 Chong Peng
Chong Peng Yan Lin
Yan Lin Hao Luo
Hao Luo Feng Gao
Feng Gao- 1Department of Physics, School of Science, Tianjin University, Tianjin, China
- 2Key Laboratory of Systems Bioengineering (Ministry of Education), Tianjin University, Tianjin, China
- 3SynBio Research Platform, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin University, Tianjin, China
Genes critical for the survival or reproduction of an organism in certain circumstances are classified as essential genes. Essential genes play a significant role in deciphering the survival mechanism of life. They may be greatly applied to pharmaceutics and synthetic biology. The continuous progress of experimental method for essential gene identification has accelerated the accumulation of gene essentiality data which facilitates the study of essential genes in silico. In this article, we present some available online resources related to gene essentiality, including bioinformatic software tools for transposon sequencing (Tn-seq) analysis, essential gene databases and online services to predict bacterial essential genes. We review several computational approaches that have been used to predict essential genes, and summarize the features used for gene essentiality prediction. In addition, we evaluate the available online bacterial essential gene prediction servers based on the experimentally validated essential gene sets of 30 bacteria from DEG. This article is intended to be a quick reference guide for the microbiologists interested in the essential genes.
Introduction
Essential genes are those that play a decisive role in the survival and development of an organism under general conditions. Even though the genome sizes and gene compositions differ dramatically, all so far sequenced genomes contain a set of essential genes that sustain key cellular functions. However, the phrase “essential gene” is highly context-dependent. Only when the environment in which organisms live is clearly defined can a gene be classified as essential gene or not. Another closely linked concept is the minimal gene set. A minimal gene set is defined as the minimal set of genes needed for a cell to carry out basic metabolism and reproduction under the most favorable conditions, in which all essential nutrients are available and there is no environmental stress (Koonin, 2000, 2003; Gil et al., 2004). Research on essential genes, with important theoretical as well as practical values, is quite appealing. Identification of essential genes can help a lot in deciphering the survival mechanisms of life. Moreover, because the deletion or inactivation of essential genes confer lethal phenotypes to microorganisms, essential genes or proteins encoded by essential genes form logical targets for new antibiotics in the pharmaceutical industry (Galperin and Koonin, 1999; Juhas et al., 2011; Mobegi et al., 2014). In the emerging scientific field of synthetic biology, devising a minimal genome is a desirable research direction (Pei et al., 2011; Juhas et al., 2012). For example, researchers at the J. Craig Venter Institute (JCVI) produced the first self-replicating synthetic cell Mycoplasma mycoides JCVI-syn1.0 in 2010 (Gibson et al., 2010). By the design-build-test (DBT) cycle, they removed non-essential genes in JCVI-syn1.0 genome and produced JCVI-syn3.0. Containing 531,560 base pairs and only 473 genes, JCVI-syn3.0 has smaller genome than that of any free-living organism found in nature (Hutchison et al., 2016).
Since 1999, when the first global transposon mutagenesis was performed on Mycoplasma genitalium to experimentally confirm the minimal gene set for a living organism (Hutchison et al., 1999), the attempt to search for essential genes has been persistently carried out in a wide range of species. The experimental approaches used to identify essential genes include single-gene knockout (Kobayashi et al., 2003), transposon mutagenesis (Hutchison et al., 1999), and antisense RNA inhibition (Ji et al., 2001). In the past decade, the integration of transposon mutagenesis and high-throughput sequencing has facilitated many methods in the recognition of essential genes. These development lead to a significant increase in the number of species involved in gene essentiality screens. Apart from bacteria, essential genes in archaea (Sarmiento et al., 2013) and eukaryotes such as Saccharomyces cerevisiae (Giaever et al., 2002), Schizosaccharomyces pombe (Kim et al., 2010), Arabidopsis thaliana (Meinke et al., 2008), Mus musculus (Liao and Zhang, 2007) and Homo sapiens (Blomen et al., 2015; Wang et al., 2015) are all identified. Based on these abundant data, researchers have constructed many essential gene databases. The bioinformatic resources greatly promote the investigation of essential genes (Lin et al., 2010; Gao and Zhang, 2011; Peng and Gao, 2014; Luo et al., 2015; Zhang et al., 2015; Zheng et al., 2015).
Except the development of experimental approaches, researchers also tried in many ways to computationally recognize the essential genes. In fact, computational approach to search for the minimal gene set was performed as early as 1996. Supposing that genes conserved between organisms are likely to be essential, Mushegian and Koonin compared genomes of Haemophilus influenzae and Mycoplasma genitalium to determine the minimal gene set (Mushegian and Koonin, 1996). In the past few years, the accumulation of completely sequenced bacterial genomes and the establishment of essential gene database greatly facilitated the identification of bacterial gene essentiality in silico. Computational methods are becoming more important in essential gene study because they can dramatically save time and efforts. This article is a comprehensive overview of online resources to identify and predict bacterial essential genes. We present some available web resources related to gene essentiality, including the bioinformatic tools and databases. We also summarize several features used in essential gene prediction. In the final part, the currently available online bacterial essential gene prediction servers are listed and tried based on the experimentally validated essential gene sets of 30 bacteria for evaluation. Figure 1 shows the outline of this article.
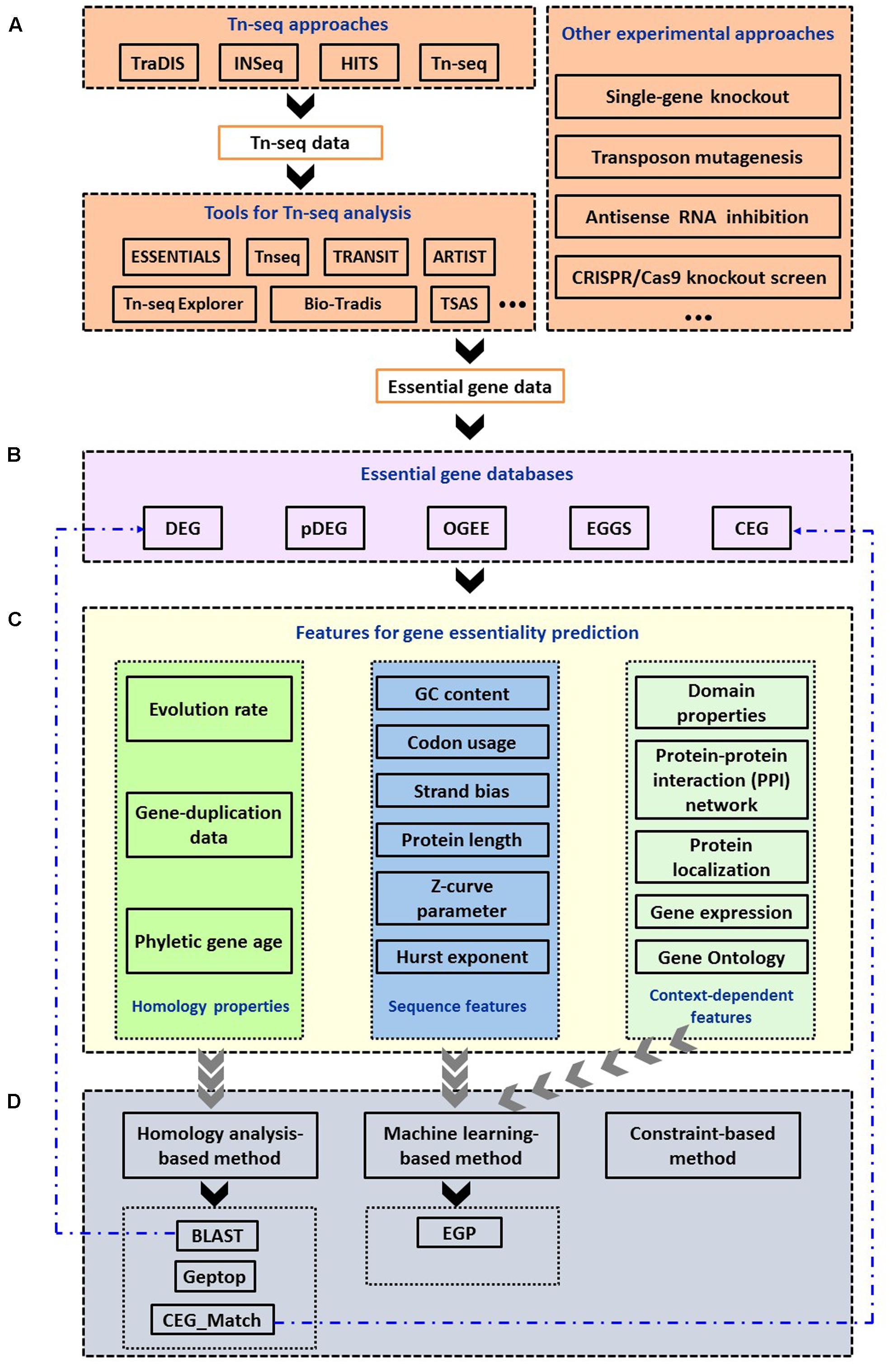
FIGURE 1. Summary of computational methods used in the identification and prediction of bacterial essential genes. (A) Experimental approaches and bioinformatic tools to identify essential genes. (B) Essential gene databases. (C) Features used for gene essentiality prediction. (D) Computational approaches to predict essential genes and online services.
Experimental Approaches and Bioinformatic Tools to Identify Essential Genes
Previous experimental approaches used to identify essential genes include the systematic inactivation of each individual gene present in a genome, the use of antisense RNA to inhibit gene expression and massive transposon mutagenesis (the most widely used approach) (Gil et al., 2004). Briefly, the single-gene knockout strategy is designed to insert a non-replicating plasmid into the target gene via a single crossover recombination, which is able to disrupt the function of the target gene and generate knockout mutations. The gene that could not be inactivated by insertion is deemed essential (Kobayashi et al., 2003). Antisense RNA inhibition method decreases the expression level of a target gene through binding by double-stranded RNA (dsRNA) (Ji et al., 2001). Another method, transposon mutagenesis is used to identify essential genes by constructing a random transposon-insertion library, then determining the insertion sites by DNA hybridization (Hensel et al., 1995) or microarray (Mazurkiewicz et al., 2006). However, these experimental methods have limitations more or less. The single-gene knockout strategy requires detailed genome annotation. The use of antisense RNA is limited to the genes for which an adequate expression of the inhibitory RNA can be obtained in the organism under study. Shortcomings of transposon mutagenesis method include missing low-abundance transcripts, low resolution in locating insertion sites, and narrow ranges in counting probe density. Therefore, these methods have only been performed in a limited number of organisms and identified their essential genes with low throughput (Gil et al., 2004).
In recent years, technologies that use a random transposon mutant library followed by next-generation sequencing such as transposon-directed insertion site sequencing (TraDIS) (Langridge et al., 2009), insertion sequencing (INSeq) (Goodman et al., 2009), high-throughput insertion tracking by deep sequencing (HITS) (Gawronski et al., 2009) and transposon insertion site sequencing (Tn-seq) (van Opijnen et al., 2009; van Opijnen and Camilli, 2013) are becoming powerful tools to facilitate high-throughput identification of essential genes. Currently, several bioinformatic software tools have been built and maintained by different research groups, which help researchers to analyze the data from transposon insertion sequencing experiments. A list of Tn-seq data analysis software tools related to essential genes is presented in Table 1. Most of these tools are included in the manually curated meta-database OMICtools (Henry et al., 2014).
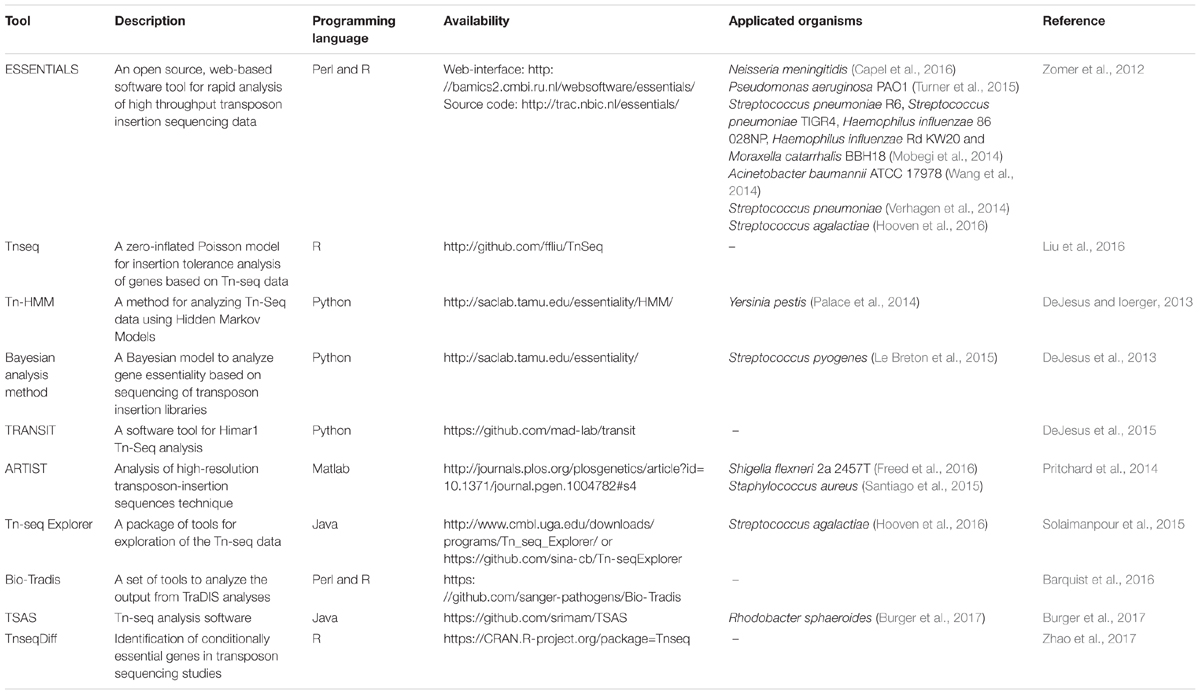
TABLE 1. Software tools to analyze transposon insertion sequencing data for identifying essential genes.
Table 1 shows that several software tools, especially ESSENTIALS (Zomer et al., 2012), have been successfully applied to the genome-wide essential genes screens in many microorganisms. ESSENTIALS uses the Negative Binomial distribution statistical model to quantify the statistical significance of essential regions. It adopts many data preprocessing steps such as data filtering and normalization as well as post-processing steps to optimize the gene essentiality prediction. ESSENTIALS provides both source code and web-interface, so that researchers with no previous computational experience can analyze the Tn-seq data (Chao et al., 2016). Tn-seq Explorer utilizes a sliding window approach which counts insertions in overlapping windows of a specific size. Regions that are significantly underrepresented in read counts compared with the rest of the genome are identified as essential genes or possibly other essential genomic segments (Solaimanpour et al., 2015). This approach can identify essential genes with a high-resolution. However, when the window size decreases and the number of windows increases, the operational quantity will be magnified (Chao et al., 2016). Algorithms in other software include models using the Poisson distribution (Liu et al., 2016), Bayesian analysis method (DeJesus et al., 2013) and Hidden Markov Models (DeJesus and Ioerger, 2013; Pritchard et al., 2014). TRANSIT, a pipeline for analyzing Himar1 Tn-seq data was developed in 2015. This tool provides two different statistical methods (Bayesian/Gumbel Method and Hidden Markov Model) to identify essential genes in individual datasets and a resampling method to identify conditionally essential genes between different growth conditions (DeJesus et al., 2015). The various statistical methods and the graphical interface make TRANSIT an effective and convenient Tn-seq data analysis tool. However, TRANSIT only offers automatic observation on libraries generated by using the Himar1 transposon. When analyzing other TnSeq libraries, a pre-processor is needed to modify the format of data files. TnseqDiff is a parametric method which uses insertion-level data to identify conditionally essential genes. This method is able to deal with data with multiple experimental conditions (Zhao et al., 2017). Bio-Tradis is a novel software tool for analyzing the output of TraDIS analyses. The provided service is similar to that in Tn-seq Explorer and TRANSIT. Better yet, this is a command-line driven approach which allows the simultaneous processing of many sequencing libraries (Barquist et al., 2016).
More recently, the CRISPR-Cas9 technology has also been used to identify essential genes (Wang et al., 2015; Morgens et al., 2016). Clustered regularly interspaced short palindromic repeats (CRISPRs), together with CRISPR-associated (Cas) proteins, provide bacteria with adaptive immunity to viruses and plasmids (Barrangou and Doudna, 2016). In the CRISPR-Cas9 system, single guide RNAs (sgRNAs), which retain a sequence complementary to the targeted region, direct Cas9 endonucleases to induce a site-specific double-strand break in the DNA. Then the double-strand break is repaired by non-homologous end-joining (NHEJ). Thus, the CRISPR system is able to knockout genes at DNA level (Doudna and Charpentier, 2014). Compared with other methods, CRISPR-based methods have features of low noise, minimal off-target effects and consistent activity across reagents (Evers et al., 2016). Currently, this method is mainly adapted to mammalian cell lines. Therefore, we have not discussed its details in this article.
Bioinformatic Databases About Essential Genes
By utilizing the experimental approaches and bioinformatic tools, researchers are able to quickly and accurately identify essential genes in a wide range of microorganisms under different experimental conditions. Experimentally screened essential gene data are constantly accumulating. These dramatically increasing data form the foundation of the development of secondary databases about essential genes. In the following part, we list some available web resources and servers related to essential genes and discuss them in detail.
DEG (a database of essential genes) is a comprehensive platform for essential genes. This database was constructed in 2004 and has been updated constantly. The newly released DEG 10 contains a considerable number of essential and non-essential genes in archaeal, bacterial and eukaryotic organisms determined under different environments. Non-essential genes can also be determined in many genome-wide essentiality screens. For the genes whose essentialities are undefined due to the limitation of the experiments, they can neither be classified as essential genes nor as non-essential genes. So non-essential genes are not always the complementary set of essential genes and vice versa. Other essential genomic elements such as essential non-coding RNAs, regulatory sequences, essential promoters and even replication origins are also included. In addition, users are allowed to perform homology searches with the embedded BLAST tool provided in the database. Single genes, multiple genes, annotated genomes and even unannotated genomes can be submitted to DEG for BLAST searches (Zhang et al., 2004; Zhang and Lin, 2009; Luo et al., 2014). The timely updated information and practical tool in DEG make it the most widely used database about essential genes.
Lin and Zhang (2011) developed an essential gene prediction algorithm by integrating the information of biased distribution of essential genes in leading and lagging strands, homologous search and codon adaptation index (CAI) values. The algorithm takes 310 and 379 essential genes in Mycoplasma pulmonis UAB CTIP and Mycoplasma genitalium G37 contained in DEG as training set. The prediction accuracy in self-consistence and cross-validation tests are 80.8 and 78.9% respectively. 5880 essential genes were predicted by this prediction algorithm in 16 Mycoplasma genomes. The predicted genes were then stored in a database of predicted Essential Genes (pDEG). Many detailed information of the predicted essential genes are provided in the database, and the records can be freely accessed and downloaded (Lin and Zhang, 2011).
OGEE is an Online GEne Essentiality database. Both essential and non-essential genes obtained from large-scale experiments are openly accessible in this database. The developers also complement their data with text-mining results. For each gene, a list of associated gene properties, such as gene duplication status, evolutionary origins of the gene, expression profiles and conservation across species, is also collected. It has been proved in a series of studies that these gene properties can affect gene essentiality. The database offers an integrated online tool. Genes can be divided into different groups according to gene properties including whether a gene is a duplicate or singleton and whether a gene is involved in development. Then the proportion of essential genes in each group can be visualized by this tool. In 2016, a new version of OGEE was developed, and new species as well as new datasets were added. Moreover, as DEG the developers reorganized 16 essential gene datasets from 9 human cancers. Users can know whether a gene is shared within different cancer types or is essential in one particular cancer type with OGEE. OGEE is a useful tool for researchers to study the essentiality of genes (Chen et al., 2012a, 2017).
EGGS (Essential Genes on Genome Scale) is a database that holds microbial gene essentiality data which are acquired from genome-wide essential gene selections. Microbial genes are classified into three categories: essential (E) genes, non-essential (N) genes and ‘undefined’ (U) for all other genes. Essentiality data of each gene can be browsed in a gene/protein page. In the EGGS database, users can also visualize and analyze essentiality data in the context of a Subsystem spreadsheet or on a Subsystem diagram. The collection of annotated Subsystems makes the comparative analysis of these data possible, which greatly facilitates the interpretation and application of essentiality data (Overbeek et al., 2005; Gerdes et al., 2006).
CEG is a database of essential gene clusters. This database is available at http://cefg.cn/ceg/. The developers obtained the data of essential genes from DEG. The difference is that essential genes with the same functions are stored in one orthologous cluster. The size of an essential gene cluster can show whether the gene is shared among many species or is species-specific. These cluster properties are of great help in evolutionary research and drug target discovery. The CEG database also provides a prediction tool CEG_Match to predict essential genes based on standard gene names, which is discussed in detail later (Ye et al., 2013).
Table 2 shows the basic information about the above four databases that store essential genes in the form of single genes. DEG and OGEE contain more species and are updated periodically. It is advised to use these resources as primary ones.
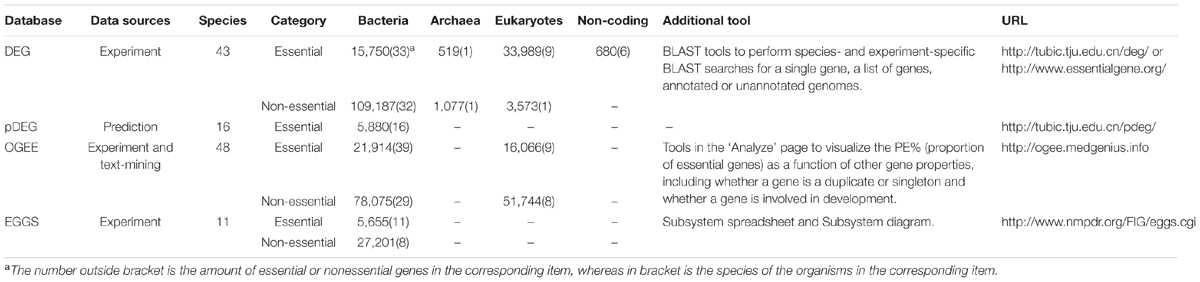
TABLE 2. The basic information of essential gene databases.
Computational Methods for the Prediction of Essential Genes
Homology Search and Evolutionary Analysis-Based Methods
Primal efforts to computationally identify essential genes adopted comparative genomic analysis based on sequence homology. Researchers tried to predict the minimal gene set by comparing the first sequenced genomes of Haemophilus influenzae and Mycoplasma genitalium, and identified 256 candidate essential genes (Mushegian and Koonin, 1996). The ideology for homology mapping methods is simple, i.e., genes shared by distantly related organisms are likely to be essential (Koonin, 2003). With the completion of more bacterial genomes’ sequencing, researchers tried to analyze bacterial genome data in different strains of a single species. Comparative genomic analysis including core genes identification (Zafar et al., 2002) has been successfully implemented to infer the essential genes from the pan-genome of bacterial species such as Mycoplasma (Liu et al., 2012), Liberibacter (Fagen et al., 2014), Plasmodium falciparum (Rout et al., 2015) and Brucella spp. (Yang et al., 2016). The evolutionary rate of essential genes is slower than that of non-essential genes. So essential genes are more evolutionarily conserved in bacteria (Jordan et al., 2002; Luo et al., 2015). Other homology properties such as gene-duplication data and phyletic gene age have also been used in the prediction of essential genes. Duplicated genes are also called paralogs. Function and expression of these paralogs often overlap with each other. Duplicated genes are less likely to be essential than singletons because deleting one of the duplicates is not lethal to an organism (Jordan et al., 2002; Chen and Xu, 2005). Genes with more recent phyletic origins (younger genes) are less likely to be essential than that with earlier phyletic origin (older genes). For genes of the same age, singletons are more likely to be essential than duplicates (Chen et al., 2012b). Homology mapping can be used to predict essential genes based solely on genomic sequences. However, this method is limited to conserved orthologs between different species, which often make up only a small percentage of the genomes (Bruccoleri et al., 1998). Moreover, although essential genes tend to be highly conserved, the conserved genes across species are not always essential.
Machine Learning-Based Methods
Machine learning-based method is another widely used approach to predict essential genes. This method identifies essential genes by constructing and training a classifier according to the features of known essential and non-essential genes. Then the classifiers are applied to the same or other genomes (Zhang et al., 2016). For example, Chen and Xu found the significant correlation between the gene essentiality and its evolutionary rate, gene-duplication rate, its connectivity in protein-protein interaction network and gene-expression cooperativity. By methods of neural network and support vector machine, they predicted gene essentiality of high-throughput data in yeast Saccharomyces cerevisiae (Chen and Xu, 2005). Machine-learning algorithms used to train the classifier include support vector machine (SVM), neural network, decision tree, Naïve Bayes model, feature-based weighted Naïve Bayes model (FWM) (Cheng et al., 2013; Ning et al., 2014), and so on. With the advancement in research, a variety of genomic and protein features have been analyzed and used in gene essentiality prediction studies. Generally, the features can be broadly classified into two groups: sequence derived features and context-dependent features (Wang et al., 2013; Mobegi et al., 2016).
Sequence Derived Features of Essential Genes
(1) GC content. DNA with high GC content is believed to be more robust and stable (Seringhaus et al., 2006).
(2) Codon usage. The codon usage of essential genes suffers from more evolutionary constraints than non-essential genes (Jordan et al., 2002).
(3) Strand bias. Essential genes tend to be encoded on the leading strand of the chromosome (Lin et al., 2010; Rocha and Danchin, 2003).
(4) Protein length. Although protein length tends to become longer through evolution, essential genes, compared to non-essential genes, have a significantly higher proportion of large and small proteins relative to medium-sized proteins (Lipman et al., 2002; Gong et al., 2008).
(5) Z-curve parameter. The Z-curve theory is a bioinformatic algorithm to display base composition distributions along DNA sequences (Zhang and Zhang, 1994; Zhang, 1997; Gao and Zhang, 2004). All the information that a given DNA sequence carries is included in the corresponding Z-curve. So Z-curve features can be used as sequence derived features for essential gene prediction (Song et al., 2014; Lin et al., 2017). Based on the Z-curve theory, Guo et al. (2017) created a λ-interval Z-curve, which considered the interval range association. They then built a support vector machine-based model to predict human gene essentiality with the λ-interval Z-curve, and obtained excellent performance (Guo et al., 2017).
(6) Hurst exponent. The Hurst exponent is a characteristic parameter which describes the degree of self-similarity of a data set. For genes of similar length, the average Hurst exponent of essential genes is smaller than that of non-essential genes (Zhou and Yu, 2014).
Context-Dependent Features of Essential Proteins
(1) Domain properties. Protein essentiality is not likely to be conserved through the conservation of overall proteins but through the function of protein domains or domain combinations (Deng et al., 2011).
(2) Protein-protein interaction (PPI) network. Genes or their protein products are connected rather than isolated. Compared with non-essential genes, essential genes tend to be more highly connected in protein interaction networks. Network topology features, such as degree centrality (DC), betweenness centrality (BC), closeness centrality (CC), eigenvector centrality (EC), subgraph centrality (SC) have been used for detecting essential proteins (Estrada, 2006; Acencio and Lemke, 2009; Hwang et al., 2009; Wang et al., 2013; Xiao et al., 2015).
(3) Protein localization. Essential proteins exist in cytoplasm with a higher proportion, while locate in cell envelope such as cytoplasm membrane, periplasm, cell wall and extracellular with a much lower proportion compared with non-essential proteins (Seringhaus et al., 2006; Peng and Gao, 2014).
(4) Gene expression. Genes whose expression levels are higher and stabler under given conditions are more likely to be essential (Jansen et al., 2002).
(5) Gene Ontology. The Gene Ontology (GO) project provides a set of hierarchical controlled vocabularies for describing the biological process, molecular function, and cellular component of gene products (Ashburner et al., 2000). GO terms related to cellular localization and biological process are shown to be reliable predictors of essential genes (Acencio and Lemke, 2009).
Compared with homology mapping, the supervised machine learning-based methods use more genomic and protein features to construct the predicting model. The prediction performance can be improved by selecting appropriate features (Deng et al., 2011; Lu et al., 2014). However, multiple available gene features lead to complexity as well. Different combinations of features may influence the prediction performance. The prediction results in different organisms with the same feature combinations could also be different. How to select suitable features for the organism under study to accurately predict essential genes is still a question (Mobegi et al., 2016). Another limitation of machine learning-based methods is that they may not be suitable for conditionally essential genes prediction.
Constraint-Based Approaches
Genome-scale metabolic networks, which help to understand the systems biology of metabolic pathways within an organism, have been reconstructed based on the genomic sequencing and annotations (Thiele and Palsson, 2010). The structure and function of these networks can be studied by constraint-based modeling methods. Constraint-based modeling uses a series of constraints to describe a biological system and characterize its possible behavior under specific environmental conditions (Edwards et al., 2002; Price et al., 2003; Orth et al., 2010). Constraint-based models have been reconstructed in organisms across all three domains of life. These models have promoted the investigation of gene essentiality (Joyce and Palsson, 2008).
Flux balance analysis (FBA) is the most widely used constraint-based approach to analyze the properties of metabolic networks. This approach allows the prediction the metabolite fluxes at steady state by applying mass balance constraints to a stoichiometric model (Kauffman et al., 2003; Raman and Chandra, 2009; Orth et al., 2010). The basic idea of applying FBA to predict essential genes is to simulate the knockout of a gene, and then evaluate the associated lethality on the system (Basler, 2015). Usually, the building and analysis procedure of FBA model can be divided into three steps. First, reconstruct the metabolic network and compile the stoichiometric matrix. Second, identify and apply appropriate constraints to the network. Finally, find the optimal flux distribution by linear programming and assess the essentiality of a gene through analysis of the optimal flux distribution in the network (Joyce and Palsson, 2008; Lu et al., 2014; Basler, 2015). FBA is less computationally expensive because it does not require kinetic parameters. FBA can be used to perform the simulation of large numbers of perturbations to the network. This approach is suitable for conditionally essential gene studies. However, it cannot be used to predict metabolite concentrations or transient dynamic states because it does not use kinetic parameters. Furthermore, the predictions sometimes disagree with experimental data, because FBA does not account for regulatory effects such as regulation of gene expression (Orth et al., 2010; Basler, 2015). Nevertheless, FBA has obvious limitations because it could only predict the essentiality of a metabolic gene.
Evaluation of Online Essential Gene Prediction Servers
The CEG_Match is developed based on the CEG database. It is a gene essentiality prediction tool based on their functions. The CEG_Match predicts essential genes by matching the standard gene names and the cluster names stored in the CEG database. Compared with direct blast search against CEG database, this methodology is more accurate because there are no obvious similarities between two genes with different functions, while two genes without obvious similarities may have the same function. Users should input gene names in a one name per line format or gene sequences in fasta format. They are also required to adjust the minimum matching number before executing the tool. Generally, it’s more likely for the gene to be essential if the matching number is larger. However, the CEG_Match tool has its limitations. It works only when the gene name is known (Guo et al., 2010, 2015; Ye et al., 2013).
Geptop is a gene essentiality prediction tool for sequenced bacterial genomes based on orthology and phylogeny. A gene is more likely to be essential if it is conserved during the long-term evolution, especially in similar species. The reciprocal best hit (RBH) method was used for estimating orthology. The distance of phylogeny between species was computed with the Composition Vector (CV) method. An open source standalone package version is also offered on the website. Any bacterial species with sequenced genome can get essential gene searched by Geptop. Moreover, the website stored essential genes in 968 bacterial genomes predicted by Geptop. Users can browse and download the data for further research (Wei et al., 2013).
ZCURVE (Guo et al., 2003) is a program that predicts genes in bacterial or archaeal genomes. It is developed based on the Z-curve theory. Its latest version ZCURVE 3.0 has an embedded Geptop program, which has an extended function of searching for essential genes in bacterial or archaeal genomes. However, different from the previous Geptop, predicted genes are used here as the input rather than annotated genes. Once the essential genes output option is selected, users can get an output file showing whether each predicted gene is essential or not (Hua et al., 2015).
EGP (Essential Gene Prediction) is an online tool for essential gene prediction of bacteria genomes. It is a support vector machine (SVM)-based method which only uses sequence compositional features. Five groups of features, including amino acid usage, codon usage, nucleotide usage of 3 codon positions, di-nucleotide usage, and CodonW features are independently and jointly input into the SVM to construct the predicting model. The training dataset consists essential genes in 16 bacterial genomes. For large-scale genome sequences, the accuracy of EGP can reach 75%. Users only need to provide nucleotide sequences of genes to make a prediction. The predicted result will be presented on the jumping window or be sent to users by e-mail (Ning et al., 2014).
The basic information of the online essential gene prediction servers including CEG_Match, Geptop, ZCURVE 3.0, EGP and BLAST tool in DEG are presented in Table 3. The differences in the use of each tool are also listed. Researchers can choose the suitable servers according to actual conditions. We test the prediction performance of BLAST tool, Geptop, CEG_Match and EGP by 30 bacteria, whose experimentally validated essential gene sets are collected in DEG. Protein sequences of both essential and non-essential genes in the 30 genomes are independently uploaded to DEG for homologous searching. At the selecting organism step, all the organisms are selected except the one the query proteins belong to, which enable it to be a cross-organism test. Geptop has the same issue. We abandon the web server and use the standalone version to perform the test. When the tested genome is included in the reference species, the other 18 proteomes are used as the training set. A limitation with CEG_Match is that we can only perform the prediction to the genes with known name. We use the AUC [area under the receiver operating characteristic (ROC) curve] score as the standard method to assess the accuracy of the four predictive tools. The AUC scores are shown in Figures 2A,B. The phylogenetic tree was constructed to elucidate the evolutionary relationship among the organisms. The black lines in Figure 2A are the phylogenetic tree of the 30 organisms used in the prediction. The tree was constructed by the MEGA6 program (Tamura et al., 2013) with the sequences of 16S ribosomal RNA of the 30 organisms, which are downloaded from the NCBI website. In Figures 2A,B, we can see that the prediction accuracy of EGP is lower than the other three tools. Figure 2C shows that the prediction accuracy of BLAST tool, Geptop and CEG_Match show positive correlation. For these three tools, if the input species belongs to the same phylogenetic lineage with any of the reference species, the prediction accuracy of this organism is higher. From this we can infer that the accumulation of the experimental data can improve the tools to get better predictions.
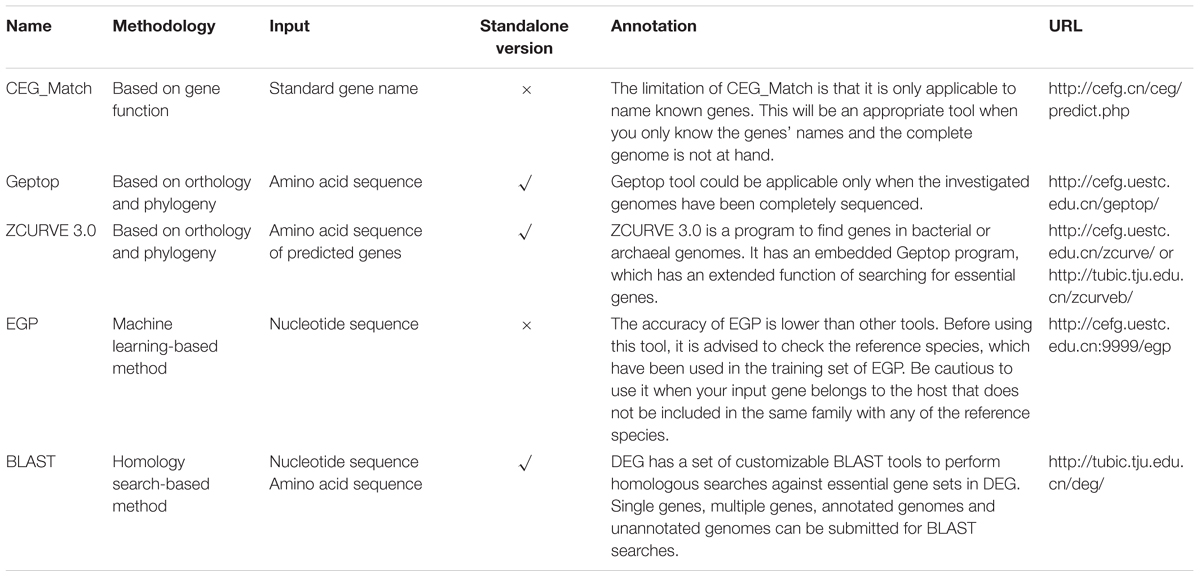
TABLE 3. Summary of the online essential gene prediction servers.
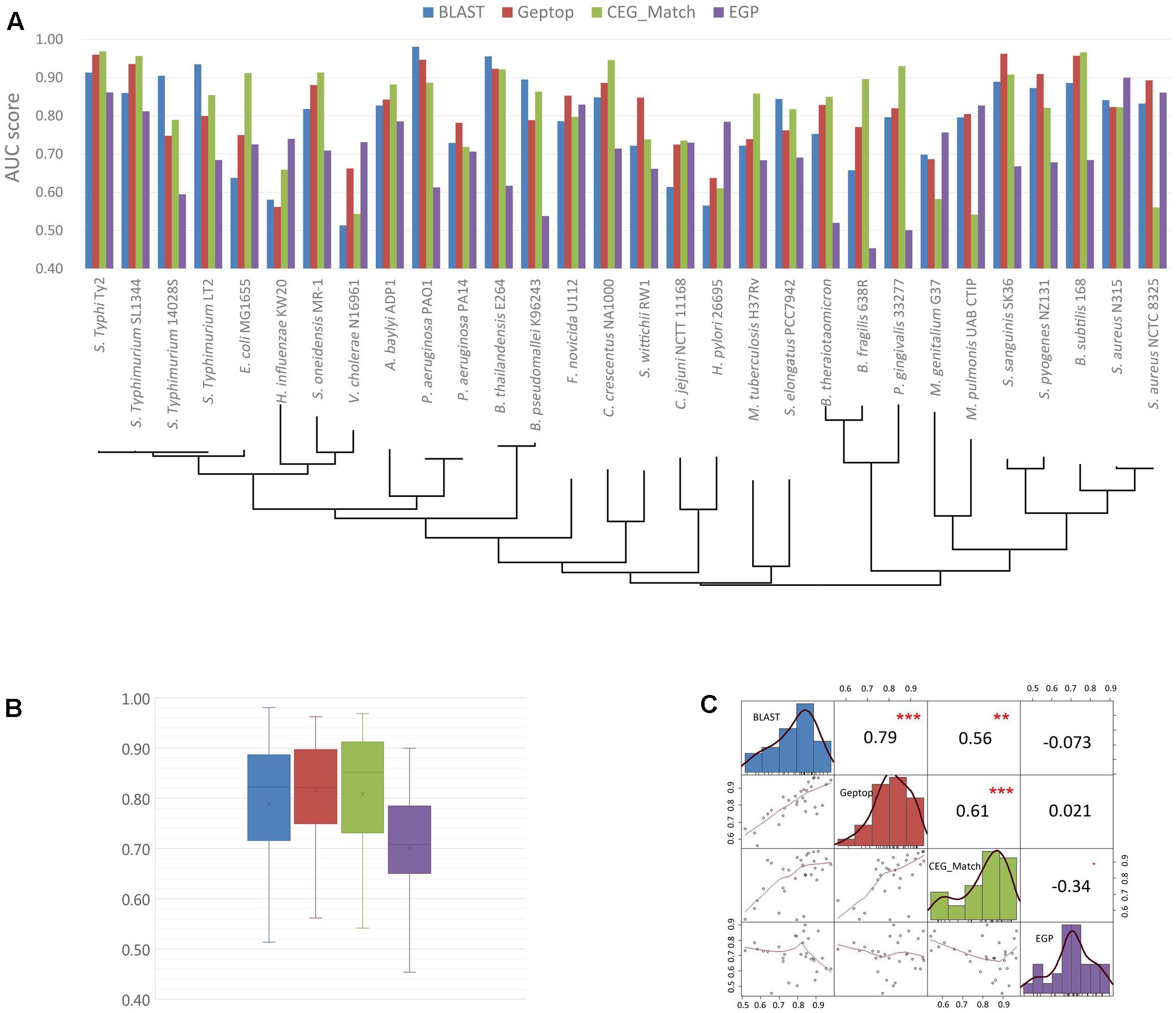
FIGURE 2. Prediction performance of BLAST tool, cross-organism Geptop, CEG_Match and EGP in the 30 genomes. (A) AUC scores of the gene essentiality prediction by BLAST tool, cross-organism Geptop, CEG_Match and EGP incorporating the phylogenetic information of the 30 genomes. (B) Box plot of AUC scores from the prediction of the four tools for the 30 genomes. (C) Correlation analysis of AUC scores from the prediction of the four tools.
Conclusion and Perspectives
Studies on essential genes are gradually becoming popular and can promote our understanding of biology. They may also be applied to pharmaceutical as well as synthetic biology. Predicting essential genes in silico will become more important because computational methods are helpful in reducing the research space for essential gene identification. The computational approaches can be performed only when enough experimental essential genes data are available. The development of many bioinformatic software tools has facilitated the identification of essential genes. The gene essentiality databases have collected such data and contributed a lot in the characterization of essential genes. Multiple computational approaches have been established based on the features proven to be related to gene essentiality, and have made significant advancement in essential gene prediction. In this review, with an emphasis on the online resources, we summarized several computational methods of predicting bacterial essential genes. However, challenges still remain. For example, diverse gene features have been proven to be related to gene essentiality, but finding out true essentiality related features for a given genome is quite complicated. When the prediction methods are applied to a few model organisms, we may usually get favorable results, but when involving more organisms, the results are not so satisfactory. Besides, it is difficult to predict essential genes under different living conditions. For such scenarios, more and better experimental data can trigger the development of enhanced prediction tools.
Author Contributions
FG conceived and designed the study. CP performed the study and drafted the manuscript. YL and HL took part in the data analysis. All the authors edited the manuscript and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 31571358, 21621004, and 31171238) and the National High-Tech Research and Development Program (863) of China (2015AA020101).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Prof. Chun-Ting Zhang and Dr. Ren Zhang for the invaluable assistance and inspiring discussions. They also thank Yi-Zhou Gao in Prof. Feng-Biao Guo’s laboratory for providing the standalone version of Geptop.
References
Acencio, M. L., and Lemke, N. (2009). Towards the prediction of essential genes by integration of network topology, cellular localization and biological process information. BMC Bioinformatics 10:290. doi: 10.1186/1471-2105-10-290
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29.
Barquist, L., Mayho, M., Cummins, C., Cain, A. K., Boinett, C. J., Page, A. J., et al. (2016). The TraDIS toolkit: sequencing and analysis for dense transposon mutant libraries. Bioinformatics 32, 1109–1111. doi: 10.1093/bioinformatics/btw022
Barrangou, R., and Doudna, J. A. (2016). Applications of CRISPR technologies in research and beyond. Nat. Biotechnol. 34, 933–941. doi: 10.1038/nbt.3659
Basler, G. (2015). Computational prediction of essential metabolic genes using constraint-based approaches. Methods Mol. Biol. 1279, 183–204. doi: 10.1007/978-1-4939-2398-4_12
Blomen, V. A., Majek, P., Jae, L. T., Bigenzahn, J. W., Nieuwenhuis, J., Staring, J., et al. (2015). Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096. doi: 10.1126/science.aac7557
Bruccoleri, R. E., Dougherty, T. J., and Davison, D. B. (1998). Concordance analysis of microbial genomes. Nucleic Acids Res. 26, 4482–4486. doi: 10.1093/nar/26.19.4482
Burger, B. T., Imam, S., Scarborough, M. J., Noguera, D. R., and Donohue, T. J. (2017). Combining genome-scale experimental and computational methods to identify essential genes in Rhodobacter sphaeroides. mSystems 2:e00015-17. doi: 10.1128/mSystems.00015-17
Capel, E., Zomer, A. L., Nussbaumer, T., Bole, C., Izac, B., Frapy, E., et al. (2016). Comprehensive identification of meningococcal genes and small noncoding RNAs required for host cell colonization. mBio 7:e01173-16. doi: 10.1128/mBio.01173-16
Chao, M. C., Abel, S., Davis, B. M., and Waldor, M. K. (2016). The design and analysis of transposon insertion sequencing experiments. Nat. Rev. Microbiol. 14, 119–128. doi: 10.1038/nrmicro.2015.7
Chen, W. H., Lu, G., Chen, X., Zhao, X. M., and Bork, P. (2017). OGEE v2: an update of the online gene essentiality database with special focus on differentially essential genes in human cancer cell lines. Nucleic Acids Res. 45, D940–D944. doi: 10.1093/nar/gkw1013
Chen, W. H., Minguez, P., Lercher, M. J., and Bork, P. (2012a). OGEE: an online gene essentiality database. Nucleic Acids Res. 40, D901–D906. doi: 10.1093/nar/gkr986
Chen, W. H., Trachana, K., Lercher, M. J., and Bork, P. (2012b). Younger genes are less likely to be essential than older genes, and duplicates are less likely to be essential than singletons of the same age. Mol. Biol. Evol. 29, 1703–1706. doi: 10.1093/molbev/mss014
Chen, Y., and Xu, D. (2005). Understanding protein dispensability through machine-learning analysis of high-throughput data. Bioinformatics 21, 575–581. doi: 10.1093/bioinformatics/bti058
Cheng, J., Wu, W., Zhang, Y., Li, X., Jiang, X., Wei, G., et al. (2013). A new computational strategy for predicting essential genes. BMC Genomics 14:910. doi: 10.1186/1471-2164-14-910
DeJesus, M. A., Ambadipudi, C., Baker, R., Sassetti, C., and Ioerger, T. R. (2015). TRANSIT - a software tool for Himar1 TnSeq analysis. PLOS Comput. Biol. 11:e1004401. doi: 10.1371/journal.pcbi.1004401
DeJesus, M. A., and Ioerger, T. R. (2013). A Hidden Markov Model for identifying essential and growth-defect regions in bacterial genomes from transposon insertion sequencing data. BMC Bioinformatics 14:303. doi: 10.1186/1471-2105-14-303
DeJesus, M. A., Zhang, Y. J. J., Sassetti, C. M., Rubin, E. J., Sacchettini, J. C., and Ioerger, T. R. (2013). Bayesian analysis of gene essentiality based on sequencing of transposon insertion libraries. Bioinformatics 29, 695–703. doi: 10.1093/bioinformatics/btt043
Deng, J. Y., Deng, L., Su, S. C., Zhang, M. L., Lin, X. D., Wei, L., et al. (2011). Investigating the predictability of essential genes across distantly related organisms using an integrative approach. Nucleic Acids Res. 39, 795–807. doi: 10.1093/nar/gkq784
Doudna, J. A., and Charpentier, E. (2014). The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096. doi: 10.1126/science.1258096
Edwards, J. S., Covert, M., and Palsson, B. (2002). Metabolic modelling of microbes: the flux-balance approach. Environ. Microbiol. 4, 133–140. doi: 10.1046/j.1462-2920.2002.00282.x
Estrada, E. (2006). Virtual identification of essential proteins within the protein interaction network of yeast. Proteomics 6, 35–40. doi: 10.1002/pmic.200500209
Evers, B., Jastrzebski, K., Heijmans, J. P. M., Grernrum, W., Beijersbergen, R. L., and Bernards, R. (2016). CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat. Biotechnol. 34, 631–633. doi: 10.1038/nbt.3536
Fagen, J. R., Leonard, M. T., Mccullough, C. M., Edirisinghe, J. N., Henry, C. S., Davis, M. J., et al. (2014). Comparative genomics of cultured and uncultured strains suggests genes essential for free-living growth of liberibacter. PLOS ONE 9:e84469. doi: 10.1371/journal.pone.0084469
Freed, N. E., Bumann, D., and Silander, O. K. (2016). Combining Shigella Tn-seq data with gold-standard E. coli gene deletion data suggests rare transitions between essential and non-essential gene functionality. BMC Microbiol. 16:203. doi: 10.1186/s12866-016-0818-0
Galperin, M. Y., and Koonin, E. V. (1999). Searching for drug targets in microbial genomes. Curr. Opin. Biotechnol. 10, 571–578. doi: 10.1016/s0958-1669(99)00035-x
Gao, F., and Zhang, C. T. (2004). Comparison of various algorithms for recognizing short coding sequences of human genes. Bioinformatics 20, 673–U232. doi: 10.1093/bioinformatics/btg467
Gao, F., and Zhang, R. R. (2011). Enzymes are enriched in bacterial essential genes. PLOS ONE 6:e21683. doi: 10.1371/journal.pone.0021683
Gawronski, J. D., Wong, S. M. S., Giannoukos, G., Ward, D. V., and Akerley, B. J. (2009). Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc. Natl. Acad. Sci. U.S.A. 106, 16422–16427. doi: 10.1073/pnas.0906627106
Gerdes, S., Edwards, R., Kubal, M., Fonstein, M., Stevens, R., and Osterman, A. (2006). Essential genes on metabolic maps. Curr. Opin. Biotechnol. 17, 448–456. doi: 10.1016/j.copbio.2006.08.006
Giaever, G., Chu, A. M., Ni, L., Connelly, C., Riles, L., Veronneau, S., et al. (2002). Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391. doi: 10.1038/nature00935
Gibson, D. G., Glass, J. I., Lartigue, C., Noskov, V. N., Chuang, R. Y., Algire, M. A., et al. (2010). Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329, 52–56. doi: 10.1126/science.1190719
Gil, R., Silva, F. J., Pereto, J., and Moya, A. (2004). Determination of the core of a minimal bacterial gene set. Microbiol. Mol. Biol. Rev. 68, 518–537. doi: 10.1128/mmbr.68.3.518-537.2004
Gong, X. D., Fan, S. H., Bilderbeck, A., Li, M. K., Pang, H. X., and Tao, S. H. (2008). Comparative analysis of essential genes and nonessential genes in Escherichia coli K12. Mol. Genet. Genomics 279, 87–94. doi: 10.1007/s00438-007-0298-x
Goodman, A. L., Mcnulty, N. P., Zhao, Y., Leip, D., Mitra, R. D., Lozupone, C. A., et al. (2009). Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6, 279–289. doi: 10.1016/j.chom.2009.08.003
Guo, F. B., Dong, C., Hua, H. L., Liu, S., Luo, H., Zhang, H. W., et al. (2017). Accurate prediction of human essential genes using only nucleotide composition and association information. Bioinformatics 33, 1758–1764. doi: 10.1093/bioinformatics/btx055
Guo, F. B., Ning, L. W., Huang, J., Lin, H., and Zhang, H. X. (2010). Chromosome translocation and its consequence in the genome of Burkholderia cenocepacia AU-1054. Biochem. Biophys. Res. Commun. 403, 375–379. doi: 10.1016/j.bbrc.2010.11.039
Guo, F. B., Ou, H. Y., and Zhang, C. T. (2003). ZCURVE: a new system for recognizing protein-coding genes in bacterial and archaeal genomes. Nucleic Acids Res. 31, 1780–1789. doi: 10.1093/nar/gkg254
Guo, F. B., Ye, Y. N., Ning, L. W., and Wei, W. (2015). Three computational tools for predicting bacterial essential genes. Methods Mol. Biol. 1279, 205–217. doi: 10.1007/978-1-4939-2398-4_13
Henry, V. J., Bandrowski, A. E., Pepin, A. S., Gonzalez, B. J., and Desfeux, A. (2014). OMICtools: an informative directory for multi-omic data analysis. Database 2014:bau069. doi: 10.1093/database/bau069
Hensel, M., Shea, J. E., Gleeson, C., Jones, M. D., Dalton, E., and Holden, D. W. (1995). Simultaneous identification of bacterial virulence genes by negative selection. Science 269, 400–403. doi: 10.1126/science.7618105
Hooven, T. A., Catomeris, A. J., Akabas, L. H., Randis, T. M., Maskell, D. J., Peters, S. E., et al. (2016). The essential genome of Streptococcus agalactiae. BMC Genomics 17:406. doi: 10.1186/s12864-016-2741-z
Hua, Z. G., Lin, Y., Yuan, Y. Z., Yang, D. C., Wei, W., and Guo, F. B. (2015). ZCURVE 3.0: identify prokaryotic genes with higher accuracy as well as automatically and accurately select essential genes. Nucleic Acids Res. 43, W85–W90. doi: 10.1093/nar/gkv491
Hutchison, C. A. III, Chuang, R. Y., Noskov, V. N., Assad-Garcia, N., Deerinck, T. J., Ellisman, M. H., et al. (2016). Design and synthesis of a minimal bacterial genome. Science 351:aad6253. doi: 10.1126/science.aad6253
Hutchison, C. A., Peterson, S. N., Gill, S. R., Cline, R. T., White, O., Fraser, C. M., et al. (1999). Global transposon mutagenesis and a minimal mycoplasma genome. Science 286, 2165–2169. doi: 10.1126/science.286.5447.2165
Hwang, Y. C., Lin, C. C., Chang, J. Y., Mori, H., Juan, H. F., and Huang, H. C. (2009). Predicting essential genes based on network and sequence analysis. Mol. Biosyst. 5, 1672–1678. doi: 10.1039/b900611g
Jansen, R., Greenbaum, D., and Gerstein, M. (2002). Relating whole-genome expression data with protein-protein interactions. Genome Res. 12, 37–46. doi: 10.1101/gr.205602
Ji, Y. D., Zhang, B., Van Horn, S. F., Warren, P., Woodnutt, G., Burnham, M. K. R., et al. (2001). Identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA. Science 293, 2266–2269. doi: 10.1126/science.1063566
Jordan, I. K., Rogozin, I. B., Wolf, Y. I., and Koonin, E. V. (2002). Essential genes are more evolutionarily conserved than are nonessential genes in bacteria. Genome Res. 12, 962–968. doi: 10.1101/gr.87702
Joyce, A. R., and Palsson, B. O. (2008). Predicting gene essentiality using genome-scale in silico models. Methods Mol. Biol. 416, 433–457. doi: 10.1007/978-1-59745-321-9_30
Juhas, M., Eberl, L., and Church, G. M. (2012). Essential genes as antimicrobial targets and cornerstones of synthetic biology. Trends Biotechnol. 30, 601–607. doi: 10.1016/j.tibtech.2012.08.002
Juhas, M., Eberl, L., and Glass, J. I. (2011). Essence of life: essential genes of minimal genomes. Trends Cell Biol. 21, 562–568. doi: 10.1016/j.tcb.2011.07.005
Kauffman, K. J., Prakash, P., and Edwards, J. S. (2003). Advances in flux balance analysis. Curr. Opin. Biotechnol. 14, 491–496. doi: 10.1016/j.copbio.2003.08.001
Kim, D.-U., Hayles, J., Kim, D., Wood, V., Park, H.-O., Won, M., et al. (2010). Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 28, 617–623. doi: 10.1038/nbt.1628
Kobayashi, K., Ehrlich, S. D., Albertini, A., Amati, G., Andersen, K. K., Arnaud, M., et al. (2003). Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. U.S.A. 100, 4678–4683. doi: 10.1073/pnas.0730515100
Koonin, E. V. (2000). How many genes can make a cell: the minimal-gene-set concept. Annu. Rev. Genomics Hum. Genet. 1, 99–116. doi: 10.1146/annurev.genom.1.1.99
Koonin, E. V. (2003). Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat. Rev. Microbiol. 1, 127–136. doi: 10.1038/nrmicro751
Langridge, G. C., Phan, M.-D., Turner, D. J., Perkins, T. T., Parts, L., Haase, J., et al. (2009). Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res. 19, 2308–2316. doi: 10.1101/gr.097097.109
Le Breton, Y., Belew, A. T., Valdes, K. M., Islam, E., Curry, P., Tettelin, H., et al. (2015). Essential genes in the core genome of the human pathogen Streptococcus pyogenes. Sci. Rep. 5:9838. doi: 10.1038/srep09838
Liao, B. Y., and Zhang, J. (2007). Mouse duplicate genes are as essential as singletons. Trends Genet. 23, 378–381. doi: 10.1016/j.tig.2007.05.006
Lin, Y., Gao, F., and Zhang, C. T. (2010). Functionality of essential genes drives gene strand-bias in bacterial genomes. Biochem. Biophys. Res. Commun. 396, 472–476. doi: 10.1016/j.bbrc.2010.04.119
Lin, Y., Zhang, F. Z., Xue, K., Gao, Y. Z., and Guo, F. B. (2017). Identifying bacterial essential genes based on a feature-integrated method. IEEE/ACM Trans. Comput. Biol. Bioinform. doi: 10.1109/tcbb.2017.2669968 [Epub ahead of print].
Lin, Y., and Zhang, R. R. (2011). Putative essential and core-essential genes in Mycoplasma genomes. Sci. Rep. 1:53. doi: 10.1038/srep00053
Lipman, D. J., Souvorov, A., Koonin, E. V., Panchenko, A. R., and Tatusova, T. A. (2002). The relationship of protein conservation and sequence length. BMC Evol. Biol. 2:20. doi: 10.1186/1471-2148-2-20
Liu, F. F., Wang, C., Wu, Z. W., Zhang, Q. J., and Liu, P. (2016). A zero-inflated Poisson model for insertion tolerance analysis of genes based on Tn-seq data. Bioinformatics 32, 1701–1708. doi: 10.1093/bioinformatics/btw061
Liu, W., Fang, L., Li, M., Li, S., Guo, S., Luo, R., et al. (2012). Comparative genomics of Mycoplasma: analysis of conserved essential genes and diversity of the pan-genome. PLOS ONE 7:e35698. doi: 10.1371/journal.pone.0035698
Lu, Y., Deng, J., Carson, M. B., Lu, H., and Lu, L. J. (2014). Computational methods for the prediction of microbial essential genes. Curr. Bioinform. 9, 89–101. doi: 10.2174/1574893608999140109113434
Luo, H., Gao, F., and Lin, Y. (2015). Evolutionary conservation analysis between the essential and nonessential genes in bacterial genomes. Sci. Rep. 5:13210. doi: 10.1038/srep13210
Luo, H., Lin, Y., Gao, F., Zhang, C. T., and Zhang, R. (2014). DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 42, D574–D580. doi: 10.1093/nar/gkt1131
Mazurkiewicz, P., Tang, C. M., Boone, C., and Holden, D. W. (2006). Signature-tagged mutagenesis: barcoding mutants for genome-wide screens. Nat. Rev. Genet. 7, 929–939. doi: 10.1038/nrg.1984
Meinke, D., Muralla, R., Sweeney, C., and Dickerman, A. (2008). Identifying essential genes in Arabidopsis thaliana. Trends Plant Sci. 13, 483–491. doi: 10.1016/j.tplants.2008.06.003
Mobegi, F. M., Van Hijum, S., Burghout, P., Bootsma, H. J., De Vries, S. P. W., Van Der Gaast-De Jongh, C. E., et al. (2014). From microbial gene essentiality to novel antimicrobial drug targets. BMC Genomics 15:958. doi: 10.1186/1471-2164-15-958
Mobegi, F. M., Zomer, A., De Jonge, M. I., and Van Hijum, S. A. (2016). Advances and perspectives in computational prediction of microbial gene essentiality. Brief. Funct. Genomics 16, 70–79. doi: 10.1093/bfgp/elv063
Morgens, D. W., Deans, R. M., Li, A., and Bassik, M. C. (2016). Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat. Biotechnol. 34, 634–636. doi: 10.1038/nbt.3567
Mushegian, A. R., and Koonin, E. V. (1996). A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc. Natl. Acad. Sci. U.S.A. 93, 10268–10273. doi: 10.1073/pnas.93.19.10268
Ning, L. W., Lin, H., Ding, H., Huang, J., Rao, N., and Guo, F. B. (2014). Predicting bacterial essential genes using only sequence composition information. Genet. Mol. Res. 13, 4564–4572. doi: 10.4238/2014.June.17.8
Orth, J. D., Thiele, I., and Palsson, B. O. (2010). What is flux balance analysis? Nat. Biotechnol. 28, 245–248. doi: 10.1038/nbt.1614
Overbeek, R., Begley, T., Butler, R. M., Choudhuri, J. V., Chuang, H. Y., Cohoon, M., et al. (2005). The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33, 5691–5702. doi: 10.1093/nar/gki866
Palace, S. G., Proulx, M. K., Lu, S., Baker, R. E., and Goguen, J. D. (2014). Genome-wide mutant fitness profiling identifies nutritional requirements for optimal growth of Yersinia pestis in deep tissue. mBio 5:e01385-14. doi: 10.1128/mBio.01385-14
Pei, L., Schmidt, M., and Wei, W. (2011). Synthetic biology: an emerging research field in China. Biotechnol. Adv. 29, 804–814. doi: 10.1016/j.biotechadv.2011.06.008
Peng, C., and Gao, F. (2014). Protein localization analysis of essential genes in prokaryotes. Sci. Rep. 4:6001. doi: 10.1038/srep06001
Price, N. D., Papin, J. A., Schilling, C. H., and Palsson, B. O. (2003). Genome-scale microbial in silico models: the constraints-based approach. Trends Biotechnol. 21, 162–169. doi: 10.1016/s0167-7799(03)00030-1
Pritchard, J. R., Chao, M. C., Abel, S., Davis, B. M., Baranowski, C., Zhang, Y. J. J., et al. (2014). ARTIST: high-resolution genome-wide assessment of fitness using transposon-insertion sequencing. PLOS Genet. 10:e1004782. doi: 10.1371/journal.pgen.1004782
Raman, K., and Chandra, N. (2009). Flux balance analysis of biological systems: applications and challenges. Brief. Bioinform. 10, 435–449. doi: 10.1093/bib/bbp011
Rocha, E. P. C., and Danchin, A. (2003). Essentiality, not expressiveness, drives gene-strand bias in bacteria. Nat. Genet. 34, 377–378. doi: 10.1038/ng1209
Rout, S., Warhurst, D. C., Suar, M., and Mahapatra, R. K. (2015). In silico comparative genomics analysis of Plasmodium falciparum for the identification of putative essential genes and therapeutic candidates. J. Microbiol. Methods 109, 1–8. doi: 10.1016/j.mimet.2014.11.016
Santiago, M., Matano, L. M., Moussa, S. H., Gilmore, M. S., Walker, S., and Meredith, T. C. (2015). A new platform for ultra-high density Staphylococcus aureus transposon libraries. BMC Genomics 16:252. doi: 10.1186/s12864-015-1361-3
Sarmiento, F., Mrazek, J., and Whitman, W. B. (2013). Genome-scale analysis of gene function in the hydrogenotrophic methanogenic archaeon Methanococcus maripaludis. Proc. Natl. Acad. Sci. U.S.A. 110, 4726–4731. doi: 10.1073/pnas.1220225110
Seringhaus, M., Paccanaro, A., Borneman, A., Snyder, M., and Gerstein, M. (2006). Predicting essential genes in fungal genomes. Genome Res. 16, 1126–1135. doi: 10.1101/gr.5144106
Solaimanpour, S., Sarmiento, F., and Mrazek, J. (2015). Tn-Seq explorer: a tool for analysis of high-throughput sequencing data of transposon mutant libraries. PLOS ONE 10:e0126070. doi: 10.1371/journal.pone.0126070
Song, K., Tong, T., and Wu, F. (2014). Predicting essential genes in prokaryotic genomes using a linear method: ZUPLS. Integr. Biol. 6, 460–469. doi: 10.1039/c3ib40241j
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Thiele, I., and Palsson, B. O. (2010). A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 5, 93–121. doi: 10.1038/nprot.2009.203
Turner, K. H., Wessel, A. K., Palmer, G. C., Murray, J. L., and Whiteley, M. (2015). Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc. Natl. Acad. Sci. U.S.A. 112, 4110–4115. doi: 10.1073/pnas.1419677112
van Opijnen, T., Bodi, K. L., and Camilli, A. (2009). Tn-seq; high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 6, 767–772. doi: 10.1038/nmeth.1377
van Opijnen, T., and Camilli, A. (2013). Transposon insertion sequencing: a new tool for systems-level analysis of microorganisms. Nat. Rev. Microbiol. 11, 435–442. doi: 10.1038/nrmicro3033
Verhagen, L. M., De Jonge, M. I., Burghout, P., Schraa, K., Spagnuolo, L., Mennens, S., et al. (2014). Genome-wide identification of genes essential for the survival of Streptococcus pneumoniae in human saliva. PLOS ONE 9:e89541. doi: 10.1371/journal.pone.0089541
Wang, J., Peng, W., and Wu, F.-X. (2013). Computational approaches to predicting essential proteins: a survey. Proteomics Clin. Appl. 7, 181–192. doi: 10.1002/prca.201200068
Wang, N. D., Ozer, E. A., Mandel, M. J., and Hauser, A. R. (2014). Genome-wide identification of Acinetobacter baumannii genes necessary for persistence in the lung. mBio 5:e01163-14. doi: 10.1128/mBio.01163-14
Wang, T., Birsoy, K., Hughes, N. W., Krupczak, K. M., Post, Y., Wei, J. J., et al. (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. doi: 10.1126/science.aac7041
Wei, W., Ning, L. W., Ye, Y. N., and Guo, F. B. (2013). Geptop: a gene essentiality prediction tool for sequenced bacterial genomes based on orthology and phylogeny. PLOS ONE 8:e72343. doi: 10.1371/journal.pone.0072343
Xiao, Q., Wang, J., Peng, X., Wu, F.-X., and Pan, Y. (2015). Identifying essential proteins from active PPI networks constructed with dynamic gene expression. BMC Genomics 16:S1. doi: 10.1186/1471-2164-16-s3-s1
Yang, X., Li, Y., Zang, J., Li, Y., Bie, P., Lu, Y., et al. (2016). Analysis of pan-genome to identify the core genes and essential genes of Brucella spp. Mol. Genet. Genomics 291, 905–912. doi: 10.1007/s00438-015-1154-z
Ye, Y. N., Hua, Z. G., Huang, J., Rao, N., and Guo, F. B. (2013). CEG: a database of essential gene clusters. BMC Genomics 14:769. doi: 10.1186/1471-2164-14-769
Zafar, N., Mazumder, R., and Seto, D. (2002). CoreGenes: a computational tool for identifying and cataloging “core” genes in a set of small genomes. BMC Bioinformatics 3:12. doi: 10.1186/1471-2105-3-12
Zhang, C. T. (1997). A symmetrical theory of DNA sequences and its applications. J. Theor. Biol. 187, 297–306. doi: 10.1006/jtbi.1997.0401
Zhang, R., and Lin, Y. (2009). DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Res. 37, D455–D458. doi: 10.1093/nar/gkn858
Zhang, R., Ou, H. Y., and Zhang, C. T. (2004). DEG: a database of essential genes. Nucleic Acids Res. 32, D271–D272. doi: 10.1093/nar/gkh024
Zhang, R., and Zhang, C. T. (1994). Z curves, an intutive tool for visualizing and analyzing the DNA sequences. J. Biomol. Struct. Dyn. 11, 767–782. doi: 10.1080/07391102.1994.10508031
Zhang, X., Acencio, M. L., and Lemke, N. (2016). Predicting essential genes and proteins based on machine learning and network topological features: a comprehensive review. Front. Physiol. 7:75. doi: 10.3389/fphys.2016.00075
Zhang, X., Peng, C., Zhang, G., and Gao, F. (2015). Comparative analysis of essential genes in prokaryotic genomic islands. Sci. Rep. 5:12561. doi: 10.1038/srep12561
Zhao, L., Anderson, M. T., Wu, W., Mobley, H. L. T., and Bachman, M. A. (2017). TnseqDiff: identification of conditionally essential genes in transposon sequencing studies. BMC Bioinformatics 18:326. doi: 10.1186/s12859-017-1745-2
Zheng, W. X., Luo, C. S., Deng, Y. Y., and Guo, F. B. (2015). Essentiality drives the orientation bias of bacterial genes in a continuous manner. Sci. Rep. 5:16431. doi: 10.1038/srep16431
Zhou, Q., and Yu, Y. M. (2014). Comparative analysis of bacterial essential and nonessential genes with Hurst exponent based on chaos game representation. Chaos Solitons Fractals 69, 209–216. doi: 10.1016/j.chaos.2014.10.003
Keywords: essential gene, minimal gene set, gene essentiality prediction, synthetic biology, Tn-seq analysis
Citation: Peng C, Lin Y, Luo H and Gao F (2017) A Comprehensive Overview of Online Resources to Identify and Predict Bacterial Essential Genes. Front. Microbiol. 8:2331. doi: 10.3389/fmicb.2017.02331
Received: 20 July 2017; Accepted: 13 November 2017;
Published: 27 November 2017.
Edited by:
John R. Battista, Louisiana State University, United StatesReviewed by:
Amit Kumar Yadav, Translational Health Science and Technology Institute, IndiaMarco Fondi, University of Florence, Italy
Copyright © 2017 Peng, Lin, Luo and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Gao, fgao@tju.edu.cn