 Joon L. Tan
Joon L. Tan Kee P. Ng2
Kee P. Ng2 Yun F. Ngeow
Yun F. Ngeow- 1Faculty of Information Science and Technology, Multimedia University, Melaka, Malaysia
- 2Department of Medical Microbiology, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
- 3Department of Pre-clinical Sciences, Faculty of Medicine and Health Sciences, Universiti Tunku Abdul Rahman, Petaling Jaya, Malaysia
Mycobacterium abscessus, a rapid-growing non-tuberculous mycobacterium, has been the cause of sporadic and outbreak infections world-wide. The subspecies in M. abscessus complex (M. abscessus, M. massiliense, and M. bolletii) are associated with different biologic and pathogenic characteristics and are known to be among the most frequently isolated opportunistic pathogens from clinical material. To date, the evolutionary forces that could have contributed to these biological and clinical differences are still unclear. We compared genome data from 243 M. abscessus strains downloaded from the NCBI ftp Refseq database to understand how the microevolutionary processes of homologous recombination and positive selection influenced the diversification of the M. abscessus complex at the subspecies level. The three subspecies are clearly separated in the Minimum Spanning Tree. Their MUMi-based genomic distances support the separation of M. massiliense and M. bolletii into two subspecies. Maximum Likelihood analysis through dN/dS (the ratio of number of non-synonymous substitutions per non-synonymous site, to the number of synonymous substitutions per synonymous site) identified distinct genes in each subspecies that could have been affected by positive selection during evolution. The results of genome-wide alignment based on concatenated locally-collinear blocks suggest that (a) recombination has affected the M. abscessus complex more than mutation and positive selection; (b) recombination occurred more frequently in M. massiliense than in the other two subspecies; and (c) the recombined segments in the three subspecies have come from different intra-species and inter-species origins. The results lead to the identification of possible gene sets that could have been responsible for the subspecies-specific features and suggest independent evolution among the three subspecies, with recombination playing a more significant role than positive selection in the diversification among members in this complex.
Introduction
Mycobacterium abscessus is a rapidly growing species of non-tuberculous mycobacterium (NTM). In 2011, this species was divided into two subspecies, M. abscessus subsp. abscessus and M. abscessus subsp. bolletii (Leao et al., 2011) but subsequent comparative studies including those based on whole genome sequencing (Cho et al., 2013; Tan et al., 2015) supported division of the M. abscessus complex into three subspecies, M. abscessus subsp. abscessus, M. abscessus subsp. bolletii, and M. abscessus subsp. massiliense. In this paper, the subspecies are referred to as M. abscessus, M. bolletii, and M. massiliense. All three subspecies are opportunistic pathogens associated with significant morbidity, particularly in patients with cystic fibrosis (Olivier et al., 2003; Jönsson et al., 2007; Levy et al., 2008; Bryant et al., 2013; Davidson et al., 2013). Although genetically closely related, they show differences in drug resistance (Koh et al., 2011, 2013; Mitra et al., 2012; Griffith, 2014), association with clinical disorders (Koh et al., 2011; Stout and Floto, 2012; Roux et al., 2015), and mode of transmission (Bryant et al., 2013; Tettelin et al., 2014). To date, the evolutionary forces that could have contributed to these biological and clinical differences are still unclear.
Recombination, an important evolutionary force for the diversification of bacterial core genomes (Ehrlich et al., 2005; Redfield et al., 2006), has been reported in the M. abscessus complex (Sassi et al., 2014). Gene acquisition at the hypothetical common ancestor apparently played an important role in the evolution of this species complex and the enhanced virulence in some members could have come from their ability to acquire genes from different bacterial genera via horizontal transfer.
Positive selection is another evolutionary force that contributes to significant selective advantage in organisms, varying from prokaryotes to eukaryotes (Petersen et al., 2007; Lefébure and Stanhope, 2009; Cagliani and Sironi, 2013). For instance, the insensitivity of M. tuberculosis to most common antibiotics has been hypothesized to be due to convergent evolution as a result of positive selection on drug resistance-associated genes (Casali et al., 2012; Farhat et al., 2013). In some cases, positive selection acts together with horizontal transfer and homologous recombination to bring about bacterial diversification (Donati et al., 2010).
In this study, we used bioinformatic analysis on 243 genomes to understand how the microevolutionary processes of homologous recombination and positive selection influenced the diversification of the M. abscessus complex at the subspecies level.
Materials and Methods
Genome Sequences
Of the 243 genomes examined, 12 were from clinical strains isolated between 2009 and 2011 in the Clinical Microbiology Laboratory of University of Malaya Medical Centre (UMMC), Kuala Lumpur, Malaysia. These strains were subjected to whole genome shotgun sequencing using the Illumina Genome Analyzer IIX platform, as described previously (Tan et al., 2013). For the remaining genomes, we downloaded their whole genome sequences from the NCBI ftp Refseq database (March 2016) (Supplementary Table S1). We included strains from different parts of the world to increase the likelihood of identifying subspecies-specific characteristics instead of just clonal traits.
Genome-Wide Intra-subspecies and Inter-subspecies Comparisons
For in silico inter-subspecies and intra-subspecies comparisons, a single pseudo-contig for each of the 243 genomes was generated by using “NNNNNCACACACTTAATTAATTAAGTGTGTGNNNNN” as a linker between contigs. The insertion of this 36 bp pseudo-molecule introduces a stop codon in any of the six open reading frames, and hence will not affect the annotation of each genome (Tettelin et al., 2005; Xu et al., 2010; Peng et al., 2017). The maximal unique matches index (MUMi) was implemented to infer the genomic relatedness among the strains. The choice of MUMi over the more established average nucleotide identity (ANI) is because of its greater robustness for the inference of intra-species divergence and its inclusion of both core and accessory genome sequences for the calculation of divergence. Hence, NUCmer of MUMmer was used to perform the pairwise genome alignments (Kurtz et al., 2004). Matches present with the effect of DNA inversion events were identified and overlapping segments of unique matches were filtered. The resulting data was used as input in the MUMi perl script (Deloger et al., 2009). A MUMi value of 0 indicates identical genome sequences whereas the maximum value of 1 indicates the most distantly related genomes.
Defining Orthologous Sequences and Alignments
The genome sequences were annotated using locally installed GeneMarkS with default parameters (Besemer et al., 2001). The predicted protein sequences were clustered using ProteinOrtho (Lechner et al., 2011). Protein sequences with minimum e-value of 1 × 10-5, alignment coverage of 50% and clustering connectivity of 0.1 were categorized into a single protein family (Lechner et al., 2011). Sequences were then extracted based on the defined families. Multiple-aligned sequences were obtained in fast Fourier transform (FFT) algorithm as implemented in MAFFT, using FFT-NS-I as the iterative refinement method (Katoh et al., 2002). The alignment of corresponding codons was achieved in PAL2NAL (Suyama et al., 2006).
Identification of Genes under Positive Selection
We examined each of the non-duplicated conserved proteins for positive selection in each of the three subspecies, using Branch-site model (model = 2; NS sites = 2) in CodeML of PAML on the dataset (Yang, 2007). In the Branch-site model, foreground branches comprised the branches of interest to be compared with the rest of the branches on a phylogenetic tree (background branches) (Zhang et al., 2005). Two models (null and alternate) were compared to select a suitable mode to depict the evolution of the genes. In the null model, codons may evolve neutrally or under purifying selection (fix_omega = 1, omega = 1) in both background and foreground branches. In the alternate model, codons were assumed to be undergoing positive selection in foreground branches but not in background branches (fix_omega = 0, omega = 1). The p-value was derived from the likelihood ratio test (LRT), assuming Chi-square distribution with degree of freedom of 1 (Yang, 1998). Genes were treated as undergoing positive selection if p-value is less than 0.01, and the non-synonymous to synonymous mutation ratio (dN/dS) is greater than one. The genes with significant traces of positive selection were further analyzed with the Bayes Empirical Bayes (BEB) method, to retrieve the positions of amino acids on the genes that were substituted owing to selection forces (Yang et al., 2005). The genes supported in BEB were subjected for subcellular localization prediction in CELLO webserver (Yu et al., 2006).
To investigate whether the dN/dS of each of the identified positively selected sequences was influenced by recombination, we performed recombination detection based on the three robust general testing methods (Brown et al., 2001; Posada et al., 2002), namely Neighbor Similarity Score (NSS) (Jakobsen and Easteal, 1996), Maximum χ2 (Max χ2) (Smith, 1992), and Pairwise Homoplasy Index (PHI) (Bruen et al., 2006). NSS is based on clustering of compatibilities of the informative adjacent sites in the form of a matrix; PHI highlights the calculation of minimum number of homoplasies without the influence of the sample’s history, and Max χ2 compares putative recombination break-points in two sequences, with other sequences within an alignment.
Recombination Analysis
The genomes were aligned based on locally-collinear blocks (LCBs) as defined by the progressiveMauve algorithm (Darling et al., 2010). Owing to the computational limitation in the handling of a large dataset comprising 243 genomes with a mean size of 5.1 M base pairs, we applied the following protocol – (1) 15 genomes each from M. abscessus and M. massiliense were selected as representatives for comparisons. The selection was based on the mean MUMi (indicating the average genomic distance between paired strains in a group), with five genomes each from the strains with the highest, median and lowest mean MUMi values. The intention of the selection was to maximize the inclusion of strains with high genetic diversities. Since there were only 16 M. bolletii strains, all M. bolletii genomes were included for the genome-wide alignments (Supplementary Table S1). (2) Only the LCBs with a minimum size of 500 bp which were shared by all the strains were used for the analysis. An in-house Biopython script was written to separate each LCB from the XMFA alignment file. (3) The sequence of each LCB was then identified from the genomes that were not used in the Mauve alignment. Each of the resulted LCB was manually curated to remove artifacts, and concatenated to form super-sequences. The super-sequences and a PhyML generated super-sequences SNPs tree were subjected to recombination study in the ClonalFrameML (Didelot and Wilson, 2015).
Results
Population Structure
The genomes used in this study comprised the three known subspecies of M. abscessus complex isolated from multiple countries. The subspecies for each of the 243 strains was identified in silico using the concatenation of the 50 proposed informative genes previously reported (Tan et al., 2013). The resulting pseudo-sequence was used in the calculation of a minimum-spanning tree (MST) (Figure 1). The MST illustrates the relationships of data based on the shortest possible distance between two data points. Strains from the same subspecies would have low dissimilarities and short genetic distances from each other, and hence, are expected to form a cluster. Based on this classification, 16 strains in our study clustered as M. bolletii, 138 as M. massiliense, and 89 as M. abscessus.
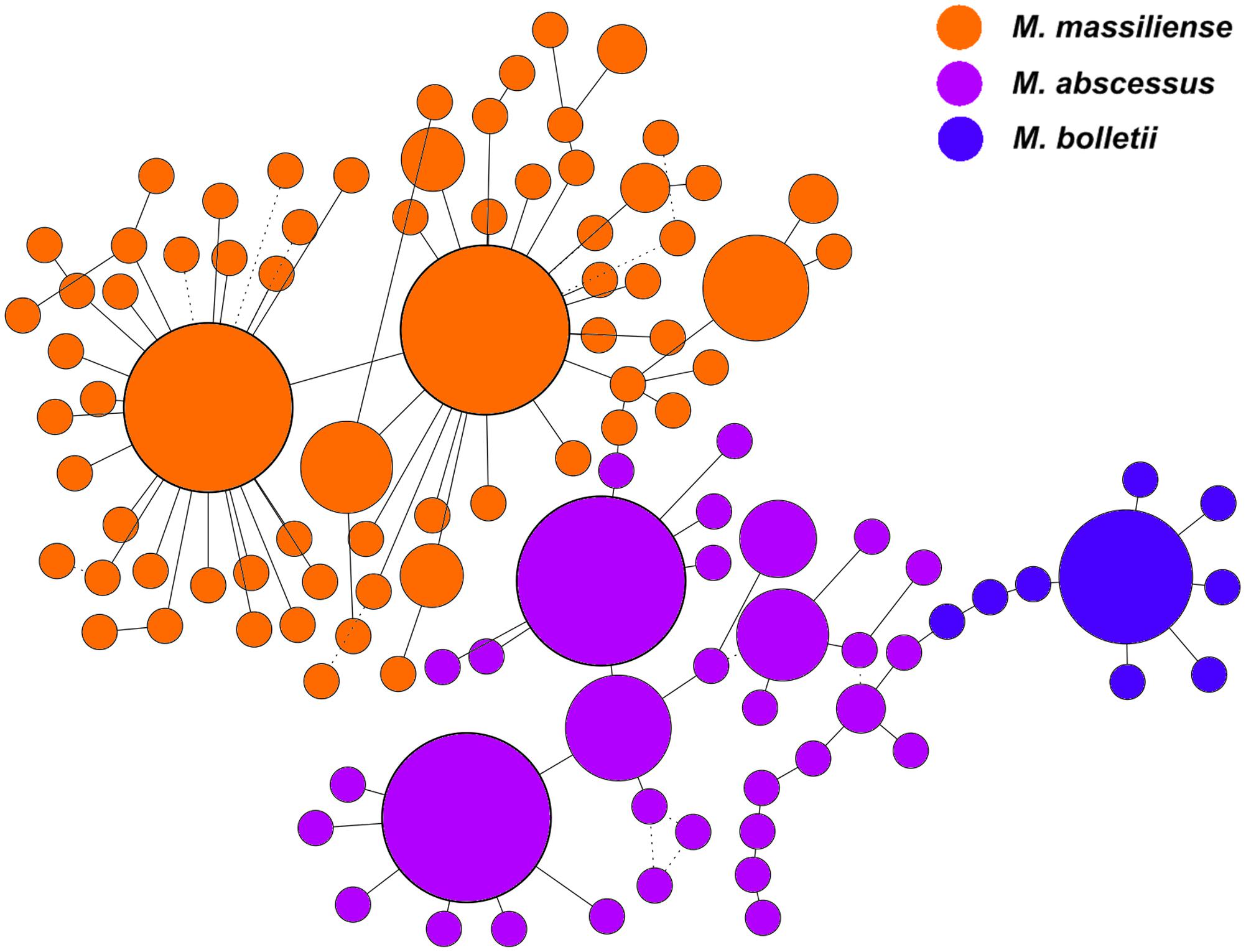
FIGURE 1. Minimum spanning tree based on 50 informative genes of the M. abscessus complex. The size of each circle corresponds to the number of genomes within respective nodes.
On the MST, the size of each circle corresponds to the number of genomes within respective nodes. Generally, strains within the same subspecies were clustered together without passing through any node from other subspecies. The MST was constructed with 100 bootstrap replications and each bootstrapping tree was consistently observed to have 110 nodes. All edges were supported with high bootstrap replication values at a minimum of 90% (represented in solid lines), except for 11 edges with lower bootstrap values of 50–70% (represented in dotted lines). The lower confidence edges were distant from the edges connecting the different subspecies. Hence, there were no strains with questionable subspecies status. No sign of geographical clustering among the strains was observed. The MST also highlighted the greater diversity among M. massiliense strains compared to M. abscessus and M. bolletii. These observations suggest that the evolution of the M. abscessus complex is not clonal but is driven by the genetic characteristics of each subspecies within the complex.
Genome-Wide Inter- and Intra-subspecies Comparisons
Using MUMi to define genomic distances, the three subspecies showed, as expected, shorter intra-subspecies distances compared to inter-subspecies distances (Supplementary Table S2). The inter-subspecies interquartile range for M. bolletii – M. massiliense, M. bolletii – M. abscessus, and M. abscessus – M. massiliense were 0.2326, 0.2072, and 0.2222, respectively. Hence, the genomic distances between M. massiliense and M. bolletii were longer than the distances between M. massiliense and M. abscessus or M. bolletii and M. abscessus. The taxonomic separation of M. massiliense from M. bolletii has been proposed by several research groups based on the analysis of core sequences with ANI as well as linear and network phylogenies (Cho et al., 2013; Tan et al., 2013, 2015). Our analysis of genomic distances lends further support, from a different perspective, for the separation of the two subspecies.
The intra-subspecies interquartile range of MUMi was 0.0998 in M. bolletii, 0.0898 in M. massiliense and 0.0407 in M. abscessus. From the boxplot constructed (Figure 2), the MUMi of M. bolletii skewed toward the higher values of the box, reflecting a relatively conserved genetic content in this subspecies. In contrast, the MUMi of M. abscessus was skewed toward the lower values in the boxplot, indicating that the majority of the M. abscessus strains have longer genomic distances from each other. The MUMi of M. massiliense, however, showed a symmetrical distribution, indicating the presence of roughly equal numbers of conserved and diverse strains.
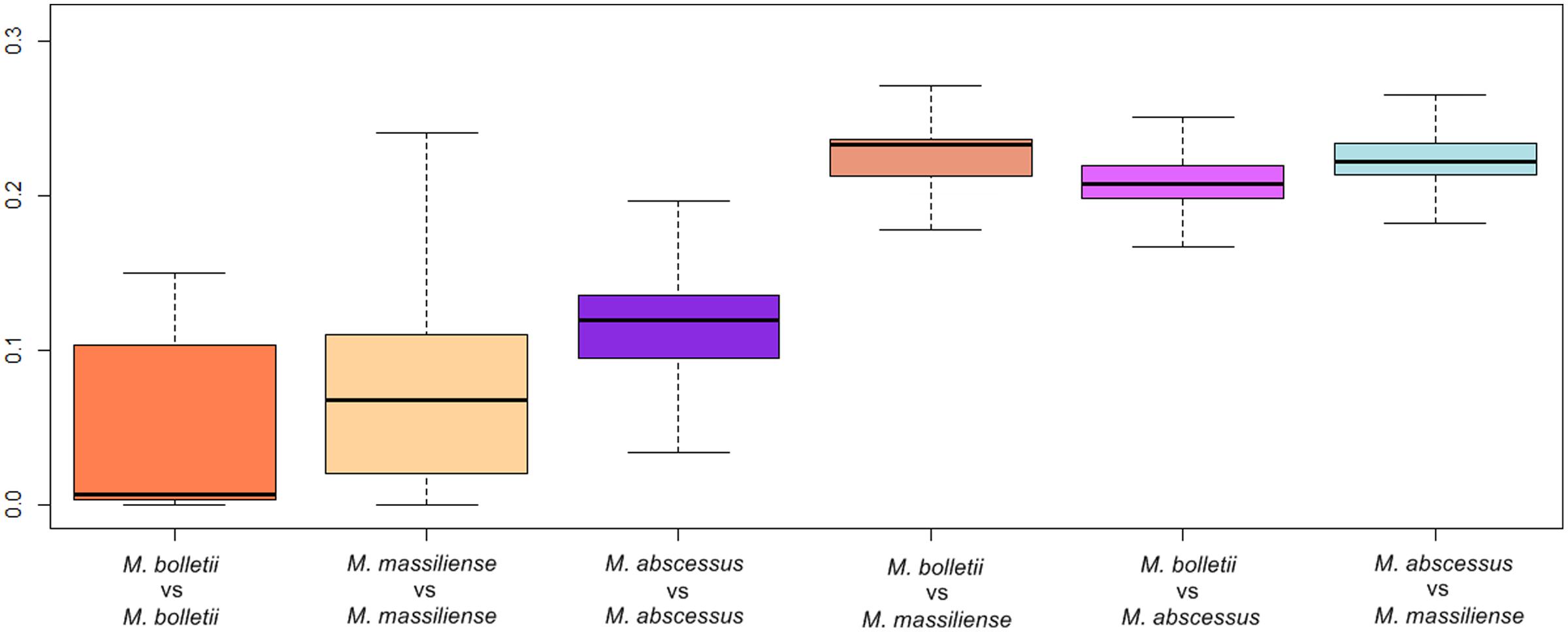
FIGURE 2. Maximum Unique Matches index (MUMi) of M. abscessus subspecies represented in boxplots. The heavy line represents the median MUMi value.
Positive Selection
Our Maximum Likelihood analysis through dN/dS calculation on each of the 1,090 non-paralogous core genes in our dataset supports the presence of genes that have undergone selective pressure. It has been said that the dN/dS ratio is not sensitive enough to detect gene sets that have undergone positive selection among closely related species (Kryazhimskiy and Plotkin, 2008). However, based on the ratios of mutations that were supported in the Likelihood Ratio test (Yang, 1998), we managed to uncover eight genes in M. massiliense, 14 in M. abscessus and 18 in M. bolletii, that could have undergone positive selection (p-value < 0.01), during the course of divergence among the three subspecies.
Based on the results of the three independent recombination methods (NSS, PHI, and Max χ2) and a p-value cutoff at 0.01, only four out of 18 positively selected genes in M. bolletii (Supplementary Table S3) and 2 out of 14 genes in M. abscessus (Supplementary Table S4) were identified as having undergone recombinations. None of the positively selected genes in M. massiliense (Supplementary Table S5) showed evidence of recombination.
Bonferroni correction is commonly applied to investigate the possibility of including genes with type I errors (false-positive) in positive selection analyses (Wong et al., 2004; Yang, 2007). On the other hand, it is also known to cause false negatives when used on a large number of tests (Perneger, 1998). In the current study, after performing Bonferroni correction for multiple testing, only two genes in M. massiliense, three genes in M. abscessus and eight genes in M. bolletii were left with a significant p-value for positive selection. However, the genes with an insignificant p-value after adjustment also failed in the recombination tests, indicating that they are likely to be false negatives in the Bonferroni test, and that the substitutions observed within them can still be the result of selection rather than random mutation or recombination.
Positive Selection in M. massiliense
The positively selected genes identified in this subspecies are an isochorismatase, a beta-lactamase-like protein, a probable deoxyribonuclease tatD, a thymidylate kinase Tmk, 30S ribosomal protein S3 and three hypothetical proteins. Metal ion acquisition is essential for bacterial invasion in the mammalian host and isochorismatase is one of the important enzymes for siderophore-based ferric iron acquisition by cells (Miethke and Marahiel, 2007). The association of this protein with virulence has been reported in Acinetobacter baumannii (Gebhardt et al., 2015). The twin-arginine translocation (tat) system has been shown to play an important role in the virulence of Yersinia pseudotuberculosis (Lavander et al., 2006) and has been associated with E. coli meningitis (Siddiqui et al., 2012). Although the function of the tatD component is not fully known, it has been reported to be the virulence factor in Plasmodium and a potential vaccine candidate for malaria (Chang et al., 2016). We had previously reported thymidylate kinase and 30S ribosomal protein S3 as being suitable for the distinction of M. massiliense from other mycobacteria including other subspecies of the M. abscessus complex (Tan et al., 2015). The positive selection of these genes implies that they may have advantageous biological functions besides being suitable markers for classification.
Positive Selection in M. bolletii
Of the 11 positively selected proteins with known functions, two affect bacterial growth, i.e., phosphoribosylaminoimidazole-succinocarboxamide synthase purC which has been described to affect the growth of M. tuberculosis (Brown et al., 2005) and Fructose-bisphosphate aldolase, frequently reported to be important for growth in pathogens such as M. tuberculosis and multidrug-resistant Staphylococcus aureus (Capodagli et al., 2014; Puckett et al., 2014). The remaining nine proteins are associated with cell membrane entry (mce), transcription regulation (IclR family), reaction to heat shock (Hsp20), metabolic reactions (trans-aconitate methyl transferase, 5-methyltetrahydropteroyltriglutamate–homocysteine methyltransferase), and probable endonuclease activity (as described for protein S16 in the 30S ribosomal subunit, by Oberto et al., 1996).
Despite its reputation as a multiple antibiotic resistant NTM, we could only find one positively selected gene associated with antibiotic resistance in this subspecies, i.e., a transmembrane efflux protein previously reported in mycobacteria (De Rossi et al., 2006). This observation supports the concept that antibiotic resistance incurs a fitness cost in bacteria in the absence of antibiotic selective pressure, and hence, is not likely to be positively selected during bacterial speciation or subspeciation.
Positive Selection in M. abscessus
In M. abscessus, positive selection was observed in the pyridoxine5-phosphate oxidase gene shown to be involved in Vitamin B6 biosynthesis (Dick et al., 2010; Mashalidis et al., 2011), the acyl-CoA thioesterase II gene associated with drug resistance expression in mycobacteria (Danelishvili et al., 2005), transcriptional regulator proteins including the IclRfamily, putative transcriptional regulator, and tetR family transcriptional regulator (Ramos et al., 2005). Also positively selected are four hypothetical proteins, one of which was also identified together with a probable regulator protein in M. bolletii, and the tatD and 30S ribosomal protein S3 that were also found in M. massiliense.
Site-Specific Positive Selection
In each M. abscessus subspecies there is evidence of selective pressure on specific positions in proteins. Manual inspection of sequence alignments and substitutions showed the involvement of 13 amino acids from two proteins in M. abscessus, 24 amino acids from five proteins in M. massiliense and 11 amino acids from four proteins in M. bolletii. The amino acids exerting site-specific positive selection were statistically evaluated with BEB probability, with 50–100% confidence (Table 1).
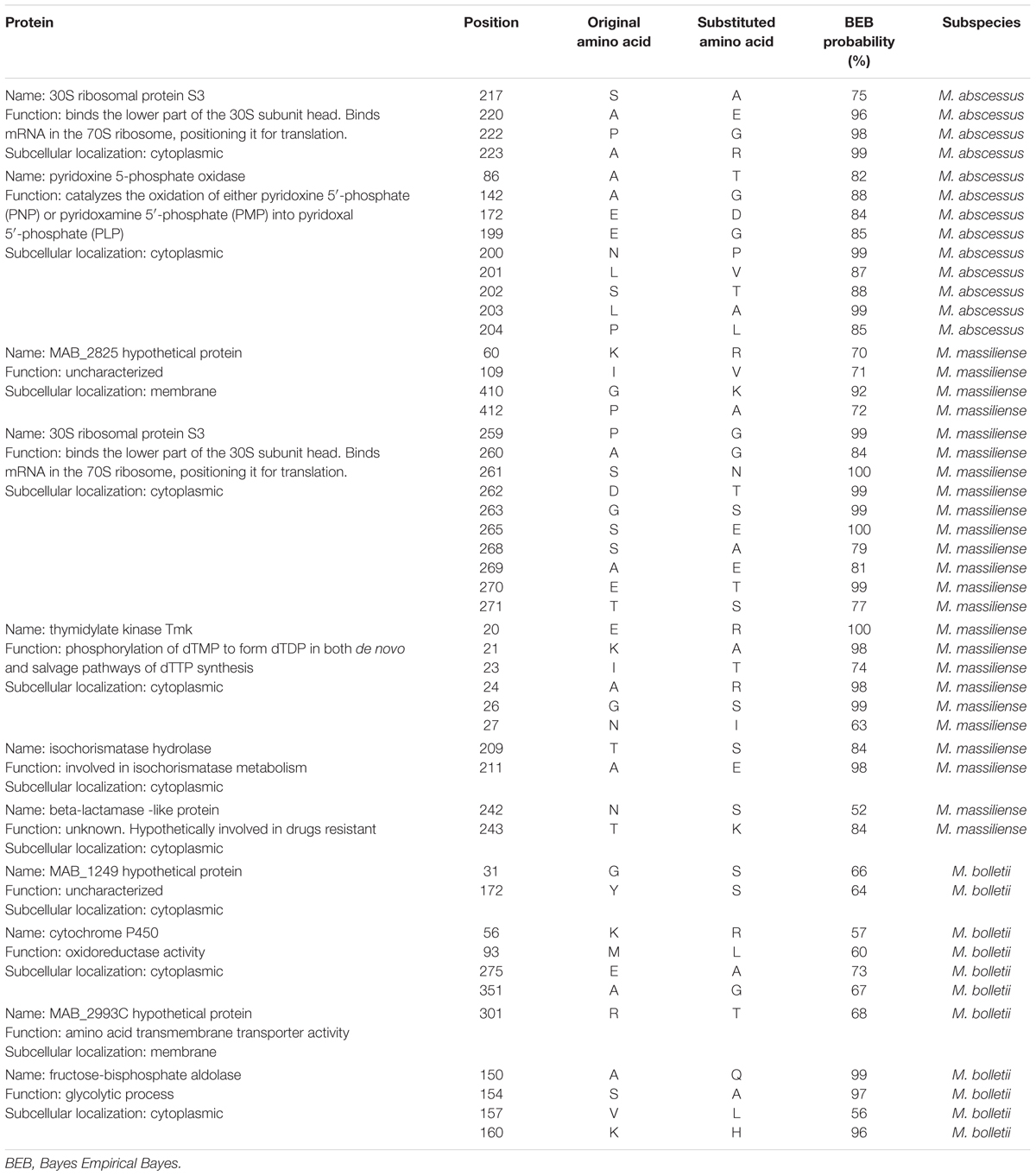
TABLE 1. List of positively selected amino acids specific to each of the subspecies.
Genome-Wide Recombinations
Apart from Darwinian evolution, recombination has been proposed as the alternative event for evolution (Livingstone and Rieseberg, 2004; Barton, 2010). Recent phylogenomics network analysis showed reticulations between and within the subspecies of M. abscessus complex, suggesting the occurrence of recombination events (Tan et al., 2015). In this study, we investigated the genome-wide recombinations that are present in both coding and non-coding regions of the M. abscessus complex, to better understand the genomic properties that could have facilitated the emergence of M. abscessus complex as a successful opportunistic pathogen. Overall, 1,468 recombination events (excluding the putative ancestral nodes) were detected from the 1,407 LCBs generated by progressiveMauve. The lengths of recombinations ranged from 2 to 34,082 bp. The ratio of rates at which polymorphisms were introduced by recombination and mutation was R/theta = 0.14, whereas the ratio of recombination and mutation (r/m) was 1.99. Hence, recombination caused two times more polymorphism than mutation. From the illustration in Figure 3, extensive recombination was observed in the three putative ancestral nodes leading to the three subspecies. Since there are long evolutionary distances separating the three putative ancestral nodes, it is not known whether the accumulation of the recombinations occurred after the subspeciation event or was responsible for the independent emergence of the three subspecies.
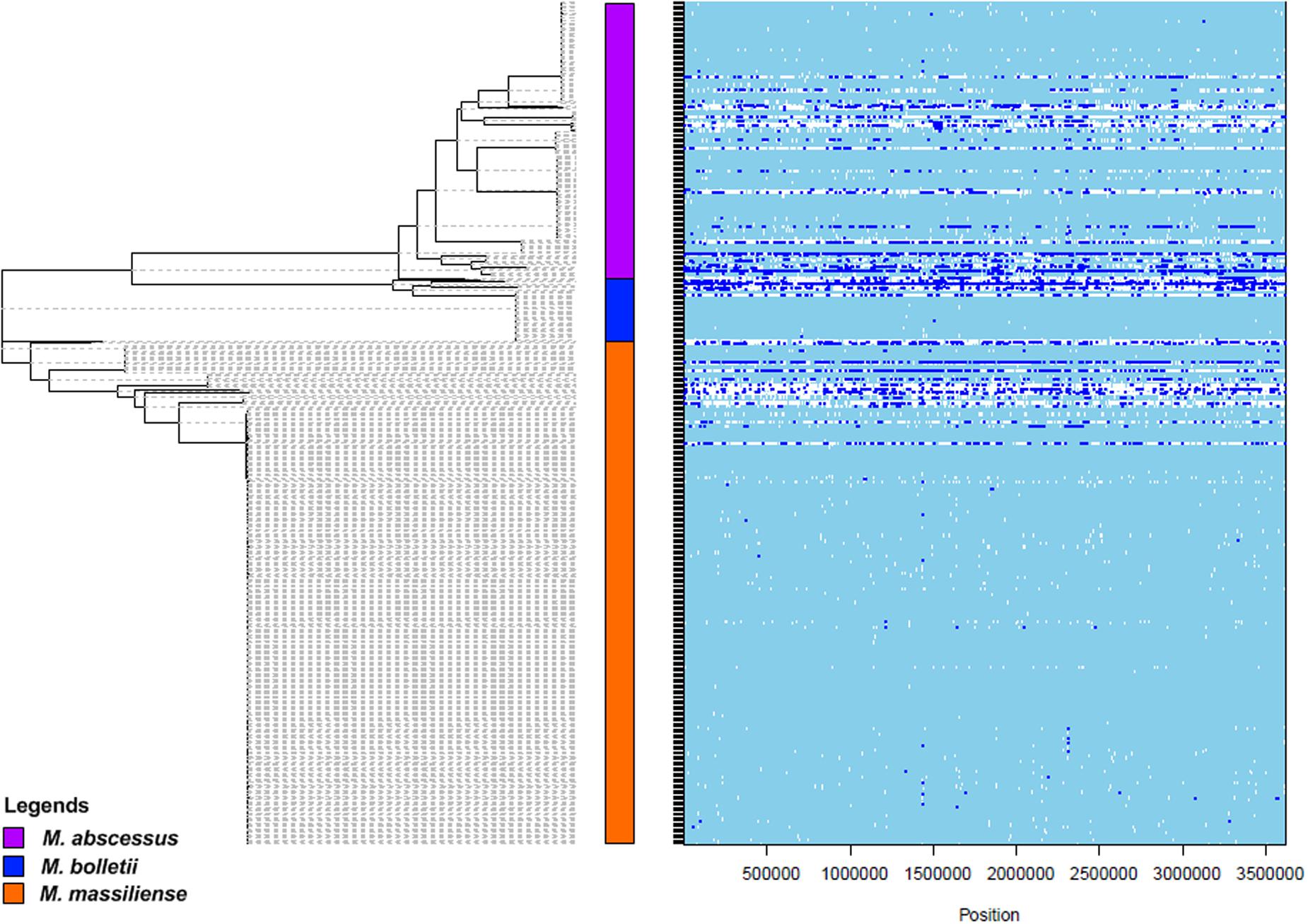
FIGURE 3. Genome wide recombination plot (obtained with the ClonalFrameML) and supersequence SNP-based phylogenetic tree (generated with PhyML) for the M. abscessus complex. The plot on the right shows the presence of recombination (in blue) and mutation (in white) throughout the genomes of the M. abscessus complex. In the phylogenetic tree, purple = M. abscessus, red = M. bolletii, blue = M. massiliense.
Besides computing the whole population genome-wide recombination structures, subspecies- specific recombinations were also investigated by r/m estimations. The results suggest a higher recombination impact among M. massiliense (r/m = 2.17), with almost equal effect of substitutions and mutations in M. abscessus (r/m = 1.41) and M. bolletii (r/m = 0.95). We predicted 675 recombination events in M. abscessus, 387 in M. bolletii and 786 in M. massiliense. The analysis on recombination segments identified the origins of approximately 23% of total recombination segments in M. abscessus (intrasubspecies origin – 103; intersubspecies origin – 54), 16% in M. bolletii (intrasubspecies origin – 16; intersubspecies origin – 44) and 17% in M. massiliense (intrasubspecies origin – 57; intersubspecies origin – 80). There were 33 segments in M. abscessus, 4 in M. bolletii and 11 in M. massiliense that could not be confirmed to be from an intra- or inter- subspecies origin. The segments that could not be found within the M. abscessus complex might have been acquired from foreign origins. However, we did not find insertion sequences (ISs) that could have contributed to the recombination events found in the M. abscessus complex. Neither did we find any CRISPRs that could have prevented the entry of foreign genetic material at the recombination sites. It is possible that novel genetic mechanisms are involved in these recombination events.
Discussion
Speciation genomics has contributed immensely to the understanding of bacterial evolution and diversity (Seehausen et al., 2014). It has enabled the identification of genetic features that are impossible to define with conventional gene-based inference analysis (Zhou et al., 2013; Librado et al., 2014), particularly in bacterial genera with very closely related members such as Salmonella variants (Desai et al., 2013) and drug-resistant Mycobacterium tuberculosis strains (Farhat et al., 2013). In this study, we focused on members of the M. abscessus complex which differ in their drug susceptibility and invasiveness, with M. massiliense reported to be the least associated with antibiotic resistance but the most frequently isolated from clinical material, and M. bolletii being the most frequently associated with multiple antibiotic resistance but the least frequently isolated from clinical specimens (Koh et al., 2011; Aitken et al., 2012; Nessar et al., 2012; Harris and Kenna, 2014; Tettelin et al., 2014). We tried to look for evidence of evolutionary trends to explain these differences.
The MST showed the strains to be clustered according to the subspecies they belong to rather than by their countries of origin. Within the M. massiliense cluster, there is a wider spread of genotypes than in the other two subspecies. This feature indicates higher allelic variability among M. massiliense genomes. In contrast, M. bolletii showed the least allelic variation. Supported by our data on MUMi evaluation of both core and accessory genomes, the low median intra-subspecies index indicated that M. bolletii is conserved compared to M. massiliense and M. abscessus. This could be reflected in its low prevalence in infections. Using the same data, the inter-species distance appears longer between M massiliense and M. bolletii than between either of them and M. abscessus. This provides another reason to consider M. massiliense and M. bolletii as distinct subspecies.
The impact of the accessory genome on the evolution of the M. abscessus complex has been discussed by other researchers (Choo et al., 2014; Sassi et al., 2014). In this analysis, we focused on the evolutionary pressure within the core genomes of the complex. We found indications of positive selection in a small proportion of genes in all three subspecies. These genes encode enzymes important for the regulation of bacterial growth, metabolism and replication, as well as proteins for virulence, drug resistance and reaction to external stimuli. There is no obvious correlation between subspecies and the function of genes positively selected. The selection of genes for pathogenicity and drug resistance in all three subspecies reflects the evolutionary adaptation of these environmental bacteria to their role as human and animal pathogens.
From the ratio of synonymous and non-synonymous mutations and the evaluation of amino-acid changes based on BEB, we speculated site-specific positive selection resulting in amino acid substitutions. The biological significance of these substitutions is not known, but two proteins involved, the 30S ribosomal protein S3 and thymidylate kinase, were previously found to contain specific signatures for the classification of M. massiliense (Tan et al., 2015). We would like to propose that the positively selected proteins that we identified in a systematic way, using sequence data from 243 genomes, would be valuable for the classification of the M. abscessus complex.
Our overall results suggest that the evolution of the M. abscessus complex is driven more by recombination than positive selections. Interestingly, our data revealed three independent recombination landscapes in the three subspecies. In a previous study (Sapriel et al., 2016), the impact of recombination in M. abscessus was the highest among the three subspecies. In the current study, M. massiliense appeared to be the most highly affected by recombination, followed by M. abscessus and M. bolletii, in descending order. The difference could be due to the use of different datasets. Sapriel et al. (2016) studied recombination in the single-targeted gene sets whereas we studied genome-wide recombination landscapes. The higher impact of recombination in M. massiliense compared to that in M. abscessus is also supported by our previous network phylogenomics analysis (Tan et al., 2015). Nevertheless, the consensus from both Sapriel et al. (2016) and our study is that recombination serves as the driver for the M. abscessus complex evolution for pathoadaptation. Lefébure and Stanhope (2007) suggested that recombination contributes to faster adaptation when positive selection is too slow. A high level of genetic similarity has been reported among clinical M. massiliense strains isolated from different countries (Davidson et al., 2013; Tettelin et al., 2014) and this similarity has been used to postulate person-to-person transmission among cystic fibrosis patients (Bryant et al., 2013). On the other hand, the genetic similarity among clinical M. massiliense strains could also be the result of horizontal transfer and recombination involving common genetic determinants for infectivity. This could illustrate the advantage conferred by a high recombination rate on the pathoadaptation of M. massiliense.
Conclusion
Our findings suggest that the diversity of the M. abscessus complex has been shaped by recombination more than Darwinian selection, and that each subspecies of the complex appears to have evolved independently. The proteins we identified to have undergone positive selection and recombination are expected to facilitate future studies on the functional significance of proteins in the M. abscessus complex.
Author Contributions
Planned and conceived the experiments: JLT and YFN. Performed the experiments: JLT and YFN. Analyzed the experiments: JLT, KPN, CSO, and YFN. Drafted the manuscript and contributed to the data interpretation: JLT, KPN, CSO, and YFN. All authors read, critically revised and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Multimedia University, Malaysia (grant number MMU/RMC/MiniFund/FIST/2016-2017/02), University of Malaya (grant number UM.C/625/1/HIR/MOHE/CHAN/14/4) and Universiti Tunku Abdul Rahman (grant number IPSR/RMC/UTARRF/2015-C2/NO3).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02042/full#supplementary-material
TABLE S1 | List of strains used in the study.
TABLE S2 | Pairwise Maximal Unique Matches Index (MUMi) of the M. abscessus complex.
TABLE S3 | List of positively selected genes in M. bolletii.
TABLE S4 | List of positively selected genes in M. abscessus.
TABLE S5 | List of positively selected genes in M. massiliense.
References
Aitken, M. L., Limaye, A., Pottinger, P., Whimbey, E., Goss, C. H., Tonelli, M. R., et al. (2012). Respiratory outbreak of Mycobacterium abscessus subspecies massiliense in a lung transplant and cystic fibrosis center. Am. J. Respir. Crit. Care Med. 185, 231–232. doi: 10.1164/ajrccm.185.2.231
Barton, N. H. (2010). Mutation and the evolution of recombination. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365, 1281–1294. doi: 10.1098/rstb.2009.0320
Besemer, J., Lomsadze, A., and Borodovsky, M. (2001). GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29, 2607–2618. doi: 10.1093/nar/29.12.2607
Brown, C. J., Garner, E. C., Dunker, K. A., and Joyce, P. (2001). The power to detect recombination using the coalescent. Mol. Biol. Evol. 18, 1421–1424. doi: 10.1093/oxfordjournals.molbev.a003927
Brown, N., Jacobs, M., Parida, S. K., Botha, T., Santos, A., Fick, L., et al. (2005). Reduced local growth and spread but preserved pathogenicity of a DeltapurC Mycobacterium tuberculosis auxotrophic mutant in gamma interferon receptor-deficient mice after aerosol infection. Infect. Immun. 73, 666–670. doi: 10.1128/IAI.73.1.666-670.2005
Bruen, T. C., Philippe, H., and Bryant, D. (2006). A simple and robust statistical test for detecting the presence of recombination. Genetics 172, 2665–2681. doi: 10.1534/genetics.105.048975
Bryant, J. M., Grogono, D. M., Greaves, D., Foweraker, J., Roddick, I., Inns, T., et al. (2013). Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet 381, 1551–1560. doi: 10.1016/S0140-6736(13)60632-7
Cagliani, R., and Sironi, M. (2013). Pathogen-driven selection in the human genome. Int. J. Evol. Biol. 2013:204240. doi: 10.1155/2013/204240
Capodagli, G. C., Lee, S. A., Boehm, K. J., Brady, K. M., and Pegan, S. D. (2014). Structural and functional characterization of methicillin-resistant Staphylococcus aureus’s class IIb fructose 1,6-bisphosphate aldolase. Biochemistry 53, 7604–7614. doi: 10.1021/bi501141t
Casali, N., Nikolayevskyy, V., Balabanova, Y., Ignatyeva, O., Kontsevaya, I., Harris, S. R., et al. (2012). Microevolution of extensively drug-resistant tuberculosis in Russia. Genome Res. 22, 735–745. doi: 10.1101/gr.128678.111
Chang, Z., Jiang, N., Zhang, Y., Lu, H., Yin, J., Wahlgren, M., et al. (2016). The TatD-like DNase of Plasmodium is a virulence factor and a potential malaria vaccine candidate. Nat. Commun. 7:11537. doi: 10.1038/ncomms11537
Cho, Y. J., Yi, H., Chun, J., Cho, S. N., Daley, C. L., Koh, W. J., et al. (2013). The genome sequence of ‘Mycobacterium massiliense’ strain CIP 108297 suggests the independent taxonomic status of the Mycobacterium abscessus complex at the subspecies level. PLOS ONE 8:e81560. doi: 10.1371/journal.pone.0081560
Choo, S. W., Wee, W. Y., Ngeow, Y. F., Mitchell, W., Tan, J. L., Wong, G. J., et al. (2014). Genomic reconnaissance of clinical isolates of emerging human pathogen Mycobacterium abscessus reveals high evolutionary potential. Sci. Rep. 4:4061. doi: 10.1038/srep04061
Danelishvili, L., Wu, M., Young, L. S., and Bermudez, L. E. (2005). Genomic approach to identifying the putative target of and mechanisms of resistance to mefloquine in mycobacteria. Antimicrob. Agents Chemother. 49, 3707–3714. doi: 10.1128/AAC.49.9.3707-3714.2005
Darling, A. C., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLOS ONE 5:e11147. doi: 10.1371/journal.pone.0011147
Davidson, R. M., Hasan, N. A., de Moura, V. C., Duarte, R. S., Jackson, M., and Strong, M. (2013). Phylogenomics of Brazilian epidemic isolates of Mycobacterium abscessus subsp. bolletii reveals relationships of global outbreak strains. Infect. Genet. Evol. 20, 292–297. doi: 10.1016/j.meegid.2013.09.012
De Rossi, E., Aínsa, J. A., and Riccardi, G. (2006). Role of mycobacterial efflux transporters in drug resistance: an unresolved question. FEMS Microbiol. Rev. 30, 36–52. doi: 10.1111/j.1574-6976.2005.00002.x
Deloger, M., El Karoui, M., and Petit, M. A. (2009). A genomic distance based on MUM indicates discontinuity between most bacterial species and genera. J. Bacteriol. 191, 91–99. doi: 10.1128/JB.01202-08
Desai, P. T., Porwollik, S., Long, F., Cheng, P., Wollam, A., Bhonagiri-Palsikar, V., et al. (2013). Evolutionary genomics of Salmonella enterica subspecies. mBio 4:e00579-12. doi: 10.1128/mBio.00579-12
Dick, T., Manjunatha, U., Kappes, B., and Gengenbacher, M. (2010). Vitamin B6 biosynthesis is essential for survival and virulence of Mycobacterium tuberculosis. Mol. Microbiol. 78, 980–988. doi: 10.1111/j.1365-2958.2010.07381.x
Didelot, X., and Wilson, D. J. (2015). ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLOS Comput. Biol. 11:e1004041. doi: 10.1371/journal.pcbi.1004041
Donati, C., Hiller, N. L., Tettelin, H., Muzzi, A., Croucher, N. J., Angiuoli, S. V., et al. (2010). Structure and dynamics of the pan-genome of Streptococcus pneumoniae and closely related species. Genome Biol. 11:R107. doi: 10.1186/gb-2010-11-10-r107
Ehrlich, G. D., Hu, F. Z., Shen, K., Stoodley, P., and Post, J. C. (2005). Bacterial plurality as a general mechanism driving persistence in chronic infections. Clin. Orthop. Relat. Res. 437, 20–24. doi: 10.1097/00003086-200508000-00005
Farhat, M. R., Shapiro, B. J., Kieser, K. J., Sultana, R., Jacobson, K. R., Victor, T. C., et al. (2013). Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nat. Genet. 45, 1183–1189. doi: 10.1038/ng.2747
Gebhardt, M. J., Gallagher, L. A., Jacobson, R. K., Usacheva, E. A., Peterson, L. R., Zurawski, D. V., et al. (2015). Joint transcriptional control of virulence and resistance to antibiotic and environmental stress in Acinetobacter baumannii. mBio 6:e01660-15. doi: 10.1128/mBio.01660-15
Griffith, D. E. (2014). Mycobacterium abscessus subsp abscessus lung disease: ‘trouble ahead, trouble behindldots...’. F1000Prime Rep. 6:107. doi: 10.12703/P6-107
Harris, K. A., and Kenna, D. T. (2014). Mycobacterium abscessus infection in cystic fibrosis: molecular typing and clinical outcomes. J. Med. Microbiol. 63, 1241–1246. doi: 10.1099/jmm.0.077164-0
Jakobsen, I. B., and Easteal, S. (1996). A program for calculating and displaying compatibility matrices as an aid in determining reticulate evolution in molecular sequences. Comput. Appl. Biosci. 12, 291–295. doi: 10.1093/bioinformatics/12.4.291
Jönsson, B. E., Gilljam, M., Lindblad, A., Ridell, M., Wold, A. E., and Welinder-Olsson, C. (2007). Molecular epidemiology of Mycobacterium abscessus, with focus on cystic fibrosis. J. Clin. Microbiol. 45, 1497–1504. doi: 10.1128/JCM.02592-06
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Koh, W. J., Jeon, K., Lee, N. Y., Kim, B. J., Kook, Y. H., Lee, S. H., et al. (2011). Clinical significance of differentiation of Mycobacterium massiliense from Mycobacterium abscessus. Am. J. Respir. Crit. Care Med. 183, 405–410. doi: 10.1164/rccm.201003-0395OC
Koh, W. J., Jeon, K., and Shin, S. J. (2013). Successful treatment of Mycobacterium massiliense lung disease with oral antibiotics only. Antimicrob. Agents Chemother. 57, 1098–1100. doi: 10.1128/AAC.02016-12
Kryazhimskiy, S., and Plotkin, J. B. (2008). The population genetics of dN/dS. PLOS Genet. 4:e1000304. doi: 10.1371/journal.pgen.1000304
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Lavander, M., Ericsson, S. K., Bröms, J. E., and Forsberg, A. (2006). The twin arginine translocation system is essential for virulence of Yersinia pseudotuberculosis. Infect. Immun. 74, 1768–1776. doi: 10.1128/IAI.74.3.1768-1776.2006
Leao, S. C., Tortoli, E., Euzeby, J. P., and Garcia, M. J. (2011). Proposal that Mycobacterium massiliense and Mycobacterium bolletii be united and reclassified as Mycobacterium abscessus subsp. bolletii comb. nov., designation of Mycobacterium abscessus subsp. abscessus subsp. nov. and emended description of Mycobacterium abscessus. Int. J. Syst. Evol. Microbiol. 61, 2311–2313. doi: 10.1099/ijs.0.023770-0
Lechner, M., Findeiss, S., Steiner, L., Marz, M., Stadler, P. F., and Prohaska, S. J. (2011). Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics 12:124. doi: 10.1186/1471-2105-12-124
Lefébure, T., and Stanhope, M. J. (2007). Evolution of the core and pan-genome of Streptococcus: positive selection, recombination, and genome composition. Genome Biol. 8:R71. doi: 10.1186/gb-2007-8-5-r71
Lefébure, T., and Stanhope, M. J. (2009). Pervasive, genome-wide positive selection leading to functional divergence in the bacterial genus Campylobacter. Genome Res. 19, 1224–1232. doi: 10.1101/gr.089250.108
Levy, I., Grisaru-Soen, G., Lerner-Geva, L., Kerem, E., Blau, H., Bentur, L., et al. (2008). Multicenter cross-sectional study of nontuberculous mycobacterial infections among cystic fibrosis patients. Israel. Emerg. Infect. Dis. 14, 378–384. doi: 10.3201/eid1403.061405
Librado, P., Vieira, F. G., Sánchez-Gracia, A., Kolokotronis, S. O., and Rozas, J. (2014). Mycobacterial phylogenomics: an enhanced method for gene turnover analysis reveals uneven levels of gene gain and loss among species and gene families. Genome Biol. Evol. 6, 1454–1465. doi: 10.1093/gbe/evu117
Livingstone, K., and Rieseberg, L. (2004). Chromosomal evolution and speciation: a recombination-based approach. New Phytol. 161, 107–112. doi: 10.1046/j.1469-8137.2003.00942.x
Mashalidis, E. H., Mukherjee, T., Sledź, P., Matak-Vinković, D., Boshoff, H., Abell, C., et al. (2011). Rv2607 from Mycobacterium tuberculosis is a pyridoxine 5′-phosphate oxidase with unusual substrate specificity. PLOS ONE 6:e27643. doi: 10.1371/journal.pone.0027643
Miethke, M., and Marahiel, M. A. (2007). Siderophore-based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 71, 413–451. doi: 10.1128/MMBR.00012-07
Mitra, S., Tapadar, S. R., Banerjee, D., Bhattacharjee, S., Dey, S., and Kundu, S. (2012). Pulmonary disease due to Mycobacterium massiliense. Indian J. Chest Dis. Allied Sci. 54, 53–57.
Nessar, R., Cambau, E., Reyrat, J. M., Murray, A., and Gicquel, B. (2012). Mycobacterium abscessus: a new antibiotic nightmare. J. Antimicrob. Chemother. 67, 810–818. doi: 10.1093/jac/dkr578
Oberto, J., Bonnefoy, E., Mouray, E., Pellegrini, O., Wikström, P. M., and Rouvière-Yaniv, J. (1996). The Escherichia coli ribosomal protein S16 is an endonuclease. Mol. Microbiol. 19, 1319–1330. doi: 10.1111/j.1365-2958.1996.tb02476.x
Olivier, K. N., Weber, D. J., Wallace, RJ Jr, Faiz, A. R., Lee, J. H., Zhang, Y., et al. (2003). Nontuberculous mycobacteria. I: multicenter prevalence study in cystic fibrosis. Am. J. Respir. Crit. Care Med. 167, 828–834. doi: 10.1164/rccm.200207-678OC
Peng, Z., Liu, S., Meng, X., Liang, W., Xu, Z., Tang, B., et al. (2017). Genome characterization of a novel binary toxin-positive strain of Clostridium difficile and comparison with the epidemic 027 and 078 strains. Gut Pathog. 9, 42. doi: 10.1186/s13099-017-0191-z
Perneger, T. V. (1998). What’s wrong with Bonferroni adjustments. BMJ 316, 1236–1238. doi: 10.1136/bmj.316.7139.1236
Petersen, L., Bollback, J. P., Dimmic, M., Hubisz, M., and Nielsen, R. (2007). Genes under positive selection in Escherichia coli. Genome Res. 17, 1336–1343. doi: 10.1101/gr.6254707
Posada, D., Crandall, K. A., and Holmes, E. C. (2002). Recombination in evolutionary genomics. Annu. Rev. Genet. 36, 75–97. doi: 10.1146/annurev.genet.36.040202.111115
Puckett, S., Trujillo, C., Eoh, H., Marrero, J., Spencer, J., Jackson, M., et al. (2014). Inactivation of fructose-1,6-bisphosphate aldolase prevents optimal co-catabolism of glycolytic and gluconeogenic carbon substrates in Mycobacterium tuberculosis. PLOS Pathog. 10:e1004144. doi: 10.1371/journal.ppat.1004144
Ramos, J. L., Martínez-Bueno, M., Molina-Henares, A. J., Terán, W., Watanabe, K., Zhang, X., et al. (2005). The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 69, 326–356. doi: 10.1128/MMBR.69.2.326-356.2005
Redfield, R. J., Findlay, W. A., Bossé, J., Kroll, J. S., Cameron, A. D., and Nash, J. H. (2006). Evolution of competence and DNA uptake specificity in the Pasteurellaceae. BMC Evol. Biol. 6:82. doi: 10.1186/1471-2148-6-82
Roux, A. L., Catherinot, E., Soismier, N., Heym, B., Bellis, G., Lemonnier, L., et al. (2015). Comparing Mycobacterium massiliense and Mycobacterium abscessus lung infections in cystic fibrosis patients. J. Cyst. Fibros. 14, 63–69. doi: 10.1016/j.jcf.2014.07.004
Sapriel, G., Konjek, J., Orgeur, M., Bouri, L., Frézal, L., Roux, A. L., et al. (2016). Genome-wide mosaicism within Mycobacterium abscessus: evolutionary and epidemiological implications. BMC Genomics 17:118. doi: 10.1186/s12864-016-2448-1
Sassi, M., Gouret, P., Chabrol, O., Pontarotti, P., and Drancourt, M. (2014). Mycobacteriophage-drived diversification of Mycobacterium abscessus. Biol. Direct 9:19. doi: 10.1186/1745-6150-9-19
Seehausen, O., Butlin, R. K., Keller, I., Wagner, C. E., Boughman, J. W., Hohenlohe, P. A., et al. (2014). Genomics and the origin of species. Nat. Rev. Genet. 15, 176–192. doi: 10.1038/nrg3644
Siddiqui, R., Beattie, R., and Khan, N. A. (2012). The role of the twin-arginine translocation pathway in Escherichia coli K1 pathogenicity in the African migratory locust, Locusta migratoria. FEMS Immunol. Med. Microbiol. 64, 162–168. doi: 10.1111/j.1574-695X.2011.00870.x
Smith, J. M. (1992). Analyzing the mosaic structure of genes. J. Mol. Evol. 34, 126–129. doi: 10.1007/BF00182389
Stout, J. E., and Floto, R. A. (2012). Treatment of Mycobacterium abscessus: all macrolides are equal, but perhaps some are more equal than others. Am. J. Respir. Crit. Care Med. 186, 822–823. doi: 10.1164/rccm.201208-1500ED
Suyama, M., Torrents, D., and Bork, P. (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612. doi: 10.1093/nar/gkl315
Tan, J. L., Khang, T. F., Ngeow, Y. F., and Choo, S. W. (2013). A phylogenomic approach to bacterial subspecies classification: proof of concept in Mycobacterium abscessus. BMC Genomics 14:879. doi: 10.1186/1471-2164-14-879
Tan, J. L., Ngeow, Y. F., and Choo, S. W. (2015). Support from phylogenomic networks and subspecies signatures for separation of Mycobacterium massiliense from Mycobacterium bolletii. J. Clin. Microbiol. 53, 3042–3046. doi: 10.1128/JCM.00541-15
Tettelin, H., Davidson, R. M., Agrawal, S., Aitken, M. L., Shallom, S., Hasan, N. A., et al. (2014). High-level relatedness among Mycobacterium abscessus subsp. massiliense strains from widely separated outbreaks. Emerg. Infect. Dis. 20, 364–371. doi: 10.3201/eid2003.131106
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Wong, W. S., Yang, Z., Goldman, N., and Nielsen, R. (2004). Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics 168, 1041–1051. doi: 10.1534/genetics.104.031153
Xu, Z., Chen, X., Li, L., Li, T., Wang, S., Chen, H., et al. (2010). Comparative genomic characterization of Actinobacillus pleuropneumoniae. J. Bacteriol. 192, 5625–5636. doi: 10.1128/JB.00535-10
Yang, Z. (1998). Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 15, 568–573. doi: 10.1093/oxfordjournals.molbev.a025957
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, Z., Wong, W. S., and Nielsen, R. (2005). Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22, 1107–1118. doi: 10.1093/molbev/msi097
Yu, C. S., Chen, Y. C., Lu, C. H., and Hwang, J. K. (2006). Prediction of protein subcellular localization. Proteins 64, 643–651. doi: 10.1002/prot.21018
Zhang, J., Nielsen, R., and Yang, Z. (2005). Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 22, 2472–2479. doi: 10.1093/molbev/msi237
Keywords: non-tuberculous mycobacterium, Mycobacterium abscessus subspecies, microevolution, recombination, positive selection
Citation: Tan JL, Ng KP, Ong CS and Ngeow YF (2017) Genomic Comparisons Reveal Microevolutionary Differences in Mycobacterium abscessus Subspecies. Front. Microbiol. 8:2042. doi: 10.3389/fmicb.2017.02042
Received: 07 July 2017; Accepted: 06 October 2017;
Published: 23 October 2017.
Edited by:
John R. Battista, Louisiana State University, United StatesReviewed by:
Zhuofei Xu, Huazhong Agricultural University, ChinaBaojun Wu, Clark University, United States
Copyright © 2017 Tan, Ng, Ong and Ngeow. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yun F. Ngeow, yunngeow@yahoo.com Joon L. Tan, tjl2309@hotmail.com