 Zhi-Luo Deng
Zhi-Luo Deng Helena Sztajer1
Helena Sztajer1 Michael Jarek
Michael Jarek Sabin Bhuju
Sabin Bhuju Irene Wagner-Döbler
Irene Wagner-Döbler- 1Research Group Microbial Communication, Department of Molecular Infection Biology, Helmholtz Centre for Infection Research, Braunschweig, Germany
- 2Genome Analytics, Helmholtz Centre for Infection Research, Braunschweig, Germany
Periodontitis is a worldwide prevalent oral disease which results from dysbiosis of the periodontal microbiome. Some of the most active microbial players, e.g., Porphyromonas gingivalis, Treponema denticola, and Fusobacterium nucleatum, have extensively been studied in the laboratory, but it is unclear to which extend these findings can be transferred to in vivo conditions. Here we show that the transcriptional profiles of P. gingivalis, T. denticola, and F. nucleatum in the periodontal niche are distinct from those in single laboratory culture and exhibit functional similarities. GO (gene ontology) term enrichment analysis showed up-regulation of transporters, pathogenicity related traits and hemin/heme uptake mechanisms for all three species in vivo. Differential gene expression analysis revealed that cysteine proteases, transporters and hemin/heme-binding proteins were highly up-regulated in the periodontal niche, while genes involved in DNA modification were down-regulated. The data suggest strong interactions between those three species regarding protein degradation, iron up-take, and mobility in vivo, explaining their enhanced synergistic pathogenicity. We discovered a strikingly high frequency of Single Nucleotide Polymorphisms (SNPs) in vivo. For F. nucleatum we discovered a total of 127,729 SNPs in periodontal niche transcripts, which were found in similar frequency in health and disease and covered the entire genome, suggesting continuous evolution in the host. We conclude that metabolic interactions shape gene expression in vivo. Great caution is required when inferring pathogenicity of microbes from laboratory data, and microdiversity is an important adaptive trait of natural communities.
Introduction
Periodontitis is a chronic inflammation of the periodontium leading to destruction of the alveolar bone and finally tooth loss, for which it is the most important reason worldwide (Darveau, 2010). Periodontitis additionally increases the risk for systemic diseases like artherovascular disease, diabetes, rheumatoid arthritis, and certain forms of cancer (Genco and Van Dyke, 2010; Lundberg et al., 2010; Lalla and Papapanou, 2011; Barton, 2017; Michaud et al., 2017). Although periodontitis is the most prevalent infectious disease and dental plaque the most thoroughly studied microbiota of humans, its etiology is still unsolved (Darveau, 2010). Current understanding implies that the periodontal pocket microbiota form a polymicrobial biofilm which continuously interacts with the human host cells, leading to a symbiotic relationship in health; in disease, the microbiota shift to a dysbiotic stage and disrupt host homeostasis by evading immune responses and triggering inflammatory reactions (Kilian et al., 2016; Van der Velden, 2017). Commensal bacteria like Prevotella nigrescens, which are present in both health and disease, turn into additional pathogens in the dysbiotic community (Szafranski et al., 2015a).
Three periodontal pathogens are consistently found in periodontal pockets of individuals suffering from chronic periodontitis, namely Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia and were named “red complex pathogens” (Socransky et al., 1998). P. gingivalis, which is a minor constituent of the periodontal community in health, belongs to the phylum Bacteroidetes and is considered a “keystone pathogen” that can initiate the shift towards a dysbiotic microbial community by evading host defense, triggering an inflammatory response, and inhibiting IL-8 synthesis, which delays the recruitment of neutrophils; as a result, colonization of the periodontium by commensal bacteria is facilitated, additional nutrients become available (degraded protein, hemin/heme) and the community shifts towards dysbiosis (Hajishengallis et al., 2012). Among the traits of P. gingivalis important for these mechanisms are gingipains (arginine-specific cysteine proteases) (Imamura, 2003), an atypical lipopolysaccharide which is a potent antagonist of TLR4 (Darveau, 2010), serine phosphatases (SerB) (Hajishengallis et al., 2012; How et al., 2016), peptidyl-arginine deiminase (PPAD) (Maresz et al., 2013) and neuraminidase (Amano et al., 2014).
The second “red-complex” pathogen, T. denticola, belongs into the phylum Spirochaetes (Dashper et al., 2011), and can become extremely abundant in periodontitis (up to 50% of the polymicrobial plaque) while it is almost absent in health (Kilian et al., 2016). T. denticola is the only motile member of the “red-complex” pathogens and can invade host cells by means of periplasmic flagella. A flagella protein of T. denticola was identified as a highly predictive functional biomarker for periodontitis (Szafranski et al., 2015a). T. denticola possess many virulence traits, including adhesins, proteases (dentilisin, dentipain), pore forming toxins (dentilisin, cytalysin), proteins for immune activation (Msp, peptidoglycan, lipoprotein) and immune evasion (resistance to defensins, TLR inhibition, Msp) and metal transport (haemin binding protein, lactoferrin binding protein) (Dashper et al., 2011; Visser and Ellen, 2011).
The fact that P. gingivalis and T. denticola often co-occur in subgingival plaque in periodontitis suggests that they might interact and form a symbiotic relationship. By comparing gene expression of P. gingivalis and T. denticola in single culture with that in co-culture it was shown that P. gingivalis might provide thiamin and glycine to T. denticola resulting in improved growth of both (Tan et al., 2014).
Fusobacterium nucleatum is one of the most abundant species in the oral cavity in both diseased and healthy individuals (Han, 2015). It is an anaerobic bacterium which has a Gram-negative cell wall and belongs to the phylum Fusobacteria (Bolstad et al., 1996). F. nucleatum is a key constituent in the periodontal microbiota due to its abundance in periodontal plaque biofilms and its capability to coaggregate with other species (Kapatral et al., 2002; Signat et al., 2011). Animal studies support a causative role of F. nucleatum in periodontal infections (Han, 2015). Additionally, F. nucleatum is associated with and potentially causative for a wide spectrum of conditions, including adverse pregnancy outcomes, colorectal cancer, inflammatory bowel disease, and cardiovascular disease, and has been isolated from all body sites, including the placenta (Han, 2015). In colorectal cancer, it causes resistance to chemotherapy (Yu et al., 2017). The most important virulence mechanism of F. nucleatum mediating such diverse pathogenicities is the adhesin FadA, which binds to cadherins, the cell-junction molecules, and in such a way it can directly invade host cells and the pericellular space (Rubinstein et al., 2013; Han, 2015). We previously showed that in the periodontal pocket, genes for the synthesis of butyrate, a cytotoxic short-chain fatty acid, are expressed by F. nucleatum both in health and disease; in chronic periodontitis, however, additional taxa and additional pathways for synthesis of butyrate were recruited (Szafranski et al., 2015a).
Microbiological investigations rely on pure cultures of the species in question which are grown on artificial media at defined cultivation conditions, thus they reveal the potential of a microorganism, but not its actual behavior in vivo. In the periodontal pocket of humans, bacteria encounter an entirely different, highly complex environment, where they compete and interact with hundreds of co-occurring bacterial species and are under continuous attack by the immune system of the human host (Kilian et al., 2016). Large differences in gene expression in vivo compared to laboratory culture are therefore expected, but until recently, it was technically impossible to exactly determine them. Using next generation sequencing (NGS) the transcriptome of all members of a sample can now be profiled, and such metatranscriptome data can be interrogated for the behaviors of microbes of interest in vivo.
The available metatranscriptome studies of the periodontal niche compared gene expression in health and periodontitis. Duran-Pinedo et al. (2014) discovered that commensals expressed virulence factors in disease and identified GO terms associated with disease progression (Yost et al., 2015). Jorth et al. (2014) compared microbial communities in healthy and diseased periodontal pockets in the same individual; they suggested that although the species composition in periodontal pockets varies widely, the metabolic networks operating in disease are conserved. We had previously investigated the taxonomic composition of periodontal pocket bacterial communities in health and disease using 16S rRNA gene sequencing (Szafranski et al., 2015b). The metatranscriptome analysis of those samples resulted in functional biomarkers and showed that Prevotella nigrescens turns into an additional pathogen in disease (Szafranski et al., 2015a). Those metatranscriptome data were then analyzed further to identify KEGG pathway enrichment in disease, and to study the activities of Archaea, virus and protozoa as well as the human host (Deng et al., 2017). Here we now focused on transcripts from our three key periodontal pathogens and extracted them from the metatranscriptomes. To obtain an understanding of the response of P. gingivalis, T. denticola, and F. nucleatum to in vivo conditions we compared their gene expression in single culture on laboratory media with that in human periodontal pockets in chronic periodontitis. Our data reveal large similarities in the functional adaptations to in vivo conditions for the three pathogens; moreover, they suggest strong interactions between them with respect to protein degradation, iron uptake and mobility, which explain their synergistic pathogenicity. Unexpectedly, we found an enormous microdiversity of all three pathogens in vivo in comparison to the laboratory culture of a clonal isolate.
Materials and Methods
Bacterial Strains and Cultivation Conditions
The in vivo data were derived from periodontal pocket metatranscriptomes of four individuals with periodontitis and 10 without and details regarding sampling and metatranscriptome sequencing have been described in our previous study (Szafranski et al., 2015a). Three single culture RNA-seq datasets from P. gingivalis were derived from a study from Hovik et al. (2012) in which P. gingivalis strain W83 was cultured in three different media. Those sequencing data contained about 15 million single end reads per sample with a length of 50 bp.
Treponema denticola ATCC 35405 was cultivated in DSM medium 9091 at 37°C in an anaerobic chamber (Don Whitley Scientific, Shipley, England) which provided an atmosphere of 80% N2, 10% H2, and 10% CO2. For RNA isolation, after 3 h of growth, 5 ml culture was harvested in the log phase, and after 8 h of growth, 5 ml was sampled in the stationary phase, respectively, from two replicate cultures.
Fusobacterium nucleatum ATCC 25586 was cultivated in modified (resazurin was omitted, since it interferes with OD measurements; vitamin K1 concentration was 10-fold increased; hemin concentration was 100-fold reduced) DSMZ medium 1041 at 37°C in an anaerobic chamber (Don Whitley Scientific, Shipley, England) which provided an atmosphere of 80% N2, 10% H2, and 10% CO2. For RNA isolation, 5 ml culture was withdrawn at log phase (3 h) and stationary phase (7 and 10 h) respectively from two replicate cultures.
RNA Extraction and Sequencing
Total RNA was isolated from 5 ml of bacterial culture using the RNeasy RNA Isolation Kit (Qiagen, Germany). mRNA enrichment was carried out with the Ribo-Zero rRNA Removal Kit according to the manufacturer’s instructions, using 1,2 μg of total RNA solved in 27 μl of nuclease free water (Qiagen, Germany). Enriched mRNA was further purified by ethanol precipitation and analyzed using capillary gel electrophoresis (Bioanalyser Agilent, Germany) to verify the depletion of 16S rRNA and 23S rRNA. Integrity of RNA was evaluated using a Bioanalyzer 2100 (Agilent, Germany). The mRNA enrichment yielded about 100 ng mRNA in 20 μl of water. Paired-end mRNA-seq strand specific libraries were prepared with the Script Seq Illumina Kit. Illumina HiSeq 2500 Sequencer (Illumina, Germany) was utilized to produce paired-end reads with a length of 110 base pairs.
Sequencing Data Preprocessing
Primers and sequencing adaptors were removed from raw sequencing data, followed by clipping the bases with a quality score below 20 from the reads to achieve cleaned reads with Fastq-Mcf (Aronesty, 2011). Reads shorter than 50 bp after trimming were eliminated. The cleaned transcriptome sequencing data of T. denticola and F. nucleatum in single laboratory culture were submitted to the European Nucleotide Archive database (ENA) and with BioProject ID: PRJEB23061. For the single culture transcriptomic data of P. gingivalis, which were downloaded from SRA and had a read length of 50 bp, reads shorter than 20 bp after trimming were discarded. Thereafter, the remaining rRNA reads were eliminated by SortMeRNA v2.0 (Kopylova et al., 2012).
Short Reads Mapping and Extraction of Species-Specific Reads
For determining the expression level of genes in vivo, we extracted P. gingivalis, T. denticola, and F. nucleatum reads from the metatranscriptomes (Szafranski et al., 2015a) using Kraken (Wood and Salzberg, 2014) and BBMAP (Bushnell, 2014). Kraken uses the K-mer strategy and the lowest common ancestor (LCA) algorithm to determine the taxon for a given read. The detailed workflow is as follows: first, the metatranscriptomic data were mapped onto a reference database consisting of prokaryote genomes (2786), virus genomes (4418), and the human genome (ver. GRCh38) downloaded from NCBI, in which the genomes of P. gingivalis, T. denticola, and F. nucleatum were included. Subsequently, the reads originating from P. gingivalis, T. denticola, and F. nucleatum based on the taxonomy were retrieved from the metatranscriptome using BBMAP. The extracted reads were then mapped against the corresponding reference genome using BWA with the BWA-MEM (Li, 2013) algorithm followed by read counting for each gene with FeatureCounts (Liao et al., 2014). For paired end reads with 110 bp, a seed length of 31 was applied, while for RNA-seq data (single end 50 bp) of single culture from P. gingivalis, the default seed length of 19 was adopted.
Differential Expression Analysis and GO Term Enrichment Analysis
The differential expression (DE) analysis was performed using edgeR (Robinson et al., 2010). The raw counts of the genes were normalized to make the in vivo and laboratory culture data comparable using the trimmed mean of M value (TMM) method which is offered by edgeR. After DE analysis, genes with FDR <=0.05 were considered as significantly differentially expressed. Those significantly up- and down-regulated genes were then used as the gene lists of interests for GO term enrichment analysis performed by topGO (Alexa and Rahnenfuhrer, 2010). Before the enrichment analysis, GO terms were assigned to each gene using UniProt (UniProt Consortium, 2014). As GO terms contain many redundant functions, we reduced the redundancy of the enriched GO terms and visualized the results using REVIGO (Supek et al., 2011).
Variants Calling
To investigate differences in the presence of variants on the transcriptome level between in vivo conditions and laboratory culture, we utilized SAMtools (Li et al., 2009), BCFtools (Narasimhan et al., 2016), VCFtools (Danecek et al., 2011), and BEDtools (Quinlan and Hall, 2010) to accomplish the variants calling based on the reads alignment files (SAM files). All sample files were pooled to identify the variants. The resulting variants were filtered in terms of the read depth, mapping quality and base quality of sequencing to achieve more confident variants (sites with mapping quality <20 or read depth >100 were marked as low-quality variants). The diversity of the SNPs per gene was calculated based on Shannon diversity defined as:
where n is the number of SNPs in a given gene, Ai is the frequency of the altered allele at the ith SNP locale relative to the reference sequence. Hence pi is the probability of the presence of a given SNP among all detected SNPs. For the calculation of the diversity, only the SNP with A/(A+R) ≥ 0.1 were taken into account.
Statistics
The PCoA was performed using R based on Bray–Curtis dissimilarity. DE analysis was conducted by edgeR with exact test, the P-values in the DE analysis were corrected to FDR with the “Benjamin Hochberg” method for multiple comparisons. The genes with FDR of DE analysis ≤0.05 were considered as differentially regulated. GO term enrichment analysis was carried out with Fisher’s exact test based on hypergeometric distribution using the R package topGO. The FDR of each GO term was also calculated and listed in the Supplementary Table S1. GO terms with enrichment analysis P-value ≤ 0.05 were taken as input for REVIGO.
Results
Study Design and Summary of Sequencing Data
The metatranscriptome samples (in vivo samples) have been described previously (Deng et al., 2017). They were derived from 14 individuals, four of which had been diagnosed with chronic periodontitis. Supplementary Figure S1 shows the relative abundance of the three-species studied here in these samples. F. nucleatum was present both in health and disease and comprised up to 25% of all reads. By contrast, T. denticola and P. gingivalis transcripts were barely detectable in health. Therefore, their mRNA reads were only extracted from the four patients with periodontitis. After quality control and rRNA removal we extracted 224,669 ± 321,128 putative mRNA reads per sample (14 samples) for F. nucleatum, 269,739 ± 161,109 reads per sample for P. gingivalis (four samples), and 72,791 ± 32,740 reads per sample for T. denticola (four samples).
For P. gingivalis laboratory culture data we utilized a published dataset based on cultivation on three different media (Hovik et al., 2012). F. nucleatum and T. denticola were cultivated on the media suggested by the culture collection under anaerobic conditions at 37°C. Samples were taken during exponential and stationary phase of growth with two replicas each and mRNA was sequenced as described (see methods for details). After quality control and rRNA removal, 55,385,310 ± 4,605,386 reads per sample were obtained for F. nucleatum, (four samples), 40,558,785 ± 6,092,565 for T. denticola (six samples), and 8,628,943 ± 1,567,710 for P. gingivalis (three samples). Details regarding the sequencing data can be seen in Supplementary Table S1 Sheets 1, 2.
Gene Expression of P. gingivalis in Periodontitis Compared to Laboratory Culture
PCoA (principal coordinate analysis) showed (Figure 1A), that the expression profile of P. gingivalis in vivo was completely distinct from that on all three laboratory media. We determined enrichment of GO terms based on differentially expressed genes. Functions involved in protein metabolism, translation, cell adhesion and pathogenesis were more active in vivo (Figure 1B and Supplementary Table S1 Sheet 5), while DNA methylation, thiamine biosynthesis and cell wall organization were up-regulated in laboratory culture (Figure 1C).
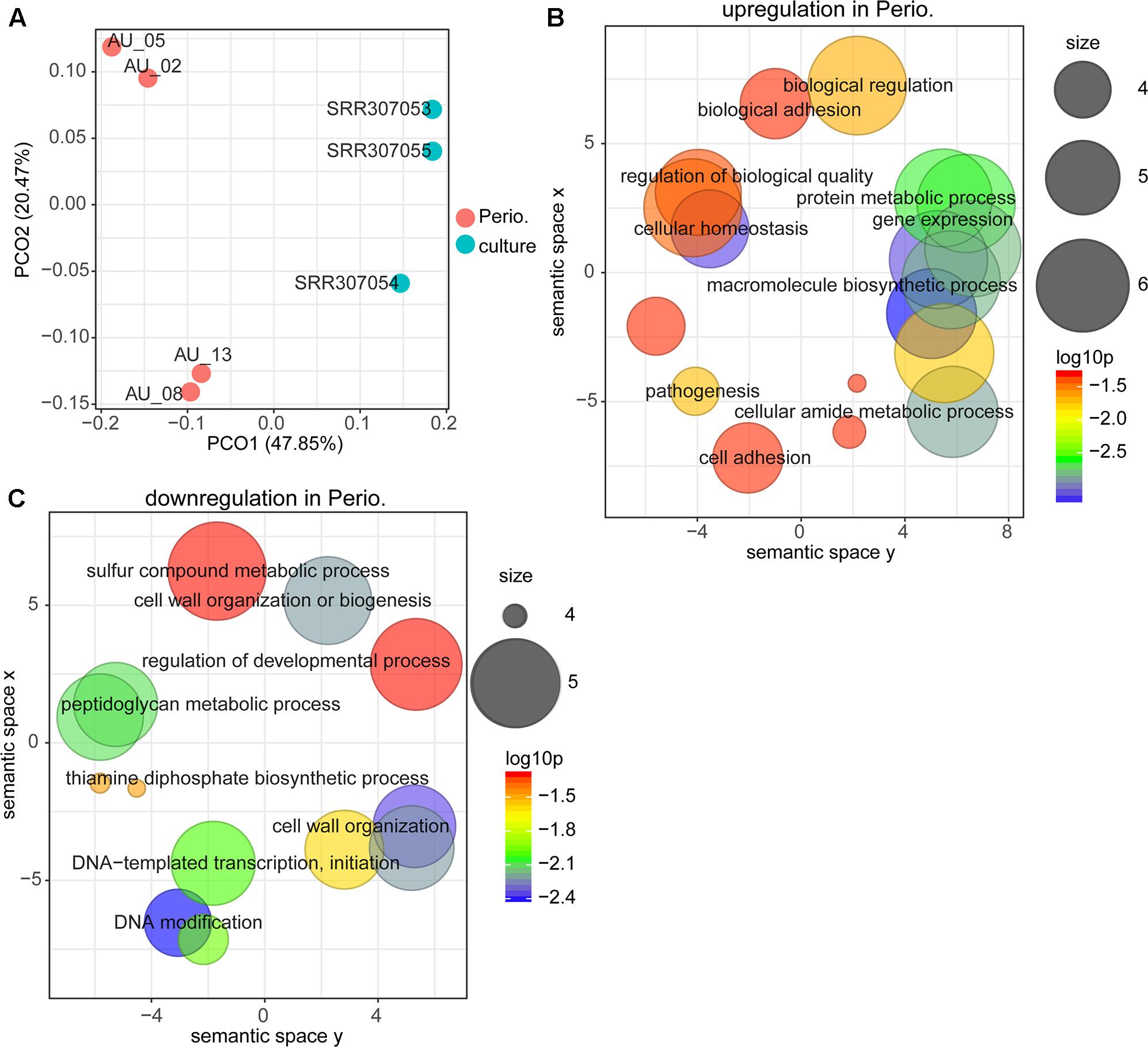
FIGURE 1. Comparison between gene expression in periodontitis and laboratory culture for Porphyromonas gingivalis. (A) Principal coordinates analysis (PCoA) of transcriptional profiles from four periodontal pocket samples (chronic periodontitis) and laboratory cultures on three different media. (B,C) Gene ontology (GO) terms up-regulated (B) and down-regulated (C) in periodontitis. All GO terms with enrichment P-value ≤ 0.05 were summarized and visualized by REVIGO (Supek et al., 2011), and the size of the bubble indicates the number of merged terms.
We then had a closer look at the differentially expressed genes. We observed that 125 genes were significantly up-regulated in vivo, whereas 166 were significantly down-regulated (Supplementary Table S1 Sheet 4 and Table 1). A fimbrilin gene and two cysteine protease genes were highly expressed in periodontitis. Among the most prominent upregulation (average fold change of 70) was the hmu gene cluster (PG1551-PG1556, hmuY, hmuR, hmuS, hmuT, hmuU, hmuV). A large variety of transporters were upregulated in vivo, including a multi-antimicrobial extrusion transporter (MATE), numerous ABC transporters and TonB-dependent receptors. The number of genes that were significantly down-regulated was even larger. Among them were genes encoding metabolic enzymes (e.g., glycerol dehydrogenase, glycosyl transferase, phosphoribosyltransferase), DNA modifying enzymes (transposases, integrases, restriction-modification system enzymes) and transcriptional regulators. Intriguingly, the clustered regularly interspaced short palindromic repeats (CRISPR) associated proteins were considerably down-regulated and the most strongly down-regulated gene in vivo was the surface protein PgaA.
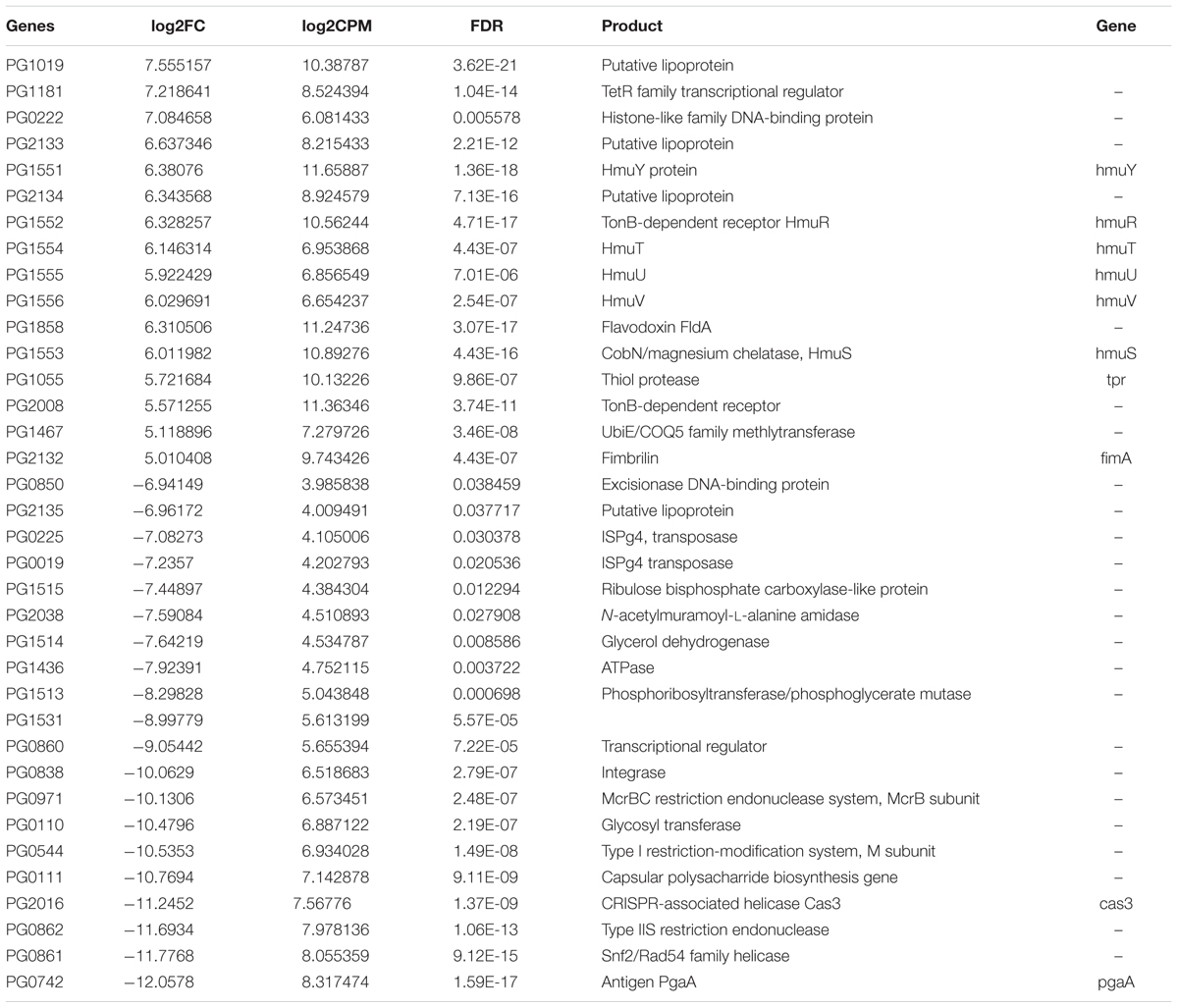
TABLE 1. Differentially expressed genes (|log2FC| ≥ 5) between periodontitis and laboratory culture in Porphyromonas gingivalis.
We then looked at those genes that were most highly expressed (log2 CPM ≥ 12) both in periodontitis and laboratory culture (Table 2). Hemagglutinin protein HagA, HagE, gingipain Kgp/HagD, receptor antigen RagA, and arginine-specific cysteine proteinase prtRII were among the most highly expressed genes under both conditions, confirming their prominent role for the pathogenesis of periodontitis.
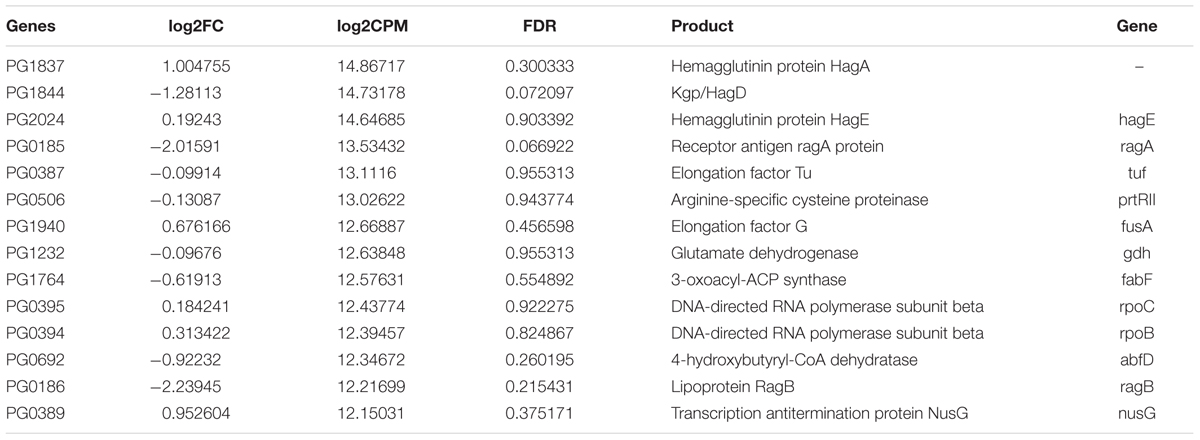
TABLE 2. Highly expressed genes (log2CPM ≥ 12) in both periodontitis and laboratory culture in P. gingivalis.
Gene Expression of T. denticola in Periodontitis Compared to Laboratory Culture
For T. denticola, too, the transcriptional profile in vivo was massively different from that in laboratory culture (Figure 2). The GO term enrichment analysis based on up-regulated or down-regulated genes suggests that gene expression, cell adhesion, protein metabolism and ion transporters were enriched in vivo, whereas GO terms of DNA methylation and cell motility were enriched in laboratory culture (Figures 2A,B and Supplementary Table S1 Sheet 8).
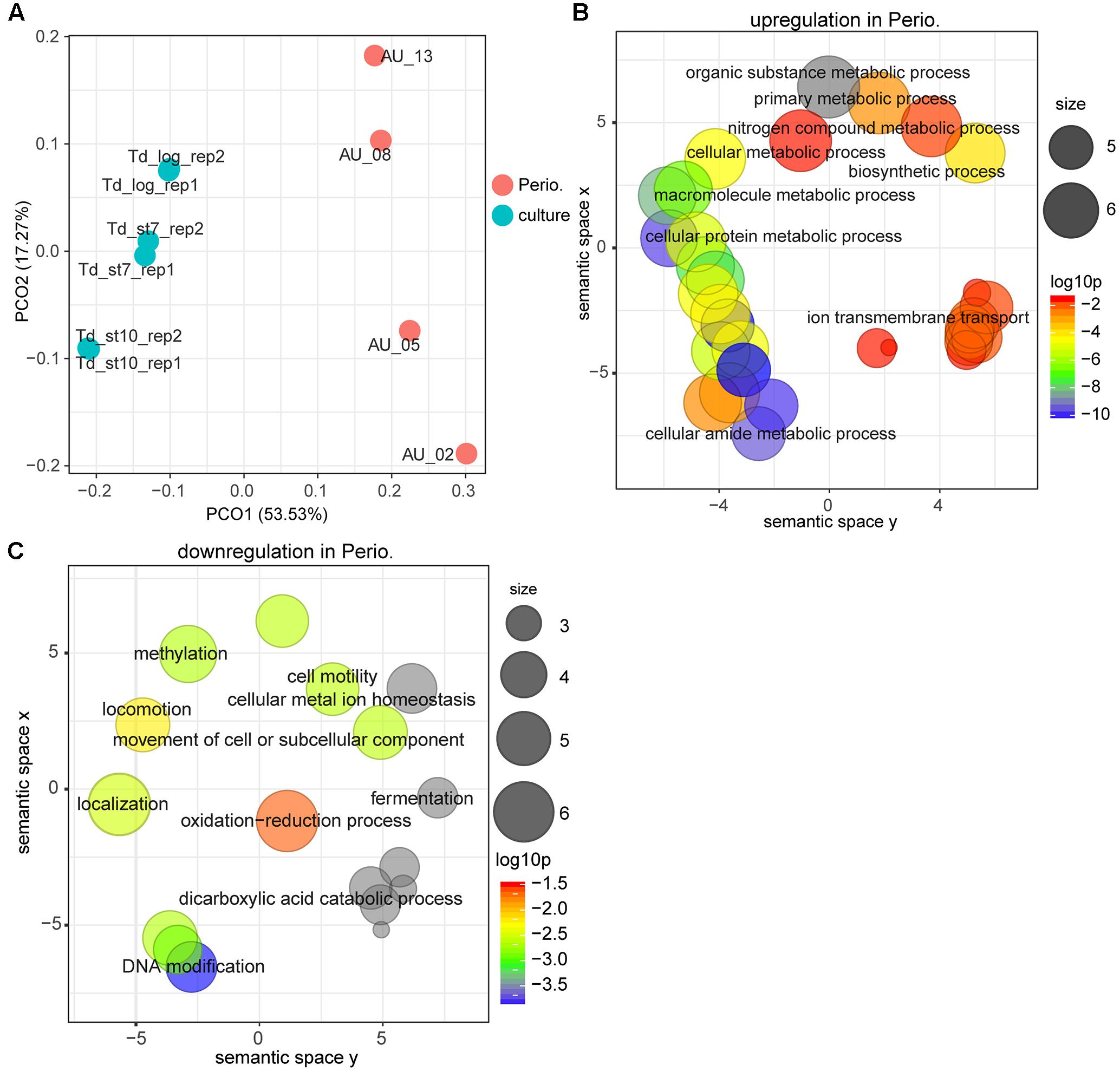
FIGURE 2. Comparison of the transcriptional profiles between periodontitis and laboratory culture for Treponema denticola. (A) PCoA of transcriptional profiles from four periodontal pocket samples (chronic periodontitis) and laboratory cultures at log or stationary phase. (B,C) GO terms up-regulated (B) and down-regulated (C) in periodontitis. All GO terms with enrichment P-value ≤ 0.05 were summarized and visualized by REVIGO, and the size of the bubble indicates the number of merged terms.
The differential expression analysis identified 257 genes that were up-regulated whereas 730 genes were down-regulated (Supplementary Table S1 Sheet 7 and Table 3). Thus, the number of genes up-regulated in periodontal pocket was two times less than that in laboratory culture, indicating strong selective pressure. As in P. gingivalis, ABC transporters, iron uptake transporters and oligopeptide/dipeptide transporter as well as the MATE transporters were the most strongly up-regulated genes in the periodontal pocket. Notably, a pathogen-specific surface antigen of T. denticola was considerably up-regulated in vivo indicating a strong interaction with the host immune system and with other bacteria. This gene was annotated based on a characterized homologous gene tpd in T. pallidum encoding Tp34 which is a 34 kDa membrane antigen (Deka et al., 2007). This protein is probably involved in iron acquisition via its propensity to bind lactoferrin (Deka et al., 2007).
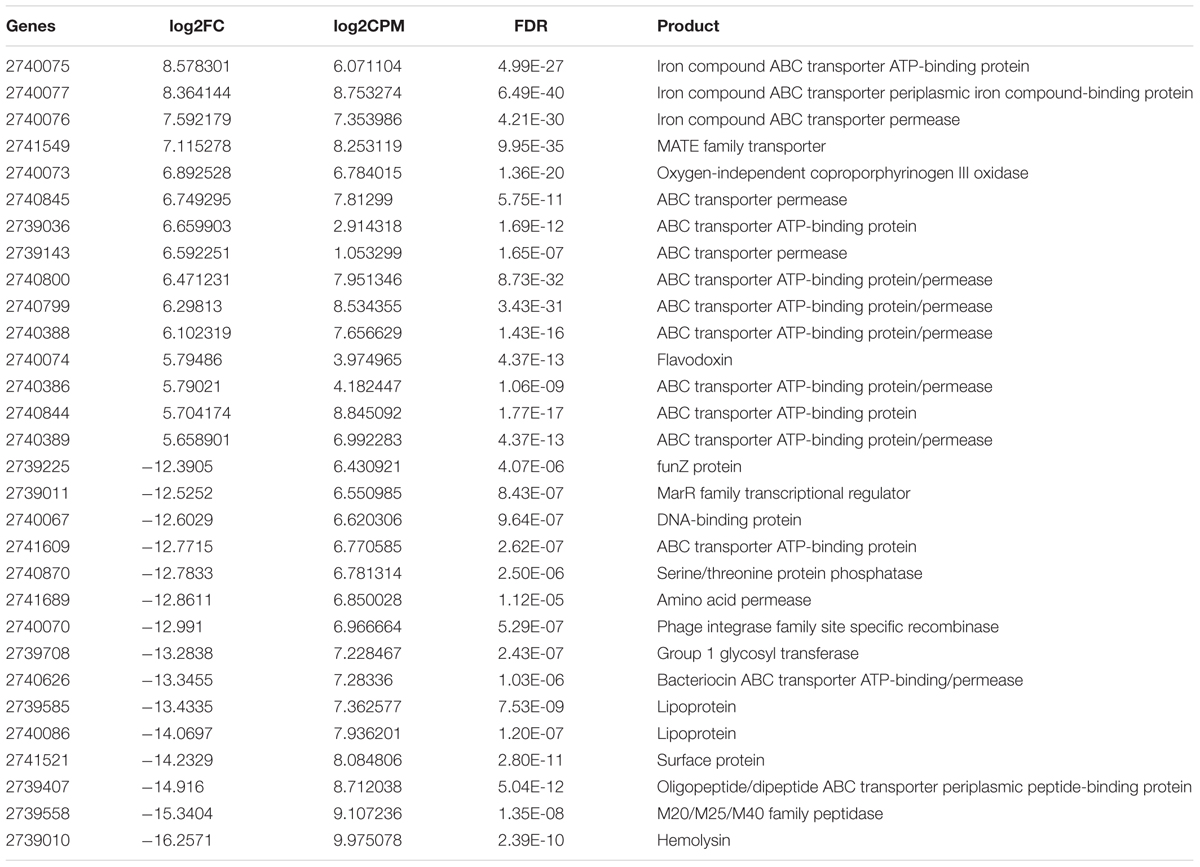
TABLE 3. Differentially expressed genes (15 most up- and down-regulated in periodontal pocket) between periodontitis and laboratory culture in Treponema denticola.
As in P. gingivalis the list of down-regulated genes comprised all cellular functions, but was much longer and more diverse. Five type I restriction modification system related genes, one type II restriction endonuclease gene and one CRISPR associated Cas1 protein gene were strongly down-regulated in vivo similar as in P. gingivalis. Phage related genes were also down-regulated (e.g., phage minor structural protein, phage integrase, phage terminase). The most strongly down-regulated gene in vivo was a hemolysin. This down-regulation was caused by the fact that its expression was not detectable in vivo using the sequencing depth of the metatranscriptome samples, while its expression in laboratory culture was high.
The most highly expressed genes of T. denticola both in vivo and in laboratory culture are shown in Table 4. Flagellar filament proteins, major outer sheath protein (Msp), glycine reductases and dentilisin were highly expressed under both conditions.
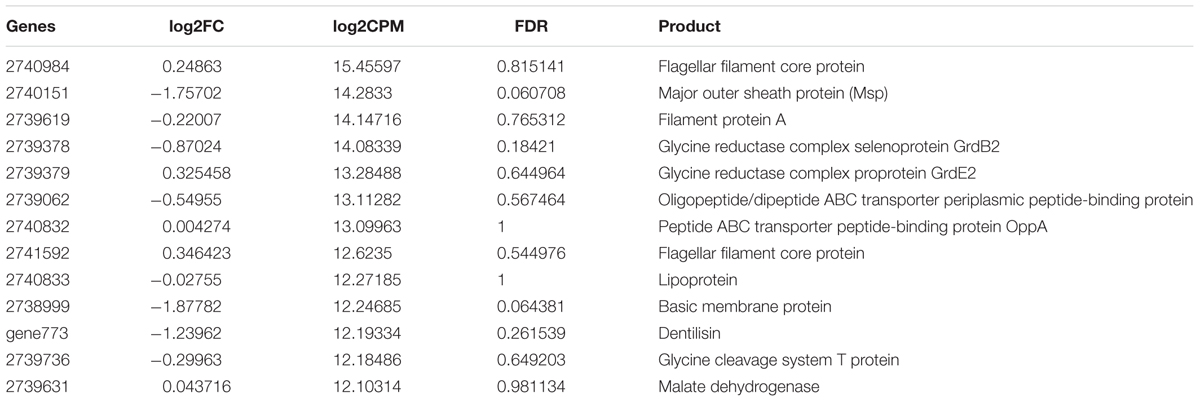
TABLE 4. Highly expressed genes (log2CPM ≥ 12) in both periodontitis and laboratory culture in T. denticola.
Gene Expression of F. nucleatum in Periodontitis and Health Compared to Laboratory Culture
Figure 3A shows that the transcriptional profile of F. nucleatum was highly diverse. Most in vivo transcriptional profiles clustered together and were distinct from those on laboratory media, irrespective of disease status. However, there were two samples of individuals with periodontitis which were intermediate between in vivo conditions and those in the laboratory, and one sample from a healthy individual which was completely distinct. In this individual, the abundance of F. nucleatum was very low. Accordingly, the overlap between those three conditions was small (Figure 3B). By using GO term enrichment analysis, we found that GO terms related to protein metabolism, pathogenesis and transport were up-regulated in periodontitis compared with laboratory culture (Figure 3C and Supplementary Table S1 Sheet 13). On the other hand, serine family amino acid metabolic process was down-regulated in periodontitis (Figure 3D).
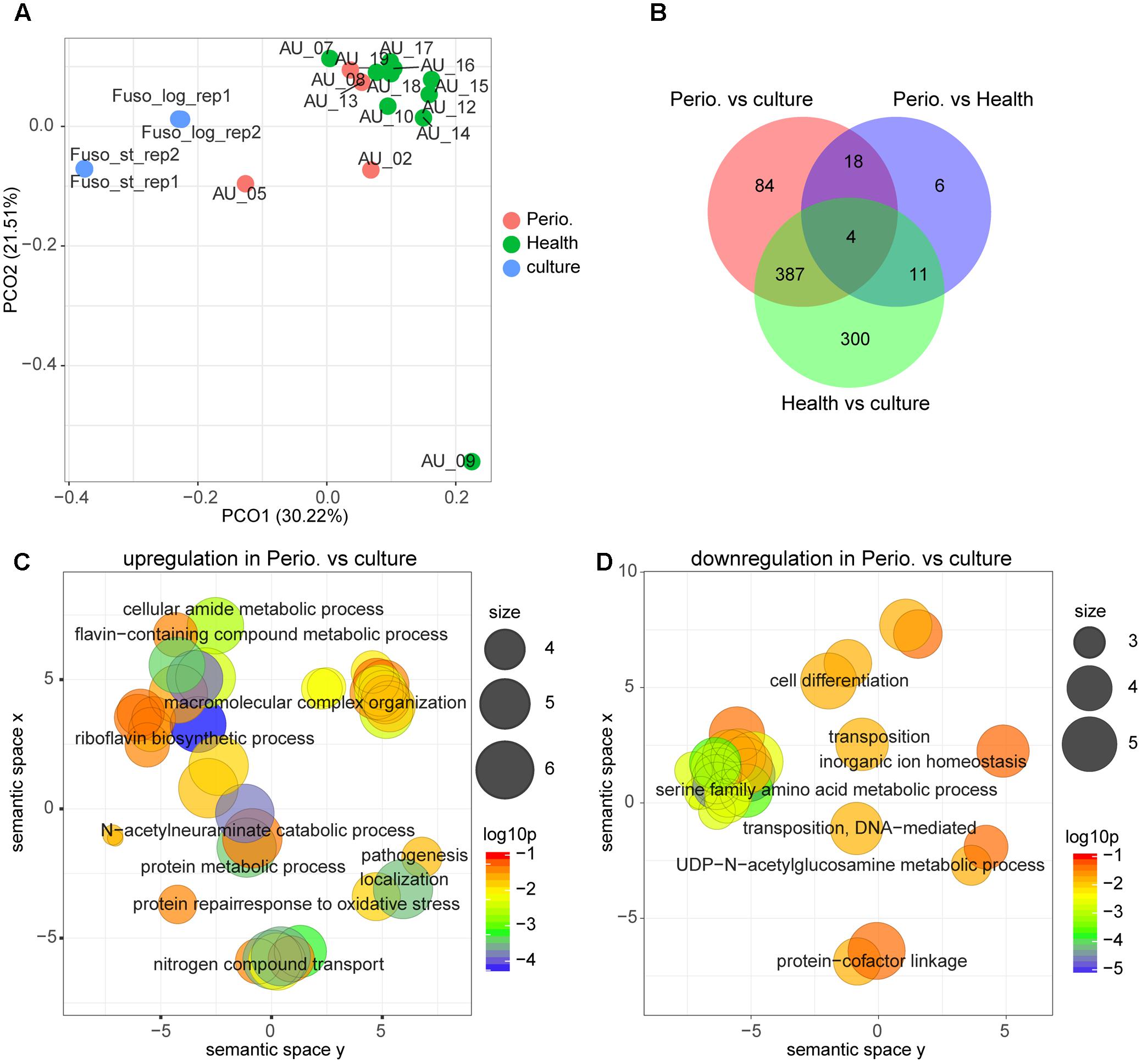
FIGURE 3. Comparison of the transcriptional profiles between periodontitis, health and laboratory culture for F. nucleatum. (A) PCoA of transcriptional profiles of Fusobacterium nucleatum in four periodontal pocket samples from patients with chronic periodontitis, 10 periodontal pocket samples from healthy individuals and four laboratory cultures at log or stationary phase. (B) Venn diagram showing the number of differentially expressed genes in each comparison. (C,D) GO terms up-regulated (C) and down-regulated (D) in periodontitis in comparison to laboratory culture. All GO terms with enrichment P-value ≤ 0.05 were summarized and visualized by REVIGO, and the size of the bubble indicates the number of merged terms.
When comparing gene expression in periodontitis with laboratory culture, we found 493 differentially expressed genes, of which 143 genes were up-regulated and 350 genes were down-regulated in periodontitis (Supplementary Table S1 Sheet 10 and Table 5). A hemin receptor was the most strongly up-regulated gene. Peptide transporter genes, Na+ driven multidrug efflux pump genes, and cell surface protein genes were highly expressed in periodontitis. On the other hand, numerous genes encoding methyltransferase, two lipopolysaccharide biosynthesis related genes and the gene encoding CRISPR-associated protein Cas5 were down-regulated in periodontitis compared with laboratory culture. The most strongly down-regulated gene in periodontitis was the LPS biosynthesis protein WbpG. The adhesin FadA was strongly expressed both in laboratory culture and in vivo (Supplementary Table S1 Sheet 10 and Table 6).
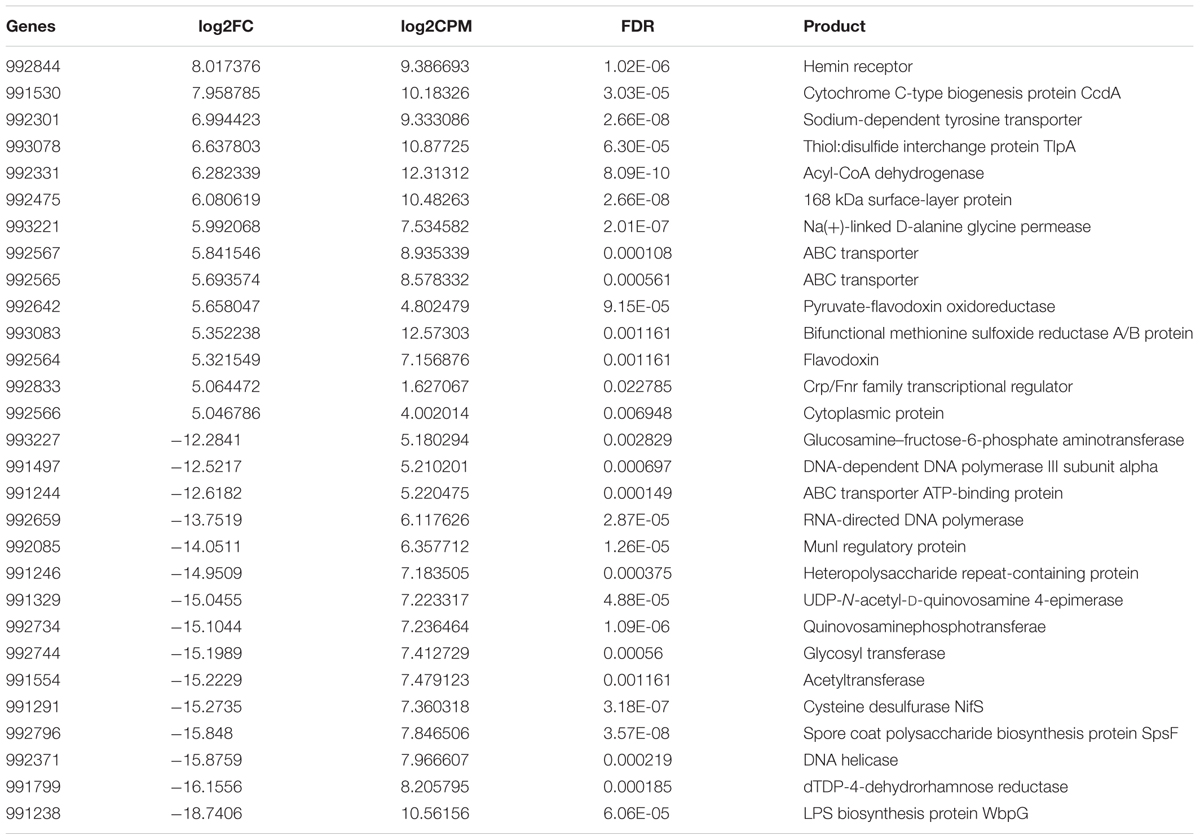
TABLE 5. Differentially expressed genes (15 most up- and down-regulated in periodontal pocket) between periodontitis and laboratory culture in Fusobacterium nucleatum.
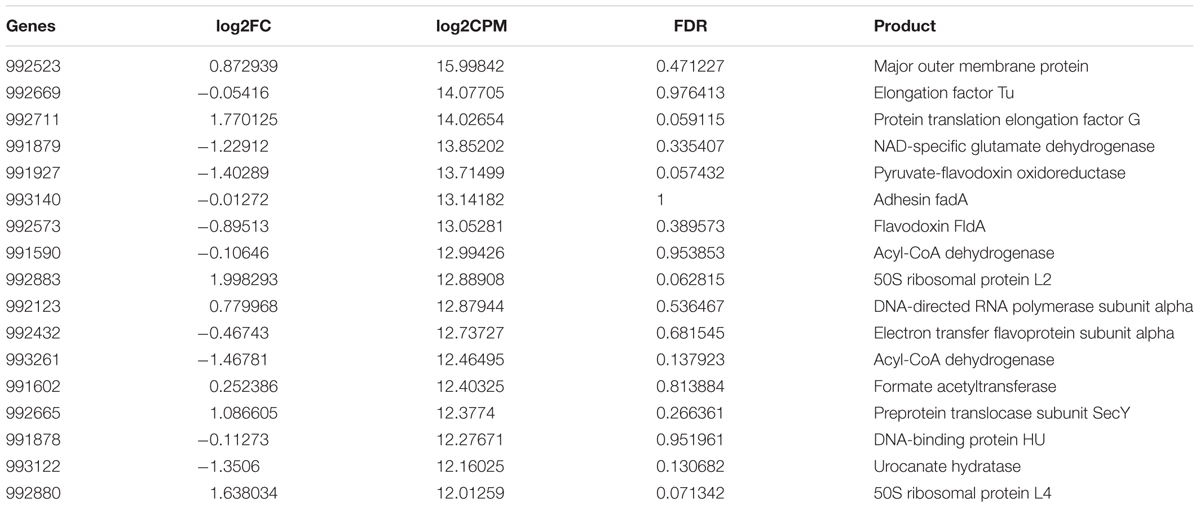
TABLE 6. Highly expressed genes (log2CPM ≥ 12) in both periodontitis and laboratory culture in F. nucleatum.
Microdiversity of Gene Expression in the Periodontal Community
To investigate the intra-species diversity of these three species in the periodontal communities, we identified SNPs (single nucleotide polymorphism) by performing variants calling analysis. For P. gingivalis we detected 29 variants of transcripts when matching the reads to the genome in single laboratory culture. However, 23,783 variants were discovered in the communities from patients with periodontitis. Only 16 variants were shared by both conditions (Supplementary Table S1 Sheet 15). Similar findings were observed for T. denticola. In laboratory culture, 314 variants were detected, whereas 62,145 variants were found in communities from patients with periodontitis, and only one was shared between both conditions (Supplementary Table S1 Sheet 15). The CRISPR-Cas genes and ABC transporters were relatively more variable in P. gingivalis and T. denticola than the other genes (Supplementary Table S1 Sheets 16, 17).
For F. nucleatum, the large number of transcripts allowed to map SNPs to the genome (Figure 4A). In health, the complete genome was covered by SNPs. A total of 127,729 SNPS were discovered in vivo, while only 35 were found in laboratory culture and a mere two shared between both conditions. Figure 4B shows variant diversity calculated as Shannon index of diversity per gene. This value was slightly larger in health, but this was due to the larger number of samples from healthy individuals (8 compared to 4). Iron uptake related genes of F. nucleatum were more variable in periodontitis (Supplementary Table S1 Sheet 18). The number of SNPs was correlated with the coverage of the gene in question, so that the genes with higher coverage tended to hold more variants for all three species (Supplementary Table S1 Sheets 16–18).
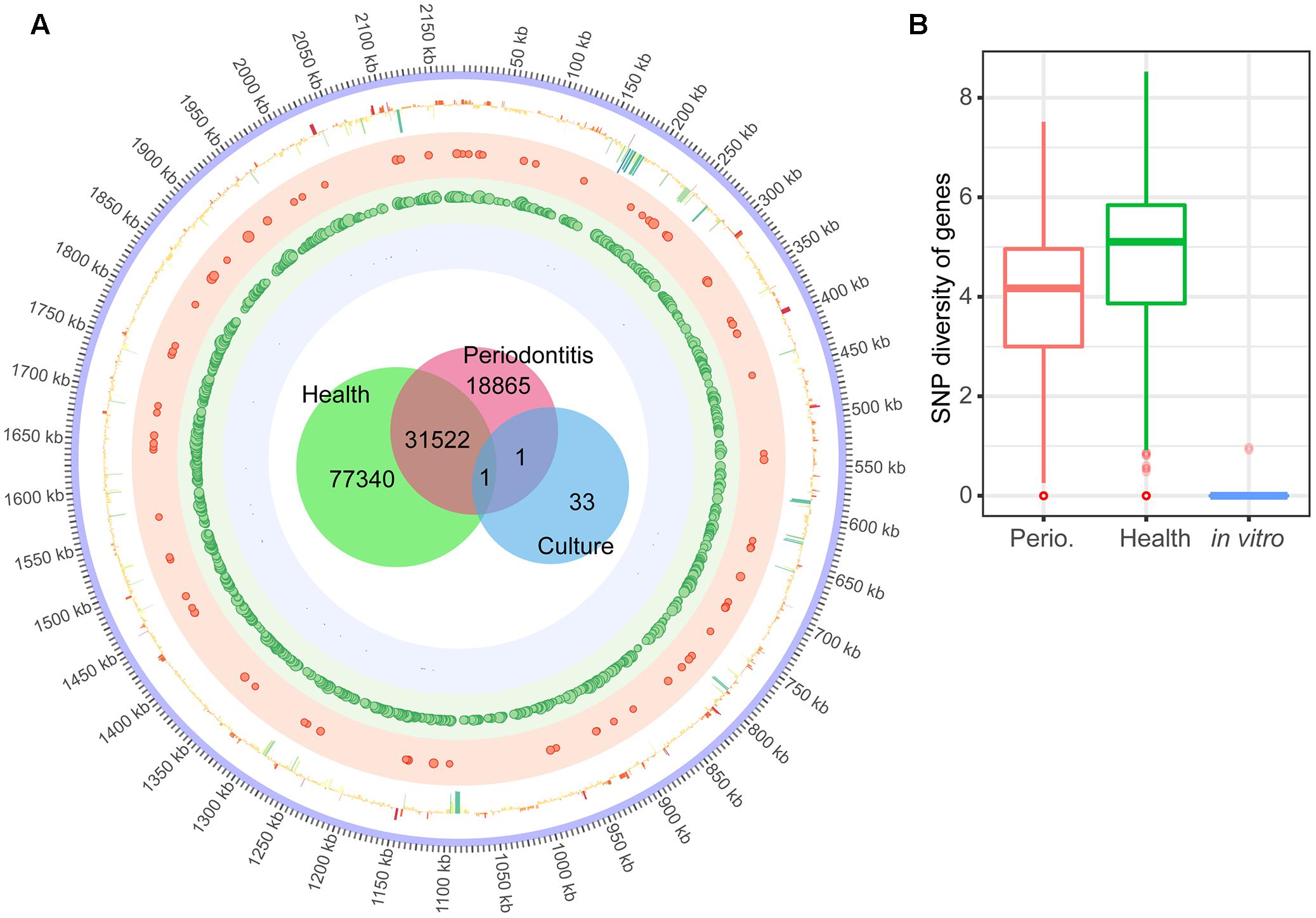
FIGURE 4. Single Nucleotide Polymorphisms (SNPs) in the transcriptome of F. nucleatum in the periodontal niche and in laboratory culture. (A) The innermost Venn diagram shows the number of SNPs of F. nucleatum transcripts in the periodontal community of individuals with periodontitis, without periodontitis, and laboratory culture. The circle shows the SNPs mapped to the genome of F. nucleatum. Size of the dots in the shaded red, green and blue circles illustrates the frequency (>50) of variants in a 1 kb window of F. nucleatum in periodontitis, health and laboratory culture, respectively. The outermost circle shows the log2FC of each gene in periodontitis compared to laboratory culture. (B) Frequency of SNPs per gene calculated as Shannon diversity (see Section “Materials and Methods” for details). The median, inter quantile range and outliers are shown in the boxplot.
Discussion
The data clearly show that gene expression of all three bacterial pathogens studied here was fundamentally different in periodontitis (in vivo) from that found on laboratory media. Among the factors that can be expected to shape in vivo gene expression are the type of carbon source used, the availability of micronutrients like iron and vitamins, competition, cross-feeding and co-aggregation with other bacterial species present in the periodontal microbiota, phages, and interactions with the human host. Examples for each of these influences on in vivo gene expression were found in the data presented here. We will discuss the most striking findings below.
For all three pathogens, more genes were down- than up-regulated in the natural niche. This may be because the periodontal community is a more competitive environment and it is not as nutrient rich as an optimized culture medium. This was most pronounced in T. denticola and F. nucleatum. Of the total number of 987 differentially expressed genes of T. denticola, 74% were down-regulated, and from the 493 differentially expressed genes of F. nucleatum in the periodontal pockets from individuals with periodontitis compared to laboratory culture, 71% were down-regulated in vivo. By contrast, only 291 genes significantly changed their gene expression in P. gingivalis, and of those 57% were down-regulated in vivo. The laboratory data for P. gingivalis covered a broad physiological range, since they were derived from three different cultivation media, of which two were complex media and one was a chemically defined minimal medium (Hovik et al., 2012). The pair-wise differences in gene expression between these media were 218, 223, and 267 genes in total at a level of more than 1.5-fold change in mean RNA-seq read count (Hovik et al., 2012), thus these differences were much smaller than those observed here between in vivo and laboratory conditions which were in total 784 differentially expressed genes with at least 2.46-fold changes. We conclude that the natural niche has a much stronger influence on gene expression of a pathogen than the chosen cultivation medium, and that in vivo only a small subset of its genes are actually highly expressed, which may reflect distinct selective pressure which is not present during mono-culture cultivation on a laboratory medium.
To get an overview of the functions that were differentially expressed in vivo we analyzed GO term enrichment. Some similarities between the three pathogens were observed, i.e., GO terms for protein metabolism, translation, cell adhesion, iron transporters, and pathogenesis were up-regulated in at least two of them, indicating that the cells were highly active in vivo and most probably more pathogenic than on laboratory media.
We then inspected those genes that were highly expressed both in vivo and in the laboratory, and those that were differentially expressed. For P. gingivalis we observed consistent expression of several of its virulence factors under all conditions, namely the hemagglutinin proteins HagA and HagE, the receptor antigen protein RagA, and the arginin-specific cystein proteinase (gingipain). Among the genes up-regulated in vivo were a fimbrilin gene and two cystein proteinase genes, required for adhering to other bacterial species and host tissue (Nelson et al., 2003). The most strongly up-regulated genes (average fold change of 70) belonged to the hmu hemin/heme uptake locus (PG1551-PG1556, hmuY, hmuR, hmuS, hmuT, hmuU, hmuV) which has been shown to be important for virulence in an animal model (Kesavalu et al., 2003; Lewis et al., 2006). Here we confirm that it is indeed the major route to obtain iron for P. gingivalis in the human periodontal pocket. The up-regulation of ABC transporters (Schneider and Hunke, 1998) and TonB-dependent receptors (Noinaj et al., 2010) in vivo suggests competition for nutrient uptake. Strikingly CRISPR-Cas3 as well as restriction-modification enzymes, transposases, integrases and helicases were down-regulated in vivo. Since phage attack, extracellular DNA, and stress conditions requiring DNA re-arrangement should be rather more frequent under in vivo conditions than in the laboratory, the contrary would have been expected. The most strongly down-regulated gene was pgaA enoding a surface protein.
Treponema denticola showed the most dramatic differences between gene expression in vivo and in laboratory mono-culture, possibly reflecting the fact that this spirochaete invades host cells which is not mimicked in culture. 730 genes were significantly down-regulated in the periodontal niche, covering all aspects of bacterial physiology, including, as in P. gingivalis, CRISPR-related genes, DNA-re-organization, and phage related genes, but also many other metabolic functions, including lipoproteins, proteases, cobalamin-biosynthesis, and many more. The most strongly differentially expressed gene was hemolysin which was down-regulated (log2FC = 16.251) in the periodontal pocket indicating that T. denticola does not obtain its iron by lysing erythrocytes in vivo. By contrast, it uptakes iron directly from the environment using specific iron transporters and ABC transporters, which were the most strongly up-regulated genes in vivo. Under both, in vivo conditions and laboratory culture, the most highly expressed genes of T. denticola were flagella filament proteins, the Msp protein that interacts with the immune system, glycine metabolism enzymes, and dentilisin, a surface protease (Dashper et al., 2011) that can degrade interleukins (Miyamoto et al., 2006) and can bind to P. gingivalis fimbriae (Hashimoto et al., 2003). The fimbrilin gene of P. gingivalis was strongly up-regulated in the periodontal pocket (see above). Our data thus confirm that those two red-complex pathogens can indeed co-aggregate in vivo so that the non-motile P. gingivalis may take advantage from binding to the motile spirochaete.
Fusobacterium nucleatum is of tremendous medical importance, yet it is relatively understudied, possibly because of its obligate anaerobic lifestyle which requires specialized equipment for culturing. The most strongly up-regulated gene in the periodontal pocket was a hemin receptor, thus F. nucleatum obtains its iron from hemin in vivo like T. denticola and P. gingivalis. Genes encoding enzymes of the primary metabolism were down-regulated in the periodontal pocket, confirming that laboratory culture media offer more substrates than are actually utilized in vivo. The most important virulence factor of F. nucleatum, the adhesin Fad (Rubinstein et al., 2013), was highly expressed in the periodontal pocket in health and disease as well as in laboratory mono-culture. It can therefore be expected to be a reliable biomarker for F. nucleatum pathogenicity.
Recent animal studies indicate that co-infection with P. gingivalis, T. denticola, and F. nucleatum can significantly enhance tissue damage during periodontitis compared with mono-infection with these species (Kesavalu et al., 2007; Polak et al., 2009; Dashper et al., 2011). In our data, we found clues for their synergistic pathogenicity. Based on the transcriptional profiles of these key members of the periodontal community in vivo, a schematic interaction model can be established which shows two-way synergistic interactions, i.e., each species gives and takes (Figure 5). The upregulation of many surface proteins in vivo suggests enhanced adhesion in the periodontal niche (Lee et al., 2005). The data suggest that P. gingivalis attached to T. denticola or other bacteria via hemagglutinin and fimbriae to improve its mobility (Figure 5). As F. nucleatum exhibits no or weak intrinsic proteolytic activity, it will profit from the coexistence with other species with strong proteolytic activities such as P. gingivalis (Bolstad et al., 1996). Shah and co-workers observed that F. nucleatum preferentially uses peptides instead of free amino acids (Shammas et al., 1993). Here we found that P. gingivalis and T. denticola up-regulated cysteine proteases while F. nucleatum massively up-regulated peptide transporters, thus it could take advantage of the proteolytic activity of other species especially P. gingivalis. A distinct form of shared labor was observed for iron, one of the key elements in pathogenicity (Skaar, 2010). F. nucleatum engaged in lysis of erythrocytes by strong upregulation of hemolysins; accordingly, T. denticola down-regulated hemolysins. Both T. denticola and P. gingivalis likely obtained their iron by binding heme or uptaking free iron, thus they profited from the hemolytic activity of F. nucleatum.
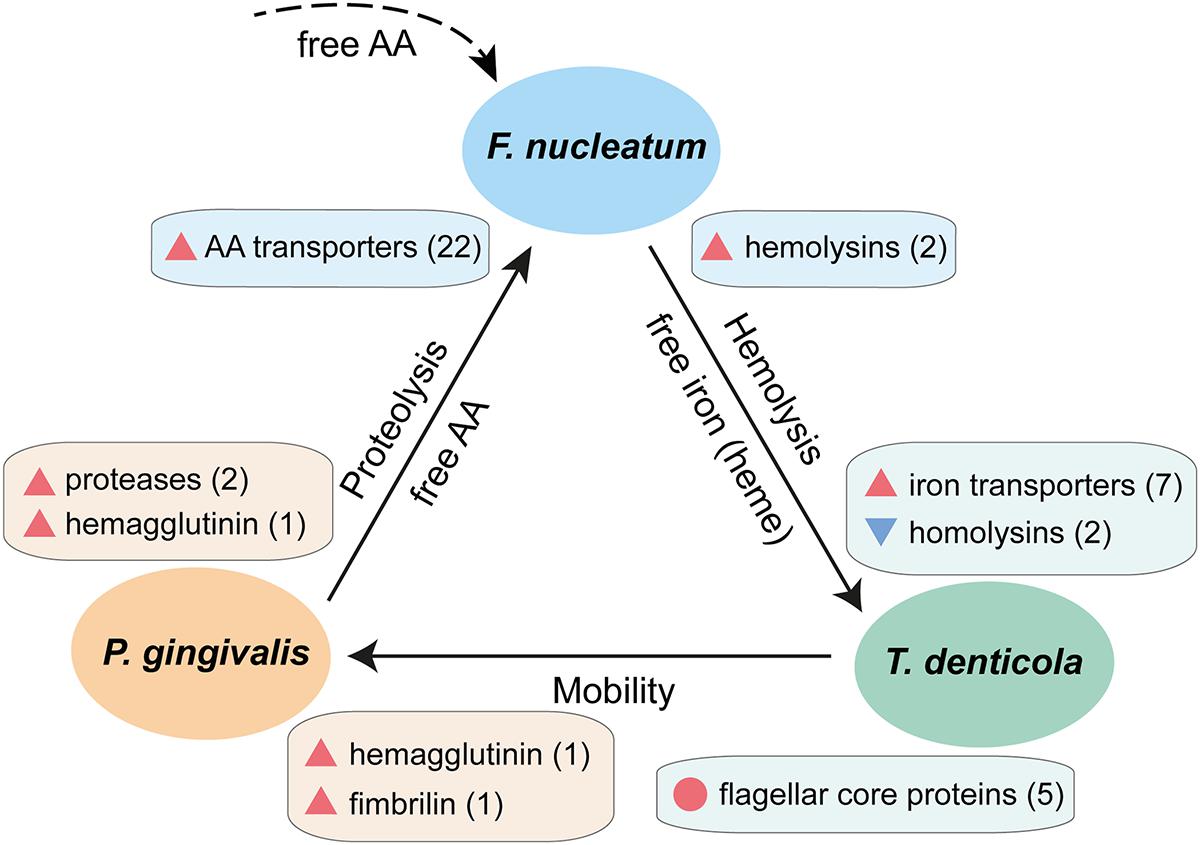
FIGURE 5. Schematic interaction model showing synergistic pathogenicity of F. nucleatum, P. gingivalis, and T. denticola based on their transcriptional profiles in chronic periodontitis. Blue triangles denote down-regulation, red triangles up-regulation in vivo, the number in parentheses indicate the number of genes of this particular function, and the red circle denotes a set of genes which are highly expressed both in vivo and in laboratory culture. The dashed line indicates contribution of free amino acids by other members of the periodontal microbiota.
The in vivo metatranscriptomes showed an unexpectedly high level of microdiversity for all three pathogens. We observed 23,783 single nucleotide variants (SNPs) for P. gingivalis, 62,145 for T. denticola and 127,729 for F. nucleatum transcripts. In F. nucleatum, SNPs were found in all chromosomal genes. The number of SNPs increased with the number of periodontal samples analyzed, and it tended to be higher for highly expressed genes. In an attempt to compare the number of SNPs per gene in health and periodontitis, we calculated the Shannon Index of diversity per gene. It was slightly higher in health, reflecting the larger number of samples available in health.
The data show that microdiversity is a common trait of pathogens in the periodontal pocket and affects every gene. Therefore, it likely reflects continuous micro-evolution in vivo. The average age of the subjects in our study was 52 (health) and 56 years (chronic periodontitis) so the microbial communities had co-evolved for a long time. Such SNPs could occur through the mutagenic impact of DNA damaging agents like reactive oxygen species, antibiotics or toxins, but also through horizontal gene transfer (HGT). Dental plaque is a hot spot of HGT due to the close physical contact between the microorganisms and their fast growth. S. mutans, for example, is genetically competent in a density dependent fashion and has integrated the regulation of competence and the synthesis of mutacins through the alternative sigma factor sigX (Reck et al., 2015).
Such microdiversity has been observed in metagenomics studies of various habitats before (Larkin and Martiny, 2017) resulting in the concept of “ecotypes” (Farrant et al., 2016). The functional importance of population microdiversity could be demonstrated for the colonization of premature babies, where polymorphism of Citrobacter koseri genomes at 47 sites was found and it could actually be shown that a specific subpopulation was restricted to the gut (Olm et al., 2017). The adaptive importance of microdiversity in the periodontal pocket remains to be explored.
Conclusion
In the natural environment of the periodontal niche, bacteria need to fight for their survival due to the shortage of essential nutrients and clearance of the host immune system. Under such conditions, bacteria upregulate the genes which they need the most and cooperate with each other to improve their fitness. By comparing the transcriptional profiles from in vivo conditions to gene expression in single culture, we were able to identify those genes which help them to obtain essential nutrients, evade the immune system and cooperate. When we analyzed transcriptional profiles on different culture media for P. gingivalis, we found that there were differences but they were very small in comparison to in vivo expression.
Interfering with the genes upregulated by key microbial members (such as cysteine proteases and heme binding proteins of P. gingivalis, peptide transporters of F. nucleatum, iron transporters of T. denticola) may influence their abundance in the community and help to shift the dysbiosis toward eubiosis. Therefore, the findings in this study may provide insights for the development of novel therapeutic strategies specifically reducing certain key-stone pathogens and bringing back the community to an ecological equilibrium, rather than wiping it out completely using standard antibiotics.
Author Contributions
HS performed the optimization of culture conditions for growth, the RNA extraction, quality checking, and mRNA enrichment. SB and MJ prepared the cDNA libraries and performed the Illumina sequencing. Z-LD designed the data analysis pipeline and analyzed the data. IW-D supported and supervised the research. Z-LD and IW-D wrote the manuscript, all authors reviewed the manuscript.
Funding
This work was funded by COMBACTE grant and Deutsche Forschungsgemeinschaft (DFG) in Transregio-SFB TR51.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Juergen Tomasch for stimulating discussions and constructive suggestions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00124/full#supplementary-material
FIGURE S1 | Relative abundance of transcripts from Porphyromonas gingivalis, Treponema denticola, and Fusobacterium nucleatum in the periodontal pocket metatranscriptomes. The red dots on the top of the bar indicate the communities in periodontitis.
TABLE S1 | Legend for each sheet.
SHEET 1 | The number of reads for P. gingivalis, T. denticola and F. nucleatum from laboratory culture.
SHEET 2 | The number of reads for P. gingivalis, T. denticola and F. nucleatum from periodontal metatranscriptome.
SHEET 3 | The gene expression of P. gingivalis in periodontal and mono-culture.
SHEET 4 | The differential expression of P. gingivalis in periodontal niche vs. mono-culture.
SHEET 5 | Gene ontology (GO) term enrichment analysis of P. gingivalis.
SHEET 6 | The gene expression of T. denticola in periodontal and mono-culture.
SHEET 7 | The differential expression of T. denticola in periodontal niche vs. mono-culture.
SHEET 8 | Gene ontology term enrichment analysis of T. denticola.
SHEET 9 | The gene expression of F. nucleatum in periodontal niche and mono-culture.
SHEET 10 | The differential expression of F. nucleatum in periodontitis vs. mono-culture.
SHEET 11 | The differential expression of F. nucleatum in health vs. mono-culture.
SHEET 12 | The differential expression of F. nucleatum in periodontitis vs. health.
SHEET 13 | Gene ontology term enrichment analysis of F. nucleatum in periodontitis compared to mono-culture.
SHEET 14 | Gene ontology term enrichment analysis of F. nucleatum in periodontitis compared to health.
SHEET 15 | The number of variants detected in the transcriptomes of P. gingivalis, T. denticola, and F. nucleatum from periodontal niche and laboratory culture.
SHEET 16 | The variants per gene in the transcriptome of P. gingivalis.
SHEET 17 | The variants per gene in the transcriptome of T. denticola.
SHEET 18 | The variants per gene in the transcriptome of F. nucleatum.
Footnotes
References
Alexa, A., and Rahnenfuhrer, J. (2010). topGO: Enrichment Analysis for Gene Ontology. R Package Version 2. Available at: http://www.bioconductor.org/packages/release/bioc/html/topGO.html
Amano, A., Chen, C., Honma, K., Li, C., Settem, R. P., and Sharma, A. (2014). Genetic characteristics and pathogenic mechanisms of periodontal pathogens. Adv. Dent. Res. 26, 15–22. doi: 10.1177/0022034514526237
Aronesty, E. (2011). ea-utils: Command-line Tools for Processing Biological Sequencing Data. Durham: Expression Analysis.
Barton, M. K. (2017). Evidence accumulates indicating periodontal disease as a risk factor for colorectal cancer or lymphoma. CA Cancer J. Clin. 67, 173–174. doi: 10.3322/caac.21367
Bolstad, A. I., Jensen, H. B., and Bakken, V. (1996). Taxonomy, biology, and periodontal aspects of Fusobacterium nucleatum. Clin. Microbiol. Rev. 9, 55–71.
Bushnell, B. (2014). BBMap: A Fast, Accurate, Splice-Aware Aligner. Berkeley, CA: Ernest Orlando Lawrence Berkeley National Laboratory.
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Darveau, R. P. (2010). Periodontitis: a polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 8, 481–490. doi: 10.1038/nrmicro2337
Dashper, S. G., Seers, C. A., Tan, K. H., and Reynolds, E. C. (2011). Virulence factors of the oral spirochete Treponema denticola. J. Dent. Res. 90, 691–703. doi: 10.1177/0022034510385242
Deka, R. K., Brautigam, C. A., Tomson, F. L., Lumpkins, S. B., Tomchick, D. R., Machius, M., et al. (2007). Crystal structure of the Tp34 (TP0971) lipoprotein of Treponema pallidum: implications of its metal-bound state and affinity for human lactoferrin. J. Biol. Chem. 282, 5944–5958. doi: 10.1074/jbc.M610215200
Deng, Z. L., Szafranski, S. P., Jarek, M., Bhuju, S., and Wagner-Dobler, I. (2017). Dysbiosis in chronic periodontitis: key microbial players and interactions with the human host. Sci. Rep. 7:3703. doi: 10.1038/s41598-017-03804-8
Duran-Pinedo, A. E., Chen, T., Teles, R., Starr, J. R., Wang, X., Krishnan, K., et al. (2014). Community-wide transcriptome of the oral microbiome in subjects with and without periodontitis. ISME J. 8, 1659–1672. doi: 10.1038/ismej.2014.23
Farrant, G. K., Dore, H., Cornejo-Castillo, F. M., Partensky, F., Ratin, M., Ostrowski, M., et al. (2016). Delineating ecologically significant taxonomic units from global patterns of marine picocyanobacteria. Proc. Natl. Acad. Sci. U.S.A. 113, E3365–E3374. doi: 10.1073/pnas.1524865113
Genco, R. J., and Van Dyke, T. E. (2010). Prevention: reducing the risk of CVD in patients with periodontitis. Nat. Rev. Cardiol. 7, 479–480. doi: 10.1038/nrcardio.2010.120
Hajishengallis, G., Darveau, R. P., and Curtis, M. A. (2012). The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 10, 717–725. doi: 10.1038/nrmicro2873
Han, Y. W. (2015). Fusobacterium nucleatum: a commensal-turned pathogen. Curr. Opin. Microbiol. 23, 141–147. doi: 10.1016/j.mib.2014.11.013
Hashimoto, M., Ogawa, S., Asai, Y., Takai, Y., and Ogawa, T. (2003). Binding of Porphyromonas gingivalis fimbriae to Treponema denticola dentilisin. FEMS Microbiol. Lett. 226, 267–271. doi: 10.1016/S0378-1097(03)00615-3
Hovik, H., Yu, W. H., Olsen, I., and Chen, T. (2012). Comprehensive transcriptome analysis of the periodontopathogenic bacterium Porphyromonas gingivalis W83. J. Bacteriol. 194, 100–114. doi: 10.1128/JB.06385-11
How, K. Y., Song, K. P., and Chan, K. G. (2016). Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Front. Microbiol. 7:53. doi: 10.3389/fmicb.2016.00053
Imamura, T. (2003). The role of gingipains in the pathogenesis of periodontal disease. J. Periodontol. 74, 111–118. doi: 10.1902/jop.2003.74.1.111
Jorth, P., Turner, K. H., Gumus, P., Nizam, N., Buduneli, N., and Whiteley, M. (2014). Metatranscriptomics of the human oral microbiome during health and disease. mBio 5:e01012-14. doi: 10.1128/mBio.01012-14
Kapatral, V., Anderson, I., Ivanova, N., Reznik, G., Los, T., Lykidis, A., et al. (2002). Genome sequence and analysis of the oral bacterium Fusobacterium nucleatum strain ATCC 25586. J. Bacteriol. 184, 2005–2018. doi: 10.1128/JB.184.7.2005-2018.2002
Kesavalu, L., Holt, S. C., and Ebersole, J. L. (2003). In vitro environmental regulation of Porphyromonas gingivalis growth and virulence. Oral Microbiol. Immunol. 18, 226–233. doi: 10.1034/j.1399-302X.2003.00071.x
Kesavalu, L., Sathishkumar, S., Bakthavatchalu, V., Matthews, C., Dawson, D., Steffen, M., et al. (2007). Rat model of polymicrobial infection, immunity, and alveolar bone resorption in periodontal disease. Infect. Immun. 75, 1704–1712. doi: 10.1128/IAI.00733-06
Kilian, M., Chapple, I. L., Hannig, M., Marsh, P. D., Meuric, V., Pedersen, A. M., et al. (2016). The oral microbiome - an update for oral healthcare professionals. Br. Dent. J. 221, 657–666. doi: 10.1038/sj.bdj.2016.865
Kopylova, E., Noe, L., and Touzet, H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217. doi: 10.1093/bioinformatics/bts611
Lalla, E., and Papapanou, P. N. (2011). Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat. Rev. Endocrinol. 7, 738–748. doi: 10.1038/nrendo.2011.106
Larkin, A. A., and Martiny, A. C. (2017). Microdiversity shapes the traits, niche space, and biogeography of microbial taxa. Environ. Microbiol. Rep. 9, 55–70. doi: 10.1111/1758-2229.12523
Lee, S. H., Kim, K. K., and Choi, B. K. (2005). Upregulation of intercellular adhesion molecule 1 and proinflammatory cytokines by the major surface proteins of Treponema maltophilum and Treponema lecithinolyticum, the phylogenetic group IV oral spirochetes associated with periodontitis and endodontic infections. Infect. Immun. 73, 268–276. doi: 10.1128/IAI.73.1.268-276.2005
Lewis, J. P., Plata, K., Yu, F., Rosato, A., and Anaya, C. (2006). Transcriptional organization, regulation and role of the Porphyromonas gingivalis W83 hmu haemin-uptake locus. Microbiology 152, 3367–3382. doi: 10.1099/mic.0.29011-0
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Liao, Y., Smyth, G. K., and Shi, W. (2014). Featurecounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi: 10.1093/bioinformatics/btt656
Lundberg, K., Wegner, N., Yucel-Lindberg, T., and Venables, P. J. (2010). Periodontitis in RA-the citrullinated enolase connection. Nat. Rev. Rheumatol. 6, 727–730. doi: 10.1038/nrrheum.2010.139
Maresz, K. J., Hellvard, A., Sroka, A., Adamowicz, K., Bielecka, E., Koziel, J., et al. (2013). Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD). PLOS Pathog. 9:e1003627. doi: 10.1371/journal.ppat.1003627
Michaud, D. S., Fu, Z., Shi, J., and Chung, M. (2017). Periodontal disease, tooth loss, and cancer risk. Epidemiol. Rev. 39, 49–58. doi: 10.1093/epirev/mxx006
Miyamoto, M., Ishihara, K., and Okuda, K. (2006). The treponema denticola surface protease dentilisin degrades interleukin-1 beta (IL-1 beta), IL-6, and tumor necrosis factor alpha. Infect. Immun. 74, 2462–2467. doi: 10.1128/IAI.74.4.2462-2467.2006
Narasimhan, V., Danecek, P., Scally, A., Xue, Y., Tyler-Smith, C., and Durbin, R. (2016). BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 32, 1749–1751. doi: 10.1093/bioinformatics/btw044
Nelson, K. E., Fleischmann, R. D., DeBoy, R. T., Paulsen, I. T., Fouts, D. E., Eisen, J. A., et al. (2003). Complete genome sequence of the oral pathogenic Bacterium Porphyromonas gingivalis strain W83. J. Bacteriol. 185, 5591–5601. doi: 10.1128/JB.185.18.5591-5601.2003
Noinaj, N., Guillier, M., Barnard, T. J., and Buchanan, S. K. (2010). TonB-dependent transporters: regulation, structure, and function. Annu. Rev. Microbiol. 64, 43–60. doi: 10.1146/annurev.micro.112408.134247
Olm, M. R., Brown, C. T., Brooks, B., Firek, B., Baker, R., Burstein, D., et al. (2017). Identical bacterial populations colonize premature infant gut, skin, and oral microbiomes and exhibit different in situ growth rates. Genome Res. 27, 601–612. doi: 10.1101/gr.213256.116
Polak, D., Wilensky, A., Shapira, L., Halabi, A., Goldstein, D., Weiss, E. I., et al. (2009). Mouse model of experimental periodontitis induced by Porphyromonas gingivalis/Fusobacterium nucleatum infection: bone loss and host response. J. Clin. Periodontol. 36, 406–410. doi: 10.1111/j.1600-051X.2009.01393.x
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Reck, M., Tomasch, J., and Wagner-Dobler, I. (2015). The alternative sigma factor SigX controls bacteriocin synthesis and competence, the two quorum sensing regulated traits in Streptococcus mutans. PLOS Genet. 11:e1005353. doi: 10.1371/journal.pgen.1005353
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Rubinstein, M. R., Wang, X., Liu, W., Hao, Y., Cai, G., and Han, Y. W. (2013). Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe 14, 195–206. doi: 10.1016/j.chom.2013.07.012
Schneider, E., and Hunke, S. (1998). ATP-binding-cassette (ABC) transport systems: functional and structural aspects of the ATP-hydrolyzing subunits/domains. FEMS Microbiol. Rev. 22, 1–20. doi: 10.1111/j.1574-6976.1998.tb00358.x
Shammas, N. W., Murphy, G. W., Eichelberger, J., Klee, D., Schwartz, R., and Bachman, W. (1993). Infective endocarditis due to Fusobacterium nucleatum: case report and review of the literature. Clin. Cardiol. 16, 72–75. doi: 10.1002/clc.4960160116
Signat, B., Roques, C., Poulet, P., and Duffaut, D. (2011). Fusobacterium nucleatum in periodontal health and disease. Curr. Issues Mol. Biol. 13, 25–36.
Skaar, E. P. (2010). The battle for iron between bacterial pathogens and their vertebrate hosts. PLOS Pathog. 6:e1000949. doi: 10.1371/journal.ppat.1000949
Socransky, S. S., Haffajee, A. D., Cugini, M. A., Smith, C., and Kent, R. L Jr. (1998). Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25, 134–144. doi: 10.1111/j.1600-051X.1998.tb02419.x
Supek, F., Bosnjak, M., Skunca, N., and Smuc, T. (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLOS ONE 6:e21800. doi: 10.1371/journal.pone.0021800
Szafranski, S. P., Deng, Z.-L., Tomasch, J., Jarek, M., Bhuju, S., Meisinger, C., et al. (2015a). Functional biomarkers for chronic periodontitis and insights into the roles of Prevotella nigrescens and Fusobacterium nucleatum; a metatranscriptome analysis. NPJ Biofilms Microbiomes 57, 10–13. doi: 10.1038/npjbiofilms.2015.17
Szafranski, S. P., Wos-Oxley, M. L., Vilchez-Vargas, R., Jauregui, R., Plumeier, I., Klawonn, F., et al. (2015b). High-resolution taxonomic profiling of the subgingival microbiome for biomarker discovery and periodontitis diagnosis. Appl. Environ. Microbiol. 81, 1047–1058. doi: 10.1128/AEM.03534-14
Tan, K. H., Seers, C. A., Dashper, S. G., Mitchell, H. L., Pyke, J. S., Meuric, V., et al. (2014). Porphyromonas gingivalis and Treponema denticola exhibit metabolic symbioses. PLOS Pathog. 10:e1003955. doi: 10.1371/journal.ppat.1003955
UniProt Consortium (2014). UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212. doi: 10.1093/nar/gku989
Van der Velden, U. (2017). What exactly distinguishes aggressive from chronic periodontitis: is it mainly a difference in the degree of bacterial invasiveness? Periodontol 2000 75, 24–44. doi: 10.1111/prd.12202
Visser, M. B., and Ellen, R. P. (2011). New insights into the emerging role of oral spirochaetes in periodontal disease. Clin. Microbiol. Infect. 17, 502–512. doi: 10.1111/j.1469-0691.2011.03460.x
Wood, D. E., and Salzberg, S. L. (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15:R46. doi: 10.1186/gb-2014-15-3-r46
Yost, S., Duran-Pinedo, A. E., Teles, R., Krishnan, K., and Frias-Lopez, J. (2015). Functional signatures of oral dysbiosis during periodontitis progression revealed by microbial metatranscriptome analysis. Genome Med. 7:27. doi: 10.1186/s13073-015-0153-3
Keywords: periodontitis, Porphyromonas gingivalis, Treponema denticola, Fusobacterium nucleatum, metatranscriptome, inter-species interaction, SNPs
Citation: Deng Z-L, Sztajer H, Jarek M, Bhuju S and Wagner-Döbler I (2018) Worlds Apart – Transcriptome Profiles of Key Oral Microbes in the Periodontal Pocket Compared to Single Laboratory Culture Reflect Synergistic Interactions. Front. Microbiol. 9:124. doi: 10.3389/fmicb.2018.00124
Received: 17 October 2017; Accepted: 18 January 2018;
Published: 06 February 2018.
Edited by:
Fumito Maruyama, Kyoto University, JapanReviewed by:
Ulvi Kahraman Gürsoy, University of Turku, FinlandTakahiko Shiba, Tokyo Medical and Dental University, Japan
Copyright © 2018 Deng, Sztajer, Jarek, Bhuju and Wagner-Döbler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi-Luo Deng, zhiluo.deng@helmholtz-hzi.de