol (HPLC grade) was used as a mobile phase at a flow rate of 1 mL per minute. Elemental sulfur was detected at 230 nm wavelength with an MDL of 10 μmol kg-1 (wet sediment). In two samples of surface sediments at site RS-V-21, the elemental sulfur content was high enough to allow analysis of isotopic composition. Elemental sulfur was preconcentrated by partial evaporation of methanol on a rotary evaporator, extraction with dichloromethane from the residual methanol-water mixture and evaporation of dichloromethane under gentle flow of nitrogen. Elemental sulfur was reduced to hydrogen sulfide by Cr(II) reduction in the ethanol-water medium according to Gröger et al. (2009). Evolving hydrogen sulfide was trapped in AgNO3/HNO3 trapping solution.
Acid-volatile sulfur and CRS distillations were performed on the sediment sample after it had been subjected to methanol extraction, so that elemental sulfur was not accounted for in the CRS distillation. Although greigite (Fe3S4) is an important precursor for pyrite, its content in sediments is usually low (Morse and Cornwell, 1987). Thus, the main sulfur mineral recovered by CRS was pyrite sulfur, which will be referred to as pyrite-S (or Py-S) hereafter. The sediments were subjected to a two-step distillation. AVS was extracted over two hours by boiling in 5 M HCl, which was followed by 3 h boiling with 1 mol L-1 acidic CrCl2 solution (extracting CRS) (Fossing and Jørgensen, 1989). The MDL was 10 μmol kg-1 wet sediment or 50 nmol total pyrite-S.
Total iron and manganese concentrations were measured according to Aller et al. (1986). For quantification of iron and manganese SpectrAA 300/400 Series Varian AAS was used. Results are presented in moles of metal per kilogram of wet sediment after correction based on sediment porosity. The MDL was 2 μmol kg-1 (wet sediment) for iron and 0.6 μmol kg-1 (wet sediment) for manganese. For quantification of reactive iron species (FeHR) a sequential extraction scheme was used according to Poulton and Canfield (2005). Quantification was performed by AAS, and the MDL was 2 μmol kg-1 Fe (wet sediment). Pyritic iron was measured on a subsample by the CRS protocol.
For porosity analysis, the weight and volume of wet sediment and of sediment dried at 50°C for one week were measured. The porosity was calculated as the fraction of pore-water volume in the wet sediment. Total organic carbon (TOC) was quantified by an elemental analyzer (LECO SC632). The MDL was 0.005% TOC with precision of 1%.
Isotopic Analysis
For preparation for sulfur isotopic analysis of AVS and pyrite-S, zinc sulfide that was formed during hydrogen sulfide distillation was converted to silver sulfide by addition of an excess of silver nitrate solution. Each Ag2S sample was aged for at least one week and cleaned with sequential washes of 4 × 50 mL Milli-Q water, rinsed in 50 mL of 1 M NH4OH overnight, then washed with 3 × 50 mL Milli-Q water and dried at 55°C. For the isotopic analysis of sulfate, sediments were centrifuged, the supernatant was filtered, and an excess of barium chloride solution was added to the filtrate in order to precipitate BaSO4. The precipitate was washed with 3 × 50 mL Milli-Q water and dried at 55°C.
For the analysis of δ18OSO4, barium sulfate was pyrolyzed at 1450°C in a temperature conversion element analyzer (TC/EA), producing carbon monoxide. Carbon monoxide was measured by continuous helium flow in a GS-IRMS (Thermo Finnegan Delta V Plus, at the Godwin Laboratory, University of Cambridge). Analyses of δ18OSO4 were performed in replicates (n = 3–5) and the standard deviation of the replicate analysis is reported (∼0.4‰ 1σ). To analyze the δ34SSO4, δ34SPy-S, δ34SAV S, δ34SS0 values, barite or silver sulfide with an excess of vanadium pentoxide were combusted at 1030°C in a flash element analyzer (EA), and the resulting sulfur dioxide (SO2) was measured by continuous helium flow on a GS-IRMS (Thermo Finnegan Delta V Plus Godwin Laboratory, University of Cambridge). The analytical error for the δ34S analysis was determined using the standard deviation of standards run at the beginning and the end of each run (long term reproducibility of our standards is ∼0.3‰ 1σ). δ18OSO4 is reported relative to the Vienna Standard Mean Ocean Water (VSMOW) and δ34S is reported with respect to Vienna Canyon Diablo Troilite (VCDT).
Results
Sediment Description
In the vicinity of the north shore of the Gulf of Aqaba (sites RS-II-19 and RS-V-21), sediments contain grayish sandy particles, suggesting the source of the sediment is terrigenous material derived from adjacent rocks by erosion and/or weathering and transported by seasonal floods. The sediments from near the fish farm contained black fragments which are likely residual material from fish cages, consistent with the composition of sediment cores collected during the operation period of Ardag fish farm (Katz et al., 2002). The bulk composition of the north shore sediments is dominated by siliciclastic material with lower carbonate content (6.17%; Al-Rousan et al., 2006) compared to coral reef sediments (74.8%; Al-Rousan, 1998). The sediment from the intermediate and deep-water sites (RS-IV-306, RS-III-420, and RS-I-694) is characterized by a much finer grain size with a higher content of light brown clay and mud. These textural properties at the deeper-water sites reflect the aeolian source of particles derived from desert soils (Garrison et al., 2003).
Pore-Water Chemistry
Dissolved iron was present in the sediments of the deep-water sites (RS-IV-306, RS-III-420, and RS-I-694) at all depths below seafloor, while at the shallow water site that were not influenced by the fish farms (RS-V-21) no dissolved iron was detected below 3 cm below seafloor (Figure 2A). Dissolved manganese concentrations increase with overlying water column depth. Concentrations above 10 μmol L-1 were detectd only at the deepest site RS-I-694 (Figure 2B).
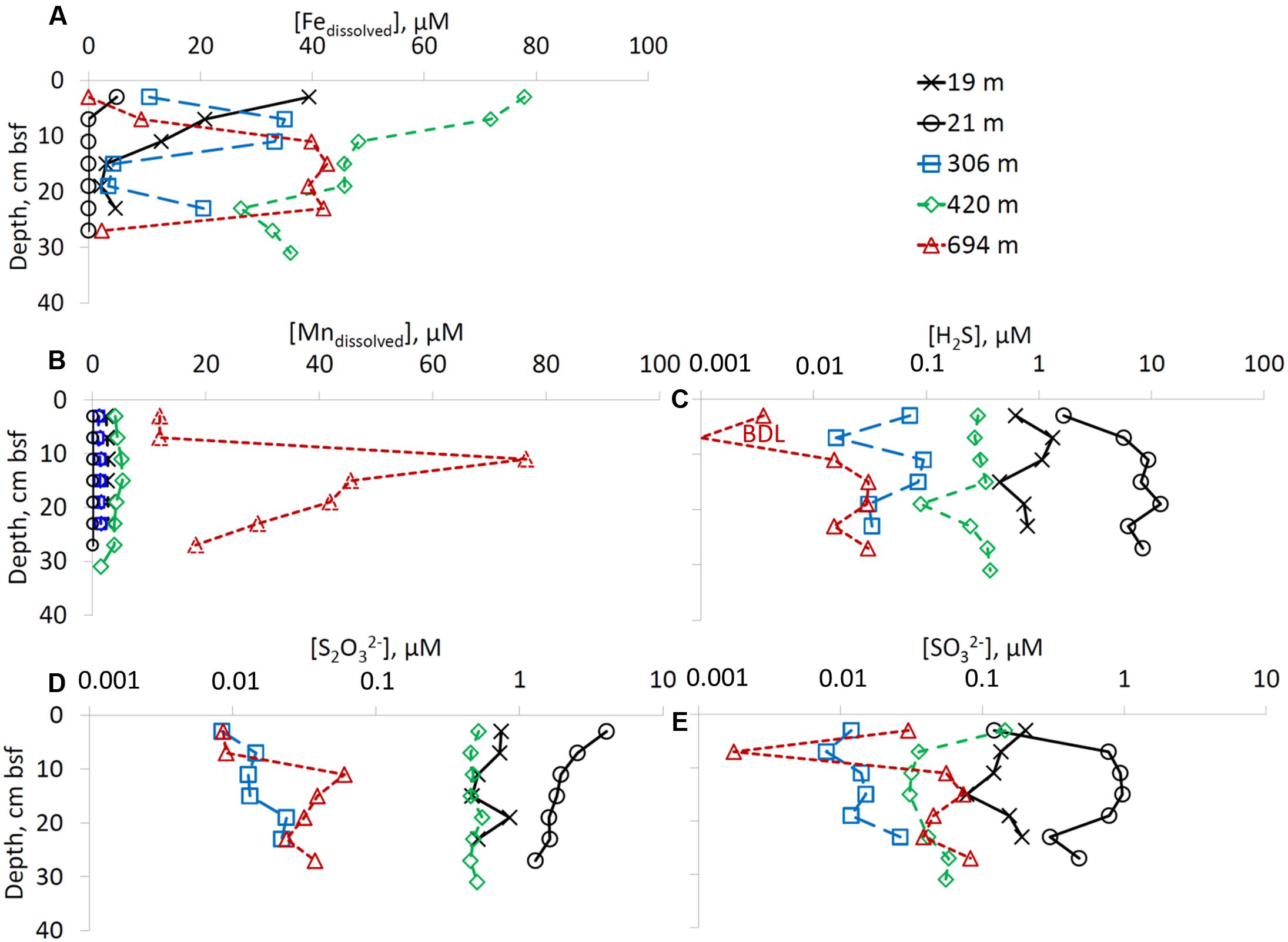
FIGURE 2. Pore-water chemistry. Profiles of (A) dissolved iron, (B) dissolved manganese, (C) hydrogen sulfide (pay attention to logarithmic x-axis), (D) thiosulfate (pay attention to logarithmic x-axis), and (E) sulfite (pay attention to logarithmic x-axis). Right upper panel presnets a color codes for sampling sites.
Sulfate concentrations were constant with depth at all the sampling sites, in the range of 32–35 mmol L-1 (data not shown). Hydrogen sulfide concentrations were below 0.34 μmol L-1 at the deep-water sites (RS-IV-306, RS-III-420 and RS-I-694), up to 1.3 μmol L-1 at former fish farm site RS-II-19, and up to 12 μmol L-1 at site RS-V-21 (Figure 2C). Zero valent sulfur (ZVS) was below detection limit at sites RS-II-19 and RS-IV-306, below 200 nmol L-1 at sites RS-III-420 and RS-I-694 and up to 301 nmol L-1 at site RS-V-21 (data not shown). The highest thiosulfate concentrations were detected at the shallow water sites (up to 4.05 μmol L-1 at site RS-V-21). At sites RS-IV-306 and RS-I-694, thiosulfate concentrations were <0.1 μmol L-1 (Figure 2D). Sulfite concentrations in the pore-waters were lower than thiosulfate concentrations. At the shallow sites (RS-II-19 and RS-V-21), sulfite concentrations were 78–972 nmol L-1. Sulfite concentrations at the deeper sites were <150 nmol L-1 (Figure 2E).
Solid Phase Chemistry
The porosity of the sediments increased with increasing water depth as well as with depth below the sediment-water interface (Figure 3A). TOC content was in the range of 0.1-0.6 wt% at all sites. The AVS and pyrites sulfur contents of the sediment decreased with an increase in water column depth. At the shallow site not influenced by anthropogenic pollution (RS-V-21), AVS content decreased with depth below the seafloor and pyrite sulfur increased with depth below the seafloor. At site RS-II-19 (the site impacted by the fish farm) the lowest AVS concentration was detected in the upper 10 cm of sediments (Figures 3B,C). In the upper 10 cm of the sediment, elemental sulfur content was higher at the shallow water sites than at the deep-water sites (Figure 3D).
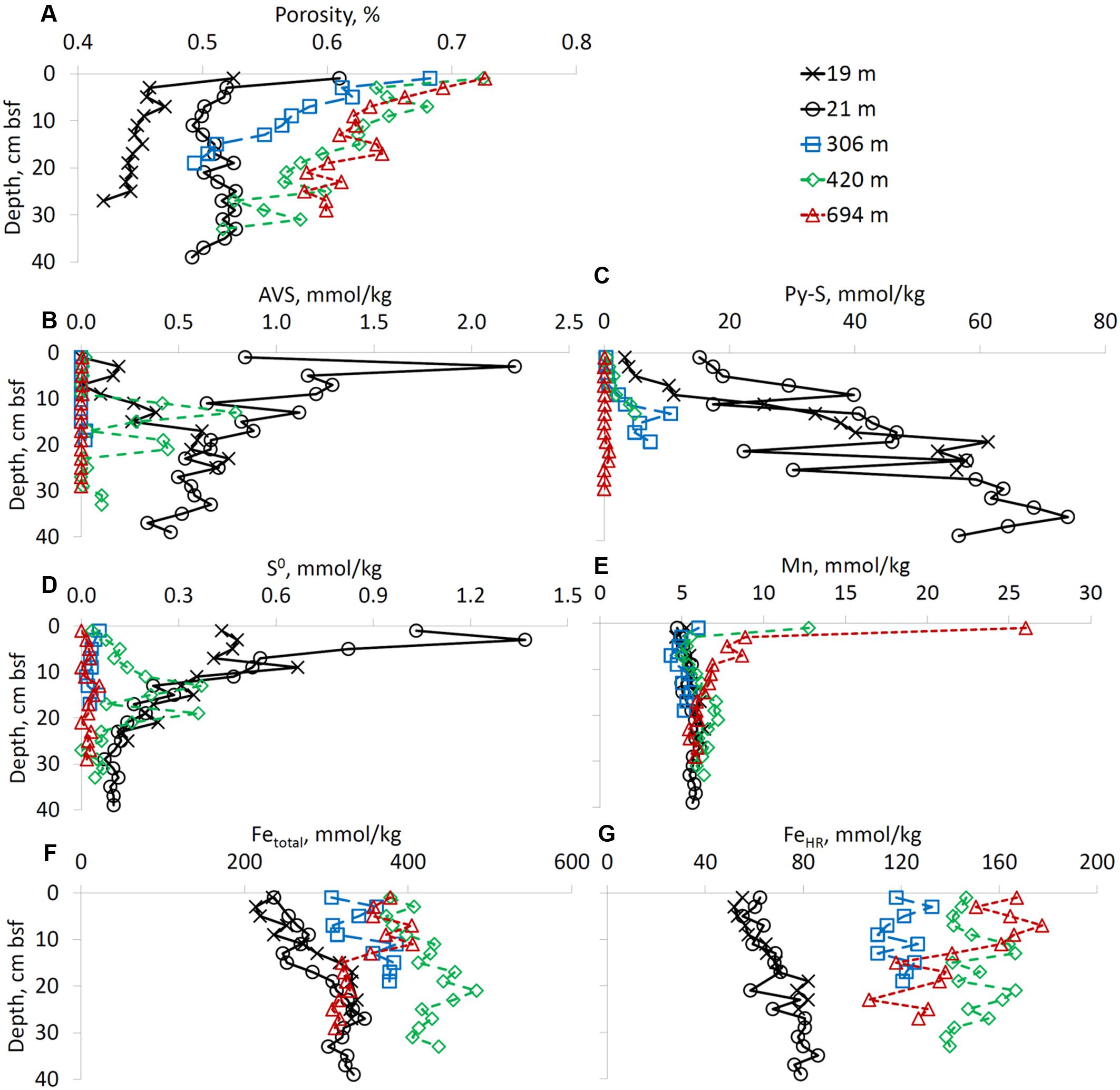
FIGURE 3. Sediment porosity and solid-phase composition (except iron speciation). Profiles of (A) porosity, (B) AVS, (C) pyrite sulfur, (D) elemental sulfur, (E) manganese, (F) total iron, and (G) sum of “highly reactive” iron species. Right upper panel presnets a color codes for sampling sites. All concentrations are given in mmol kg-1 of wet sediment.
Sediments from the deep-water sites were enriched in total manganese in the upper two centimeters of the core. At RS-I-694 site total manganese content reached 26.0 mmol kg-1 of wet sediment. In cores taken from shallow-water sites (RS-II-19 and RS-V-21), the total manganese content was below 7 mmol kg-1 of wet sediment (Figure 3E). Sediments in the deep-water sites were enriched in iron compared to shallow-water sites, especially in the upper 14 cm of the sediment. The highest total iron content was detected at site RS-II-420 (Figure 3F). Traditionally, highly reactive iron is defined as the sum of iron species which are reactive toward hydrogen sulfide and iron which has already reacted with sulfide (e.g., pyrite iron). The content of highly reactive iron, which in our study includes pyrite iron, acetate-, hydroxylamine-, dithionite-, and oxalate-extractable iron, increases with water depth (Figure 3G). As concentrations of pyrite are higher at the shallow-water sites (Figure 3C), concentrations of iron species which are still reactive toward hydrogen sulfide increase further with water depth. At the shallow-water sites (RS-II-19 and RS-V-21) concentrations of highly reactive iron slightly increased with depth (Figure 3G). Sodium acetate-extractable iron, which accounts for Fe(II) carbonate minerals such as siderite and ankerite represented less than 10% of the highly reactive iron at all water depths. Dithionite-extractable iron (goethite + hematite + akaganéite) represented the most abundant fraction of highly reactive iron in the sediments of the Gulf of Aqaba (data not shown).
Isotopic Composition of Sulfur Species
The isotopic composition of the Gulf of Aqaba seawater sulfate was measured in five samples. Three samples were taken at the surface at various locations and two samples were retrieved from 20 and 700 m depths. The isotope composition of sulfate at various locations and water depths within the Gulf of Aqaba is similar (δ34S = 20.3 ± 0.2‰, δ18O = 8.6 ± 0.2‰), and does not differ more than the error of the analytical method. At all sites, except at the former fish farm (site RS-II-19), the δ34S of sulfate near the top of the sediment was similar to the overlying water (20.3–20.5‰). At these sites, the δ34S increased with depth below the seafloor to 21.2–23.1‰. At the former fish farm site, sulfate sulfur was isotopically lower than seawater sulfate, possibly due to re-oxidation of isotopically light reduced sulfur minerals formed during fish farm operation (Figure 4A). At all depths bsf, the δ18O of sulfate was higher than seawater sulfate (9.4–10.4‰ in the upper 2 cm of sediment vs. 8.6% in the water column). For the deep-water sites, a slight increase in δ18O of sulfate was observed with depth (Figure 4B).
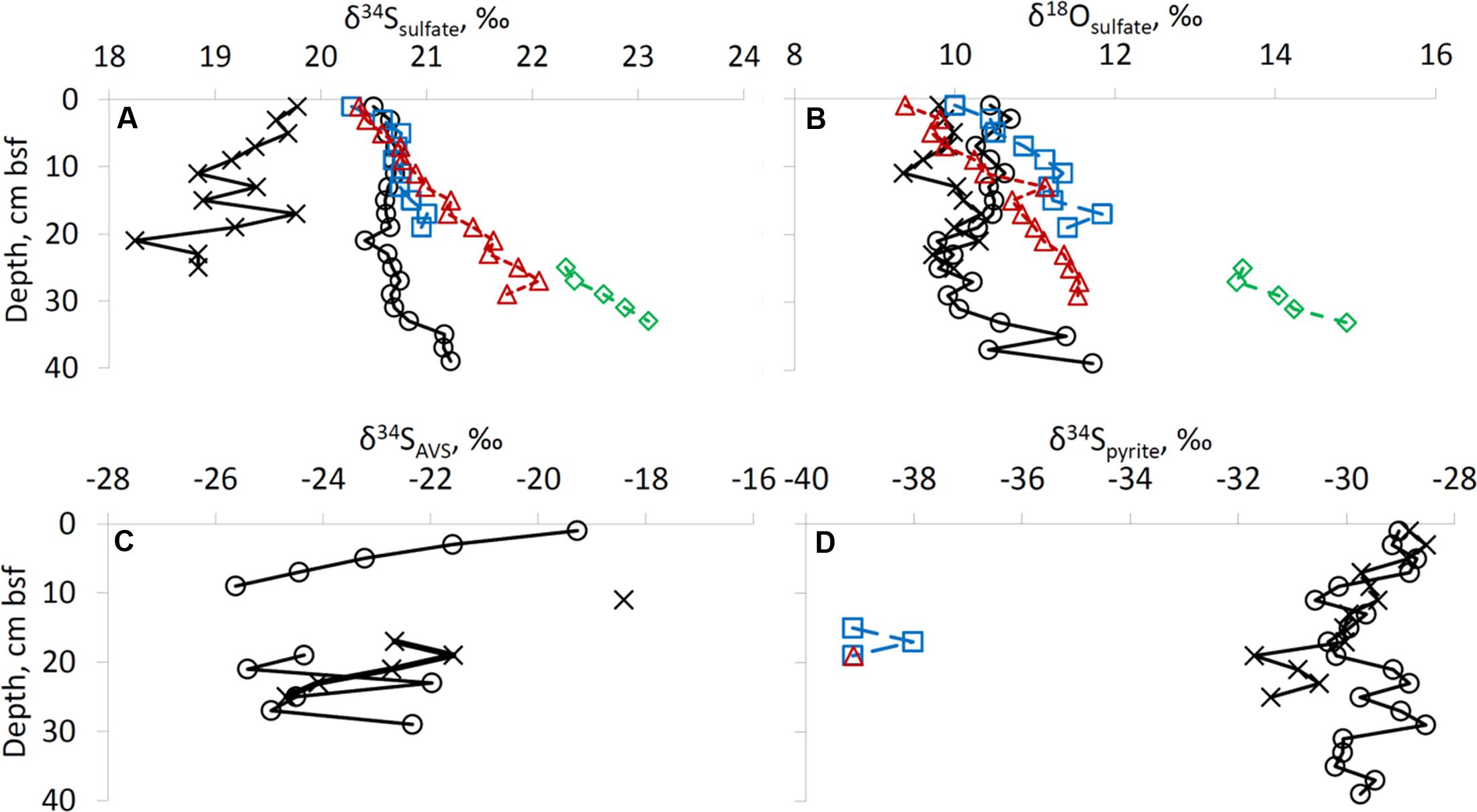
FIGURE 4. Isotopic composition of sulfur species. Profiles of (A) δ34S of sulfate, (B) δ18O of sulfate, (C) δ34S of AVS, (D) δ34S of pyrite. For legend see right upper panel of Figure 2.
The sulfur isotopic composition of AVS is only reported for the shallow-water sites, as at the deep-water sites the AVS content was too low to perform sulfur isotopic analysis. The most 34S-enriched AVS was detected at both shallow-water sites in the uppermost samples: δ34S = –18.4‰ to –19.3‰. At 17–29 cm bsf, the δ34S of AVS was in the range of –21.6‰ to –25.4‰ (Figure 4C). Enough pyrite-S for sufur isotopic analysis was retrieved from all sediments at the two shallow-water sites, three samples from site RS-IV-306 from 14 to 20 cm, and one sample from site RS-I-694 at 18–20 cm (this point cannot be seen on the figure as it coincides with the point for RS-IV-306 profile). Pyrite sulfur was isotopically lower in δ34S than AVS sulfur. At deepwater sites RS-IV-306 and RS-I-694, pyrite was isotopically lower in δ34S than at the shallow-water sites (Figure 4D). The isotopic composition of elemental sulfur was measured in the 0–2 cm and 2–4 cm intervals in the sediments from site RS-V-21and the δ34S was –20.9‰ and –22.4‰, respectively.
Discussion
Manganese and Iron Cycles
The concentration of total manganese in the solid sediment increases with the depth of the overlying water column (Figure 3E). This increase is likely caused by greater contribution of aeolian dust to the sediment further from the shore. The atmospherically transported material from the Sahara and adjacent deserts has a high manganese content, as much as 880 ppm (16 mmol kg-1) in Saharan dust (Mendez et al., 2010). In contrast, the shallow sediment has a dominance of fluvial input with lower manganese content. An enrichment in solid manganese in the upper 2 cm of the sediment was observed in the deep-water sites (Figure 3E). This solid manganese enrichment results from the oxidation of reduced aqueous manganese (Mn2+), which is produced by microbial manganese reduction coupled to organic carbon oxidation. This reduced Mn2+ produced deeper in the sediment diffuses toward the water-sediment interface where it is oxidized to manganese oxides. The only site at which concentrations of dissolved manganese were higher than concentrations of dissolved iron was deepwater site RS-I-694 suggesting a dominance of the manganese cycle at this location.
The concentration of total sedimentary solid iron, similar to total manganese, increases as the depth of the overlying water column increases, except for the deepest site (RS-I-694) (Figure 3F). As with manganese, this increase may be explained by greater contribution of aeolian dust away from the shore. The fine-grain sediment that is transported from the Sahara and adjacent deserts by wind has a high iron content (Chase et al., 2006). The lack of a significant increase of solid total iron in the surface sediment layer is likely due to low iron sulfide solubility, which causes retention of iron in the sediment even at low concentrations of hydrogen sulfide in the pore-waters (Figures 2A,C).
Highly reactive iron is traditionally defined as the fraction of iron in the sediment that can react with hydrogen sulfide to produce FeS or FeS2 (Berner, 1970). There are different pools of iron that are defined based on their reactivity toward hydrogen sulfide. Ferrihydrite and lepidocrocite react with hydrogen sulfide within hours. The most abundant iron minerals in dust from the Saharan Desert, such as hematite and goethite, react with hydrogen sulfide on time scales of days (Poulton et al., 2004) and years (magnetite) (Canfield et al., 1992). For iron in sheet-silicate minerals it may take up to millions of years to react with hydrogen sulfide (Canfield et al., 1992; Raiswell and Canfield, 1996). Highly reactive iron concentrations are lower at the shallow water sites (RS-II-19 and RS-V-21) than at the sites overlain by deeper waters. We suggest that the difference in the reactive iron content could be due to the difference in the highly reactive iron content of dust (relatively high) and desert soil and rock material transported by flash floods (relatively low), which predominantly impact shallow-water sediments. Although detailed studies of reactive iron speciation of minerals and soils in the various locations in arid Arava valley are not yet published, total iron content in granite was recently reported for two locations near Eilat: Wadi Shelomo and Mt. Rehavam with 0.30–0.76% Fe2O3 and 0.12–1.60% Fe2O3, respectively (Eyal et al., 2010). The iron in these locations is mostly associated with biotite (Bogoch et al., 1997). The detailed study of iron speciation in the seasonal stream beds, which is out of scope of this study, should be performed in order to evaluate this hypothesis.
Sulfur Cycle
The concentration of hydrogen sulfide in the pore-water decreased with an increase in water column depth from 0.5 to 12 μmol L-1 at the shallow-water sites to ≤30 nmol L-1 at site RS-I-694. The latter concentration was detected by HPLC with a fluorescence detector and is a concentration we usually consider to be below the detection limit of standard spectrophotometric techniques (1 μmol L-1 – Cline, 1969). Despite extremely low concentrations of hydrogen sulfide in the deeper sediments, we use three lines of evidence to confirm the presence of a sedimentary sulfur cycle in the Gulf of Aqaba.
Pyrite Content
The first line of evidence for microbial sulfate reduction is the presence of pyrite. Pyrite is formed by two mechanisms, both involving hydrogen sulfide: the “polysulfide” mechanism (Eqs. 1–3) (Luther, 1991; Kamyshny et al., 2004) and the “sulfide” mechanism (Eqs. 1, 4) (Rickard and Luther, 1997; Gartman and Luther, 2013).
In our case, circumneutral pH and the presence of elemental sulfur in the sediment (Figure 3D), suggest that the “polysulfide” mechanism is the most feasible pathway of pyrite formation. Although concentrations of polysulfides in the pore-waters were below the detection limit of our analytical technique (Kamyshny et al., 2006), it is likely that their concentration was low due to their fast reaction during pyrite precipitation. Oxidation of polysulfides by oxygen is known to be faster than oxidation of hydrogen sulfide by oxygen (Kleinjan et al., 2005). Thus, concentrations of polysulfides in various natural aquatic systems are better explained by kinetics of their reaction rather than by thermodynamic considerations (Kamyshny and Ferdelman, 2010; Lichtschlag et al., 2013).
Sedimentary pyrite content decreases with increasing water column depth and with depth below the sediment-water interface (Figure 3C). Depth-based net pyrite formation rates were calculated from the linear approximation of the increase of pyrite concentrations with depth, and converted to time-based rates using the typical sedimentation rate for deep sites (0.054 cm year-1 – Al-Rousan et al., 2004). Net pyrite formation rates were estimated to be 0.68, 0.53, 0.11, 0.073, and 0.0063 nmol cm-3 wet sediment day-1 at sites RS-II-19, RS-V-21, RS-IV-306, RS-III-420, and RS-I-694, respectively. These rates of pyrite formation are lower than rates of microbial sulfate reduction in most marine sediments. One explanation of this observation is re-oxidation of a significant portion of hydrogen sulfide formed during microbial sulfate reduction. Reoxidation of hydrogen sulfide to sulfide oxidation intermeditates, which in turn may be microbially disproportionated to sulfate and hydrogen sulfide allows only trace amounts of pyrite to form. On the other hand, the higher highly reactive iron content at the deeper sites (Figure 3G) may give preference to iron reducing microorganisms, thus suppressing microbial sulfate reduction.
Sulfide Oxidation Intermediates
The second line of evidence for an active sulfur redox cycle in the sediments of the Gulf of Aqaba is the presence of intermediate-valence state sulfur species. Hydrogen sulfide is a precursor to sulfide oxidation intermediates such as zero-valent sulfur, thiosulfate and sulfite in sedimentary pore-waters (Figures 2D,E) as well as of zero-valent sulfur in the solid phase (Figure 3D). Thus, the presence of intermediate sulfur species in the sedimentary pore fluids may be interpreted as an argument for chemical or microbial oxidation of hydrogen sulfide.
Zopfi et al. (2004) suggested that concentrations of sulfide oxidation intermediates in marine sediments are controlled by their availability for microbial metabolism. Usually, the concentration of these intermediates decreases in the order [S0] > [S2O32-] > [SO32-] > [S4O62-], as we observe in the sediments of the Gulf of Aqaba (Figures 2D,E, 3D). As expected, the concentration of elemental sulfur in the sediments decreases with increasing water column depth, e.g., with the decrease in the rates of formation of its precursor, hydrogen sulfide. In the shallow-water sites, the concentration of elemental sulfur decreases with sediment depth as well, due to the formation of pyrite which will consume the available hydrogen sulfide. At the shallow-water sites, the concentration of elemental sulfur reaches 1 mmol kg-1 of wet sediment. Only colloidal sulfur with particles <150 nm, polysulfide sulfur, and dissolved elemental sulfur can pass the membrane of the Rhizon samplers. Sulfur is predominantly in the solid sediment, as zero-valent sulfur concentrations in pore-waters are <300 nmol L-1 at all sites and sediment depths. As the solubility of elemental sulfur in seawater at 25°C is 147 nmol L-1 (Kamyshny, 2009b), at some sites the presense of polysulfides is required to explain the observed results in the porewater, as colloidal sulfur equilibrates with hydrogen sulfide according to Eq. (2) in minutes to hours (Fossing and Jørgensen, 1990; Kamyshny and Ferdelman, 2010).
The concentration of thiosulfate in the pore-waters decreases with increasing water depth as is expected due to the corresponding decrease of hydrogen sulfide concentrations. A similar trend was observed for concentrations of sulfite. The concentrations of thiosulfate and sulfite are typical for marine sediments, although, it must be mentioned that in marine sediments, hydrogen sulfide concentrations are often much higher than our results for the Gulf of Aqaba (Thamdrup et al., 1994; Zopfi et al., 2004, 2008; Lichtschlag et al., 2013).
The co-dependences of the concentration of thiosulfate and sulfite on the concentration of their precursor - hydrogen sulfide - produces a rather unexpected relationship (Figure 5). The increase in the concentration of thiosulfate and sulfite in pore-waters is often more moderate than the increase in hydrogen sulfide concentrations: a 10-fold increase in hydrogen sulfide leads to only c.a. fivefold increase in concentration of intermediate sulfur oxyanions. The simultaneous presence of hydrogen sulfide and intermediate oxyanions in marine sediments is possible only if two conditions are fullfilled: (1) low or zero dissolved oxygen levels are present in pore-waters; (2) if oxygen is absent, other electron acceptors, capable of chemical or microbial oxidation of hydrogen sulfide (e.g., nitrate, MnO2, Fe(III) (hydr)oxides)), are present in the sediment. As nitrate concentrations in seawater of the Northern Gulf of Aqaba are low (<6 μmol L-1) (Al-Qutob et al., 2002) and it is depleted quickly in the pore-waters, manganese and iron oxides are the main electron acceptors responsible for hydrogen sulfide oxidation in these locations. Chemical oxidation of hydrogen sulfide by Mn(IV) leads to formation of zero-valence sulfur, thiosulfate, sulfite, and sulfate (Burdige and Nealson, 1986), and chemical oxidation of hydrogen sulfide by Fe(III) forms mostly zero-valence sulfur, although the formation of other sulfide oxidation intermediates has been documented (Yao and Millero, 1996; Wan et al., 2014). Therefore, the distribution of sulfide oxidation intermediates depends on the ratio of hydrogen sulfide-to-electron acceptor, where at lower ratios more oxidized products are formed. These reactions are first order with respect to both hydrogen sulfide and the concentration of electron acceptor. Microbial disproportionation and oxidation of intermediate sulfur species may lead to the formation of other sulfide oxidation intermediates as well as of the terminal oxidation product, sulfate (Bak and Cypionka, 1987; Jørgensen, 1990a,b; Zopfi et al., 2004, and references therein).
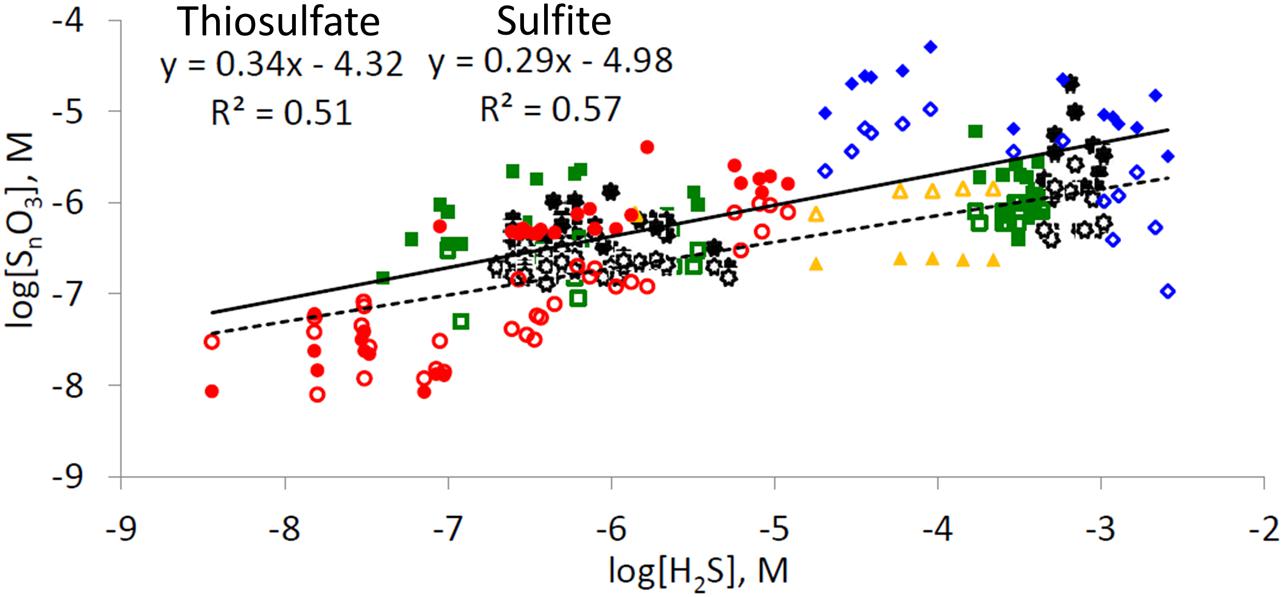
FIGURE 5. Dependence of concentrations of thiosulfate (closed symbols) and sulfite (opened symbols) on concentrations of hydrogen sulfide in marine sediments (not including salt marshes and tidal flats). Red circles – this work from the Gulf of Aqaba, orange triangles – results from Thamdrup et al. (1994) from Aarhus Bay sediments, green rectangles – results from Zopfi et al. (2004) from the Black Sea, black stars – results from Zopfi et al. (2008) from the upwelling area off central Chile coast, blue diamonds – results from Lichtschlag et al. (2013) from Dvurechenskii mud volcano, Black Sea. Solid line represents a trendline for all thiosulfate vs. hydrogen sulide data points, dashed line represents a trendline for all sulfite vs. hydrogen sulfide datapoints. Only data which were obtained by chromatographic analysis after 2,2′-dithiobis(5-nitropyridine) derivatization (Valravamurthy and Mopper, 1990) and monobromobimane derivatization (see Materials and Methods section and references therein) for points where both hydrogen sulfide and sulfur oxoanion concentrations were above the detection limit are presented.
We suggest the following explanation for the observed trends. The decrease in hydrogen sulfide concentration in pore-waters leads to a decrease in the ratio between hydrogen sulfide and the concentrations of reactive iron and manganese oxides in the sediment. This, in turn, leads to formation of more oxidized sulfide oxidation intermediates (e.g., more sulfite and thiosulfate and less zero-valent sulfur species). On the other hand, a decrease in the concentration of intermediate sulfur oxyanions, leads to a decrease in the rates of their chemical and microbial consumption. Thus, an increase in the fraction of hydrogen sulfide that is converted to sulfur oxyanions, combined with a decrease in their consumption rate, results in only a moderate decrease in their concentrations with a corresponding decrease in hydrogen sulfide concentrations. In the extreme case of the sediments of the Gulf of Aqaba, concentration ratios of up to 4 and 8 between thiosulfate and sulfite, respectively, to hydrogen sulfide were found at site RS-I-694.
Isotopic Composition of Sulfur Species
The third line of evidence for microbial sulfur cycling in the sediments is the isotopic composition of sulfur species. In this study, the sulfur isotopic fractionation between sulfate and AVS (which represents the sulfur isotopic composition of the most recently formed hydrogen sulfide) is 37–46‰ (Figure 6A). Usually, in marine sediments both AVS and pyrite become isotopically more enriched in 34S with depth due to Rayleigh distillation of sulfur isotopes during microbial sulfate reduction and the diffusion of sulfate within pore-fluids. In our case, Rayleigh distillation cannot be invoked to explain the observed trends as (1) sulfate concentrations do not decrease significantly with depth, (2) AVS becomes lower in δ34S with sediment depth, and (3) pyrite sulfur becomes isotopically lower in δ34S with sediment depth and near the sediment surface it is isotopically lower in δ34S than AVS by approximately 10‰. An increase in sulfur isotope fractionation with depth may be explained by a decrease in microbial sulfate reduction rates (Chambers et al., 1975; Canfield, 2001; Sim et al., 2011b), which results from preferential mineralization of more bioavailable organic matter in the uppermost sediments. The difference of approximately 10‰ in the sulfate-pyrite sulfur isotope fractionation between shallow-water and deep-water sites (Figure 6B) supports an increase in sulfur isotope fractionation with a decrease in the rate of formation of hydrogen sulfide. A possible explanation for the relatively constant δ34S of pyrite in the surface sediments is the impact of bioturbation (Figure 3C). Rates of bioturbation in the shallow-water site near the fish farm (derived from chlorophyll profiles) were previously shown to be in the range of 0.013–0.069 cm2 d-1 (Black et al., 2012). The sulfur isotopic composition and concentrations in the upper 6 cm of sediments at shallow-water sites are thus possibly homogenized by bioturbation (Figures 3C, 4D) which exponentially decreases with depth (Steiner et al., 2016).
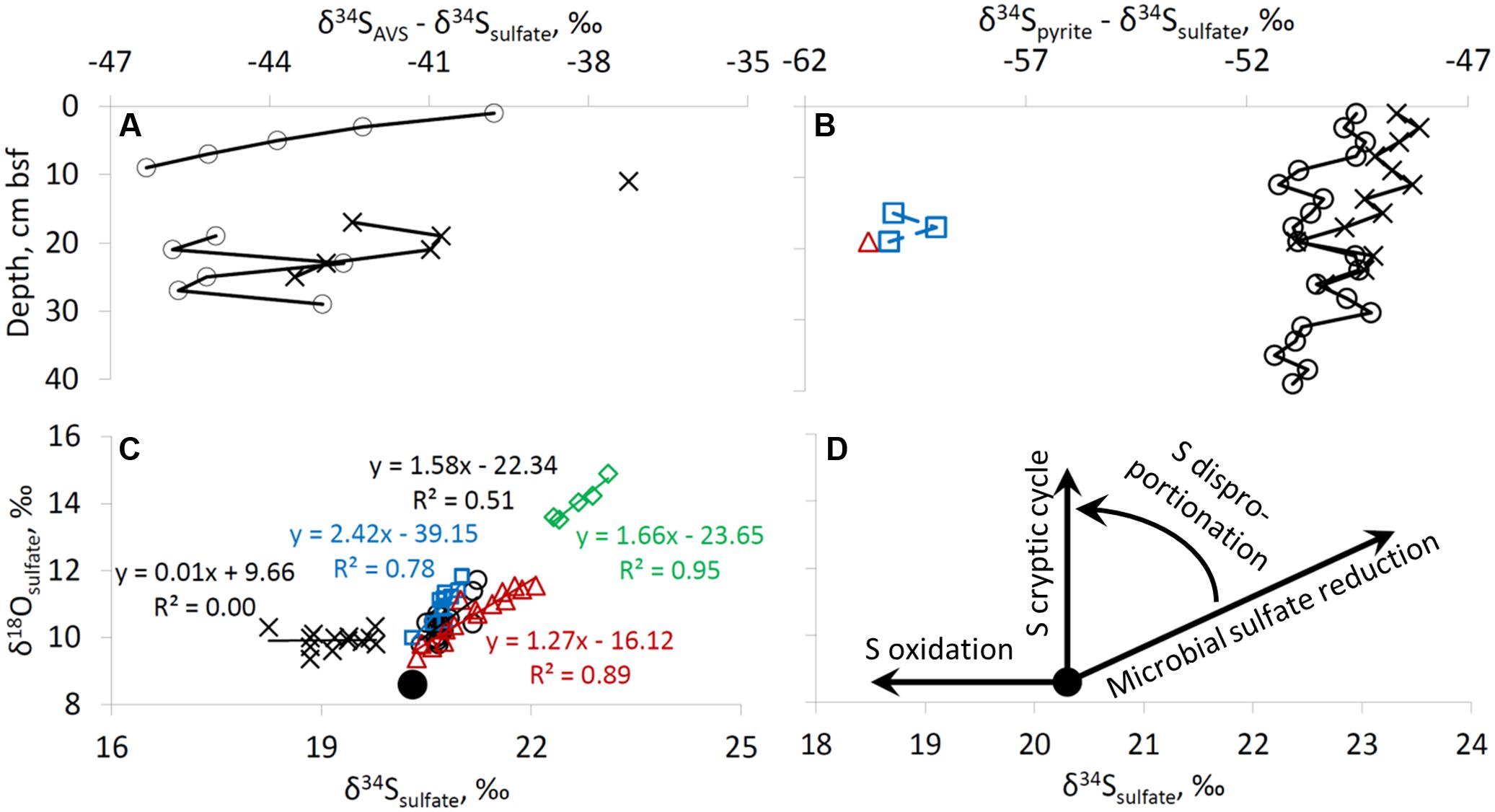
FIGURE 6. Difference between δ34S values of AVS and sulfate (A), and of pyrite and sulfate (B). Plot of δ18O of sulfate vs. δ34S of sulfate is presented in plate (C). Schematic pathways of dependence of δ34S and δ18O values of sulfate on microbial sulfur transformation process is presented in the panel (D). Large black circle in panels (C,D) represents isotopic composition of sulfate in the water column of the Red Sea. For legend see right upper panel of Figure 2.
In two analyzed samples (0–2 cm and 2–4 cm bsf at site RS-V-21) elemental sulfur was lower in δ34S by 0.8–1.6‰ than AVS. These results suggest that equilibrium in the H2S-polysulfide-S0 system is not achieved, as at the equilibrium zero-valent sulfur is higher in δ34S than hydrogen sulfide by up to 4‰ (Amrani et al., 2006; Kamyshny et al., 2014).
The relative change in the δ34S and δ18O of sulfate during microbial sulfate reduction has been shown to yield insight into the sulfur cycle in marine sediments and in laboratory cultures. At all the sites, except for RS-V-21, there is a coupled increase in δ18O and δ34S in sulfate as expected during microbial sulfate reduction. The relative change in δ18O vs. δ34S has been shown to correlate with the rate of microbial sulfate reduction (Böttcher et al., 1999; Aharon and Fu, 2003; Antler et al., 2013): the slower the sulfate reduction rate, the steeper the slope. The slopes in our sampling sites range between 1.2 ± 0.1 (at 694 m) to 2.4 ± 0.5 (at 306 m). It seems, however, that there is a discrepancy in the correlation between the slope and the sulfate reduction rates, as the most moderate slope is in the site where sulfate reduction rate is expected to be the lowest (694 m). We suggest that the disproportionation of intermediates of hydrogen sulfide oxidation increases the measured slope between δ18O and δ34S (Böttcher et al., 2005) (Figures 6C,D). At the former fish farm contaminated site (RS-II-19), a decrease in δ34S was observed together with a scattered change in the δ18O of sulfate (±0.6 ‰). This observation may be explained by more intense oxidation of reduced sulfur species at this site (Figures 6C,D). Combined with other evidence, the oxygen isotope composition of sulfate provides strong evidence for a presence of sulfur cycling in the sediments of the Gulf of Aqaba, including a near quantitative cycling of sulfur between oxidized and reduced or intermediate valence states with low net-consumption, which has been termed a ‘cryptic’ sulfur cycle.
Impact of Aeolian Deposition on the Sulfur Cycle in the Sediments of the Gulf of Aqaba
An integrated scheme of processes affecting cycling of redox-sensitive elements in the Gulf of Aqaba is presented in Figure 7. Sediments overlain by deep water receive predominantly input of aeolian dust, which has high manganese and reactive iron content. At these water depths, microbial iron and manganese reduction is preferred over sulfate reduction. In spite of a microbial cycle dominated by iron and manganese cycling, there is microbial sulfate reduction and trace amounts of pyrite accumulating. However, the vast majority of the hydrogen sulfide produced is reoxidized in the presence of the high concentration of iron and manganese oxides to sulfide oxidation intermediates, which are further oxidized or disproportionated to form the terminal oxidation product, sulfate. These processes induce the high concentration of sulfide oxidation intermediates in pore waters and leave fingerprints in oxygen isotope composition of the sulfate. Thus, at these deep water sites the sulfur cycle is mostly cryptic.
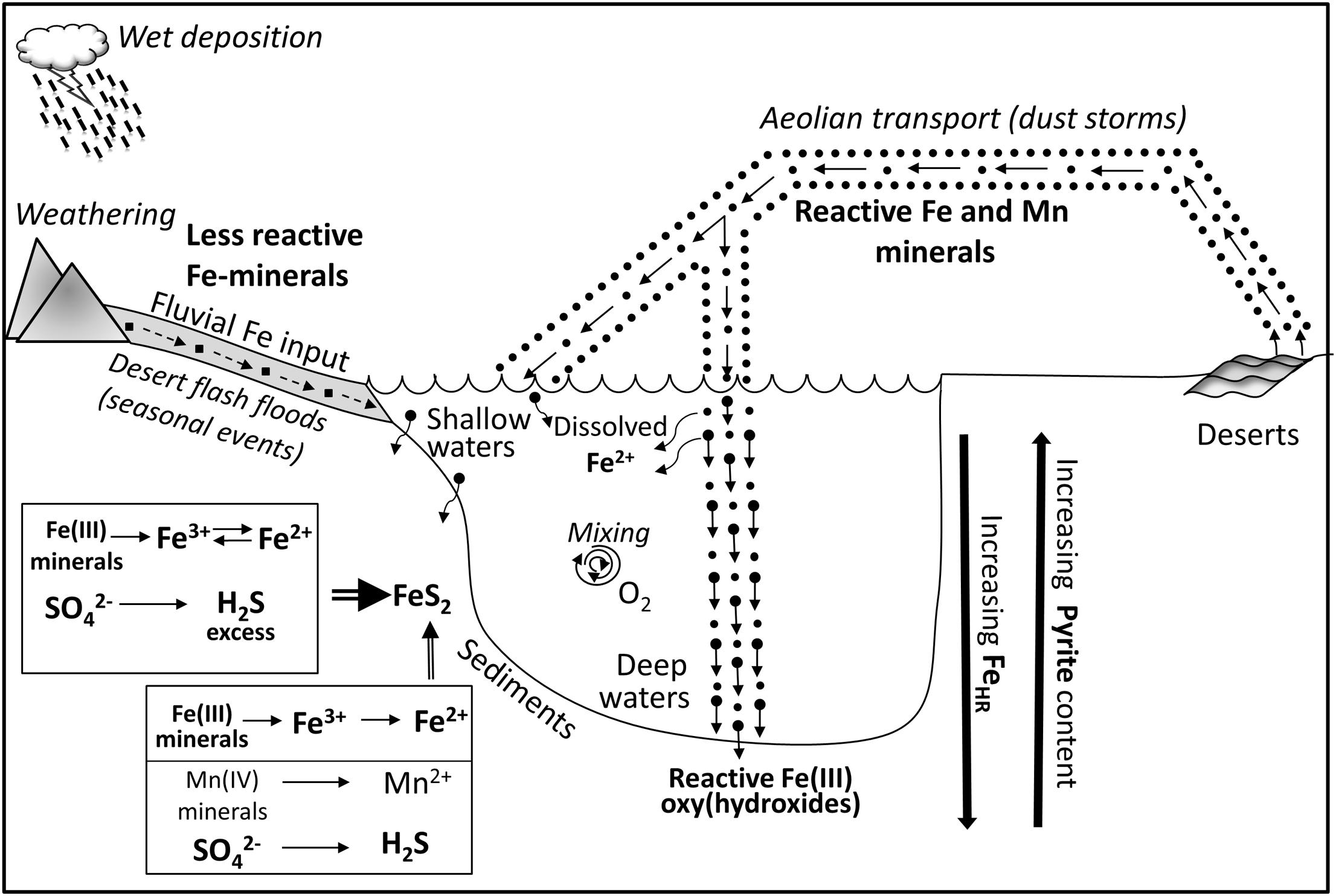
FIGURE 7. Schematic illustration of impacts of aeolian and fluvial sedimentary inputs on biogeochemistry redox-sensitive elements in the sediments of the Gulf of Aqaba. Aeolian input of highly reactive iron minerals is presented as a sum of fluxes of mineral dust which is derived from adjacent areas (rock weathering, secondary airflow) and dust which is transported from deserts by long distance aeolian transport (dust events, dust storms). Fluvial iron input is presented as sediment transport from adjacent areas (mainly Arava desert) by seasonal flashfloods which take place during winter rainfall events. The majority of wind-blown iron particles rapidly settle throughout the water column to the sediment, while minor fraction of reactive iron (III) oxyhydroxides dissolves in the water column. See text for detailed explanation of biogeochemical processes in the sediments.
Shallow water sites are influenced by a combination of aeolian dust input and fluvial iron and manganese inputs. At these sites, the manganese (especially close to the sediment-water interface) and reactive iron contents are lower than at the deep-water sites. Lower reactive iron content allows relatively high amounts of hyrdogen sulfide to be present in the pore-waters and to be further preserved in the form of pyrite.
In summary, this work shows that in arid environments, such as the Gulf of Aqaba, aeolian dry deposition from the surrounding deserts results in an increased flux of reactive iron and manganese to the sediments. Such increase leads to fast reoxidation of hydrogen sulfide and prevents pyrite formation in the sediments. Future work should concentrate on the detailed study of speciation of iron and manganese in the aeolian and fluvial sedimentary sources. Another direction of future research should include the study of impact of aeolian dry deposition on sedimentary sulfur cycle in other marine systems affected by aeolian dust deposition. Such research efforts are required in order to understand whether these considerations are unique to the Gulf of Aqaba or may be applied to other marine systems situated in arid regions.
Author Contributions
BB and AK designed research; BB, VB, AT, GA, NK, RK and AK performed analyses; BB, VB, AT, GA, US, NK, RK and AK interpreted data, wrote the paper and approved the final version to be published.
Funding
This work was funded by Marie Curie Actions CIG Program, project number 303740. The isotope analyses were funded by ERC StG 307582 CARBONSINK to AT.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Itamar Pelly, Jiwchar Ganor, Shimon Feinstein, and Orit Sivan (BGU) for an opportunity to use their analytical equipment. The sulfur isotope analyses were partially funded by ERC-StG 307582 (CARBONSINK) to AT. We are grateful to Sarit Melamed, Bibi Kerpel, Tamir Buchshtav, Peter Rendel, and Edna Danon (BGU) for assistance with sample analyses. Additionally, we would like to thank Timor Katz, Asaph Rivlin, Sefi Baruch and Moty Ohevia (IUI, Eilat) for assistance with the field work. We would like to thank Alyssa Findlay and Adi Torfstein for comments on the draft of the manuscript.
Footnotes
References
Aharon, P., and Fu, B. (2003). Sulfur and oxygen isotopes of coeval sulfate–sulfide in pore fluids of cold seep sediments with sharp redox gradients. Chem. Geol. 195, 201–218. doi: 10.1016/S0009-2541(02)00395-9
Aller, R. C., Mackin, J. E., and Cox, R. T. Jr. (1986). Diagenesis of Fe and S in Amazon inner shelf muds: apparent dominance of Fe reduction and implications for the genesis of ironstones. Cont. Shelf Res. 6, 263–289. doi: 10.1016/0278-4343(86)90064-6
Al-Qutob, M., Häse, C., Tilzer, M. M., and Lazar, B. (2002). Phytoplankton drives nitrite dynamics in the Gulf of Aqaba. Red Sea. Mar. Ecol. Prog. Ser. 239, 233–239. doi: 10.3354/meps239233
Al-Rousan, S. (1998). Sediment Role in Nutrient Cycle within Coral Reefs of the Gulf of Aqaba, Red Sea. Master thesis, Yarmouk University, Irbid.
Al-Rousan, S., Patzold, J., Al-Moghrabi, S., and Wefer, G. (2004). Invasion of anthropogenic CO2 recorded in planktonic foraminifera from the northern Gulf of Aqaba. Int. J. Earth Sci. 93, 1066–1076. doi: 10.1007/s00531-004-0433-4
Al-Rousan, S., Rasheed, M., Al-Horani, F., and Manasrah, R. (2006). Geochemical and textural properties of carbonate and terrigenous sediments along the Jordanian coast of the Gulf of Aqaba. J. Oceanogr. 62, 839–849. doi: 10.1007/s10872-006-0102-2
Al-Taani, A. A., Rashdan, M., and Khashashneh, S. (2015). Atmospheric dry deposition of mineral dust to the Gulf of Aqaba, Red Sea: rate and trace elements. Mar. Pollut. Bull. 92, 252–258. doi: 10.1016/j.marpolbul.2014.11.047
Amrani, A. (2014). Organosulfur compounds: molecular and isotopic evolution from biota to oil and gas. Annu. Rev. Earth Planet. Sci. 42, 733–768. doi: 10.1146/annurev-earth-050212-124126
Amrani, A., Kamyshny, A. Jr., Lev, O., and Aizenshtat, Z. (2006). Sulfur stable isotope distribution of polysulfide anions in (NH4)2Sn aqueous solution. Inorg. Chem. 45, 1427–1429. doi: 10.1021/ic051748r
Antler, G., Turchyn, A., Rennie, V., Herut, B., and Sivan, O. (2013). Coupled sulfur and oxygen isotope insight into bacterial sulfate reduction in the natural environment. Geochim. Cosmochim. Acta 118, 98–117. doi: 10.1016/j.gca.2013.05.005
Badran, M. I. (2001). Dissolved oxygen, chlorophyll a and nutrient: seasonal cycles in waters of the Gulf of Aqaba, Red Sea. Aquat. Ecosyst. Health Manag. 4, 139–150. doi: 10.1080/14634980127711
Bak, F., and Cypionka, H. (1987). A novel type of energy metabolism involving fermentation of inorganic sulphur compounds. Nature 326, 891–892. doi: 10.1038/326891a0
Ben-Avraham, Z., Almagor, G., and Garfunkel, Z. (1979). Sediments and structure of the Gulf of Elat (Aqaba) – Northern Red Sea. Sediment. Geol. 23, 239–267. doi: 10.1016/0037-0738(79)90016-2
Bishop, T., Turchyn, A. V., and Sivan, O. (2013). Fire and brimstone: the microbially mediated formation of elemental sulfur nodules from an isotope and major element study in the Paleo-Dead Sea. PLoS ONE 8:e75883. doi: 10.1371/journal.pone.0075883
Biton, E., and Gildor, H. (2011). The general circulation of the Gulf of Eilat/Aqaba revisited: the interplay between the exchange flow through the Straits of Tiran and surface fluxes. J. Geophys. Res. 116:C08020. doi: 10.1029/2010jc006860
Black, K. D., Calder, L. A., Nickell, T. D., Sayer, M. D. J., Orr, H., Brand, T., et al. (2012). Chlorophyll, lipid profiles and bioturbation in sediments around a fish cage farm in the Gulf of Eilat. Israel. Aquaculture 35, 317–327. doi: 10.1016/j.aquaculture.2012.04.049
Bogoch, R., Bourne, J., Shirav, M., and Harnois, L. (1997). Petrochemistry of a late Precambrian garnetiferous granite, pegmatite and aplite, southern Israel. Mineral. Mag. 61, 111–122. doi: 10.1180/minmag.1997.061.404.11
Böttcher, M., Thamdrup, B., Gehre, M., and Theune, A. (2005). 34S/ 32S and 18O/ 16O fractionation during sulfur disproportionation by Desulfobulbus propionicus. Geomicrobiol. J. 22, 219–226. doi: 10.1080/01490450590947751
Böttcher, M. E., Bernasconi, S. M., and Brumsack, H.-J. (1999). Carbon, sulfur, and oxygen isotope geochemistry of interstitial waters from the western Mediterranean. Proc. Ocean Drill. Prog. Sci. Results 161, 413–421.
Böttcher, M. E., Oelschläger, B., Höpner, T., Brumsack, H.-J., and Rullkötter, J. (1998). Sulfate reduction related to the early diagenetic degradation of organic matter and “black spot” formation in tidal sandflats of the German Wadden Sea (southern North Sea): stable isotope (13C, 34S, 18O) and other geochemical results. Org. Geochem. 29, 1517–1530. doi: 10.1016/S0146-6380(98)00124-7
Boudreau, B. P. (1992). A kinetic model for microbic organic-matter decomposition in marine sediments. FEMS Microbiol. Ecol. 102, 1–14. doi: 10.1111/j.1574-6968.1992.tb05789.x
Brunner, B., Bernasconi, S., Kleikemper, J., and Schroth, M. (2005). A model for oxygen and sulfur isotope fractionation in sulfate during bacterial sulfate reduction processes. Geochim. Cosmochim. Acta 69, 4773–4785. doi: 10.1186/1467-4866-15-7
Brunner, B., and Bernasconi, S. M. (2005). A revised isotope fractionation model for dissimilatory sulfate reduction in sulfate reducing bacteria. Geochim. Cosmochim. Acta 69, 4759–4771. doi: 10.1016/j.gca.2005.04.015
Brunner, B., Einsiedl, F., Arnold, G., Müller, I., Templer, S., and Bernasconi, S. (2012). The reversibility of dissimilatory sulphate reduction and the cell-internal multi-step reduction of sulphite to sulphide: insights from the oxygen isotope composition of sulphate. Isotopes Environ. Health Stud. 48, 33–54. doi: 10.1080/10256016.2011.608128
Burdige, D. J., and Nealson, K. H. (1986). Chemical and microbiological studies of sulfide-mediated manganese reduction. Geochem. J. 4, 361–387.
Canfield, D. (2001). Biogeochemistry of sulfur isotopes. Rev. Mineral. Geochem. 43, 607–636. doi: 10.2138/gsrmg.43.1.607
Canfield, D. E., Raiswell, R., and Bottrell, S. (1992). The reactivity of sedimentary iron minerals toward sulfide. Am. J. Sci. 292, 659–683. doi: 10.2475/ajs.292.9.659
Canfield, D. E., Thamdrup, B., and Hansen, J. W. (1993). The anaerobic degradation of organic-matter in Danish coastal sediments – iron reduction, manganese reduction, and sulfate reduction. Geochim. Cosmochim. Acta 57, 3867–3883. doi: 10.1016/0016-7037(93)90340-3
Chambers, L. A., Trudinger, P. A., Smith, J. W., and Burns, M. S. (1975). Fractionation of sulfur isotopes by continuous cultures of Desulfovibrio desulfuricans. Can. J. Microbiol. 21, 1602–1607. doi: 10.1139/m75-234
Chase, Z., Paytan, A., Johnson, K. S., Street, J., and Chen, Y. (2006). Input and cycling of iron in the Gulf of Aqaba. Red Sea. Glob. Biogeochem. Cycles 20:GB3017.
Chen, K. Y., and Morris, J. C. (1972). Kinetics of oxidation of aqueous sulfide by oxygen. Environ. Sci. Technol. 6, 529–537. doi: 10.1021/es60065a008
Chen, Y., Paytan, A., Chase, Z., Measures, C., Beck, A. J., Sañudo-Wilhelmy, S. A., et al. (2008). Sources and fluxes of atmospheric trace elements to the Gulf of Aqaba. Red Sea. J. Geophys. Res. 113:D5. doi: 10.1016/j.marpolbul.2014.11.047
Chen, Y., Street, J., and Paytan, A. (2006). Comparison between pure-water- and seawater-soluble nutrient concentrations of aerosols in the Gulf of Aqaba. Mar. Chem. 101, 141–152. doi: 10.1016/j.marchem.2006.02.002
Cline, J. (1969). Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol. Oceanogr. 14, 454–458. doi: 10.4319/lo.1969.14.3.0454
Duce R. A., Liss P. S., Merrill J. T., Atlas E. L., Buat-Menard P., Hicks B. B. (1991). The atmospheric input of trace species to the world ocean. Glob. Biogeochem. Cycles 5:193–259. doi: 10.1029/91GB01778
Duce, R. A., and Tindale, N. W. (1991). Atmospheric transport of iron and its deposition in the ocean. Limnol. Oceanogr. 36, 1715–1726. doi: 10.4319/lo.1991.36.8.1715
Eyal, M., Litvinovsky, B., Jahn, B. M., Zanvilevich, A., and Katzir, Y. (2010). Origin and evolution of post-collisional magmatism: coeval neoproterozoic calc-alkaline and alkaline suites of the Sinai Peninsula. Chem. Geol. 269, 153–179. doi: 10.1016/j.chemgeo.2009.09.010
Fahey, R. C., and Newton, G. L. (1987). Determination of low-molecular-weight thiols using monobromobimane fluorescent labeling and high-performance liquid chromatography. Methods Enzymol. 143, 85–96. doi: 10.1016/0076-6879(87)43016-4
Farquhar, J., Johnston, D. T., Wing, B. A., Habicht, K. S., Canfield, D. E., Airieau, S., et al. (2003). Multiple sulphur isotopic interpretations of biosynthetic pathways: implications for biological signatures in the sulphur isotope record. Geobiology 1, 27–36. doi: 10.1046/j.1472-4669.2003.00007.x
Fossing, H., and Jørgensen, B. B. (1989). Measurement of bacterial sulfate reduction in sediments - evaluation of a single-step chromium reduction method. Biogeochemistry 8, 205–222. doi: 10.1007/BF00002889
Fossing, H., and Jørgensen, B. B. (1990). Oxidation and reduction of radiolabeled inorganic sulfur compounds in an estuarine sediment, Kysing Fjord, Denmark. Geochim. Cosmochim. Acta 54, 2731–2742. doi: 10.1016/0016-7037(90)90008-9
Fritz, P., Basharmal, G., Drimmie, R., Ibsen, J., and Qureshi, R. (1989). Oxygen isotope exchange between sulphate and water during bacterial reduction of sulphate. Chem. Geol. Isot. Geosci. 79, 99–105. doi: 10.1080/10256016.2011.622443
Froelich, P., Klinkhammer, G., Bender, M., Luedtke, N., Heath, G., Cullen, D., et al. (1979). Early oxidation of organic-matter in pelagic sediments of the eastern equatorial Atlantic: suboxic diagenesis. Geochim. Cosmochim. Acta 43, 1075–1090. doi: 10.1016/0016-7037(79)90095-4
Garrison, V. H., Shinn, E. A., Foreman, W. T., Griffin, D. W., Holmes, C. W., Kellogg, C. A., et al. (2003). African and Asian dust: from desert soils to coral reefs. BioScience 53, 469–480. doi: 10.1641/0006-3568(2003)053[0469:AAADFD]2.0.CO;2
Gartman, A., and Luther, G. W. (2013). Comparison of pyrite (FeS2) synthesis mechanism to reproduce natural FeS2 nanoparticles found at hydrothermal vents. Geochim. Cosmochim. Acta 120, 447–458. doi: 10.1016/j.gca.2013.06.016
Genin, A., Lazar, B., and Brenner, S. (1995). Atmospheric cooling, unusual vertical mixing and coral mortality following the eruption of Mt. Pinatubo. Nature 377, 507–510. doi: 10.1038/377507a0
Goto, K., Taguchi, S., Fukue, Y., Ohta, K., and Watanabe, H. (1977). Spectrophotometric determination of manganese with 1-(2-pyridylazo)-2-naphthol and a non-ionic surfactant. Talanta 24, 752–753. doi: 10.1016/0039-9140(77)80206-3
Gröger, J., Franke, J., Hamer, K., and Schulz, H. D. (2009). Quantitative recovery of elemental sulfur and improved selectivity in a chromium-reducible sulfur distillation. Geostand. Geoanal. Res. 33, 17–27. doi: 10.1111/j.1751-908X.2009.00922.x
Habicht, K. S., Gade, M., Thamdrup, B., Berg, P., and Canfield, D. E. (2002). Calibration of sulfate levels in the Archean Ocean. Science 298, 2372–2374. doi: 10.1126/science.1078265
Hansel, C. M., Ferdelman, T. G., and Tebo, B. M. (2015). Cryptic cross-linkages among biogeochemical cycles: novel insights from reactive intermediates. Elements 11, 409–414. doi: 10.2113/gselements.11.6.409
Harrison, A. G., and Thode, H. G. (1958). Mechanism of the bacterial reduction of sulphate from isotope fractionation studies. Chem. Soc. Faraday Trans. 54, 84–92. doi: 10.1039/tf9585400084
Holmkvist, L., Ferdelman, T. G., and Jørgensen, B. B. (2011). A cryptic sulfur cycle driven by iron in the methane zone of marine sediment (Aarhus Bay. Denmark). Geochim. Cosmochim. Acta 75, 3581–3599. doi: 10.1016/j.gca.2011.03.033
Holmkvist, L., Kamyshny, A. Jr., Brüchert, V., Ferdelman, T. G., and Jørgensen, B. B. (2014). Sulfidization of lacustrine glacial clay upon Holocene marine transgression (Arkona Basin. Baltic Sea). Geochim. Cosmochim. Acta 142, 75–94. doi: 10.1016/j.gca.2014.07.030
Jickells, T. D. (1995). Atmospheric inputs of metals and nutrients to the oceans: their magnitude and effects. Mar. Chem. 48, 199–214. doi: 10.1016/0304-4203(95)92784-P
Johnston, D. T., Gill, B. C., Masterson, A., Beirne, E., Casciotti, K. L., Knapp, A. N., et al. (2014). Placing an upper limit on cryptic marine sulphur cycling. Nature 513, 530–533. doi: 10.1038/nature13698
Jørgensen, B. B. (1990a). A thiosulfate shunt in the sulfur cycle of marine sediments. Science 249, 152–154.
Jørgensen, B. B. (1990b). The sulfur cycle of freshwater sediments: role of thiosulfate. Limnol. Oceanogr. 35, 1329–1342. doi: 10.1099/ijs.0.004986-0
Kamyshny, A. Jr. (2009a). Improved cyanolysis protocol for detection of zero-valent sulfur in natural aquatic systems. Limnol. Oceanogr. Methods 7, 442–448. doi: 10.4319/lom.2009.7.442
Kamyshny, A. Jr. (2009b). Solubility of cyclooctasulfur in pure water and sea water at different temperatures. Geochim. Cosmochim. Acta 73, 6022–6028. doi: 10.1016/j.gca.2009.07.003
Kamyshny, A. Jr., Druschel, G., Mansaray, Z. F., and Farquhar, J. (2014). Multiple sulfur isotopes fractionations associated with abiotic sulfur transformations in Yellowstone National Park geothermal springs. Geochem. Trans. 15:7. doi: 10.1186/1467-4866-15-7
Kamyshny, A. Jr., and Ferdelman, T. G. (2010). Dynamics of zero-valent sulfur species including polysulfides at seep sites on intertidal sand flats (Wadden Sea. North Sea). Mar. Chem. 121, 17–26. doi: 10.1016/j.marchem.2010.03.001
Kamyshny, A., Ekeltchik, I., Gun, J., and Lev, O. (2006). Method for the determination of inorganic polysulfide distribution in aquatic systems. Anal. Chem. 78, 2631–2639. doi: 10.1021/ac051854a
Kamyshny, A., Goifman, A., Gun, J., Rizkov, D., and Lev, O. (2004). Equilibrium distribution of polysulfide ions in aqueous solutions at 25 degrees C: a new approach for the study of polysulfides equilibria. Environ. Sci. Technol. 38, 6633–6644. doi: 10.1021/es049514e
Katz, T., Ginat, H., Eyal, G., Steiner, Z., Braun, Y., Shalev, S., et al. (2015). Desert flash floods form hyperpycnal flows in the coral-rich Gulf of Aqaba. Red Sea. Earth Planet. Sci. Lett. 417, 87–98. doi: 10.1016/j.epsl.2015.02.025
Katz, T., Herut, B., Genin, A., and Angel, D. L. (2002). Gray mullets ameliorate organically enriched sediments below a fish farm in the oligotrophic Gulf of Aqaba (Red Sea). Mar. Ecol. Prog. Ser. 234, 205–214. doi: 10.3354/meps234205
Kleinjan, W. E., de Keizer, A., and Janssen, A. J. H. (2005). Kinetics of chemical oxidation of polysulfide anions in aqueous solutions. Water Res. 39, 4093–4100. doi: 10.1016/j.watres.2005.08.006
Kosower, N., Kosower, E., Newton, G., and Ranney, H. (1979). Bimane fluorescent labels - labeling of normal human red-cells under physiological conditions. Proc. Natl. Acad. Sci. U.S.A. 76, 3382–3386. doi: 10.1073/pnas.76.7.3382
Labiosa, R. G., Arrigo, K. R., Genin, A., Monismith, S. G., and van Dijken, G. (2003). The interplay between upwelling and deep convective mixing in determining the seasonal phytoplankton dynamics in the Gulf of Aqaba: evidence from SeaWiFS and MODIS. Limnol. Oceanogr. 48, 2355–2368. doi: 10.4319/lo.2003.48.6.2355
Larsen, A. R. (1978). A note on the estimation of sedimentation rates in the Gulf of Elat (Aqaba), by use of Foraminifera. Sedimentology 25, 583–585. doi: 10.1111/j.1365-3091.1978.tb02082.x
Levanon-Spanier, I., Padan, E., and Reiss, Z. (1979). Primary production in a desert-enclosed sea: the Gulf of Elat (Aqaba). Red Sea. Deep Sea Res. 26, 673–686. doi: 10.1016/0198-0149(79)90040-2
Li, H., Veldhuis, M. J. W., and Post, A. F. (1998). Alkaline phosphatase activities among planktonic communities in the northern Red Sea. Mar. Ecol. Prog. Ser. 173, 107–115. doi: 10.3354/meps173107
Lichtschlag, A., Kamyshny, A. Jr., Ferdelman, T. G., and de Beer, D. (2013). Intermediate sulfur oxidation state compounds in the euxinic surface sediments of the Dvurechenskii mud volcano (Black Sea). Geochim. Cosmochim. Acta 105, 130–145. doi: 10.1016/j.gca.2012.11.025
Luther, G. W. III. (1991). Pyrite synthesis via polysulfide compounds. Geochim. Cosmochim. Acta 55, 2839–2849. doi: 10.1016/0016-7037(91)90449-F
Mahowald, N. M., Baker, A. R., Bergametti, G., Brooks, N., Duce, R. A., Jickells, T. D., et al. (2005). Atmospheric global dust cycle and iron inputs to the ocean. Glob. Biogeochem. Cycles 19, GB4025. doi: 10.1029/2004gb002402
Mendez, J., Guieu, C., and Adkins, J. (2010). Atmospheric input of manganese and iron to the ocean: seawater dissolution experiments with Saharan and North American dusts. Mar. Chem. 120, 34–43. doi: 10.1016/j.marchem.2008.08.006
Mills, J. V., Antler, G., and Turchyn, A. V. (2016). Geochemical evidence for cryptic sulfur cycling in salt marsh sediments. Earth Planet. Sci. Lett. 453, 23–32. doi: 10.1016/j.epsl.2016.08.001
Mizutani, Y., and Rafter, T. (1973). Isotopic behaviour of sulphate oxygen in the bacterial reduction of sulphate. Geochem. J. 6, 183–191. doi: 10.2343/geochemj.6.183
Morse, J., and Cornwell, J. C. (1987). Analysis and distribution of iron sulfide minerals in recent anoxic marine sediments. Mar. Chem. 22, 55–69. doi: 10.1016/0304-4203(87)90048-X
Orcutt, B. N., Sylvan, J. B., Knab, N. J., and Edwards, K. J. (2011). Microbial ecology of the dark ocean above at and below the seafloor. Microbiol. Mol. Biol. Rev. 75, 361–422. doi: 10.1128/MMBR.00039-10
Poulton, S. W., and Canfield, D. E. (2005). Development of a sequential extraction procedure for iron: implications for iron partitioning in continentally derived particulates. Chem. Geol. 214, 209–221. doi: 10.1016/j.chemgeo.2004.09.003
Poulton, S. W., Krom, M. D., and Raiswell, R. (2004). A revised scheme for the reactivity of iron (oxyhydr)oxide minerals towards dissolved sulfide. Geochim. Cosmochim. Acta 68, 3703–3715. doi: 10.1016/j.gca.2004.03.012
Raiswell, R., and Canfield, D. (1996). Rates of reaction between silicate iron and dissolved sulfide in Peru Margin sediments. Geochim. Cosmochim. Acta 60, 2777–2787. doi: 10.1016/0016-7037(96)00141-X
Rasheed, M., Badran, M. I., and Huettel, M. (2003). Influence of sediment permeability and mineral composition on organic matter degradation in three sediments from the Gulf of Aqaba. Red Sea. Estuar. Coast. Shelf Sci. 57, 369–384. doi: 10.1016/S0272-7714(02)00362-1
Reiss, Z., and Hottinger, L. (1984). The Gulf of Aqaba: Ecological Micropaleontology: Ecological Studies, Vol. 50. Berlin: Springer, 1–354. doi: 10.1007/978-3-642-69787-6
Reiss, Z., Luz, B., Almogi-Labin, A., Halicz, E., Winter, A., Wolf, M., et al. (1980). Late quaternary paleoceanography of the Gulf of Aqaba (Elat). Red Sea. Quat. Res. 14, 294–308. doi: 10.1016/0033-5894(80)90013-7
Rickard, D. T., and Luther, G. W. (1997). Kinetics of pyrite formation by H2S oxidation of iron(II) monosulfide in aqueous solutions between 25 and 125 degrees C: the mechanism. Geochim. Cosmochim. Acta 61, 135–147. doi: 10.1016/S0016-7037(96)00322-5
Rong, L., Lim, L., and Takeuchi, T. (2005). Determination of iodide and thiocyanate in seawater by liquid chromatography with poly(ethylene glycol) stationary phase. Chromatographia 61, 371–374. doi: 10.1365/s10337-005-0501-3
Rudnicki, M. D., Elderfield, H., and Spiro, B. (2001). Fractionation of sulfur isotopes during bacterial sulfate reduction in deep ocean sediments at elevated temperatures. Geochim. Cosmochim. Acta 65, 777–789. doi: 10.1016/S0016-7037(00)00579-2
Ryan, W. B. F., Carbotte, S. M., Coplan, J. O., O’Hara, S., Melkonian, A., Arko, R., et al. (2009). Global multi-resolution topography synthesis. Geochem. Geophys. Geosyst. 10:Q03014. doi: 10.1029/2008GC002332
Schumacher, H., Kiene, W., and Dullo, W. C. (1995). Factors controlling Holocene reef growth: an interdisciplinary approach. Facies 32, 145–188. doi: 10.1007/BF02536867
Seeberg-Elverfeldt, J., Schluter, M., Feseker, T., and Kolling, M. (2005). Rhizon sampling of porewaters near the sediment-water interface of aquatic systems. Limnol. Oceanogr. Methods 3, 361–371. doi: 10.4319/lom.2005.3.361
Sim, M. S., Bosak, T., and Ono, S. (2011a). Large sulfur isotope fractionation does not require disproportionation. Science 333, 74–77. doi: 10.1126/science.1205103
Sim, M. S., Ono, S., Donovan, K., Templer, S. P., and Bosak, T. (2011b). Effect of electron donors on the fractionation of sulfur isotopes by a marine Desulfovibrio sp. Geochim. Cosmochim. Acta 75, 4244–4259. doi: 10.1016/j.gca.2011.05.021
Stambler, N. (2006). Light and picophytoplankton in the Gulf of Eilat (Aqaba). J. Geophys. Res. 111:C11009. doi: 10.1029/2005jc003373
Steiner, Z., Lazar, B., Levi, S., Tsroya, S., Pelled, O., Bookman, R., et al. (2016). The effect of bioturbation in pelagic sediments: lessons from radioactive tracers and planktonic foraminifera in the Gulf of Aqaba (Eilat). Red Sea. Geochim. Cosmochim. Acta 194, 139–152. doi: 10.1016/j.gca.2016.08.037
Stookey, L. (1970). Ferrozine - a new spectrophotometric reagent for iron. Anal. Chem. 42, 779–781. doi: 10.1021/ac60289a016
Street, J., and Paytan, A. (2005). Iron, phytoplankton growth, and the carbon cycle. Met. Ions Biol. Syst. 43, 153–185.
Thamdrup, B. (2000). “Bacterial manganese and iron reduction in aquatic sediments,” in Advances in Microbial Ecology, Vol. 16, ed. B. Schink (New York, NY: Springer), 41–84. doi: 10.1007/978-1-4615-4187-5_2
Thamdrup, B., Finster, K., Fossing, H., Hansen, J. W., and Jørgensen, B. B. (1994). Thiosulfate and sulfite distributions in porewater of marine sediments related to manganese, iron, and sulfur geochemistry. Geochim. Cosmochim. Acta 58, 67–73. doi: 10.1016/0016-7037(94)90446-4
Turchyn, A., Brüchert, V., Lyons, T., Engel, G., Balci, N., Schrag, D., et al. (2010). Kinetic oxygen isotope effects during dissimilatory sulfate reduction: a combined theoretical and experimental approach. Geochim. Cosmochim. Acta 74, 2011–2024. doi: 10.1016/j.gca.2010.01.004
Turchyn, A., Sivan, O., and Schrag, D. (2006). Oxygen isotopic composition of sulfate in deep sea pore fluid: evidence for rapid sulfur cycling. Geobiology 4, 191–201. doi: 10.1111/j.1472-4669.2006.00079.x
Valravamurthy, A., and Mopper, K. (1990). Determination of sulfite and thiosulfate in aqueous samples including anoxic seawater by liquid chromatography after derivatization with 2,2′-dithiobis(5-nitropyridine). Environ. Sci. Technol. 24, 333–337. doi: 10.1021/es00073a007
Wan, M., Shchukarev, A., Lohmayer, R., Planer-Friedrich, B., and Peiffer, S. (2014). Occurrence of surface polysulfides during the interaction between ferric (hydr)oxides and aqueous sulfide. Environ. Sci. Technol. 48, 5076–5084. doi: 10.1021/es405612f
Werne, J. P., Lyons, T. W., Hollander, D. J., Schouten, S., Hopmans, E. C., and Sinninghe Damsté, J. S. (2008). Investigating pathways of diagenetic organic matter sulfurization using compound-specific sulfur isotope analysis. Geochim. Cosmochim. Acta 72, 3489–3502. doi: 10.1016/j.gca.2008.04.033
Yao, W., and Millero, F. J. (1996). Oxidation of hydrogen sulfide by hydrous Fe(III) oxides in seawater. Mar. Chem. 52, 1–16. doi: 10.1016/0304-4203(95)00072-0
Zopfi, J., Böttcher, M. E., and Jørgensen, B. B. (2008). Biogeochemistry of sulfur and iron in Thioploca-colonized surface sediments in the upwelling area off central Chile. Geochim. Cosmochim. Acta 72, 827–843. doi: 10.1016/j.gca.2007.11.031
Zopfi, J., Ferdelman, T. G., and Fossing, H. (2004). “Distribution and fate of sulfur intermediates – sulfite, tetrathionate, thiosulfate, and elemental sulfur – in marine sediments,” in Sulfur Biogeochemistry – Past and Present, eds J. P. Amend, K. J. Edwards, and T. W. Lyons (Denver, CO: The Geological Society of America), 97–116.
Keywords: Red Sea, Gulf of Aqaba, aeolian dust deposition, highly reactive iron, manganese, sulfide oxidation intermediates, cryptic sulfur cycle
Citation: Blonder B, Boyko V, Turchyn AV, Antler G, Sinichkin U, Knossow N, Klein R and Kamyshny A Jr. (2017) Impact of Aeolian Dry Deposition of Reactive Iron Minerals on Sulfur Cycling in Sediments of the Gulf of Aqaba. Front. Microbiol. 8:1131. doi: 10.3389/fmicb.2017.01131
Received: 23 March 2017; Accepted: 02 June 2017;
Published: 20 June 2017.
Edited by:
Brian T. Glazer, Hawaii University, United StatesReviewed by:
Kai Waldemar Finster, Aarhus University, DenmarkDirk De Beer, Max Planck Society (MPG), Germany
Copyright © 2017 Blonder, Boyko, Turchyn, Antler, Sinichkin, Knossow, Klein and Kamyshny. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexey Kamyshny Jr., alexey93@gmail.com