 Jordan M. Meyers1,2†
Jordan M. Meyers1,2† Miranda Grace
Miranda Grace Aayushi Uberoi
Aayushi Uberoi Karl Munger
Karl Munger- 1Program in Virology, Harvard Medical School, Boston, MA, United States
- 2Department of Developmental, Molecular and Chemical Biology, Tufts University School of Medicine, Boston, MA, United States
- 3McArdle Laboratory for Cancer Research, Department of Oncology, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, WI, United States
Infections with cutaneous papillomaviruses have been linked to cutaneous squamous cell carcinomas that arise in patients who suffer from a rare genetic disorder, epidermodysplasia verruciformis, or those who have experienced long-term, systemic immunosuppression following organ transplantation. The E6 proteins of the prototypical cutaneous human papillomavirus (HPV) 5 and HPV8 inhibit TGF-β and NOTCH signaling. The Mus musculus papillomavirus 1, MmuPV1, infects laboratory mouse strains and causes cutaneous skin warts that can progress to squamous cell carcinomas. MmuPV1 E6 shares biological and biochemical activities with HPV8 E6 including the ability to inhibit TGF-β and NOTCH signaling by binding the SMAD2/SMAD3 and MAML1 transcription factors, respectively. Inhibition of TGF-β and NOTCH signaling is linked to delayed differentiation and sustained proliferation of differentiating keratinocytes. Furthermore, the ability of MmuPV1 E6 to bind MAML1 is necessary for wart and cancer formation in experimentally infected mice. Hence, experimental MmuPV1 infection in mice will be a robust and valuable experimental system to dissect key aspects of cutaneous HPV infection, pathogenesis, and carcinogenesis.
Introduction
As of January 2018, more than 300 human papillomavirus (HPV) genotypes are listed in the PAVE database1 (Van Doorslaer et al., 2017). HPVs have ∼8 kb circular double stranded DNA genomes, do not generally infect heterologous hosts and exhibit a preference for infecting either mucosal or cutaneous epithelia. Mucosal HPVs are generally transmitted by sexual contact and mucosal infections with α-genus HPVs are the most common sexually transmitted disease with a prevalence of 70 million cases and an annual incidence of 14 million new transmissions in the United States (CDC, 2015). Mucosal HPVs mostly include members of the α-genus. Clinically, α-HPVs can be designated either as “low-risk” or “high-risk.” Low-risk α-HPV infections trigger benign genital warts, whereas high-risk α-HPVs cause intraepithelial lesions that can undergo malignant progression. Almost 100% of cervical carcinoma are caused by high-risk HPVs and infections with these viruses also contribute to a large percentage of anogenital tract as well as oral cancers (Schiffman et al., 2007). Globally, high-risk α-HPVs infections cause approximately 5% of all human cancers. Despite the availability of efficacious prophylactic vaccines for the most abundant high-risk α-HPVs, there will be approximately 4,170 cervical cancer deaths in 2018 in the United States alone, which means that approximately every 2 h one woman will succumb to cervical cancer (Siegel et al., 2018). Because cancers arise years to decades after the initial infection, a significant impact of these prophylactic vaccines on cervical cancer rates is not expected for another 20 years (Frazer, 2004; Schiller and Lowy, 2012). Unfortunately, there are no antiviral compounds to combat HPV infections (Hellner and Munger, 2011).
Cutaneous HPVs include the β-, γ-, μ-, and ν-genera and some α-HPVs also have a preference for infecting cutaneous epithelia. Cutaneous HPV infections are very frequent and have been linked to development of warts, actinic keratosis, keratoacanthoma, psoriasis, and non-melanoma skin cancers. In Australia, up to 24% of 16- to 18-year-olds have skin warts (Kilkenny et al., 1998). The prevalence of actinic keratosis in the United States is ∼20% and resulted in 47 million dermatology visits in 2006 (Del Rosso et al., 2014). Keratoacanthoma are low grade, rapidly growing skin tumors that, unlike squamous cell carcinomas, are derived from cells located in the hair follicle. Cutaneous HPV genomes are readily detected in hair follicle cells harvested by plucking hair, and a high viral load is a predictor of cancer development (Neale et al., 2013). Keratoacanthoma mostly occur in sun-exposed skin in middle aged patients. The incidence has been estimated at approximately 1:1,000 (Chuang et al., 1993). Cutaneous HPVs have also been frequently detected in psoriasis patients (Mahe et al., 2003). In patients suffering from a rare inherited disease, epidermodysplasia verruciformis (EV), and in the rapidly increasing population of chronically immune-suppressed individuals, such as organ transplant patients, lesions caused by cutaneous HPV infections can progress to cutaneous squamous cell carcinomas (cSCCs) (Pass et al., 1977; Orth et al., 1978; Neale et al., 2013). Indeed, cSCCs arising in EV patients were the first human tumors that were conclusively linked to HPV infections. More recently, β- and γ-HPV genomes have also been detected at mucosal sites, suggesting the possibility that these viruses may also infect mucosal epithelia (Bottalico et al., 2011; Hampras et al., 2017; Smelov et al., 2017).
The incidence of cSCCs in the general population has been linked to UV exposure and is rising rapidly with more than 700,000 cases and 8,000 deaths yearly in the United States (Alam and Ratner, 2001; Deady et al., 2014). The estimated annual costs for cSCC treatments in the United States are at $3.8 billion (Guy et al., 2015). Whether and how HPVs may be involved in cSCCs development in immunocompetent individuals, however, remains a matter of debate (Feltkamp et al., 2008), even though a recent meta-analysis provided evidence for a modestly increased overall risk of 1.42 (1.18–1.72) for β-HPV infection and cSCC development (Chahoud et al., 2016).
Hence, β-HPV infections are highly associated with cSCC development in EV patients, correlated in the case of cSCCs arising in immune suppressed patients, and only slightly or sporadically associated with cSCCs in immune competent patients. HPV sequences are not retained and expressed in every cSCC cell. This indicates that, unlike what has been shown for high-risk α-HPV-associated cancers, cutaneous HPVs may not be necessary for tumor maintenance. Cutaneous HPV genomes are almost universally detected in asymptomatic patients (Antonsson et al., 2003a; Kohler et al., 2007), but it is not clear whether this is reflective of active and/or persistent infection. Detection of HPV RNA or protein expression may provide a better estimation of the frequency of active HPV infection in non-neoplastic skin (Borgogna et al., 2012, 2014) and it has been debated whether cutaneous HPVs function as carcinogenic drivers, or if they appear as innocuous passengers in cSCCs that arise in immunocompetent individuals (Quint et al., 2015). A recent study with a transgenic mouse model, has provided evidence for the alternative model that some cutaneous HPVs are drivers of cancer initiation but are not necessary for tumor maintenance (Viarisio et al., 2017).
Carcinogenic Activities of β-HPVs
Because of their isolation from cSCCs in EV patients, much of the early research efforts with the β-HPVs focused on HPV5 and HPV8. Somewhat surprisingly, HPV5 and HPV8 scored as only weakly transforming or non-transforming in standard cell-based assays (Yamashita et al., 1993; Tommasino, 2017). However, experiments with genetically engineered mice where HPV8 sequences were expressed in basal epithelial cells, impressively demonstrated the oncogenic activity of this HPV. While these mice spontaneously develop skin cancer without the requirement of any additional treatment, they also recapitulated the co-carcinogenic effect of UV exposure (reviewed in Howley and Pfister, 2015). These experiments showed that HPV8 E6, E7 and, uniquely, also E2 could each independently contribute to skin cancer formation when ectopically expressed in basal epithelial cells of a transgenic mouse (Schaper et al., 2005; Pfefferle et al., 2008; Marcuzzi et al., 2009).
The β-HPVs are phylogenetically diverse and as of January 2018, 64 β-HPVs are listed in the PAVE database (see text footnote 1; Van Doorslaer et al., 2017). These are further classified into phylogenetic species. HPV5 and HPV8 together with 19 additional HPVs are classified as species 1, there are 22 members classified as species 2, four are species 3, one is species 4, three are species 5, and 13 have not yet been classified by the HPV reference center.
The E6 and E7 proteins of other β-HPVs, including HPV types 20, 38, and 48 also exhibit carcinogenic activities in transgenic mouse models. Mice expressing β1-HPV20 E6 or E7 from keratin K10 promoter and mice with β2-HPV38 E6/E7 expression from a keratin K14 promoter developed skin cancers when exposed to UV. In contrast, mice expressing the β3-HPV49 E6/E7 genes from a keratin 14 promoter were not susceptible to skin carcinogenesis after UV exposure but developed upper digestive tract carcinomas when treated with the chemical carcinogen 4-nitroquinoline 1-oxide (Michel et al., 2006; Viarisio et al., 2011, 2016). Some of these more recently detected β-HPVs also exhibit transforming activities in cell-based assays. Ectopic expression of the β1 HPV24 or HPV36 E6/E7 proteins causes life span extension of primary human foreskin keratinocytes (HFKs), whereas expression of the β2 HPV38 and β3 HPV49 E6/E7 proteins can trigger HFK immortalization (Cornet et al., 2012).
Despite similar genomic organization, there are some differences between the β- and high-risk α-HPVs (McLaughlin-Drubin et al., 2012). Most strikingly, β-HPVs do not encode E5 proteins, which are oncogenic in high-risk α-HPVs (Genther et al., 2003; Maufort et al., 2007, 2010). Moreover, while high-risk α-HPV-associated cervical carcinomas are generally non-productive, cSCCs arising in EV patients are productive infections. Despite these differences, early mechanistic analyses of the oncogenic activities of β-HPVs were modeled after studies with high-risk α-HPV E6 and E7 proteins. Like high-risk α-HPV E6 and E7 proteins, β-HPV E6 and E7s are low molecular weight, cysteine-rich, metal binding proteins of approximately 150 and 100 amino acids, respectively. They lack intrinsic enzymatic activities, are not known to directly bind specific DNA sequences and presumably exert their biological activities by interacting with and functionally modifying cellular regulatory proteins (Munger et al., 2004; Roman and Munger, 2013; Vande Pol and Klingelhutz, 2013). The best studied cellular targets of the high-risk α-HPV E6 and E7 proteins are the TP53 and RB1 tumor suppressors, respectively. The transforming activities of papillomaviruses are a reflection of their life cycles, specifically their need to secure the availability of cellular replication factors in terminally differentiated epithelial cells that would have normally withdrawn from the cell division cycle (Harden and Munger, 2017). Papillomaviruses need to uncouple cell-cycle withdrawal from epithelial differentiation, effectively allowing for unlicensed proliferation in suprabasal keratinocytes. Aberrant DNA synthesis can be sensed by cells and triggers cell abortive responses, which will need to be inhibited by the virus. The strategies that specific papillomaviruses have evolved to enable their productive replication cycles largely determine their oncogenic potential (McLaughlin-Drubin and Munger, 2009; Moody and Laimins, 2010).
The high-risk HPV E7 proteins cause S-phase competence in differentiating cells by targeting the retinoblastoma tumor suppressor, RB1, for degradation. Uncontrolled S-phase entry as a consequence of RB1 inactivation causes activation of the TP53 tumor suppressor, which triggers a transcriptional program to eliminate such rogue cells by apoptosis. This simple, albeit likely not entirely correct, model (Munger et al., 2004; Munger and Jones, 2015) also served as a blueprint for many studies with the EV-associated β-HPVs, even though it became clear very quickly that it was unlikely to apply to these viruses (McLaughlin-Drubin et al., 2012; Meyers and Munger, 2014; Tommasino, 2017). HPV8 E6 does not detectably interact with TP53 or the UBE3A (E6AP) E3 ubiquitin ligase that is necessary for high-risk α-HPV E6 mediated TP53 degradation (Elbel et al., 1997; White et al., 2012a; Brimer et al., 2017). HPV5 and HPV8 E7s weakly bind and do not degrade RB1, similar to the low-risk α-HPV E7s (Yamashita et al., 1993).
All known β-HPV E7 proteins contain canonical LXCXE (L, leucine; C, cysteine; E, glutamate; X, any amino acid)-based binding sites for the retinoblastoma tumor suppressor family of proteins and many have been documented to bind RB1, RBL1 (p107), and RBL2 (p130). Some β-HPV E6 and E7 in fact interfere with RB1 and TP53 stability, reminiscent of cervical cancer associated, high-risk α-HPVs (Caldeira et al., 2003; Accardi et al., 2006; Cornet et al., 2012; White et al., 2014). The interactions described for β-HPV E6 proteins with TP53 are particularly complex as some have been reported to stabilize TP53 (HPV17, HPV38, and HPV92), while others destabilize it (HPV49) and some have been reported to interfere with TP53 activation and/or TP53 mediated activation of certain target genes (HPV5, HPV8, and HPV38) (Cornet et al., 2012; Wallace et al., 2014; White et al., 2014). Whether humans infected with cutaneous HPVs that target RB1 and TP53 similar to the high-risk α-HPVs are at a particularly high risk for developing cSCC, remains to be determined.
Given the fact that most β-HPV-associated tumors arise in sun exposed areas, it was investigated whether these viruses might interfere with UV-induced apoptosis and/or DNA damage repair. Consistent with expectations, several groups reported that β-HPV E6 proteins cause degradation of BAK, a pro-apoptotic BCL2 family member that is released from the mitochondria following UV exposure (Jackson et al., 2000). BAK degradation by β-HPV E6 proteins is predicted to inhibit apoptosis and allow for survival of UV-damaged cells that may have possibly acquired oncogenic mutations (Leverrier et al., 2007; Hufbauer et al., 2015).
Other studies showed that the repair of UV-damaged DNA damage is inhibited in HPV8 E6 expressing cells. HPV5 and HPV8 E6 proteins were reported to bind the EP300/CREBBP acetyltransferases and target them for degradation thereby inhibiting the Ataxia Telangiectasia Mutated (ATM) and Ataxia Telangiectasia and Rad3-Related (ATR) kinases, key sensors, and mediators of double strand DNA break repair. By abrogating ATM/ATR mediated sensing and/or repair of double strand DNA breaks, cellular mutations may accumulate in β-HPV infected cells, thereby driving carcinogenesis (Howie et al., 2011; Wallace et al., 2012; Wendel and Wallace, 2017). These findings support a “hit-and-run” mechanism for β-HPV carcinogenesis where viral gene expression contributes to cancer initiation by increasing the mutational burden caused by UV exposure but is not necessary for tumor maintenance. This would explain why viral gene expression is not maintained in all tumor cells (Arron et al., 2011). Such a model was recently recapitulated with HPV38 transgenic mice (Viarisio et al., 2018). Interestingly, unbiased proteomic analyses of the HPV38 E6 associated proteins provided no evidence for EP300/CREBBP binding to HPV38 E6 (White et al., 2012a), suggesting that inhibition of DNA break repair by β-HPV E6 proteins may not be necessarily linked to EP300/CREBBP degradation.
As mentioned previously, β-HPV genomes have been detected in hair follicles, which contain epithelial stem cells and HPV8 has been reported to increase the number of keratinocytes with stem cell characteristics both in cell- and animal-based model systems (Hufbauer et al., 2013; Lanfredini et al., 2017; Marthaler et al., 2017). Several studies have shown that the TP53 family member and epithelial stem cell marker TP63 is dysregulated in HPV8 E6 expressing keratinocytes. Specifically, higher levels of the amino terminally truncated ΔNp63α and lower levels of full-length TAp63 isoforms were detected in HPV8 E6 expressing human and murine keratinocytes (Lanfredini et al., 2017; Marthaler et al., 2017). NOTCH inhibition decreased TP63 levels in HPV8 E6 expressing undifferentiated keratinocytes, but TP63 expression remained high in differentiating HPV8 E6 expressing keratinocytes, suggesting that there are NOTCH independent pathways that cause deregulated TP63 expression (Meyers et al., 2013). One of these pathways involves down-regulation of microRNA-203 (miR-203) by HPV8 E6 by inhibiting EP300 which in turn affects CCAAT/enhancer-binding protein a (CEBPA) transcription factor activity that controls miR-203 expression. Significantly, up-regulation of miR-203 during calcium-induced differentiation is abrogated in HPV8 E6 expressing keratinocytes (Marthaler et al., 2017). High-level TP63 and low levels of miR-203 were also detected throughout the lesional tissue of an HPV8 positive EV patient (Marthaler et al., 2017). Since EP300 binding is not shared by all β-HPV E6 (White et al., 2012a) it remains to be determined whether this pathway is universally targeted by β-HPV E6 proteins.
The γ-HPVs; Benign Flora or Dangerous Villains?
As of January 2018, a total of 171 γ-HPVs have been detected, which are classified into 27 different species. The number of species may increase, since 93 γ-HPVs have not yet been classified into species by the HPV reference center (see text footnote 1; Van Doorslaer et al., 2017). The enormous phylogenetic diversity of γ-HPVs is also reflected in their genome organization; members of the γ6 species, for example, do not encode recognizable E6 proteins and some γ-HPVs encode E7 proteins that lack the canonical LXCXE binding site for the RB1 tumor suppressor and the related p107 (RBL1) and p130 (RBL2) proteins. The γ-HPVs were initially isolated from cutaneous epithelia and are often regarded as components of the normal skin viral flora (Antonsson et al., 2003a,b; Forslund, 2007; Chen et al., 2008; Foulongne et al., 2012). More recent studies also detected γ-HPV DNA at mucosal sites (Bottalico et al., 2011; Smelov et al., 2017).
A “deep sequencing” study of human skin cancers and matched normal tissues showed that HPV197, a γ24-HPV, was the most frequently detected HPV genome in cSCCs and was not present in normal skin (Arroyo Muhr et al., 2015). HPV197 could not have been previously detected because the genome is not recognized by the “consensus” PCR primers that are frequently used for HPV analysis of clinical specimens (Arroyo Muhr et al., 2015). To this date, however, there is no evidence that HPV197 sequences are actively expressed in skin cancers and/or that HPV197 or potentially other γ-HPVs contribute to cSCC development in immunosuppressed or immunocompetent individuals.
Analysis of potential cellular targets by affinity purification followed by mass spectrometry (AP/MS) revealed that HPV197 E6 shares many cellular targets with β-HPV E6 proteins, particularly the β1 HPVs that include HPV5 and HPV8 (Grace and Munger, 2017). The HPV197 E7 protein lacks an LXCXE-based RB1 binding site, yet, consistent with earlier work with the γ1 HPV4 E7 protein (Wang et al., 2010), HPV197 E7 interacts with RB1 (Grace and Munger, 2017). Interestingly, however, HPV197 E7 does not bind to RBL1 or RBL2 (Grace and Munger, 2017). The detection of HPV197 DNA in human skin cancers but not normal skin, and the interactions of the E6 and E7 proteins with human tumor suppressor proteins are consistent with the possibility that HPV197 and potentially other γ-HPVs may be associated with skin cancer development. Currently, however, only very few studies have explored the biological activities of γ-HPVs in cell-based or animal models.
MmuPV1, An Animal Model for Cutaneous HPV Infections
Papillomaviruses are species specific and generally do not infect heterologous hosts (Knipe and Howley, 2013). This has severely hindered the study of PV pathogenesis in vivo. Research has also been stymied by the lack of a papillomavirus that naturally infects and replicates in a genetically tractable laboratory animal. The isolation of Mus musculus papillomavirus 1, MmuPV1, from skin warts on immunodeficient nude mice promises to revolutionize PV pathogenesis studies (Ingle et al., 2011; Joh et al., 2011). The ability to cause wart formation upon experimental infection with quasivirus or by naked DNA inoculation was originally observed in mouse strains that have T-cell deficiencies or are temporarily immune suppressed (Handisurya et al., 2014; Joh et al., 2016; Uberoi et al., 2016). Lesions arising due to MmuPV1 infection have malignant potential and can progress to cSCCs. Upon UVB-exposure, experimental MmuPV1 infection also causes skin warts that can progress to cSCCs in immunocompetent, standard laboratory mouse strains (Uberoi and Lambert, 2017). Importantly, there is a correlation between immunosuppression due to UV irradiation and onset of MmuPV1 dependent cutaneous papillomas (Uberoi et al., 2016).
MmuPV1 is a member of the Pi genus, which encompasses rodent PVs (Van Doorslaer et al., 2017) and is related to γ- and β-HPVs (Joh et al., 2011). Consistent with these phylogenetic relationships, the MmuPV1 E6 protein has similar cellular targets as the β-HPV8 E6 protein (Meyers et al., 2017). The MmuPV1 E7 protein is related to the γ-HPV197 E7 protein in that it lacks the canonical LXCXE-based RB1 binding site (Joh et al., 2011; Grace and Munger, 2017). Moreover, transcriptomic analyses revealed that similar to cutaneous HPVs, MmuPV1 expresses E6 and E7 from two separate early promoters (Xue et al., 2017).
Because of its ability to infect cutaneous epithelia and cause lesions capable of malignant progression upon UV exposure, MmuPV1 may be an important model for determining the potential roles of β- and some γ-HPVs in skin cancer development.
Interestingly, MmuPV1 can productively infect both cutaneous and mucosal tissues (Cladel et al., 2013, 2016; Hu et al., 2015) and thus may also model aspects of mucosal pathogenesis. In any case, results from infection experiments with mutant MmuPV1 genomes can be combined with infection studies of relevant, genetically engineered mouse models. This will enable studies to define mechanisms of action of viral proteins and to determine how viral targeting of specific cellular signaling pathways contribute to the life cycle, pathogenesis and carcinogenicity of a papillomavirus in a natural host.
Subversion of TGF-β and Notch Signaling by Cutaneous Papillomaviruses
While inhibition of UV-mediated apoptosis and/or DNA damage sensing/repair provides a satisfying model to mechanistically support a hit-and-run mechanism of cutaneous HPV mediated carcinogenesis, it does not explain how these viruses may cause warts, a necessary precursor to cSCCs. Moreover, studies with MmuPV1 showed that UV-mediated immune suppression rather than mutagenesis was important for pathogenesis and tumor development (Uberoi et al., 2016).
Inhibition of TGF-β Signaling
Multiple developmental and cellular processes are regulated by TGF-β signaling. The core components of this signal transduction pathway consist of the ligand, TGF-β, heterotetrameric receptors with serine/threonine kinase activity, receptor associated SMAD (R-SMADs) proteins, and co-activator SMADs (co-SMADs). The tetrameric receptor consists of one dimer of the ligand interacting TGF-β type II receptor (TGFBR2), and one dimer of TGF-β type I receptor (TGFBR1). TGF-β binding triggers a phosphorylation cascade from TGFBR2 to TGFBR1 to R-SMADs (Chaikuad and Bullock, 2016). Activin/Nodal and TGFBR1 receptors phosphorylate the R-SMAD pair SMAD2 and SMAD3, whereas the Bone Morphogenetic Protein (BMP) type I receptors activate the R-SMADs SMAD1, SMAD5, and SMAD8 (Miyazawa et al., 2002). After phosphorylation, R-SMADs complex with the co-SMAD, SMAD4, enter the nucleus, bind specific DNA sequences, and activate target gene transcription (Figure 1A). Transcriptional activation of the CDK2 inhibitors p21CIP1 (CDKN1A) and p27KIP1 (CDKN1B), and the CDK4/CDK6 inhibitor p15INK4B (CDKN2B) have been linked to TGF-β mediated G1 cell cycle arrest (Datto et al., 1995; Reynisdottir et al., 1995). Hence, TGF-β functions as a tumor suppressor in epithelial cells, and TGF-β pathway loss of function mutations are frequent in epithelial tumors (Wang et al., 2000; Qiu et al., 2007; Fleming et al., 2013). Importantly, however, TGF-β signaling also induces expression of factors involved in epithelial to mesenchymal transition (EMT) and causes increased production of matrix metalloproteases, which results in enhanced migration and invasion (Valcourt et al., 2016). This is consistent with an oncogenic activity of TGF-β as was originally discovered in fibroblasts (Roberts et al., 1985). Hence, the NOTCH and TGF-β signaling pathways each have tumor suppressor activities at early stages of carcinogenesis and can function as oncogenes during later stages of carcinogenesis.
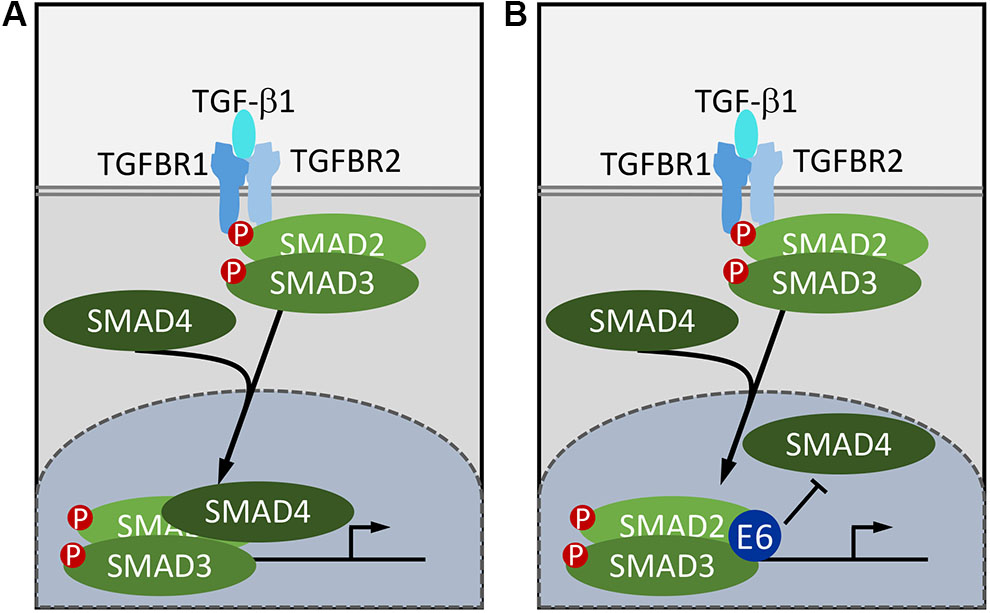
FIGURE 1. Subversion of TGF-β signaling by HPV8 and MmuPV1 E6. In normal cells, TGF-β binding to the heterotetrameric serine/threonine kinase receptor complex triggers a phosphorylation cascade resulting in phosphorylation of receptor the SMADs, SMAD2, and SMAD3. Phosphorylated receptor SMADs disassociate from the receptor, form a complex with the co-SMAD, SMAD4, and enter the nucleus. The SMAD2/SMAD3/SMAD4 complex then binds to SMAD binding sites to stimulate target gene transcription (A). HPV8 and MmuPV1 E6 proteins bind SMAD2 and SMAD3 and inhibit TGF-β signaling by preventing the formation of the active DNA bound SMAD2/SMAD3/SMAD4 complex (B).
The first evidence that cutaneous HPVs inhibit TGF-β signaling was provided by Michel Favre’s group who reported that HPV5 E6 could target SMAD3 for proteasomal degradation (Mendoza et al., 2006). Consistent with this report, AP/MS experiments provided evidence for SMAD2 and SMAD3 association with HPV8 E6 (Meyers et al., 2017). Similarly, the MmuPV1 E6 protein was also shown to bind SMAD2 and SMAD3 (Meyers et al., 2017). Unlike many other papillomavirus E6 binding cellular proteins, SMAD2 and SMAD3 do not contain helical LXXLL (L, leucine; X, any amino acid) motifs (Chen et al., 1998). There is no evidence for SMAD2, SMAD3, or SMAD4 destabilization by HPV8 E6 or MmuPV1 E6 either under basal or TGF-β stimulated conditions (Meyers et al., 2017). Moreover, there is no defect in SMAD2/SMAD3 phosphorylation and nuclear translocation following TGF-β stimulation in HPV8 or MmuPV1 E6 expressing human keratinocytes. The HPV8 and MmuPV1 E6 proteins, however, were shown to reduce SMAD4 association with SMAD2 and SMAD3 after TGF-β stimulation and to inhibit SMAD2/3 and SMAD4 binding to target DNA. Hence cutaneous papillomavirus E6 proteins inhibit TGF-β signaling by blocking productive binding of the SMAD2/3/4 complex to TGF-β responsive promoters (Meyers et al., 2017; Figure 1B). It will be interesting to determine whether these E6 proteins can also inhibit signaling by TGF-β related cytokines. Interestingly, TGF-β signaling is also inhibited by α-HPVs and high-risk HPV16 has been reported to inhibit TGF-β through E7 (Pietenpol et al., 1990; Lee et al., 2002) as well as E5 (French et al., 2013), but not through E6. Hence the cutaneous and mucosal HPVs share the ability to inhibit TGF-β signaling although they have evolved different mechanisms.
Inhibition of NOTCH Signaling
Similar to TGF-β, NOTCH regulates multiple processes during embryogenesis and in the adult organism. Of particular interest to papillomavirus biology, NOTCH signaling drives epithelial differentiation and is required for proper formation of the skin barrier (Rangarajan et al., 2001). NOTCH signaling is triggered through cell-to-cell contact where membrane anchored ligands of the Delta or Jagged family bind to NOTCH family receptors. In humans there are four NOTCH receptors (NOTCH1–4), which exhibit some overlapping functions, though this may be context specific (Kojika and Griffin, 2001). Receptor–ligand binding induces conformational changes in NOTCH that result in a series of proteolytic cleavages ultimately releasing an intracellular fragment of the NOTCH receptor, termed Intracellular NOTCH (ICN; De Strooper et al., 1999). ICN translocates to the nucleus where it associates with Recombination Signal Binding Protein for Immunoglobulin Kappa J Region (RBPJ), which binds to specific DNA sequences. In the absence of ICN, RBPJ is bound to the DNA in association with co-repressors, thus repressing target gene expression (Oswald et al., 2002; Figure 2A). ICN displaces these co-repressors and the ICN–RBPJ complex recruits a member of the transcriptional co-activators mastermind-like (MAML) family (Nam et al., 2006). The ICN/RBPJ/MAML complex represents the core NOTCH transcriptional activator complex and MAML then recruits additional co-activators (Figure 2B; Saint Just Ribeiro and Wallberg, 2009). Classical NOTCH targets include the Hairy and Enhancer of Split (HES) gene, which belongs to a family of basic helix-loop-helix transcriptional repressors, which are particularly important for lateral inhibition and patterning during development (Iso et al., 2003). NOTCH also regulates other targets such as c-myc (MYC; Weng et al., 2006), cyclin D1 (CCND1; Cohen et al., 2010), and the p21CIP1 (CDKN1A) cyclin dependent kinase inhibitor (Rangarajan et al., 2001). Due to the variety of cell types where NOTCH signaling is important, NOTCH target genes are lineage specific and critical NOTCH targets that drive keratinocyte differentiation have not been well-characterized.
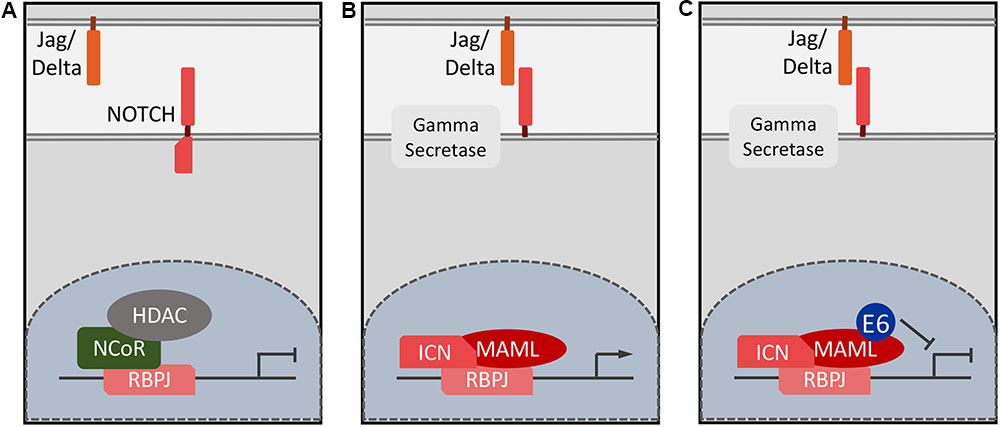
FIGURE 2. Schematic of NOTCH target gene expression. In the absence of ligand binding, NOTCH responsive genes remain repressed and RBPJ is in a DNA bound, nuclear complex with repressors such as the N-CoR complex (A). Ligand binding leads to NOTCH cleavage and ICN enters the nucleus, displaces repressors and recruits MAML and other co-activators such EP300/CREBBP (B). HPV8 and MmuPV1 E6 proteins inhibit NOTCH signaling by forming a complex with the DNA bound ICN/MAML1 complex thereby preventing formation of a functional transcriptional activator complex (C).
Human cSCCs frequently harbor loss of function mutations in the NOTCH pathway, implicating NOTCH activity as tumor suppressive in epithelial cells (Nicolas et al., 2003; Agrawal et al., 2011; Stransky et al., 2011; Wang et al., 2011; South et al., 2014). The tumor suppressive effects of NOTCH in keratinocytes are largely thought to be due to a differentiation-induced block of cell proliferation and the activation of keratinocyte cell death (Zhang et al., 2016; Aster et al., 2017). NOTCH, however, also functions as an oncogenic driver in a number of human tumors, particularly hematopoietic cancers such as T-cell acute lymphoblastic leukemia (T-ALL) where gain of function mutations are commonly observed (Demarest et al., 2008).
MAML1 was originally identified in a yeast-two hybrid screen with HPV16 E6 as a bait (Wu et al., 2000). Like many cellular E6 binding proteins, MAML1 contains LXXLL sequence motifs and several groups detected MAML1 as a cellular interactor of β-HPV E6 proteins in AP/MS experiments (Brimer et al., 2012; Rozenblatt-Rosen et al., 2012; Tan et al., 2012; White et al., 2012a). Similarly, the bovine papillomavirus type 1 (BPV1) E6 protein was also shown to bind MAML1 (Brimer et al., 2012; Tan et al., 2012). Despite the original identification as an HPV16 E6 associated cellular protein (Wu et al., 2000), there is no evidence that mucosal α-HPV E6 proteins detectably associate with MAML1 in mammalian cells (Brimer et al., 2017).
Biological Consequences of TGF-β and NOTCH Inhibition by Cutaneous Papillomaviruses
Given that the TGF-β and NOTCH pathways are critical in regulating epithelial differentiation and function as tumor suppressors in a variety of epithelial cancers, inhibition of these two pathways by cutaneous papillomavirus E6 proteins may be key to the transforming activities of these viruses.
Consistent with this model, it was shown that keratinocyte differentiation is inhibited in HPV8 E6 expressing epithelial cells. Chromatin immunoprecipitation experiments showed that E6 binds to and inhibits the DNA bound RBPJ/ICN/MAML1 activator complex (Meyers et al., 2013). HPV8 E6 binds to an LXXLL motif within the C-terminal transactivation domain (TAD) 2 of MAML1 (Tan et al., 2012; Meyers et al., 2017). The mechanism of action of TAD2 is unknown; EP300 and related co-activators associate with MAML through its TAD1 domain (Saint Just Ribeiro and Wallberg, 2009; Gerhardt et al., 2014). The MmuPV1 E6 protein also interacts with MAML1 and inhibits NOTCH signaling thereby inhibiting keratinocyte differentiation (Meyers et al., 2017; Figure 2C). MAML1 binding defective HPV8 and MmuPV1 E6 mutants were identified using a structure guided approach (Meyers et al., 2017). Of note, many of the HPV8 E6 mutants that were previously used to characterize the biological relevance of the interaction between cutaneous HPV E6 protein and EP300/CREBBP, were found to be MAML1 binding defective, thus further complicating the interpretation of published studies investigating the biological consequences of the EP300/CREBBP interactions with HPV5 and HPV8 E6 (Meyers et al., 2017). Association of E6 with MAML and inhibition of NOTCH signaling is independent of EP300/CREBBP binding, however, since MAML1 binding defective HPV8 E6 mutants retained EP300/CREBBP association and MmuPV1 E6 does not detectably bind EP300/CREBBP (Meyers et al., 2017). Indeed, the notable absence of EP300/CREBBP peptides in AP/MS experiments with MmuPV1 E6 is consistent with a model whereby E6 binding to TAD2 of MAML1 may inhibit expression of NOTCH transcriptional targets by interfering with recruitment or by displacing EP300/CREBBP from MAML1 TAD1. This model, however, awaits experimental validation.
The presence of an intact binding site for MAML1 in MmuPV1 E6 was shown to be necessary for formation of papillomas and carcinomas in experimentally infected mice (Meyers et al., 2017). While these studies cannot definitively exclude the possibility that association of E6 with other LXXLL domain proteins may also contribute to this phenotype, this is unlikely given a recent study that showed that UBE3A and MAML1 are the major LXXLL targets of papillomavirus E6 proteins and, consistent with the published E6/LXXLL structures (Zanier et al., 2013), the various papillomavirus E6 proteins displayed mutually exclusive binding to either UBE3A or MAML1 (Brimer et al., 2017).
Despite the fact that the E6 proteins of mucosal, high-risk α-HPVs have a strong preference for UBE3A binding and do not detectably interact with MAML1 (White et al., 2012a; Brimer et al., 2017), they have also been reported to inhibit NOTCH. HPV16 E6 can inhibit NOTCH signaling indirectly through degradation of the TP53 tumor suppressor (Dotto, 2009) and/or the TAp63β protein (Ben Khalifa et al., 2011). More recent studies showed that NOTCH inhibition is relevant to the biology of these viruses as HPV16 E6 mediated NOTCH inhibition was shown to decrease the commitment of keratinocytes to differentiation (Kranjec et al., 2017).
Hence, similar to TGF-β, inhibition of keratinocyte differentiation by targeting NOTCH signaling by cutaneous HPVs is shared with mucosal HPVs but is mediated through different mechanisms. Conceptually this is rather satisfying as papillomaviruses all had to evolve to render normally postmitotic, terminally differentiated epithelial cells conducive for supporting the viral life cycle (Figure 3). Interestingly however, different papillomaviruses have evolved mechanistically distinct strategies to solve this problem.
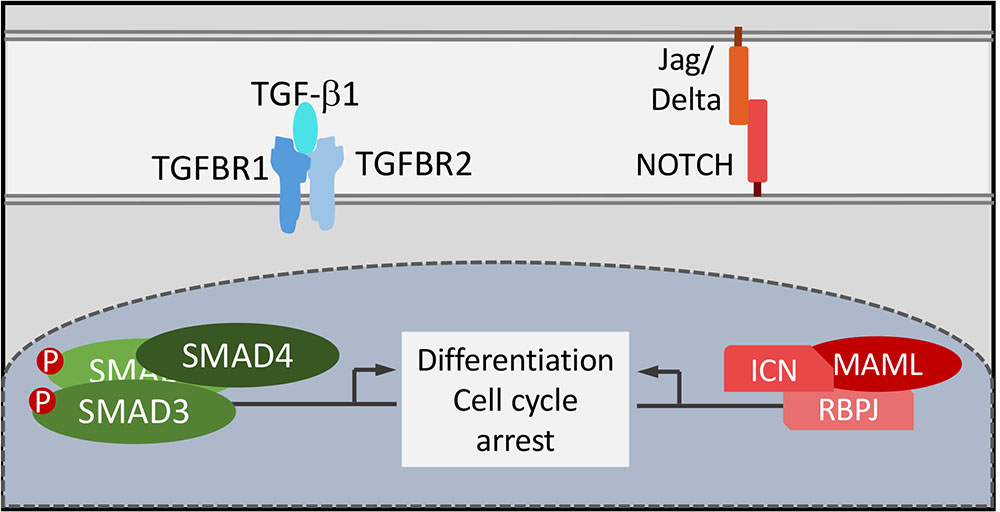
FIGURE 3. Functional consequences of NOTCH and TGF-β signaling by HPV8 E6 and MmuPV1 E6. TGF-β and NOTCH signaling are critical for cell cycle arrest and keratinocyte differentiation that are tightly coupled in normal squamous epithelial cells. Functional uncoupling of these two processes is a necessary prerequisite for papillomavirus progeny formation in terminally differentiated epithelial cells.
“Hit and Run” Carcinogenesis: A Consequence of E6 Mediated NOTCH and TGF-β Inhibition?
Hit and run carcinogenesis is a concept that historically has generated some skepticism, also with the authors of this article. The failure to detect viral gene expression in the majority of cancer cells may also imply that β- and γ-HPVs are biologically innocuous passengers that do not drive the carcinogenic process. Given, however, that at least some of these papillomaviruses are carcinogenic in transgenic mouse models and inhibit TGF-β and NOTCH signaling as well as other tumor suppressor pathways, it is more likely that these viruses provide oncogenic hits that drive at least some early aspects of epithelial carcinogenesis. In addition to many other biological activities of cutaneous HPVs, which include stem cell expansion, abrogating apoptosis, sensing and repair of UV-induced double strand DNA breaks, inhibition of the TGF-β and NOTCH tumor suppressors provide plausible oncogenic “hits” during early phases of carcinogenesis. Since the NOTCH and TGF-β pathways are oncogenic at later stages and drive malignant progression (Massague, 2008; Aster et al., 2017) loss of papillomavirus gene expression will re-activate NOTCH and TGF-β. This may drive malignant progression, thereby providing the mechanistic underpinnings for the “run” component of hit and run carcinogenesis.
It is important to note that the β- and γ-HPVs are phylogenetically and biologically diverse (Cornet et al., 2012; Van Doorslaer et al., 2017) and that this diversity is also reflected in the cellular interactomes of their E6 and E7 proteins (White et al., 2012a,b). Hence, findings with one specific β- or γ-HPV type may not be applicable to other HPVs, even within the same genus. The accumulation of UV-induced mutations in a recently published HPV38 transgenic skin cancer model that recapitulates hit-and-run carcinogenesis, for example, is unlikely to be caused through an EP300/CREBBP dependent mechanism as no EP300/CREBBP binding has been detected in unbiased AP/MS studies with HPV38 E6 (White et al., 2012a; Viarisio et al., 2018).
The wide application of AP/MS to define cellular interactomes of papillomavirus proteins has already generated interesting hypotheses, many of which await rigorous validation (White and Howley, 2013; McBride, 2017). Next generation sequencing experiments may detect HPV DNA and/or RNA sequences in unexpected anatomic locations and tentatively link them to unanticipated pathologies (Chen et al., 2017). Such studies may also lead to the identification of HPVs that evaded detection by standard PCR-based techniques. The frequent detection of the novel γ-HPV197 genome in a deep sequencing study of human skin cancers, but not in normal skin, while not implying an etiologic link, should provide at least an impetus to study the biology of γ-HPVs in greater detail (Arroyo Muhr et al., 2015). Given that the HPV197 E7 protein, similar to MmuPV1, lacks an LXCXE-based canonical RB1 binding domain, and binds RB1 through a different sequence, suggest that some aspects of HPV197 E7 can be studied with the MmuPV1 animal model. These and other studies might also reveal whether HPV197, and potentially other γ-HPVs, have oncogenic activities.
Author Contributions
JM and KM drafted the manuscript. AU, PL, and MG provided editorial and substantive input. KM wrote the final version of the manuscript.
Funding
This work was supported by grants from the National Institutes of Health under CA066980 (KM), CA022443 (PL), and AR066524 (PL). JM was a Ryan Fellow.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnote
References
Accardi, R., Dong, W., Smet, A., Cui, R., Hautefeuille, A., Gabet, A. S., et al. (2006). Skin human papillomavirus type 38 alters p53 functions by accumulation of deltaNp73. EMBO Rep. 7, 334–340. doi: 10.1038/sj.embor.7400615
Agrawal, N., Frederick, M. J., Pickering, C. R., Bettegowda, C., Chang, K., Li, R. J., et al. (2011). Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 333, 1154–1157. doi: 10.1126/science.1206923
Alam, M., and Ratner, D. (2001). Cutaneous squamous-cell carcinoma. N. Engl. J. Med. 344, 975–983. doi: 10.1056/NEJM200103293441306
Antonsson, A., Erfurt, C., Hazard, K., Holmgren, V., Simon, M., Kataoka, A., et al. (2003a). Prevalence and type spectrum of human papillomaviruses in healthy skin samples collected in three continents. J. Gen. Virol. 84, 1881–1886.
Antonsson, A., Karanfilovska, S., Lindqvist, P. G., and Hansson, B. G. (2003b). General acquisition of human papillomavirus infections of skin occurs in early infancy. J. Clin. Microbiol. 41, 2509–2514.
Arron, S. T., Ruby, J. G., Dybbro, E., Ganem, D., and Derisi, J. L. (2011). Transcriptome sequencing demonstrates that human papillomavirus is not active in cutaneous squamous cell carcinoma. J. Invest. Dermatol. 131, 1745–1753. doi: 10.1038/jid.2011.91
Arroyo Muhr, L. S., Hultin, E., Bzhalava, D., Eklund, C., Lagheden, C., Ekstrom, J., et al. (2015). Human papillomavirus type 197 is commonly present in skin tumors. Int. J. Cancer 136, 2546–2555. doi: 10.1002/ijc.29325
Aster, J. C., Pear, W. S., and Blacklow, S. C. (2017). The varied roles of notch in cancer. Annu. Rev. Pathol. 12, 245–275. doi: 10.1146/annurev-pathol-052016-100127
Ben Khalifa, Y., Teissier, S., Tan, M. K., Phan, Q. T., Daynac, M., Wong, W. Q., et al. (2011). The human papillomavirus E6 oncogene represses a cell adhesion pathway and disrupts focal adhesion through degradation of TAp63beta upon transformation. PLoS Pathog. 7:e1002256. doi: 10.1371/journal.ppat.1002256
Borgogna, C., Lanfredini, S., Peretti, A., De Andrea, M., Zavattaro, E., Colombo, E., et al. (2014). Improved detection reveals active beta-papillomavirus infection in skin lesions from kidney transplant recipients. Mod. Pathol. 27, 1101–1115. doi: 10.1038/modpathol.2013.240
Borgogna, C., Zavattaro, E., De Andrea, M., Griffin, H. M., Dell’oste, V., Azzimonti, B., et al. (2012). Characterization of beta papillomavirus E4 expression in tumours from Epidermodysplasia Verruciformis patients and in experimental models. Virology 423, 195–204. doi: 10.1016/j.virol.2011.11.029
Bottalico, D., Chen, Z., Dunne, A., Ostoloza, J., Mckinney, S., Sun, C., et al. (2011). The oral cavity contains abundant known and novel human papillomaviruses from the Betapapillomavirus and Gammapapillomavirus genera. J. Infect. Dis. 204, 787–792. doi: 10.1093/infdis/jir383
Brimer, N., Drews, C. M., and Vande Pol, S. B. (2017). Association of papillomavirus E6 proteins with either MAML1 or E6AP clusters E6 proteins by structure, function, and evolutionary relatedness. PLoS Pathog. 13:e1006781. doi: 10.1371/journal.ppat.1006781
Brimer, N., Lyons, C., Wallberg, A. E., and Vande Pol, S. B. (2012). Cutaneous papillomavirus E6 oncoproteins associate with MAML1 to repress transactivation and NOTCH signaling. Oncogene 31, 4639–4646. doi: 10.1038/onc.2011.589
Caldeira, S., Zehbe, I., Accardi, R., Malanchi, I., Dong, W., Giarre, M., et al. (2003). The E6 and E7 proteins of the cutaneous human papillomavirus type 38 display transforming properties. J. Virol. 77, 2195–2206. doi: 10.1128/JVI.77.3.2195-2206.2003
CDC (2015). Epidemiology and Prevention of Vaccine-Preventable Diseases. Washington, DC: Public Health Foundation.
Chahoud, J., Semaan, A., Chen, Y., Cao, M., Rieber, A. G., Rady, P., et al. (2016). Association between betα-genus human papillomavirus and cutaneous squamous cell carcinoma in immunocompetent individuals-A meta-analysis. JAMA Dermatol. 152, 1354–1364. doi: 10.1001/jamadermatol.2015.4530
Chaikuad, A., and Bullock, A. N. (2016). Structural basis of intracellular TGF-β signaling: receptors and smads. Cold Spring Harb. Perspect. Biol. 8:a022111. doi: 10.1101/cshperspect.a022111
Chen, A. A., Gheit, T., Stellin, M., Lupato, V., Spinato, G., Fuson, R., et al. (2017). Oncogenic DNA viruses found in salivary gland tumors. Oral Oncol. 75, 106–110. doi: 10.1016/j.oraloncology.2017.11.005
Chen, A. C., Mcmillan, N. A., and Antonsson, A. (2008). Human papillomavirus type spectrum in normal skin of individuals with or without a history of frequent sun exposure. J. Gen. Virol. 89, 2891–2897. doi: 10.1099/vir.0.2008/003665-0
Chen, J. J., Hong, Y., Rustamzadeh, E., Baleja, J. D., and Androphy, E. J. (1998). Identification of an alpha helical motif sufficient for association with papillomavirus E6. J. Biol. Chem. 273, 13537–13544. doi: 10.1074/jbc.273.22.13537
Chuang, T. Y., Reizner, G. T., Elpern, D. J., Stone, J. L., and Farmer, E. R. (1993). Keratoacanthoma in Kauai, Hawaii. The first documented incidence in a defined population. Arch. Dermatol. 129, 317–319. doi: 10.1001/archderm.1993.01680240057005
Cladel, N. M., Budgeon, L. R., Balogh, K. K., Cooper, T. K., Hu, J., and Christensen, N. D. (2016). Mouse papillomavirus MmuPV1 infects oral mucosa and preferentially targets the base of the tongue. Virology 488, 73–80. doi: 10.1016/j.virol.2015.10.030
Cladel, N. M., Budgeon, L. R., Cooper, T. K., Balogh, K. K., Hu, J., and Christensen, N. D. (2013). Secondary infections, expanded tissue tropism, and evidence for malignant potential in immunocompromised mice infected with Mus musculus papillomavirus 1 DNA and virus. J. Virol. 87, 9391–9395. doi: 10.1128/JVI.00777-13
Cohen, B., Shimizu, M., Izrailit, J., Ng, N. F., Buchman, Y., Pan, J. G., et al. (2010). Cyclin D1 is a direct target of JAG1-mediated Notch signaling in breast cancer. Breast Cancer Res. Treat. 123, 113–124. doi: 10.1007/s10549-009-0621-9
Cornet, I., Bouvard, V., Campo, M. S., Thomas, M., Banks, L., Gissmann, L., et al. (2012). Comparative analysis of transforming properties of E6 and E7 from different beta human papillomavirus types. J. Virol. 86, 2366–2370. doi: 10.1128/JVI.06579-11
Datto, M. B., Li, Y., Panus, J. F., Howe, D. J., Xiong, Y., and Wang, X. F. (1995). Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. U.S.A. 92, 5545–5549. doi: 10.1073/pnas.92.12.5545
De Strooper, B., Annaert, W., Cupers, P., Saftig, P., Craessaerts, K., Mumm, J. S., et al. (1999). A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522. doi: 10.1038/19083
Deady, S., Sharp, L., and Comber, H. (2014). Increasing skin cancer incidence in young, affluent, urban populations: a challenge for prevention. Br. J. Dermatol. 171, 324–331. doi: 10.1111/bjd.12988
Del Rosso, J. Q., Kircik, L., Goldenberg, G., and Brian, B. (2014). Comprehensive management of actinic keratoses: practical integration of available therapies with a review of a newer treatment approach. J. Clin. Aesthet. Dermatol. 7, S2–S12.
Demarest, R. M., Ratti, F., and Capobianco, A. J. (2008). It’s T-ALL about Notch. Oncogene 27, 5082–5091. doi: 10.1038/onc.2008.222
Dotto, G. P. (2009). Crosstalk of Notch with p53 and p63 in cancer growth control. Nat. Rev. Cancer 9, 587–595. doi: 10.1038/nrc2675
Elbel, M., Carl, S., Spaderna, S., and Iftner, T. (1997). A comparative analysis of the interactions of the E6 proteins from cutaneous and genital papillomaviruses with p53 and E6AP in correlation to their transforming potential. Virology 239, 132–149. doi: 10.1006/viro.1997.8860
Feltkamp, M. C., De Koning, M. N., Bavinck, J. N., and Ter Schegget, J. (2008). Betapapillomaviruses: innocent bystanders or causes of skin cancer. J. Clin. Virol. 43, 353–360. doi: 10.1016/j.jcv.2008.09.009
Fleming, N. I., Jorissen, R. N., Mouradov, D., Christie, M., Sakthianandeswaren, A., Palmieri, M., et al. (2013). SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. 73, 725–735. doi: 10.1158/0008-5472.CAN-12-2706
Forslund, O. (2007). Genetic diversity of cutaneous human papillomaviruses. J. Gen. Virol. 88, 2662–2669. doi: 10.1099/vir.0.82911-0
Foulongne, V., Sauvage, V., Hebert, C., Dereure, O., Cheval, J., Gouilh, M. A., et al. (2012). Human skin microbiota: high diversity of DNA viruses identified on the human skin by high throughput sequencing. PLoS One 7:e38499. doi: 10.1371/journal.pone.0038499
Frazer, I. H. (2004). Prevention of cervical cancer through papillomavirus vaccination. Nat. Rev. Immunol. 4, 46–54. doi: 10.1038/nri1260
French, D., Belleudi, F., Mauro, M. V., Mazzetta, F., Raffa, S., Fabiano, V., et al. (2013). Expression of HPV16 E5 down-modulates the TGFbeta signaling pathway. Mol. Cancer 12:38. doi: 10.1186/1476-4598-12-38
Genther, S. M., Sterling, S., Duensing, S., Munger, K., Sattler, C., and Lambert, P. F. (2003). Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. J. Virol. 77, 2832–2842. doi: 10.1128/JVI.77.5.2832-2842.2003
Gerhardt, D. M., Pajcini, K. V., D’altri, T., Tu, L., Jain, R., Xu, L., et al. (2014). The Notch1 transcriptional activation domain is required for development and reveals a novel role for Notch1 signaling in fetal hematopoietic stem cells. Genes Dev. 28, 576–593. doi: 10.1101/gad.227496.113
Grace, M., and Munger, K. (2017). Proteomic analysis of the gamma human papillomavirus type 197 E6 and E7 associated cellular proteins. Virology 500, 71–81. doi: 10.1016/j.virol.2016.10.010
Guy, G. P. Jr., Machlin, S. R., Ekwueme, D. U., and Yabroff, K. R. (2015). Prevalence and costs of skin cancer treatment in the U.S., 2002-2006 and 2007-2011. Am. J. Prev. Med. 48, 183–187. doi: 10.1016/j.amepre.2014.08.036
Hampras, S. S., Rollison, D. E., Giuliano, A. R., Mckay-Chopin, S., Minoni, L., Sereday, K., et al. (2017). Prevalence and concordance of cutaneous beta human papillomavirus infection at mucosal and cutaneous sites. J. Infect. Dis. 216, 92–96. doi: 10.1093/infdis/jix245
Handisurya, A., Day, P. M., Thompson, C. D., Bonelli, M., Lowy, D. R., and Schiller, J. T. (2014). Strain-specific properties and T cells regulate the susceptibility to papilloma induction by Mus musculus papillomavirus 1. PLoS Pathog. 10:e1004314. doi: 10.1371/journal.ppat.1004314
Harden, M. E., and Munger, K. (2017). Human papillomavirus molecular biology. Mutat. Res. Rev. Mutat. Res. 772, 3–12. doi: 10.1016/j.mrrev.2016.07.002
Hellner, K., and Munger, K. (2011). Human papillomaviruses as therapeutic targets in human cancer. J. Clin. Oncol. 29, 1785–1794. doi: 10.1200/JCO.2010.28.2186
Howie, H. L., Koop, J. I., Weese, J., Robinson, K., Wipf, G., Kim, L., et al. (2011). Betα-HPV 5 and 8 E6 promote p300 degradation by blocking AKT/p300 association. PLoS Pathog. 7:e1002211. doi: 10.1371/journal.ppat.1002211
Howley, P. M., and Pfister, H. J. (2015). Beta genus papillomaviruses and skin cancer. Virology 479–480, 290–296. doi: 10.1016/j.virol.2015.02.004
Hu, J., Budgeon, L. R., Cladel, N. M., Balogh, K., Myers, R., Cooper, T. K., et al. (2015). Tracking vaginal, anal and oral infection in a mouse papillomavirus infection model. J. Gen. Virol. 96, 3554–3565. doi: 10.1099/jgv.0.000295
Hufbauer, M., Biddle, A., Borgogna, C., Gariglio, M., Doorbar, J., Storey, A., et al. (2013). Expression of Betapapillomavirus oncogenes increases the number of keratinocytes with stem cell-like properties. J. Virol. 87, 12158–12165. doi: 10.1128/JVI.01510-13
Hufbauer, M., Cooke, J., Van Der Horst, G. T., Pfister, H., Storey, A., and Akgul, B. (2015). Human papillomavirus mediated inhibition of DNA damage sensing and repair drives skin carcinogenesis. Mol. Cancer 14:183. doi: 10.1186/s12943-015-0453-7
Ingle, A., Ghim, S., Joh, J., Chepkoech, I., Bennett Jenson, A., and Sundberg, J. P. (2011). Novel laboratory mouse papillomavirus (MusPV) infection. Vet. Pathol. 48, 500–505. doi: 10.1177/0300985810377186
Iso, T., Kedes, L., and Hamamori, Y. (2003). HES and HERP families: multiple effectors of the Notch signaling pathway. J. Cell. Physiol. 194, 237–255. doi: 10.1002/jcp.10208
Jackson, S., Harwood, C., Thomas, M., Banks, L., and Storey, A. (2000). Role of Bak in UV-induced apoptosis in skin cancer and abrogation by HPV E6 proteins. Genes Dev. 14, 3065–3073. doi: 10.1101/gad.182100
Joh, J., Ghim, S. J., Chilton, P. M., Sundberg, J. P., Park, J., Wilcher, S. A., et al. (2016). MmuPV1 infection and tumor development of T cell-deficient mice is prevented by passively transferred hyperimmune sera from normal congenic mice immunized with MmuPV1 virus-like particles (VLPs). Exp. Mol. Pathol. 100, 212–219. doi: 10.1016/j.yexmp.2016.01.003
Joh, J., Jenson, A. B., King, W., Proctor, M., Ingle, A., Sundberg, J. P., et al. (2011). Genomic analysis of the first laboratory-mouse papillomavirus. J. Gen. Virol. 92, 692–698. doi: 10.1099/vir.0.026138-0
Kilkenny, M., Merlin, K., Young, R., and Marks, R. (1998). The prevalence of common skin conditions in Australian school students: 1. Common, plane and plantar viral warts. Br. J. Dermatol. 138, 840–845. doi: 10.1046/j.1365-2133.1998.02222.x
Kohler, A., Forschner, T., Meyer, T., Ulrich, C., Gottschling, M., Stockfleth, E., et al. (2007). Multifocal distribution of cutaneous human papillomavirus types in hairs from different skin areas. Br. J. Dermatol. 156, 1078–1080. doi: 10.1111/j.1365-2133.2007.07809.x
Kojika, S., and Griffin, J. D. (2001). Notch receptors and hematopoiesis. Exp. Hematol. 29, 1041–1052. doi: 10.1016/S0301-472X(01)00676-2
Kranjec, C., Holleywood, C., Libert, D., Griffin, H., Mahmood, R., Isaacson, E., et al. (2017). Modulation of basal cell fate during productive and transforming HPV-16 infection is mediated by progressive E6-driven depletion of Notch. J. Pathol. 242, 448–462. doi: 10.1002/path.4917
Lanfredini, S., Olivero, C., Borgogna, C., Calati, F., Powell, K., Davies, K. J., et al. (2017). HPV8 field cancerization in a transgenic mouse model is due to Lrig1+ keratinocyte stem cell expansion. J. Invest. Dermatol. 137, 2208–2216. doi: 10.1016/j.jid.2017.04.039
Lee, D. K., Kim, B. C., Kim, I. Y., Cho, E. A., Satterwhite, D. J., and Kim, S. J. (2002). The human papilloma virus E7 oncoprotein inhibits transforming growth factor-beta signaling by blocking binding of the Smad complex to its target sequence. J. Biol. Chem. 277, 38557–38564. doi: 10.1074/jbc.M206786200
Leverrier, S., Bergamaschi, D., Ghali, L., Ola, A., Warnes, G., Akgul, B., et al. (2007). Role of HPV E6 proteins in preventing UVB-induced release of pro-apoptotic factors from the mitochondria. Apoptosis 12, 549–560. doi: 10.1007/s10495-006-0004-1
Mahe, E., Bodemer, C., Descamps, V., Mahe, I., Crickx, B., De Prost, Y., et al. (2003). High frequency of detection of human papillomaviruses associated with epidermodysplasia verruciformis in children with psoriasis. Br. J. Dermatol. 149, 819–825. doi: 10.1046/j.1365-2133.2003.05587.x
Marcuzzi, G. P., Hufbauer, M., Kasper, H. U., Weissenborn, S. J., Smola, S., and Pfister, H. (2009). Spontaneous tumour development in human papillomavirus type 8 E6 transgenic mice and rapid induction by UV-light exposure and wounding. J. Gen. Virol. 90, 2855–2864. doi: 10.1099/vir.0.012872-0
Marthaler, A. M., Podgorska, M., Feld, P., Fingerle, A., Knerr-Rupp, K., Grasser, F., et al. (2017). Identification of C/EBPalpha as a novel target of the HPV8 E6 protein regulating miR-203 in human keratinocytes. PLoS Pathog. 13:e1006406. doi: 10.1371/journal.ppat.1006406
Maufort, J. P., Shai, A., Pitot, H. C., and Lambert, P. F. (2010). A role for HPV16 E5 in cervical carcinogenesis. Cancer Res. 70, 2924–2931. doi: 10.1158/0008-5472.CAN-09-3436
Maufort, J. P., Williams, S. M., Pitot, H. C., and Lambert, P. F. (2007). Human papillomavirus 16 E5 oncogene contributes to two stages of skin carcinogenesis. Cancer Res. 67, 6106–6112. doi: 10.1158/0008-5472.CAN-07-0921
McBride, A. A. (2017). Perspective: the promise of proteomics in the study of oncogenic viruses. Mol. Cell Proteomics 16, S65–S74. doi: 10.1074/mcp.O116.065201
McLaughlin-Drubin, M. E., Meyers, J., and Munger, K. (2012). Cancer associated human papillomaviruses. Curr. Opin. Virol. 2, 459–466. doi: 10.1016/j.coviro.2012.05.004
McLaughlin-Drubin, M. E., and Munger, K. (2009). Oncogenic activities of human papillomaviruses. Virus Res. 143, 195–208. doi: 10.1016/j.virusres.2009.06.008
Mendoza, J. A., Jacob, Y., Cassonnet, P., and Favre, M. (2006). Human papillomavirus type 5 E6 oncoprotein represses the transforming growth factor beta signaling pathway by binding to SMAD3. J. Virol. 80, 12420–12424. doi: 10.1128/JVI.02576-05
Meyers, J. M., and Munger, K. (2014). The viral etiology of skin cancer. J. Invest. Dermatol. 134, E29–E32. doi: 10.1038/skinbio.2014.6
Meyers, J. M., Spangle, J. M., and Munger, K. (2013). The human papillomavirus type 8 E6 protein interferes with NOTCH activation during keratinocyte differentiation. J. Virol. 87, 4762–4767. doi: 10.1128/JVI.02527-12
Meyers, J. M., Uberoi, A., Grace, M., Lambert, P. F., and Munger, K. (2017). Cutaneous HPV8 and MmuPV1 E6 proteins target the NOTCH and TGF-β tumor suppressors to inhibit differentiation and sustain keratinocyte proliferation. PLoS Pathog. 13:e1006171. doi: 10.1371/journal.ppat.1006171
Michel, A., Kopp-Schneider, A., Zentgraf, H., Gruber, A. D., and De Villiers, E. M. (2006). E6/E7 expression of human papillomavirus type 20 (HPV-20) and HPV-27 influences proliferation and differentiation of the skin in UV-irradiated SKH-hr1 transgenic mice. J. Virol. 80, 11153–11164. doi: 10.1128/JVI.00954-06
Miyazawa, K., Shinozaki, M., Hara, T., Furuya, T., and Miyazono, K. (2002). Two major Smad pathways in TGF-β superfamily signalling. Genes Cells 7, 1191–1204. doi: 10.1046/j.1365-2443.2002.00599.x
Moody, C. A., and Laimins, L. A. (2010). Human papillomavirus oncoproteins: pathways to transformation. Nat. Rev. Cancer 10, 550–560. doi: 10.1038/nrc2886
Munger, K., Baldwin, A., Edwards, K. M., Hayakawa, H., Nguyen, C. L., Owens, M., et al. (2004). Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 78, 11451–11460. doi: 10.1128/JVI.78.21.11451-11460.2004
Munger, K., and Jones, D. L. (2015). Human papillomavirus carcinogenesis: an identity crisis in the retinoblastoma tumor suppressor pathway. J. Virol. 89, 4708–4711. doi: 10.1128/JVI.03486-14
Nam, Y., Sliz, P., Song, L., Aster, J. C., and Blacklow, S. C. (2006). Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell 124, 973–983. doi: 10.1016/j.cell.2005.12.037
Neale, R. E., Weissenborn, S., Abeni, D., Bavinck, J. N., Euvrard, S., Feltkamp, M. C., et al. (2013). Human papillomavirus load in eyebrow hair follicles and risk of cutaneous squamous cell carcinoma. Cancer Epidemiol. Biomarkers Prev. 22, 719–727. doi: 10.1158/1055-9965.EPI-12-0917-T
Nicolas, M., Wolfer, A., Raj, K., Kummer, J. A., Mill, P., Van Noort, M., et al. (2003). Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 33, 416–421. doi: 10.1038/ng1099
Orth, G., Jablonska, S., Favre, M., Croissant, O., Jarzabek-Chorzelska, M., and Rzesa, G. (1978). Characterization of two types of human papillomaviruses in lesions of epidermodysplasia verruciformis. Proc. Natl. Acad. Sci. U.S.A. 75, 1537–1541. doi: 10.1073/pnas.75.3.1537
Oswald, F., Kostezka, U., Astrahantseff, K., Bourteele, S., Dillinger, K., Zechner, U., et al. (2002). SHARP is a novel component of the Notch/RBP-Jkappa signalling pathway. EMBO J. 21, 5417–5426. doi: 10.1093/emboj/cdf549
Pass, F., Reissig, M., Shah, K. V., Eisinger, M., and Orth, G. (1977). Identification of an immunologically distinct papillomavirus from lesions of epidermodysplasia verruciformis. J. Natl. Cancer Inst. 59, 1107–1112. doi: 10.1093/jnci/59.4.1107
Pfefferle, R., Marcuzzi, G. P., Akgul, B., Kasper, H. U., Schulze, F., Haase, I., et al. (2008). The human papillomavirus type 8 E2 protein induces skin tumors in transgenic mice. J. Invest. Dermatol. 128, 2310–2315. doi: 10.1038/jid.2008.73
Pietenpol, J. A., Stein, R. W., Moran, E., Yaciuk, P., Schlegel, R., Lyons, R. M., et al. (1990). TGF-β 1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell 61, 777–785. doi: 10.1016/0092-8674(90)90188-K
Qiu, W., Schonleben, F., Li, X., and Su, G. H. (2007). Disruption of transforming growth factor beta-Smad signaling pathway in head and neck squamous cell carcinoma as evidenced by mutations of SMAD2 and SMAD4. Cancer Lett. 245, 163–170. doi: 10.1016/j.canlet.2006.01.003
Quint, K. D., Genders, R. E., De Koning, M. N., Borgogna, C., Gariglio, M., Bouwes Bavinck, J. N., et al. (2015). Human Beta-papillomavirus infection and keratinocyte carcinomas. J. Pathol. 235, 342–354. doi: 10.1002/path.4425
Rangarajan, A., Talora, C., Okuyama, R., Nicolas, M., Mammucari, C., Oh, H., et al. (2001). Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 20, 3427–3436. doi: 10.1093/emboj/20.13.3427
Reynisdottir, I., Polyak, K., Iavarone, A., and Massague, J. (1995). Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-β. Genes Dev. 9, 1831–1845. doi: 10.1101/gad.9.15.1831
Roberts, A. B., Anzano, M. A., Wakefield, L. M., Roche, N. S., Stern, D. F., and Sporn, M. B. (1985). Type beta transforming growth factor: a bifunctional regulator of cellular growth. Proc. Natl. Acad. Sci. U.S.A. 82, 119–123. doi: 10.1073/pnas.82.1.119
Roman, A., and Munger, K. (2013). The papillomavirus E7 proteins. Virology 445, 138–168. doi: 10.1016/j.virol.2013.04.013
Rozenblatt-Rosen, O., Deo, R. C., Padi, M., Adelmant, G., Calderwood, M. A., Rolland, T., et al. (2012). Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 487, 491–495. doi: 10.1038/nature11288
Saint Just Ribeiro, M., and Wallberg, A. E. (2009). Transcriptional mechanisms by the coregulator MAML1. Curr. Protein Pept. Sci. 10, 570–576. doi: 10.2174/138920309789630543
Schaper, I. D., Marcuzzi, G. P., Weissenborn, S. J., Kasper, H. U., Dries, V., Smyth, N., et al. (2005). Development of skin tumors in mice transgenic for early genes of human papillomavirus type 8. Cancer Res. 65, 1394–1400. doi: 10.1158/0008-5472.CAN-04-3263
Schiffman, M., Castle, P. E., Jeronimo, J., Rodriguez, A. C., and Wacholder, S. (2007). Human papillomavirus and cervical cancer. Lancet 370, 890–907. doi: 10.1016/S0140-6736(07)61416-0
Schiller, J. T., and Lowy, D. R. (2012). Understanding and learning from the success of prophylactic human papillomavirus vaccines. Nat. Rev. Microbiol. 10, 681–692. doi: 10.1038/nrmicro2872
Siegel, R. L., Miller, K. D., and Jemal, A. (2018). Cancer statistics, 2018. CA Cancer J. Clin. 68, 7–30. doi: 10.3322/caac.21442
Smelov, V., Hanisch, R., Mckay-Chopin, S., Sokolova, O., Eklund, C., Komyakov, B., et al. (2017). Prevalence of cutaneous beta and gamma human papillomaviruses in the anal canal of men who have sex with women. Papillomavirus Res. 3, 66–72. doi: 10.1016/j.pvr.2017.02.002
South, A. P., Purdie, K. J., Watt, S. A., Haldenby, S., Den Breems, N. Y., Dimon, M., et al. (2014). NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J. Invest. Dermatol. 134, 2630–2638. doi: 10.1038/jid.2014.154
Stransky, N., Egloff, A. M., Tward, A. D., Kostic, A. D., Cibulskis, K., Sivachenko, A., et al. (2011). The mutational landscape of head and neck squamous cell carcinoma. Science 333, 1157–1160. doi: 10.1126/science.1208130
Tan, M. J., White, E. A., Sowa, M. E., Harper, J. W., Aster, J. C., and Howley, P. M. (2012). Cutaneous beta-human papillomavirus E6 proteins bind Mastermind-like coactivators and repress Notch signaling. Proc. Natl. Acad. Sci. U.S.A. 109, E1473–E1480. doi: 10.1073/pnas.1205991109
Tommasino, M. (2017). The biology of beta human papillomaviruses. Virus Res. 231, 128–138. doi: 10.1016/j.virusres.2016.11.013
Uberoi, A., and Lambert, P. F. (2017). Rodent Papillomaviruses. Viruses 9:E362. doi: 10.3390/v9120362
Uberoi, A., Yoshida, S., Frazer, I. H., Pitot, H. C., and Lambert, P. F. (2016). Role of ultraviolet radiation in papillomavirus-induced disease. PLoS Pathog. 12:e1005664. doi: 10.1371/journal.ppat.1005664
Valcourt, U., Carthy, J., Okita, Y., Alcaraz, L., Kato, M., Thuault, S., et al. (2016). Analysis of epithelial-mesenchymal transition induced by transforming growth factor beta. Methods Mol. Biol. 1344, 147–181. doi: 10.1007/978-1-4939-2966-5_9
Van Doorslaer, K., Li, Z., Xirasagar, S., Maes, P., Kaminsky, D., Liou, D., et al. (2017). The papillomavirus episteme: a major update to the papillomavirus sequence database. Nucleic Acids Res. 45, D499–D506. doi: 10.1093/nar/gkw879
Vande Pol, S. B., and Klingelhutz, A. J. (2013). Papillomavirus E6 oncoproteins. Virology 445, 115–137. doi: 10.1016/j.virol.2013.04.026
Viarisio, D., Gissmann, L., and Tommasino, M. (2017). Human papillomaviruses and carcinogenesis: well-established and novel models. Curr. Opin. Virol. 26, 56–62. doi: 10.1016/j.coviro.2017.07.014
Viarisio, D., Muller-Decker, K., Accardi, R., Robitaille, A., Durst, M., Beer, K., et al. (2018). Beta HPV38 oncoproteins act with a hit-and-run mechanism in ultraviolet radiation-induced skin carcinogenesis in mice. PLoS Pathog. 14:e1006783. doi: 10.1371/journal.ppat.1006783
Viarisio, D., Mueller-Decker, K., Kloz, U., Aengeneyndt, B., Kopp-Schneider, A., Grone, H. J., et al. (2011). E6 and E7 from beta HPV38 cooperate with ultraviolet light in the development of actinic keratosis-like lesions and squamous cell carcinoma in mice. PLoS Pathog. 7:e1002125. doi: 10.1371/journal.ppat.1002125
Viarisio, D., Muller-Decker, K., Zanna, P., Kloz, U., Aengeneyndt, B., Accardi, R., et al. (2016). Novel ß-HPV49 transgenic mouse model of upper digestive tract cancer. Cancer Res. 76, 4216–4225. doi: 10.1158/0008-5472.CAN-16-0370
Wallace, N. A., Robinson, K., and Galloway, D. A. (2014). Beta human papillomavirus E6 expression inhibits stabilization of p53 and increases tolerance of genomic instability. J. Virol. 88, 6112–6127. doi: 10.1128/JVI.03808-13
Wallace, N. A., Robinson, K., Howie, H. L., and Galloway, D. A. (2012). HPV 5 and 8 E6 abrogate ATR activity resulting in increased persistence of UVB induced DNA damage. PLoS Pathog. 8:e1002807. doi: 10.1371/journal.ppat.1002807
Wang, D., Kanuma, T., Mizunuma, H., Takama, F., Ibuki, Y., Wake, N., et al. (2000). Analysis of specific gene mutations in the transforming growth factor-beta signal transduction pathway in human ovarian cancer. Cancer Res. 60, 4507–4512.
Wang, J., Zhou, D., Prabhu, A., Schlegel, R., and Yuan, H. (2010). The canine papillomavirus and gamma HPV E7 proteins use an alternative domain to bind and destabilize the retinoblastoma protein. PLoS Pathog. 6:e1001089. doi: 10.1371/journal.ppat.1001089
Wang, N. J., Sanborn, Z., Arnett, K. L., Bayston, L. J., Liao, W., Proby, C. M., et al. (2011). Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. U.S.A. 108, 17761–17766. doi: 10.1073/pnas.1114669108
Wendel, S. O., and Wallace, N. A. (2017). Loss of genome fidelity: Beta HPVs and the DNA damage response. Front. Microbiol. 8:2250. doi: 10.3389/fmicb.2017.02250
Weng, A. P., Millholland, J. M., Yashiro-Ohtani, Y., Arcangeli, M. L., Lau, A., Wai, C., et al. (2006). c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 20, 2096–2109. doi: 10.1101/gad.1450406
White, E. A., and Howley, P. M. (2013). Proteomic approaches to the study of papillomavirus-host interactions. Virology 435, 57–69. doi: 10.1016/j.virol.2012.09.046
White, E. A., Kramer, R. E., Tan, M. J., Hayes, S. D., Harper, J. W., and Howley, P. M. (2012a). Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J. Virol. 86, 13174–13186. doi: 10.1128/JVI.02172-12
White, E. A., Sowa, M. E., Tan, M. J., Jeudy, S., Hayes, S. D., Santha, S., et al. (2012b). Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proc. Natl. Acad. Sci. U.S.A. 109, E260–E267. doi: 10.1073/pnas.1116776109
White, E. A., Walther, J., Javanbakht, H., and Howley, P. M. (2014). Genus beta human papillomavirus E6 proteins vary in their effects on the transactivation of p53 target genes. J. Virol. 88, 8201–8212. doi: 10.1128/JVI.01197-14
Wu, L., Aster, J. C., Blacklow, S. C., Lake, R., Artavanis-Tsakonas, S., and Griffin, J. D. (2000). MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nat. Genet. 26, 484–489. doi: 10.1038/82644
Xue, X. Y., Majerciak, V., Uberoi, A., Kim, B. H., Gotte, D., Chen, X., et al. (2017). The full transcription map of mouse papillomavirus type 1 (MmuPV1) in mouse wart tissues. PLoS Pathog. 13:e1006715. doi: 10.1371/journal.ppat.1006715
Yamashita, T., Segawa, K., Fujinaga, Y., Nishikawa, T., and Fujinaga, K. (1993). Biological and biochemical activity of E7 genes of the cutaneous human papillomavirus type 5 and 8. Oncogene 8, 2433–2441.
Zanier, K., Charbonnier, S., Sidi, A. O., Mcewen, A. G., Ferrario, M. G., Poussin-Courmontagne, P., et al. (2013). Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science 339, 694–698. doi: 10.1126/science.1229934
Keywords: viral oncogenesis, squamous cell carcinoma, epidermodysplasia verruciformis, keratinocyte differentiation, hit-and-run carcinogenesis
Citation: Meyers JM, Grace M, Uberoi A, Lambert PF and Munger K (2018) Inhibition of TGF-β and NOTCH Signaling by Cutaneous Papillomaviruses. Front. Microbiol. 9:389. doi: 10.3389/fmicb.2018.00389
Received: 21 January 2018; Accepted: 20 February 2018;
Published: 08 March 2018.
Edited by:
Herbert Johannes Pfister, Universität zu Köln, GermanyReviewed by:
Nicholas A. Wallace, Kansas State University, United StatesMarco De Andrea, University of Turin, Italy
Copyright © 2018 Meyers, Grace, Uberoi, Lambert and Munger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karl Munger, karl.munger@tufts.edu
†Present address: Jordan M. Meyers, Program in Epithelial Biology, Stanford University School of Medicine, Stanford, CA, United States