Response: Commentary: Probing Genomic Aspects of the Multi-Host Pathogen Clostridium perfringens Reveals Significant Pangenome Diversity, and a Diverse Array of Virulence Factors
Commentary: Probing Genomic Aspects of the Multi-Host Pathogen Clostridium perfringens Reveals Significant Pangenome Diversity, and a Diverse Array of Virulence Factors
 Raymond Kiu
Raymond Kiu Shabhonam Caim
Shabhonam Caim Sarah Alexander
Sarah Alexander Purnima Pachori
Purnima Pachori Lindsay J. Hall
Lindsay J. Hall- 1Gut Health and Food Safety, Quadram Institute Bioscience, Norwich Research Park, Norwich, United Kingdom
- 2Norwich Medical School, University of East Anglia, Norwich Research Park, Norwich, United Kingdom
- 3Public Health England, London, United Kingdom
- 4Earlham Institute, Norwich Research Park, Norwich, United Kingdom
Clostridium perfringens is an important cause of animal and human infections, however information about the genetic makeup of this pathogenic bacterium is currently limited. In this study, we sought to understand and characterise the genomic variation, pangenomic diversity, and key virulence traits of 56 C. perfringens strains which included 51 public, and 5 newly sequenced and annotated genomes using Whole Genome Sequencing. Our investigation revealed that C. perfringens has an “open” pangenome comprising 11667 genes and 12.6% of core genes, identified as the most divergent single-species Gram-positive bacterial pangenome currently reported. Our computational analyses also defined C. perfringens phylogeny (16S rRNA gene) in relation to some 25 Clostridium species, with C. baratii and C. sardiniense determined to be the closest relatives. Profiling virulence-associated factors confirmed presence of well-characterised C. perfringens-associated exotoxins genes including α-toxin (plc), enterotoxin (cpe), and Perfringolysin O (pfo or pfoA), although interestingly there did not appear to be a close correlation with encoded toxin type and disease phenotype. Furthermore, genomic analysis indicated significant horizontal gene transfer events as defined by presence of prophage genomes, and notably absence of CRISPR defence systems in >70% (40/56) of the strains. In relation to antimicrobial resistance mechanisms, tetracycline resistance genes (tet) and anti-defensins genes (mprF) were consistently detected in silico (tet: 75%; mprF: 100%). However, pre-antibiotic era strain genomes did not encode for tet, thus implying antimicrobial selective pressures in C. perfringens evolutionary history over the past 80 years. This study provides new genomic understanding of this genetically divergent multi-host bacterium, and further expands our knowledge on this medically and veterinary important pathogen.
Introduction
Clostridium perfringens is a Gram-positive spore-forming anaerobe, best known as the causative agent for the tissue necrotic disease gas gangrene (also known as Clostridial myonecrosis). Notably, C. perfringens has also been widely associated with various intestinal diseases across human and animal species including: broiler necrotic enteritis (Keyburn et al., 2008), human food poisoning (Scallan et al., 2011), and preterm necrotising enterocolitis (Sim et al., 2015; Heida et al., 2016). Remarkably, it is reported to secrete >20 degradative toxins which constitute its primary arsenal to initiate histotoxic pathogenesis in both humans and animals (Uzal et al., 2014; Revitt-Mills et al., 2015). Intestinal-associated disease aetiology is characterised by rapid anaerobic proliferation in host tissue accompanied by in vivo production of several key pore-forming toxins including α-toxin, β-toxin, and β2-toxin, that consequently disrupt epithelial barrier function and induce histotoxic infections or tissue necrosis (Li and Mcclane, 2006). Common disease symptoms include pronounced diarrhoea (human adult food poisoning) (Mcclung, 1945), gaseous tissue necrosis (neonatal humans and animals) (Kosloske et al., 1978), and a haemorrhagic gut (neonatal horses) (Mehdizadeh Gohari et al., 2015).
C. perfringens was first isolated in the 1890s and named Clostridium welchii after its discoverer William Henry Welch (Welch and Nuttall, 1891). This gut pathogen is serotyped (or, specifically toxinotyped) clinically from A to E based upon the combination of “major toxins” (namely, α-toxin, β-toxin, ι-toxin, and ε-toxin) it encodes (Hassan et al., 2015). Multiplex PCR on the key toxin genes and Matrix Assisted Laser Desorption/Ionization time-of-flight mass (MALDI-TOF) spectrometry are the most common typing/identification methods in medical laboratories alongside conventional culture-based biochemical identification (Van Asten et al., 2009; Nagy et al., 2012).
Although this widespread gut pathogen has been studied and experimentally characterised over the past century, these studies have primarily focused on C. perfringens-associated toxins (Nagler, 1939; Ohno-Iwashita et al., 1986; Gibert et al., 1997; Stevens and Bryant, 2002). Notably, information on the entire pangenome [or, “the entire collection of gene families that are found in a given species” (Mcinerney et al., 2017)] of this pathogen is limited; to the date of analysis (April 2017) only 51 strains have been sequenced and made available publicly (Geer et al., 2010).
Previous comparative genomic studies on C. perfringens (up to 12 strains) indicated substantial genome variation within the pangenome (Hassan et al., 2015). Hence, we sought to construct the latest and largest pangenome of C. perfringens to probe evolutionary relationships and understand the genomic makeup of this bacterium. The C. perfringens genomes publicly available represent strains isolated from various diseased hosts including gas gangrene (human), food poisoning (human), necrotic enteritis (poultry), enterotoxaemia (sheep), and haemorrhagic enteritis (dogs and foals) from the past 100 years. This collection also represents the full range of C. perfringens toxinotypes (A–E). In this study, we also sequenced an additional 5 C. perfringens strains (as part of the NCTC3000 project1), for their historical significance (mostly isolated before 1960s), and thus included 56 genomes (including 51 genomes in NCBI databases) in our in silico investigation. Here we characterise genomic aspects of this medically and veterinary important pathogen, and through bioinformatic analysis of Whole Genome Sequencing (WGS) data identify virulence traits, define evolutionary relationships and functional annotations of the pangenome. Our analysis indicates that C. perfringens has a surprisingly diverse pangenome (core genes ~12.6%), potentially driven by horizontal gene transfer (HGT).
Results
Probing Evolutionary Relationships
Initially we sought to determine taxonomy and phylogenetic relatedness between C. perfringens and a subset of pathogenic/environmental Clostridium species (25) using 16S rRNA approach (Supplementary Table 1) (Woese et al., 1990) as previous earlier studies had reported that Clostridium pasteurianum, Clostridium baratii, and Clostridium absonum were the closest relatives (Canard et al., 1992; Collins et al., 1994). Using the 16S rRNA gene predictor Barrnap (>800 bp sequences used, 38/56 strains) we determined that the 16S rRNA regions appear to be highly conserved (identity >99.1%; Supplementary Table 2) across all selected 38 C. perfringens strains as they form a monophyletic lineage in the 16S Clostridium phylogenetic tree. C. perfringens cluster was clearly separated from other Clostridium species, with closest relatives identified as C. baratii (toxin producer associated with infant botulism) and Clostridium sardiniense (α-toxin producers isolated from gas gangrene cases) (Figure 1) (Masaki et al., 1988; Harvey et al., 2002). Clinically important toxin-generating bacteria C. botulinum, potentially pathogenic Clostridium paraputrificum, Clostridium tertium, scavenger Clostridium cadaveris, and deadly-toxin producer Clostridium tetani all fall in the same sub-lineage (cluster 2) as C. perfringens, with Clostridium difficile (known for nosocomial antibiotic-associated diarrhea), Clostridium sordellii and C. bifermentans (uncommon environmental species infrequently linked to human diseases) diverging earlier from their common ancestor (cluster 1), suggesting an ancient divergence in evolutionary history (Edagiz et al., 2015).
FIGURE 1
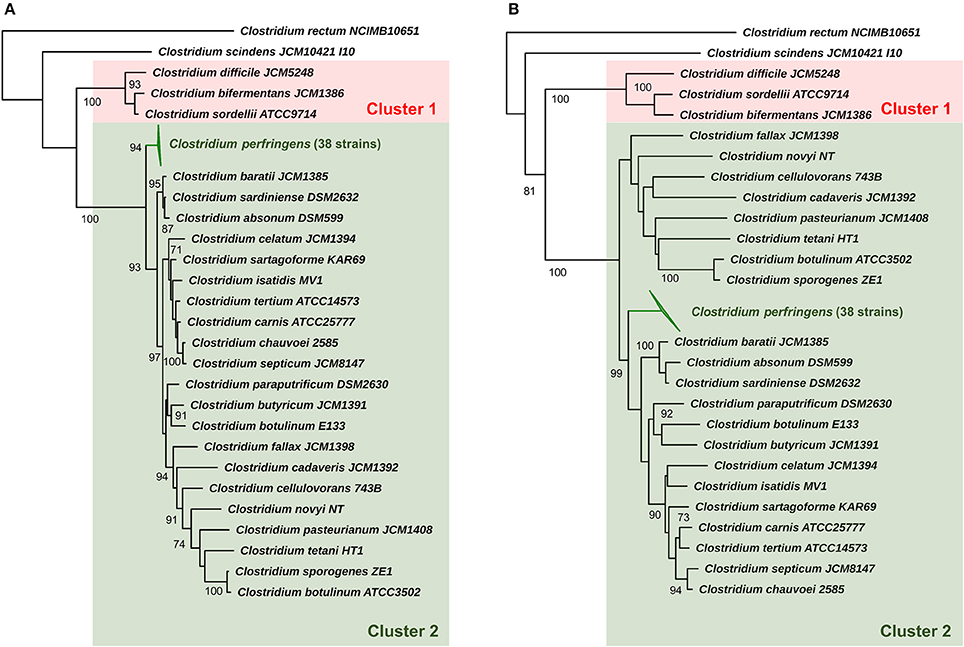
Figure 1. 16S rRNA phylogeny of representative Clostridium species (25) and C. perfringens. (A) Maximum-likelihood phylogenetic tree of C. perfringens strains and representative Clostridium species (25) based on 16S rRNA genes with aLRT branch support values displayed. (B) Neighbour-joining phylogeny based on 16S rRNA genes supported by 1000 bootstrap replicates based on 1585 sites. Clusters 1 and 2 were assigned for description purposes. Branch support values >70 are shown on the nodes.
Evolutionary distances based on near full-length 16S rRNA gene sequence (Table 1) also indicates that C. baratii and C. sardiniense are the closest relatives of C. perfringens in this dataset with sequence identities of ~95%, followed by C. absonum (94%), C. paraputrificum (93%), C. septicum (93%), C. butyricum (93%), C. tetani (91%), and C. pasteurianum (90%). These data support the 16S rRNA ML tree (Figure 1) as strains with higher sequence identities all appear to be more closely related to C. perfringens as indicated by tree topology.
TABLE 1
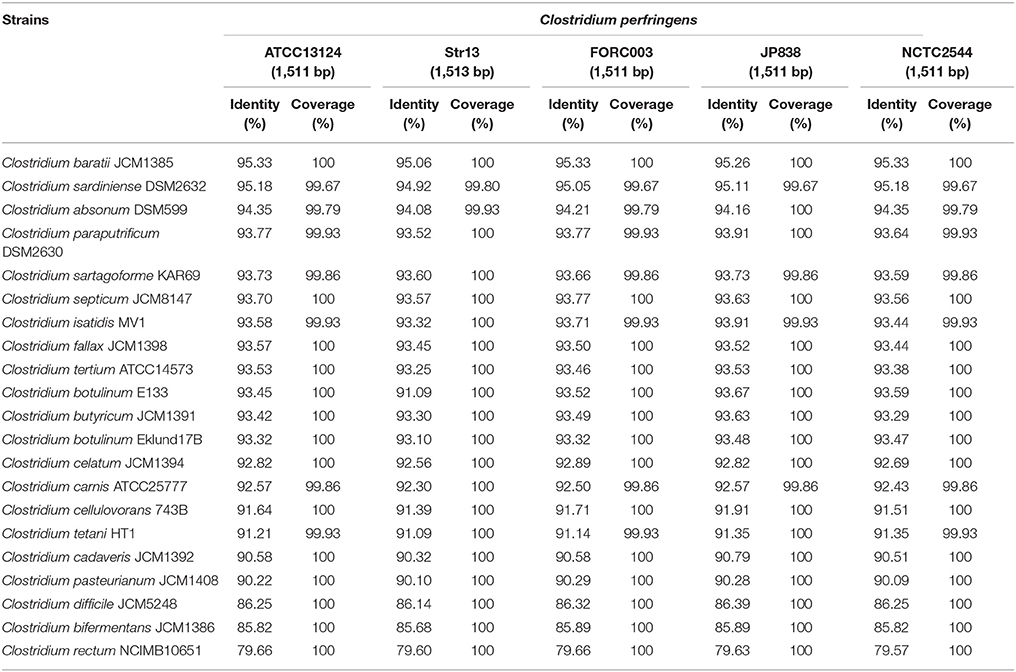
Table 1. 16S rRNA sequence pair-wise comparison between five representative C. perfringens strains and 20 selected Clostridium species.
Genome Description
A total of 51 genomes were retrieved in preassembled nucleotide FASTA files from NCBI database for further analysis (March 2017) and 5 NCTC strains were subjected to optimised DNA extraction procedures and sequenced (PacBio) as part of NCTC 3000 project (www.sanger.ac.uk/resources/downloads/bacteria/nctc/) with genomes annotated using Prokka. Whilst, contig number was variable (1–274) for NCBI-associated genomes (sequenced by different platforms and assembled using different methods), NCTC13170 and NCTC2544 reads assembled into 1 unitig, and NCTC8503, NCTC8797, and NCTC8678 genomes <10 contigs. Genome size of C. perfringens isolates ranged from 2.9 to 4.1 million bases, with an average GC content between 27.7 and 28.7% (Table 2). Predicted genes range from 2,600 to 3,800 with a median of 3304 genes.
TABLE 2
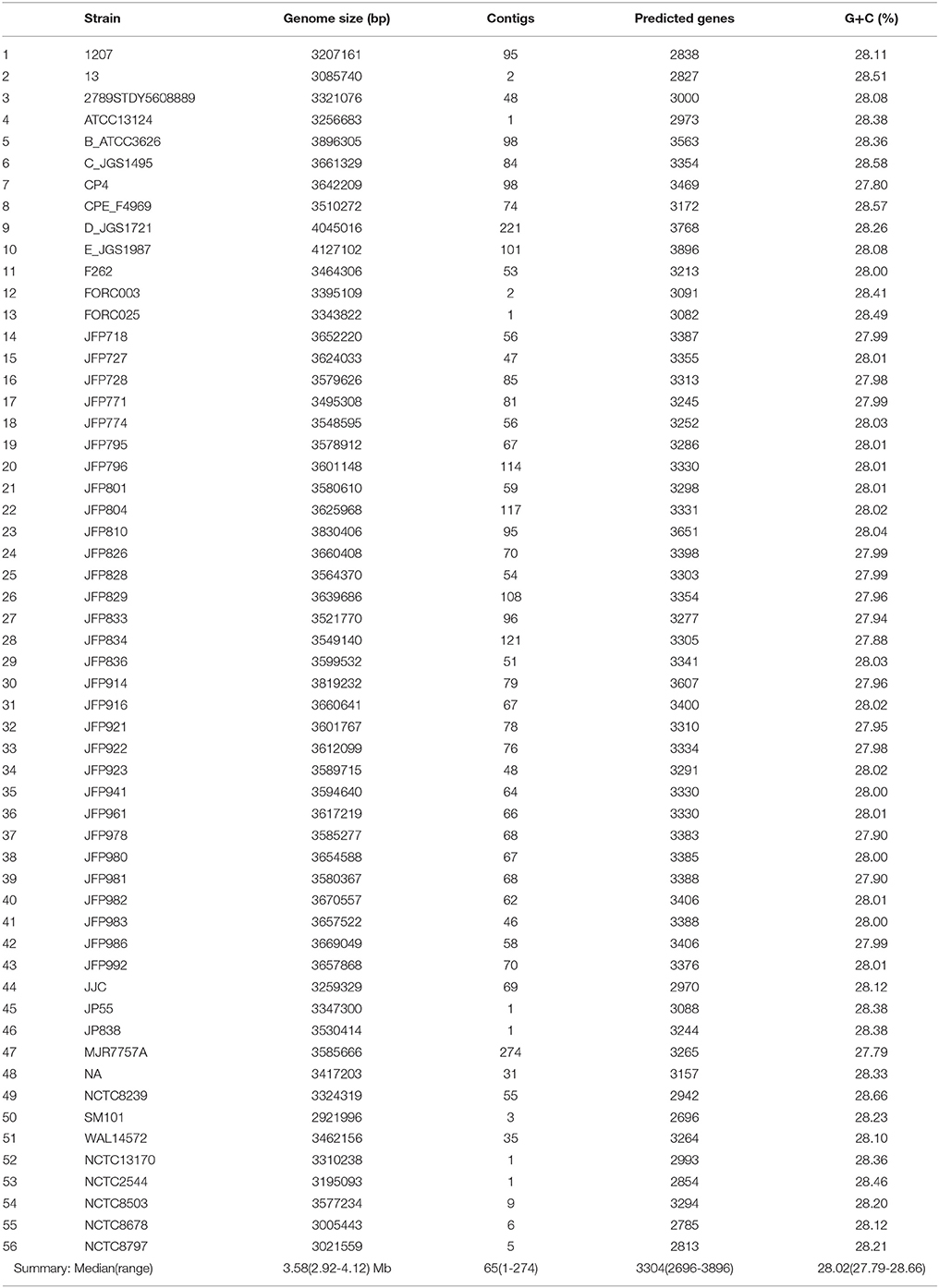
Table 2. Genome description of 56 C. perfringens isolates included in this study.
Pangenome Construction and Analysis
To define key genomic components, we next constructed the pangenome of C. perfringens encompassing 56 isolates, which represents the largest analysis of this type to date. This pangenome comprises 11,667 genes; 1470 core genes and 10,197 accessory genes (Figure 2A). Remarkably, only 12.6% represented core genes and a surprisingly high 44% (5139/11667) of unique genes (defined as genes only present in 1 strain in this pangenome; Figures 2B–E). This analysis implies very high genome plasticity in this pathogen, more than any other known prokaryotes currently published and studied (Mcinerney et al., 2017).
FIGURE 2
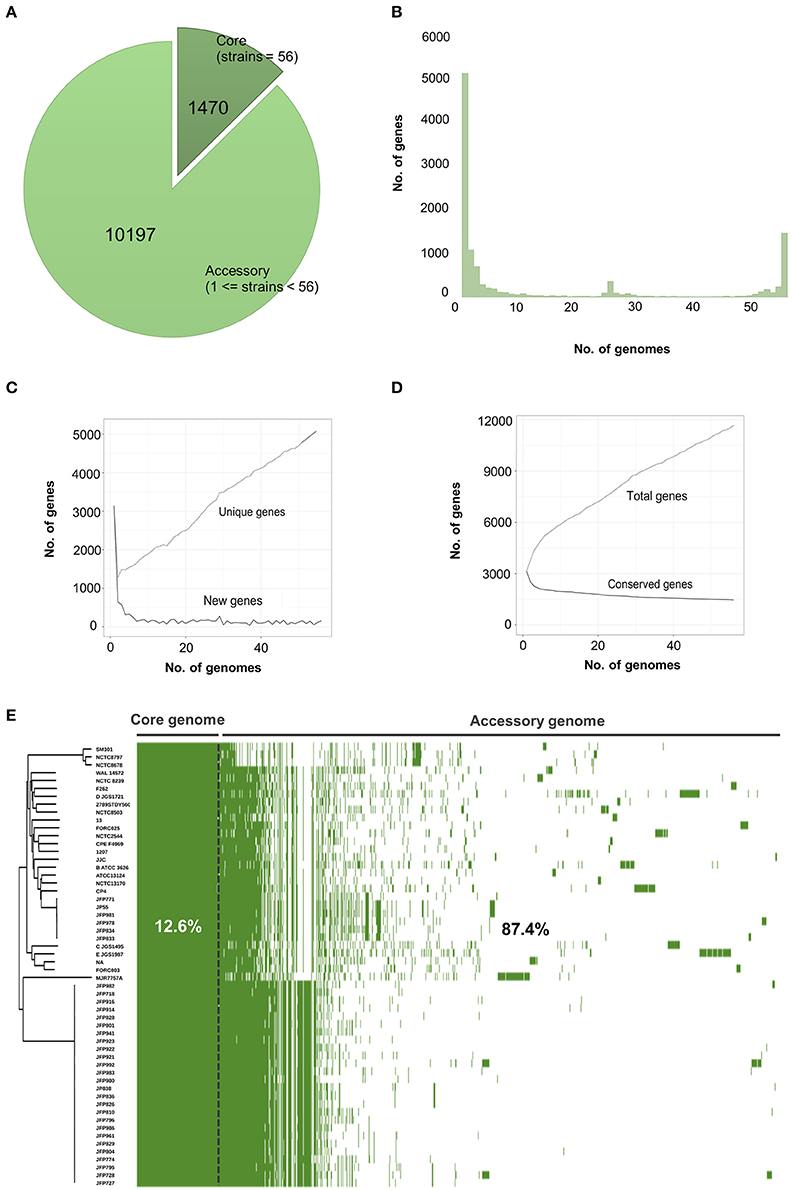
Figure 2. Visualisation and statistics of C. perfringens pangenome. (A) Core and accessory genes statistics. (B) Frequency bar graphs of number of identical genes against number of genomes. (C) Number of new genes and unique genes along pangenome computation. (D) Number of conserved genes and total genes along pangenome calculation. (E) Linearised pangenomic view of 56 C. perfringens isolates. Green cells represent syntenies aligned next to Neighbour-Joining core-genome based phylogenetic tree.
Phylogeny and Genome Comparison
A core-genome phylogenetic approach was used for inference of C. perfringens genetic relatedness of these 56 isolates (Figure 3). The generated phylogenetic tree indicated four main clades, which correlated with pertinent metadata (Table 3). Food poisoning isolates SM101, NCTC8797, and NCTC8678 (collected between1940–60 from diseased human adults) clustered in a single lineage (clade 1), whereas human, sheep, chicken, dog, horse, and soil isolates intermingled in clade 2 or clade 3, suggesting potential spread and transmission between hosts. In clade 4 (clearly split from clade 1–3), most isolates were collected from a North American dog and horse C. perfringens haemorrhagic enteritis study (JFP isolates) indicating these isolates might be host- and disease-specific. This supports previous work where these JFP netF-positive isolates were found to cluster closely in a cgMLST tree (Mehdizadeh Gohari et al., 2017). Furthermore, the presence of 2 JFP isolates of Swiss origin also clustered in clade 4, which again suggests disease- and host- specificity, but exclusion of geographical linkages. We also observed that different toxinotypes grouped within the same clade (Figure 3 and Table 3). These findings are similar to those previously reported by Hassan et al. and Mehdizadeh Gohari et al. and suggest that certain toxin genes are encoded within the accessory genome (Li et al., 2013; Hassan et al., 2015; Park et al., 2016; Mehdizadeh Gohari et al., 2017). Our analysis also demonstrates the presence of several major toxins encoded within C. perfringens plasmids (sequences individually retrieved from NCBI nucleotide database), highlighting the potential for toxin gene transfer via HGT (Supplementary Figure 1). Interestingly, whole-genome alignment-free phylogeny (Supplementary Figure 2) shows the clustering of toxinotypes (B-E in 1 lineage), in contrast to the core-genome based phylogeny (Figure 3), which further supports the hypothesis that several specific major toxins obtained via HGT are present in accessory genome.
FIGURE 3
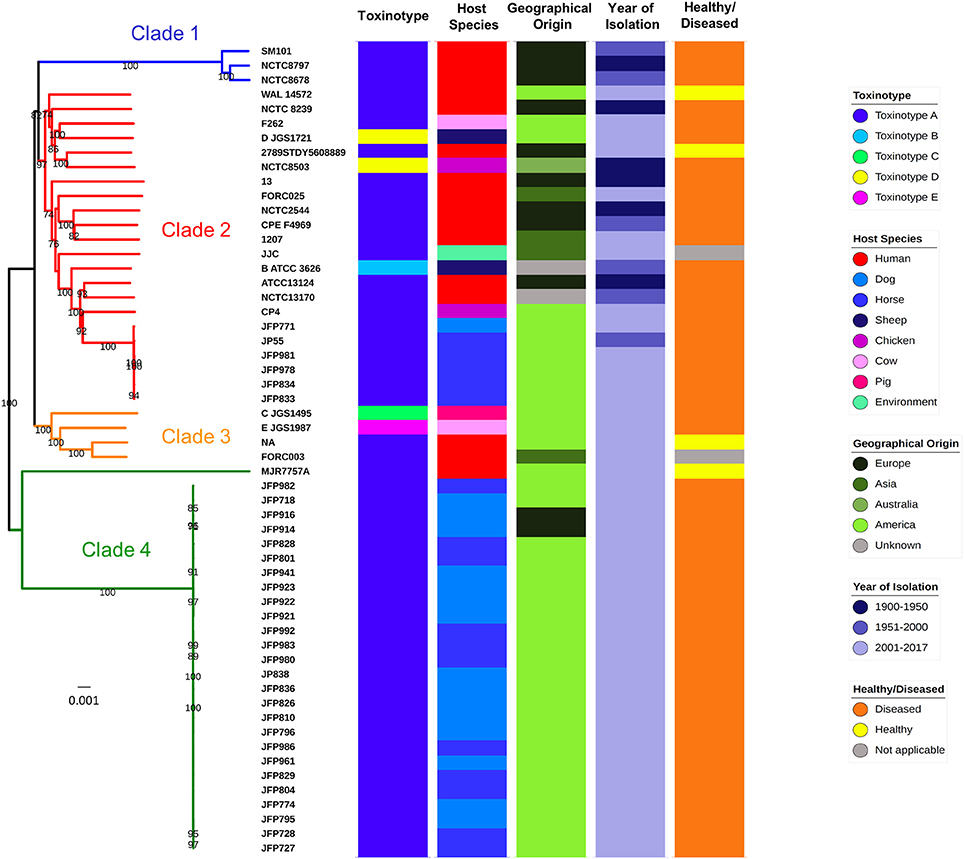
Figure 3. Genome comparison and core-genome phylogeny of C. perfringens. Core-genome based Neighbour-Joining (NJ) phylogeny of 56 isolates. Branch support values >70 (based on 1,000 bootstrap replicates) are shown on the nodes. Categories of metadata were displayed for comparison. MJR7757A is assigned to Clade 4 although it is distant from the rest of JFP isolates.
TABLE 3
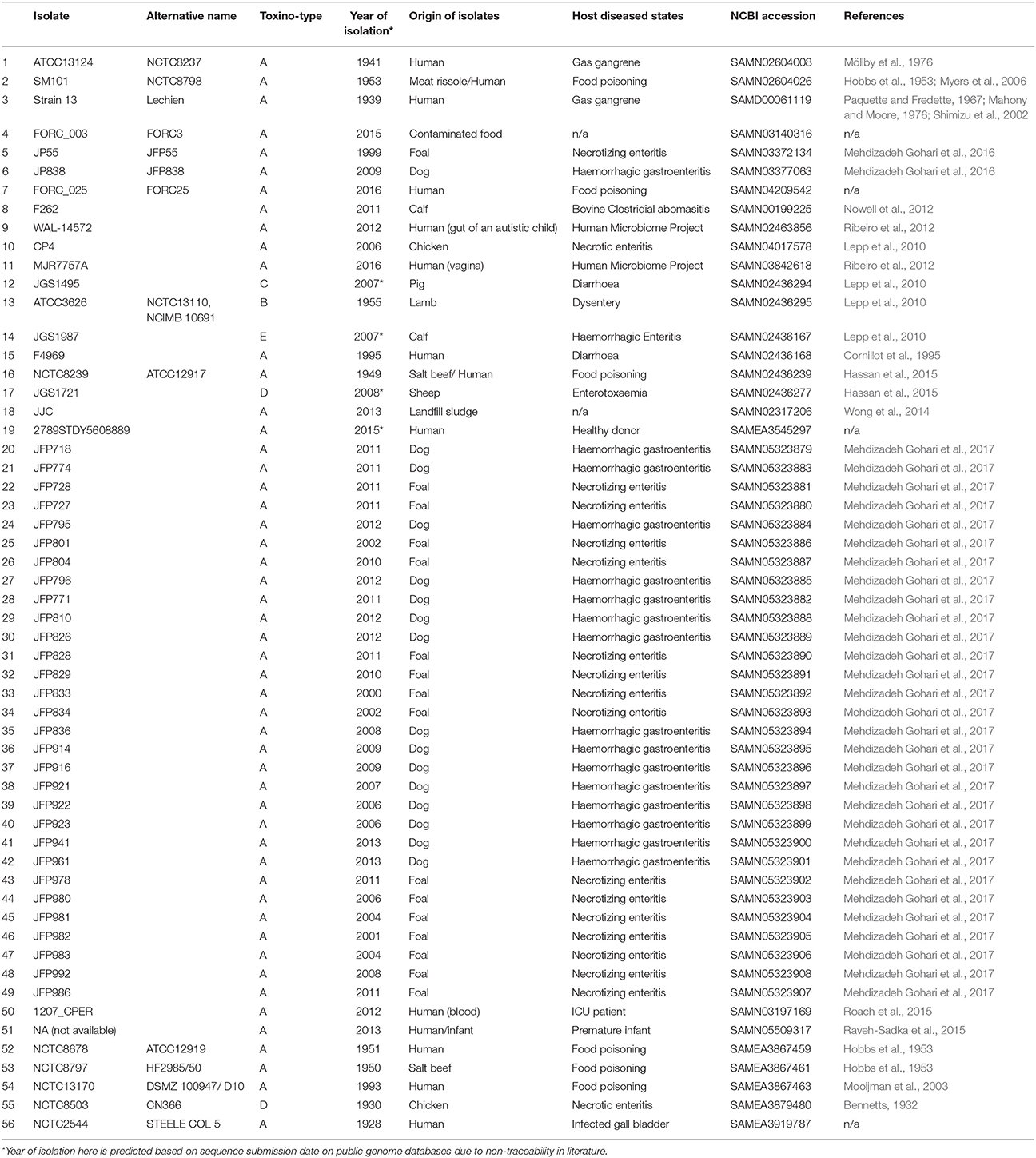
Table 3. Metadata of the C. perfringens genomes used for analysis in this study.
Multi-dimensional scaling (MDS) investigation (Supplementary Figure 3) based on whole-genome gene-absence-presence phylogeny (Supplementary Figure 4) also mirrors the core-genome phylogeny findings (Figure 3); food-poisoning isolates cluster tightly, JFP isolates nest in 2 distinct lineages (clades 2 and 4) and remaining isolates form another cluster. Using JP838 as our reference genome (as it contains a large 3.5 Mbp chromosome for nucleotide comparison) we generated a circular genome comparison figure, based on 1–3 strains from each of the 4 core-genome clades (Supplementary Figure 5). This comparison supports our results from the pangenomic analysis highlighting significant variation; faded areas (<80% sequence similarity) exist in several regions as well as regions (>10) with differences in GC content, which may indicate genomic island insertion sites via HGT. Interestingly, an additional whole-genome search on strain JP838 indicated that most coding sequences predicted in divergent GC regions encode for hypothetical protein genes (via BLAST NR database) of unknown function.
Functional Annotation of Core and Accessory Genomes
We next analysed both the core and accessory genomes at a functional level using “Clusters of Orthologous Groups of proteins” (COG) and “Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups” (eggNOG) databases (Galperin et al., 2015; Huerta-Cepas et al., 2016), to probe potential C. perfringens host adaption and/or pathogenesis traits (Figure 4).
FIGURE 4
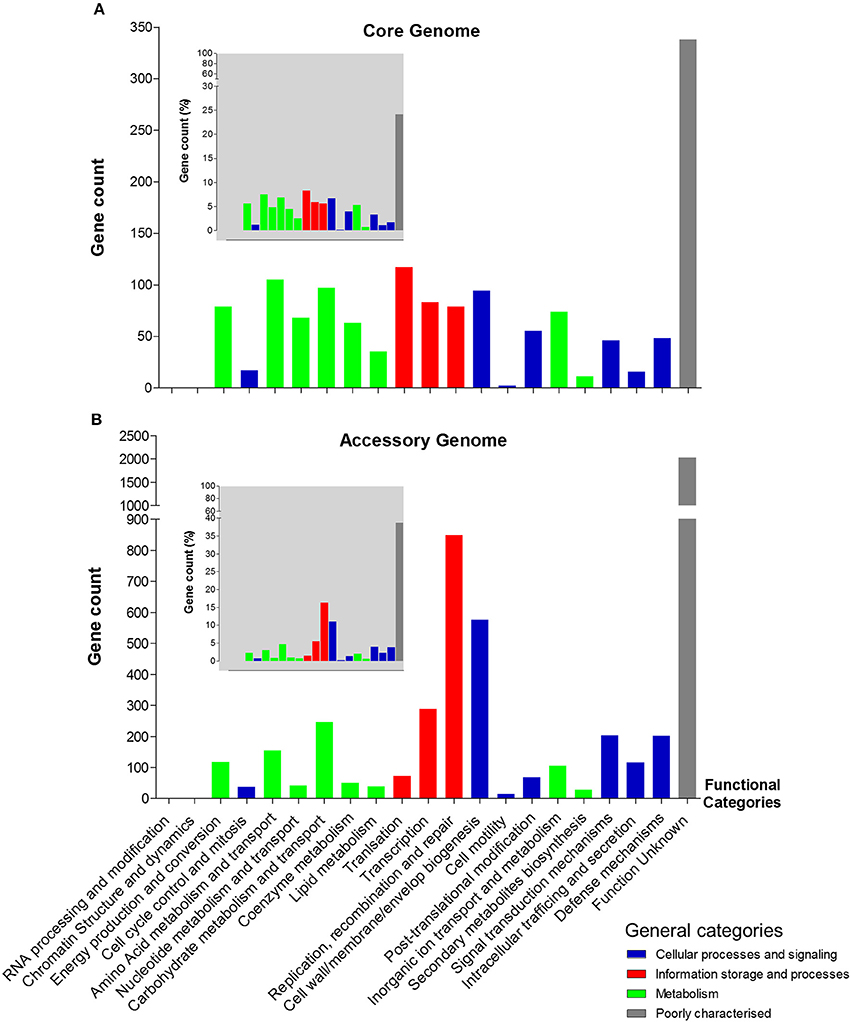
Figure 4. Functional annotation (COG) of C. perfringens core and accessory genomes. (A) Core genome functional annotation. (B) Accessory genome functional annotation. The smaller-scale grey-background charts show the percentage of gene count of each functional class in both core and accessory genomes corresponding to the functional categories. Non-matching genes (unclassified) are not included in the statistics.
Interestingly, 849 genes were assigned to protein families associated with “replication, recombination, and repair” comprising mainly transposases (n = 254), integrases (n = 31), and phage proteins (n = 7) within the accessory genome. Whereas, only 79 genes encoded within the core genome were assigned to this functional class, suggesting a key difference between both genomes at a functional level (849 vs. 79; 16.2 vs. 5.6%). Genes associated with “defense mechanisms” were higher within the accessory genome (n = 202), when compared to the core genome (n = 48), which includes genes encoding efflux pumps, restriction enzymes and ABC transporters (linked with iron uptake systems). Genes involved in metabolism (including carbohydrate, amino acid, and lipid) are proportionately more represented within the core genome (16.8%) vs. the accessory genome (8.3%), whilst recombination and cell-wall biogenesis related genes dominate the functionality of the accessory genome (27.1% vs. core genome: 12.2%). Notably, as indicated above there is a high proportion of unknown-function genes observed in both genomes (core: 24% vs. accessory: 38.7%; Figure 4).
In Silico Profiling of Virulence Traits
Toxins secreted by C. perfringens have long been considered key virulence factors and are implicated in multiple clinical disease phenotypes (Welch and Nuttall, 1891; Rood, 1998). Primary toxins produced include α-toxin, β-toxin, ι-toxin, and ε-toxins, which are used to classify this pathogen into different toxinotypes (Petit et al., 1999). These toxins, as well as additional toxins β2-toxin and perfringolysin O, potentially initiate pathogenesis through pore-forming and cytolysis (Supplementary Table 4). Therefore, we analysed the toxin profiles of the 56 isolates, and additional virulence-associated factors such as antimicrobial resistance (AMR) and prophage content (Figure 5).
FIGURE 5
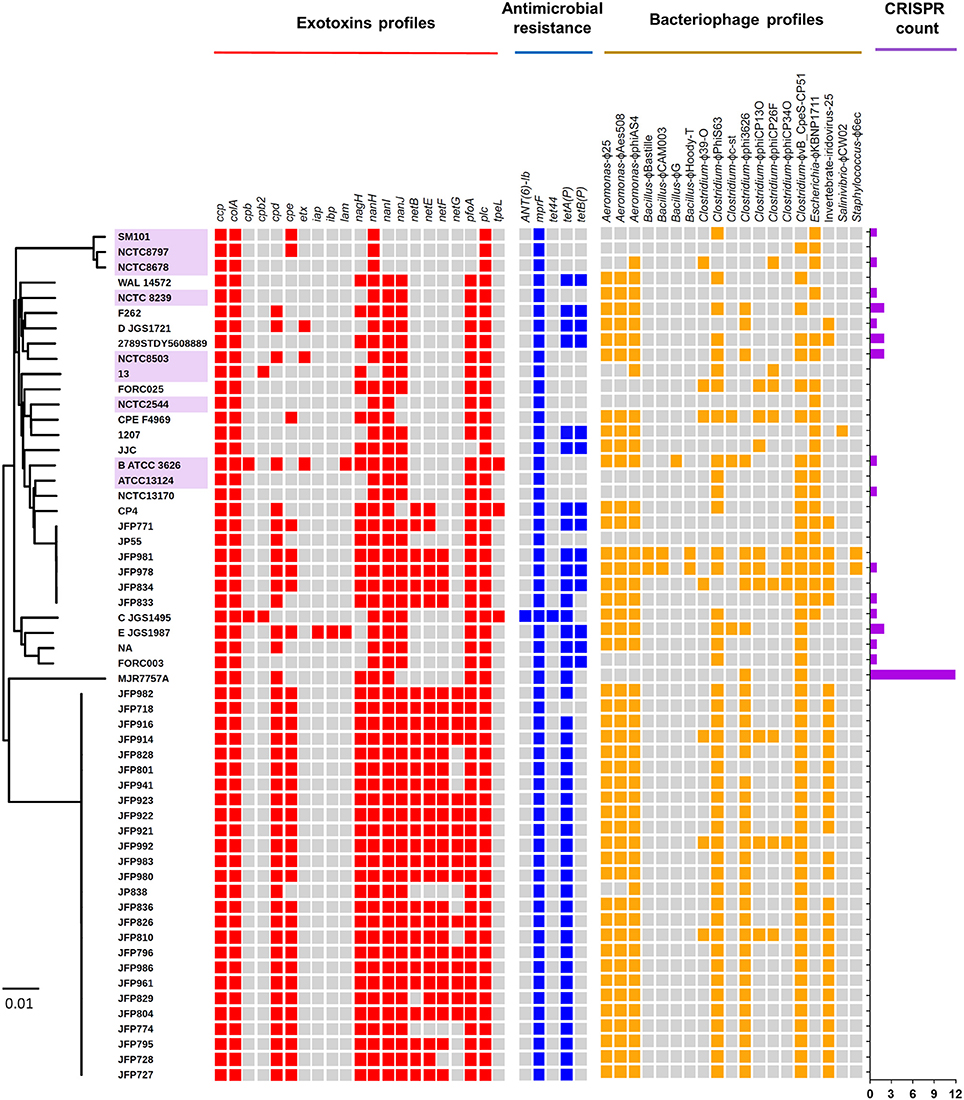
Figure 5. Virulence-associated potentials in C. perfringens genomes and plasmids. Heat-map visualising exotoxins, antimicrobial resistance, prophage profiles, and CRISPR counts of 56 C. perfringens genomes. Presence of genes are indicated by cell colours: grey (absence); red, blue, and yellow (presence). Purple-labelled isolates indicate historical isolates (pre-antibiotic era).
Several toxins, including α-toxin (plc), α-clostripain (ccp), and microbial collagenase (colA) genes are conserved in all isolates (Figure 5). Other toxins, such as enterotoxin (cpe), a food-poisoning-linked toxin, was found to be encoded in food-poisoning isolates SM101 and NCTC8797, however this gene is also present in all JFP isolates (haemorrhagic enteritis isolates). Perfringolysin O (pfoA or pfo), a well-characterised pore-forming toxin, is encoded in most genomes except food-poisoning and JJC environmental isolates and presence of sialidase genes (nan and nag) vary across the isolates while netB, netE, netF, and netG genes were found to be uniquely encoded within JFP isolates (associated with haemorrhagic enteritis in dogs and foals), as reported previously (Mehdizadeh Gohari et al., 2016).
Antimicrobial-peptide (AMP) resistance gene multi-peptide resistance factor (mprF) was detected in all isolates. This gene is highly-conserved in certain Gram-positive and Gram-negative bacteria (Figure 5) and is known to confer resistance through modifying surface molecules of AMPs, including host defensins, and potentially also aminoglycoside antibiotics (Ernst and Peschel, 2011; Cole and Nizet, 2016). Tetracycline-resistant efflux protein tetA(P) gene (Bannam et al., 2004) were detected in 75% of the isolates (n = 42), and 23% of the isolates (n = 14) harboured double tet-resistance genes—tetA(P) and tetB(P). The only other AMR-associated gene detected was specific aminoglycosides resistance gene (ANT(6)-Ib), which was encoded within 1 toxinotype C porcine isolate JGS1495.
Finally, genome scanning for prophage elements indicated that 19 in total were encoded, potentially derived from a variety of bacterial genera including Aeromonas, Bacillus, Clostridium, Escherichia, and Staphylococcus (Figure 5). Notably, JFP isolates also displayed unique prophage profiles including Clostridium phage ϕvB_cpes_CP51 (n = 48), Aeromonas phage ϕphiAS4 (n = 47), ϕaes508 and ϕ25 (n = 45), and Clostridium phage ϕphiS63 (n = 44). Interestingly, 40/56 (71.5%) isolates do not encode CRISPR system at all (in silico prediction = 0; Figure 5).
Discussion
Although C. perfringens is an important causative agent of animal and human infections; our knowledge on the genome content and phylogenic characteristics of this pathogenic bacterium is currently limited. Our current study, analysing the largest number of C. perfringens genomes to date, highlights significant genomic plasticity, and an arsenal of key virulence factors, including toxins and AMR genes, that are encoded by this pathogen.
Using the 16S rRNA gene to initially probe C. perfringens phylogenic relationships (Koch et al., 1994), we observed clear deviation of 16S alignment from other Clostridium species (Figure 1), with C. baratii and C. sardiniense as the closest relatives of C. perfringens (16S sequence similarity ~95.3 and 95.1%), which agrees with previous 16S phylogenetic studies (Collins et al., 1994; Vos et al., 2009). We also performed alignment-free whole-genome-based phylogenetics, which has been reported to represent a more comprehensive and robust method for clustering genetically variable prokaryotes including Helicobacter pylori (Van Vliet and Kusters, 2015; Bernard et al., 2016). Our analysis indicates that this approach can also be used for C. perfringens, as we observed near-identical phylogeny when compared to the core-genome based method (Supplementary Figure 2), and may prove useful in epidemiology and outbreak management.
Previous limited genomic studies have indicated that C. perfringens is a genetically diverse organism, and our larger scale study probing 56 genomes also supports this notion. We determined that C. perfringens has a highly variable open/infinite pangenome, with new genes potentially being added at an average of 106 genes for each new genome sequenced (Supplementary Figure 6), which is in comparison to other pathogens, including Yersinia pestis (Eppinger et al., 2010) and Bacillus anthracis (Rouli et al., 2015), which have closed pangenomes. The “lifestyle” of the organism is proposed to reflect the nature of the pangenome, and C. perfringens' ability to thrive in multiple host species and environmental niches, including the human and animal gut and soil, allows active interaction with other bacterial species, and links to our open pangenome data (Rouli et al., 2015). To our knowledge, only a handful of bacterial pangenome studies have reported extreme pan-genomic variation. This trait (12.5% core genes) is an uncommon observation in other bacteria and is significantly lower than other reported prokaryotic species including C. difficile (30.3%) (Knight et al., 2016), Streptococcus pneumoniae (46.5%) (Hiller et al., 2007), Salmonella enterica (16%), and Klebsiella pneumoniae (26%) (Mcinerney et al., 2017). It should be noted that this pangenome analysis may be limited in terms of capturing total C. perfringens diversity due to the fact that 30 isolates in our analysis are from a single study (Mehdizadeh Gohari et al., 2017). These netF-positive isolates were sequenced and assembled for plasmid pathogenicity analysis, and were reported to have tight clonal relationship using a MLST approach that could potentially bias the analysis of this dataset. Thus, we expect that due to C. perfringens' wide host and environmental range, additional genetic variation potentially exists, and that a significantly larger-scale study, encompassing a greater variety and number of isolates from multiple ecological niches, would be required to obtain higher-resolution evolutionary insights.
C. perfringens is recognised to be the most prolific toxin-producing organism currently characterised, and is reported to produce >20 exotoxins. However, not all strains produce all toxins, and certain toxins are encoded on plasmids rather than chromosomes (Supplementary Figure 1). In our analysis, we showed that individual strains produce a diverse profile of toxins, with the exception of the JFP isolates, which display a unique pattern of toxin gene profiles (netE, netF, and netG), inferring that these encoded toxins may be associated with specific hosts (i.e., dogs and horses), and/or disease (i.e., haemorrhagic enteritis) (Mehdizadeh Gohari et al., 2016). Although poultry necrotic enteritis (NE) is thought to be mediated through the netB toxin, the NE isolate NCTC8503 was not found to encode this gene, which confirms a previous study by Brady et al. who determined that netB (via PCR testing) was present in both healthy and diseased chickens (Brady et al., 2010). Interestingly, netB was detected in 87.5% JFP isolates (28/32), suggesting that netB toxin might be involved in haemorrhagic enteritis in dogs and horses. Whist our analysis provides interesting linkages with toxin-associated genes with disease and host phenotypes, determination of specific associations will require further WGS data from a wider variety of isolates (including from healthy hosts), and confirmation from experimental studies.
As C. perfringens produces this vast array of toxins, a combination of several “key toxins” are traditionally used to type this species. However, our understanding behind the disease-toxinotype link is limited at present, in part due to lack of in-depth WGS studies. We observed that most of the disease-causing isolates in this dataset are toxinotype A (88%; 44/50), and are associated with diverse diseases in different hosts, suggesting the need to review current toxinotyping methods to reflect C. perfringens genetic and pathogenesis relationships more closely. From a public health standpoint, a reliable typing method is also necessary to track the spread of bacterial pathogens, and outbreaks. Currently, there is a move from traditional serotyping and MLST approaches to more recent WGS core-genome based methods, which are employed by national-level health department including Public Health England in the UK (Ashton et al., 2016). Notably, phylogenomic clustering (using CVTree) resembles the core-genome tree, indicating the possibility of employing phylogenomic approaches for large dataset epidemic analysis, as it uses significantly less computational resources (Bernard et al., 2016).
The extreme pan-genomic variation of C. perfringens, appears to be driven by HGT events, as indicated by the high number of genes associated with transposases, integrases, and phages, which infers the high potential for gene gain or loss events (Figure 4). Notably, C. perfringens is found in multiple ecological niches, which are associated with diverse microbial (including viral) communities. Genomic integration of prophages seemingly represents a key source of genetic diversity in C. perfringens, and prophage diversity in these isolates surpass the phage contents of C. difficile; 7 prophages were found in a total of 44 isolates (Knight et al., 2016). Notably, phages in the human gut are reported to outnumber bacteria by at least a factor of 10, and their lysogenic lifestyle has a powerful biological impact on its hosts such as modifying the fitness and metabolism of bacterial hosts (Brüssow and Hendrix, 2002). Bacteriophages were reported to boost the sporulation capacity of C. perfringens in several studies, which reflects the survival strategy employed by these viral particles (Stewart and Johnson, 1977). Some prophage-like genes were also associated with toxin secretion in C. difficile (Goh et al., 2005), Salmonella enterica (Thomson et al., 2004), and E. coli (Zhang et al., 2000). Thus, diverse prophage-associated gene integration in C. perfringens genomes may be central for our understanding of the pathogenicity of C. perfringens in terms of human and animal health. This high prophage genome plasticity is further supported by our CRISPR (Clustered regularly interspaced short palindromic repeats) array investigation on these 56 isolates as 71.5% (40/56) are predicted to possess no CRISPR system (count = 0), which encodes systems to defend against viral/phage invasion (Figure 5). Interestingly, genes involved in “defense mechanisms” are encoded at higher numbers within the accessory genome (n = 202), than the core genome (n = 48), again suggesting constant genomic adaptation to environmental change. These include genes encoding multidrug efflux pumps (associated with antibiotic resistance; n = 11) and ABC transporters (linked with iron-uptake mechanisms and multi-drug resistance; n = 83) (Köster, 2001; Andersen et al., 2015). Notably, genes encoding functions expected to be conserved, such as metabolism (i.e., carbohydrate, amino acid, and lipid metabolism) are more represented within the core (16.8%) rather than the accessory genome (8.3%), indicating their critical functionality in terms of colonisation and growth in different nutritional environments.
There is an ever-increasing incidence of multi-drug resistance pathogens that now pose a critical risk to human and animal health. AMR within C. perfringens strains is a serious concern due to its ability to encode multiple virulence associated factors e.g. toxins, that are linked to severe disease. Previous phenotyping-based studies have reported C. perfringens as a multidrug resistant pathogen (resist against >1 antimicrobial drug) (Tansuphasiri et al., 2005). However, our genomic analysis indicates that tetracycline resistance genes are the only AMR-specific genes that are wide-spread in this pathogen, which corresponds to previous findings of phenotypically observed tetracycline resistance in C. perfringens strains isolated from pigs, human, and environments (Rood et al., 1978). Resistance genes tetA(P) and tetB(P) are both tetracycline efflux proteins most commonly known to be encoded in C. perfringens since the 1980s (Lyras and Rood, 1996). Notably, our data indicates that strains isolated prior to the 1950s do not encode any tet genes, which may correlate with the fact that tetracycline did not come into commercial use until 1978, supporting the emergence of antimicrobial resistance under selective pressure in the post-antibiotic era. Although C. perfringens does encode tetracycline resistance-associated genes, this is a limited AMR profile when compared to other Gram-positive enteric pathogens like C. difficile (Knight et al., 2016). Novel/uncharacterised AMR genes and transmission route (directly from environments such as soil) may account for these findings, however a larger dataset from hospital and/or farming environments and phenotypic testing is required to confirm this hypothesis. Nevertheless, it is suggestive that efflux pumps families including efflux pumps (e.g., mepA) and ATP-Binding Cassette (ABC) transporters were in silico detected as these could potentially play a significant role in multidrug resistance as observed in previous clinical cases (Andersen et al., 2015). Furthermore, the environment in which C. perfringens resides (e.g., the gut) could also potentially play a significant role in the acquisition of new AMR genes via the “resistome,” and as such warrants further investigations (Browne et al., 2017). The host also has specific mechanisms for directly killing gut-associated bacteria including via AMPs. Interestingly, the mprF gene (known to be involved in resistance to human defensins) was found to be conserved in all C. perfringens genomes, which may link to its ability to evade host-associated defenses for gut colonisation. Indeed, previous studies in Staphylococcus aureus have indicated that mprF mutant strains are more prone to mammalian AMP killing including neutrophil defensins (Peschel et al., 2001), and mutants are less virulent in animal infection models (Kristian et al., 2003). Importantly, homologues of mprF are also reported to dampen the effectiveness of certain clinical antimicrobials (aminoglycosides) such as vancomycin and gentamicin (Nishi et al., 2004), and this may link to previous studies showing resistance of C. perfringens to these antibiotics (Traub and Raymond, 1971; Tyrrell et al., 2006; Osman and Elhariri, 2013).
Conclusion
High-throughput WGS has paved the way for in-depth pangenomic studies into bacterial species, however previous studies on C. perfringens have utilised a maximum of 12 strains for analysis, and thus genomic information on this pathogen is currently limited. Our analysis of 56 strains indicates a surprisingly diverse pangenome, and in silico functional analysis reveals frequent exchange of genes in its evolutionary history, associated with prophages, toxins and AMR genes, that potentially influence pathogenicity. Interestingly, this study did not correlate any toxinotype to a specific disease in phylogenetic clustering, however a larger dataset may be needed to establish any causative relationships. As most of the isolates in our study are from diseased patients/animals, inclusion of isolates from healthy samples may enable us to understand the genomic signatures that separate commensal from pathogenic C. perfringens (Nagpal et al., 2015). Whilst these genomic studies are of central importance it is vital to also determine the phenotypic disease-causing mechanisms, thus experimental studies are essential to probe our understanding of C. perfringens pathogenesis in tandem with in silico exploration.
Materials and Methods
Bacterial Isolates and Metadata
C. perfringens isolates previously sequenced and deposited in NCBI were used in this study (n = 51; retrieved in March 2017), a further 5 C. perfringens isolates (NCTC8678, NCTC8797, NCTC13170, NCTC2544, and NCTC8503) were sequenced as part of the NCTC 3000 sequencing project to be included in this study. Metadata of all 56 isolates were retrieved manually online (March 2017) either from published literature, National Center of Biological Information (NCBI) databases or European Bioinformatics Institute (EBI) databases. Year of isolation and geographical origins of isolates were taken from published research papers, otherwise estimated using dates/locations on public databases as indicated. WGS accessions, references and metadata for all genomes are provided in the Table 3.
Genomic DNA Extraction and Whole Genome Sequencing
For genomic DNA extraction, C. perfringens strains (NCTC8678, NCTC8797, NCTC13170, NCTC2544, and NCTC8503) were propagated on blood agar at 37°C for 36 h under anaerobic conditions. Bacterial lysates were prepared in buffer B1 (containing additional RNase A, protease, and Lysozyme) and the lysates were incubated at 80°C for 24 h. Post-incubation high Molecular weight DNA was isolated from C. perfringens strains using the Qiagen midi kit. DNA quality (>60 kb) and quantity (>3 μg) was assessed using the Agilent 2200 TapeStation and Qubit® dsDNA BR Assay Kit respectively. WGS was performed using the PacBio SMRT® DNA Sequencing technology. Appropriate biosafety procedures were in place for both bacterial culturing and DNA extraction.
De Novo Assembly and Annotation
Assembled genomes (51) previously sequenced were retrieved online on NCBI Genomes database (Geer et al., 2010). All 56 genomes were annotated using Prokka v1.10 (Seemann, 2014). HGAP.3 from SMRTpipe-2.3.0 was used to perform de novo assembly of the 5 strains of C. perfringens sequenced on PacBio RS I obtained from ENA, with one SMRT cell per strain assembled into unitigs. The minlongreadlength (assembly seed length) parameter was set such that there was 30X genome coverage above the seed length. Two strains (NCTC13170 and NCTC2544) were assembled into one single unitig of 3,310,238 bp and the rest three strains (NCTC8678, NCTC8503, and NCTC8797) were assembled into 5–9 unitigs with an N50 of over 2,969,557 bp. These assemblies were run on a High-Performance Computing Cluster using SLURM, utilizing 50 Gb of memory and running parallel on four computing nodes taking ~634.93 min to finish.
Pangenome Assembly and Visualisation
The annotation files (general feature format, GFF) of 56 C. perfringens genomes were submitted to Roary pangenome pipeline v3.6.1 to perform pangenomic analysis (Page et al., 2015). Roary was run with default parameters. A gene-absence-presence data matrix was subsequently derived and visualised in Phandango genome visualizer (v0.87) (Hadfield and Harris, 2017). Fripan2 was used to visualised MDS clustering of isolates (Supplementary Figure 3) and gene-presence-absence tree (Supplementary Figure 4). Recommended R scripts from Roary package were used to generate pangenome stats using R studio(R Development Core Team, 2010; Rstudio Team, 2015).
Core-Genome Alignment and Construction of Phylogenetic Trees
Core-genome alignment generated using MAFFT v7.305b (Katoh and Standley, 2013) was used as input for Seaview v4.0 (Gouy et al., 2010) to construct core-genome phylogeny. Neighbour-Joining tree (NJ) was built using Jukes-Cantor model of DNA evolution on a set of 1,000 bootstrap replicates; Maximum-Likelihood (ML) tree was constructed using PhyML v3.0 (Guindon et al., 2010), GTR model and supported by Approximate Likelihood Ratio Test (aLRT). Interactive Tree of Life (iTOL) was used as a tree editor to assign clade and annotate metadata (Letunic and Bork, 2016).
Alignment-Free Whole-Genome Phylogeny
CVTree standalone v5.0 was used for constructing alignment-free whole-genome phylogeny with parameters k = 6 on amino acid sequences as recommended for prokaryotes and ATCC13124 was selected as the reference sequence (Qi et al., 2004). All genomes were subject to Prokka annotation prior to building phylogeny to obtain annotated FASTA files (genes in amino acid sequences) (Seemann, 2014). Webtool iTOL was used for phylogeny annotation and editing (Letunic and Bork, 2016).
16S rRNA Gene Alignment, Phylogeny, and Pair-Wise Blast
This study employs BAsic Rapid Ribosomal RNA Predictor (Barrnap) v0.5 as 16S rRNA gene predictor (Seemann, 2013). In-house Perl scripts were utilised for extracting the maximum-length 16S rRNA sequence on the positive strands predicted by Barrnap on the WGS data in each isolate for optimal consistency and comparison (Supplementary Table 1). Sequence were aligned using MUSCLE v3.8.31 (Edgar, 2004). Phylogenetic trees were generated as described in previous section. Clostridium species 16S rRNA gene sequences were retrieved from NCBI databases (Supplementary Table 3). Pair-wise BLASTn was performed using BLAST+ v2.2.30 and in-house Perl scripts (Camacho et al., 2009).
Functional Annotation of Genomes (COG)
Core and accessory genes were extracted using in-house Perl scripts based on pangenome data. The Clusters of Orthologous Groups (COG) classification was done on both the extracted datasets using eggnog-mapper (v0.99.3) based on COG and EggNOG databases, and HMMER was used for sequence homology search and COG class assignment on default parameters (Huerta-Cepas et al., 2016, 2017). COG classes assigned to each gene were then extracted using Shell script3 and were plotted using GraphPad Prism v5.04. In-house bash scripts and Linux commands were used to explore annotated data on different functional classes.
In Silico Analysis of Virulence Potentials
Exotoxin profiles: 23 presently identified toxins and virulent enzymes sequence data from NCBI database were built into database for BLAST query (Supplementary Table 4). BLAST+ v2.2.30 was employed for sequence similarity search (BLASTn) using in-house Perl and bash scripts (Camacho et al., 2009). Heat maps were generated using R heatmap2 package (R Development Core Team, 2010). Individual C. perfringens plasmids were retrieved from NCBI database for toxin profiling (Supplementary Table 5). Antimicrobial resistance: sequence similarity search was performed as described above using Comprehensive Antimicrobial Resistance Database (CARD) (Jia et al., 2017). Prophage profiles: sequence similarity search was performed as described above using viral databases retrieved from NCBI (March 2017). Percentage identity of 90% was applied for secondary BLASTn filter. BLAST parameters: Double-filter parameters were implemented for screening—expect value of 1e-10 and percentage identity of >80% for sequence homology inference unless otherwise indicated.
CRISPR Array Detection
CRISPR were predicted using MinCED v0.1.6, a tool derived from CRT (Bland et al., 2007; Angly and Skennerton, 2011). Default parameters were implemented as follows: minimum number of repeats a CRISPR must contain–3, minimum length of CRISPR repeats–23, maximum CRISPR spacers–26, maximum length of CRISPR spaces–50. Counts were parsed in custom bash scripts and visualised using GraphPad Prism v5.04.
Multiple Circular Genome Comparison
Circular whole genome comparisons were generated and visualised using BLAST Ring Image Generator (BRIG) v0.95 (Alikhan et al., 2011). Percentage identity cut-offs were indicated in the graph legends.
Author Contributions
LH conceived the study and RK, SC, and LH conceptualised the study. RK and SC designed and performed all bioinformatics analysis, visualised all data, and co-wrote the manuscript. PP and SA contributed to the sequencing and assembly work, and co-wrote the manuscript. LH co-wrote and revised the manuscript. All authors have read and approved the final manuscript.
Funding
This work was supported by a Wellcome Trust Investigator Award to LH (100974/C/13/Z) and support of the Biotechnology and Biological Sciences Research Council (BBSRC) Institute Strategic Programme grant for Gut Health and Food Safety BB/J004529/1 (LH). RK is a PhD student at the Norwich Medical School of University of East Anglia (UEA), partially funded by UEA international bursary (Faculty of Medical and Health Sciences). NCTC3000 project is funded by the Wellcome Trust (101503/Z/13/Z). Funders had no input into the design of the study, analysis, or interpretation of the data, or in writing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02485/full#supplementary-material
Footnotes
1. ^NCTC 3000 Project website: www.sanger.ac.uk/resources/downloads/bacteria/nctc/
2. ^Fripan website: drpowell.github.io/FriPan/
3. ^Public repository: github.com/raymondkiu/eggnog-mapper_COGextraction
References
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Andersen, J. L., He, G. X., Kakarla, P. K. C. R., Kumar, S., Lakra, W. S., et al. (2015). Multidrug efflux pumps from Enterobacteriaceae, Vibrio cholerae and Staphylococcus aureus bacterial food pathogens. Int. J. Environ. Res. Public Health 12, 1487–1547. doi: 10.3390/ijerph120201487
Angly, F., and Skennerton, C. (2011). Mining CRISPRs in Environmental Datasets [Online]. Available online at: https://github.com/ctSkennerton/minced/tree/master (Accessed on June, 1 2017).
Ashton, P. M., Nair, S., Peters, T. M., Bale, J. A., Powell, D. G., Painset, A., et al. (2016). Identification of Salmonella for public health surveillance using whole genome sequencing. PeerJ 4:e1752. doi: 10.7717/peerj.1752
Bannam, T. L., Johanesen, P. A., Salvado, C. L., Pidot, S. J., Farrow, K. A., and Rood, J. I. (2004). The Clostridium perfringens TetA(P) efflux protein contains a functional variant of the Motif A region found in major facilitator superfamily transport proteins. Microbiology 150, 127–134. doi: 10.1099/mic.0.26614-0
Bernard, G., Chan, C. X., and Ragan, M. A. (2016). Alignment-free microbial phylogenomics under scenarios of sequence divergence, genome rearrangement and lateral genetic transfer. Sci. Rep. 6:28970. doi: 10.1038/srep28970
Bland, C., Ramsey, T. L., Sabree, F., Lowe, M., Brown, K., Kyrpides, N. C., et al. (2007). CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8:209. doi: 10.1186/1471-2105-8-209
Brady, J., Hernandez-Doria, J. D., Bennett, C., Guenter, W., House, J. D., and Rodriguez-Lecompte, J. C. (2010). Toxinotyping of necrotic enteritis-producing and commensal isolates of Clostridium perfringens from chickens fed organic diets. Avian Pathol. 39, 475–481. doi: 10.1080/03079457.2010.521141
Browne, H. P., Neville, B. A., Forster, S. C., and Lawley, T. D. (2017). Transmission of the gut microbiota: spreading of health. Nat. Rev. Microbiol. 15, 531–543. doi: 10.1038/nrmicro.2017.50
Brüssow, H., and Hendrix, R. W. (2002). Phage genomics: small is beautiful. Cell 108, 13–16. doi: 10.1016/S0092-8674(01)00637-7
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Canard, B., Garnier, T., Lafay, B., Christen, R., and Cole, S. T. (1992). Phylogenetic analysis of the pathogenic anaerobe Clostridium perfringens using the 16S rRNA nucleotide sequence. Int. J. Syst. Bacteriol. 42, 312–314. doi: 10.1099/00207713-42-2-312
Cole, J. N., and Nizet, V. (2016). Bacterial evasion of host antimicrobial peptide defenses, in Virulence Mechanisms of Bacterial Pathogens, Fifth Edition, eds I. Kudva, N. Cornick, P. Plummer, Q. Zhang, T. Nicholson, J. Bannantine, and B. Bellaire (Washington, DC: ASM Press), 413–443. doi: 10.1128/microbiolspec.VMBF-0006-2015
Collins, M. D., Lawson, P. A., Willems, A., Cordoba, J. J., Fernandez-Garayzabal, J., Garcia, P., et al. (1994). The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int. J. Syst. Bacteriol. 44, 812–826. doi: 10.1099/00207713-44-4-812
Cornillot, E., Saint-Joanis, B., Daube, G., Katayama, S., Granum, P. E., Canard, B., et al. (1995). The enterotoxin gene (cpe) of Clostridium perfringens can be chromosomal or plasmid-borne. Mol. Microbiol. 15, 639–647. doi: 10.1111/j.1365-2958.1995.tb02373.x
Edagiz, S., Lagace-Wiens, P., Embil, J., Karlowsky, J., and Walkty, A. (2015). Empyema caused by Clostridium bifermentans: a case report. Can. J. Infect. Dis. Med. Microbiol. 26, 105–107.
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eppinger, M., Worsham, P. L., Nikolich, M. P., Riley, D. R., Sebastian, Y., Mou, S., et al. (2010). Genome sequence of the deep-rooted Yersinia pestis strain angola reveals new insights into the evolution and pangenome of the plague bacterium. J. Bacteriol. 192, 1685–1699. doi: 10.1128/JB.01518-09
Ernst, C. M., and Peschel, A. (2011). Broad-spectrum antimicrobial peptide resistance by MprF-mediated aminoacylation and flipping of phospholipids. Mol. Microbiol. 80, 290–299. doi: 10.1111/j.1365-2958.2011.07576.x
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. doi: 10.1093/nar/gku1223
Geer, L. Y., Marchler-Bauer, A., Geer, R. C., Han, L., He, J., He, S., et al. (2010). The NCBI BioSystems database. Nucleic Acids Res. 38, D492–D496. doi: 10.1093/nar/gkp858
Gibert, M., Jolivet-Reynaud, C., and Popoff, M. R. (1997). Beta2 toxin, a novel toxin produced by Clostridium perfringens. Gene 203, 65–73. doi: 10.1016/S0378-1119(97)00493-9
Goh, S., Chang, B. J., and Riley, T. V. (2005). Effect of phage infection on toxin production by Clostridium difficile. J. Med. Microbiol. 54, 129–135. doi: 10.1099/jmm.0.45821-0
Gouy, M., Guindon, S., and Gascuel, O. (2010). SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224. doi: 10.1093/molbev/msp259
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hadfield, J., and Harris, S. (2017). Interactive Visualization of Genome Phylogenies - Phandango [Online]. Wellcome Trust Sange Institute. Available online at: https://jameshadfield.github.io/phandango/ (Accessed on April, 5 2017).
Harvey, S. M., Sturgeon, J., and Dassey, D. E. (2002). Botulism due to Clostridium baratii type F toxin. J. Clin. Microbiol. 40, 2260–2262. doi: 10.1128/JCM.40.6.2260-2262.2002
Hassan, K. A., Elbourne, L. D., Tetu, S. G., Melville, S. B., Rood, J. I., and Paulsen, I. T. (2015). Genomic analyses of Clostridium perfringens isolates from five toxinotypes. Res. Microbiol. 166, 255–263. doi: 10.1016/j.resmic.2014.10.003
Heida, F. H., van Zoonen, A. G. J. F., Hulscher, J. B. F., Te Kiefte, B. J. C., Wessels, R., Kooi, E. M. W., et al. (2016). A necrotizing enterocolitis-associated gut microbiota is present in the meconium: results of a prospective study. Clin. Infect. Dis. 62, 863–870. doi: 10.1093/cid/ciw016
Hiller, N. L., Janto, B., Hogg, J. S., Boissy, R., Yu, S., Powell, E., et al. (2007). Comparative genomic analyses of seventeen Streptococcus pneumoniae strains: insights into the pneumococcal supragenome. J. Bacteriol. 189, 8186–8195. doi: 10.1128/JB.00690-07
Hobbs, B. C., Smith, M. E., Oakley, C. L., Warrack, G. H., and Cruickshank, J. C. (1953). Clostridium welchii food poisoning. J. Hyg. 51, 75–101. doi: 10.1017/S0022172400015515
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., Von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7:improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Keyburn, A. L., Boyce, J. D., Vaz, P., Bannam, T. L., Ford, M. E., Parker, D., et al. (2008). NetB, a new toxin that is associated with avian necrotic enteritis caused by Clostridium perfringens. PLoS Pathog. 4:e26. doi: 10.1371/journal.ppat.0040026
Knight, D. R., Squire, M. M., Collins, D. A., and Riley, T. V. (2016). Genome analysis of Clostridium difficile pcr ribotype 014 lineage in australian pigs and humans reveals a diverse genetic repertoire and signatures of long-range interspecies transmission. Front. Microbiol. 7:2138. doi: 10.3389/fmicb.2016.02138
Koch, C., Rainey, F. A., and Stackebrandt, E. (1994). 16s Rdna studies on members of Arthrobacter and Micrococcus - an aid for their future taxonomic restructuring. FEMS Microbiol. Lett. 123, 167–171. doi: 10.1111/j.1574-6968.1994.tb07217.x
Kosloske, A. M., Ulrich, J. A., and Hoffman, H. (1978). Fulminant necrotising enterocolitis associated with Clostridia. Lancet 312, 1014–1016. doi: 10.1016/S0140-6736(78)92337-1
Köster, W. (2001). ABC transporter-mediated uptake of iron, siderophores, heme and vitamin B12. Res. Microbiol. 152, 291–301. doi: 10.1016/S0923-2508(01)01200-1
Kristian, S. A., Lauth, X., Nizet, V., Goetz, F., Neumeister, B., Peschel, A., et al. (2003). Alanylation of teichoic acids protects Staphylococcus aureus against Toll-like receptor 2-dependent host defense in a mouse tissue cage infection model. J. Infect. Dis. 188, 414–423. doi: 10.1086/376533
Lepp, D., Roxas, B., Parreira, V. R., Marri, P. R., Rosey, E. L., Gong, J., et al. (2010). Identification of novel pathogenicity loci in Clostridium perfringens strains that cause avian necrotic enteritis. PLoS ONE 5:e10795. doi: 10.1371/annotation/501e5656-71ac-420f-a194-80141f6381e5
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Li, J., Adams, V., Bannam, T. L., Miyamoto, K., Garcia, J. P., Uzal, F. A., et al. (2013). Toxin plasmids of Clostridium perfringens. Microbiol. Mol. Biol. Rev. 77, 208–233. doi: 10.1128/MMBR.00062-12
Li, J., and Mcclane, B. A. (2006). Further comparison of temperature effects on growth and survival of Clostridium perfringens type A isolates carrying a chromosomal or plasmid-borne enterotoxin gene. Appl. Environ. Microbiol. 72, 4561–4568. doi: 10.1128/AEM.00177-06
Lyras, D., and Rood, J. I. (1996). Genetic organization and distribution of tetracycline resistance determinants in Clostridium perfringens. Antimicrob. Agents Chemother. 40, 2500–2504.
Mahony, D. E., and Moore, T. I. (1976). Stable L-forms of Clostridium perfringens and their growth on glass surfaces. Can. J. Microbiol. 22, 953–959. doi: 10.1139/m76-138
Masaki, T., Umehashi, H., Miyazaki, H., Takano, M., Yamakawa, K., and Nakamura, S. (1988). Clostridium absonum from gas gangrene. Jpn. J. Med. Sci. Biol. 41, 27–30. doi: 10.7883/yoken1952.41.27
Mcclung, L. (1945). Human food poisoning due to growth of Clostridium perfringens (C. welchii) in freshly cooked chicken. J. Bacteriol. 50, 229–231.
Mcinerney, J. O., Mcnally, A., and O'connell, M. J. (2017). Why prokaryotes have pangenomes. Nat Microbiol 2:17040. doi: 10.1038/nmicrobiol.2017.40
Mehdizadeh Gohari, I., Parreira, V. R., Nowell, V. J., Nicholson, V. M., Oliphant, K., and Prescott, J. F. (2015). A novel pore-forming toxin in type A Clostridium perfringens is associated with both fatal canine hemorrhagic gastroenteritis and fatal foal necrotizing enterocolitis. PLoS ONE 10:e0122684. doi: 10.1371/journal.pone.0122684
Mehdizadeh Gohari, I., Kropinski, A. M., Weese, S. J., Parreira, V. R., Whitehead, A. E., Boerlin, P., et al. (2016). Plasmid characterization and chromosome analysis of two netF+ Clostridium perfringens isolates associated with foal and canine necrotizing enteritis. PLoS ONE 11:e0148344. doi: 10.1371/journal.pone.0148344
Mehdizadeh Gohari, I., Kropinski, A. M., Weese, S. J., Whitehead, A. E., Parreira, V. R., Boerlin, P., et al. (2017). NetF-producing Clostridium perfringens: clonality and plasmid pathogenicity loci analysis. Infect. Genet. Evol. 49, 32–38. doi: 10.1016/j.meegid.2016.12.028
Möllby, R., Holme, T., Nord, C. E., Smyth, C. J., and Wadström, T. (1976). Production of phospholipase C (alpha-toxin), haemolysins and lethal toxins by Clostridium perfringens types A to D. J. Gen. Microbiol. 96, 137–144. doi: 10.1099/00221287-96-1-137
Mooijman, K. A., During, M., and Nagelkerke, N. J. D. (2003). MICROCRM: Preparation and Control of Batches of Microbiological Reference Materials Consisting of Capsules. The Netherlands National Institute for Public Health and the Environment (RIVM).
Myers, G. S., Rasko, D. A., Cheung, J. K., Ravel, J., Seshadri, R., Deboy, R. T., et al. (2006). Skewed genomic variability in strains of the toxigenic bacterial pathogen, Clostridium perfringens. Genome Res. 16, 1031–1040. doi: 10.1101/gr.5238106
Nagler, F. P. O. (1939). Observations on a reaction between the lethal toxin of Cl. welchii (Type A) and human serum. Br. J. Exp. Pathol. 20, 473–485.
Nagpal, R., Ogata, K., Tsuji, H., Matsuda, K., Takahashi, T., Nomoto, K., et al. (2015). Sensitive quantification of Clostridium perfringens in human feces by quantitative real-time PCR targeting alpha-toxin and enterotoxin genes. BMC Microbiol. 15:219. doi: 10.1186/s12866-015-0561-y
Nagy, E., Becker, S., Kostrzewa, M., Barta, N., and Urbán, E. (2012). The value of MALDI-TOF MS for the identification of clinically relevant anaerobic bacteria in routine laboratories. J. Med. Microbiol. 61, 1393–1400. doi: 10.1099/jmm.0.043927-0
Nishi, H., Komatsuzawa, H., Fujiwara, T., Mccallum, N., and Sugai, M. (2004). Reduced content of lysyl-phosphatidylglycerol in the cytoplasmic membrane affects susceptibility to moenomycin, as well as vancomycin, gentamicin, and antimicrobial peptides, in Staphylococcus aureus. Antimicrob. Agents Chemother. 48, 4800–4807. doi: 10.1128/AAC.48.12.4800-4807.2004
Nowell, V. J., Kropinski, A. M., Songer, J. G., Macinnes, J. I., Parreira, V. R., and Prescott, J. F. (2012). Genome sequencing and analysis of a type A Clostridium perfringens isolate from a case of bovine clostridial abomasitis. PLoS ONE 7:e32271. doi: 10.1371/journal.pone.0032271
Ohno-Iwashita, Y., Iwamoto, M., Mitsui, K., Kawasaki, H., and Ando, S. (1986). Cold-labile hemolysin produced by limited proteolysis of theta-toxin from Clostridium perfringens. Biochemistry 25, 6048–6053. doi: 10.1021/bi00368a032
Osman, K. M., and Elhariri, M. (2013). Antibiotic resistance of Clostridium perfringens isolates from broiler chickens in Egypt. Rev. Sci. Technol. 32, 841–850. doi: 10.20506/rst.32.2.2212
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Paquette, G., and Fredette, V. (1967). Avirulent Clostridium perfringens strains obtained by euflavine treatment. J. Bacteriol. 94, 1437–1442.
Park, M., Deck, J., Foley, S. L., Nayak, R., Songer, J. G., Seibel, J. R., et al. (2016). Diversity of Clostridium perfringens isolates from various sources and prevalence of conjugative plasmids. Anaerobe 38, 25–35. doi: 10.1016/j.anaerobe.2015.11.003
Peschel, A., Jack, R. W., Otto, M., Collins, L. V., Staubitz, P., Nicholson, G., et al. (2001). Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J. Exp. Med. 193, 1067–1076. doi: 10.1084/jem.193.9.1067
Petit, L., Gibert, M., and Popoff, M. (1999). Clostridium perfringens: toxinotype and genotype. Trends Microbiol. 7, 104–110. doi: 10.1016/S0966-842X(98)01430-9
Qi, J., Luo, H., and Hao, B. (2004). CVTree: a phylogenetic tree reconstruction tool based on whole genomes. Nucleic Acids Res. 32, W45–W47. doi: 10.1093/nar/gkh362
Raveh-Sadka, T., Thomas, B. C., Singh, A., Firek, B., Brooks, B., Castelle, C. J., et al. (2015). Gut bacteria are rarely shared by co-hospitalized premature infants, regardless of necrotizing enterocolitis development. Elife 4:e05477. doi: 10.7554/eLife.05477
R Development Core Team (2010). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Revitt-Mills, S. A., Rood, J. I., and Adams, V. (2015). Clostridium perfringens extracellular toxins and enzymes: 20 and counting. Microbiol. Aust. 36, 114–117. doi: 10.1071/MA15039
Ribeiro, F. J., Przybylski, D., Yin, S., Sharpe, T., Gnerre, S., Abouelleil, A., et al. (2012). Finished bacterial genomes from shotgun sequence data. Genome Res. 22, 2270–2277. doi: 10.1101/gr.141515.112
Roach, D. J., Burton, J. N., Lee, C., Stackhouse, B., Butler-Wu, S. M., Cookson, B. T., et al. (2015). A year of infection in the intensive care unit: prospective whole genome sequencing of bacterial clinical isolates reveals cryptic transmissions and novel microbiota. PLoS Genet. 11:e1005413. doi: 10.1371/journal.pgen.1005413
Rood, J. I. (1998). Virulence genes of Clostridium perfringens. Annu. Rev. Microbiol. 52, 333–360. doi: 10.1146/annurev.micro.52.1.333
Rood, J. I., Scott, V. N., and Duncan, C. L. (1978). Identification of a transferable tetracycline resistance plasmid (pCW3) from Clostridium perfringens. Plasmid 1, 563–570. doi: 10.1016/0147-619X(78)90013-6
Rouli, L., Merhej, V., Fournier, P. E., and Raoult, D. (2015). The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect. 7, 72–85. doi: 10.1016/j.nmni.2015.06.005
Scallan, E., Hoekstra, R. M., Angulo, F. J., Tauxe, R. V., Widdowson, M. A., Roy, S. L., et al. (2011). Foodborne illness acquired in the United States–major pathogens. Emerg Infect Dis 17, 7–15. doi: 10.3201/eid1701.P11101
Seemann, T. (2013). barrnap 0.7: Rapid Ribosomal RNA Prediction [Online]. Available online at: https://github.com/Victorian-Bioinformatics-Consortium/barrnap (Accessed on April, 6 2017).
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shimizu, T., Ohtani, K., Hirakawa, H., Ohshima, K., Yamashita, A., Shiba, T., et al. (2002). Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc. Natl. Acad. Sci. U.S.A. 99, 996–1001. doi: 10.1073/pnas.022493799
Sim, K., Shaw, A. G., Randell, P., Cox, M. J., Mcclure, Z. E., Li, M. S., et al. (2015). Dysbiosis anticipating necrotizing enterocolitis in very premature infants. Clin. Infect. Dis. 60, 389–397. doi: 10.1093/cid/ciu822
Stevens, D. L., and Bryant, A. E. (2002). The role of clostridial toxins in the pathogenesis of gas gangrene. Clin. Infect. Dis. 35, S93–S100. doi: 10.1086/341928
Stewart, A. W., and Johnson, M. G. (1977). Increased numbers of heat-resistnat spores produced by two strains of Clostridium perfringens bearing temperate phage s9. J. Gen. Microbiol. 103, 45–50. doi: 10.1099/00221287-103-1-45
Tansuphasiri, U., Matra, W., and Sangsuk, L. (2005). Antimicrobial resistance among Clostridium perfringens isolated from various sources in Thailand. Southeast Asian J. Trop. Med. Public Health 36, 954–961.
Thomson, N., Baker, S., Pickard, D., Fookes, M., Anjum, M., Hamlin, N., et al. (2004). The role of prophage-like elements in the diversity of Salmonella enterica serovars. J. Mol. Biol. 339, 279–300. doi: 10.1016/j.jmb.2004.03.058
Traub, W. H., and Raymond, E. A. (1971). In vitro resistance of Clostridium perfringens type A to gentamicin sulfate, and reduced activity of the antibiotic under anaerobic atmospheric conditions. Chemotherapy 16, 162–165. doi: 10.1159/000220723
Tyrrell, K. L., Citron, D. M., Warren, Y. A., Fernandez, H. T., Merriam, C. V., and Goldstein, E. J. C. (2006). In vitro activities of daptomycin, vancomycin, and penicillin against Clostridium difficile, C. perfringens, Finegoldia magna, and Propionibacterium acnes. Antimicrob. Agents Chemother. 50, 2728–2731. doi: 10.1128/Aac.00357-06
Uzal, F. A., Freedman, J. C., Shrestha, A., Theoret, J. R., Garcia, J., Awad, M. M., et al. (2014). Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol. 9, 361–377. doi: 10.2217/fmb.13.168
Van Asten, A. J., Van Der Wiel, C. W., Nikolaou, G., Houwers, D. J., and Gröne, A. (2009). A multiplex PCR for toxin typing of Clostridium perfringens isolates. Vet. Microbiol. 136, 411–412. doi: 10.1016/j.vetmic.2008.11.024
Van Vliet, A. H., and Kusters, J. G. (2015). Use of alignment-free phylogenetics for rapid genome sequence-based typing of Helicobacter pylori virulence markers and antibiotic susceptibility. J. Clin. Microbiol. 53, 2877–2888. doi: 10.1128/JCM.01357-15
Vos, P., Garrity, G., Jones, D., Krieg, N. R., Ludwig, W., Rainey, F. A., et al. (2009). Bergey's Manual of Systematic Bacteriology. New York, NY: Springer-Verlag.
Welch, W. H., and Nuttall, G. H. F. (1891). A gas-producing bacillus (Bacillus aerogenes capsulatus, Nov, Spec.) capable of rapid development in the body after death. Bull. John Hopkins Hosp. 3, 81–91.
Woese, C. R., Kandler, O., and Wheelis, M. L. (1990). Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. U.S.A. 87, 4576–4579. doi: 10.1073/pnas.87.12.4576
Wong, Y. M., Juan, J. C., Gan, H. M., and Austin, C. M. (2014). Draft Genome Sequence of Clostridium perfringens Strain, J. J. C., a Highly Efficient Hydrogen Producer Isolated from Landfill Leachate Sludge. Genome Announc 2:e00064-14. doi: 10.1128/genomeA.00064-14
Keywords: Clostridium perfringens, pangenome, antimicrobial resistance, genomics, whole genome sequencing, Clostridial infection, exotoxins
Citation: Kiu R, Caim S, Alexander S, Pachori P and Hall LJ (2017) Probing Genomic Aspects of the Multi-Host Pathogen Clostridium perfringens Reveals Significant Pangenome Diversity, and a Diverse Array of Virulence Factors. Front. Microbiol. 8:2485. doi: 10.3389/fmicb.2017.02485
Received: 24 August 2017; Accepted: 29 November 2017;
Published: 12 December 2017.
Edited by:
Frank T. Robb, University of Maryland, Baltimore, United StatesReviewed by:
Nikolai Ravin, Research Center for Biotechnology of Russian Academy of Sciences, RussiaKarl Hassan, University of Newcastle, Australia
Copyright © 2017 Kiu, Caim, Alexander, Pachori and Hall. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lindsay J. Hall, lindsay.hall@quadram.ac.uk
†These authors have contributed equally to this work and shared first authorship.