 Skyla A. Duncan
Skyla A. Duncan Dieudonné R. Baganizi
Dieudonné R. Baganizi Rajnish Sahu
Rajnish Sahu Shree R. Singh
Shree R. Singh Vida A. Dennis
Vida A. Dennis- Center for NanoBiotechnology Research, Alabama State University, Montgomery, AL, United States
Severe bacterial infections can lead to both acute and chronic inflammatory conditions. Innate immunity is the first defense mechanism employed against invading bacterial pathogens through the recognition of conserved molecular patterns on bacteria by pattern recognition receptors (PRRs), especially the toll-like receptors (TLRs). TLRs recognize distinct pathogen-associated molecular patterns (PAMPs) that play a critical role in innate immune responses by inducing the expression of several inflammatory genes. Thus, activation of immune cells is regulated by cytokines that use the Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling pathway and microbial recognition by TLRs. This system is tightly controlled by various endogenous molecules to allow for an appropriately regulated and safe host immune response to infections. Suppressor of cytokine signaling (SOCS) family of proteins is one of the central regulators of microbial pathogen-induced signaling of cytokines, principally through the inhibition of the activation of JAK/STAT signaling cascades. This review provides recent knowledge regarding the role of SOCS proteins during bacterial infections, with an emphasis on the mechanisms involved in their induction and regulation of antibacterial immune responses. Furthermore, the implication of SOCS proteins in diverse processes of bacteria to escape host defenses and in the outcome of bacterial infections are discussed, as well as the possibilities offered by these proteins for future targeted antimicrobial therapies.
Introduction
Cytokines are signaling molecules secreted by cells to elicit a particular effect on the behavior and communication of surrounding cells (Dinarello, 2000, 2007; Zhang and An, 2007). They are known protagonists in the development and pathology of a variety of diseases, including but not limited to, autoimmune (He et al., 2016), rheumatoid arthritis (Khondker and Khan, 2014), celiac (Girard-Madoux et al., 2016), bacterial (Yilma et al., 2013), Crohn's (Smith et al., 2009), and cystic fibrosis (Dosunmu et al., 2016). Cytokines are either pro-inflammatory (e.g., IL-6, IFN-γ, TNF-α, IL-1β), anti-inflammatory (e.g., IL-10, IL-1RA, IL-4, IL-13) or chemokines (e.g., IL-8, CCL2, CCL5, CXCL1, CXCL10). While pro-inflammatory cytokines help to exacerbate disease and are algesic (Uceyler et al., 2009), anti-inflammatory cytokines are analgesic (Uceyler et al., 2009) and promote healing, while reducing inflammation. Chemokines are immune migration factors that stimulate the recruitment of leukocytes to the sites of infection. Research evidence has shown that some cytokines participate in both the initiation and persistence of pathologic pain by directly activating nociceptive sensory neurons, which respond to potentially harmful stimuli such as sprains, bruises, burns, and inflammation (Uceyler et al., 2009). Furthermore, pro-inflammatory cytokines (e.g., IL-1β, TNF-α) (Copray et al., 2001; Ozaktay et al., 2006) and chemokines (e.g., CCL2) (Oh et al., 2001; White et al., 2005) may directly modulate neuronal activity in the peripheral and central nervous systems (Zhang and An, 2007).
The breadth, persistence and robust nature of immune responses are dictated by the integration of complex immune signaling cascades mediated by TLRs along with B-cells, T-cells and cytokine receptors (Elliott and Johnston, 2004; Dinarello, 2007). During an immune response, positive signals sent to immune cells via signaling pathways get activated by effector and regulatory T-cells using their negative feedback mechanisms (Dinarello, 2007). This ability of cytokines to have both positive and adverse effects on the immune system highlights the complexity in solidifying the exact role of cytokine biology to structure and function ratio. Innate immune responses although necessary for host survival also may be associated with adverse disease pathology. For example, IFN-γ is essential for defense against several intracellular bacteria such as Listeria monocytogenes, Francisella tularensis, Mycobacteria tuberculosis, and Chlamydia trachomatis but yet bolsters the pathogenesis of several autoimmune diseases (Huang et al., 1993; Harty and Bevan, 1995; Dinarello, 2007). Also, despite the fact that IL-2 is crucial for the generation of cytotoxic T-cells (CTLs) and forms the basis for several vaccines, it drives graft vs. host disease and limits the success of bone marrow transplantation (Dinarello, 2007). Understanding when and how cytokines illicit their pleiotropic and redundant effects on immune responses are essential for designing effective drug therapies.
Suppressor of cytokine signaling (SOCS) family of proteins apparently are modulators of a variety of diseases including those with autoimmune etiologies, inflammation, allergies, bacteria, and cancer. SOCS regulate signaling pathways on an intracellular level to potently and specifically inhibit cytokine and growth factor signaling (Yoshimura et al., 2005; Linossi et al., 2013; Ushiki et al., 2016). There are eight related SOCS family of proteins [SOCS 1-7 and CIS (cytokine-inducible SH2-containing protein)] (Masuhara et al., 1997; Trengove and Ward, 2013; Hao and Sun, 2016) that regulate cytokine signaling by inhibiting JAK activity or targeting signaling components for ubiquitination. Studies have revealed that SOCS protein expression induced by cytokine stimulation can negatively impede cytokine signaling by blocking the JAK/STAT pathway (Cooney, 2002; Elliott and Johnston, 2004; Croker et al., 2008; Tamiya et al., 2011). Other stimuli, including lipopolysaccharide (LPS), bacterial products, and chemokines can also induce SOCS expression (Rakesh and Agrawal, 2005). Since cytokines primarily regulate host immune responses to infection, the tight modulation of cytokines release may hinder disease progression. This review will delve into the regulation of several key cytokines or cytokine cascades by the central action of the intracellular SOCS proteins during a bacterial-induced inflammatory response. Emphasis will be placed on the mechanisms involved in SOCS proteins induction and regulation of antibacterial immune responses. Furthermore, the implication of SOCS proteins in diverse processes of bacteria to escape host defenses and in the outcome of bacterial infections are discussed, as well as the possibilities offered by these proteins for future targeted antimicrobial therapies.
SOCS Family of Proteins and Regulation of Immune Response
Structure of the SOCS Box as Related to Function
The SOCS protein structure consists of an N-terminal domain, a central SH2 domain and a C-terminal SOCS box (Bullock et al., 2007; Hao and Sun, 2016). They all share sequence homology, but especially these pairs, CIS/SOCS1/SOCS2, SOCS3/SOCS4/SOCS5, and SOCS6/SOCS7 have unquestionable marked pair-wise homology. Specifically, the SOCS box is a small, 40- to 60-amino acid (aa) residue domain structurally similar to the domain of the von Hippel–Lindau protein and lesser to the F-box from Skp2 (Kile et al., 2002). The SOCS box interacts with Elongins (B and C) to recruit E2 ubiquitin–transferase, necessary for negative regulation of cytokine signaling (Kamizono et al., 2001). The interaction between SOCS and Elongin BC complex and Cullin 2, facilitates the ubiquitination of JAKs and their cytokine receptors, which targets them for proteasomal degradation (Rawlings et al., 2004; Kershaw et al., 2013).
Structurally, SOCS family of proteins can be subdivided based on aa residues, with the shortest N-terminal region being CIS, SOCS1-3, or longest being SOCS4-7. CIS and SOCS1-3 act in a negative feedback loop through the JAK/STAT pathway in response to cytokine signaling; whereas, SOCS4-7 mainly regulate growth factor receptor signaling (Krebs et al., 2002; Kario et al., 2005; Trengove and Ward, 2013) (Table 1). Notably, SOCS1 and SOCS3 share a similar kinase inhibitory region (KIR) at the N-terminus that is essential for JAK inhibition (Sasaki et al., 1999; Yasukawa et al., 1999; Alexander, 2002; Ushiki et al., 2016). The SH2 domain/KIR ability to inhibit the signaling cascades independently by either blocking STAT docking or directly inhibiting JAK kinase activity confers substrate specificity. Depending on the size and structure of the SOCS protein, each domain interacts directly or indirectly with JAKs or their specific cytokine receptors to inhibit signaling proteins (Hilton, 1999; Nicholson et al., 1999; Sasaki et al., 1999, 2000; Yasukawa et al., 1999; Lehmann et al., 2003). Supposedly, the SOCS box mediates signaling suppression differently by promoting the degradation of bound signaling intermediates via an interaction with the cellular ubiquitination machinery (Zhang et al., 1999, 2001; Kamizono et al., 2001; Kile et al., 2002; Rui et al., 2002; van de Geijn et al., 2004). Revealing how SOCS proteins associate and interact with other proteins or external factors may offer much-needed premise in biomedical therapy approaches.

Table 1. The functions of SOCS 1-7 and CIS proteins.
SOCS Signaling Pathway
The SOCS proteins were first identified based on their ability to suppress cytokine signaling through the JAK/STAT pathway (Dalpke et al., 2003, 2008). The mechanism of cytokines binding to their putative cell surface receptors induces receptor dimerization, which allows trans-phosphorylation of JAKs (Dalpke et al., 2008) and tyrosine phosphorylation of the intracellular receptor subunits, to be bound by STATs. Following STAT phosphorylation, there is dimerization and then translocation into the nucleus (Dalpke et al., 2001). All SOCS proteins inhibit the JAK/STAT pathway similarly, (Dalpke et al., 2001; Caballero et al., 2016) upon cytokine stimulation, which blocks further signaling in a classic feedback loop by targeting signaling intermediates for degradation (Elliott and Johnston, 2004). Moreover, SOCS proteins have been implicated in regulating inflammation and determining cell fate because their obstruction or imbalance causes a broad range of diseases (Elliott and Johnston, 2004).
Upon receiving a signal, a receptor protein changes conformation simultaneously, creating a series of biochemical reactions within the cell that are amplified by intracellular signaling pathways. The JAK/STAT pathway, which coincidentally is involved in SOCS induction, serves as the primary signaling mechanism for most cytokines in mammals (Rawlings et al., 2004). Moreover, the JAK/STAT circuitry includes a negative feedback loop that activates STATs to stimulate the transcription of SOCS genes (Alexander, 2002; Rawlings et al., 2004). JAK is first activated when various ligands, usually cytokines and growth factors bind to cell surface receptors to form a dimer that can phosphorylate each other. This phosphorylation further activates JAK, allowing it to phosphorylate the receptor. When STAT binds to the receptor, it then becomes phosphorylated by JAK. Once phosphorylated, STAT dimerization occurs followed by translocation to the nucleus, where it binds to specific sequences in the DNA. Inactivation of STATs occurs via dephosphorylating proteins along the signaling pathway. Alterations or mutations that perturb the JAK/STAT pathway will affect homeostasis, growth regulation, survival and cell migration; which are all critical functions of this pathway (Rawlings et al., 2004). Furthermore, mutations that activate or fail to regulate JAK signaling properly, cause inflammatory diseases and other etiologies (Rawlings et al., 2004). Because understanding the mechanism of signaling during SOCS-induced responses to bacteria can assist in halting or altering disease pathogenesis, this pathway is of great scientific interest for targeted therapeutics.
Bacterial Pathogenesis and Immune Response
Bacterial Pathogenesis
Despite this new era of biomedical development, the leading cause of mortality is still significantly influenced by new and pre-existing infectious diseases (O'Connor et al., 2006). Added to this for further exacerbation is the increasing incidence of antimicrobial resistant strains, the emergence of new diseases, and the re-surging of older deadly infectious diseases causing a direct negative impact on the economy and welfare in endemic areas (Peterson, 1996; Morens et al., 2004). It is well-documented that microbial pathogens use common strategies to cause infection and disease. These include adherence, invasion, and enhanced pathogenicity, while also evading host defenses (Peterson, 1996; Wilson et al., 2002; Morens et al., 2004). A common strategy employed by bacterial pathogens is the Type III secretion system (T3SS), which in some cases can be used to invade host cells and/or evade immune detection by injecting bacterial signaling proteins to manipulate host immune response for their intracellular survival. Some bacteria that employ the T3SS machinery, as well as the secreted effector proteins for their virulent functions, are C. trachomatis (Betts-Hampikian and Fields, 2010), Yersinia pestis (Nair et al., 2015), Salmonella serovar Typhi (Johnson et al., 2017), Shigella (Hu et al., 2017), Escherichia coli (Hu et al., 2017; Shaulov et al., 2017), and Pseudomonas aeruginosa (Brannon et al., 2009). Pathogens may also reside within a phagolysosome, a phagosome or within the host cell cytosol to evade host immune responses (Wilson et al., 2002). The production of virulent microbial toxins also plays a vital role in the pathogenesis of some diseases (de Sousa, 2003; Ramachandran, 2014). Highly infectious microbes such as Clostridium tetani (tetanus toxin) (Caballero et al., 2016), Corynebacterium diphtheria (diphtheria toxin) (Bermejo-Martin et al., 2016), Shigella dysenteriae (Shiga toxin) (Zadravec et al., 2016), and Clostridium botulinum (botulinum toxin) (Ozcan and Ismi, 2016) produce some of the most potent and lethal toxins.
Immune Responses to Bacterial Infections
The manifestation and severity of a disease are under the influence of the host immune response induced by a bacterial pathogen. Mediation of the host defense mechanisms occurs by its primary and secondary defense responses, respectively innate and adaptive immune responses (Chaplin, 2010). Consequently, the host inflammatory response may be the most important for dealing with microbial infections because it purposely diverts antimicrobial factors such as phagocytes and lymphocytes directly to the infection site. Mediation of inflammation occurs via central effector cells such as mast cells or blood basophils that give rise to localized or systemic responses, respectively (Chaplin, 2010; Ren and Dubner, 2010). Other effectors include phagocytes that engulf microbes, neutralization of microbial pathogens by antibodies or toxins that possess potent antimicrobial properties as well as by lymphocytes and macrophages that initiate immune responses against the pathogen (Tosi, 2005).
When bacteria, such as Neisseria meningitidis, and Salmonella spp. invade their respective hosts; complement proteins are up-regulated and assist in bacteria-killing via complement-mediated lysis (Finlay and McFadden, 2006; Lewis and Ram, 2014). Gram-positive bacteria such as Staphylococcus spp. that are resistant to this type of bacteria-killing mechanism eventually will become opsonized by acute phase proteins and destroyed by phagocytes. However, other pathogens can avoid these above-described killing mechanisms. In these cases, the host relies on cell-mediated immune responses to identify and eliminate such organisms. Macrophages are targets for intracellular bacteria (e.g., Salmonella spp.) that have evaded detection by complement or antibody (do Vale et al., 2016). When infected, these macrophages use MHC class II molecules to present bacterial peptides on their cell surface for recognition by T-helper cells (Goldman and Prabhakar, 1996). T-helper cells recognize the microbial peptides and release IFN-γ that initiates killing mechanisms for clearance of the invading intracellular bacterium (Goldman and Prabhakar, 1996). Notably, many bacteria can benefit from the stimulation of inflammatory reactions as their induced responses usually cause considerable tissue damage to the host making the host more susceptible to an infection (Mogensen, 2009). Moreover, the same cytokines and chemokines present at the inflammatory site are also very critical in regulating the immune system and inflammation (Cekici et al., 2000). Thus, dysregulation or an improper balance of cytokine signaling can cause a variety of diseases not only limited to bacterial but also including allergy, intensified inflammation, and some forms of cancer (26). It is therefore urgent that additional studies be performed with SOCS proteins as inflammatory regulators to encourage novel therapeutic approaches to eradicate bacterial diseases.
Broad Activity of SOCS Proteins in Bacterial Responses
Robust innate and adaptive immune responses against microbial pathogens are determined by the detection of the diverse repertoire of their specific PAMPs, by PRRs of the host innate immune cells such as TLRs, and nucleotide oligomerization domain proteins (NOD) (Janeway and Medzhitov, 2002; O'Riordan et al., 2002; Takeuchi and Akira, 2010). Upon bacterial infection and PAMPs recognition, the PRRs initiate highly complex intracellular signaling pathways, which trigger pro-inflammatory and antimicrobial responses allowing the host to respond promptly to the infection (Athman and Philpott, 2004; Philpott and Girardin, 2004; Kumar and Yu, 2006; Gerold et al., 2007; Mogensen, 2009; Takeuchi and Akira, 2010; Stokes et al., 2015). TLRs play a central role in recognition of PAMPs and in driving host inflammatory responses. They activate the cells of innate immunity and promote pathogen-specific adaptive immunity through their action on antigen-presenting cells (Dalpke et al., 2001; Athman and Philpott, 2004; Kumar and Yu, 2006; Tapping, 2009). Triggering of PRRs and cytokine signaling in immune effector cells induces the expression of inflammatory and antimicrobial mediators as well as regulatory factors, which coordinate the elimination of the pathogen and infected cells (Mogensen, 2009; Takeuchi and Akira, 2010; Stokes et al., 2015). This process mainly occurs via the activation of JAK/STAT signaling pathways and results in gene expression and production of a variety of molecules, including an array of cytokines, chemokines, growth factors and immune-receptors (Rawlings et al., 2004; Mogensen, 2009). These proteins, especially cytokines play essential roles as mediators of immune responses and therefore have to be tightly regulated to induce appropriate and safe antimicrobial responses (Baetz et al., 2007; Dalpke et al., 2008).
SOCS proteins, protein inhibitors of activated stats (PIAS) and protein tyrosine phosphatases (PTPs) are negative regulators that activate the JAK/STAT pathway effectors of PRRs (Rawlings et al., 2004; Abbas et al., 2012). SOCS proteins represent one of the fundamental molecular mechanisms, which regulate the level of microbial pathogen-induced signaling of cytokines employing JAK/STAT signaling cascades (Yoshimura et al., 2012; Trengove and Ward, 2013; Kyoko Inagaki-Ohara, 2014), and also they interfere with cell signaling by mediating the degradation of signaling proteins (Grutkoski et al., 2003). These proteins regulate a broad range of pro- and anti-inflammatory cytokines in immune cells and therefore determine the sensitivity of the host to bacterial infections and the outcome of various bacterial infections (Dalpke et al., 2003; Baetz et al., 2004; Takagi et al., 2004; Yoshimura et al., 2004, 2007; Chaves de Souza et al., 2013).
Gram-Negative Bacteria and SOCS Proteins
Many gram-negative bacteria of the genera Escherichia, Pseudomonas, Chlamydia, Klebsiella, Neisseria, and Salmonella can cause spectra of manifestations in humans (Kang et al., 2005; Mogensen, 2009). Their cell wall is composed of peptidoglycan surrounded by LPS, phospholipids, and proteins (Mogensen, 2009); LPS is their main immune-stimulatory component and primary PAMP (Freudenberg et al., 2008). LPS interacts with host immune cells via TLR4 in association with several co-receptors: myeloid differentiation protein-2 (MD2), CD14 and LPS-binding protein (LBP) (Dumitru et al., 2000; Kumar and Yu, 2006; Strengell et al., 2006; Freudenberg et al., 2008). Moreover, these bacteria can simultaneously activate other TLRs via alternative PAMPs, including TLR2 (peptidoglycan and bacterial membrane proteins), TLR9 (non-methylated CpG (cytosine-guanosine)-DNA), and TLR5 (flagellin) (Mogensen, 2009). The interaction of LPS with TLR4 leads to activation of NF-κB and MAPK (JNK, p38, ERK) via myeloid differentiation factor 88 (MyD88)-dependent pathway, serine/threonine kinase IL-1R-associated kinase 4 (IRAK-4), and TNFR-associated factor 6 (TRAF-6). Besides, there are MyD88-independent pathways that activate interferon regulatory factor-3 (IRF-3) and IRF-7 resulting in the induction of IFN-dependent genes to activate the JAK/STAT pathway (Qin et al., 2007; Freudenberg et al., 2008; Hu et al., 2009). These various activation machineries culminate in triggering multiple immune response genes, especially pro-inflammatory cytokines and chemokines (Nakagawa et al., 2002; Qin et al., 2007; Freudenberg et al., 2008; Hu et al., 2009) (Figure 1).
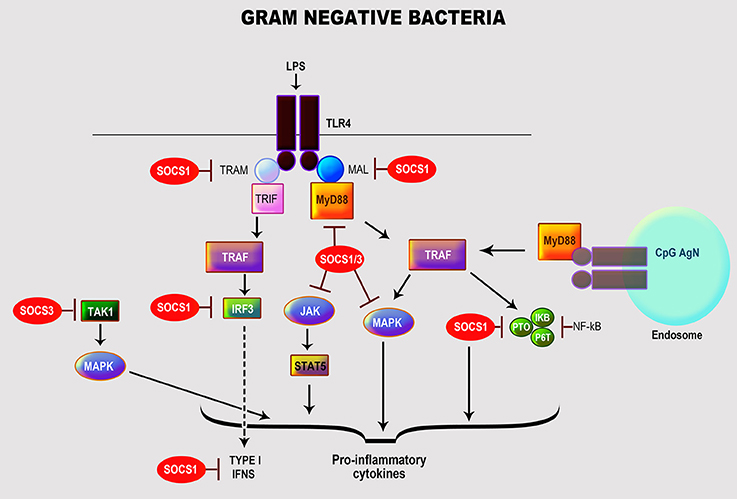
Figure 1. Role of SOCS proteins in the regulation of the signaling pathways induced by recognition of gram-negative bacteria. Recognition of gram-negative bacteria through LPS by TLR4. Activation by LPS of TLR4 leads to the activation of transcription factor NF-κB and MAP kinases (JNK, p38, ERK) by myeloid differentiation factor 88 (MyD88)-dependent pathway, serine/threonine kinase IL-1R-associated kinase 4 (IRAK-4), and TNFR-associated factor 6 (TRAF-6) resulting in the induction of essential cytokines and chemokines (Nakagawa et al., 2002; Qin et al., 2007; Freudenberg et al., 2008; Hu et al., 2009). The regulation of TLR signaling by the specific SOCS protein involved is highlighted in red.
Host immune cells have developed negative regulatory mechanisms, such as SOCS proteins, to control the exacerbated inflammatory reactions caused by prolonged exposure to LPS. Studies have shown that SOCS1 protects a host from fatal LPS responses (Kinjyo et al., 2002; Nakagawa et al., 2002; Hu et al., 2012), as underscored in SOCS1-deficient mice that exhibit a high sensitivity to LPS mediated thru MyD88-dependent and—independent pathways in association with IRAK1 (Kinjyo et al., 2002; Baetz et al., 2004; Croker et al., 2008; Manicassamy and Pulendran, 2009; Fujimoto and Naka, 2010). SOCS1 also facilitates blocking the uptake of LPS in mouse hepatocytes potentially to control sepsis (Scott et al., 2009). Experiments using SOCS1- and IFN-γ-deficient mice showed that IFN-signaling was modulated via JNK, p38, and NF-κB activations (Kinjyo et al., 2002; Croker et al., 2008) through direct interactions with NF-κB p65 and TLR/MAL (MyD88-adaptor-like protein) leading to their suppression and degradation (Nakagawa et al., 2002; Abbas et al., 2012). Others have reported that SOCS1 regulates the IFN-β-induced JAK/STAT pathway by directly inhibiting STAT1 phosphorylation and indirectly TLR4 signaling via IRF-3 (Wilson, 2014).
SOCS3 plays a vital role in regulating LPS inflammation by targeting multiple cytokine signaling cascades. Results from Qin et al. (2007) confirm that the transcriptional expression of SOCS3 by LPS in macrophages and microglia was mediated by activation of MAPK (ERK1/2, JNK, p38), STAT3 and endogenously produced IL-10. Macrophages deficient in SOCS3 expressed heightened LPS-induced STAT1, STAT3, and IL-6, but with no ensuing effect on NF-κB and ERK1/2 activation (Qin et al., 2012; Wilson, 2014). Moreover, it appears that depletion of SOCS3 in macrophages results in positively regulating TLR4 responses by, respectively suppressing STAT3- and SMAD3-mediated IL-6R and TGF-β activations, which are both necessary for negatively regulating LPS-induced IL-6 and TNF-α (Frobose et al., 2006). Also, SOCS3 has been implicated in controlling bone-associated inflammation as it inhibited LPS-induced IL-6 in osteoblasts by blocking the transcription factor, CAAT/enhancer-binding protein (C/EBPβ) (Yan et al., 2010). Paradoxically, SOCS3 positively regulated LPS/TLR4 responses by a feedback inhibition of endogenous TGFβ-1/SMAD3 signaling in macrophages (Liu et al., 2008). Others have reported that SOCS3 regulates IL-10 control of LPS-induced TNF, iNOS (inducible nitric oxide synthase) and nitric oxide (NO) in macrophages by targeting specific SOCS3 protein domains (SH2, SOCS box, and KIR) (Qasimi et al., 2006), thus associating SOCS3 with the IL-10 anti-inflammatory effects. SOCS3 inhibited STAT1 and regulated IFN-γ signaling, in response to LPS stimulation by binding to phosphorylated tyrosine sites of the JAK2 receptor domain (Stoiber et al., 1999) to control macrophage anti-bactericidal effects. Likewise, SOCS3 prevented IL-1 signaling, among others, by inactivating the TRAF-6/TAK1 complex (Posselt et al., 2011; Qin et al., 2012) to regulate LPS deleterious inflammatory responses.
Unlike SOCS1 and SOCS3, the control of LPS signaling by SOCS2 is minimal. Moreover, SOCS2 is differentially regulated in human and mouse cells (Frobose et al., 2006; Hu et al., 2009; Posselt et al., 2011), and the reason for this divergence has yet to be delineated. To promote TLR4 signaling, SOCS2 may target and mediate proteasome-dependent degradation of SOCS1 and SOCS3 (Tannahill et al., 2005; Hu et al., 2009). It is noteworthy to mention that some intact gram-negative organisms like E. coli (Qin et al., 2007; Hu et al., 2012; Demirel et al., 2013), P. aeruginosa (Ding et al., 2017), Chlamydia pneumoniae (Yang et al., 2008), Burkholderia pseudomallei (Ekchariyawat et al., 2005), Salmonella enterica (Uchiya and Nikai, 2005, 2008), Rickettsia conorii (Colonne et al., 2013), and Anaplasma phagocytophilum (Bussmeyer et al., 2010) can directly stimulate the expression of SOCS1 and SOCS3 in vitro and in vivo. These organisms exploit multiple signaling pathways including STAT1, STAT3, MAPK and NF-κB to induce the transcription and/or protein expressions of SOCS1 or SOCS3 as a feedback mechanism to control their induced inflammatory responses (Ekchariyawat et al., 2005; Uchiya and Nikai, 2005, 2008; Yang et al., 2008; Bussmeyer et al., 2010; Colonne et al., 2013; Demirel et al., 2013; Ding et al., 2017).
Gram-Positive Bacteria and SOCS Proteins
Gram-positive bacteria such as Listeria, Bacillus, Clostridium, Staphylococcus, Streptococcus, and Enterococcus cause numerous severe infections in humans (Navarre and Schneewind, 1999; Plouffe, 2000; Hessle et al., 2005; Moellering, 2009; Woodford and Livermore, 2009; van 't Veer et al., 2011). These bacteria have a high resistance to a variety of antimicrobial therapies (Plouffe, 2000; Hessle et al., 2005; Moellering, 2009; Woodford and Livermore, 2009; van 't Veer et al., 2011; Schneewind and Missiakas, 2012) as their cell wall is composed of a layer of peptidoglycan (PGN) and lipoteichoic acid (LTA), encased in the cytoplasmic membrane by diacylglycerol (Nandi et al., 2004; Hessle et al., 2005; Brown et al., 2015). PGN is their principal PAMP that is recognized through Nod-like receptors [NLRs (Nod1 and Nod2)] and cryopyrin response proteins (Plouffe, 2000; Draing et al., 2008; Brown et al., 2015). Exposure to gram-positive bacteria triggers various patterns of pro-inflammatory cytokines notably, amongst many, IL-1α/β, TNF-α, IL-6, and IL-8 (Plouffe, 2000; Draing et al., 2008; Brown et al., 2015). TLR2 is the primary receptor activated in response to PGN and LTA (Draing et al., 2008). Furthermore, both S. aureus and S. pneumoniae LTA-recognition is attained by TLR2 associated with LBP and CD14 in human monocytes to y contribute in the pathogeneses of their diseases (McDonald et al., 2005). The activation of TLR2 by these bacteria is mediated by MyD88 and Toll/interleukin-1 (IL-1)-receptor (TIR)-domain, which leads to the activation of NF-κB, MAPK (via JNK, ERK-1, and p38) and pro-inflammatory caspase-1 (Schroder et al., 2003; Draing et al., 2008) (Figure 2).
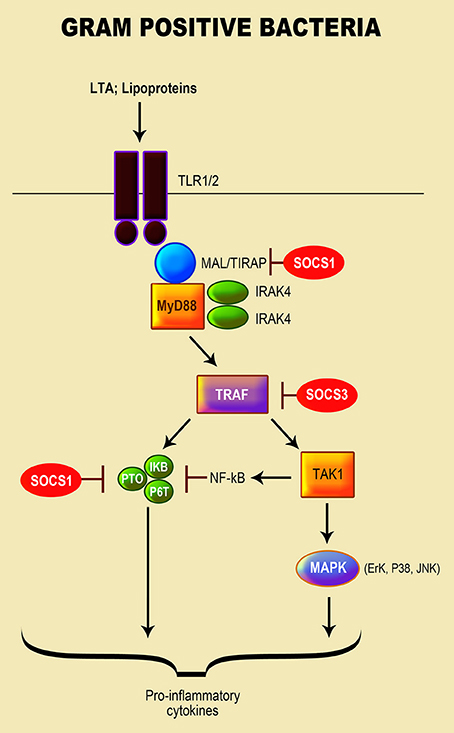
Figure 2. Role of SOCS proteins in the regulation of the signaling pathways induced by recognition of gram positive bacteria. Recognition of gram positive bacteria through their lipopeptide by TLR1 or/and 2. MyD88 and Toll/interleukin-1 (IL-1)-receptor (TIR)-domain mediates the activation of TLR2 by gram-positive bacteria, leading to the activation of the NF-κB pathway; MAPK signaling pathway via JNK, ERK-1, and p38 kinase activation; and pro-inflammatory caspase-1 (Schroder et al., 2003; Draing et al., 2008). The SOCS protein responsible for regulation of TLR signaling is highlighted in red.
The role of SOCS proteins in regulation of gram-positive bacteria-induced inflammation has not been extensively investigated in comparison to gram-negative bacteria and their LPS. Wu and colleagues (Son et al., 2015) reported that SOCS1 enhancement in macrophages infected with the pathogenic Group A Streptococcus (GAS), led to the blockage of cytokine expression. In addition to IFN-β signaling, which is involved in the GAS-induced SOCS1, the TLR4/MyD88 pathway was observed to play a crucial role in stimulating SOCS1 by forming a complex with JAK1/STAT1 (Son et al., 2015). Both Bifidobacterium (B. breve, B. longum, and B. adolescentis) and E. faecalis stimulated an increase in SOCS1 and SOCS3 mRNA transcripts in mouse macrophages by triggering NF-κB and MAPK signaling pathways to regulate the production of pro-inflammatory cytokines (Wu et al., 2015).
A study by Stoiber et al. (Okada et al., 2009) revealed that prolonged infection of macrophages with L. monocytogenes inhibited the phosphorylation of STAT1 and IFN-γ signaling with an enhancement of SOCS3 transcript and protein via the p38 MAPK pathway. Both live and heat-killed bacteria induced SOCS3; however, live bacteria induction of SOCS3 required de novo protein synthesis (Okada et al., 2009). The non-pathogenic probiotic bacterium Lactobacillus and non-pathogenic/pathogenic Streptococcus spp. induced the expression of SOCS3 mRNA in human primary macrophages by directly stimulating macrophages. Expression of SOCS3 by these bacteria was dependent on endogenously produced IL-10 and mediated through the p38 MAPK signaling pathway (Stoiber et al., 2001). Consequently, their stimulation of SOCS3 is induced directly, through at least p38 MAPK-mediated signaling pathway, and indirectly through IL-10 produced by bacterial-stimulated macrophages.
Mycobacteria and SOCS Proteins
The Mycobacterium genus includes, but not limited to, M. tuberculosis and M. avium complexes (Imai et al., 2003; Gao et al., 2006; Latvala et al., 2011), that are responsible for several pulmonary diseases in humans, in particular, Tuberculosis (TB) caused by M. tuberculosis (MTB) (Prince et al., 1989; Gao et al., 2006). Mycobacteria cell wall is composed of a thin internal layer of peptidoglycan, phosphatidyl-myo-inositol mannosides (PIMs) and arabinogalactan, and an external layer of hydrophobic mycolic acids (Nandi et al., 2004; Rottenberg and Carow, 2014). Other components include mannose-capped lipoarabinomannan (Man-LAM), a significant virulence factor; the related lipomannan (LM), and mannoglycoproteins (Rottenberg and Carow, 2014). Mycobacteria are facultative intracellular pathogens, and macrophages are their primary host cells (Gao et al., 2006; Kleinnijenhuis et al., 2011).
Several PRRs are implicated in recognition of mycobacteria by host macrophages and DCs, including TLR1, TLR2, TLR4, and TLR9, C-type lectin receptors (CLRs) (i.e., mannose receptor, DC-SIGN, Mincle, and Dectin-1) and NLRs (Rottenberg and Carow, 2014; Zhao et al., 2014; Mortaz et al., 2015). Numerous mycobacterial components activate TLRs, namely lipoproteins (LpqH, LprA, LprG), PhoS1, LAM, LM, and PIMs, which activate TLR2; glycolipoprotein and PIM6, which activate TLR2/TLR4; and mycobacterial DNA, which respond via TLR9/TLR2 (Killick et al., 2013). Mycobacteria interaction with TLRs results in the activation of NF-κB activated protein-1 (AP-1) via MyD88, MAL, and IRAK, leading to the production of chemokines and several pro-inflammatory cytokines (Rajaram et al., 2014; Rottenberg and Carow, 2014; Zhao et al., 2014) (Figure 3). Mycobacterial components also induce IL-10 via caspase recruitment domain-containing protein 9 (CARD9) or p38 MAPK and serine/threonine Akt kinases (Jo, 2008; Redford et al., 2011).
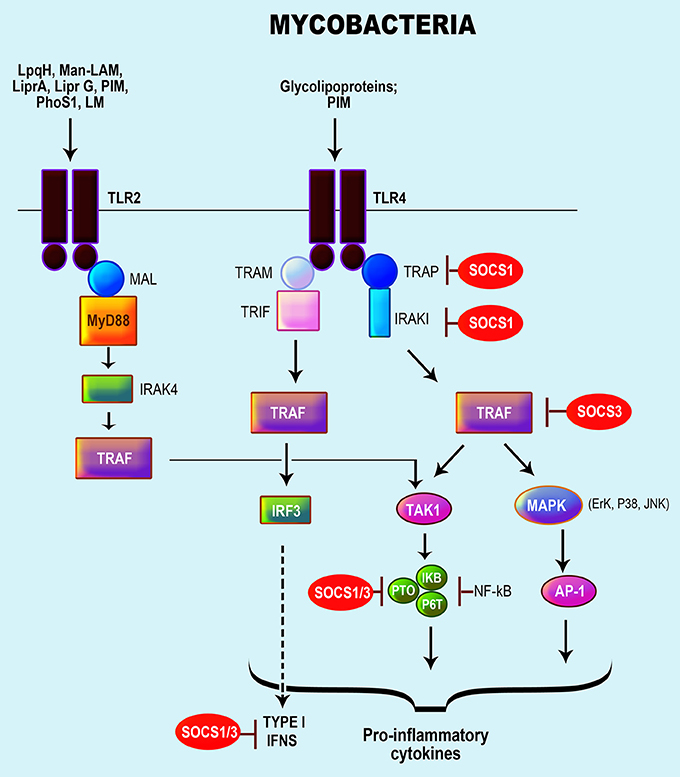
Figure 3. Role of SOCS proteins in the regulation of the signaling pathways induced by recognition of mycobacteria. Recognition of mycobacteria by the TLR 4 and TLR2. Mycobacteria activates the TLR 2/4 with a signaling cascade that results in the activation of NF-κB activated protein-1 (AP-1) via MyD88, MAL, and interleukin (IL)-1R-associated kinase (IRAK), resulting in the production of chemokines, pro-inflammatory cytokines particularly TNFα, IL-1β, IL-18, IL-12, and nitric oxide (Rajaram et al., 2014; Rottenberg and Carow, 2014; Zhao et al., 2014). Subsequently, the SOCS protein involved in the regulation of TLR signaling is accentuated in red.
Mycobacterial infections trigger the expressions of SOCS1 and SOCS3 (Gao et al., 2006; Dorhoi et al., 2010; Killick et al., 2013) along with SOCS4 and SOCS5 in mice infected with highly virulent MTB isolates (Vazquez et al., 2006). Overexpression of SOCS1 and SOCS3 results in polarizing effects permitting induction of suppressor responses, but also the survival of mycobacteria through the manipulation of cytokine responses, especially IFN-γ that is required in the resolution of mycobacterial infections (Manca et al., 2005; Dorhoi et al., 2010). Mycobacterial-induced SOCS1 and SOCS3 are dependent TLR2/MyD88 along with NF-κ and p38 MAPK activation (Manca et al., 2005). Specifically, SOCS1 suppressed STAT1 phosphorylation resulting in the inhibition of STAT1-mediated IFN-α/β signaling (Manca et al., 2005; Dorhoi et al., 2010). SOCS1 also promotes mycobacterial growth in macrophages by blocking IFN-γ secretion in response to IL-12 induced by the infection (Srivastava et al., 2009).
Mycobacteria-specific components (i.e., PIM2 and PPE protein, PPE-18) via TLR2/MyD88-activation of macrophages augment SOCS3 expression, and dislocation of the MyD88/TLR2 pathway modulated SOCS3 expression (Prince et al., 1989). Additionally, SOCS3 induced by PPE18 inhibited NF-κB activation by diminishing the phosphorylation of IκBα (Carow et al., 2011). In general, mycobacterial-induced SOCS3 inhibits STAT3 activation through cytokine receptors that activate STAT3 (Prince et al., 1989). As an example, SOCS3 binding to gp130 mediated the control of MTB infection in myeloid cells by inhibiting the IL-6/STAT3 signaling pathway (Nair et al., 2011; Carow et al., 2013).
Both SOCS2 and CIS can also play a role in regulating responses to mycobacterial infections. The expression of SOCS2 increased in macrophages infected with mycobacteria, and SOCS2-deficient mice exhibited a higher sensitivity to the inflammation induced by M. bovis infection (Carow and Rottenberg, 2014). Nonetheless, the activity of SOCS2 seems to be scarce and redundant and still requires a better understanding. CIS, on the other hand, is associated with increased susceptibility to TB (Sun et al., 2014; McCormick and Heller, 2015), likely by negatively regulating SOCS1 and SOCS3 (Trengove and Ward, 2013).
Correlation of SOCS Proteins with Bacterial Disease
Bacterial pathogens exploit SOCS proteins to manipulate cytokine receptor signaling and thereby influence infection outcomes as a strategy of evading host immune defenses (Baetz et al., 2007). Hence, the over-expression of SOCS proteins in bacterial infections supposedly is linked to the immune escape and exacerbation of disease. As SOCS1 and SOCS3 play essential roles in response to bacterial infections, they are therefore explicitly targeted for immune evasion. The reports above have therefore indicated that pathogens can induce SOCS1 and SOCS3 to evade deleterious host immune responses for their perpetuation and/or to control their induced inflammation. The most intended target is the interferon responses, mediated by STAT1 and controlled by SOCS1 and SOCS3, which play pivotal roles in the defense against bacterial infections.
The highly pathogenic bacterium, L. monocytogenes manipulates the macrophage machinery during early infection where there is heightened macrophage activation to permit its intracellular establishment. However, during persistent infections, L. monocytogenes regulates macrophage activation by inhibiting the transcription of IFN-γ and tyrosine phosphorylation of STAT1 via induction of SOCS3 (Okada et al., 2009). As stimulation of IFN-γ is necessary for macrophage activation and functions, inhibiting IFN-γ signaling is a stratagem utilized by L. monocytogenes to facilitate its intracellular survival by controlling its inflammation. Similarly, perturbations of IFN-γ and STAT1 signaling pathways by the facultative intracellular B. pseudomallei through induction of SOCS3 and CIS is a mechanistic tactic to reduce the macrophage bactericidal effect and enabled its intracellular survival (Ekchariyawat et al., 2005).
Results from studies by Uchiya and Nikai (Uchiya and Nikai, 2005, 2008) demonstrated how Salmonella pathogenicity island 2 (SPI-2) T3SS and its encoded virulence factor SpiC trigger SOCS3 up-regulation via the ERK1/2 pathway for inhibition of the JAK/STAT inflammatory signaling cascades for its continued survival in macrophages. GAS, which causes various systemic diseases induced SOCS1 that participates in the GAS' evasion of host immune responses in murine macrophages by dampening cytokine expression leading to rapid bacterial infection (Son et al., 2015). Expression of SOCS1 was shown to prevent C. pneumoniae-induced lethal inflammation through a STAT1 and IFN-α/β signaling-dependent manner, but conversely, its impact on IFN-α/β and IFN-γ impeded an efficient bacterial clearance (Yang et al., 2008). Borrelia burgdoferi (non-gram staining bacteria), the spirochetal agent of Lyme disease, stimulates the expression of SOCS1 and SOCS3 in macrophages to possibly control its inflammatory disorders (Khor et al., 2010). Additionally, B. burgdorferi via CD14 signaling induced SOCS1, SOCS3, and CIS as mediated by the p38 MAPK pathway to control the development of chronic inflammatory etiologies (Dennis et al., 2006).
Various mycobacteria manipulate IFN-γ-driven immunity by inducing SOCS1 and SOCS3 to evade the immune response or hamper the disease control. Augmentation of SOCS1 and SOCS3 levels and their subsequent inhibition of IFN-γ-induced STAT1 were found to alleviate the immune response for several mycobacterial species like M. tuberculosis, M. avium, and M. bovis (Gao et al., 2006; Srivastava et al., 2009; Dorhoi et al., 2010; Trengove and Ward, 2013). M. bovis infection stimulated SOCS1 and SOCS3 in mouse macrophages, which mediated the inhibition of IFN-γ-stimulated phosphorylation of STAT1 and thereby the subsequent inhibition of growth and activation of macrophages required for the control of this intracellular pathogen (Gao et al., 2006). Moreover, there are observations of both SOCS1 and SOCS3 association with disease progression in peripheral blood mononuclear cells and human macrophages of patients with TB (Sahay et al., 2009; Masood et al., 2012, 2013). SOCS1 and SOCS3 were up-regulated and contributed to Th2 immune polarization and down-modulation of Th1-mediated IFN-γ responses, and hence increased the disease severity by promoting the intracellular persistence of M. tuberculosis (Sahay et al., 2009; Masood et al., 2012, 2013). Infection of mice with highly virulent clinical isolates of MTB induced type I IFNs, which led to the up-regulation of SOCS1, SOCS4, SOCS5 and other negative regulators of the JAK/STAT pathway resulting in a decrease of Th1 type cytokines and decreased survival of MTB-infected mice (Vazquez et al., 2006).
Implications and Possible Requirements for Therapeutic Approaches
SOCS proteins regulate cytokine signal transduction for maintaining immune functions but still contribute to the onset of immunological diseases and inflammation (Yoshimura et al., 2005). Therefore, modulating cytokine release holds promise for minimizing disease progression. SOCS1 and SOCS3 are tightly linked to cancer cell proliferation, as well as cancer-associated inflammation. In some cancer therapy studies, SOCS proteins have been used to control or suppress cytokine signaling for an efficacious treatment. One approach is overexpressing SOCS proteins to inhibit the growth of tumors mediated by suppressing tumor-promoting STATs. Another method is enhancing anti-tumor immunity by siRNA silencing of SOCS in DCs or CTLs (Ashenafi et al., 2014). In most cases, the silencing of SOCS1 and SOCS3 exacerbated carcinogenesis; thus, overexpression of SOCS1 and SOCS3 or SOCS-mimetics can be targeted therapeutics (Zhang et al., 2012). However, SOCS1 in DCs and likely T cells suppress anti-tumor immunity; therefore, silencing SOCS1 in these cells could also be therapeutic. Silencing of the SOCS1 gene may hinder the negative feedback regulation of the JAK/STAT pathway, therefore, resulting in heightened responsiveness to cytokines, and supporting survival and expansion of myeloma myeloid cells (Inagaki-Ohara et al., 2013). Blocking of constitutive STAT3 signaling results in growth inhibition and apoptosis of STAT3-positive tumor cells in vitro and in vivo (Galm et al., 2003). Development of SOCS-targeted therapeutics based on structural analysis of the JAK/SOCS complex (Zhang et al., 2012) could thus be a highly desirable approach.
The regulation of the levels of pro- and anti-inflammatory cytokines and chemokines by the immune system is critical in limiting or modulating the host defense against invading pathogens. SOCS proteins as negative regulators of JAK/STAT represent a promising target for anti-inflammatory therapies (Turkson and Jove, 2000). Therefore, the use of recombinant forms of SOCS proteins to refill the intracellular stores of SOCS needed to control acute or protracted inflammatory disease can be viewed as a novel targeted therapy to suppress the JAK/STAT pathway and prevent cytokine-mediated lethal inflammation (Recio et al., 2014). A recombinant cell-penetrating form of SOCS1 (CP-SOCS1) and SOCS3 were indeed shown to potently inhibit the JAK/STAT signaling pathway in vitro by interacting with the IFN-γ signaling complex and functionally reducing the phosphorylation of STAT1, which further resulted in inhibiting the production of pro-inflammatory cytokines and chemokines (Jo et al., 2005; DiGiandomenico et al., 2009; Fletcher et al., 2010). Moreover, CP-SOCS3 protected mice from lethal effects of Staphylococcal Enterotoxin B and LPS by decreasing the production of inflammatory cytokines (DiGiandomenico et al., 2009).
The exploitation of host SOCS proteins and manipulation of their functions by bacterial pathogens make them particularly attractive therapeutic targets. Therapeutic approaches targeting host-directed immunomodulatory components against bacterial infections have already been described (Finlay and Hancock, 2004; Hancock et al., 2012; Hawiger and Jo, 2013). Regulator peptides of the innate immune defense, agonists of innate immune receptors and adjuvants of innate immune components, have been tested for this purpose (Finlay and Hancock, 2004; Hawiger and Jo, 2013). TLRs and NOD receptors have, in fact, been targets of several immunomodulatory therapies, of which several are approved, to inhibit or treat bacterial infections (Finlay and Hancock, 2004; Hancock and Sahl, 2006). Among them, the most notable example is CADI-05, an agonist for many TLRs, which was successfully investigated as a potential therapy for TB (Finlay and Hancock, 2004; Hancock and Sahl, 2006). Besides, vaccines formulated with small molecule immune-potentiators that trigger TLRs were shown efficient in protection against bacterial infections. A notable example is the vaccine adjuvant based on a TLR7 agonist adsorbed to alum (Alum-TLR7), which induced a high and broad protection against Staphylococcus aureus (Hennessy et al., 2010; Bagnoli et al., 2015). Furthermore, targeting STAT activity that is strictly regulated by SOCS1 and SOCS3 proteins, by inhibiting tyrosine kinases, could allow avoiding the subversion of the innate immune responses during a bacterial infection and therefore represents an attractive antibacterial therapeutic approach. For instance, dual-inhibitors of Ser/Thr protein kinases PknG/PknG, which are required for mycobacteria growth were able to prevent their replication in mice (Hennessy et al., 2010; Gil et al., 2013; Mancini et al., 2016). However, investigations of SOCS proteins as therapeutic targets have not been beyond the development of a cell-penetrating form of SOCS to compensate the loss of endogenous SOCS (DiGiandomenico et al., 2009; Recio et al., 2014). Nonetheless, since bacteria target several intracellular pathways, many of which are linked to the SOCS proteins, it is clear that SOCS proteins represent unnavigable therapeutic targets in the control or eradication of bacterial infections.
Conclusion and Future Directions
A better understanding of the control mechanisms involved in SOCS modulation of immune responses and inflammation is key to developing effective targeted therapeutics and vaccines. Each SOCS protein contributes either to the negative regulation of cytokine signaling or the regulation of many biological processes. The expression of SOCS proteins can define host susceptibility to infection by facilitating accelerated bacterial growth or protecting the host from severe inflammation. However, the direct action of the SOCS-mediated inhibition in inflammatory response is yet to be fully elucidated, thus limiting the progress being made scientifically and clinically by microbiologist and immunologists. Additionally, factors such as increased virulence, mutations and antibiotic resistance over time pose recurring challenges in the control of epidemic bacterial diseases worldwide. Admittedly, SOCS-targeted therapy for bacterial-induced inflammation is provocative, but yet one that should be considered exploring. So far, inhibiting the action of JAKs by small composites or drugs show reparative potential (DiGiandomenico et al., 2009). Such insight along with advances in medicine and technology may offer more efficient, and novel strategies surrounding SOCS therapy to control the inflammatory bacterial disease.
Author Contributions
SD did the literature search and exploration and wrote the manuscript. DB and RS helped with writing and reading the manuscript. SS read and edited the manuscript. VD edited the manuscript and coordinated the project. All authors read and approved the final manuscript.
Funding
This research was supported by funding from the National Science Foundation (NSF)-CREST (HRD-1241701), NSF-HBCU-RISE (HRD-1646729) and the National Institutes of Health (NIH)-MBRS-RISE (1R25GM106995-01) grants.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Yvonne Williams, LaShaundria Lucas, and Juwana Smith-Henderson of CNBR for their excellent administrative assistance and Golden Muse (http://www.golden-muse.com/) for the illustrations used in this review.
References
Abbas, A. K., Lichtman, A. H., and Pillai, S. (2012). Cellular and Molecular Immunology. Philadelphia, PA: Elsevier/Saunders.
Alexander, W. S. (2002). Suppressors of cytokine signalling (SOCS) in the immune system. Nat. Rev. Immunol. 2, 410–416. doi: 10.1038/nri818
Ashenafi, S., Aderaye, G., Bekele, A., Zewdie, M., Aseffa, G., Hoang, A. T. N., et al. (2014). Progression of clinical tuberculosis is associated with a Th2 immune response signature in combination with elevated levels of SOCS3. Clin. Immunol. 151, 84–99. doi: 10.1016/j.clim.2014.01.010
Athman, R., and Philpott, D. (2004). Innate immunity via Toll-like receptors and Nod proteins. Curr. Opin. Microbiol. 7, 25–32. doi: 10.1016/j.mib.2003.12.013
Baetz, A., Frey, M., Heeg, K., and Dalpke, A. H. (2004). Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J. Biol. Chem. 279, 54708–54715. doi: 10.1074/jbc.M410992200
Baetz, A., Zimmermann, S., and Dalpke, A. H. (2007). Microbial immune evasion employing suppressor of cytokine signaling (SOCS) proteins. Inflamm. Allergy Drug Targets 6, 160–167. doi: 10.2174/187152807781696446
Bagnoli, F., Fontana, M. R., Soldaini, E., Mishra, R. P., Fiaschi, L., Cartocci, E., et al. (2015). Vaccine composition formulated with a novel TLR7-dependent adjuvant induces high and broad protection against Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 112, 3680–3685. doi: 10.1073/pnas.1424924112
Bermejo-Martin, J. F., Avila-Alonso, A., González-Rivera, M., Tamayo, E., Eiros, J. M., and Almansa, R. (2016). Postbooster antibodies from humans as source of diphtheria antitoxin. Emerg. Infect. Dis. 22, 1265–1267. doi: 10.3201/eid2207.151670
Betts-Hampikian, H. J., and Fields, K. A. (2010). The chlamydial type III secretion mechanism: revealing cracks in a tough nut. Front. Microbiol. 1:114. doi: 10.3389/fmicb.2010.00114
Brannon, M. K., Davis, J. M., Mathias, J. R., Hall, C. J., Emerson, J. C., Crosier, P. S., et al. (2009). Pseudomonas aeruginosa Type III secretion system interacts with phagocytes to modulate systemic infection of zebrafish embryos. Cell. Microbiol. 11, 755–768. doi: 10.1111/j.1462-5822.2009.01288.x
Brown, L., Wolf, J. M., Prados-Rosales, R., and Casadevall, A. (2015). Through the wall: extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat. Rev. Microl. 13, 620–630. doi: 10.1038/nrmicro3480
Bullock, A. N., Rodriguez, M. C., Debreczeni, J. E., Songyang, Z., and Knapp, S. (2007). Structure of the SOCS4-ElonginB/C complex reveals a distinct SOCS box interface and the molecular basis for SOCS-dependent EGFR degradation. Structure 15, 1493–1504. doi: 10.1016/j.str.2007.09.016
Bussmeyer, U., Sarkar, A., Broszat, K., Lüdemann, T., Möller, S., van Zandbergen, G., et al. (2010). Impairment of gamma interferon signaling in human neutrophils infected with Anaplasma phagocytophilum. Infect. Immun. 78, 358–363. doi: 10.1128/IAI.01005-09
Caballero, P., Troncoso, M., Patterson, S. I., López Gómez, C., Fernandez, R., and Sosa, M. A. (2016). Neurotoxins from Clostridium botulinum (serotype A) isolated from the soil of Mendoza (Argentina) differ from the A-Hall archetype and from that causing infant botulism. Toxicon 121, 30–35. doi: 10.1016/j.toxicon.2016.08.010
Carow, B., Reuschl, A.-K., Gavier-Widén, D., Jenkins, B. J., Ernst, M., Yoshimura, A., et al. (2013). Critical and independent role for SOCS3 in either myeloid or T cells in resistance to Mycobacterium tuberculosis. PLoS Pathog. 9:e1003442. doi: 10.1371/journal.ppat.1003442
Carow, B., and Rottenberg, M. E. (2014). SOCS3, a major regulator of infection and inflammation. Front. Immunol. 5:58. doi: 10.3389/fimmu.2014.00058
Carow, B., Ye, X., Gavier-Widén, D., Bhuju, S., Oehlmann, W., Singh, M., et al. (2011). Silencing suppressor of cytokine signaling-1 (SOCS1) in macrophages improves Mycobacterium tuberculosis control in an interferon-gamma (IFN-gamma)-dependent manner. J. Biol. Chem. 286, 26873–26887. doi: 10.1074/jbc.M111.238287
Cekici, A., Kantarci, A., Hasturk, H., and Van Dyke, T. E. (2000). Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontology 64, 57–80. doi: 10.1111/prd.12002
Chaplin, D. D. (2010). Overview of the immune response. J. Allergy Clin. Immunol. 125(2 Suppl. 2), S3–23. doi: 10.1016/j.jaci.2009.12.980
Chaves de Souza, J. A., Nogueira, A. V., Chaves de Souza, P. P., Kim, Y. J., Silva Lobo, C., Pimentel Lopes de Oliveira, G. J., et al. (2013). SOCS3 expression correlates with severity of inflammation, expression of proinflammatory cytokines, and activation of STAT3 and p38 MAPK in LPS-induced inflammation in vivo. Mediators Inflammol. 2013:650812. doi: 10.1155/2013/650812
Chrétien, S., Varlet, P., Verdier, F., Gobert, S., Cartron, J. P., Gisselbrecht, S., et al. (1996). Erythropoietin-induced erythroid differentiation of the human erythroleukemia cell line TF-1 correlates with impaired STAT5 activation. EMBO J. 15, 4174–4181.
Colonne, P. M., Sahni, A., and Sahni, S. K. (2013). Suppressor of cytokine signalling protein SOCS1 and UBP43 regulate the expression of type I interferon-stimulated genes in human microvascular endothelial cells infected with Rickettsia conorii. J. Med. Microbiol. 62(Pt. 7), 968–979. doi: 10.1099/jmm.0.054502-0
Cooney, R. N. (2002). Suppressors of cytokine signaling (SOCS): inhibitors of the JAK/STAT pathway. Shock 17, 83–90. doi: 10.1097/00024382-200202000-00001
Copray, J. C., Mantingh, I., Brouwer, N., Biber, K., Kust, B. M., Liem, R. S., et al. (2001). Expression of interleukin-1 beta in rat dorsal root ganglia. J. Neuroimmunol. 118, 203–211. doi: 10.1016/S0165-5728(01)00324-1
Croker, B. A., Kiu, H., and Nicholson, S. E. (2008). SOCS regulation of the JAK/STAT signalling pathway. Semin. Cell Dev. Biol. 19, 414–422. doi: 10.1016/j.semcdb.2008.07.010
Dalpke, A. H., Eckerle, S., Frey, M., and Heeg, K. (2003). Triggering of Toll-like receptors modulates IFN-gamma signaling: involvement of serine 727 STAT1 phosphorylation and suppressors of cytokine signaling. Eur. J. Immunol. 33, 1776–1787. doi: 10.1002/eji.200323621
Dalpke, A., Heeg, K., Bartz, H., and Baetz, A. (2008). Regulation of innate immunity by suppressor of cytokine signaling (SOCS) proteins. Immunobiology 213, 225–235. doi: 10.1016/j.imbio.2007.10.008
Dalpke, A. H., Opper, S., Zimmermann, S., and Heeg, K. (2001). Suppressors of cytokine signaling (SOCS)-1 and SOCS-3 are induced by CpG-DNA and modulate cytokine responses in APCs. J. Immunol. 166, 7082–7089. doi: 10.4049/jimmunol.166.12.7082
Demirel, I., Säve, S., Kruse, R., and Persson, K. (2013). Expression of suppressor of cytokine signalling 3 (SOCS3) in human bladder epithelial cells infected with uropathogenic Escherichia coli. APMIS 121, 158–167. doi: 10.1111/j.1600-0463.2012.02951.x
Dennis, V. A., Jefferson, A., Singh, S. R., Ganapamo, F., and Philipp, M. T. (2006). Interleukin-10 anti-inflammatory response to Borrelia burgdorferi, the agent of Lyme disease: a possible role for suppressors of cytokine signaling 1 and 3. Infect Immun. 74, 5780–5789. doi: 10.1128/IAI.00678-06
de Sousa, C. P. (2003). Pathogenicity mechanisms of prokaryotic cells: an evolutionary view. Braz. J. Infect. Dis. 7, 23–31. doi: 10.1590/S1413-86702003000100004
DiGiandomenico, A., Wylezinski, L. S., and Hawiger, J. (2009). Intracellular delivery of a cell-penetrating SOCS1 that targets IFN-gamma signaling. Sci. Signal. 2:ra37. doi: 10.1126/scisignal.1162191
Dinarello, C. A. (2000). Proinflammatory cytokines. Chest 118, 503–508. doi: 10.1378/chest.118.2.503
Dinarello, C. A. (2007). Historical insights into cytokines. Eur. J. Immunol. 37 (Suppl. 1), S34–S45. doi: 10.1002/eji.200737772
Ding, F. M., Liao, R. M., Chen, Y. Q., Xie, G. G., Zhang, P. Y., Shao, P., et al. (2017). Upregulation of SOCS3 in lung CD4+ T cells in a mouse model of chronic PA lung infection and suppression of Th17mediated neutrophil recruitment in exogenous SOCS3 transfer in vitro. Mol. Med. Rep. 16, 778–786. doi: 10.3892/mmr.2017.6630
Dorhoi, A., Desel, C., Yeremeev, V., Pradl, L., Brinkmann, V., Mollenkopf, H. J., et al. (2010). The adaptor molecule CARD9 is essential for tuberculosis control. J. Exp. Med. 207, 777–792. doi: 10.1084/jem.20090067
Dosunmu, E. F., Chaudhari, A. A., Bawage, S., Bakeer, M. K., Owen, D. R., Singh, S. R., et al. (2016). Novel cationic peptide TP359 down-regulates the expression of outer membrane biogenesis genes in Pseudomonas aeruginosa: a potential TP359 anti-microbial mechanism. BMC Microbiol. 16:192. doi: 10.1186/s12866-016-0808-2
do Vale, A., Cabanes, D., and Sousa, S. (2016). Bacterial toxins as pathogen weapons against phagocytes. Front. Microbiol. 7:42. doi: 10.3389/fmicb.2016.00042
Draing, C., Sigel, S., Deininger, S., Traub, S., Munke, R., Mayer, C., et al. (2008). Cytokine induction by Gram-positive bacteria. Immunobiology 213, 285–296. doi: 10.1016/j.imbio.2007.12.001
Dumitru, C. D., Ceci, J. D., Tsatsanis, C., Kontoyiannis, D., Stamatakis, K., Lin, J.-H., et al. (2000). TNF-α Induction by LPS Is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell 103, 1071–1083. doi: 10.1016/S0092-8674(00)00210-5
Ekchariyawat, P., Pudla, S., Limposuwan, K., Arjcharoen, S., Sirisinha, S., and Utaisincharoen, P. (2005). Burkholderia pseudomallei-induced expression of suppressor of cytokine signaling 3 and cytokine-inducible src homology 2-containing protein in mouse macrophages: a possible mechanism for suppression of the response to gamma interferon stimulation. Infect. Immun. 73, 7332–7339. doi: 10.1128/IAI.73.11.7332-7339.2005
Elliott, J., and Johnston, J. A. (2004). SOCS: role in inflammation, allergy and homeostasis. Trends Immunol. 25, 434–440. doi: 10.1016/j.it.2004.05.012
Finlay, B. B., and Hancock, R. E. (2004). Can innate immunity be enhanced to treat microbial infections? Nat. Rev. Microbiol. 2, 497–504. doi: 10.1038/nrmicro908
Finlay, B. B., and McFadden, G. (2006). Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell 124, 767–782. doi: 10.1016/j.cell.2006.01.034
Fletcher, T. C., DiGiandomenico, A., and Hawiger, J. (2010). Extended anti-inflammatory action of a degradation-resistant mutant of cell-penetrating suppressor of cytokine signaling 3. J. Biol. Chem. 285, 18727–18736. doi: 10.1074/jbc.M109.095216
Freudenberg, M. A., Tchaptchet, S., Keck, S., Fejer, G., Huber, M., Schütze, N., et al. (2008). Lipopolysaccharide sensing an important factor in the innate immune response to Gram-negative bacterial infections: benefits and hazards of LPS hypersensitivity. Immunobiology 213, 193–203. doi: 10.1016/j.imbio.2007.11.008
Frobøse, H., Rønn, S. G., Heding, P. E., Mendoza, H., Cohen, P., Mandrup-Poulsen, T., et al. (2006). Suppressor of cytokine signaling-3 inhibits interleukin-1 signaling by targeting the TRAF-6/TAK1 complex. Mol. Endocrinol. 20, 1587–1596. doi: 10.1210/me.2005-0301
Fujimoto, M., and Naka, T. (2010). SOCS1, a negative regulator of cytokine signals and TLR responses, in human liver diseases. Gastroenterol. Res. Pract. 2010, 1–7. doi: 10.1155/2010/470468
Galm, O., Yoshikawa, H., Esteller, M., Osieka, R., and Herman, J. G. (2003). SOCS-1, a negative regulator of cytokine signaling, is frequently silenced by methylation in multiple myeloma. Blood 101, 2784–2788. doi: 10.1182/blood-2002-06-1735
Gao, B., Paramanathan, R., and Gupta, R. S. (2006). Signature proteins that are distinctive characteristics of Actinobacteria and their subgroups. Antonie Van Leeuwenhoek 90, 69–91. doi: 10.1007/s10482-006-9061-2
Gerold, G., Zychlinsky, A., and de Diego, J. L. (2007). What is the role of Toll-like receptors in bacterial infections? Semin. Immunol. 19, 41–47. doi: 10.1016/j.smim.2006.12.003
Gil, M., Grana, M., Schopfer, F. J., Wagner, T., Denicola, A., Freeman, B. A., et al. (2013). Inhibition of Mycobacterium tuberculosis PknG by non-catalytic rubredoxin domain specific modification: reaction of an electrophilic nitro-fatty acid with the Fe-S center. Free Radic. Biol. Med. 65, 150–161. doi: 10.1016/j.freeradbiomed.2013.06.021
Gingras, S., Parganas, E., de Pauw, A., Ihle, J. N., and Murray, P. J. (2004). Re-examination of the role of suppressor of cytokine signaling 1 (SOCS1) in the regulation of toll-like receptor signaling. J. Biol. Chem. 279, 54702–54707. doi: 10.1074/jbc.M411043200
Girard-Madoux, M. J., Ober-Blobaum, J. L., Costes, L. M., Kel, J. M., Lindenbergh-Kortleve, D. J., Brouwers-Haspels, I., et al. (2016). IL-10 control of CD11c+ myeloid cells is essential to maintain immune homeostasis in the small and large intestine. Oncotarget 7, 32015–32030. doi: 10.18632/oncotarget.8337
Goldman, A. S., and Prabhakar, B. S. (1996). “Immunology overview,” in Medical Microbiology, 4th Edn., ed S. Baron (Galveston, TX: University of Texas Medical Branch at Galveston).
Grutkoski, P. S., Chen, Y., Chung, C. S., and Ayala, A. (2003). Sepsis-induced SOCS-3 expression is immunologically restricted to phagocytes. J. Leukoc. Biol. 74, 916–922. doi: 10.1189/jlb.0303108
Hancock, R. E., Nijnik, A., and Philpott, D. J. (2012). Modulating immunity as a therapy for bacterial infections. Nat. Rev. Microbiol. 10, 243–254. doi: 10.1038/nrmicro2745
Hancock, R. E., and Sahl, H. G. (2006). Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24, 1551–1557. doi: 10.1038/nbt1267
Hao, L. X., and Sun, L. (2016). Comparative analysis of the expression patterns of eight suppressors of cytokine signaling in tongue sole, Cynoglossus semilaevis. Fish Shellfish Immunol. 55, 595–601. doi: 10.1016/j.fsi.2016.06.034
Harty, J. T., and Bevan, M. J. (1995). Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity 3, 109–117. doi: 10.1016/1074-7613(95)90163-9
Hawiger, J. J., and Jo, D. (2013). Cell-Penetrating SOCS Polypeptides that Inhibit Cytokine-Induced Signaling. Google Patents.
He, X., Zhang, Y., Zhu, A., Zeng, K., Zhang, X., Gong, L., et al. (2016). Suppression of interleukin 17 contributes to the immunomodulatory effects of adipose-derived stem cells in a murine model of systemic lupus erythematosus. Immunol. Res. 64, 1157–1167. doi: 10.1007/s12026-016-8866-y
Hennessy, E. J., Parker, A. E., and O'Neill, L. A. J. (2010). Targeting Toll-like receptors: emerging therapeutics? Nat. Rev. Drug Discov. 9, 293–307. doi: 10.1038/nrd3203
Hessle, C. C., Andersson, B., and Wold, A. E. (2005). Gram-positive and Gram-negative bacteria elicit different patterns of pro-inflammatory cytokines in human monocytes. Cytokine 30, 311–318. doi: 10.1016/j.cyto.2004.05.008
Hilton, D. J. (1999). Negative regulators of cytokine signal transduction. Cell. Mol. Life Sci. 55, 1568–1577. doi: 10.1007/s000180050396
Hu, J., Winqvist, O., Flores-Morales, A., Wikström, A. C., and Norstedt, G. (2009). SOCS2 influences LPS induced human monocyte-derived dendritic cell maturation. PLoS ONE 4:e7178. doi: 10.1371/journal.pone.0007178
Hu, B., Lara-Tejero, M., Kong, Q., Galán, J. E., and Liu, J. (2017). In situ molecular architecture of the Salmonella Type III secretion machine. Cell 168, 1065–1074e10. doi: 10.1016/j.cell.2017.02.022
Hu, J., Lou, D., Carow, B., Winerdal, M. E., Rottenberg, M., Wikström, A. C., et al. (2012). LPS regulates SOCS2 transcription in a type I interferon dependent autocrine-paracrine loop. PLoS ONE 7:e30166. doi: 10.1371/journal.pone.0030166
Huang, S., Hendriks, W., Althage, A., Hemmi, S., Bluethmann, H., Kamijo, R., et al. (1993). Immune response in mice that lack the interferon-gamma receptor. Science 259, 1742–1745. doi: 10.1126/science.8456301
Imai, K., Kurita-Ochiai, T., and Ochiai, K. (2003). Mycobacterium bovis bacillus Calmette-Guerin infection promotes SOCS induction and inhibits IFN-gamma-stimulated JAK/STAT signaling in J774 macrophages. FEMS Immunol. Med. Microbiol. 39, 173–180. doi: 10.1016/S0928-8244(03)00231-1
Inagaki-Ohara, K., Kondo, T., Ito, M., and Yoshimura, A. (2013). SOCS, inflammation, and cancer. Jak-Stat. 2:e24053. doi: 10.4161/jkst.24053
Janeway, C. A. Jr., and Medzhitov, R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197–216. doi: 10.1146/annurev.immunol.20.083001.084359
Jo, D., Liu, D., Yao, S., Collins, R. D., and Hawiger, J. (2005). Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat. Med. 11, 892–898. doi: 10.1038/nm1269
Jo, E. K. (2008). Mycobacterial interaction with innate receptors: TLRs, C-type lectins, and NLRs. Curr. Opin. Infect. Dis. 21, 279–286. doi: 10.1097/QCO.0b013e3282f88b5d
Johnson, R., Byrne, A., Berger, C. N., Klemm, E., Crepin, V. F., Dougan, G., et al. (2017). The Type III Secretion system effector SptP of Salmonella enterica serovar typhi. J. Bacteriol. 199, e00647–0064716. doi: 10.1128/JB.00647-16
Kamizono, S., Hanada, T., Yasukawa, H., Minoguchi, S., Kato, R., Minoguchi, M., et al. (2001). The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2. J. Biol. Chem. 276, 12530–12538. doi: 10.1074/jbc.M010074200
Kang, C.-I., Kim, S.-H., Park, W. B., Lee, K.-D., Kim, H.-B., Kim, E.-C., et al. (2005). Bloodstream infections caused by antibiotic-resistant gram-Negative bacilli: risk factors for mortality and impact of inappropriate initial antimicrobial therapy on outcome. Antimicrob. Agents Chemother. 49, 760–766. doi: 10.1128/AAC.49.2.760-766.2005
Kario, E., Marmor, M. D., Adamsky, K., Citri, A., Amit, I., Amariglio, N., et al. (2005). Suppressors of cytokine signaling 4 and 5 regulate epidermal growth factor receptor signaling. J. Biol. Chem. 280, 7038–7048. doi: 10.1074/jbc.M408575200
Kershaw, N. J., Murphy, J. M., Lucet, I. S., Nicola, N. A., and Babon, J. J. (2013). Regulation of Janus kinases by SOCS proteins. Biochem. Soc. Trans. 41, 1042–1047. doi: 10.1042/BST20130077
Khondker, L., and Khan, S. I. (2014). Association of rheumatoid factor and uric acid with psoriatic arthritis: a review. Mymensingh Med. J. 23, 609–613.
Khor, C., Vannberg, F., Chapman, S., Guo, H., Wong, S., Walley, A., et al. (2010). CISH and susceptibility to infectious diseases. New Eng. J. Med. 362, 2092–2101. doi: 10.1056/NEJMoa0905606
Kile, B. T., Schulman, B. A., Alexander, W. S., Nicola, N. A., Martin, H. M., and Hilton, D. J. (2002). The SOCS box: a tale of destruction and degradation. Trends Biochem. Sci. 27, 235–241. doi: 10.1016/S0968-0004(02)02085-6
Killick, K. E., Ní Cheallaigh, C., O'Farrelly, C., Hokamp, K., MacHugh, D. E., and Harris, J. (2013). Receptor-mediated recognition of mycobacterial pathogens. Cell. Microbiol. 15, 1484–1495. doi: 10.1111/cmi.12161
Kinjyo, I., Hanada, T., Inagaki-Ohara, K., Mori, H., Aki, D., Ohishi, M., et al. (2002). SOCS1/JAB Is a negative regulator of LPS-induced macrophage activation. Immunity 17, 583–591. doi: 10.1016/S1074-7613(02)00446-6
Kleinnijenhuis, J., Oosting, M., Joosten, L. A. B., Netea, M. G., and Van Crevel, R. (2011). Innate immune recognition of Mycobacterium tuberculosis. Clin. Dev. Immunol. 2011:12. doi: 10.1155/2011/405310
Krebs, D. L., Uren, R. T., Metcalf, D., Rakar, S., Zhang, J. G., Starr, R., et al. (2002). SOCS-6 binds to insulin receptor substrate 4, and mice lacking the SOCS-6 gene exhibit mild growth retardation. Mol. Cell. Biol. 22, 4567–4578. doi: 10.1128/MCB.22.13.4567-4578.2002
Kumar, A., and Yu, F. S. (2006). Toll-like receptors and corneal innate immunity. Curr. Mol. Med. 6, 327–337. doi: 10.2174/156652406776894572
Latvala, S., Miettinen, M., Kekkonen, R. A., Korpela, R., and Julkunen, I. (2011). Lactobacillus rhamnosus GG and Streptococcus thermophilus induce suppressor of cytokine signalling 3 (SOCS3) gene expression directly and indirectly via interleukin-10 in human primary macrophages. Clin. Exp. Immunol. 165, 94–103. doi: 10.1111/j.1365-2249.2011.04408.x
Lehmann, U., Schmitz, J., Weissenbach, M., Sobota, R. M., Hortner, M., Friederichs, K., et al. (2003). SHP2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130. J. Biol. Chem. 278, 661–671. doi: 10.1074/jbc.M210552200
Lewis, L. A., and Ram, S. (2014). Meningococcal disease and the complement system. Virulence 5, 98–126. doi: 10.4161/viru.26515
Linossi, E. M., Babon, J. J., Hilton, D. J., and Nicholson, S. E. (2013). Suppression of cytokine signaling: the SOCS perspective. Cytokine Growth Factor Rev. 24, 241–248. doi: 10.1016/j.cytogfr.2013.03.005
Liu, X., Zhang, Y., Yu, Y., Yang, X., and Cao, X. (2008). SOCS3 promotes TLR4 response in macrophages by feedback inhibiting TGF-beta1/Smad3 signaling. Mol. Immunol. 45, 1405–1413. doi: 10.1016/j.molimm.2007.08.018
Manca, C., Tsenova, L., Freeman, S., Barczak, A. K., Tovey, M., Murray, P. J., et al. (2005). Hypervirulent, M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J. Interferon Cytokine Res. 25, 694–701. doi: 10.1089/jir.2005.25.694
Mancini, F., Monaci, E., Lofano, G., Torre, A., Bacconi, M., Tavarini, S., et al. (2016). One Dose of Staphylococcus aureus 4C-staph vaccine formulated with a novel TLR7-dependent adjuvant rapidly protects mice through antibodies, effector CD4+ T Cells, and IL-17A. PLoS ONE 11:e0147767. doi: 10.1371/journal.pone.0147767
Manicassamy, S., and Pulendran, B. (2009). Modulation of adaptive immunity with Toll-like receptors. Semin. Immunol. 21, 185–193. doi: 10.1016/j.smim.2009.05.005
Masood, K. I., Rottenberg, M. E., Carow, B., Rao, N., Ashraf, M., Hussain, R., et al. (2012). SOCS1 gene expression is increased in severe pulmonary tuberculosis. Scand. J. Immunol. 76, 398–404. doi: 10.1111/j.1365-3083.2012.02731.x
Masood, K. I., Rottenberg, M. E., Salahuddin, N., Irfan, M., Rao, N., Carow, B., et al. (2013). Expression of M. tuberculosis-induced suppressor of cytokine signaling (SOCS) 1, SOCS3, FoxP3 and secretion of IL-6 associates with differing clinical severity of tuberculosis. BMC Infect. Dis. 13:13. doi: 10.1186/1471-2334-13-13
Masuhara, M., Sakamoto, H., Matsumoto, A., Suzuki, R., Yasukawa, H., Mitsui, K., et al. (1997). Cloning and characterization of novel CIS family genes. Biochem. Biophys. Res. Commun. 239, 439–446. doi: 10.1006/bbrc.1997.7484
Matsumoto, A., Masuhara, M., Mitsui, K., Yokouchi, M., Ohtsubo, M., Misawa, H., et al. (1997). CIS, a cytokine inducible SH2 protein, is a target of the JAK-STAT5 pathway and modulates STAT5 activation. Blood 89, 3148–3154.
McCormick, S. M., and Heller, N. M. (2015). Regulation of macrophage, dendritic cell, and microglial phenotype and function by the SOCS proteins. Front. Immunol. 6:549. doi: 10.3389/fimmu.2015.00549
McDonald, C., Inohara, N., and Nuñez, G. (2005). Peptidoglycan signaling in innate immunity and inflammatory disease. J. Biol. Chem. 280, 20177–20180. doi: 10.1074/jbc.R500001200
Moellering, R. C. (2009). New treatments for multiply drug-resistant gram-positive bacteria. J. Infect. 59, S1–S3. doi: 10.1016/S0163-4453(09)60002-5
Mogensen, T. H. (2009). Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 22, 240–273. doi: 10.1128/CMR.00046-08
Morens, D. M., Folkers, G. K., and Fauci, A. S. (2004). The challenge of emerging and re-emerging infectious diseases. Nature. 430, 242–249. doi: 10.1038/nature02759
Mortaz, E., Adcock, I. M., Tabarsi, P., Masjedi, M. R., Mansouri, D., Velayati, A. A., et al. (2015). Interaction of pattern recognition receptors with Mycobacterium Tuberculosis. J. Clin. Immunol. 35, 1–10. doi: 10.1007/s10875-014-0103-7
Mui, A. L., Wakao, H., Kinoshita, T., Kitamura, T., and Miyajima, A. (1996). Suppression of interleukin-3-induced gene expression by a C-terminal truncated Stat5: role of Stat5 in proliferation. EMBO J. 15, 2425–2433.
Nair, M. K., De Masi, L., Yue, M., Galván, E. M., Chen, H., Wang, F., et al. (2015). Adhesive properties of YapV and paralogous autotransporter proteins of Yersinia pestis. Infect. Immun. 83, 1809–1819. doi: 10.1128/IAI.00094-15
Nair, S., Pandey, A. D., and Mukhopadhyay, S. (2011). The PPE18 protein of Mycobacterium tuberculosis inhibits NF-kappaB/rel-mediated proinflammatory cytokine production by upregulating and phosphorylating suppressor of cytokine signaling 3 protein. J. Immunol. 186, 5413–5424. doi: 10.4049/jimmunol.1000773
Nakagawa, R., Naka, T., Tsutsui, H., Fujimoto, M., Kimura, A., Abe, T., et al. (2002). SOCS-1 Participates in negative regulation of LPS responses. Immunity 17, 677–687. doi: 10.1016/S1074-7613(02)00449-1
Nandi, S., Maurer, J. J., Hofacre, C., and Summers, A. O. (2004). Gram-positive bacteria are a major reservoir of Class 1 antibiotic resistance integrons in poultry litter. Proc. Natl. Acad. Sci. U.S.A. 101, 7118–7122. doi: 10.1073/pnas.0306466101
Navarre, W. W., and Schneewind, O. (1999). Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 63, 174–229.
Nicholson, S. E., Willson, T. A., Farley, A., Starr, R., Zhang, J. G., Baca, M., et al. (1999). Mutational analyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction. EMBO J. 18, 375–385. doi: 10.1093/emboj/18.2.375
O'Connor, S. M., Taylor, C. E., and Hughes, J. M. (2006). Emerging infectious determinants of chronic diseases. Emerg. Infect. Dis. 12, 1051–1057. doi: 10.3201/eid1207.060037
Oh, S. B., Tran, P. B., Gillard, S. E., Hurley, R. W., Hammond, D. L., and Miller, R. J. (2001). Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J. Neurosci. 21, 5027–5035. doi: 10.1523/JNEUROSCI.3588-08.2008
Okada, Y., Tsuzuki, Y., Hokari, R., Komoto, S., Kurihara, C., Kawaguchi, A., et al. (2009). Anti-inflammatory effects of the genus Bifidobacterium on macrophages by modification of phospho-I kappaB and SOCS gene expression. Int. J. Exp. Pathol. 90, 131–140. doi: 10.1111/j.1365-2613.2008.00632.x
O'Riordan, M., Yi, C. H., Gonzales, R., Lee, K. D., and Portnoy, D. A. (2002). Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc. Natl. Acad. Sci. U.S.A. 99, 13861–13866. doi: 10.1073/pnas.202476699
Ozaktay, A. C., Kallakuri, S., Takebayashi, T., Cavanaugh, J. M., Asik, I., DeLeo, J. A., et al. (2006). Effects of interleukin-1 beta, interleukin-6, and tumor necrosis factor on sensitivity of dorsal root ganglion and peripheral receptive fields in rats. Eur. Spine J. 15, 1529–1537. doi: 10.1007/s00586-005-0058-8
Ozcan, C., and Ismi, O. (2016). Botulinum toxin for rhinitis. Curr. Allergy Asthma Rep. 16:58. doi: 10.1007/s11882-016-0636-3
Peterson, J. W. (1996). “Bacterial pathogenesis,” in Medical Microbiology, 4th Edn, ed S. Baron (Galveston, TX: University of Texas Medical Branch at Galveston).
Philpott, D. J., and Girardin, S. E. (2004). The role of Toll-like receptors and Nod proteins in bacterial infection. Mol. Immunol. 41, 1099–1108. doi: 10.1016/j.molimm.2004.06.012
Plouffe, J. F. (2000). Emerging therapies for serious gram-positive bacterial infections: a focus on linezolid. Clin. Infect. Dis. 31(Suppl. 4), S144–S149. doi: 10.1086/314080
Posselt, G., Schwarz, H., Duschl, A., and Horejs-Hoeck, J. (2011). Suppressor of cytokine signaling 2 is a feedback inhibitor of TLR-induced activation in human monocyte-derived dendritic cells. J. Immunol. 187, 2875–2884. doi: 10.4049/jimmunol.1003348
Prince, D. S., Peterson, D. D., Steiner, R. M., Gottlieb, J. E., Scott, R., Israel, H. L., et al. (1989). Infection with Mycobacterium avium complex in patients without predisposing conditions. N. Engl. J. Med. 321, 863–868. doi: 10.1056/NEJM198909283211304.
Qasimi, P., Ming-Lum, A., Ghanipour, A., Ong, C. J., Cox, M. E., Ihle, J., et al. (2006). Divergent mechanisms utilized by SOCS3 to mediate interleukin-10 inhibition of tumor necrosis factor alpha and nitric oxide production by macrophages. J. Biol. Chem. 281, 6316–6324. doi: 10.1074/jbc.M508608200
Qin, H., Holdbrooks, A. T., Liu, Y., Reynolds, S. L., Yanagisawa, L. L., and Benveniste, E. N. (2012). SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J. Immunol. 189, 3439–3448. doi: 10.4049/jimmunol.1201168
Qin, H., Roberts, K. L., Niyongere, S. A., Cong, Y., Elson, C. O., and Benveniste, E. N. (2007). Molecular mechanism of lipopolysaccharide-induced SOCS-3 gene expression in macrophages and microglia. J. Immunol. 179, 5966–5976. doi: 10.4049/jimmunol.179.9.5966
Rajaram, M. V., Ni, B., Dodd, C. E., and Schlesinger, L. S. (2014). Macrophage immunoregulatory pathways in tuberculosis. Semin. Immunol. 26, 471–485. doi: 10.1016/j.smim.2014.09.010
Rakesh, K., and Agrawal, D. K. (2005). Controlling cytokine signaling by constitutive inhibitors. Biochem. Pharmacol. 70, 649–657. doi: 10.1016/j.bcp.2005.04.042
Ramachandran, G. (2014). Gram-positive and gram-negative bacterial toxins in sepsis: a brief review. Virulence 5, 213–218. doi: 10.4161/viru.27024
Rawlings, J. S., Rosler, K. M., and Harrison, D. A. (2004). The JAK/STAT signaling pathway. J. Cell Sci. 117(Pt 8), 1281–1283. doi: 10.1242/jcs.00963
Recio, C., Oguiza, A., Lazaro, I., Mallavia, B., Egido, J., and Gomez-Guerrero, C. (2014). Suppressor of cytokine signaling 1-derived peptide inhibits Janus kinase/signal transducers and activators of transcription pathway and improves inflammation and atherosclerosis in diabetic mice. Arterioscler. Thromb. Vasc. Biol. 34, 1953–1960. doi: 10.1161/ATVBAHA.114.304144
Redford, P. S., Murray, P. J., and O'Garra, A. (2011). The role of IL-10 in immune regulation during M. tuberculosis infection. Mucosal Immunol. 4, 261–270. doi: 10.1038/mi.2011.7
Ren, K., and Dubner, R. (2010). Interactions between the immune and nervous systems in pain. Nat. Med. 16, 1267–1276. doi: 10.1038/nm.2234
Rottenberg, M. E., and Carow, B. (2014). SOCS3 and STAT3, major controllers of the outcome of infection with Mycobacterium tuberculosis. Semin. Immunol. 26, 518–532. doi: 10.1016/j.smim.2014.10.004
Rui, L., Yuan, M., Frantz, D., Shoelson, S., and White, M. F. (2002). SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J. Biol. Chem. 277, 42394–42398. doi: 10.1074/jbc.C200444200
Sahay, B., Patsey, R. L., Eggers, C. H., Salazar, J. C., Radolf, J. D., and Sellati, T. J. (2009). CD14 signaling restrains chronic inflammation through induction of p38-MAPK/SOCS-dependent tolerance. PLoS Pathog. 5:e1000687. doi: 10.1371/journal.ppat.1000687
Sasaki, A., Yasukawa, H., Shouda, T., Kitamura, T., Dikic, I., and Yoshimura, A. (2000). CIS3/SOCS-3 suppresses erythropoietin (EPO) signaling by binding the EPO receptor and JAK2. J. Biol. Chem. 275, 29338–29347. doi: 10.1074/jbc.M003456200
Sasaki, A., Yasukawa, H., Suzuki, A., Kamizono, S., Syoda, T., Kinjyo, I., et al. (1999). Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells 4, 339–351. doi: 10.1046/j.1365-2443.1999.00263.x
Sasi, W., Sharma, A. K., and Mokbel, K. (2014). The role of suppressors of cytokine signalling in human neoplasms. Mol. Biol. Int. 2014:630797. doi: 10.1155/2014/630797
Schneewind, O., and Missiakas, D. M. (2012). Protein secretion and surface display in Gram-positive bacteria. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 1123–1139. doi: 10.1098/rstb.2011.0210
Schroder, N. W., Morath, S., Alexander, C., Hamann, L., Hartung, T., Zahringer, U., et al. (2003). Lipoteichoic acid (LTA) of Streptococcus pneumoniae and Staphylococcus aureus activates immune cells via Toll-like receptor (TLR)-2, lipopolysaccharide-binding protein (LBP), and CD14, whereas TLR-4 and MD-2 are not involved. J. Biol. Chem. 278, 15587–15594. doi: 10.1074/jbc.M212829200
Scott, M. J., Liu, S., Shapiro, R. A., Vodovotz, Y., and Billiar, T. R. (2009). Endotoxin uptake in mouse liver is blocked by endotoxin pretreatment through a suppressor of cytokine signaling-1-dependent mechanism. Hepatology 49, 1695–1708. doi: 10.1002/hep.22839
Shaulov, L., Gershberg, J., Deng, W., Finlay, B. B., and Sal-Man, N. (2017). The ruler protein EscP of the enteropathogenic Escherichia coli Type III secretion system is involved in calcium sensing and secretion hierarchy regulation by interacting with the gatekeeper protein SepL. MBio 8:e01733–16. doi: 10.1128/mBio.01733-16
Smith, A. M., Rahman, F. Z., Hayee, B., Graham, S. J., Marks, D. J., Sewell, G. W., et al. (2009). Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn's disease. J. Exp. Med. 206, 1883–1897. doi: 10.1084/jem.20091233
Son, A., Shin, D. M., and Hong, J. H. (2015). Peptidoglycan induces the production of interleukin-8 via calcium signaling in human gingival epithelium. Korean J. Phys. Pharmacol. 19, 51–57. doi: 10.4196/kjpp.2015.19.1.51
Srivastava, V., Manchanda, M., Gupta, S., Singla, R., Behera, D., Das, G., et al. (2009). Toll-like receptor 2 and DC-SIGNR1 differentially regulate suppressors of cytokine signaling 1 in dendritic cells during Mycobacterium tuberculosis infection. J. Biol. Chem. 284, 25532–25541. doi: 10.1074/jbc.M109.006221
Stoiber, D., Kovarik, P., Cohney, S., Johnston, J. A., Steinlein, P., and Decker, T. (1999). Lipopolysaccharide induces in macrophages the synthesis of the suppressor of cytokine signaling 3 and suppresses signal transduction in response to the activating factor IFN-gamma. J. Immunol. 163, 2640–2647.
Stoiber, D., Stockinger, S., Steinlein, P., Kovarik, J., and Decker, T. (2001). Listeria monocytogenes modulates macrophage cytokine responses through STAT serine phosphorylation and the induction of suppressor of cytokine signaling 3. J. Immunol. 166, 466–472. doi: 10.4049/jimmunol.166.1.466
Stokes, B. A., Yadav, S., Shokal, U., Smith, L. C., and Eleftherianos, I. (2015). Bacterial and fungal pattern recognition receptors in homologous innate signaling pathways of insects and mammals. Front. Microbiol. 6:19. doi: 10.3389/fmicb.2015.00019
Strengell, M., Lehtonen, A., Matikainen, S., and Julkunen, I. (2006). IL-21 enhances SOCS gene expression and inhibits LPS-induced cytokine production in human monocyte-derived dendritic cells. J. Leukoc. Biol. 79, 1279–1285. doi: 10.1189/jlb.0905503
Sun, L., Jin, Y. Q., Shen, C., Qi, H., Chu, P., Yin, Q. Q., et al. (2014). Genetic contribution of CISH promoter polymorphisms to susceptibility to tuberculosis in Chinese children. PLoS ONE 9:e92020. doi: 10.1371/journal.pone.0092020
Takagi, H., Sanada, T., Minoda, Y., and Yoshimura, A. (2004). [Regulation of cytokine and toll-like receptor signaling by SOCS family genes]. Nihon Rinsho 62, 2189–2196.
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820. doi: 10.1016/j.cell.2010.01.022
Tamiya, T., Kashiwagi, I., Takahashi, R., Yasukawa, H., and Yoshimura, A. (2011). Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: regulation of T-cell inflammation by SOCS1 and SOCS3. Arterioscler. Thromb. Vasc. Biol. 31, 980–985. doi: 10.1161/ATVBAHA.110.207464
Tannahill, G. M., Elliott, J., Barry, A. C., Hibbert, L., Cacalano, N. A., and Johnston, J. A. (2005). SOCS2 can enhance interleukin-2 (IL-2) and IL-3 signaling by accelerating SOCS3 degradation. Mol. Cell. Biol. 25, 9115–9126. doi: 10.1128/MCB.25.20.9115-9126.2005
Tapping, R. I. (2009). Innate immune sensing and activation of cell surface Toll-like receptors. Semin. Immunol. 21, 175–184. doi: 10.1016/j.smim.2009.05.003
Tobelaim, W. S., Beaurivage, C., Champagne, A., Pomerleau, V., Simoneau, A., Chababi, W., et al. (2015). Tumour-promoting role of SOCS1 in colorectal cancer cells. Sci. Rep. 5:14301. doi: 10.1038/srep14301
Tosi, M. F. (2005). Innate immune responses to infection. J. Allergy Clin. Immunol. 116, 241–249; quiz 50. doi: 10.1016/j.jaci.2005.05.036
Trengove, M. C., and Ward, A. C. (2013). SOCS proteins in development and disease. Am. J. Clin. Exp. Immunol. 2, 1–29.
Turkson, J., and Jove, R. (2000). STAT proteins: novel molecular targets for cancer drug discovery. Oncogene 19, 6613–6626. doi: 10.1038/sj.onc.1204086
Uçeyler, N., Schäfers, M., and Sommer, C. (2009). Mode of action of cytokines on nociceptive neurons. Exp. Brain Res. 196, 67–78. doi: 10.1007/s00221-009-1755-z
Uchiya, K., and Nikai, T. (2005). Salmonella pathogenicity island 2-dependent expression of suppressor of cytokine signaling 3 in macrophages. Infect. Immun. 73, 5587–5594. doi: 10.1128/IAI.73.9.5587-5594.2005
Uchiya, K., and Nikai, T. (2008). Salmonella virulence factor SpiC is involved in expression of flagellin protein and mediates activation of the signal transduction pathways in macrophages. Microbiology 154(Pt. 11), 3491–3502. doi: 10.1099/mic.0.2008/021667-0
Ushiki, T., Huntington, N. D., Glaser, S. P., Kiu, H., Georgiou, A., Zhang, J. G., et al. (2016). Rapid Inflammation in mice lacking Both SOCS1 and SOCS3 in hematopoietic Cells. PLoS ONE 11:e0162111. doi: 10.1371/journal.pone.0162111
van de Geijn, G. J., Gits, J., and Touw, I. P. (2004). Distinct activities of suppressor of cytokine signaling (SOCS) proteins and involvement of the SOCS box in controlling G-CSF signaling. J. Leukoc. Biol. 76, 237–244. doi: 10.1189/jlb.0104041
Vázquez, N., Greenwell-Wild, T., Rekka, S., Orenstein, J. M., and Wahl, S. M. (2006). Mycobacterium avium-induced SOCS contributes to resistance to IFN-gamma-mediated mycobactericidal activity in human macrophages. J. Leukoc. Biol. 80, 1136–1144. doi: 10.1189/jlb.0306206
van 't Veer, C., van den Pangaart, P. S., Kruijswijk, D., Florquin, S., de Vos, A. F., and van der Poll, T. (2011). Delineation of the role of Toll-like receptor signaling during peritonitis by a gradually growing pathogenic Escherichia coli. J. Biol. Chem. 286, 36603–36618. doi: 10.1074/jbc.M110.189126
White, F. A., Sun, J., Waters, S. M., Ma, C., Ren, D., Ripsch, M., et al. (2005). Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc. Natl. Acad. Sci. U.S.A. 102, 14092–14097. doi: 10.1073/pnas.0503496102
Wilson, H. M. (2014). SOCS Proteins in Macrophage Polarization and Function. Front. Immunol. 5:357. doi: 10.3389/fimmu.2014.00357
Wilson, J. W., Schurr, M. J., LeBlanc, C. L., Ramamurthy, R., Buchanan, K. L., and Nickerson, C. A. (2002). Mechanisms of bacterial pathogenicity. Postgrad. Med. J. 78, 216–224. doi: 10.1136/pmj.78.918.216
Woodford, N., and Livermore, D. M. (2009). Infections caused by Gram-positive bacteria: a review of the global challenge. J. Infect. 59(Suppl. 1), S4–16. doi: 10.1016/S0163-4453(09)60003-7
Wu, J., Ma, C., Wang, H., Wu, S., Xue, G., Shi, X., et al. (2015). A MyD88-JAK1-STAT1 complex directly induces SOCS-1 expression in macrophages infected with Group A Streptococcus. Cell. Mol. Immunol. 12, 373–383. doi: 10.1038/cmi.2014.107
Yan, C., Cao, J., Wu, M., Zhang, W., Jiang, T., Yoshimura, A., et al. (2010). Suppressor of cytokine signaling 3 inhibits LPS-induced IL-6 expression in osteoblasts by suppressing CCAAT/enhancer-binding protein {beta} activity. J. Biol. Chem. 285, 37227–37239. doi: 10.1074/jbc.M110.132084
Yang, T., Stark, P., Janik, K., Wigzell, H., and Rottenberg, M. E. (2008). SOCS-1 protects against Chlamydia pneumoniae-induced lethal inflammation but hampers effective bacterial clearance. J. Immunol. 180, 4040–4049. doi: 10.4049/jimmunol.180.6.4040
Yasukawa, H., Misawa, H., Sakamoto, H., Masuhara, M., Sasaki, A., Wakioka, T., et al. (1999). The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. 18, 1309–1320. doi: 10.1093/emboj/18.5.1309
Yilma, A.N., Singh, S. R., Dixit, S., and Dennis, V. A. (2013). Anti-inflammatory effects of silver-polyvinyl pyrrolidone (Ag-PVP) nanoparticles in mouse macrophages infected with live Chlamydia trachomatis. Int. J. Nanomed. 8, 2421–2432. doi: 10.2147/IJN.S44090
Yoshimura, A. (1998). The CIS family: negative regulators of JAK-STAT signaling. Cytokine Growth Factor Rev. 9, 197–204. doi: 10.1016/S1359-6101(98)00019-7
Yoshimura, A., Naka, T., and Kubo, M. (2007). SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 7, 454–465. doi: 10.1038/nri2093
Yoshimura, A., Nishinakamura, H., Matsumura, Y., and Hanada, T. (2005). Negative regulation of cytokine signaling and immune responses by SOCS proteins. Arthritis Res. Ther. 7, 100–110. doi: 10.1186/ar1741
Yoshimura, A., Ohishi, H. M., Aki, D., and Hanada, T. (2004). Regulation of TLR signaling and inflammation by SOCS family proteins. J. Leukoc. Biol. 75, 422–427. doi: 10.1189/jlb.0403194
Yoshimura, A., Suzuki, M., Sakaguchi, R., Hanada, T., and Yasukawa, H. (2012). SOCS, Inflammation, and Autoimmunity. Front. Immunol. 3:20. doi: 10.3389/fimmu.2012.00020
Zadravec, P., Marecková, L., Petroková, H., Hodnik, V., Perišić Nanut, M., Anderluh, G., et al. (2016). Development of recombinant Lactococcus lactis displaying albumin-binding domain variants against shiga toxin 1 B subunit. PLoS ONE 11:e0162625. doi: 10.1371/journal.pone.0162625
Zhang, J. G., Farley, A., Nicholson, S. E., Willson, T. A., Zugaro, L. M., Simpson, R. J., et al. (1999). The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc. Natl. Acad. Sci. U.S.A. 96, 2071–2076. doi: 10.1073/pnas.96.5.2071
Zhang, J. G., Metcalf, D., Rakar, S., Asimakis, M., Greenhalgh, C. J., Willson, T. A., et al. (2001). The SOCS box of suppressor of cytokine signaling-1 is important for inhibition of cytokine action in vivo. Proc. Natl. Acad. Sci. U.S.A. 98, 13261–13265. doi: 10.1073/pnas.231486498
Zhang, J., Li, H., Yu, J. P., Wang, S. E., and Ren, X. B. (2012). Role of SOCS1 in tumor progression and therapeutic application. Int. J. Cancer 130, 1971–1980. doi: 10.1002/ijc.27318
Zhang, J. M., and An, J. (2007). Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 45, 27–37. doi: 10.1097/AIA.0b013e318034194e
Zhao, Q., Li, W., Chen, T., He, Y., Deng, W., Luo, H., et al. (2014). Mycobacterium tuberculosis serine protease Rvc can manipulate the host-pathogen interaction via Erk-NF-kappaB axis-mediated cytokine differential expression. J. Interferon Cytokine Res. 34, 686–698. doi: 10.1089/jir.2013.0071
Keywords: SOCS, immune response, inflammation, bacteria, cytokines, JAK/STAT, therapy, signaling
Citation: Duncan SA, Baganizi DR, Sahu R, Singh SR and Dennis VA (2017) SOCS Proteins as Regulators of Inflammatory Responses Induced by Bacterial Infections: A Review. Front. Microbiol. 8:2431. doi: 10.3389/fmicb.2017.02431
Received: 10 September 2017; Accepted: 23 November 2017;
Published: 12 December 2017.
Edited by:
Amy Rasley, Lawrence Livermore National Laboratory (DOE), United StatesReviewed by:
Elizabeth Hong-Geller, Los Alamos National Laboratory (DOE), United StatesErguang Li, Nanjing University, China
Copyright © 2017 Duncan, Baganizi, Sahu, Singh and Dennis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vida A. Dennis, vdennis@alasu.edu