 Ravinder Nagpal1*†
Ravinder Nagpal1*† Hirokazu Tsuji1,2
Hirokazu Tsuji1,2 Takuya Takahashi1,2Koji Nomoto1,2†Kazunari Kawashima3Satoru Nagata4
Takuya Takahashi1,2Koji Nomoto1,2†Kazunari Kawashima3Satoru Nagata4 Yuichiro Yamashiro1
Yuichiro Yamashiro1- 1Probiotics Research Laboratory, Juntendo University Graduate School of Medicine, Tokyo, Japan
- 2Yakult Central Institute, Kunitachi, Japan
- 3Gonohashi Obstetrics and Gynecology Hospital, Koto, Japan
- 4Department of Pediatrics, School of Medicine, Tokyo Women’s Medical University, Shinjuku, Japan
Early-life intestinal microbiota development is crucial for host’s long-term health and is influenced by many factors including gestational age, birth and feeding modes, birth environment, ethnic/geographical background, etc. However, ‘quantitative’ data on the actual population levels of gut bacterial communities when these influences are controlled for is relatively rare. Herein, we demonstrate a quantitative perspective of microbiota development in natural and healthy milieus, i.e., in healthy, full-term, vaginally born and breast-fed infants (n = 19) born at same clinic. Fecal microbiota at age 1 and 7 days, 1, 3, and 6 months and 3 years is quantified using highly sensitive reverse-transcription-quantitative-PCR assays targeting bacterial rRNA molecules. At day 1, we detect one or more bacteria in all (100%) of the babies, wherein the microbiota is composed mainly of enterobacteria (35%), Bacteroides fragilis group (23%), enterococci (18%), staphylococci (13%), and bifidobacteria (9%). Altogether, facultative anaerobes predominate during first few weeks whereafter obligate anaerobes including bifidobacteria, B. fragilis group, Clostridium coccoides group, and Clostridium leptum subgroup gradually start prevailing. At 3 years, the composition is represented almost entirely (99%) by obligate anaerobes including C. leptum subgroup (34%), bifidobacteria (22%), B. fragilis group (21%), C. coccoides group (17%), Atopobium cluster (4%), and Prevotella (1%). The overall obligate/facultative proportion is 32/68, 37/63, 54/46, 70/30, 64/36, and 99/1% at 1 and 7 days, 1, 3, and 6 months and 3 years, respectively. However, interestingly, considerable individual-specific variations in the obligate/facultative ratios as well as in the proportions of Firmicutes, Bacteroides, Actinobacteria, and Proteobacteria communities are seen among these babies. This disparity even within this highly homogenous cohort manifests the magnitude of diverse patterns of gut microbiota configuration and hence underpins the importance of considering not only the gestational age, birth, and feeding modes, and ethnic/geographical background but also other potential outstanding factors when investigating the elements shaping the early microbiota development. In summary, the data demonstrate a quantitative bird’s-eye view of the ontogenesis of early-life gut microbiota in typically natural and healthy milieus and should be informative and facilitative for future studies exploring various aspects of the human gut microbiota.
Introduction
The past decade has been remarkable in revealing the fundamental role of human gut microbiota in various aspects of host health and disease. It is now well established that the microbial colonization in human gut starts immediately at birth (Penders et al., 2006; Dominguez-Bello et al., 2010; Tsuji et al., 2012; Backhed et al., 2015; Bokulich et al., 2016; Nagpal et al., 2016) or perhaps even before that, i.e., in utero (Aagaard et al., 2014; Collado et al., 2016; Chu et al., 2017); and the magnitude and array of this early-life microbiota acquisition may affect several aspects of the newborn’s long-term health (Nagpal and Yamashiro, 2015; Tamburini et al., 2016). However, majority of the available knowledge has been acquired using bacterial 16S rRNA gene-based sequencing methods which, while being remarkably elaborative and comprehensive, are largely qualitative and may not provide precise data on fecal bacterial counts. In addition, the data acquired from DNA-based PCR or sequencing methods may also include unsought DNA entities (e.g., DNA of dead bacteria) and hence may not discriminate between viable and dead bacteria. As a result, quantitative longitudinal data on the actual population levels (i.e., the bacterial count) of various important bacterial clades dwelling in the infant gut during early life remains limited and disparate. Undeniably, even in this golden age of revolutionary sequence tools, the quantitative data denoting the ‘gold standard’ viable bacterial count still remains of indispensable worth. In this context, we have previously established a novel and sensitive analytical system for human intestinal microbiota analysis based on reverse-transcription-quantitative-PCR (RT-qPCR) assays targeting bacterial 16S rRNA ‘molecules,’ wherein we had methodologically validated that this RT-qPCR approach is relatively highly sensitive (detection limit: 103–104 bacterial cells/g feces; approximately 100- to 1000-fold higher than that of other molecular methods including qPCR and t-RFLP) and provides fecal bacterial enumeration comparable to the viable cell counts as enumerated by culturing and fluorescent in situ hybridization methods (Matsuda et al., 2007, 2009; Kubota et al., 2010; Kurakawa et al., 2015).
Utilizing this sensitive quantitative approach in longitudinal studies to examine the development of early life gut microbiota composition in a cohort of more than 150 Japanese infants, we have recently demonstrated that the mode of delivery (vaginal vs. cesarean birth) and the feeding type (breast-milk vs. formula-feed) have a strong influence on the intestinal carriage of various important bacterial clades and that the effects of these influences on gut microbiota start building up as early as the first day or week of life (Tsuji et al., 2012; Nagpal et al., 2016). Hence, these findings as well as the similar results from several other recent studies (Dominguez-Bello et al., 2010; Backhed et al., 2015; Dogra et al., 2015; Bokulich et al., 2016) instigated us to investigate afresh – and with a different perspective – the data of early life microbiota development when these influences are controlled for. Therefore, in this addendum, with an aim to manifest a quantitative bird’s-eye view of the ontogenesis of early gut microbiota configuration in natural and healthy milieus, we demonstrate the fecal microbiota composition in a selected cohort of 19 healthy, full-term, vaginally born and exclusively breast-fed infants prospectively from birth to 3 years of age. Unlike previous reports wherein we presented the data largely in the form of bacterial count and prevalence (Tsuji et al., 2012; Nagpal et al., 2016), this addendum focuses mainly on the relative proportions of various important gut bacterial groups and the ratios between obligate and facultative anaerobes as well as between Firmicutes, Bacteroides, Actinobacteria, and Proteobacteria, thereby outlining a characteristic profile of the early-life gut microbiota structure in a ‘healthy’ infant cohort that may be reflective of characteristics common to the present-day healthy Japanese infant population.
Materials and Methods
The study includes 19 healthy Japanese infants enrolled at the Gonohashi Obstetrics and Gynecology Hospital, Tokyo. As mentioned beforehand, these infants are part of a large cohort of over 150 Japanese babies wherein fecal carriage of various gut microbes during the first 3 years of age has previously been reported (Nagpal et al., 2016). The general information about these infants is provided in Table 1, and the detailed information about the cohort can be found elsewhere (Nagpal et al., 2016). Briefly, all these 19 babies were full-term and vaginally born and remained exclusively breast-fed until 3 to 6 months of age. All the babies as well as their mothers remained apparently healthy with no indication of any major illness or any considerable exposure to antibiotics/drugs during the study period. The study design was approved by the ethical committees of the Juntendo University and the Yakult Central Institute, Tokyo. In accordance with the Declaration of Helsinki, prior written informed consent was obtained from all the parents or legal representatives.
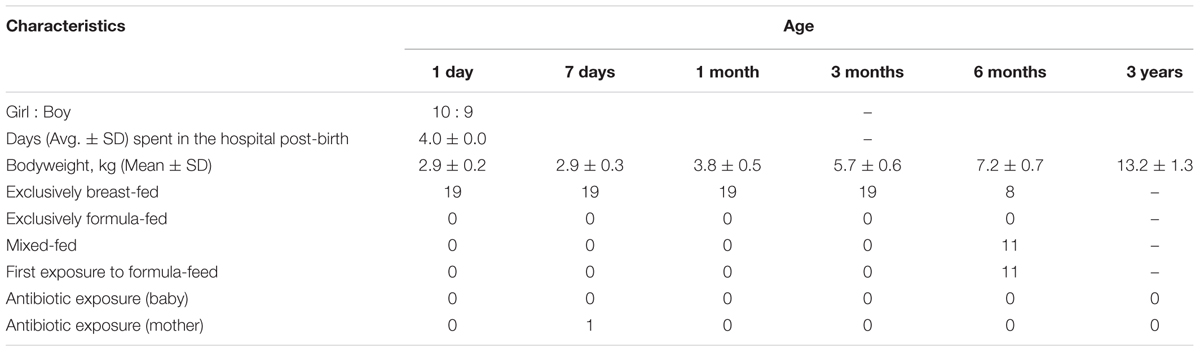
TABLE 1. General characteristics of the 19 infants enrolled in the study.
Details of sample collection procedure, RNA extraction, RT-qPCR reactions, and primer specifics have been provided elsewhere (Nagpal et al., 2016). Briefly, to obtain an accurate description of the population levels of specific gut bacterial groups and to precisely enumerate changes in their fecal carriage, we used a highly sensitive analytical approach based on RT-qPCR assays targeting bacterial 16S rRNA molecules (Matsuda et al., 2009; Nagpal et al., 2016). As specified elsewhere (Nagpal et al., 2016), the minimum detection limit of these assays ranged from 103 to 104 cells/g feces. Using this analytical approach, we aimed to cover a wide-range of the human gut microbiota by targeting a wide array of anaerobic, facultative anaerobic, Gram-positive, Gram-negative, predominant, subdominant, indigenous pathobiont, and opportunistic pathogenic bacterial clades etc. that are frequently and prevalently associated with human gut microbiota. We herein quantified 11 different major gut bacterial groups including Clostridium coccoides group, Clostridium leptum subgroup, Bacteroides fragilis group, Prevotella, Bifidobacterium, Atopobium cluster, Lactobacillus, Enterobacteriaceae, Enterococcus, Staphylococcus, and Clostridium perfringens in the fecal specimens of these infants at six time-points viz. age 1 and 7 days, 1, 3, and 6 months, and 3 years. The bacterial counts enumerated by RT-qPCR assays are expressed as log10 cells per gram of feces (mean ± standard deviation), and the carriage rate or prevalence of colonization is expressed as the percentage of infants in which the specific bacterium was detected. The total bacterial count was estimated as the sum of the counts of these 11 bacterial clades. The count of genus Bifidobacterium was estimated as the sum of the counts of one group and seven species viz. B. catenulatum group (including B. catenulatum and B. pseudocatenulatum), B. longum, B. infantis, B. bifidum, B. breve, B. adolescentis, B. angulatum, and B. dentium. The count of genus Lactobacillus was estimated as the sum of the counts of eight subgroups/species, i.e., L. gasseri subgroup, L. casei subgroup, L. plantarum subgroup, L. reuteri subgroup, L. ruminis subgroup, L. sakei subgroup, L. brevis, and L. fermentum.
The proportions of each bacterial group were calculated with regard to the total bacterial count. Normalization was done by dividing the values of each bacterial group by the value obtained for the ‘total bacterial count.’ The proportions of obligate and facultative anaerobes were estimated by the sum of the corresponding bacterial groups (C. coccoides group, C. leptum subgroup, B. fragilis group, Bifidobacterium, Atopobium cluster, Prevotella and C. perfringens for obligate anaerobes; and Enterobacteriaceae, Enterococcus, Lactobacillus and Staphylococcus for facultative anaerobes). The proportion of total Firmicutes, Bacteroides, Actinobacteria, and Proteobacteria were estimated by adding the corresponding bacterial values obtained from (i) C. coccoides group, C. leptum subgroup, C. perfringens, Enterococcus, Lactobacillus and Staphylococcus (total Firmicutes); (ii) Bacteroides fragilis group and Prevotella (total Bacteroides); (iii) Bifidobacterium and Atopobium cluster (total Actinobacteria); and (iv) Enterobacteriaceae (total Proteobacteria).
Results and Discussion
Gestational age, birth mode, feeding type, birth environment, and the geographical background are known, among various other factors, as the most prominent elements shaping the neonatal gut microbiome (Penders et al., 2006; Dominguez-Bello et al., 2010; Echarri et al., 2011; Backhed et al., 2015; Bokulich et al., 2016; Tamburini et al., 2016; Hill et al., 2017). We herein aim to demonstrate the development of gut microbiota composition when these potential influences are controlled for. All 19 babies in this study are born at full-term, via vaginal delivery, and remain exclusively breast-fed until 3 to 6 months of age. Notably, all these babies also share same ethnicity (Japanese), geographical setting (Tokyo-born) as well as the birth environment (i.e., born at same clinic and spent same time, i.e., 4 days at the hospital post-birth). Hence, this highly homogenous cohort, although small in sample size, might manifest the pattern of gut microbiota development in typical natural and healthy milieus, particularly in Japanese birth population.
Our data outlining a typical infant profile, along the lines of previous reports (Aagaard et al., 2014; Ardissone et al., 2014; Collado et al., 2016; Martin et al., 2016; Chu et al., 2017; Hill et al., 2017), shows that various bacterial groups, predominantly the facultative anaerobes such as Enterobacteriaceae, Enterococcus and Staphylococcus, are already dwelling in the first intestinal discharge (i.e., meconium) of these babies (Figure 1 and Supplementary Table S1). We noted that all (100%) of the babies harbor one or more bacteria in their meconium samples (Figure 1B). Since previous studies have largely used DNA-based methods, it remains debatable whether the prenatal niches encounter live bacteria or only contain dead (non-viable) bacteria that might have seeped in from maternal circulation for immune priming of the neonate. In this context, our RNA-based data of the meconium bacterial flora corroborates and fortifies the mounting body of evidence that supports the notion of in utero live microbial exposures (Aagaard et al., 2014; Ardissone et al., 2014; Collado et al., 2016; Chu et al., 2017), although the clear-cut routes and sources of these bacteria and their significance in context to the fetal programming and infant’s long-term health still remain largely unclear. Nevertheless, we found that at day 1, the prevalence (detection rate, %) was higher for enterobacteria, enterococci, staphylococci followed by B. fragilis group, bifidobacteria and lactobacilli (Figure 1B). Concordant with previous reports, this observation clearly suggests that the facultative anaerobes are the first settlers in the human gut whereas obligate anaerobes such as clostridia, bacteroides, and bifidobacteria thrive at later stages (Solís et al., 2010; Ardissone et al., 2014; Backhed et al., 2015; Bokulich et al., 2016; Collado et al., 2016; Hill et al., 2017). The data further demonstrate a clear predominance of these facultative anaerobes during the first few weeks of life whereafter the predominance gradually transitions toward obligate anaerobes including bifidobacteria, B. fragilis group, C. coccoides group, and C. leptum subgroup (Figure 2). At day 1, the microbiota is composed mainly of facultative anaerobes with Enterobacteriaceae (35%), enterococci (18%), and staphylococci (13%) dominating the overall composition. Among obligates, B. fragilis group (23%) and bifidobacteria (9%) represent the main components. However, this configuration is gradually shifted toward the predominance of obligate anaerobes, especially bifidobacteria, at the cost of facultative anaerobes viz. enterococci and staphylococci during subsequent stages. At 6 months, the microbiota is dominated by bifidobacteria (42%) followed by enterobacteria (34%) and B. fragilis group (19%). At 3 years, the composition is represented almost entirely (99%) by obligate anaerobes including C. coccoides group (17%), C. leptum subgroup (34%), B. fragilis group (21%), bifidobacteria (22%), Atopobium cluster (4%), and Prevotella (1%).
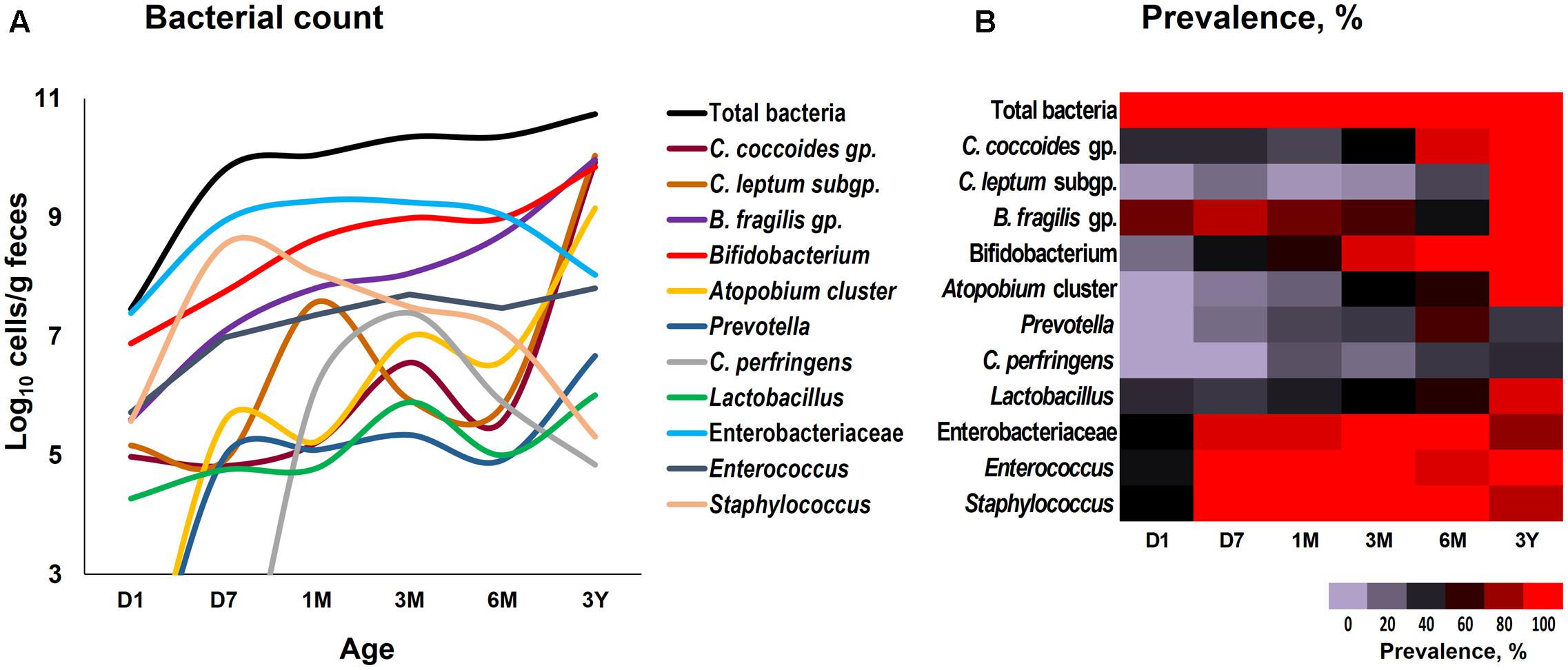
FIGURE 1. Fecal count (A) and prevalence (B) of various bacterial groups in healthy Japanese infants (n = 19) at different time-points during the first 3 years of life. Bacterial count is expressed as the mean of log10 cells/g feces. Prevalence (detection rate, %) was expressed as the percentage of infants in which the specific bacterium was detected. Age (x-axis): 1 day, 7 days, 1 month, 3 months, 6 months, 3 years. Numerical values on these data have been provided in Supplementary Table S1.
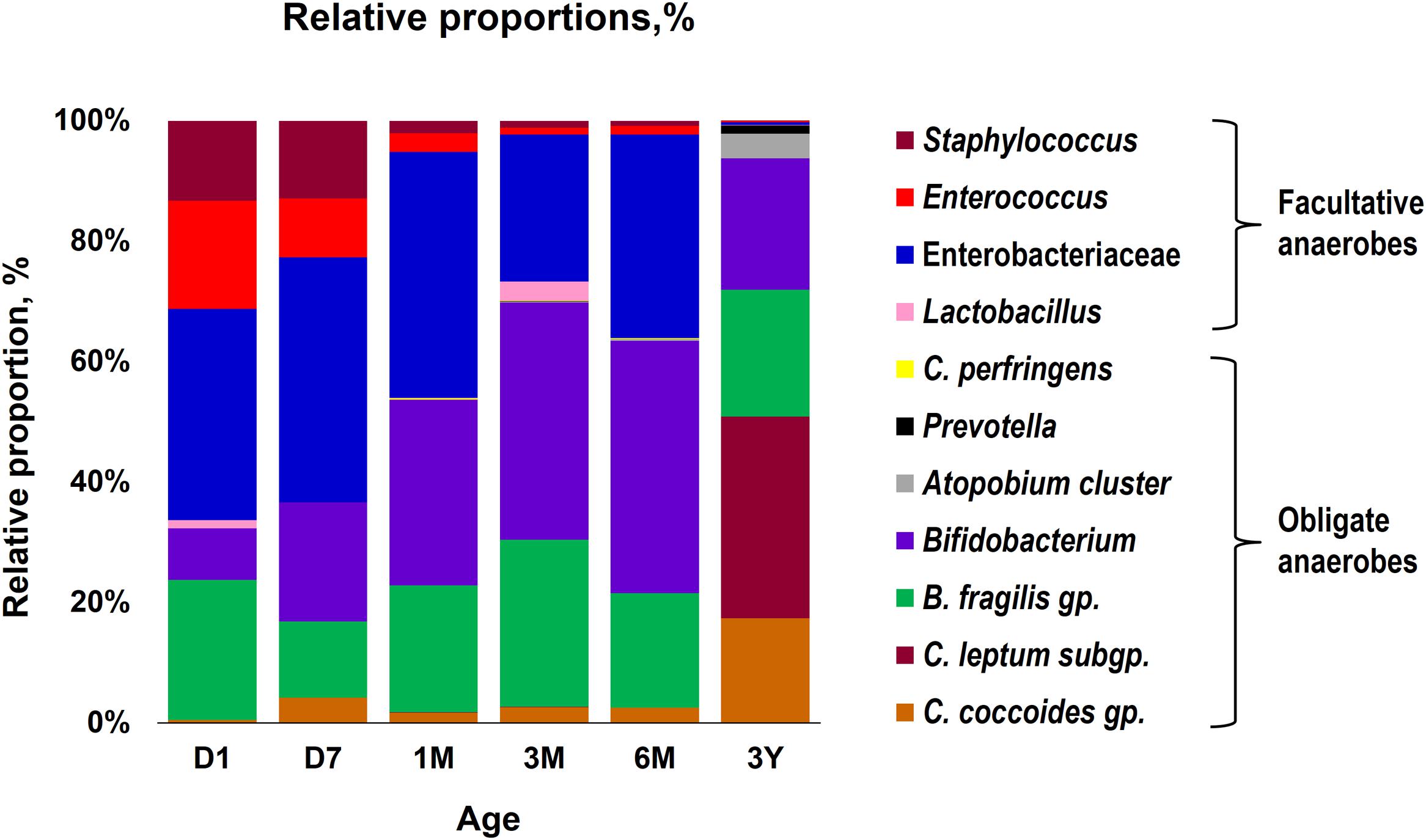
FIGURE 2. Proportional composition of the gut bacterial microbiota in healthy Japanese infants (n = 19) at different time-points during the first 3 years of life. Relative proportions of different bacteria were calculated by using the original arithmetical number of the bacterial count and are expressed as the percent of the total fecal bacterial count. Age (x-axis): 1 day, 7 days, 1 month, 3 months, 6 months, 3 years.
Overall, the obligate/facultative proportion is 32/68, 37/63, 54/46, 70/30, 64/36, and 99/1 at age 1 and 7 days, 1, 3, and 6 months, and 3 years, respectively (Figure 3). These proportions as well as the overall patterns are consistent with other studies (Arboleya et al., 2012; Backhed et al., 2015; Odamaki et al., 2016); however, interestingly, considerable variations can be seen at the individual level. For instance, several babies are dominated by obligate anaerobes even on day 1 whereas several babies are still dominated by facultative anaerobes even at 6 months of age (Figure 3). Interestingly, compared to full-term babies, premature babies are found to have significantly higher facultative/obligate ratio until 3 months of age or perhaps even longer (Arboleya et al., 2012); however, we noted a similar predominance of facultative anaerobes from day 1 through 6 months in several babies even in this healthy full-term cohort (Figure 3). Although geographical and/or methodological differences may underlie this disparity (Echarri et al., 2011), this observation of notable variations within this small cohort also clearly hints that the microbiota configuration is highly individual-specific and hence the mean values of a particular cohort might not always extrapolate the true picture of a given element or parameter. Similar to the facultative-obligate ratios, the overall mean proportions of Firmicutes, Bacteroides, Actinobacteria, and Proteobacteria communities in this cohort (Supplementary Figure S1) also seem to be consistent with previous reports (Backhed et al., 2015; Odamaki et al., 2016; Murphy et al., 2017); however, at individual-level, there are noticeable differences in the magnitude of these signatures particularly during the first 6 months. For example, at each time-point, some babies appear to be of Proteobacteria- or Actinobacteria-type while several babies show Bacteroides- or Firmicutes-predominance. Moreover, even at individual level, these signatures vary at different time-points. Nevertheless, this variation appear to diminish by age 3 years. Similar trends in inter-individual variability in the gut microbiota composition during early life have been reported in several previous studies (Roger and McCartney, 2010; Echarri et al., 2011). Interestingly, Echarri et al. (2011), while reporting considerable inter-individual variability, also observed significant differences in the gut carriage of several bacterial groups such as Bacteroides, staphylococci, enterobacteria in infants from two different Spanish locations just about 1000 km apart from each other. In this context, although all the infants in our study were born at the same clinic, the effect of differences in their residential locations (e.g., different wards and cities of Tokyo) on the gut microbiota composition cannot be ruled out completely. In a recent study on children from different Asian countries, our colleagues clustered the gut microbiome signatures into two groups, P-type (predominated by Prevotella) and BB-type (predominated by Bifidobacterium and Bacteroides), and reported that Japanese children are mostly of BB-type (Nakayama et al., 2015). In the present study, the data at 3 years seem to concord with this notion because many babies are dominated by Actinobacteria and Bacteroides and hence might arbitrarily be envisaged to be of BB-type (Supplementary Figure S1). The overall composition at 3 years – wherein B. fragilis group and Bifidobacterium together make up for about 43% of the total fecal bacterial count – also seem to corroborate this notion (Figure 2). Nevertheless, we notice slight variation in these arrays even at 3 years of age since some babies are still dominated by Firmicutes community (Supplementary Figure S1). This disparity with the previous report by Nakayama et al. (2015) may be due to the methodological (RT-qPCR vs. pyrosequencing) or cohort-age (3 years vs. 7–11 years) differences; or maybe the microbiota is still under transition state at age 3 years and hence attains an adult-like configuration at a later age than previously suspected. Nonetheless, the data do hint that even at 3 years of age, there are considerable individual-level differences in the microbiota composition within this small but controlled cohort.
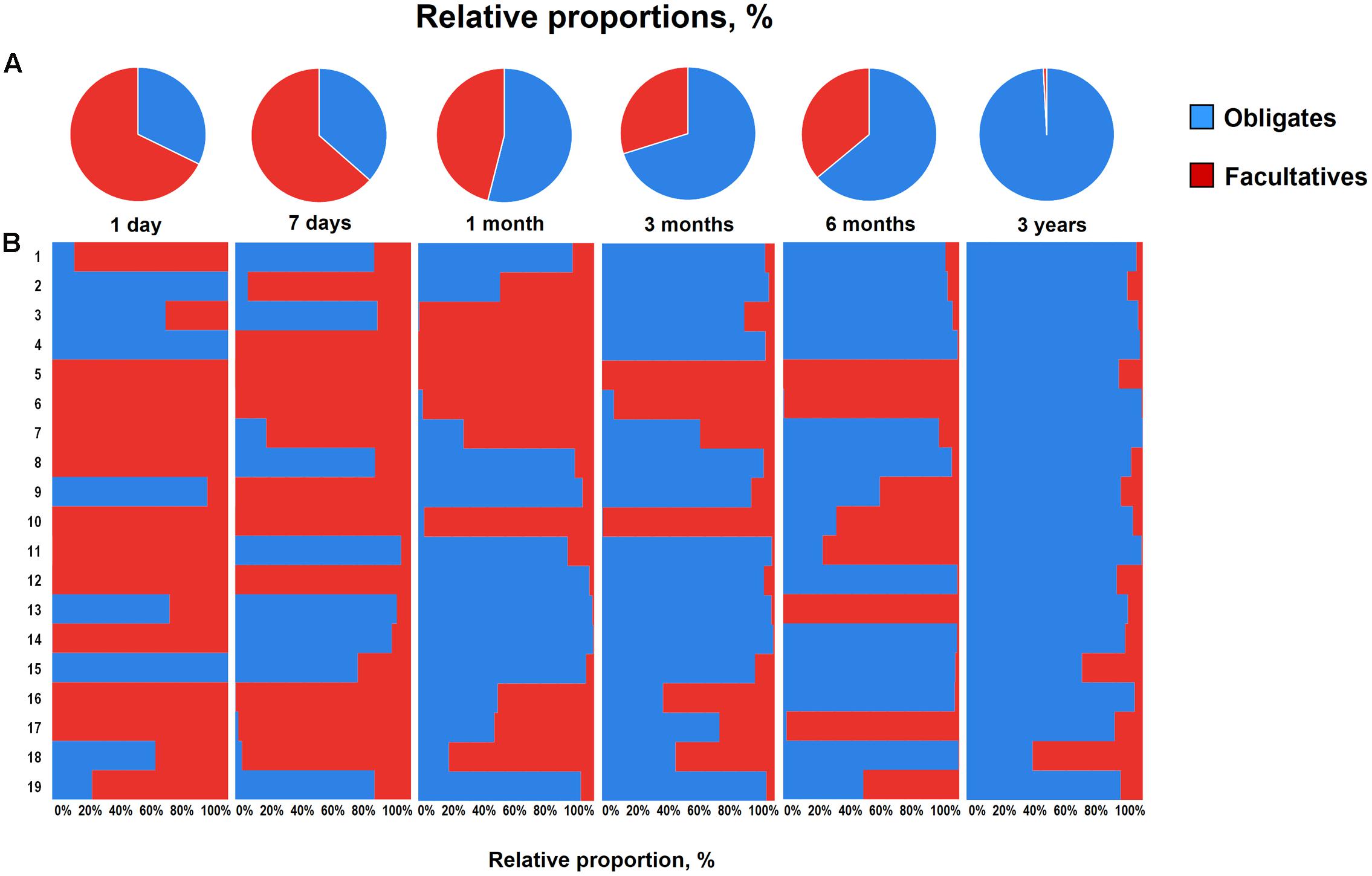
FIGURE 3. Proportional ratios of obligatory anaerobic vs. facultative anaerobic bacteria averaged (A) and at individual-level (B) in healthy Japanese infants (n = 19) at different time-points during the first 3 years of life. Proportions were calculated by using the original arithmetical number of the bacterial count and are expressed as the percent of the total fecal bacterial count. Obligates: Clostridium coccoides group, Clostridium leptum subgroup, Bacteroides fragilis group, Bifidobacterium, Atopobium cluster, Prevotella and Clostridium perfringens. Facultatives: Enterobacteriaceae, Enterococcus, Lactobacillus, and Staphylococcus. Age: 1 day, 7 days, 1 month, 3 months, 6 months, 3 years.
Similar individual-specific differences were also reflected when we estimated the bacterial predominance by evaluating the counts of different bacterial groups and tagging the most abundant clade, i.e., the one with highest bacterial count at a given time-point (Figure 4). We noted that at first day of life, the microbiota is dominated mainly by Enterobacteriaceae, staphylococci, or enterococci, or by B. fragilis group in some babies, but this dominance gradually shifts toward B. fragilis group and bifidobacteria during subsequent phases. Finally, by age 3 years, the predominance switches completely toward obligate anaerobes, possibly in conjunction with the natural process of transition toward adult-microbiota configuration. Weaning, i.e., the introduction (and type) of solid foods, may also be one of the key factors driving this compositional shift between 6 months and 3 years. Nevertheless, even at 3 years, the spectrum of this predominance is diverse with most babies being dominated by C. coccoides group or C. leptum subgroup whereas few babies are dominated by B. fragilis group or bifidobacteria. This observation of considerable inter-individual disparity even within this healthy homogenous cohort underpins the importance of assessing not only the birth mode, feeding type, gestational age and ethnic/geographical background but also other potential factors (which we were unfortunately not able to peruse due to information paucity) such as maternal microbiome and gestational diet, breast-milk’s microbial/biochemical composition, prior drug exposures, household environment, host genetics, intestinal physiology and hormonal status etc. when investigating the elements that shape and drive the early life gut microbiota dynamics (Echarri et al., 2011; Koenig et al., 2011; Yatsunenko et al., 2012; Blekhman et al., 2015; Bokulich et al., 2016; Charbonneau et al., 2016; Chu et al., 2016, 2017; Matsuki et al., 2016).
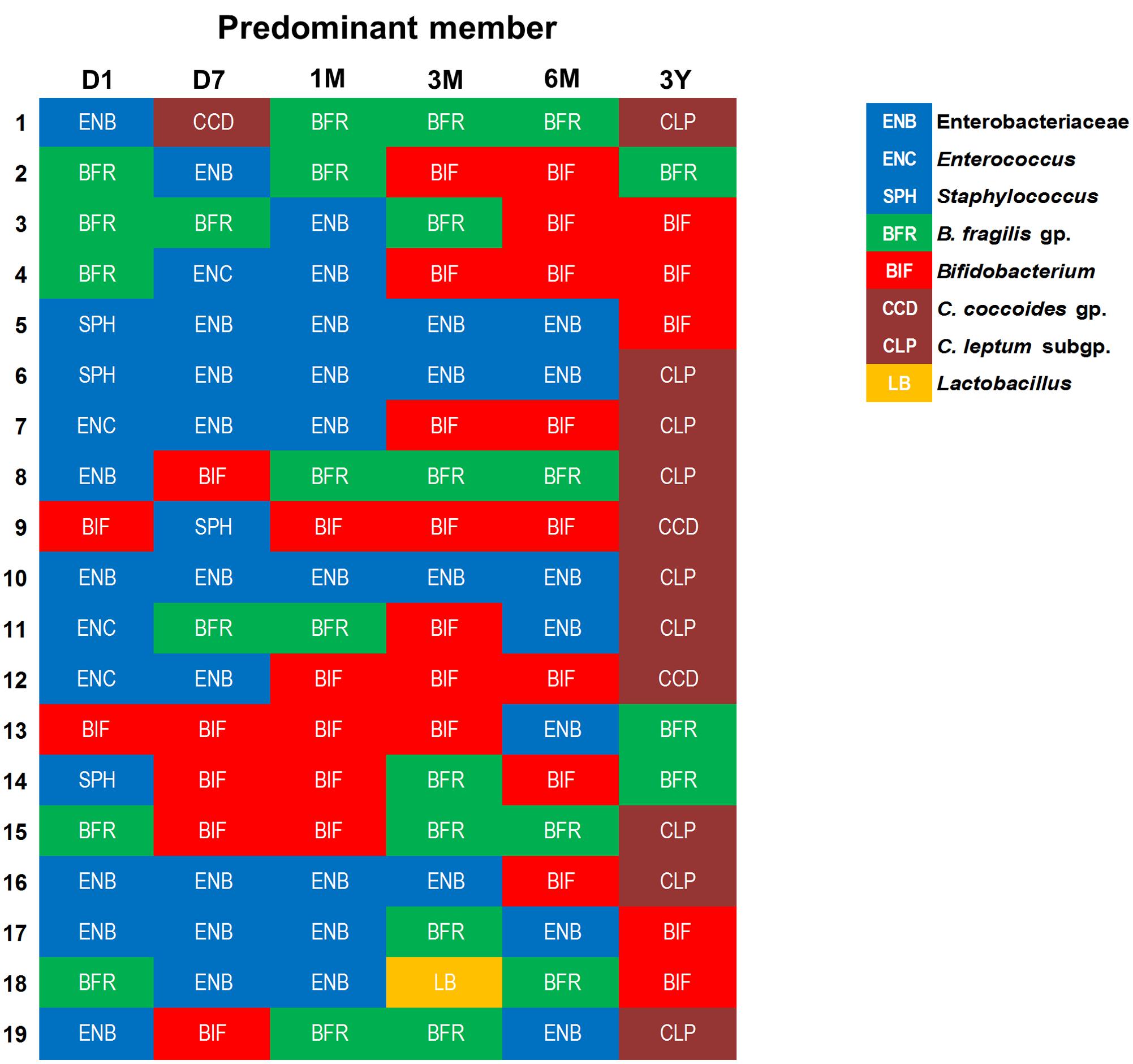
FIGURE 4. An arbitrary overview of the bacterial predominance in healthy Japanese infants (n = 19) at different time-points during the first 3 years of life. The abundance was estimated based on the fecal bacterial counts (log10 cells/g feces) wherein (out of various bacteria analyzed in this study) the bacterial clade with highest fecal count at a given time-point was considered as the most abundant clade among all the targeted bacterial groups. Age: 1 day, 7 days, 1 month, 3 months, 6 months, 3 years.
This small study has several strengths and limitations. The main strength is the homogenous nature of the cohort controlled for gestational age, birth mode, feeding type, place of birth, ethnic and geographical background etc. Also, all the babies remained apparently healthy (without any serious illness or considerable exposure to antibiotics/drugs) during the study period. In addition, the cohort was also quite homogenous in terms of the babies’ weight during different time-points (Table 1), which actually did not allow us to statistically examine the correlation if any of gut microbiota with host bodyweight. The main limitation of the study is the lack of data on multiple outstanding elements (e.g., maternal microbiome and diet during pregnancy, maternal disease history and drug exposures, familial/household environment, siblings, pets, etc.) that may influence the gut microbiota development. Small samples size is another limitation that merits validation of similar analyses in further larger cohort studies. Another limitation is that the bacteria were quantified by RT-qPCR assays using specific primer sets for major gut bacterial groups and genera and hence the microbiota data presented in here may not truly represent the whole array of the gut microbiota; although we have previously validated that the sum of the fecal counts of all the bacterial groups enumerated herein using RT-qPCR assays correspond to 71.3 ± 9.4% (mean ± SD) of total intestinal bacterial count as estimated by in situ hybridization using a universal probe (Hasegawa et al., 2015). Nevertheless, as mentioned beforehand, this quantitative approach could also be considered as a strength of this study, especially considering the high detection sensitivity of these assays and the lack of availability of such numeric and longitudinal data on major human gut bacterial clades.
Conclusion
In summary, this addendum demonstrates a quantitative bird’s-eye view of the development of the gut microbiota community composition during the first 3 years of life in typically natural and healthy milieus, i.e., full-term, vaginal-birth, and exclusive-breast-feeding (and same geographical setting). The findings from this small but homogenous cohort of babies continue to expand our understanding of the maturation of human intestinal microbiota during neonatal and early childhood period, a phase where the microbiota is in most fluctuating stages of its lifespan. The data provide important numeric information about the population levels of various important dominant and subdominant gut bacterial groups inhabiting the early infant gut and hence should prove to be informative and facilitative for future studies exploring various aspects of the human gut microbiota. The data illustrate how the gut microbiota configuration evolves during early life and hint that there are certainly many more known or yet-to-be-known intrinsic and extrinsic factors (other than the gestation age, birth-mode, feeding type, place of birth and geographical background) that may impact the early-life gut microbiome even in healthy infants and hence may underlie the inter-individual disparities in the microbiota composition. Studies deciphering these inter-individual dynamics and elements of the gut microbiota composition would certainly offer new hypotheses, insights and avenues for improving the infant’s intestinal as well as overall long-term health while refining our understanding of this complex gut microbial ecosystem.
Author Contributions
RN, HT, and YY: conceived and designed the study; RN and HT: performed the experiments; RN: analyzed the data; HT and KK: coordinated the sample collection; RN: wrote the manuscript; HT, TT, KN, and YY: checked and revised the manuscript; RN, HT, TT, KN, KK, SN, and YY: approved the final version of manuscript.
Funding
This work was supported by grants from Juntendo Medical University, Tokyo, and Yakult Honsha Co. Ltd., Japan. The funding agency had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all the babies and parents for participating in this study. We also thank all the participating clinicians, researchers, and technicians for their consistent helps during this study.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01388/full#supplementary-material
References
Aagaard, K., Ma, J., Antony, K. M., Ganu, R., Petrosino, J., and Versalovic, J. (2014). The placenta harbors a unique microbiome. Sci. Transl. Med. 6, 237ra65. doi: 10.1126/scitranslmed.3008599
Arboleya, S., Solis, G., Fernandez, N., de los Reyes-Gavilan, C. G., and Gueimonde, M. (2012). Facultative to strict anaerobes ratio in the preterm infant microbiota: a target for intervention? Gut Microbes 3, 583–588. doi: 10.4161/gmic.21942
Ardissone, A. N., de la Cruz, D. M., Davis-Richardson, A. G., Rechcigl, K. T., Li, N., Drew, J. C., et al. (2014). Meconium microbiome analysis identifies bacteria correlated with premature birth. PLoS ONE 9:e90784. doi: 10.1371/journal.pone.0090784
Backhed, F., Roswall, J., Peng, Y., Feng, Q., Jia, H., Kovatcheva-Datchary, P., et al. (2015). Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17, 690–703. doi: 10.1016/j.chom.2015.04.004
Blekhman, R., Goodrich, J. K., Huang, K., Sun, Q., Bukowski, R., Bell, J. T., et al. (2015). Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 16:191. doi: 10.1186/s13059-015-0759-1
Bokulich, N. A., Chung, J., Battaglia, T., Henderson, N., Jay, M., Li, H., et al. (2016). Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med. 8, 343ra82. doi: 10.1126/scitranslmed.aad7121
Charbonneau, M. R., Blanton, L. V., DiGiulio, D. B., Relman, D. A., Lebrilla, C. B., Mills, D. A., et al. (2016). A microbial perspective of human developmental biology. Nature 535, 48–55. doi: 10.1038/nature18845
Chu, D. M., Antony, K. M., Ma, J., Prince, A. L., Showalter, L., Moller, M., et al. (2016). The early infant gut microbiome varies in association with a maternal high-fat diet. Genome Med. 8:77. doi: 10.1186/s13073-016-0330-z
Chu, D. M., Ma, J., Prince, A. L., Antony, K. M., Seferovic, M. D., and Aagaard, K. M. (2017). Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat. Med. 23, 314–326. doi: 10.1038/nm.4272
Collado, M. C., Rautava, S., Aakko, J., Isolauri, E., and Salminen, S. (2016). Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 6:23129. doi: 10.1038/srep23129
Dogra, S., Sakwinska, O., Soh, S. E., Ngom-Bru, C., Brück, W. M., Berger, B., et al. (2015). Dynamics of infant gut microbiota are influenced by delivery mode and gestational duration and are associated with subsequent adiposity. mBio 6:e02419-14. doi: 10.1128/mBio.02419-14
Dominguez-Bello, M. G., Costello, E. K., Contreras, M., Magris, M., Hidalgo, G., Fierer, N., et al. (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. U.S.A. 107, 11971–11975. doi: 10.1073/pnas.1002601107
Echarri, P. P., Graciá, C. M., Berruezo, G. R., Vives, I., Ballesta, M., Solís, G., et al. (2011). Assessment of intestinal microbiota of full-term breast-fed infants from two different geographical locations. Early Hum. Dev. 87, 511–513. doi: 10.1016/j.earlhumdev.2011.03.013
Hasegawa, S., Goto, S., Tsuji, H., Okuno, T., Asahara, T., Nomoto, K., et al. (2015). Intestinal dysbiosis and lowered serum lipopolysaccharide-binding protein in Parkinson’s Disease. PLoS ONE 10:e0142164. doi: 10.1371/journal.pone.0142164
Hill, C. J., Lynch, D. B., Murphy, K., Ulaszewska, M., Jeffery, I. B., O’Shea, C. A., et al. (2017). Evolution of gut microbiota composition from birth to 24 weeks in the INFANTMET Cohort. Microbiome 5:4. doi: 10.1186/s40168-016-0213-y
Koenig, J. E., Spor, A., Scalfone, N., Fricker, A. D., Stombaugh, J., Knight, R., et al. (2011). Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. U.S.A. 108, 4578–4585. doi: 10.1073/pnas.1000081107
Kubota, H., Tsuji, H., Matsuda, K., Kurakawa, T., Asahara, T., and Nomoto, K. (2010). Detection of human intestinal catalase-negative, Gram-positive cocci by rRNA-targeted reverse transcription-PCR. Appl. Environ. Microbiol. 76, 5440–5451. doi: 10.1128/AEM.03132-09
Kurakawa, T., Ogata, K., Tsuji, H., Kado, Y., Takahashi, T., Kida, Y., et al. (2015). Establishment of a sensitive system for analysis of human vaginal microbiota on the basis of rRNA-targeted reverse transcription-quantitative PCR. J. Microbiol. Methods 111, 93–104. doi: 10.1016/j.mimet.2015.01.021
Martin, R., Makino, H., Cetinyurek-Yavuz, A., Ben-Amor, K., Roelofs, M., Ishikawa, E., et al. (2016). Early-life events, including mode of delivery and type of feeding, siblings and gender, shape the developing gut microbiota. PLoS ONE 11:e0158498. doi: 10.1371/journal.pone.0158498
Matsuda, K., Tsuji, H., Asahara, T., Kado, Y., and Nomoto, K. (2007). Sensitive quantitative detection of commensal bacteria by rRNA-targeted reverse transcription-PCR. Appl. Environ. Microbiol. 73, 32–39. doi: 10.1128/AEM.01224-06
Matsuda, K., Tsuji, H., Asahara, T., Matsumoto, K., Takada, T., and Nomoto, K. (2009). Establishment of an analytical system for the human fecal microbiota, based on reverse transcription-quantitative PCR targeting of multicopy rRNA molecules. Appl. Environ. Microbiol. 75, 1961–1969. doi: 10.1128/AEM.01843-08
Matsuki, T., Yahagi, K., Mori, H., Matsumoto, H., Hara, T., Tajima, S., et al. (2016). A key genetic factor for fucosyllactose utilization affects infant gut microbiota development. Nat. Commun. 7:11939. doi: 10.1038/ncomms11939
Murphy, K., Curley, D., O’Callaghan, T. F., O’Shea, C. A., Dempsey, E. M., O’Toole, P. W., et al. (2017). The composition of human milk and infant faecal microbiota over the first three months of life: a pilot study. Sci. Rep. 7:40597. doi: 10.1038/srep40597
Nagpal, R., Tsuji, H., Takahashi, T., Kawashima, K., Nagata, S., Nomoto, K., et al. (2016). Sensitive quantitative analysis of the meconium bacterial microbiota in healthy term infants born vaginally or by cesarean section. Front. Microbiol. 7:1997. doi: 10.3389/fmicb.2016.01997
Nagpal, R., and Yamashiro, Y. (2015). Early-life gut microbial composition. J. Pediatr. Biochem. 5, 41–50. doi: 10.1055/s-0035-1564573
Nakayama, J., Watanabe, K., Jiang, J., Matsuda, K., Chao, S. H., Haryono, P., et al. (2015). Diversity in gut bacterial community of school-age children in Asia. Sci. Rep. 5:8397. doi: 10.1038/srep08397
Odamaki, T., Kato, K., Sugahara, H., Hashikura, N., Takahashi, S., Xiao, J. Z., et al. (2016). Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol. 16:90. doi: 10.1186/s12866-016-0708-5
Penders, J., Thijs, C., Vink, C., Stelma, F. F., Snijders, B., Kummeling, I., et al. (2006). Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118, 511–521. doi: 10.1542/peds.2005-2824
Roger, L. C., and McCartney, A. L. (2010). Longitudinal investigation of the faecal microbiota of healthy full-term infants using fluorescence in situ hybridization and denaturing gradient gel electrophoresis. Microbiology 156, 3317–3328. doi: 10.1099/mic.0.041913-0
Solís, G., de Los Reyes-Gavilan, C. G., Fernández, N., Margolles, A., and Gueimonde, M. (2010). Establishment and development of lactic acid bacteria and bifidobacteria microbiota in breast-milk and the infant gut. Anaerobe 16, 307–310. doi: 10.1016/j.anaerobe.2010.02.004
Tamburini, S., Shen, N., Wu, H. C., and Clemente, J. C. (2016). The microbiome in early life: implications for health outcomes. Nat. Med. 22, 713–722. doi: 10.1038/nm.4142
Tsuji, H., Oozeer, R., Matsuda, K., Matsuki, T., Ohta, T., Nomoto, K., et al. (2012). Molecular monitoring of the development of intestinal microbiota in Japanese infants. Benef. Microbes 3, 113–125. doi: 10.3920/BM2011.0038
Keywords: bacterial quantification, gut bacterial communities, gut microbiome, intestinal microbiota development, RT-qPCR
Citation: Nagpal R, Tsuji H, Takahashi T, Nomoto K, Kawashima K, Nagata S and Yamashiro Y (2017) Ontogenesis of the Gut Microbiota Composition in Healthy, Full-Term, Vaginally Born and Breast-Fed Infants over the First 3 Years of Life: A Quantitative Bird’s-Eye View. Front. Microbiol. 8:1388. doi: 10.3389/fmicb.2017.01388
Received: 28 April 2017; Accepted: 10 July 2017;
Published: 21 July 2017.
Edited by:
M. Pilar Francino, Foundation for the Promotion of Health and Biomedical Research of Valencia Region (FISABIO), SpainReviewed by:
Miguel Gueimonde, Instituto de Productos Lácteos de Asturias (CSIC), SpainJosef Neu, University of Florida, United States
Copyright © 2017 Nagpal, Tsuji, Takahashi, Nomoto, Kawashima, Nagata and Yamashiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ravinder Nagpal, nagpal511@gmail.com
†Present address: Ravinder Nagpal, Gut Microbiome and Metabolic Diseases Group, Center for Diabetes, Obesity and Metabolism, Wake Forest School of Medicine, Winston-Salem, NC, United States; Koji Nomoto, Division of Molecular Microbiology, Tokyo University of Agriculture, Setagaya, Japan