 Nathalie L. van der Mee-Marquet1
Nathalie L. van der Mee-Marquet1 Lucie Bénéjat2
Lucie Bénéjat2 Seydina M. Diene3
Seydina M. Diene3 Adrien Lemaignen4
Adrien Lemaignen4 Nadia Gaïa5
Nadia Gaïa5 Annemieke Smet6
Annemieke Smet6 Freddy Haesebrouck7
Freddy Haesebrouck7 Abdessalam Cherkaoui8Astrid Ducournau2Sabrina Lacomme9Etienne Gontier9Louis Bernard4
Abdessalam Cherkaoui8Astrid Ducournau2Sabrina Lacomme9Etienne Gontier9Louis Bernard4 Francis Mégraud2Alain Goudeau1
Francis Mégraud2Alain Goudeau1 Philippe Lehours2*
Philippe Lehours2* Patrice François5*
Patrice François5*- 1Service de Bactériologie, Virologie et Hygiène, Hôpital Trousseau, Réseau des Hygiénistes du Centre, CPIAS Centre Val de Loire, Centre Hospitalier Régional Universitaire, and UMR 1282 Infectiologie Santé Publique, Université François-Rabelais, Tours, France
- 2Laboratoire de Bactériologie, Centre National de Référence des Campylobacters et des Hélicobacters, Bordeaux, France
- 3Faculté de Médecine et de Pharmacie, URMITE, Aix-Marseille Université, UMR 63, Centre National de la Recherche Scientifique 7278, IRD 198, Institut National de la Santé et de la Recherche Médicale, IHU-Méditerranée Infection, Marseille, France
- 4Service de Médecine Interne et des Maladies Infectieuses, Centre Hospitalier Régional Universitaire, Hôpital Bretonneau, Tours, France
- 5Genomic Research Laboratory, University Hospital of Geneva, Geneva, Switzerland
- 6Laboratory Experimental Medicine and Pediatrics, Faculty of Medicine and Health Sciences, University of Antwerp, Antwerp, Belgium
- 7Department of Pathology, Bacteriology and Avian Diseases, Faculty of Veterinary Medicine, Ghent University, Merelbeke, Belgium
- 8Bacteriology Laboratory, Division of Laboratory Medicine, Geneva University Hospitals, Geneva, Switzerland
- 9Bordeaux Imaging Center, Imagerie Electronique, UMS 3420 Centre National de la Recherche Scientifique US4 Institut National de la Santé et de la Recherche Médicale Université de Bordeaux, Bordeaux, France
We isolated from aerobic and anaerobic blood culture bottles from a febrile patient, a Helicobacter-like Gram negative, rod-shaped bacterium that MALDI-TOF MS failed to identify. Blood agar cultures incubated in a microaerobic atmosphere revealed a motile Gram negative rod, which was oxidase, catalase, nitrate reductase, esterase, and alkaline phosphatase positive. It grew at 42°C with no detectable urease activity. Antimicrobial susceptibility testing showed that the organism was susceptible to beta-lactams, gentamicin, erythromycin, and tetracycline but resistant to ciprofloxacin. Electronic microscopy analysis revealed a 3 × 0.5 μm curved rod bacterium harboring two sheathed amphitrichous flagella. Whole genome sequencing revealed a genome 1,708,265 base-pairs long with a GC content of 37.80% and a total of 1,697 coding sequences. The genomic analyses using the nucleotide sequences of the 16S rRNA gene, hsp60 and gyrB genes, as well as the GyrA protein sequence, and the results of Average Nucleotide Identity and in silico DNA-DNA hybridization suggest evidence for a novel Helicobacter species close to Helicobacter equorum and belonging to the group of enterohepatic Helicobacter species. As soon as the particular peptide mass fingerprint of this pathogen is added to the spectral databases, MALDI-TOF MS technology will improve its identification from clinical specimens, especially in case of “sterile infection”. We propose to associate the present strain with the Latin name of the place of isolation; Caesarodunum (Tours, France) and suggest “Helicobacter caesarodunensis” for further description of this new bacterium.
Introduction
The Helicobacter genus of the Helicobacteraceae family is composed of Gram-negative, helical-shaped rods (Euzéby, 1997), distributed into 37 different gastric and non-gastric species, also called enterohepatic Helicobacters. Helicobacter pylori is the main species found in human gastric mucosa. It infects a large part of the human population and is associated with chronic gastric inflammation that can evolve toward an ulcer or gastric cancer (such as gastric adenocarcinoma or gastric MALT lymphoma) (Mégraud et al., 2015; Floch et al., 2017). Other Helicobacters from the “heilmannii group” can also be detected in human gastric mucosa, probably by cross-contamination from pets or consumption of raw pork meat products (Baele et al., 2009; Ménard et al., 2014). Gastric Helicobacters produce urease to help them survive in the acidic gastric environment. Enterohepatic Helicobacters found in humans are usually associated with diarrheal diseases (such as Helicobacter pullorum) (Borges et al., 2015) or systemic infection in immunocompromised hosts (such as Helicobacter cinaedi) (Kawamura et al., 2014). They are urease negative and most of them produce a cytolethal distending toxin (CDT) responsible for symptoms of infection such as inflammation (Varon et al., 2014; Péré-Védrenne et al., 2016). Recent data also suggest a potential role for CDT in intestinal carcinogenesis (Ge et al., 2017).
Helicobacter species cannot be easily identified when the tools used are based on the study of enzymatic activities, sugar fermentation or assimilation. Today, in clinical laboratories, routine bacterial identification is improved by MALDI-TOF MS technology (Cherkaoui et al., 2011; Angeletti, 2016). Databases associated with MALDI-TOF MS systems are regularly updated with novel spectra in order to increase the accuracy of identification. Nevertheless, this technology fails when the bacterial protein spectrum is absent or too different from those in the database. Fortunately, whole genome sequencing has become popular and easy to access for many clinical laboratories, and many free-access bioinformatics tools can be quickly run to analyze genomic data.
In the present study, we report the identification and characterization of a strain that MALDI-TOF MS failed to identify, belonging to the Helicobacter genus, and recovered from a patient suffering a bloodstream infection. Based on the results of routine identification tests, electron microscopy and whole genome sequence analysis, this strain differed from all previously described Helicobacter species and was named S15.
Materials and Methods
Bacterial Strain Isolation
The S15 strain was isolated from a Bactec blood culture bottle grown aerobically (Becton Dickinson, Le Pont de Claix, France), detected 4 days and 22 h after inoculation. Further culture of strains S15 was performed in trypcase soy agar (Difco, Becton Dickinson) enriched with 5% horse blood for 2–3 days at 37°C or 2 days at 42° under microaerophilic conditions. Plate cultures were incubated in jars using an Anoxomat microprocessor (Mart Microbiology, B.V. Lichtenvoorde, The Netherlands) which creates an atmosphere of 80–90% N2, 5–10% CO2, and 5–10% H2. The strain S15 has been assigned to the CCUG, CIP, and DSMZ collections under the identification numbers 68986, 111406 and 105791, respectively.
Phenotypic Testing
Enzymatic activities were assessed by using the API Campy gallery (bioMérieux, Marcy L'Etoile, France). The presence of catalase and oxidase was investigated. Antibiotic susceptibilities were assessed by the disk diffusion method (+ Etest strip for ciprofloxacin) following the CASFM/EUCAST 2017 recommendations for Campylobacter (in-house Mueller–Hinton (MH) agar supplemented with 5% defibrinated horse blood (MH-F) and 20 mg/L β-NAD; inoculum: McFarland 0.5; incubation during 48 h in a microaerobic environment).
MALDI-TOF MS Assessment
Colonies were subjected to a Bruker Daltonics biotyper device using the Microflex platform (Bruker Daltonics, Bremen, Germany) as previously described (van Veen et al., 2010). Single colonies were transferred onto a target plate and overlaid with matrix solution. FlexControl software (version 3.4) was used for measurements; this version includes eight Helicobacter species (Helicobacter canadensis, Helicobacter canis, Helicobacter cholecystus, H. cinaedi, Helicobacter fenneliae, Helicobacter mustelae, Helicobacter pullorum, and H. pylori with 1–9 spectra for each species). The spectra were analyzed using BioTyper software (version 3.1.66; Bruker) and MBT-Compass and MBT-RUO databases, and extracted profiles were compared to that of H. pylori.
Electronic Microscopy
The morphology, cell size, and presence of flagella were determined by transmission electron microscopy. S15 cells were scraped and introduced into a fixative solution of 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) and incubated 1.5 h at room temperature. After centrifugation for 3 min at 5,000 rpm, pellets were mixed/suspended in 500 μl of 0.1 M cacodylate buffer (pH 7.4). A volume of 10 μL of bacterial suspension was adsorbed on carbon grids (Delta Microscopy, Toulouse, France) and negatively stained with freshly prepared 1% (p/v) aqueous uranyl acetate solution. Grids were examined with a transmission electron microscope (H7650, HITACHI, Elexience, France) at 80 kV using high contrast mode, equipped with an Orius 11Mpixel camera (Roper Scientific, Lisses, France).
Genome Sequencing and Analysis
Genomic DNA obtained from colonies was purified using the DNeasy kit (Qiagen, Courtaboeuf, France). DNA was subjected to whole genome sequencing on the Illumina HiSeq 2500 (Illumina, San Diego, CA, USA) using 200 base-pair reads with paired-ends according to the Truseq protocol (Illumina), following the manufacturer's recommendations. The quality of sequence reads was assessed with the Fastqc program (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and reads were quality filtered using the fastq-mcf program (https://expressionanalysis.github.io/ea-utils/). Genome assembly was performed using the Edena v3 assembler (Hernandez et al., 2014). Assembled genomes were annotated using the Prokka v1.10 program (Seemann, 2014). The “Get_homologues.pl” script was used for core proteome comparisons (Contreras-Moreira and Vinuesa, 2013). The CVTree3 Web Server was used to perform the phylogenetic analysis (Xu and Hao, 2009). Prokka annotation and blastP analysis were performed to identify specific genes involved in the phenotype, evolution, and virulence of the strain. The predicted gene function is based on protein similarity of at least 40% and an E-value of 10e-6. Phylogenetic tree topologies of nucleotide (16S rRNA gene, gyrB and hsp60 genes) and predicted amino acid alignments (for GyrA) were constructed with the neighbor-joining method using the MEGA (Molecular Evolutionary Genetics Analysis) software version 6.06 (Trespalacios et al., 2015). The sequences previously created by Ménard et al. were used for that purpose (Ménard et al., 2016) (The accession numbers are provided in Supplemental Table S2); the sequences from strain S15 were obtained after genome sequencing. Four phylogenetic trees were constructed based on the 16S rRNA gene, hsp60 and gyrB genes and the GyrA protein sequence according to phylogenetic analyses previously described by Ménard et al. (2016). The evolutionary distances were computed using the Kimura 2-parameter method for the 16S rRNA, hsp60 and gyrB genes, and the Poisson correction method for GyrA.
Determination of Average Nucleotide Identity (ANI) and in Silico DNA-DNA Hybridization (DDH)
ANI- and DDH-values were assessed in silico using online tools. An assembled genome of strain S15 was uploaded in GGDC (http://ggdc.dsmz.de/ggdc.php#) with the recommended local alignment tool BLAST+ and compared with the closest genomes available in public databases identified in the different phylogenetic trees, to obtain DDH-values. The accession numbers of the strains included in the phylogenetic analyses are provided in Supplemental Table S1. The statistic comparison (logistic regression) used a significant probability value of DDH >79%. Accession numbers of genomes for these comparisons are;: NC_004917.1: Helicobacter hepaticus ATCC 51449; NZ_CP014991.1: H. himalayensis YS1; NC_020555.1: H. cinaedi ATCC BAA-847; NZ_CM000776.2: H. canadensis MIT 98-5491; NC_013949.1: H. mustelae 12198; NC_014810.2: Helicobacter felis ATCC 49179; NZ_CP019645.1: Helicobacter bilis AAQJH;, JNOB01000034.1: H. pullorum 229313/12 and Helicobacter equorum QF1 (EMBL accession number: FZPO00000000). Pairwise ANI-values were obtained using JSpeciesWS (http://jspecies.ribohost.com/jspeciesws/#analyse) with BLAST.
Results
Clinical Data and Bacterial Isolation
An 83-year-old man was admitted to the emergency unit complaining of painful ankles. His main medical history included an aortic prosthetic valve replacement, ischemic heart disease, post-operative ischemic colitis, osteosynthesis of the right hip after a fracture, a moderate chronic renal failure and a prostatic adenocarcinoma under hormonotherapy. He was febrile (maximum fever 39.5°C) following the diagnosis of a urinary tract infection treated with ofloxacin. Upon examination he had a painful swelling of the left ankle suggesting arthritis, and arthritis of the right knee. He had purple infiltrated plaques on the skin of his inner thigh, suggesting panniculitis, and a 3/6 systolic murmur at heart auscultation. Blood analysis showed an inflammatory response with CRP at 211 mg/L and a leukocyte count of 17,000/mm3 with a predominance of neutrophils, associated with acute renal failure (creatinine level of 200 μmol/L with an estimated clearance of 29 mL/min and blood urea nitrogen at 20.1 mmol/L). Ankle joint aspiration was purulent and culturing only resulted in growth of Staphylococcus epidermidis, which is considered a contaminant. His arthritis was first treated as a microcrystalline arthropathy using colchicine with no improvement. After 3 days, he became feverish again, with no chills. Blood cultures were positive after 5 days of growth and showed small Gram-negative helical-shaped rods, suggestive of the Campylobacter genus. Bacterial colonies were obtained in trypcase soy agar (Difco, Becton Dickinson) enriched with 5% horse blood for 2–3 days at 37°C or 2 days at 42° under microaerophilic conditions (Figure 1). The patient was prescribed with piperacillin-tazobactam and ciprofloxacin for 4 weeks (potential endocarditis and the polyarthritis). The patient quickly showed clinical and general improvement. Antibiotics were stopped after 3 weeks because of an agranulocytosis, but by that time, the patient was clinically cured of this infection. MALDI-TOF MS performed in the context of routine analysis failed to identify the isolate and showed poor scores (unacceptable score for identification of 1.1 with H. pullorum with the Bruker Daltonics Biotyper). Profiles obtained with six different colonies from the isolation plate were superimposed and showed a unique profile (Supplemental Figure S1A). The two highest scores obtained were Staphylococcus capitis and Lactobacillus paracasei; these profiles were then extracted from the database and compared with that of strain S15. However, as shown in Supplemental Figures S1B,C, these two profiles were totally different from strain S15, suggesting distant organisms not phylogenetically related.

Figure 1. Aspect of S15 colonies on trypcase soy agar. Culture of an identified organism on trypcase soy agar enriched with 5% horse blood for 2–3 days at 37°C.
Bacterial cells were motile, curved and Gram-negative, evoking a Campylobacter. Fuzzy colonies were visible on trypcase soy agar plates, at 37 and 42°C after a 48 h incubation under microaerophilic conditions. There was no visible growth in a CO2 enriched or anaerobic atmosphere. Bacterial cells underwent transformation to coccoidal forms upon exposure to air and in cultures after prolonged incubation. Tests performed on the colonies revealed the presence of catalase and oxidase activities. Gallery API Campy showed that the isolate was positive for nitrate reductase, esterase, and alkaline phosphatase. The isolate also reduced triphenyl-tetrazolium chloride. There was no production of H2S, nor any detectable activity of urease, γ -glutamyl transpeptidase, pyrrolidonyl arylamidase, L-arginine arylamidase, L-aspartate arylamidase, or hippuricase. Glucose was also not assimilated (data not shown). Microscopic examinations revealed a rod-shaped bacterium with a size of 3 μm in length and 0.5 μm in width (Figure 2A). Two amphitrichous sheathed flagella with a diameter around 60 nm (Figure 2B) were visible. According to antibiotic sensitivity testing, the strain was susceptible to the β-lactams, ampicillin and amoxicillin-clavulanate, but resistant to cephalothin (diameter: 6 mm), gentamicin, erythromycin and tetracycline. It was also resistant to ciprofloxacin (MIC at 2 μg/mL) and intermediate to nalidixic acid (diameter: 12 mm).
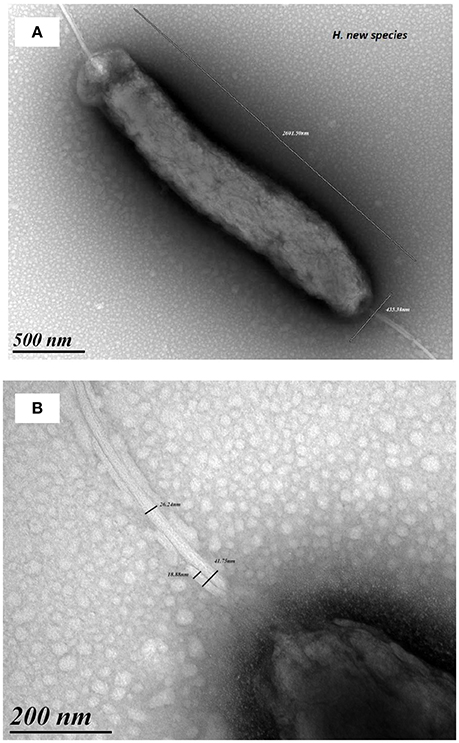
Figure 2. Electron microscopy pictures of strain S15. Microscopic examinations revealed a rod-shaped bacterium 3 μm in length and 0.5 μm width (A). Pictures show amphitrichous sheathed flagella with a diameter around 60 nm (B).
Genomic Content
The genome of the S15 isolate was assembled into 43 contigs, with an N50 of 102,565 bp and the largest contig being 214,427 bp. The total size of the genome was evaluated around 1,708,265 base-pairs and contained 1,697 coding sequences, two copies of rRNA genes, three putative miscellaneous RNAs, 1 tmRNA, and 38 tRNAs, for a total of 1,741 genes. The GC content of S15 is 37.80%. Among the genes homologous to previously described sequences, we identified one putative toxin-/anti-toxin system, similar to YwqK found in H. bilis. Regarding resistance genes, we detected at least 4 multidrug resistance loci (AcrB and SecD protein sequences were 69–70% identical to Helicobacter himalayensis proteins; a MATE-family multidrug export protein showing 61% similarity with a similar system identified in H. himalayensis, and a system that showed 67% homology with QacE of Campylobacter helveticus; and one locus encoding heavy metal resistance). Other putative genes involved in virulence were a type II secretion system showing 50% similarity with the Eps system of H. hepaticus and a type VI secretion system IcmF identified in Helicobacter trogontum and H. bilis that is involved in survival within macrophages. Analysis of genome content confirmed enzyme activities for catalase, thiol-oxidase, and esterase and phosphatases but failed to identify genes encoding urease activity. Overall, 30% of CDSs (n = 509) detected in the genome were classified as hypothetical proteins. Comparison of important house-keeping genes (ATPase subunits, DNA polymerase, purA, gro genes) showed maximum similarity for protein sequences between 60 and 91%. The predicted protein sequence of GyrA contained an isoleucine at amino acid position 81 known to contribute to the quinolone resistance phenotype in H. pylori; this confirms the in vitro ciprofloxacin resistance result described earlier. The flagellar operon contained 32 ORFs showing limited similarity to all publicly available sequences, between 25 and 65% for FliK and FliL, respectively. S15 does not encode homologs of CDT and γ-glutamyltranspeptidase, the main toxins of the Helicobacter genus.
Phylogenetic Analysis
The sequences of several genes (16S rRNA gene, hsp60, gyrA and gyrB) were compared between strain S15 and closely related Helicobacter spp.; sequence identities are shown in Table 1. Based on these data, the two most genetically similar species were H. pullorum for the 16S rRNA gene and H. equorum for the three other genes. Phylogenetic trees were constructed from nucleotide (16S rRNA gene, hsp60 and gyrB genes) or predicted amino acid alignments (for GyrA). The sequences obtained by Ménard et al., were used for all Helicobacter spp. except for the S15 sequences, which were obtained from genome sequencing (Ménard et al., 2016). The tree obtained with the 16S rRNA gene showed that S15 clustered in a subgroup of enterohepatic Helicobacter species close to H. pullorum (Figure 3A). The phylogenetic position of S15 was then assessed using the hsp60 (Supplemental Figure S2A) and gyrB nucleotide sequences (Supplemental Figure S2B), and GyrA protein sequence (Figure 3B), a relevant marker for determining Helicobacter relatedness. In our case, the rRNA 16S sequence of strain S15 was 97% with H. equorum and only 96% with the sequence of H. bilis. Information extracted from these trees suggests that the S15 strain most likely corresponds to a new enterohepatic sheathed Helicobacter species close to H. equorum. The closest relatives of S15, identified using the phylogenetic trees, were used to calculate in silico values of DDH and ANI. Pairwise ANI-values of the closest strains identified by the algorithm in the JSpeciesWS database are depicted in Supplementary Tables S3A,B, respectively. The highest DDH-values were 39.40% obtained for the comparison with the genome of H. canadensis MIT98-5491 and 26.5% for H. equorum QF1. Comparison with the other species from the 16s RNA and GyrA phylogenetic trees (H. bilis and H. pullorum) yields a distance of ≥0.975 and a probability that DDH is >70% between these species and S15 of 0%. The highest ANI-value was 93% between S15 strain and H. equorum QF1. A comparison with similarly distant species was around 70% concerning H. canadensis MIT98-5491 and H. himalayensis YS1, thus below the cut-off of 95%.

Table 1. Best BLAST hits for the target genes used in the present study.
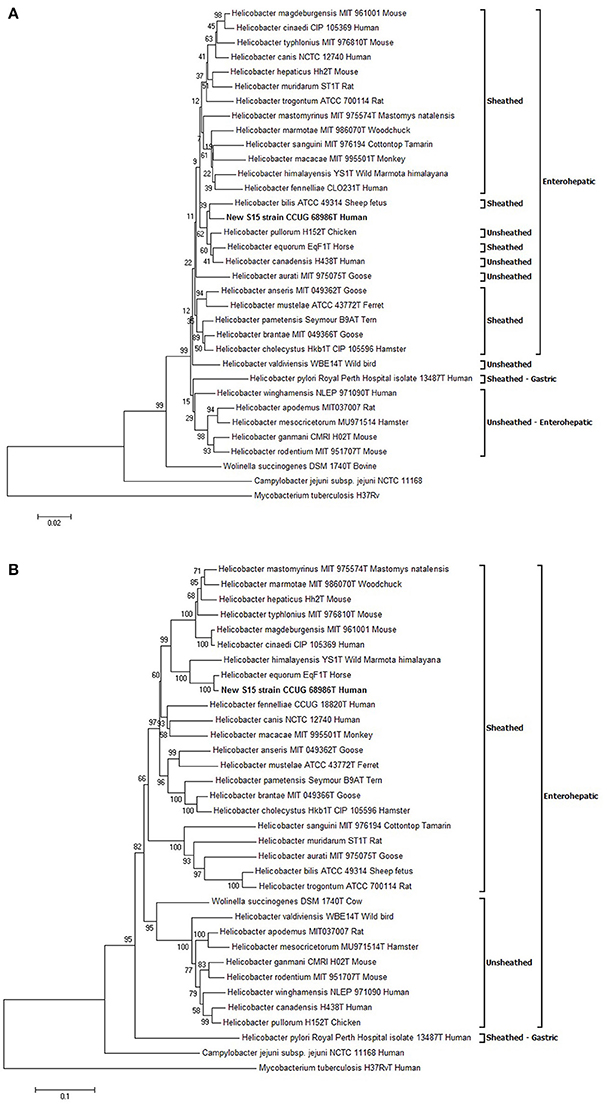
Figure 3. Phylogenetic trees. Evolutionary relationships of Helicobacter taxa based on the 16S rRNA gene (A) and GyrA protein encoded gene (B). The phylogeny presented is based on the alignment of approximately 1,400 nucleotides of the 16S rRNA gene and 700 residues of the GyrA protein. The phylogenetic analyses were generated with the neighbor-joining method. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to the branches. The trees are not rooted and drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Kimura 2-parameter method for 16S rRNA gene, the Poisson correction method for GyrA and are represented in the units of the number of base substitutions per site. The analysis included 34 sequences. All ambiguous positions were removed for each sequence pair. Evolutionary analyses were conducted using MEGA6. All sequences are labeled according to species, strain name, collection number in brackets. Mycobacterium tuberculosis was used as the outgroup sequence. All accession numbers are provided in Supplementary Table S2.
Discussion and Conclusion
We identified in the blood cultures from a febrile patient a Gram-negative rod-shaped mobile bacterium requiring a prolonged time/incubation (almost 5 days) for detectable growth. MALDI-TOF MS used in the routine identification process failed to reliably identify this bacterium (named S15 strain), because of the bacterial protein spectrum absent in the current database. Its belonging to the Helicobacter genus was suggested by the phenotypic characteristics of the strain.
The whole genome sequencing data strongly provided evidence for a strain close to, but distinct from, several Helicobacter species. A clear distinction can be made between S15 and the major well-characterized Helicobacter species based on the phenotypic characteristics (e.g., ability to grow at 42°C, alkaline phosphate hydrolysis, number and disposition of flagella; Supplementary Table S3). WGS analysis provided evidence that the S15 strain is close to H. equorum, a bacterium that does not commonly infect humans (Moyaert et al., 2009) and is rarely found in human samples. It should be noted that the number of nucleotide sequences in public databases has evolved rapidly facilitating genome comparison. Public databases already contain an impressive number of 16S rRNA gene sequences, a lower number of gyrA/B sequences and only a limited diversity of Helicobacter genomes. Based on all available sequences, comparison at the whole genome level showed a sharp distance between our S15 strain and H. equorum but clearly suggest that our strain belongs to a likely novel Helicobacter species. We propose to associate the present strain with the Latin name of the place of isolation; Caesarodunum (Tours, France) and suggest “Helicobacter caesarodunensis” for further description of this new bacterium.
Septicemia associated with a Helicobacter that is not characterized at the species level is not scarce (Schwarze-Zander et al., 2010). As soon as the databases associated with MALDI-TOF MS systems are updated with the peptide mass spectra of strain S15, the identification of S15-like Helicobacters will be improved in routine clinical laboratory settings. Further isolations of this bacterium in patient clinics will enable a better understanding of bacterial pathogenicity, origin, and potential routes of transmission.
Genome Sequence Accession
The whole genome sequence of the S15 isolate has been deposited at DDBJ/ENA/GenBank under the accession number NESU00000000. The version described in this paper is version NESU01000000. The 16S rRNA gene has been also deposited under the accession number KX057485.
Ethics Statement
Considering that the infection was not trivial, the patient was informed that specific investigations will be needed as the organism responsible for infection was not common. The patient gave oral consent and the strain identified in the blood cultures was shipped to the National Reference Center for Campylobacters and Helicobacters following further macroscopic visualization on blood agar. The current study was carried out in accordance with national and local recommendations. The patient gave his consent to publish the report.
Author Contributions
NvdM-M was involved in strain isolation, maintenance, and manuscript writing. LuB: performed the phenotypic experiments, analyzed the data, and wrote the manuscript. SD: performed genome assembly, annotation, and submission of the whole genome sequence to the NCBI database. AL and LoB: contributed to the clinical follow-up of the patient. NG, AS, and FH: were involved in sequence and genomic analyses. AC: performed MALDI-TOF MS analysis, protein profile comparisons and was involved in strain preservation and cultivation for culture collections. AS and FH: contributed to the sequencing and assembly of the H. equorum genome used for ANI and DDH. AD, SL, and EG: performed strain characterization experiments. FM was involved in writing the manuscript. AG: contributed to manuscript writing and correction and performed ANI and DDH determinations. PL: designed the experiments, analyzed the data, and wrote the manuscript. PF: involved in the analysis of genome annotation and writing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Armelle Ménard for her advice on the phylogenetics analyses, and Lindsay Mégraud for english revision of the manuscript. Electron microscopy studies were conducted at the Bordeaux Imaging Center—Bordeaux University, a Core facility of the national infrastructure “France BioImaging” (ANR-10-INBS-04 France BioImaging).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02533/full#supplementary-material
References
Angeletti, S. (2016). Matrix assisted laser desorption time of flight mass spectrometry (MALDI-TOF MS) in clinical microbiology. J. Microbiol. Methods 138, 20–29. doi: 10.1016/j.mimet.2016.09.003
Baele, M., Pasmans, F., Flahou, B., Chiers, K., Ducatelle, R., and Haesebrouck, F. (2009). Non-Helicobacter pylori helicobacters detected in the stomach of humans comprise several naturally occurring Helicobacter species in animals. FEMS Immunol. Med. Microbiol. 55, 306–313. doi: 10.1111/j.1574-695X.2009.00535.x
Borges, V., Santos, A., Correia, C. B., Saraiva, M., Ménard, A., Vieira, L., et al. (2015). Helicobacter pullorum isolated from fresh chicken meat: antibiotic resistance and genomic traits of an emerging foodborne pathogen. Appl. Environ. Microbiol. 81, 8155–8163. doi: 10.1128/AEM.02394-15
Cherkaoui, A., Emonet, S., Fernandez, J., Schorderet, D., and Schrenzel, J. (2011). Evaluation of matrix-assisted laser desorption ionization-time of flight mass spectrometry for rapid identification of Beta-hemolytic streptococci. J. Clin. Microbiol. 49, 3004–3005. doi: 10.1128/JCM.00240-11
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Euzéby, J. P. (1997). List of bacterial names with standing in nomenclature: a folder available on the internet. Int. J. Syst. Bacteriol. 47, 590–592. doi: 10.1099/00207713-47-2-590
Floch, P., Mégraud, F., and Lehours, P. (2017). Helicobacter pylori strains and gastric MALT Lymphoma. Toxins 9:132. doi: 10.3390/toxins9040132
Ge, Z., Feng, Y., Ge, L., Parry, N., Muthupalani, S., and Fox, J. G. (2017). Helicobacter hepaticus cytolethal distending toxin promotes intestinal carcinogenesis in 129Rag2-deficient mice. Cell. Microbiol. 19. doi: 10.1111/cmi.12728
Hernandez, D., Tewhey, R., Veyrieras, J. B., Farinelli, L., Osteras, M., Francois, P., et al. (2014). De novo finished 2.8 Mbp Staphylococcus aureus genome assembly from 100 bp short and long range paired-end reads. Bioinformatics 30, 40–49. doi: 10.1093/bioinformatics/btt590
Kawamura, Y., Tomida, J., Morita, Y., Fujii, S., Okamoto, T., and Akaike, T. (2014). Clinical and bacteriological characteristics of Helicobacter cinaedi infection. J. Infect. Chemother. 20, 517–526. doi: 10.1016/j.jiac.2014.06.007
Mégraud, F., Bessede, E., and Varon, C. (2015). Helicobacter pylori infection and gastric carcinoma. Clin. Microbiol. Infect. 21, 984–990. doi: 10.1016/j.cmi.2015.06.004
Ménard, A., Buissonnière, A., Prouzet-Mauléon, V., Sifré, E., and Megraud, F. (2016). The GyrA encoded gene: a pertinent marker for the phylogenetic revision of Helicobacter genus. Syst. Appl. Microbiol. 39, 77–87. doi: 10.1016/j.syapm.2015.09.008
Ménard, A., Pere-Vedrenne, C., Haesebrouck, F., and Flahou, B. (2014). Gastric and enterohepatic helicobacters other than Helicobacter pylori. Helicobacter 19(Suppl. 1), 59–67. doi: 10.1111/hel.12162
Moyaert, H., Pasmans, F., Decostere, A., Ducatelle, R., and Haesebrouck, F. (2009). Helicobacter equorum: prevalence and significance for horses and humans. FEMS Immunol. Med. Microbiol. 57, 14–16. doi: 10.1111/j.1574-695X.2009.00583.x
Péré-Védrenne, C., Cardinaud, B., Varon, C., Mocan, I., Buissonnière, A., Izotte, J., et al. (2016). The cytolethal distending toxin subunit CdtB of Helicobacter induces a Th17-related and antimicrobial signature in intestinal and hepatic cells in vitro. J. Infect. Dis. 213, 1979–1989. doi: 10.1093/infdis/jiw042
Schwarze-Zander, C., Becker, S., Wenzel, J., Rockstroh, J. K., Spengler, U., and Yassin, A. F. (2010). Bacteremia caused by a novel Helicobacter species in a 28-year-old man with X-linked agammaglobulinemia. J. Clin. Microbiol. 48, 4672–4676. doi: 10.1128/JCM.01350-10
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Trespalacios, A. A., Rimbara, E., Otero, W., Reddy, R., and Graham, D. Y. (2015). Improved allele-specific PCR assays for detection of clarithromycin and fluoroquinolone resistant of Helicobacter pylori in gastric biopsies: identification of N87I mutation in GyrA. Diagn. Microbiol. Infect. Dis. 81, 251–255. doi: 10.1016/j.diagmicrobio.2014.12.003
van Veen, S. Q., Claas, E. C., and Kuijper, E. J. (2010). High-throughput identification of bacteria and yeast by matrix-assisted laser desorption ionization-time of flight mass spectrometry in conventional medical microbiology laboratories. J. Clin. Microbiol. 48, 900–907. doi: 10.1128/JCM.02071-09
Varon, C., Mocan, I., Mihi, B., Péré-Védrenne, C., Aboubacar, A., Morate, C., et al. (2014). Helicobacter pullorum cytolethal distending toxin targets vinculin and cortactin and triggers formation of lamellipodia in intestinal epithelial cells. J. Infect. Dis. 209, 588–599. doi: 10.1093/infdis/jit539
Keywords: Helicobacter sp., human infection, phylogeny, electron microscopy, genome content
Citation: van der Mee-Marquet NL, Bénéjat L, Diene SM, Lemaignen A, Gaïa N, Smet A, Haesebrouck F, Cherkaoui A, Ducournau A, Lacomme S, Gontier E, Bernard L, Mégraud F, Goudeau A, Lehours P and François P (2017) A Potential New Human Pathogen Belonging to Helicobacter Genus, Identified in a Bloodstream Infection. Front. Microbiol. 8:2533. doi: 10.3389/fmicb.2017.02533
Received: 30 May 2017; Accepted: 05 December 2017;
Published: 18 December 2017.
Edited by:
Axel Cloeckaert, Institut National de la Recherche Agronomique (INRA), FranceReviewed by:
Luis Collado, Universidad Austral de Chile, ChileJames G. Fox, Massachusetts Institute of Technology (MIT), United States
Maarten Gilbert, Utrecht University, Netherlands
Copyright © 2017 van der Mee-Marquet, Bénéjat, Diene, Lemaignen, Gaïa, Smet, Haesebrouck, Cherkaoui, Ducournau, Lacomme, Gontier, Bernard, Mégraud, Goudeau, Lehours and François. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philippe Lehours, philippe.lehours@u-bordeaux.fr
Patrice François, patrice.francois@genomic.ch