 Byoung-Jun Kim
Byoung-Jun Kim Bo-Ram Kim
Bo-Ram Kim Yoon-Hoh Kook
Yoon-Hoh Kook Bum-Joon Kim
Bum-Joon Kim- Department of Microbiology and Immunology, Biomedical Sciences, Liver Research Institute and Cancer Research Institute, College of Medicine, Seoul National University, Seoul, South Korea
We recently showed that Mycobacterium yongonense could be divided into two genotypes: Type I, in which the rpoB gene has been transferred from Mycobacterium parascrofulaceum, and Type II, in which the rpoB gene has not been transferred. Comparative genome analysis of three M. yongonense Type I, two M. yongonense Type II and M. parascrofulaceum type strains were performed in this study to gain insight into gene transfer from M. parascrofulaceum into M. yongonense Type I strains. We found two genome regions transferred from M. parascrofulaceum: one contained 3 consecutive genes, including the rpoBC operon, and the other contained 57 consecutive genes that had been transferred into M. yongonense Type I genomes via homologous recombination. Further comparison between the M. yongonense Type I and II genomes revealed that Type I, but not Type II has a distinct DNA mismatch repair gene (MutS4 subfamily) that was possibly transferred via non-homologous recombination from other actinomycetes. We hypothesized that it could facilitate homologous recombination from the M. parascrofulaceum to the M. yongonense Type I genomes. We therefore generated recombinant Mycobacterium smegmatis containing a MutS4 operon of M. yongonense. We found that the M. tuberculosis rpoB fragment with a rifampin resistance-conferring mutation was more frequently inserted into recombinant M. smegmatis than the wild type, suggesting that MutS4 is a driving force in the gene transfer from M. parascrofulaceum to M. yongonense Type I strains via homologous recombination. In conclusion, our data indicated that MutS4 in M. yongonense Type I genomes may drive gene transfer from M. parascrofulaceum via homologous recombination, resulting in division of M. yongonense into two genotypes, Type I and II.
Introduction
Recombination is defined as process leading to the exchange of information between DNA or RNA and is a fundamental process with important implications for the evolution of the cell (Achtman and Wagner, 2008; Sheppard et al., 2008; Fraser et al., 2009). Recombination is typically classified as being homologous or non-homologous recombination, based upon the presence or absence of nucleotide sequence homology between the parental sites, respectively: homologous recombination, in which a fragment of a genome is replaced by one of sequence homology within another genome (Didelot and Maiden, 2010), and non-homologous recombination, which causes genetic additions and is often referred to as lateral gene transfer (LGT) (Ochman et al., 2000). Both homologous and non-homologous types of recombination are key elements in the evolution of bacteria and can be linked to variations in fitness and the consequent changes in ecologies and lifestyles (Didelot et al., 2012).
In most organisms, the mismatch repair system (MMR) pathway is highly conserved and enhances replication fidelity 50- to 1000-folds by repairing nucleotide mismatches and small insertions and deletions (Modrich and Lahue, 1996; Umar and Kunkel, 1996; Iyer et al., 2006). Members of the MutS and MutL protein families normally play a pivotal role in mismatch correction. The MMR system also prevents recombination between not identical DNA sequences (homeologous recombination) (Reenan and Kolodner, 1992). Defects in the MMR system could therefore lead to highly elevated mutation rates (hypermutability), meiotic defects and infertility (Harfe and Jinks-Robertson, 2000; Surtees et al., 2004). The genus Mycobacterium has no homologs of MutS or MutL (Mizrahi and Andersen, 1998; Sachadyn, 2010; Banasik and Sachadyn, 2014). Instead, its genome stability is maintained via an alternative NucS pathway that appears in many Archaea (Castaneda-Garcia et al., 2017).
Homologous recombination and homeologous recombination are important mechanisms that contribute to the genomic diversity of various bacteria. To restrict recombination between moderately divergent (up to ∼10%) DNA sequences at the DNA hybridization step, prokaryotes and eukaryotes utilize a canonical MutS–MutL-based MMR system that facilitates gene transfer via homologous recombination during eukaryotic meiosis in eukaryotes or during genome acquisition from foreign bacterial DNA (Modrich and Lahue, 1996; Vulic et al., 1997). Previous reports that genes acquired from other bacteria are rarely found in the genomes of M. tuberculosis or M. leprae, both lacking the highly conserved MutS-based MMR system (Vulic et al., 1997; Cole et al., 1998, 2001), strongly support the above notion.
We recently found that Mycobacterium yongonense can be divided into two genotypes: Type I, in which the rpoB gene has been transferred from Mycobacterium parascrofulaceum, and Type II, in which the rpoB gene has not been a subject of gene transfer (Kim et al., 2016). Comparative genome analysis between three M. yongonense Type I (DSM 45126T, MOTT-12 and MOTT-27) and two M. yongonense Type II (MOTT-36Y and MOTT-H4Y) strains and an M. parascrofulaceum type strain (ATCC BAA-614T) was performed to gain insight into the gene transfer from M. parascrofulaceum to M. yongonense Type I strains. We found for the first time in mycobacteria a distinct DNA mismatch repair gene that belonged to the MutS4 subfamily in the genome of M. yongonense Type I strains and that served as a putative driving force of homologous recombination between the M. parascrofulaceum and M. yongonense Type I genomes.
Results
Identification of Two Putative Regions in the M. yongonense Type I Genome That Were Transferred from M. parascrofulaceum
As described in a previous report, M. yongonense Type I strains (DSM 45126T, MOTT-12 and MOTT-27) have an rpoB gene that may have been transferred from the distantly related scotochromogenic species M. parascrofulaceum (Kim et al., 2013a,b,c, 2016). The rpoB gene was also found to differ between the M. yongonense Type I (DSM 45126T, MOTT-12 and MOTT-27) and Type II strains (MOTT-36Y and MOTT-H4Y) used in this study. The complete genome sequences of two M. yongonense Type I strains, MOTT-12 (GenBank accession No, CP015964) and MOTT-27 (GenBank accession No, CP015965), were analyzed in this study to gain further insight into gene transfer from M. parascrofulaceum to M. yongonense Type I (Table 1). All the ORFs of seven strains (three M. yongonense Type I strains, two M. yongonense Type II, strains and one M. parascrofulaceum strain) were compared and analyzed using the BLASTN and BLASTP programs. Two loci that showed higher sequence similarities to sequences in M. parascrofulaceum than to those in the phylogenetically related M. yongonense Type II strains were found in the genomes of the three M. yongonense Type I strains. The first region includes three consecutive ORFs, an ABC transporter and the rpoB and rpoC genes [designated as TR1 (Transfer Region 1), OEM_44170∼44190 in M. yongonense DSM 45126T], and the second region contains 57 consecutive ORFs, including genes corresponding to dehydrogenase, MCE family, which could enable mycobacteria to enter into and survive inside the mammalian macrophage (Arruda et al., 1993; Kumar et al., 2003; Zhang and Xie, 2011), and fatty acid biosynthesis [designated as TR2 (Transfer Region 2), OEM_08030∼08590 in M. yongonense DSM 45126T] (Figure 1 and Supplementary Table S1). All 60 transferred ORFs of the M. yongonense Type I strains (DMS 45126T, MOTT-12, and MOTT-27) were always more closely related to sequences in M. parascrofulaceum than those in M. intracellulare and M. yongonense Type II strains [in TR1 (3 ORFs), sequences were 97–99% similar to their counterparts in M. parascrofulaceum, and in TR2 (57 ORFs), sequences were 95–100% similar to their counterparts in M. parascrofulaceum] (Figure 1 and Supplementary Table S1). The ABC transporter (OEM_44190) and rpoC (OEM_44170), which correspond to the two ends (5′ and 3′) of TR1, of M. yongonense Type I strains (DSM 45126T, MOTT-12 and MOTT-27) were clustered with their homologs from M. parascrofulaceum (92 or 100% bootstrap values) (Supplementary Figures S1B,C). However, the outer neighboring ORFs (OEM_44200; sim14 and OEM_44160; endonuclease IV) of M. yongonense Type I strains were more closely grouped with those of M. intracellulare (ATCC 13950T, MOTT-02 and MOTT-64) or M. yongonense Type II (MOTT-36Y and MOTT-H4Y) strains than those of the M. parascrofulaceum strain (Supplementary Figures S1A,D). A similar trend was also found in TR2 (Supplementary Figure S2).
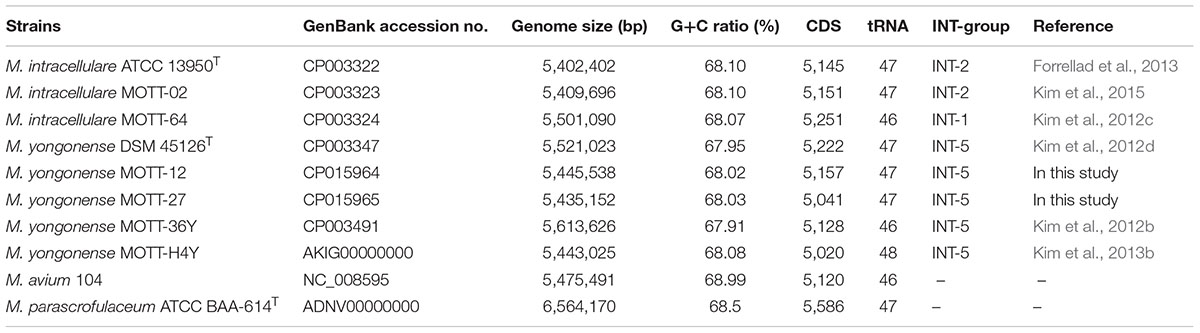
TABLE 1. Genome sequences used in this study.
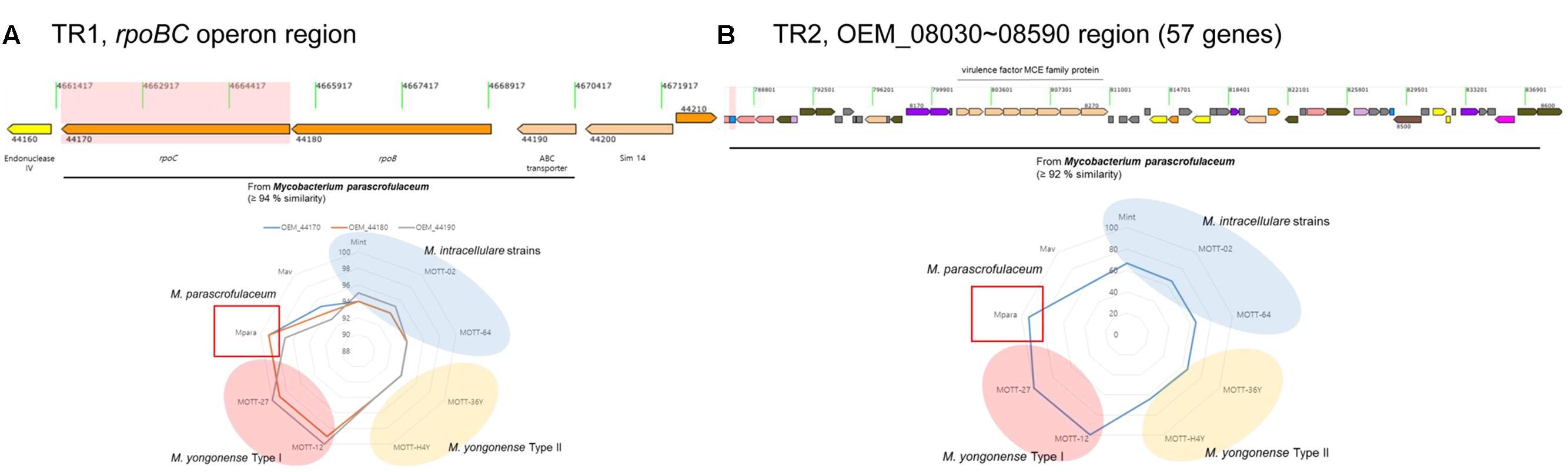
FIGURE 1. Visualization of two putative lateral gene transferred regions within the M. yongonense DSM 45126T genome. (A) First region (rpoBC operon, OEM_44170∼44190) (TR1). (B) Second region (57 ORFs, OEM_08030∼08590) (TR2). Sequence similarities between M. yongonense DSM 45126T and related strains were also visualized in each lower image. Red region, M. yongonense Type II strains; yellow region, M. yongonense Type I strains; blue region, M. intracellulare.
Identification of Homologous Recombination Sites in the Two Putative Transferred Regions, TR1 and TR2 of the M. yongonense Type I Genome
To identify potential breakpoints for gene transfer within the two putative transferred regions, TR1 and TR2, of the M. yongonense Type I genome, we applied BootScan analysis to the TR1 and TR2 sequences of nine mycobacterial strains (three M. yongonense Type I, two M. yongonense Type II and M. parascrofulaceum and M. intracellulare type strains). Potential locations of the recombination breakpoints of the 5′ and 3′ ends were found in TR1 at the 261st nucleotide (nt) of OEM_44190 (ABC transporter) and the 3,852nd nt of OEM_44170 (rpoC), respectively. Aligned sequences showed 44-bp sequences flanking the potential breakpoints of the 5′ end (nt 229–272 of the ABC transporter) and 26-bp sequences flanking the potential breakpoints of the 3′ end (nt 3920–3945 of rpoC), which were conserved in all nine aligned mycobacterial strains. The aligned and phylogenetic profiles clearly differed in the sequences located at the 5′ and 3′ ends of these conserved regions. The potential locations of the recombination breakpoints of the 5′ and 3′ ends in TR2 were found at the 351st nt of OEM_08020 and the 408th nt of OEM_08590, respectively (Figure 2 and Supplementary Figure S3). Aligned sequences showed 29-bp sequences flanking the potential breakpoints of the 5′ end (nt 337–365 of OEM_08020) and 34-bp sequences flanking the potential breakpoints of the 3′ end (nt 410–443 of OEM_08590), which were conserved in almost all seven aligned mycobacterial strains despite minor differences. Our data suggest that TR1 and TR2 of the M. yongonense Type I genome may have been transferred from M. parascrofulaceum via homologous recombination.
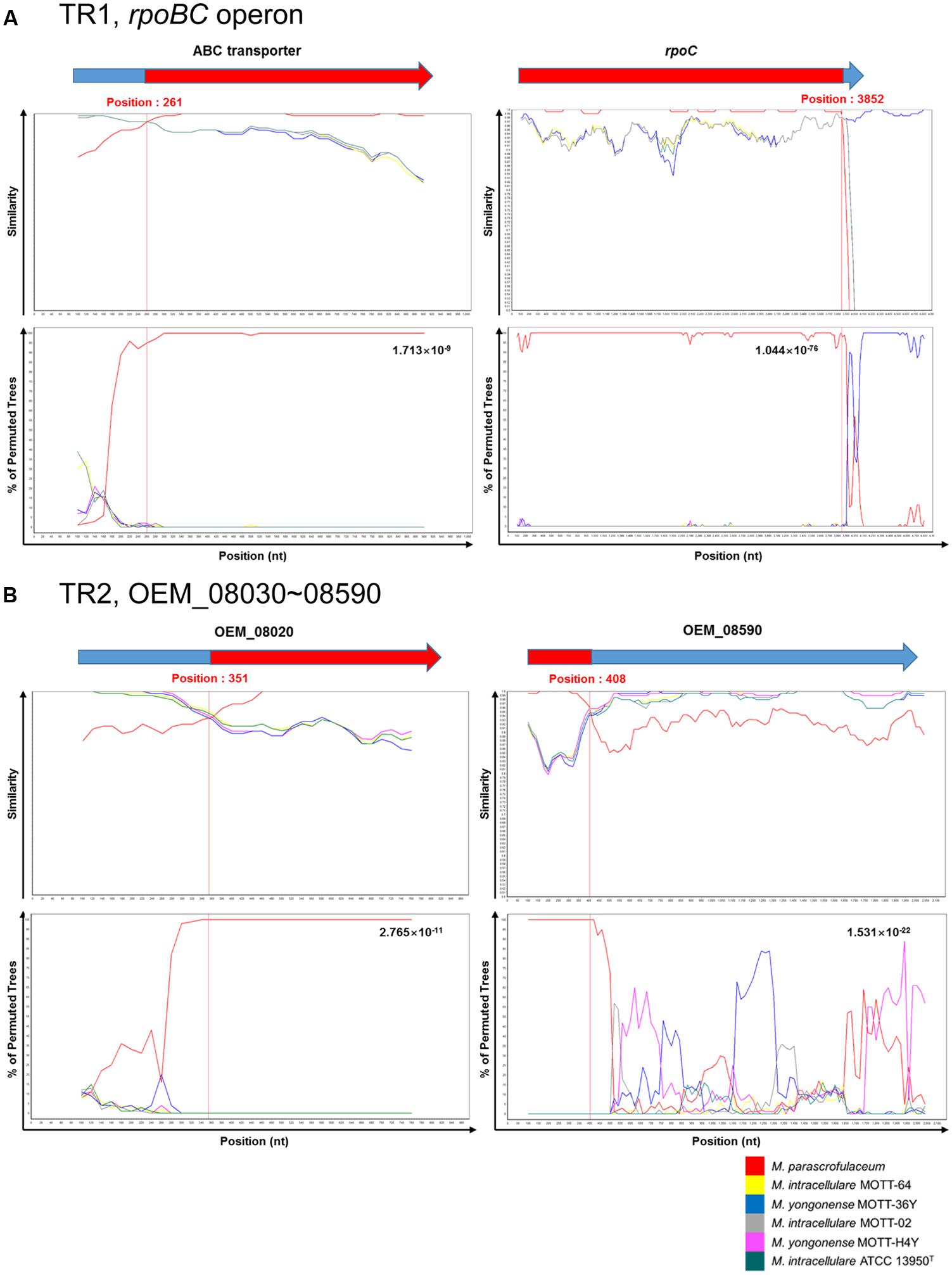
FIGURE 2. Plots of similarity in a putative recombination site from M. intracellulare (ATCC 13950T, MOTT-02 and MOTT-64) or M. yongonense Type II (MOTT-36Y and MOTT-H4Y) and M. parascrofulaceum strains to M. yongonense Type I (DSM 45126T, MOTT-12 and MOTT-27) strains. (A) First recombination site (from OEM_44170 to 44190) (TR1). (B) Second recombination site (from OEM_08030 to 08590) (TR2). Each figure indicate SimPlot and BootScan results of M. yongonense Type I strains compared to M. intracellulare, M. yongonense Type II and M. parascrofulaceum strains. Each point plotted is percent identity within a sliding window 200 bp wide centered on the position plotted, with a 20-bp step size between points. Detailed parameters used for analysis are as follows; Window: 200 bp, Step: 20 bp, GapStrip: On, Reps: 100, Kimura (2-parameter), T/t: 2.0, Neighbor-joining.
Identification of Distinct MutS4-Related DNA Mismatch Repair Genes in the Genome of M. yongonense Type I Strains
Although M. yongonense Type I and Type II strains are members of the same species, only M. yongonense Type I strains have unique gene regions, TR1 and TR2, that were transferred from M. parascrofulaceum via homologous recombination. This finding prompted us to hypothesize that there may be distinct ORFs in the M. yongonense Type I genome that drive gene acquisition via homologous recombination. To address this issue, we analyzed the putative genomic islands in the M. yongonense genome by web-based program, “IslandViewer 4”1 (Didelot and Maiden, 2010; Zhang and Xie, 2011). The result showed that the 9 putative genomic islands were identified from genome of the M. yongonense Type I (DSM 45126T) (Supplementary Figure S4). Among these putative genomic islands, a distinct region composed of 13 consecutive ORFs that was possibly transferred from non-mycobacterial actinomycetes was found in the genome of only M. yongonense Type I strains (DSM 45126T, MOTT-12 and MOTT-27), but not in the genome of M. yongonense Type II strains (MOTT-36Y and MOTT-H4Y) (Figure 3, Supplementary Table S2, and Supplementary Figure S5). This region contains enolase (OEM_51290), NADH dehydrogenase complex (OEM_51300∼51330 and 51350), hydrogenase subunit (OEM_51340) and DNA mismatch repair genes (OEM_51400 and 51410) (Figure 3, Supplementary Table S2, and Supplementary Figure S5). Since the DNA mismatch repair gene has been reported to result from homologous recombination (Lin et al., 2007), we hypothesized that the two consecutive ORFs (OEM_51400 and 51410) encoding DNA mismatch repair genes, which are distinct in M. yongonense Type I strains, could drive gene transfer from M. parascrofulaceum to M. yongonense Type I strains via homologous recombination. Notably, the two ORFs of the DNA mismatch repair genes were MutS4A and MutS4B homologs of the MutS4 subfamily (Figure 4). Furthermore, detailed sequence inspection showed that these two ORFs (OEM_51400 and 51410) also have a signature structure from the MutS4 subfamily; MutS4A and MutS4B are adjacent, and the stop codon of MutS4A overlaps with the initiation codon of MutS4B (Supplementary Figure S6).
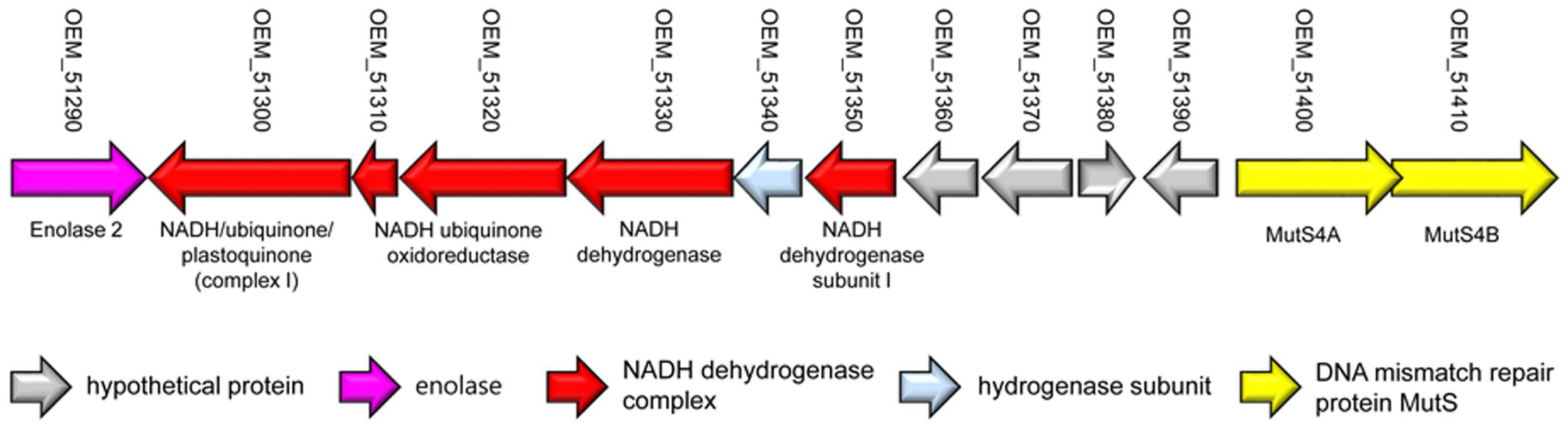
FIGURE 3. Presentation of a locus containing non-mycobacterial genes in M. yongonense Type I strains, but not Type II strains.
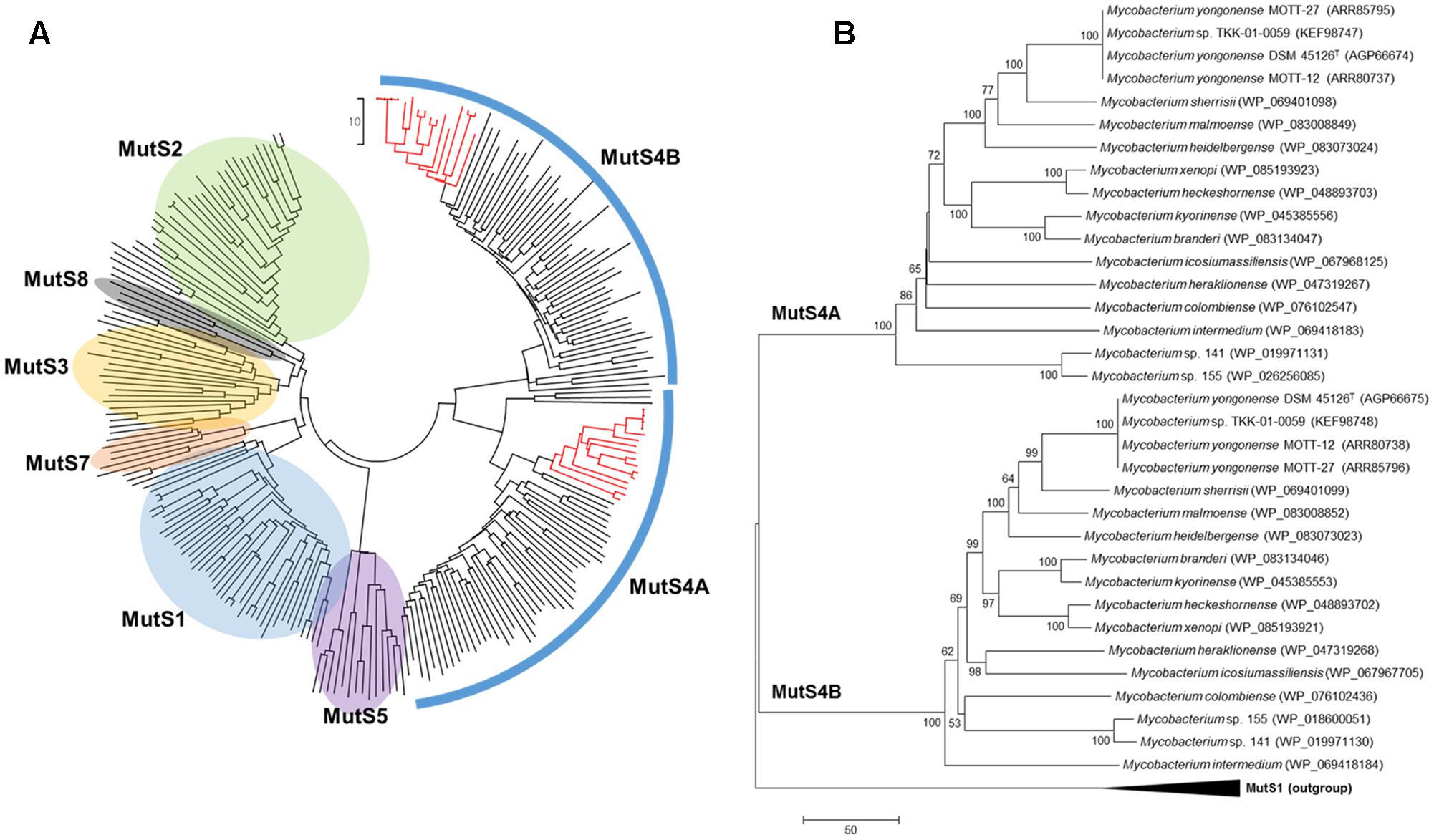
FIGURE 4. Neighbor-Joining phylogenetic trees of MutS family proteins. (A) Phylogenetic tree covering MutS homologs from diverse bacterial and archaeal strains. Red branches indicate MutS4 homologs of Mycobacterium strains including M. yongonense Type I strains. (B) Phylogenetic tree based on MutS4 homologs among Mycobacterium strains and rooted with MutS1 sequences as an out-group. Evolutionary distances are in units of numbers of amino acid differences per sequence.
Phylogenetic Analysis of Mycobacterial MutS4 Orthologs
To confirm the presence of MutS4 genes in mycobacterial species other than M. yongonense Type I strains, the amino acid sequences of M. yongonense Type I MutS4 were subject to BLAST analysis against mycobacterial genome databases. We found MutS4 orthologs in another 14 of the 109 mycobacterial species whose complete or draft genomes have been introduced (Supplementary Table S3). We confirmed that all 14 strains have two MutS4 homologs, MutS4A and MutS4B, that share the signature structure of the MutS4 subfamily. Global phylogenetic analysis using the MutS amino acid sequences of eubacteria, archaebacteria and eukaryotes showed that 17 mycobacteria were located in a distinct cluster based on similarities in MutS4A or MutS4B (Figure 4). A difference in mycobacterial phylogenetic topology between MutS4A and MutS4B was not observed. Notably, two MutS4 ORFs of the Mycobacterium sp. TKK-01-0059 strain isolated from Ngwelezane, South Africa shared 100% sequence similarities with those of M. yongonense Type I strains, suggesting that this strain may be a member of the M. yongonense Type I family. Further hsp65 and genome sequence-based phylogenetic analyses also support this hypothesis (Supplementary Figures S7, S8).
To address the origin of mycobacterial MutS4 genes and determine whether they were present because of an LGT mechanism, we compared the topology between the phylogenetic trees of MutS4 genes, the hsp65 as a chronometer gene and whole genome sequences. Incongruence between the tree topologies of MutS4 and hsp65 or whole genome sequences was found. For example, M. colombiense, a member of the Mycobacterium avium complex (MAC), was the closest related to Mycobacterium intermedium (MutS4A tree) or to Mycobacterium sp. 141 and 155 strains (MutS4B tree) in MutS4 based trees; but, this species was closely located to M. yongonense strains both in the hsp65-based tree and in the whole genome-based tree (Supplementary Figures S7, S8). This result strongly supports an evolutionary scenario that includes the distribution of the MutS4 gene into several mycobacterial species via LGT.
Increased Frequency of Homologous Recombination in Recombinant M. smegmatis Harboring a M. yongonense Type I MutS4 Operon
To examine the role of M. yongonense Type I MutS4 in homologous recombination, we amplified the region (3,838 bp) including MutS4A (OEM_51400), MutS4B (OEM_51410) and their promoter from a M. yongonense Type I strain (DSM 45126T) as described in the Methods (Supplementary Figure S6). This amplicon was cloned into the integrative pMV306 vector and transformed into M. smegmatis to generate a recombinant M. smegmatis harboring a M. yongonense Type I MutS4 operon or an empty vector (rSmeg-D6 or rSmeg-pMV306). The recombinant M. smegmatis strains were confirmed by colony PCR and RT-PCR (Supplementary Figure S9). To confirm the role of the MutS4 operon in homologous recombination, we created a pSE100-317 vector with a M. tuberculosis partial rpoB sequence (684 bp) containing a mutation in codon 522 (TCG → TTG; 317); this mutation confers resistance to rifampin. The constructed pSE100-317 was then transformed into rSmeg-D6 or rSmeg-pMV306 (rSmeg-D6-p317 or rSmeg-pMV306-p317) (Figure 5A). After the transformed strains were plated onto 7H10 agar medium with 100 μg/ml of rifampin. Colonies grown on the rifampin medium (100 μg/ml) were judged as potential recombinants and final authenticity of their recombination were confirmed via checking the presence of M. tuberculosis specific SNPs related to rifampin resistance (mutation at codon 522) by PCR-sequencing protocol targeting the rpoB region. Also, the break point between the M. smegmatis-distinct and M. tuberculosis-distinct sequences was considered to be a potential recombination site. From the three independent trials, total of 22 (rSmeg-pMV306-p317; 7.33 ± 2.52 colonies/trial) and 55 (rSmeg-D6-p317; 18.33 ± 3.06 colonies/trial) colonies were grown on the rifampin medium and identified as putative recombinants. Among them, 16 colonies of rSmeg-pMV306-p317 and 36 colonies of rSmeg-D6-p317 were randomly selected and used for sequencing the rpoB region. Sequence analysis of rSmeg-D6-p317 showed that 27 colonies (75%) of 36 selected colonies grown in rifampin 7H10 agar have a SNP at codon 522 (TCG → TTG) that confers resistance to rifampin. In the case of rSmeg-pMV306-p317, from the 16 selected colonies, only five colonies (31.3%) have changed SNP at codon 522 (Figure 5B, Supplementary Figure S10, and Supplementary Table S4). This result means that the rifampin resistance of these colonies is due to the recombined resistance-conferring M. tuberculosis rpoB gene and is not induced by the mutation of the M. smegmatis rpoB. The average length of the recombined M. tuberculosis rpoB gene in rSmeg-D6-p317 strains (121.3 ± 31.0 nt) is significantly longer than that in the control strain, rSmeg-pMV306-p317 (44.0 ± 0.0 nt) (Figure 5C and Supplementary Table S4), suggesting that the MutS4 gene of M. yongonense Type I strains plays a pivotal role in homologous recombination in M. smegmatis.
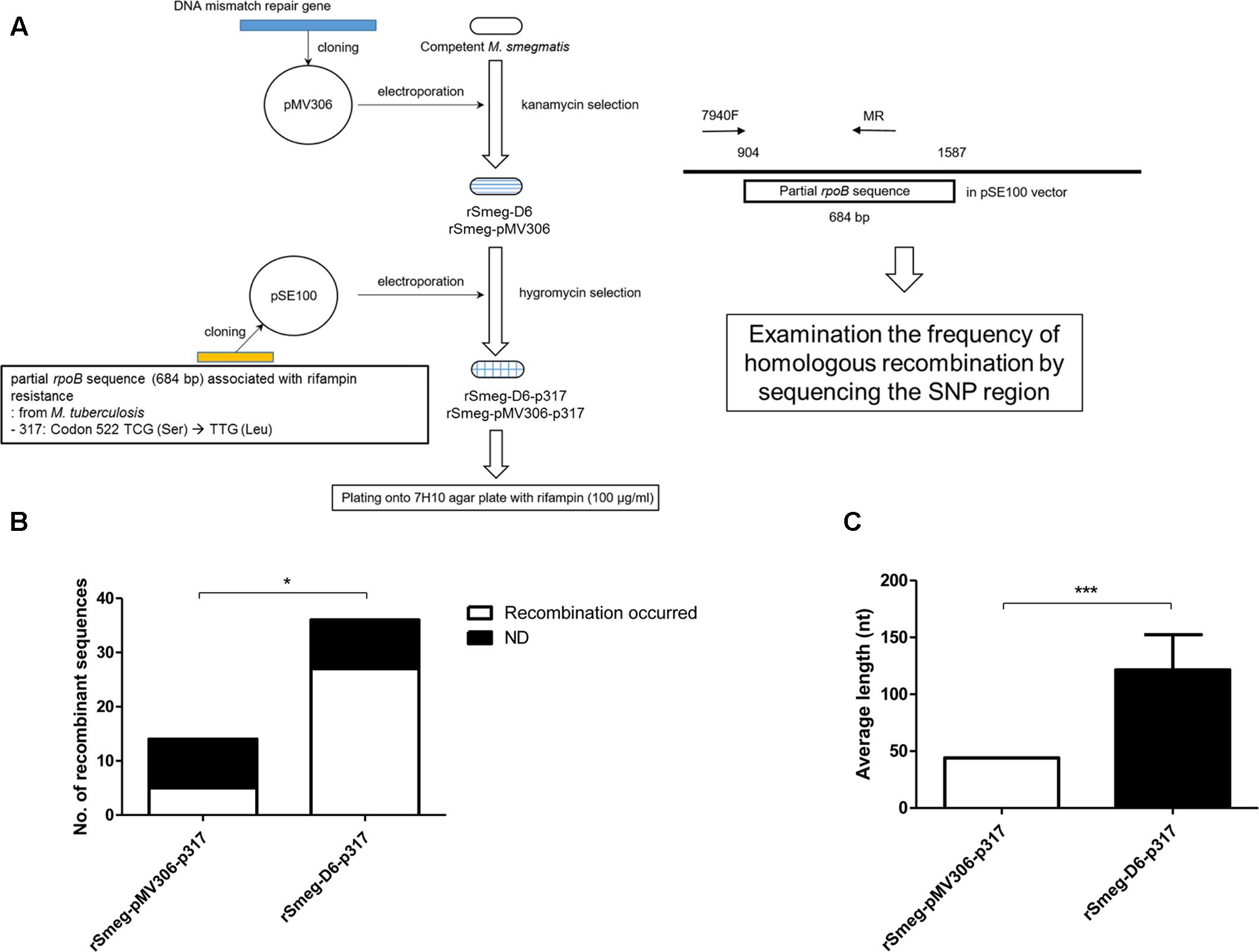
FIGURE 5. Analysis the putative homologous recombination event in recombinant M. smegmatis harboring DNA mismatch repair genes and partial rpoB gene of M. tuberculosis. (A) Schematic flow for analyzing frequency of homologous recombination using recombinant M. smegmatis harboring DNA mismatch repair genes and partial rpoB gene of M. tuberculosis. (B) Comparison the numbers of putative homologous recombined rpoB sequences in the transformed recombinant strains which were grown on 7H10 agar plate supplemented with rifampin. From the transformed colonies, partial rpoB was analyzed whether the sequence was recombined with rifampin resistant M. tuberculosis partial rpoB gene sequence. ND means the colonies where recombination has not been occurred. P-value was calculated by Chi-square test. ∗P < 0.05. (C) Comparison the average length of putative homologous recombined sequences in the transformed recombinant M. smegmatis strains. P-value was calculated by student’s-t test. ∗∗∗P < 0.001.
Discussion
In this study, we found the first MutS homologs in Mycobacterium genomes via genome analysis of three M. yongonense Type I strains (Figures 3, 4). Most unexpectedly, our BLAST analysis indicated that another 14 of 109 mycobacterial species whose whole genomes are currently available, also have MutS4 orthologs. Notably, all 17 mycobacterial strains with MutS4, including the three M. yongonense type I strains, are slowly and not rapidly growing mycobacteria (Figure 4B and Supplementary Table S3). However, the incongruence between phylogenetic analyses based on MutS4 homologs and the hsp65 gene or whole genome sequences, strongly supports our hypothesis that MutS4 distribution between slow-growing mycobacterial strains may also be due to LGT (Figure 4B and Supplementary Figures S7, S8). For example, both M. yongonense and M. colombiense belong to members of the same MAC (Kim et al., 2013c). However, they are phylogenetically separated in MutS4 gene-based phylogenetic analysis, suggesting that the acquisition of the MutS gene may have recently occurred via LGT. MutS4 is present only in several distantly related bacter4ial species (Lin et al., 2007), and most strains contain two copies, MutS4A and MutS4B, whose sequences are phylogenetically closely related to each other, suggesting their generation by duplication in an ancestral bacterial strain (Lin et al., 2007). The signature gene structure present between MutS4A and MutS4B in a bacterial genome was also found, i.e., they are adjacent and the stop codon of MutS4A overlaps with the initiation codon of MutS4B (Lin et al., 2007). We also confirmed that all mycobacterial strains contain two copies, MutS4A and MutS4B, and have the conserved signature gene structure in their genomes.
A MutSac domain in the MutS4 gene is expected to be involved in yet to be defined functions related to DNA metabolism in bacteria (Lin et al., 2007). However, its absence from most bacteria suggests that its functions are not essential and are gradually lost during evolution (Lin et al., 2007). Nevertheless, recent acquisition of MutS4 homologs by several mycobacterial species during evolution is more or less unusual. Two different genotypes of M. yongonense differ in the presence of a MutS4 homolog, leading to speculating that comparative genome analysis of Type I and II strains can provide a clue to the putative role of the MutS4 gene in mycobacterial evolution (Figure 6). In fact, our detailed inspection of genome sequences indicated that there are three distinctly different regions between the genomes of two different genotypes of M. yongonense, Type I and Type II. The first region is present only in the genome of Type I strains, not that of Type II, and includes 13 consecutive ORFs (OEM_51290 to 51410) (Figure 3 and Supplementary Table S2), including a MutS4 gene that may have been acquired from non-mycobacterial actinobacteria via LGT (Figure 6). Of these genes, the enolase-coding ORF (OEM 51290) and six consecutive ORFs related to the NADH dehydrogenase complex (OEM 51290-51350) appear to be related to mycobacterial pathogenesis (Velmurugan et al., 2007; Miller et al., 2010). The second and third regions, which are also distinct in the Type I genome, consists of three consecutive genes, including the rpoBC operon (OEM_44170 to 44190) (TR1) (Figure 1A and Supplementary Table S1), and 57 consecutive genes (OEM_08030 to 08590) (TR2) (Figure 1B and Supplementary Table S1), respectively, that may have been acquired from M. parascrofulaceum via homologous recombination, respectively. We hypothesized that the LGT of MutS4 to an ancestor of M. yongonense Type I strains facilitated the transfer of approximately 60 genes from M. parascrofulaceum into the genome of M. yongonense by a homologous recombination mechanism, leading to distinct evolutionary pathways between M. yongonense Type I and Type II strains. Indeed, we found that rSmeg reinforced by the MutS4 operon of M. yongonense Type I strain exhibited a significantly more frequent homologous recombination when transformed with an M. tuberculosis rpoB fragment carrying a rifampin-resistance (rifR)-related mutation than M. smegmatis reinforced by a mock plasmid (pMV306 only) (Figures 5B,C, Supplementary Figure S10, and Supplementary Table S4), suggesting that the MutS4 gene plays a central role in gene transfer by homologous recombination in mycobacteria.
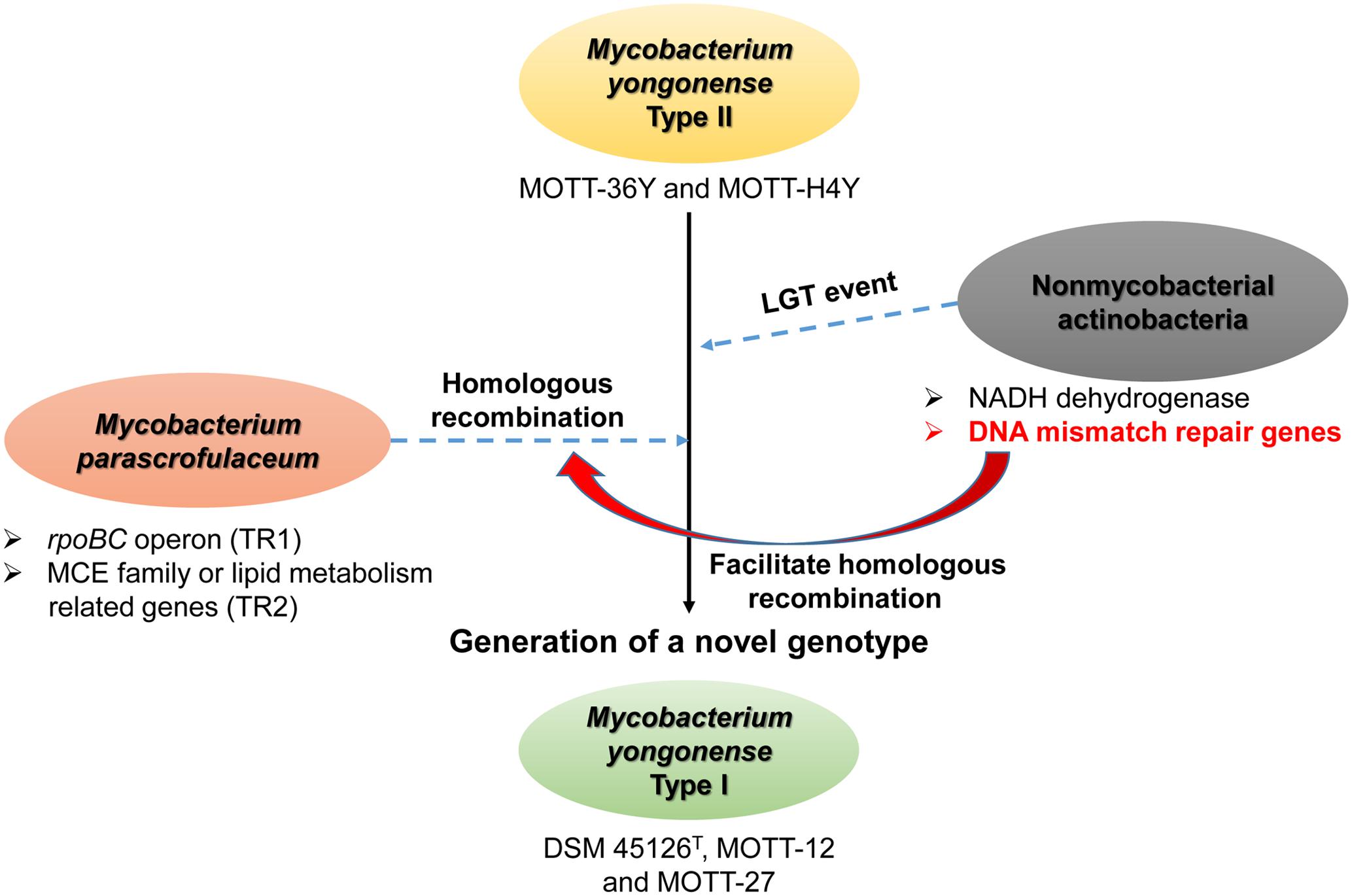
FIGURE 6. Schematic representation showing role of MutS4 gene in evolution of M. yongonense Type I via homologous recombination event of TR1 and TR2 from M. parascrofulaceum to M. yongonense Type I.
Genetic exchanges in the rpoB gene between closely related subspecies within the M. abscessus complex were recently reported to frequently occur (Macheras et al., 2011; Kim et al., 2017). In terms of clinical diagnostics, the rpoB typing method lacks power, culminating in 20% failure rates in the M. massiliense subspecies (Sapriel et al., 2016) and thus suggesting they have hybrid rpoB genes, a part of which were from M. abscessus subspecies. Since only a small portion of rpoB in the M. abscessus complex is exchanged between the same species, the function of the resulting hybrid products may be almost the same as that of the original. However, M. yongonense Type I strains differ because almost all of their rpoBC operon (but not its major interacting partners, such as rpoA and rpoD) are transferred from a distantly related species, M. parascrofulaceum (data not shown). This may be the first report of LGT of the entire rpoBC operon. A comparison of the deduced amino acid sequences showed a marked difference in the rpoBC operons of M. yongonense Type I and II strains, possibly indicating changes in function. The issue of evolutionary merit in the LGT of M. parascrofulaceum rpoBC to the M. yongonense Type I genome remains a mystery. No significant difference in rifampin resistance between M. yongonense Type I and II strains was found (data not shown). One plausible explanation is that transferred M. parascrofulaceum rpoBC contributed to the facilitated expression of the 57 consecutive genes (OEM_08030 to 08590) in TR2 that were acquired in M. yongonense Type I strains from M. parascrofulaceum via homologous recombination.
TR2, within the M. yongonense Type I genome, was composed of 57 consecutive genes (OEM_08030 to 08590), was putatively transferred from M. parascrofulaceum and includes several virulence-related mce family genes and fatty acid biosynthesis-related genes capable of affecting mycobacterial cell wall structure, possibly leading to a change in host-pathogen interactions (Zhang and Xie, 2011; Forrellad et al., 2013). Phylogenetic analysis of every single gene indicated that 53 of the 57 consecutive genes within TR2 closely clustered with M. parascrofulaceum but not M. yongonense Type II genes (Figure 1B and Supplementary Table S1), with most showing 100% sequence similarity with their M. parascrofulaceum orthologs. The four genes not clustered into M. parascrofulaceum (OEM_08190, OEM_08520 to 08540) have no corresponding orthologs in the M. parascrofulaceum or M. yongonense Type II genomes, suggesting that their presence may be due to intragenomic gene transfer by mobile genetic elements within the M. yongonense Type I strain’s own genome, rather than to gene loss followed by gene transfer from M. parascrofulaceum. Indeed, the three consecutive genes (OEM_08520 to 08540) proved to be insertion sequence (IS) elements that are frequently reported to be found in M. yongonense (Kim et al., 2015), and supporting the above hypothesis.
In conclusion, our genome sequence-based phylogenetic analysis and gain-of-function experiment using rSmeg indicated that the MutS4 gene of M. yongonense Type I could play a pivotal role in mycobacterial evolution via increasing genetic transfer through homologous recombination from other distantly related mycobacteria.
Materials and Methods
Genome Sequences Used in This Study
Ten mycobacterial genome sequences, from strains belonging to the M. avium complex [3 M. intracellulare (Mint) strains: ATCC 13950T, MOTT-02, and MOTT-64; 5 M. yongonense strains: DSM 45126T, MOTT-12, -27, -36Y and -H4Y; one M. avium (Mav) strain: M. avium 104; and one M. parascrofulaceum strain: M. parascrofulaceum ATCC BAA-614T] (Kim et al., 2012a,b,c,d, 2013b; Lee et al., 2013) and were retrieved from the GenBank database (Table 1) and used for comparative genome analysis.
Identification of Putative Lateral Gene Transferred Regions in M. yongonense Type I Strains from M. parascrofulaceum or Other Genus Strains
To identify putative lateral gene transferred regions of M. yongonense, all ORFs were compared and analyzed using BLASTN and BLASTP programs. The ORFs with high sequence similarities to M. parascrofulaceum (compared length > 80% and sequence similarities > 80% in nucleotide sequence) were selected and analyzed for possible recombination events. Also, webserver based program, IslandViewer 42 (Langille and Brinkman, 2009; Bertelli et al., 2017) was used to identify and visualize the putative genomic islands in the genome of M. yongonense DSM 45126T with three prediction methods: IslandPick (Langille et al., 2008), IslandPath (Hsiao et al., 2003) and SIGI-HMM (Langille and Brinkman, 2009). Among the selected putative regions, ORFs with high sequence similarities to other genus in the genome of M. yongonense DMS 45126T (M. yongonense Type I) were selected and compared to two other M. yongonense Type II (MOTT-36Y and MOTT-H4Y), three M. intracellulare (ATCC 13950T, MOTT-02 and MOTT-64) and M. parascrofulaceum strains. Finally, ORFs that are specific for M. yongonense Type I strains, and not found in other comparative strains were identified, multiply aligned and visualized by Mauve multiple genome alignment system.3
Construction of Phylogenetic Trees and SimPlot Analysis
All identified ORFs from the genome of M. yongonense DSM 45126T were compared to other M. intraceullulare (ATCC 13950T, MOTT-02 and -64), M. yongonense Type I (MOTT-12 and -27), M. yongonense Type II (MOTT-36Y and -H4Y), M. avium and M. parascrofulaceum strains (Table 1). MutS proteins in the M. yongonense Type I strains (DSM 45126T, MOTT-12, and -27) were compared to MutS homologs from other bacteria or viruses (Lin et al., 2007; Ogata et al., 2011) and additional MutS4 family sequences which were retrieved from the GenBank database are listed in Supplementary Table S1. Amino acid or nucleotide sequences were aligned by the ClustalW method, and phylogenetic trees were constructed using the neighbor-joining method (Saitou and Nei, 1987) in MEGA 7.0 software (Kumar et al., 2016). In the case of genome-based phylogenetic tree, all the compared genome sequences were subjected to whole-genome multiple sequence alignments using the neighbor-joining method (Saitou and Nei, 1987) by the Mauve Multiple Genome Alignments software.3 A phylogenetic tree was generated using the aligned genome sequences and visualized by the TreeViewX program4. To visualize the putative recombination site in the genome of M. yongonense Type I strains, identified ORFs with high sequence similarities to M. parascrofulaceum were aligned with other M. intracellulare (ATCC 13950T, MOTT-02 and MOTT-64), M. yongonense Type II (MOTT-36Y and MOTT-H4Y), M. avium and M. parascrofulaceum strains using the MegAlign program in the DNASTAR package. The possibility of recombination event in the genome of M. yongonense Type I strains from M. parascrofulaceum was examined using SimPlot program6 and boot scanning analysis (Lole et al., 1999). The used parameters are as follows: Window: 200 bp, Step: 20 bp, GapStrip: on, Reps: 100, Kimura (2-parameter), T/t: 2.0, Neighbor-Joining.
Construction of Recombinant M. smegmatis Harboring DNA Mismatch Repair Genes from M. yongonense DSM 45126T
To generate recombinant M. smegmatis harboring DNA mismatch repair genes from M. yongonense DSM 45126T, approximately 3.8 kb of DNA fragment containing the DNA mismatch repair gene (3,069 bp) and a putative promoter (770 bp) was amplified using a primer set as follows: forward primer; 5′ – TTGCGGCCGCCGACCGAGTTGGCGTGG – 3′ and reverse primer; 5′ – GCTCTAGACCTTTAGACGGCAGTCAG – 3′. The underlined sequence of the forward and reverse primer indicates NotI and XbaI restriction enzyme sites, respectively. Genomic DNA for M. yongonense DSM 45126T was used as a template, and the DNA repair mismatch gene was amplified with i-MAXTM II DNA polymerase (iNtRON Biotechnology, Gyeonggi-do, Korea) and a primer set as described above. The PCR amplification condition was as follows: 5 min at 95°C; 40 cycles of 30 s at 95°C, 30 s at 68°C, and 3 min at 72°C; 5 min at 72°C. The PCR amplicon was digested with NotI and XbaI restriction enzymes and ligated into the pMV306 vector (Blokpoel et al., 2005; Murry et al., 2005; Andreu et al., 2010), which was also digested with the same enzyme.
The pMV306 vector comprising the DNA mismatch repair gene was electroporated into competent M. smegmatis mc2 155 using the GenePulser II electroporation apparatus (Bio-Rad, Hercules, CA, United States) (Snapper et al., 1990). Transformants were cultured in Middlebrook 7H9 broth (Difco, Detroit, MI, United States) containing 10% ADC (albumin-dextrose-catalase; Difco) for 3 h and plated onto Middlebrook 7H10 agar plate (supplemented with OADC, oleic acid-albumin-dextrose-catalase; Difco) containing 100 μg/ ml of kanamycin. To check the MutS4 expression in transformants, the mRNA was purified from recombinant M. smegmatis carrying MutS4 and empty vector, and RT-PCR was performed using One-step RT-PCR kit (iNtRON Biotechnology, Gyeonggi-do, Korea) with primer sets as follows: forward primer; 5′ – TCC AGG TCC GGC GCA AGG TGT T – 3′ and reverse primer; 5′ – CGC GGG CGG CTG ATG AAG AAG ATA – 3′.
Examination of the Frequency of Homologous Recombination in Recombinant M. smegmatis Harboring DNA Mismatch Repair Gene
A partial RNA polymerase β-subunit gene (rpoB) from M. tuberculosis was amplified by PCR using genomic DNA from M. tuberculosis that had mutations at codon 522 TCG (Ser) → TTG (Leu), designated with ‘317’ related with rifampin resistance (Mariam et al., 2004). The DNA fragment of ‘317’ was provided by the Korean Institute of Tuberculosis (KIT). The partial rpoB gene fragments (684 bp) were amplified with a primer set as follows: forward primer, 5′ – CGGGATCCCGTCGGTCGCTATAAGGTCAACA – 3′ and reverse primer, 5′ – CCCAAGCTTCTCGTCGGCGGTCAGGTA – 3′. The underlined sequence of the forward and reverse primers indicates cut sites for BamHI and HindIII, respectively. The PCR amplification conditions were as follows: 5 min at 95°C; 30 cycles of 30 s at 95°C, 30 s at 63°C, and 45 s at 72°C; 5 min at 72°C. The amplified fragment was cloned into the BamHI and HindIII sites of pSE100 (Guo et al., 2007) to construct pSE100-317.
Constructed pSE100-317 vector was then electroporated into each M. smegmatis strain transformed with the present DNA mismatch repair gene or an empty vector pMV306, as described above. The transformants were then plated onto 7H10 agar plates with 50 μg/ml of hygromycin and incubated for 72 h at 37°C. After that, colonies were picked and suspended in 7H9 broth with 50 μg/ml of hygromycin and cultured for 72 h at 37°C. The cultured bacterial sample was adjusted to 0.2 OD (optical density at 600 nm) and plated onto the 7H10 agar plate with 100 μg/ml of rifampin. Colonies grown on the rifampin 7H10 agar plates were judged as potential recombinants. The number of colonies was counted after 3 days incubation and the rpoB gene was amplified by PCR using the primers 7940F (forward, 5′ – TCAAGGAGAAGCGCTACGACC – 3′) and MR (reverse, 5′ – TCGATCGGGCACATCCGG – 3′) from the randomly selected colonies. PCR amplicons were then sequenced using the 7940F and MR primers. Recombination-emerging colonies were identified by having M. tuberculosis-specific SNPs, especially rifampin resistant related SNPs in their rpoB sequences. Also, the lengths of the recombined rpoB gene was calculated by determination the boundary between the M. smegmatis- and M. tuberculosis-specific SNP.
Author Contributions
By-JK and B-RK performed the experiments and sequence analyses. Bu-JK and Y-HK designed and interpreted the experiments. Bu-JK wrote the manuscript.
Funding
This research was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Education (Grant No. NRF-2016R1A2B4011847).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
B-RK received a scholarship from the BK21-plus education program provided by the National Research Foundation of Korea.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02578/full#supplementary-material
Footnotes
- ^ http://www.pathogenomics.sfu.ca/islandviewer/browse/
- ^ http://www.pathogenomics.sfu.ca/islandviewer/browse/
- ^ http://darlinglab.org/mauve/mauve.html
- ^ http://darwin.zoology.gla.ac.uk/~rpage/treeviewx/
References
Achtman, M., and Wagner, M. (2008). Microbial diversity and the genetic nature of microbial species. Nat. Rev. Microbiol. 6, 431–440. doi: 10.1038/nrmicro1872
Andreu, N., Zelmer, A., Fletcher, T., Elkington, P. T., Ward, T. H., Ripoll, J., et al. (2010). Optimisation of bioluminescent reporters for use with Mycobacteria. PLOS ONE 5:e10777. doi: 10.1371/journal.pone.0010777
Arruda, S., Bomfim, G., Knights, R., Huima-Byron, T., and Riley, L. W. (1993). Cloning of an M. tuberculosis DNA fragment associated with entry and survival inside cells. Science 261, 1454–1457. doi: 10.1126/science.8367727
Banasik, M., and Sachadyn, P. (2014). Conserved motifs of MutL proteins. Mutat. Res. 769, 69–79. doi: 10.1016/j.mrfmmm.2014.07.006
Bertelli, C., Laird, M. R., Williams, K. P., Lau, B. Y., Hoad, G., Winsor, G. L., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. doi: 10.1093/nar/gkx343 [Epub ahead of print].
Blokpoel, M. C., Murphy, H. N., O’Toole, R., Wiles, S., Runn, E. S., Stewart, G. R., et al. (2005). Tetracycline-inducible gene regulation in mycobacteria. Nucleic Acids Res. 33:e22. doi: 10.1093/nar/gni023
Castaneda-Garcia, A., Prieto, A. I., Rodriguez-Beltran, J., Alonso, N., Cantillon, D., Costas, C., et al. (2017). A non-canonical mismatch repair pathway in prokaryotes. Nat. Commun. 8:14246. doi: 10.1038/ncomms14246
Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., et al. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. doi: 10.1038/31159
Cole, S. T., Eiglmeier, K., Parkhill, J., James, K. D., Thomson, N. R., Wheeler, P. R., et al. (2001). Massive gene decay in the leprosy bacillus. Nature 409, 1007–1011. doi: 10.1038/35059006
Didelot, X., and Maiden, M. C. (2010). Impact of recombination on bacterial evolution. Trends Microbiol. 18, 315–322. doi: 10.1016/j.tim.2010.04.002
Didelot, X., Meric, G., Falush, D., and Darling, A. E. (2012). Impact of homologous and non-homologous recombination in the genomic evolution of Escherichia coli. BMC Genomics 13:256. doi: 10.1186/1471-2164-13-256
Forrellad, M. A., Klepp, L. I., Gioffre, A., Garcia, J. S. Y., Morbidoni, H. R., Santangelo, M. D., et al. (2013). Virulence factors of the Mycobacterium tuberculosis complex. Virulence 4, 3–66. doi: 10.4161/viru.22329
Fraser, C., Alm, E. J., Polz, M. F., Spratt, B. G., and Hanage, W. P. (2009). The bacterial species challenge: making sense of genetic and ecological diversity. Science 323, 741–746. doi: 10.1126/science.1159388
Guo, X. V., Monteleone, M., Klotzsche, M., Kamionka, A., Hillen, W., Braunstein, M., et al. (2007). Silencing essential protein secretion in Mycobacterium smegmatis by using tetracycline repressors. J. Bacteriol. 189, 4614–4623. doi: 10.1128/Jb.00216-07
Harfe, B. D., and Jinks-Robertson, S. (2000). DNA mismatch repair and genetic instability. Annu. Rev. Genet. 34, 359–399. doi: 10.1146/annurev.genet.34.1.359
Hsiao, W., Wan, I., Jones, S. J., and Brinkman, F. S. L. (2003). IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics 19, 418–420. doi: 10.1093/bioinformatics/btg004
Iyer, R. R., Pluciennik, A., Burdett, V., and Modrich, P. L. (2006). DNA mismatch repair: functions and mechanisms. Chem. Rev. 106, 302–323. doi: 10.1021/cr0404794
Kim, B. J., Choi, B. S., Choi, I. Y., Lee, J. H., Chun, J., Hong, S. H., et al. (2012a). Complete genome sequence of Mycobacterium intracellulare clinical strain MOTT-36Y, belonging to the INT5 genotype. J. Bacteriol. 194, 4141–4142. doi: 10.1128/JB.00752-12
Kim, B. J., Choi, B. S., Lim, J. S., Choi, I. Y., and Kook, Y. H. (2012b). Complete genome sequence of Mycobacterium intracellulare clinical strain MOTT-64, belonging to the INT1 genotype. J. Bacteriol. 194:3268. doi: 10.1128/JB.00471-12
Kim, B. J., Choi, B. S., Lim, J. S., Choi, I. Y., Lee, J. H., Chun, J., et al. (2012c). Complete genome sequence of Mycobacterium intracellulare strain ATCC 13950(T). J. Bacteriol. 194:2750. doi: 10.1128/JB.00295-12
Kim, B. J., Choi, B. S., Lim, J. S., Choi, I. Y., Lee, J. H., Chun, J., et al. (2012d). Complete genome sequence of Mycobacterium intracellulare clinical strain MOTT-02. J. Bacteriol. 194:2771. doi: 10.1128/JB.00365-12
Kim, B. J., Hong, S. H., and Kook, Y. H. (2013a). Molecular evidence of lateral gene transfer in rpoB gene of Mycobacterium yongonense strains via multilocus sequence analysis. PLOS ONE 8:e51846. doi: 10.1371/journal.pone.0051846
Kim, B. J., Kim, B. R., Lee, S. Y., Seok, S. H., and Kook, Y. H. (2013b). Whole-genome sequence of a novel species, Mycobacterium yongonense DSM 45126T. Genome Announc. 1:e00604-13. doi: 10.1128/genomeA.00604-13
Kim, B. J., Math, R. K., Jeon, C. O., Yu, H. K., Park, Y. G., and Kook, Y. H. (2013c). Mycobacterium yongonense sp. nov., a slow-growing non-chromogenic species closely related to Mycobacterium intracellulare. Int. J. Syst. Evol. Microbiol. 63(Pt 1), 192–199. doi: 10.1099/ijs.0.037465-0
Kim, B. J., Kim, B. R., Lee, S. Y., Kim, G. N., and Kook, Y. H. (2016). Molecular taxonomic evidence for two distinct genotypes of Mycobacterium yongonense via genome-based phylogenetic analysis. PLOS ONE 11:e0152703. doi: 10.1371/journal.pone.0152703
Kim, B. J., Kim, G. N., Kim, B. R., Shim, T. S., Kook, Y. H., and Kim, B. J. (2017). Phylogenetic analysis of Mycobacterium massiliense strains having recombinant rpoB gene laterally transferred from Mycobacterium abscessus. PLOS ONE 12:e0179237. doi: 10.1371/journal.pone.0179237
Kim, B. J., Kim, K., Kim, B. R., and Kook, Y. H. (2015). Identification of ISMyo2, a novel insertion sequence element of IS21 family and its diagnostic potential for detection of Mycobacterium yongonense. BMC Genomics 16:794. doi: 10.1186/s12864-015-1978-2
Kumar, A., Bose, M., and Brahmachari, V. (2003). Analysis of expression profile of mammalian cell entry (mce) operons of Mycobacterium tuberculosis. Infect. Immun. 71, 6083–6087. doi: 10.1128/IAI.71.10.6083-6087.2003
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Langille, M. G. I., and Brinkman, F. S. L. (2009). IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics 25, 664–665. doi: 10.1093/bioinformatics/btp030
Langille, M. G. I., Hsiao, W. W. L., and Brinkman, F. S. L. (2008). Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinformatics 9:329. doi: 10.1186/1471-2105-9-329
Lee, H., Kim, B. J., Kim, K., Hong, S. H., and Kook, Y. H. (2013). Whole-genome sequence of Mycobacterium intracellulare clinical strain MOTT-H4Y, belonging to INT5 genotype. Genome Announc. 1:e00006-13. doi: 10.1128/genomeA.00006-13
Lin, Z., Nei, M., and Ma, H. (2007). The origins and early evolution of DNA mismatch repair genes–multiple horizontal gene transfers and co-evolution. Nucleic Acids Res. 35, 7591–7603. doi: 10.1093/nar/gkm921
Lole, K. S., Bollinger, R. C., Paranjape, R. S., Gadkari, D., Kulkarni, S. S., Novak, N. G., et al. (1999). Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73, 152–160.
Macheras, E., Roux, A. L., Bastian, S., Leao, S. C., Palaci, M., Sivadon-Tardy, V., et al. (2011). Multilocus sequence analysis and rpoB sequencing of Mycobacterium abscessus (sensu lato) strains. J. Clin. Microbiol. 49, 491–499. doi: 10.1128/JCM.01274-10
Mariam, D. H., Mengistu, Y., Hoffner, S. E., and Andersson, D. I. (2004). Effect of rpoB mutations conferring rifampin resistance on fitness of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 48, 1289–1294. doi: 10.1128/AAC.48.4.1289-1294.2004
Miller, J. L., Velmurugan, K., Cowan, M. J., and Briken, V. (2010). The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell apoptosis. PLOS Pathog. 6:e1000864. doi: 10.1371/journal.ppat.1000864
Mizrahi, V., and Andersen, S. J. (1998). DNA repair in Mycobacterium tuberculosis. What have we learnt from the genome sequence? Mol. Microbiol. 29, 1331–1339. doi: 10.1046/j.1365-2958.1998.01038.x
Modrich, P., and Lahue, R. (1996). Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 65, 101–133. doi: 10.1146/annurev.bi.65.070196.000533
Murry, J., Sassetti, C. M., Moreira, J., Lane, J., and Rubin, E. J. (2005). A new site-specific integration system for mycobacteria. Tuberculosis 85, 317–323. doi: 10.1016/j.tube.2005.08.016
Ochman, H., Lawrence, J. G., and Groisman, E. A. (2000). Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304. doi: 10.1038/35012500
Ogata, H., Ray, J., Toyoda, K., Sandaa, R. A., Nagasaki, K., Bratbak, G., et al. (2011). Two new subfamilies of DNA mismatch repair proteins (MutS) specifically abundant in the marine environment. ISME J. 5, 1143–1151. doi: 10.1038/ismej.2010.210
Reenan, R. A., and Kolodner, R. D. (1992). Characterization of insertion mutations in the Saccharomyces cerevisiae MSH1 and MSH2 genes: evidence for separate mitochondrial and nuclear functions. Genetics 132, 975–985.
Sachadyn, P. (2010). Conservation and diversity of MutS proteins. Mutat. Res. 694, 20–30. doi: 10.1016/j.mrfmmm.2010.08.009
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Sapriel, G., Konjek, J., Orgeur, M., Bouri, L., Frezal, L., Roux, A. L., et al. (2016). Genome-wide mosaicism within Mycobacterium abscessus: evolutionary and epidemiological implications. BMC Genomics 17:118. doi: 10.1186/s12864-016-2448-1
Sheppard, S. K., McCarthy, N. D., Falush, D., and Maiden, M. C. (2008). Convergence of Campylobacter species: implications for bacterial evolution. Science 320, 237–239. doi: 10.1126/science.1155532
Snapper, S. B., Melton, R. E., Mustafa, S., Kieser, T., and Jacobs, W. R. Jr. (1990). Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 4, 1911–1919. doi: 10.1111/j.1365-2958.1990.tb02040.x
Surtees, J. A., Argueso, J. L., and Alani, E. (2004). Mismatch repair proteins: key regulators of genetic recombination. Cytogenet. Genome Res. 107, 146–159. doi: 10.1159/000080593
Umar, A., and Kunkel, T. A. (1996). DNA-replication fidelity, mismatch repair and genome instability in cancer cells. Eur. J. Biochem. 238, 297–307. doi: 10.1111/j.1432-1033.1996.0297z.x
Velmurugan, K., Chen, B., Miller, J. L., Azogue, S., Gurses, S., Hsu, T., et al. (2007). Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. PLOS Pathog. 3:e110. doi: 10.1371/journal.ppat.0030110
Vulic, M., Dionisio, F., Taddei, F., and Radman, M. (1997). Molecular keys to speciation: DNA polymorphism and the control of genetic exchange in enterobacteria. Proc. Natl. Acad. Sci. U.S.A. 94, 9763–9767. doi: 10.1073/pnas.94.18.9763
Keywords: Mycobacterium yongonense, lateral gene transfer, DNA mismatch repair gene, MutS4, homologous recombination
Citation: Kim B-J, Kim B-R, Kook Y-H and Kim B-J (2017) Role of the DNA Mismatch Repair Gene MutS4 in Driving the Evolution of Mycobacterium yongonense Type I via Homologous Recombination. Front. Microbiol. 8:2578. doi: 10.3389/fmicb.2017.02578
Received: 12 September 2017; Accepted: 11 December 2017;
Published: 20 December 2017.
Edited by:
Peng Luo, South China Sea Institute of Oceanology (CAS), ChinaReviewed by:
Tanya Parish, Infectious Disease Research Institute, United StatesUlisses Padua Pereira, Universidade Estadual de Londrina, Brazil
Copyright © 2017 Kim, Kim, Kook and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bum-Joon Kim, kbumjoon@snu.ac.kr