 Yanfen Cheng
Yanfen Cheng Ying Wang1
Ying Wang1 Weiyun Zhu
Weiyun Zhu- 1Jiangsu Key Laboratory of Gastrointestinal Nutrition and Animal Health, Laboratory of Gastrointestinal Microbiology, Nanjing Agricultural University, National Center for International Research on Animal Gut Nutrition, Nanjing, China
- 2Departments of Microbiology and Statistics, Oregon State University, Corvallis, OR, United States
The aim of this study was to improve the utilization of rice straw as forage in ruminants by investigating the degradation pattern of rice straw in the dairy cow rumen. Ground up rice straw was incubated in situ in the rumens of three Holstein cows over a period of 72 h. The rumen fluid at 0 h and the rice straw at 0.5, 1, 2, 4, 6, 12, 24, 48, and 72 h were collected for analysis of the bacterial community and the degradation of the rice straw. The bacterial community and the carbohydrate-active enzymes in the rumen fluid were analyzed by metagenomics. The diversity of bacteria loosely and tightly attached to the rice straw was investigated by scanning electron microscopy and Miseq sequencing of 16S rRNA genes. The predominant genus in the rumen fluid was Prevotella, followed by Bacteroides, Butyrivibrio, unclassified Desulfobulbaceae, Desulfovibrio, and unclassified Sphingobacteriaceae. The main enzymes were members of the glycosyl hydrolase family, divided into four categories (cellulases, hemicellulases, debranching enzymes, and oligosaccharide-degrading enzymes), with oligosaccharide-degrading enzymes being the most abundant. No significant degradation of rice straw was observed between 0.5 and 6 h, whereas the rice straw was rapidly degraded between 6 and 24 h. The degradation then gradually slowed between 24 and 72 h. A high proportion of unclassified bacteria were attached to the rice straw and that Prevotella, Ruminococcus, and Butyrivibrio were the predominant classified genera in the loosely and tightly attached fractions. The composition of the loosely attached bacterial community remained consistent throughout the incubation, whereas a significant shift in composition was observed in the tightly attached bacterial community after 6 h of incubation. This shift resulted in a significant reduction in numbers of Bacteroidetes and a significant increase in numbers of Firmicutes. In conclusion, the degradation pattern of rice straw in the dairy cow rumen indicates a strong contribution by tightly attached bacteria, especially after 6 h incubation, but most of these bacteria were not taxonomically characterized. Thus, these bacteria should be further identified and subjected to functional analysis to improve the utilization of crop residues in ruminants.
Introduction
The global demand for animal-source foods is estimated to undergo a dramatic increase by 2030, due to the predicted growth in the human population (FAO, 2011). One challenge facing increased animal production in China is the limited availability of high-quality forage such as alfalfa (for example, China needed to import approximately 0.9 million tons of alfalfa in 20141). By contrast, China generates large annual amounts of cellulosic crop residues, such as rice straw and wheat straw, with 0.7 billion tons of crop residues produced in 2009 alone (Cai et al., 2011). Elimination of these crop residues by combustion pollutes the air and results in smog, as well as being a waste of energy. Therefore, the use of these residues as a livestock feed for ruminants could represent a beneficial and more environmentally friendly disposal route for this cellulosic material.
Weimer et al. (2009) described the rumen as the most elegant and highly evolved cellulose-digesting system in nature. The microbiota in the rumen is responsible for the degradation of cellulose by colonization of ingested roughage and excretion of fiber-degrading enzymes. The colonization of ingested roughage has been investigated in several publications. Edwards et al. (2007), who investigated the colonization of perennial ryegrass by rumen bacteria, found colonization by a diverse and consistent population of ruminal bacteria, whose numbers increased rapidly within 5 min of roughage incubation. Huws et al. (2013, 2016), who examined degradation of perennial ryegrass in the cow rumen by denaturing gradient gel electrophoresis and 16S rRNA gene sequencing, revealed the development of a distinct primary bacterial community (Butyrivibrio, Prevotella, Selenomonas, and Fibrobacter) within 2 h of incubation and a secondary bacterial community (Butyrivibrio, Fibrobacter, Pseudobutyrivibrio, and Selenomonas) after 4 h of incubation. Piao et al. (2014) reported two major shifts in bacterial community composition during in situ incubation of switchgrass: The first shift was the colonization of Bacteroidia and Clostridia within 30 min and the second was an enrichment of Spirochaete and Fibrobacteria classes after 16 h of incubation. Liu et al. (2016) demonstrated a significant shift after 6 h incubation of both rice straw and alfalfa and significant difference in the bacterial community between the two forages. Koike et al. (2014), who investigated the colonization patterns of bacteria between untreated rice straw and sodium hydroxide-treated rice straw, found that the population sizes of fibrolytic species were significantly higher in sodium hydroxide-treated rice straw. Wang et al. (2016), who investigated the shifts in rumen fermentation and microbial community fed with different types of carbohydrates, found that rice straw increased acetate and hydrogen production and decreased the ratio of acetate propionate. For the excretion of fiber-degrading enzymes, Patel et al. (2014) reported that the carbohydrate-active enzymes (CAZymes) in the buffalo rumen included carbohydrate binding modules (CBM), carbohydrate esterases (CE), glycosyl hydrolases (GH), glycosyl transferases (GT), and polysaccharide lyases (PL), with the dominant enzymes being those of the GH family, including cellulases, hemicellulases, oligosaccharide-degrading enzyme, and debranching enzymes. Brulc et al. (2009) demonstrated that GH2 and GH3 were predominant in the rumen of bovine. The authors also suggested that the initial colonization of fiber was by microbes possessing enzymes, which attacked the easily available side chains of complex plant polysaccharides. Hess et al. (2011) investigated the biomass-degrading genes and genomes in the rumen of cow by metagenomic analysis and demonstrated most of identified sequences were belonging to endoglucanase, glucosidase, and cellobiohydrolase.
The aim of the present study was to investigate the degradation pattern of rice straw by ruminal bacteria and to identify the bacteria that colonize rice straw during its digestion in the cow rumen. A better understanding of ruminal digestion of rice crop residues will have dual benefits for China, as diversion of these residues to roughage feed for ruminants will improve animal husbandry while reducing air pollution from combustion.
Materials and Methods
Sample Collection
The rice straw was dried at 65°C, ground into ∼0.5 mm pieces, and weighed into individual nylon bags (2.5 g/bag). A total of 54 bags were prepared and placed into the rumens of three cannulated Holstein cows (18 bags per cow). The cow diet consisted of 15% corn, 2% wheat bran, 10% soybean meal, 0.7% calcium carbonate, 0.8% calcium orthophosphate, 0.5% sodium chloride, 1% premix, 20% corn silage, 30% Chinese wildrye, and 20% alfalfa hay [on a dry matter (DM) basis]. The premix contained, per kilogram: Fe 5, 500 mg, Cu 4, 080 mg, Zn 17, 500 mg, Mn 4, 980 mg, Se 110 mg, I 180 mg, Co 88.5 mg, VA >2,000,000 IU, VD3 600,000 IU, and VE 10,800 mg. The animals were fed three times per day, at 4:00 a.m., 10:30 a.m., and 4:00 p.m. All animal procedures were carried out under the protocols approved by the Animal Care and Use Committee of Nanjing Agricultural University, 1999.
Two nylon bags were retrieved from each cow’s rumen at 0.5, 1, 2, 4, 6, 12, 24, 48, and 72 h (a total of six bags for each time point) and washed gently with phosphate-buffered saline (PBS) (pH 7.4) to remove the rumen contents on the outer surface of the bag. A few residue fragments were removed from each bag and fixed with 2.5% glutaraldehyde for scanning electron microscopy (SEM). Samples (1 g) of the residues were then used to isolate the associated fraction (As) from the adherent fraction (Ad), as described previously (Larue et al., 2005). These fractions were stored at -20°C until analysis of the loosely and tightly attached bacteria. The remaining residues were washed and dried at 65°C for the analysis of relative degradation of rice straw biomass. At 0, 24, 48, and 72 h, 50 ml samples of rumen fluid were collected from each cow for the analysis of bacterial community and CAZyme profiles.
Fiber Degradation Analysis
Relative biomass degradation of rice straw was determined by DM, crude fiber (CF), neutral detergent fiber (NDF), and acid detergent fiber (ADF) analysis. DM and CF were analyzed according to AOAC (2000). NDF and ADF were determined by the method of Van Soest et al. (1991).
Bacterial Community and CAZyme Analysis of Rumen Fluid by Metagenome Shotgun Sequencing
The rumen fluid samples collected from all time points for each cow were mixed and used for DNA extraction. A 10 ml volume of each mixed rumen fluid was centrifuged and the resulting pellet was ground in liquid nitrogen for DNA extraction with CTAB buffer. The DNA was purified in an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1). The final DNA pellets were washed with ice cold 70% ethanol and dissolved in TE buffer.
The metagenomic DNA libraries from the three cows were prepared following the manufacturer’s instructions (Illumina), and the samples were then pair-end sequenced using Illumina Hiseq4000 at BGI (Shenzhen, China). A total of 119,215,228 reads with a length of 150 bp was obtained after removing the reads containing N and the adapter (SRA accession number: SRP094501). The forward sequencing reads from each sample were quality controlled with meta-qc2 and the cow genome (NCBI assembly: GCA_000003205.4) was used as reference to remove the reads derived from the host. A total of 19,447,862, 19,311,185, and 19,452,969 sequences remained after quality control for each cow. MetaPhlAn (Segata et al., 2012) was then used to analyze the bacterial composition and ShotMAP (Nayfach et al., 2015) was used to annotate with the CAZy database3.
Analysis of Bacteria Loosely and Tightly Attached to Rice Straw by Illumina MiSeq Sequencing of 16S rRNA Gene
The samples from the Ad (0.25 g) and As (1 ml) fractions were used for DNA extraction using the bead-beating and phenol–chloroform extraction method described by Zoetendal et al. (1998). The universal primers (341F 5′-CCTAYGGGRBGCASCAG-3′ and 806R 5′-GGACTACNNGGGTATCTAAT-3′) (Behrendt et al., 2012) were used to obtain the PCR amplicons for paired-end sequencing on an Illumina MiSeq platform at Novogene (Beijing, China). The paired-end reads (SRA accession number: SRP066537) were assembled, trimmed, quality-filtered, and analyzed according to Li et al. (2016). Briefly, the paired-end reads were first assembled and trimmed with PANDAseq (Bartram et al., 2011) to remove primers and barcodes, as well as sequences less than 400 bp, longer than 500 bp, or with ambiguous bases. The trimmed sequences were then filtered by ea-utils (Aronesty, 2011) to remove sequences with mean quality score values less than 30. Subsequently, the quality-filtered sequences were analyzed by QIIME for open reference OTU picking, alpha and beta diversity analysis at a sequence depth of 17,900 sequences, and taxonomic assignment based on Greengenes (Caporaso et al., 2010; McDonald et al., 2012). The functional prediction and differential characterization were conducted with PICRUSt and LEfSe (Segata et al., 2011; Langille et al., 2013).
Scanning Electron Microscopy
After fixation for more than 2 h, the samples were washed three times with PBS (pH 7.4), prior to sequential dehydration using 50, 70, 80, 90, and 100% ethanol. The ethanol was then replaced with tertiary butyl alcohol before critical point drying using a Hitachi freeze dryer (ES-2030, Hitachi, Tokyo, Japan). The dried samples were then sputter coated with approximately 10 nm of Au/Pd using a Hitachi E-1010 Ion sputter instrument (Hitachi, Tokyo, Japan). SEM imaging was performed on a Hitachi S-3000 N microscope (Hitachi, Tokyo, Japan).
Statistical Analysis
Data were presented as mean ± SEM (n = 6). The statistical analysis was carried out using General Linear Model procedure with SPSS 22 (SPSS Inc., Chicago, IL, United States). Duncan test was used to compare the means. Statistical significance was defined at P < 0.05.
Results
Bacterial Community and CAZyme Profiles in the Rumen Fluid by Metagenome Shotgun Sequencing
The metagenomic analysis of the rumen fluid showed that the relative abundances of Prevotella (16.3%), Bacteroides (15.9%), and Butyrivibrio (10.2%) were above 10%, followed by unclassified Desulfobulbaceae (8.0%), Desulfovibrio (5.1%), and unclassified Sphingobacteriaceae (5.0%) (Figure 1A). The analysis of CAZymes profiles showed that all the five enzyme classes (GHs, GTs, PLs, CEs, and AAs) were detected in the rumen fluid (data not shown). The diversity profile of the GH families (Figure 1B) showed that GH43 was the most abundant family, with a relative abundance of 8.9%, followed by GH13 (7.6%), GH3 (7.3%), and GH2 (6.9%). Among the four categories of cellulases, hemicellulases, debranching enzymes, and oligosaccharide degrading enzymes, the oligosaccharide degrading enzymes from the 16 GH families were the most abundant (43.8%), followed by hemicellulases (13.0%), cellulases (6.6%), and debranching enzymes (4.2%).
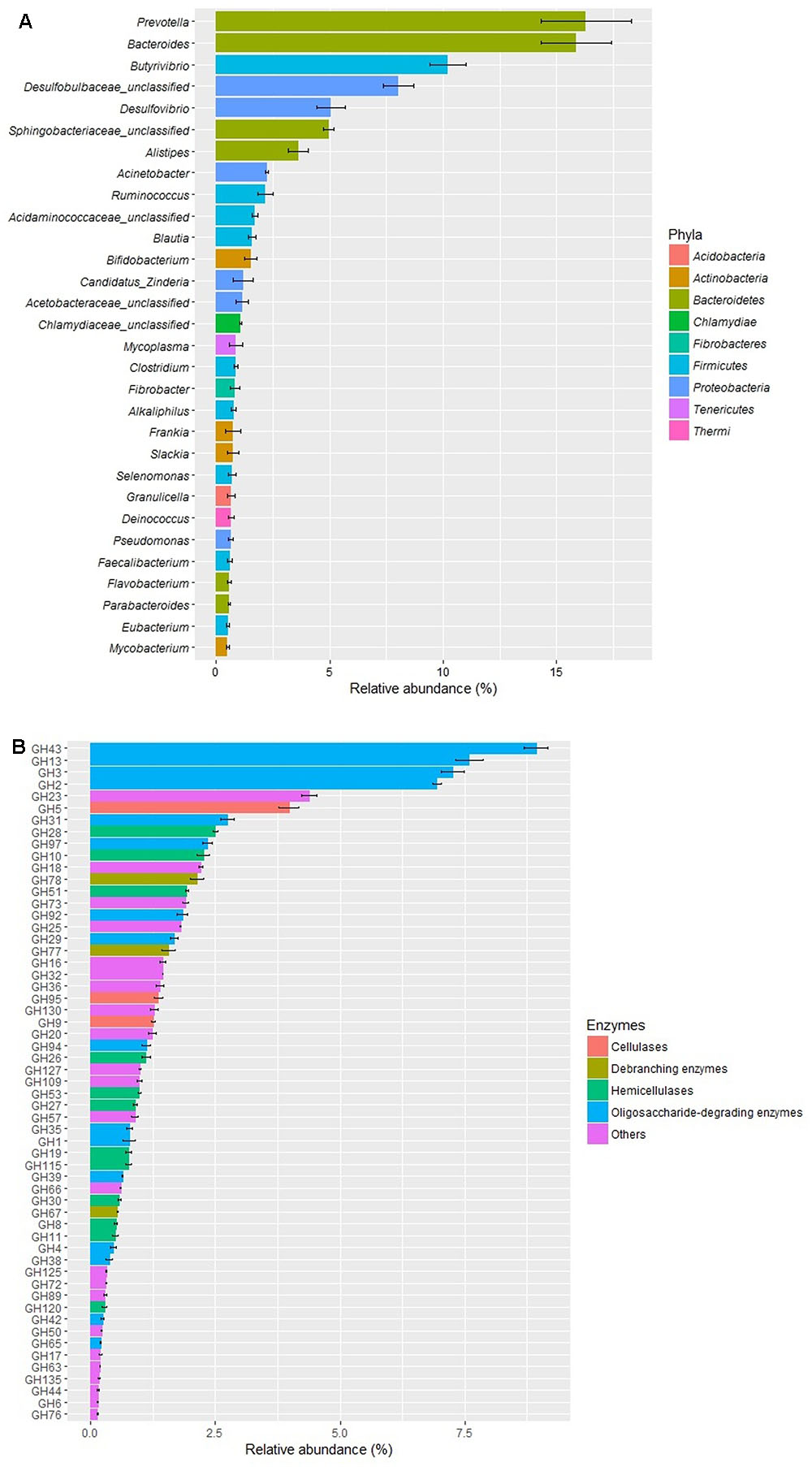
FIGURE 1. Relative abundances of predominant genera (>0.5% relative abundance, A) and GH families (B) in the rumen fluid of cows by metagenome shotgun sequencing (n = 3).
Biomass Degradation of Rice Straw in the Rumen
Approximately 13% of the DM was degraded within 0.5 h (Supplementary Figure 1). And then, no significant change of DM digestibility was observed between 0.5 and 6 h. Subsequently, the rice straw was rapidly degraded between 6 and 24 h with ∼17% of DM being degraded. After 24 h of incubation, the degradation of rice straw gradually slowed down with ∼10% of DM being degraded between 24 and 48 h and ∼7% of DM being degraded between 48 and 72 h. The digestibility of CF, NDF, and ADF was similar to the digestibility of DM (data not shown). The SEM photos (Supplementary Figure 2) showed the dynamics of the degradation of rice straw over the 72 h incubation period in the rumen.
Temporal Changes in the Community of Bacteria Loosely and Tightly Attached to Rice Straw by Illumina MiSeq Sequencing of 16S rRNA Gene
The temporal digestibility profiles of DM, CF, NDF, and ADF were used as a basis for selecting samples collected at 0.5, 6, 24, and 72 h for bacterial community analysis. A total of 1,946,473 sequences remained from 48 samples after quality-filtering with a mean of 40,551 ± 11,108 sequences per sample. A total of 4820 OTUs were identified at the 97% similarity level.
Alpha diversity analysis of loosely and tightly attached bacteria (Table 1) showed that the PD_whole_tree, Chao1, observed species, and Shannon and Simpson indexes were significantly different between the two fractions (P < 0.05). The incubation time significantly (P < 0.05) affected the PD_whole_tree, observed species, and Shannon index, while the interaction between fraction and time significantly (P < 0.05) affected all of the aforementioned metrics.
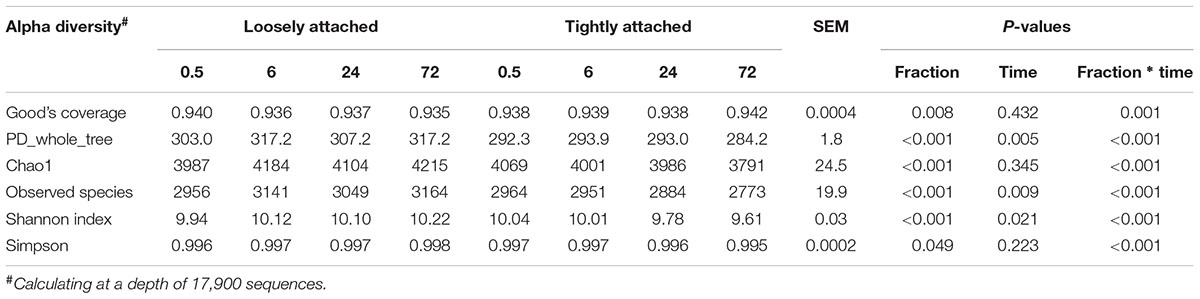
TABLE 1. Alpha diversity of bacteria loosely and tightly attached to rice straw after incubation for 0.5, 6, 24, and 72 h in the rumen of cows.
Taxonomic assignment of the OTUs showed that Bacteroidetes and Firmicutes were the dominant phyla in all samples, accounting for ∼92% and ∼97% in the loosely and tightly attached fractions, respectively (Supplementary Figure 3). Principal component analysis of the bacteria attached to rice straw (Figure 2) showed that all the samples from the loosely attached fraction clustered together in a separate group. By contrast, the samples from the tightly attached fraction separated into two groups according to the incubation time.
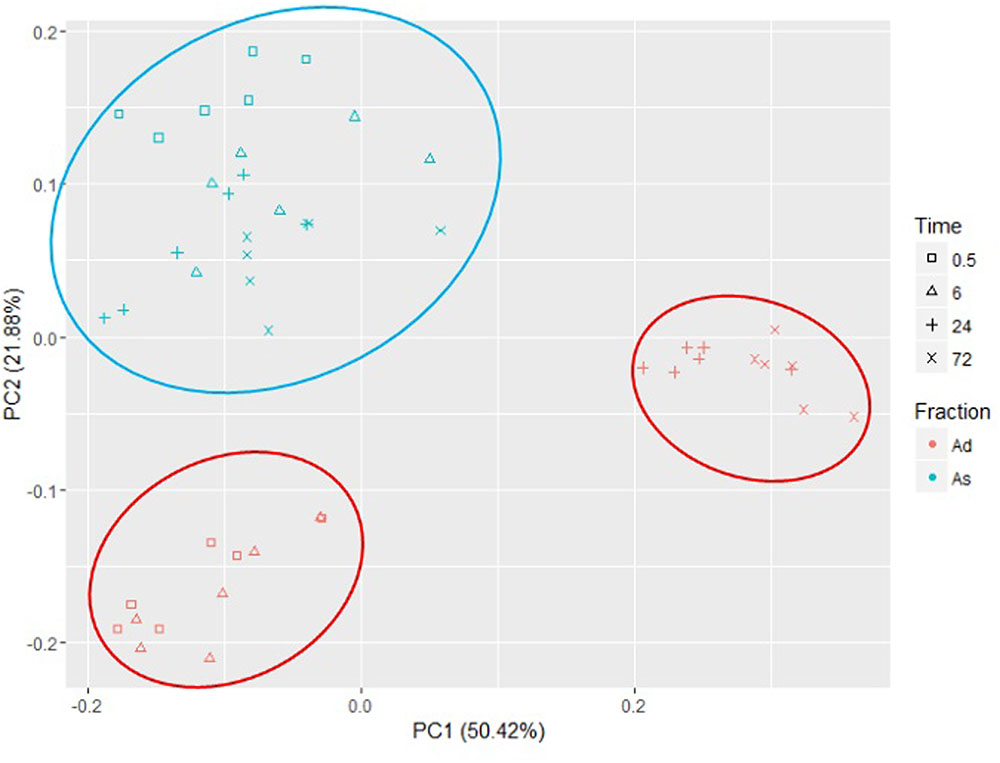
FIGURE 2. Principal component analysis of bacteria loosely (As) and tightly (Ad) attached to rice straw after 0.5, 6, 24, and 72 h incubation in the rumen of cows by Illumina MiSeq sequencing of 16S rRNA gene (n = 6).
The relative abundances of 36 genera were above 0.1% in at least one sample in the loosely attached fraction. A total of 23 genera were unidentified and their accumulative relative abundances were 67.0, 66.7, 60.3, and 65.4% at 0.5, 6, 24, and 72 h, respectively. The remaining 13 genera were classified as Prevotella, Fibrobacter, Butyrivibrio, Ruminococcus, Succiniclasticum, Clostridium, Dehalobacterium, Coprococcus, Moryella, Pseudobutyrivibrio, Oscillospira, Treponema, and Anaeroplasma. Prevotella was the predominant classified genus, accounting for 18.1, 19.2, 21.7, and 20.4% at 0.5, 6, 24, and 72 h, respectively. None of the other classified genera accounted for more than 3% in the samples (Figure 3A). The predicted function of the loosely attached bacteria (Figure 3B) showed a diversity of fiber-degrading genes.
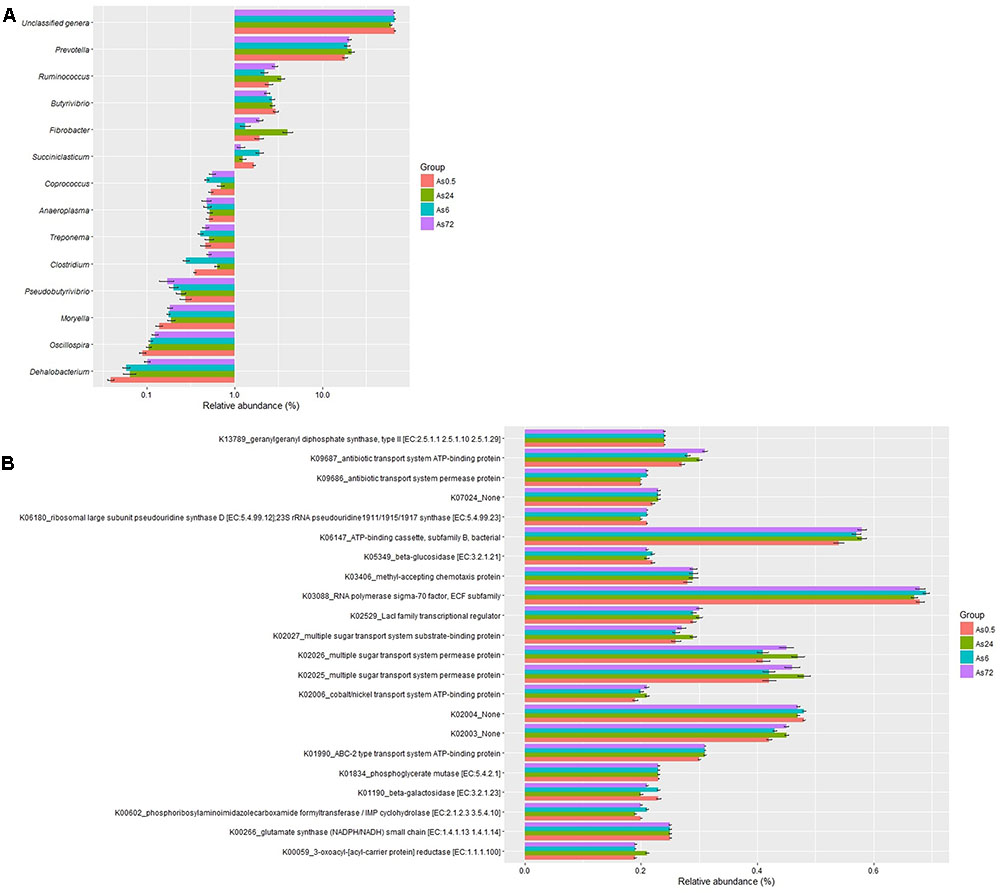
FIGURE 3. Relative abundances (A) and predicted function (B) of bacteria loosely (As) attached to rice straw after 0.5, 6, 24, and 72 h incubation in the rumen of cows by Illumina MiSeq sequencing of 16S rRNA gene. (n = 6; the relative abundances of bacteria above 0.1% or the relative abundances of KEGG Orthologies above 0.2% in at least one sample were presented.)
The relative abundances of 36 genera were also above 0.1% in at least one sample in the tightly attached fraction. A total of 21 genera were unclassified and their accumulative relative abundances were 54.6, 56.2, 75.8, and 79.3% at 0.5, 6, 24, and 72 h, respectively. The remaining 15 genera were classified as Prevotella, Fibrobacter, Butyrivibrio, Ruminococcus, Succiniclasticum, Clostridium, Dehalobacterium, Coprococcus, Moryella, Pseudobutyrivibrio, Oscillospira, Treponema, Anaeroplasma, Desulfovibrio, and Anaerostipes (Figure 4A). The predicted function of the tightly attached bacteria also showed a diversity of fiber-degrading genes (data not shown). LEfSe analysis of the microbial shift showed that Bacteroidetes, Sphaerochaetales, and TM7 clusters were significantly higher at 0.5 and 6 h, while Firmicutes, Betaproteobacteria, Synechococcophycideae, and Desulfuromonadales were significantly higher at 24 and 72 h (Figure 4B). At the genus level, the proportions of Prevotella, Ruminococcus, Succiniclasticum, Coprococcus, and Anaerostipes significantly decreased, while the proportions of Dehalobacterium, Clostridium, and unclassified genera were significantly increased at 24 and 72 h (Figure 4C).
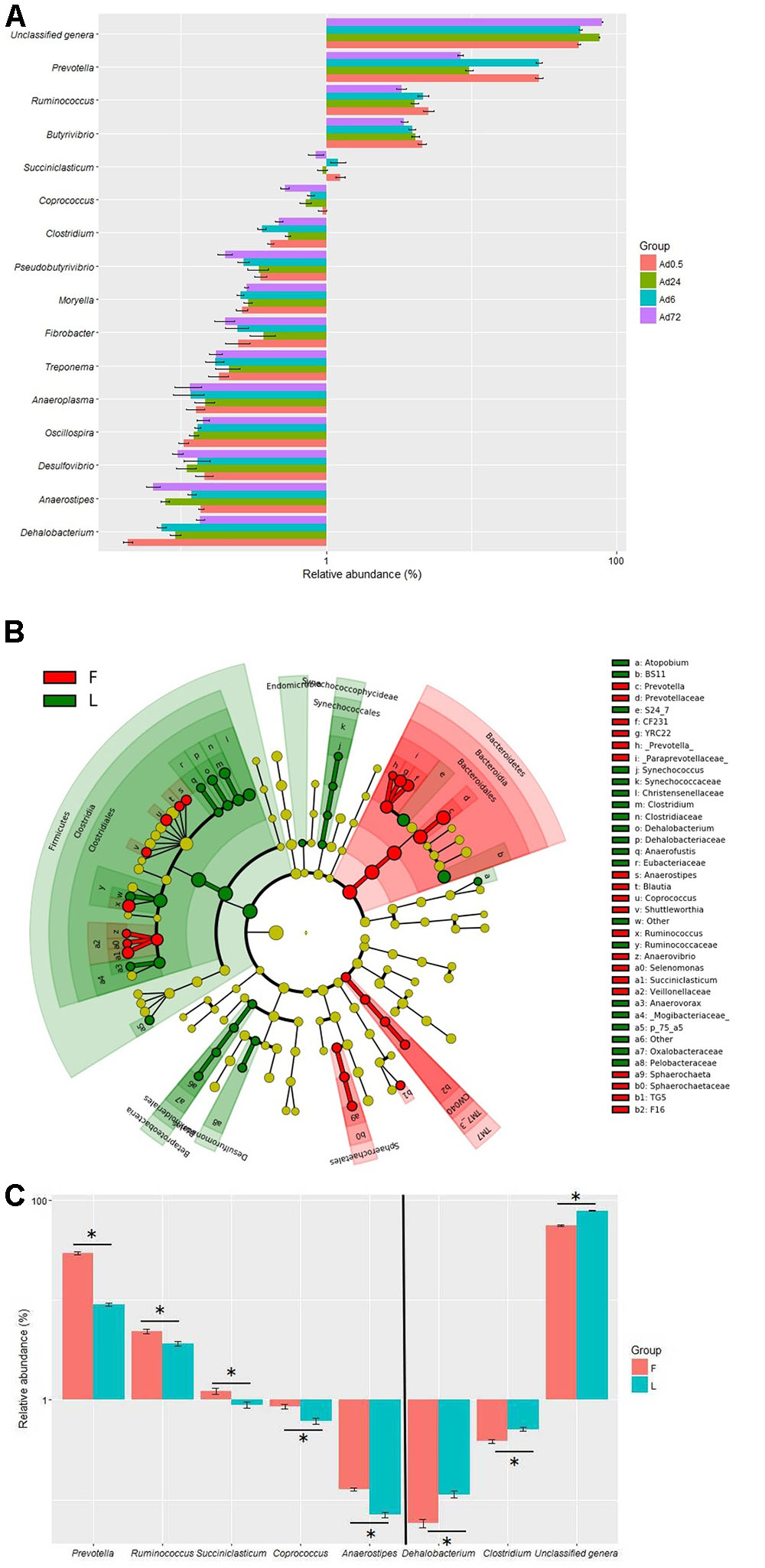
FIGURE 4. Relative abundances (A) of bacteria tightly (Ad) attached to rice straw after 0.5, 6, 24, and 72 h incubation in the rumen of cows by Illumina MiSeq sequencing of 16S rRNA gene (the relative abundances of bacteria above 0.1% in at least one sample were presented) and LEfSe analyses (B) of the bacterial community shift. (C) The significant (P < 0.05) shift genera. (n = 6; F, the bacteria community at 0.5 and 6 h; L, the bacteria community at 24 and 72 h.)
Discussion
Rice is the predominant dietary energy source in China, and its production was approximately 204 million tons in 2013 (National Bureau of Statistics of China, 2014). According to Kadam et al. (2000), about 1.35 tons of crop residues remain in the field for every ton of crop harvested, so more than 250 million tons of rice straw were produced in China in 2013. The use of rice crop residues to supplement the short supply of high-quality forages in China is therefore an attractive proposition. However, studies on the degradation of these crop residues in ruminants are still needed.
Previous work has included the study by Chen et al. (2017), who investigated the effects of Lactobacillus plantarum on the in vitro degradation of maize and rice straws. Similarly, Xin et al. (2015) investigated the effects of different treatments on the in vitro degradation of rice straw, whereas Sun et al. (2015) investigated the improvement of in vitro degradation of rice straw by silkworm excrement. Koike et al. (2014) investigated the improvement of the degradation of rice straw by sodium hydroxide treatment. Polyorach and Wanapat (2015) investigated the improvement of the rice straw quality by the addition of urea and calcium hydroxide. Liu et al. (2016) investigated the degradation pattern of rice straw and alfalfa in the rumen. Wang et al. (2016) investigated the rumen fermentation and microbial community of lactating dairy cows fed with rice straw and corn silage. However, only limited studies considered the dependence of rice straw degradation on the colonization of rice straw and excretion of fiber-degrading enzymes by the rumen microbial community.
The metagenomic analysis in the present study showed that oligosaccharide-degrading enzymes (especially GH2, GH3, GH13, and GH43) predominate in the rumen fluid, which might result from an enrichment in oligosaccharides arising from degradation of plant fiber by cellulases, debranching enzymes, and hemicellulases. Brulc et al. (2009) investigated the fiber-degrading enzymes of the fiber-adherent microbes in the rumen of bovine and found that oligosaccharide-degrading enzymes, especially GH2 and GH3, were predominant. Hess et al. (2011) investigated the biomass-degrading genes and genomes from cow rumen and also found that oligosaccharide-degrading enzymes, especially endoglucanase, glucosidase, and cellobiohydrolase, were predominant. Patel et al. (2014) conducted a metagenomics study of the microbial and CAZyme profiles of buffalo rumen and found that oligosaccharide-degrading enzymes, especially GH2 and GH3, were predominant. Similarly, the metagenomic analysis by Pitta et al. (2016) also revealed that oligosaccharide-degrading enzymes, especially GH3 and GH13, predominated in the rumen contents. Dai et al. (2015) showed that GH2 and GH3 were predominant by metagenomics as well, but metatranscriptomic analysis indicated that the relative percentage of oligosaccharide-degrading enzymes was dramatically reduced, indicating that the expression of these enzymes, and perhaps other CAZymes as well, might be induced by their substrates. Many more studies are needed to elucidate the mechanism underlying the expression of these CAZymes for improvement of the degradation of fibers in the rumen.
The dominant bacterial phyla in both loosely and tightly attached fractions were Bacteroidetes and Firmicutes. An association between bacteria belonging to the two phyla and the degradation of fiber and polysaccharides has been previously reported (Wright and Klieve, 2011; Zened et al., 2012). However, Lee et al. (2009) reported that Firmicutes often carried 4–15 rRNA operons, which implied that the 16S rRNA sequencing might lead to an overestimation of the abundance of Firmicutes.
In the present study, the predominant classified genus found attached to rice straw was Prevotella, followed by Ruminococcus and Butyrivibrio. Prevotella predominates not only in the rumen (Edwards et al., 2004; Kong et al., 2010; Mao et al., 2015), but also in the foregut of kangaroos (Li et al., 2016) and in the gut of African children (De Filippo et al., 2010). This genus was reported to have moderately saccharolytic potential (Shah and Collins, 1990) and can metabolize various sugars (including xylan) to acetate, propionate, and succinate (Sakamoto and Ohkuma, 2012). Ueki et al. (2007) isolated a novel Prevotella species from plant residue and rice roots and demonstrated that this strain had the ability to degrade xylan and hemicellulose. Ruminococcus is another well-studied rumen microorganism with high fiber-degrading ability (Latham and Wolin, 1977; Christopherson et al., 2014; Rozman et al., 2015). A recent study by Dai et al. (2015) indicated that Ruminococcus produced the largest proportion of cellulases in the rumen, as well as large amounts of hemicellulases and oligosaccharide-degrading enzymes. Another common rumen microorganism is Butyrivibrio, first reported by Bryant and Small (1956) as an anaerobic, butyric acid-producing and curved rods genus. Mao et al. (2015) reported that Butyrivibrio accounted for ∼3% in the rumen digesta and ∼12% in the mucosa of dairy cattle. Dai et al. (2015) reported that Butyrivibrio could produce ∼7% of the oligosaccharide-degrading enzymes and a limited amount of hemicellulases in the cow rumen. Liu et al. (2016) investigated the temporal dynamics of ruminal bacterial microbiota colonizing rice straw within ruminants and found that Prevotella, Ruminococcus, Butyrivibrio, Fibrobacter, and Treponema were predominant (>1% in at least one sample). Interestingly, the authors reported that the relative abundance of Fibrobacter was significantly increased after 6 h incubation (up to 5.29% at 16 h), which was not observed in the present study. Similarly, the authors reported high amount of Treponema after 6 h incubation (4.15% at 48 h), which was remained ∼0.5% over the incubation.
However, in the present study, we found that a high proportion of unclassified genera were involved in the degradation of rice straw. The relative abundance of unclassified genera, which were tightly attached to the rice straw, was ∼80% after 72 h incubation. Piao et al. (2014) also reported a relative abundance of ∼62% for unclassified genera tightly attached to switchgrass after 72 h incubation in the rumen. These findings imply that these unclassified genera might be very important in the degradation of plant fiber; therefore, the function of these microorganisms needs further investigation.
Our PCoA indicated that the group of loosely attached bacteria remained relatively stable during the 72 h incubation. However, the tightly attached bacterial community changed after 6 h of incubation. Huws et al. (2013, 2016), who investigated the successional colonization of perennial ryegrass by the bacteria in the cow rumen, identified distinct primary (0–2 h) and secondary (4 h onward) bacterial communities. These authors also demonstrated that a proportion of the primary colonizing bacteria (Succinivibrio spp.) detached after 2 h of incubation and were replaced with the population of secondary colonizing bacteria, which included Prevotella, Pseudobutyrivibrio, Roseburia, and Ruminococcus. In the present study, we found that the proportions of Anaerostipes, Coprococcus, Desulfovibrio, Prevotella, Pseudobutyribivrio, Ruminococcus, and Succiniclasticum all significantly decreased in the secondary bacterial community when compared to the primary bacterial community. For instance, the Prevotella significantly decreased to ∼9% abundance in the secondary bacterial community from ∼29% in the primary bacterial community, whereas the proportions of Clostridium, Dehalobacterium, and Oscillospira increased in the secondary bacterial community. Liu et al. (2016), who investigated the temporal dynamics of rumen bacteria colonizing rice straw and alfalfa hay, also identified distinct primary (0–6 h) and secondary (6 h onward) bacterial communities. Members of Spirochaetes and Fibrobacteria were predominant after 6 h incubation in the rumen. Piao et al. (2014) also observed a similar microbial shift when they investigated the temporal dynamics of fibrolytic microorganisms with switchgrass as substrate, again implying that the primary and secondary bacterial communities might have different functions in the degradation of plant fiber. However, the roles of these bacterial communities and their relationship to the chemical components of plant fiber require further investigation.
Conclusion
The present study documented the degradation pattern of rice straw within the cow rumen. Within 30 min of incubation, the rice straw was colonized by bacteria that included Prevotella, Ruminococcus, Butyrivibrio, and a large proportion of unclassified bacteria. After 6 h of incubation, a significant shift in the population of tightly attached bacteria was observed, resulting in decreases in Anaerostipes, Blautia, Butyrivibrio, Coprococcus, Desulfovibrio, Prevotella, Pseudobutyrivibrio, Ruminococcus, and Succiniclasticum and increases in Clostridium, Dehalobacterium, and Oscillospira. Most of the bacteria involved in the degradation of rice straw were unidentified; thus, further investigation of the function of these bacteria, and especially the secondary bacterial community, is needed to improve the utilization of crop residues in ruminants.
Author Contributions
Conceived and designed the experiments: YC and WZ. Performed the experiments: YiW, YL, YZ, TL, and YuW. Generated and analyzed the data: YiW, YL, YZ, TL, YuW, YC, and TS. Wrote the paper: YC, TS, and WZ. All authors read and approved the final manuscript.
Funding
This work was supported by National Key Research and Development Program of China (2017YFD0500505), Natural Science Foundation of Jiangsu Province (BK20141372), and the Student Research Training programming (201410307033).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We are thankful to BGI, China for metagenomic and 16S rRNA gene sequencing services.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02165/full#supplementary-material
Footnotes
References
AOAC (2000). Official Methods of Analysis of AOAC International, 17th Edn. Gaithersburg, MD: AOAC international.
Aronesty, E. (2011). ea-utils: Command-line Tools for Processing Biological Sequencing Data. Available at: http://code.google.com/p/ea-utils
Bartram, A. K., Lynch, M. D. J., Stearns, J. C., Moreno-Hagelsieb, G., and Neufeld, J. D. (2011). Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end illumine reads. Appl. Environ. Microbiol. 77, 3846–3852. doi: 10.1128/AEM.02772-10
Behrendt, L., Larkum, A. W. D., Trampe, E., Norman, A., Sorensen, S. J., and Kuhl, M. (2012). Microbial diversity of biofilm communities in microniches associated with the didemnid ascidian Lissoclinum patella. ISME J. 6, 1222–1237. doi: 10.1038/ismej.2011.181
Brulc, J. M., Antonopoulos, D. A., Miller, M. E. B., Wilson, M. K., Yannarell, A. C., Dinsdale, E. A., et al. (2009). Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc. Natl. Acad. Sci. U.S.A. 106, 1948–1953. doi: 10.1073/pnas.0806191105
Bryant, M. P., and Small, N. (1956). The anaerobic, monotrichous, butyric acid-producing, curved rod-shaped bacteria of the rumen. J. Bacteriol. 72, 16–21.
Cai, Y., Qiu, H., and Xu, Z. (2011). Evaluation on potentials of energy utilization of crop residual resources in different regions of China. J. Nat. Resour. 26, 1637–1646.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chen, L., Ren, A., Li, B., Zhou, C., and Tan, Z. (2017). Effects of Lactobacillus plantarum on in vitro rumen fermentation characteristics of maize straw and rice straw. Chin. J. Anim. Nutr. 29, 678–689.
Christopherson, M. R., Dawson, J. A., Stewenson, D. M., Cunningham, A. C., Bramhacharya, S., Weimer, P. J., et al. (2014). Unique aspects of fiber degradation by the ruminal ethanologen Ruminococcus albus 7 revealed by physiological and transcriptomic analysis. BMC Genomics 15:1066. doi: 10.1186/1471-2164-15-1066
Dai, X., Tian, Y., Li, J., Su, X., Wang, X., Zhao, S., et al. (2015). Metatranscriptomic analyses of plant cell wall polysaccharide degradation by microorganisms in the cow rumen. Appl. Environ. Microbiol. 81, 1375–1386. doi: 10.1128/AEM.03682-14
De Filippo, C., Cavalieri, D., Di Paola, M., Ramazzotti, M., Poullet, J. B., Massart, S., et al. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U.S.A. 107, 14691–14696. doi: 10.1073/pnas.1005963107
Edwards, J. E., Huws, S. A., Kim, E. J., and Kingston-Smith, A. H. (2007). Characterization of the dynamics of initial bacterial colonization of noncon served forage in the bovine rumen. FEMS Microbiol. Ecol. 62, 323–335. doi: 10.1111/j.1574-6941.2007.00392.x
Edwards, J. E., McEwan, N. R., Anthony, J. T., and Wallace, R. J. (2004). 16S rDNA library-based analysis of ruminal bacterial diversity. Antonie Van Leeuwenhoek 86, 263–281. doi: 10.1023/B:ANTO.0000047942.69033.24
FAO (2011). Mapping Supply and Demand for Animal-Source Foods to 2030. Animal Production and Health Working Paper No.2. Rome: FAO.
Hess, M., Sczyrba, A., Egan, R., Kim, T.-W., Chokhawala, H., Schroth, G., et al. (2011). Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331, 463–467. doi: 10.1126/science.1200387
Huws, S. A., Edwards, J. E., Creevey, C. J., Stevens, P. R., Lin, W., Girdwood, S., et al. (2016). Temporal dynamics of the metabolically active rumen bacteria colonising fresh perennial ryegrass. FEMS Microbiol. Ecol. 92:fiv137. doi: 10.1093/femsec/fiv137
Huws, S. A., Mayorga, O. L., Theodorou, M. K., Onime, L. A., Kim, E. J., Cookson, A. H., et al. (2013). Successional colonization of perennial ryegrass by rumen bacteria. Lett. Appl. Microbiol. 56, 186–196. doi: 10.1111/lam.12033
Kadam, K. L., Forrest, L. H., and Jacobson, W. A. (2000). Rice Straw as a lignocellulosic resource: collection, processing, transportation, and environmental aspects. Biomass Bioenergy 18, 369–389. doi: 10.1016/S0961-9534(00)00005-2
Koike, S., Yabuki, H., and Kobayashi, Y. (2014). Interaction of rumen bacteria as assumed by colonization patterns on untreated and alkali-treated rice straw. Anim. Sci. J. 85, 524–531. doi: 10.1111/asj.12176
Kong, Y., Teather, R., and Forster, R. (2010). Composition, spatial distribution, and diversity of the bacterial communities in the rumen of cows fed different forages. FEMS Microbiol. Ecol. 74, 612–622. doi: 10.1111/j.1574-6941.2010.00977.x
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Larue, R., Yu, Z., Parisi, V. A., Egan, A. R., and Morrison, M. (2005). Novel microbial diversity adherent to plant biomass in the herbivore gastrointestinal tract, as revealed by ribosomal intergenic spacer analysis and rrs gene sequencing. Environ. Microbiol. 7, 530–543. doi: 10.1111/j.1462-2920.2005.00721.x
Latham, M. J., and Wolin, M. J. (1977). Fermentation of cellulose by Ruminococcus flavefaciens in the presence and absence of Methanobacterium ruminantium. Appl. Environ. Microbiol. 34, 297–301.
Lee, Z. M., Bussema, C., and Schmidt, T. M. (2009). rrnDB: Documenting the number of rRNA and tRNA genes in bacteria and archaea. Nucleic Acids Res. 37, D489–D493. doi: 10.1093/nar/gkn689
Li, M., Jin, W., Li, Y., Zhao, L., Cheng, Y., and Zhu, W. (2016). Spatial dynamics of the bacterial community structure in the gastrointestinal tract of red kangaroo (Macropus rufus). World J. Microbiol. Biotechnol. 32, 98. doi: 10.1007/s11274-016-2054-z
Liu, J., Zhang, M., Xue, C., Zhu, W., and Mao, S. (2016). Characterization and comparison of the temporal dynamics of ruminal bacterial microbiota colonizing rice straw and alfalfa hay within ruminants. J. Dairy Sci. 99, 9668–9681. doi: 10.3168/jds.2016-11398
Mao, S., Zhang, M., Liu, J., and Zhu, W. (2015). Characterising the bacterial microbiota across the gastrointestinal tracts of dairy cattle: membership and potential function. Sci. Rep. 5:16116. doi: 10.1038/srep16116
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., DeSantis, T. Z., Probst, A., et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. doi: 10.1038/ismej.2011.139
National Bureau of Statistics of China (2014). China Statistical Yearbook. Beijing: China Statistics Press.
Nayfach, S., Bradley, P. H., Wyman, S. K., Laurent, T. J., Williams, A., Eisen, J. A., et al. (2015). Automated and accurate estimation of gene family abundance from shotgun metagenomes. PLOS Comput. Biol. 11:e1004573. doi: 10.1371/journal.pcbi.1004573
Patel, D. D., Patel, A. K., Parmar, N. R., Shah, T. M., Patel, J. B., Pandya, P. R., et al. (2014). Microbial and carbohydrate active enzyme profile of buffalo rumen metagenome and their alteration in response to variation in the diet. Gene 545, 88–94. doi: 10.1016/j.gene.2014.05.003
Piao, H., Lachman, M., Malfatti, S., Sczyrba, A., Knierim, B., Auer, M., et al. (2014). Temporal dynamics of fibrolytic and methanogenic rumen microorganisms during in situ incubation of switchgrass determined by 16S rRNA gene profiling. Front. Microbiol. 5:307. doi: 10.3389/fmicb.2014.00307
Pitta, D. W., Pinchak, W. E., Indugu, N., Vecchiarelli, B., Sinha, R., and Fulford, J. D. (2016). Metagenomic Analysis of the rumen microbiome of steers with wheat-induced frothy bloat. Front. Microbiol. 7:689. doi: 10.3389/fmicb.2016.00689
Polyorach, S., and Wanapat, M. (2015). Improving the quality of rice straw by urea and calcium hydroxide on rumen ecology, microbial protein synthesis in beef cattle. J. Anim. Physiol. Anim. Nutr. 99, 449–456. doi: 10.1111/jpn.12253
Rozman, G. I., Yin, G., Borovok, I., Berg Miller, M. E., Yeoman, C. J., Dassa, B., et al. (2015). Functional phylotyping approach for assessing intraspecific diversity of Ruminococcus albus within the rumen microbiome. FEMS Microbiol. Lett. 362, 1–10. doi: 10.1093/femsle/fnu047
Sakamoto, M., and Ohkuma, M. (2012). Reclassification of Xylanibacter oryzae Ueki et al. 2006 as Prevotella oryzae comb. nov., with an emended description of the genus Prevotella. Int. J. Syst. Evol. Microbiol. 62, 2637–2642. doi: 10.1099/ijs.0.038638-0
Segata, N., Izard, J., Walron, L., Gevers, D., Miropolsky, L., Garrett, W., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Segata, N., Waldron, L., Ballarini, A., Narasimhan, V., Jousson, O., and Huttenhower, C. (2012). Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 9, 811–814. doi: 10.1038/nmeth.2066
Shah, H. N., and Collins, D. M. (1990). Prevotella, a new genus to include Bacteroides melaninogenicus and related species formerly classified in the genus Bacteroides. Int. J. Syst. Bacteriol. 40, 205–208. doi: 10.1099/00207713-40-2-205
Sun, L., Li, H., Cui, H., Zhao, R., Wang, M., Yu, L., et al. (2015). Associative effects of different combination ratios of silkworm excrement and rice straw on the rumen microbial fermentation in vitro. Chin. J. Anim. Nutr. 27, 313–319.
Ueki, A., Akasaka, H., Satoh, A., Suzuki, D., and Ueki, K. (2007). Prevotella paludivivens sp. nov., a novel strictly anaerobic, Gram-negative, hemicelluloses-decomposing bacterium isolated from plant residue and rice roots in irrigated rice-field soil. Int. J. Syst. Evol. Microbiol. 57, 1803–1809. doi: 10.1099/ijs.0.64914-0
Van Soest, P. J., Robertson, J. B., and Lewis, B. A. (1991). Methods for dietary fiber, neutral detergent fiber, and nonstarch polysaccharides in relation to animal nutrition. J. Dairy Sci. 74, 3583–3597. doi: 10.3168/jds.S0022-0302(91)78551-2
Wang, M., Wang, R., Xie, T. Y., Janssen, P. H., Sun, X. Z., Beauchemin, K. A., et al. (2016). Shifts in rumen fermentation and microbiota are associated with dissolved ruminal hydrogen concentrations in lactating dairy cows fed different types of carbohydrates. J. Nutr. 146, 1714–1721. doi: 10.3945/jn.116.232462
Weimer, P. J., Russell, J. B., and Muck, R. E. (2009). Lessons from the cow: what the ruminant animal can teach us about consolidated bioprocessing of cellulosic biomass. Bioresour. Technol. 100, 5323–5331. doi: 10.1016/j.biortech.2009.04.075
Wright, A. D. G., and Klieve, A. V. (2011). Does the complexity of the rumen microbial ecology preclude methane mitigation? Anim. Feed Sci. Technol. 16, 248–253. doi: 10.1016/j.anifeedsci.2011.04.015
Xin, H., Liu, K., Zhang, Y., Wang, M., Li, Z., Wang, Z., et al. (2015). Effects of different treated rice straws on patterns, methane production and microflora of in vitro ruminal fermentation. Chin. J. Anim. Nutr. 27, 1632–1640.
Zened, A., Combes, S., Cauquil, L., Mariette, J., Klopp, C., Bouchez, O., et al. (2012). Microbial ecology of the rumen evaluated by 454 GS FLX pyrosequencing is affected by starch and oil supplementation of diets. FEMS Microbiol. Ecol. 83, 504–514. doi: 10.1111/1574-6941.12011
Keywords: rice straw, fiber degradation, ruminal bacteria, carbohydrate-active enzymes, metagenome
Citation: Cheng Y, Wang Y, Li Y, Zhang Y, Liu T, Wang Y, Sharpton TJ and Zhu W (2017) Progressive Colonization of Bacteria and Degradation of Rice Straw in the Rumen by Illumina Sequencing. Front. Microbiol. 8:2165. doi: 10.3389/fmicb.2017.02165
Received: 11 May 2017; Accepted: 23 October 2017;
Published: 06 November 2017.
Edited by:
Joerg Graf, University of Connecticut, United StatesReviewed by:
Shengguo Zhao, Chinese Academy of Agricultural Sciences, ChinaStephan Schmitz-Esser, Iowa State University, United States
Copyright © 2017 Cheng, Wang, Li, Zhang, Liu, Wang, Sharpton and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiyun Zhu, zhuweiyun@njau.edu.cn