 Carla López-Causapé
Carla López-Causapé Antonio Oliver
Antonio Oliver- Servicio de Microbiología y Unidad de Investigación, Hospital Universitari Son Espases, Institut d’Investigació Sanitaria Illes Balears, Palma de Mallorca, Spain
One of the most striking features of Pseudomonas aeruginosa is its outstanding capacity for developing antimicrobial resistance to nearly all available antipseudomonal agents through the selection of chromosomal mutations, leading to the failure of the treatment of severe hospital-acquired or chronic infections. Recent whole-genome sequencing (WGS) data obtained from in vitro assays on the evolution of antibiotic resistance, in vivo monitoring of antimicrobial resistance development, analysis of sequential cystic fibrosis isolates, and characterization of widespread epidemic high-risk clones have provided new insights into the evolutionary dynamics and mechanisms of P. aeruginosa antibiotic resistance, thus motivating this review. Indeed, the analysis of the WGS mutational resistome has proven to be useful for understanding the evolutionary dynamics of classical resistance pathways and to describe new mechanisms for the majority of antipseudomonal classes, including β-lactams, aminoglycosides, fluoroquinolones, or polymixins. Beyond addressing a relevant scientific question, the analysis of the P. aeruginosa mutational resistome is expected to be useful, together with the analysis of the horizontally-acquired resistance determinants, for establishing the antibiotic resistance genotype, which should correlate with the antibiotic resistance phenotype and as such, it should be useful for the design of therapeutic strategies and for monitoring the efficacy of administered antibiotic treatments. However, further experimental research and new bioinformatics tools are still needed to overcome the interpretation limitations imposed by the complex interactions (including those leading to collateral resistance or susceptibility) between the 100s of genes involved in the mutational resistome, as well as the frequent difficulties for differentiating relevant mutations from simple natural polymorphisms.
Introduction
Pseudomonas aeruginosa is one of the most frequent and severe causes of hospital-acquired infections, particularly affecting immunocompromised (especially neutropenic) and Intensive Care Unit (ICU) patients. Indeed, P. aeruginosa is the first cause of ventilator associated pneumonia (VAP) and burn wound infections, both associated with a very high mortality rate (Vincent, 2003; Bassetti et al., 2012). Likewise, P. aeruginosa is the most frequent driver of chronic respiratory infections in cystic fibrosis (CF) patients or other chronic underlying diseases (Döring et al., 2011).
One of the most striking features of P. aeruginosa is its outstanding capacity for developing antimicrobial resistance to nearly all available antipseudomonal agents through the selection of chromosomal mutations. Indeed, treatment failure caused by the development of antimicrobial resistance is a too frequent outcome of P. aeruginosa infections. The problem of mutation-mediated antibiotic resistance is further amplified in the chronic infection setting, due to the very high prevalence of hypermutable strains, showing greatly enhanced spontaneous mutation rates caused by defective DNA repair or error avoidance systems (Oliver et al., 2000; Maciá et al., 2005).
Beyond the obvious negative impact of resistance development for the treated patient, the accumulation of several of these chromosomal mutations leads to the emergence of multidrug resistant (MDR), extensively drug-resistant (XDR) or even pan-antibiotic-resistant (PDR) strains, which can be responsible for notable epidemics in the hospital setting (Deplano et al., 2005; Suarez et al., 2011). Moreover, recent studies have evidenced the existence of MDR/XDR global clones disseminated in different hospitals worldwide, and for that reason they have been denominated high-risk clones (Woodford et al., 2011; Oliver et al., 2015). Although high-risk clones are frequently associated with transferable antimicrobial resistance determinants, they also typically show a wide range of chromosomal mutations playing a major role in the resistance phenotype (Oliver et al., 2015). Likewise, recent reports have evidenced the interpatient spread of antimicrobial resistance mutations linked to the transmission of epidemic CF strains (López-Causapé et al., 2017).
Along with growing information from mechanistic studies on chromosomal resistance mechanisms and their complex regulatory pathways, involved in adaptive resistance (Lister et al., 2009; Muller et al., 2011; Skiada et al., 2011; Juan et al., 2017), the introduction of whole-genome sequencing (WGS) approaches is shaping up a new dimension for the mutational resistance landscape. The term resistome was first used to account for the set of primary antibiotic resistance genes that could be eventually transferred to human pathogens (D’Costa et al., 2006). Soon after the concept of intrinsic resistome was introduced to include all chromosomal genes that are involved in intrinsic resistance, and whose presence in strains of a bacterial species is independent of previous antibiotic exposure and is not due to horizontal gene transfer (HGT) (Fajardo et al., 2008). Finally, the term mutational resistome was more recently implemented to account for the set of mutations involved in the modulation of antibiotic resistance levels in the absence of HGT (Cabot et al., 2016b; López-Causapé et al., 2017). Recent WGS data obtained from in vitro assays on the evolution of antibiotic resistance, in vivo monitoring of antimicrobial resistance development, analysis of sequential CF isolates, and characterization of wide spread epidemic high-risk clones provide new insights into the evolutionary dynamics and mechanisms of P. aeruginosa antibiotic resistance (Cabot et al., 2016a; Feng et al., 2016; Del Barrio-Tofiño et al., 2017; Jaillard et al., 2017; López-Causapé et al., 2017). Indeed, the analysis of WGS mutational resistomes has proven to be useful for understanding the evolutionary dynamics of classical resistance mechanisms and to depict new ones for the majority of antimicrobial classes, including β-lactams, aminoglycosides, fluoroquinolones, polymixins and others, as reviewed in the following sections. Table 1 summarizes the main genes and mutations known to increase resistance levels and therefore shaping up the P. aeruginosa mutational resistome.
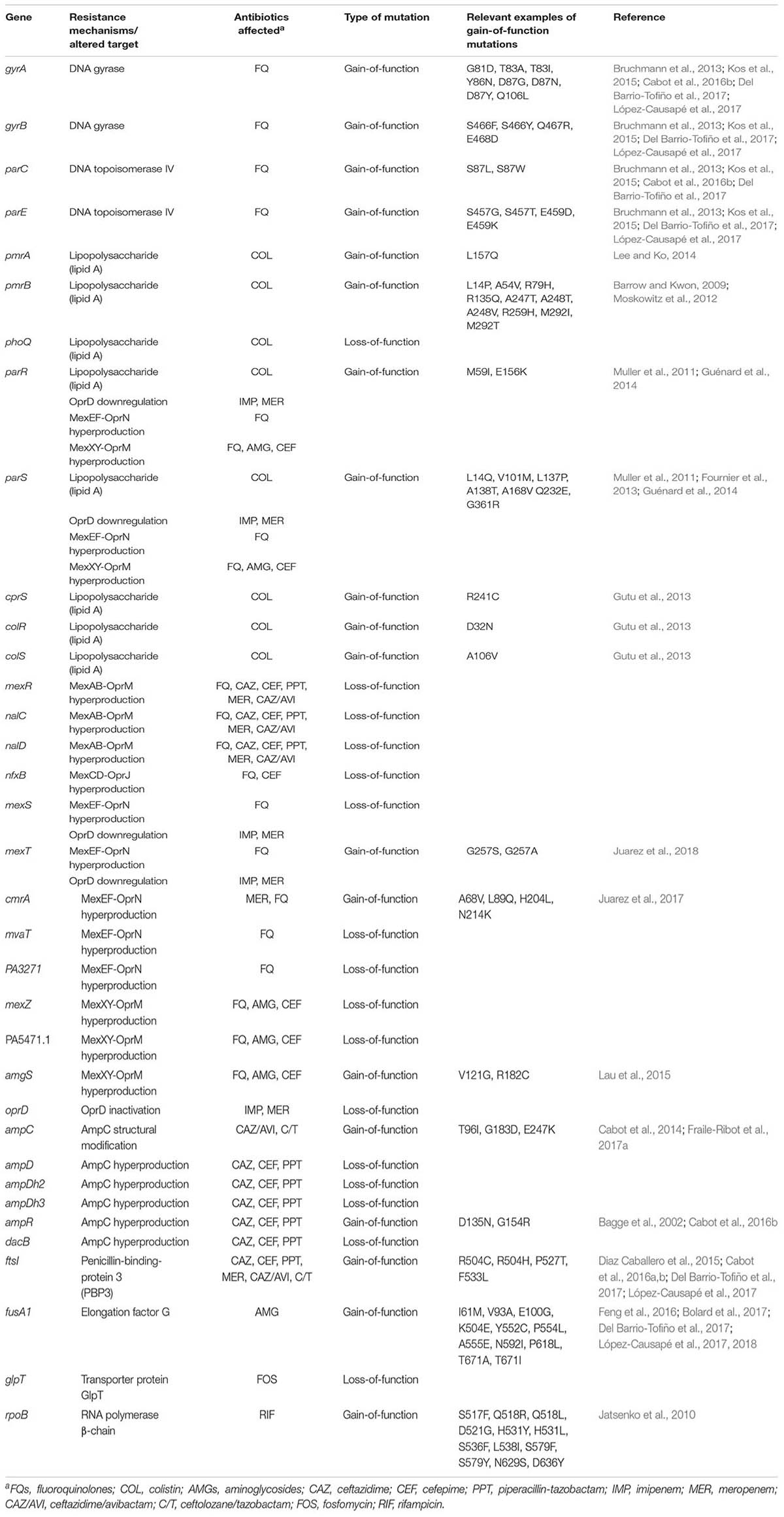
TABLE 1. Main genes and mutations known to be involved in increased P. aeruginosa antibiotic resistance.
β-Lactam Mutational Resistome
The most frequent mutation-driven β-lactam resistance mechanism is likely the overproduction of the chromosomal cephalosporinase AmpC, involving a wide range of genes belonging to complex regulatory pathways of cell-wall recycling (Juan et al., 2017). Among them, the mutational inactivation of dacB, encoding the non-essential penicillin-binding protein (PBP) PBP4, and ampD, encoding a N-acetyl-muramyl-L-alanine amidase have been found to be the most frequent cause of ampC derepression and β-lactam resistance (Juan et al., 2005; Moya et al., 2009). The inactivation of PBP4 has also been shown to activate the BlrAB/CreBC regulatory system, further increasing resistance levels (Moya et al., 2009). Additionally, specific point mutations leading to a conformation change in the transcriptional regulator AmpR, causing ampC upregulation and β-lactam resistance, have been noted among clinical strains. They include the D135N mutation, described in several species besides P. aeruginosa, including Stenotrophomonas maltophilia, Citrobacter freundii, or Enterobacter cloacae (Juan et al., 2017) or the R154H mutation, linked to the widespread MDR/XDR ST175 high-risk clone. Mutation of many other genes, including those encoding other amidases (AmpDh2 and AmpDh3), other PBPs (such as PBP5 and PBP7), lytic transglycosylases (such as SltB1 and mltB), MPL (UDP-N-acetylmuramate:Lalanyl-γ-D-glutamyl-meso-diaminopimelate ligase), or NuoN (NADH dehydrogenase I chain N) have been shown to enhance ampC expression, either alone or combined with other mutations, although their impact on β-lactam resistance among clinical strains still needs to be further analyzed (Juan et al., 2017).
In addition to ampC overexpression, recent studies have revealed that β-lactam resistance development, including the novel combinations of β-lactam-β-lactamase inhibitors ceftolozane/tazobactam and ceftazidime/avibactam, may result from mutations leading to the structural modification of AmpC (Cabot et al., 2014; Lahiri et al., 2014; Fraile-Ribot et al., 2017a; Haidar et al., 2017). Likewise, recent studies identified diverse AmpC variants associated with high-level cephalosporin resistance, including ceftolozane/tazobactam and ceftazidime/avibactam, in a small proportion (around 1%) of clinical P. aeruginosa isolates (Berrazeg et al., 2015). Over 200 Pseudomonas Derived Cephalosporinase (PDC) variants have been described so far, including those associated with enhanced ceftolozane/tazobactam and ceftazidime/avibactam resistance (Table 1). An update database of PDC variants is maintained in our laboratory and is freely available at https://arpbigidisba.com. Additionally, resistance development to ceftolozane/tazobactam and/or ceftazidime/avibactam may involve mutations leading to the structural modification of narrow spectrum OXA-2 and OXA-10 acquired oxacillinases (Fraile-Ribot et al., 2017a,b).
Besides β-lactamases, there is increasing evidence on the role of target modification in P. aeruginosa β-lactam resistance. Particularly noteworthy are the mutations in ftsI, encoding PBP3, an essential high molecular class B PBP with transpeptidase activity (Chen et al., 2016). Indeed, data from CF patients (Diaz Caballero et al., 2015; López-Causapé et al., 2017), epidemic high-risk clones (Cabot et al., 2016b; Del Barrio-Tofiño et al., 2017) as well as from in vitro studies (Cabot et al., 2016a) have recently shown that PBP3 is under strong mutational pressure, and that specific mutations in this PBP contribute to β-lactam resistance development. R504C/H and F533L mutations are likely those most commonly reported, and are located within the protein domains implicated in the formation and stabilization of the inactivating complex β-lactam-PBP3 (Han et al., 2010). Moreover, these specific mutations have been documented to emerge in vivo during chronic respiratory infection in CF patients (Diaz Caballero et al., 2015; López-Causapé et al., 2017) and upon meropenem (Cabot et al., 2016a) and aztreonam (Jorth et al., 2017) exposure in vitro. However, the precise contribution of PBP3 mutations to β-lactam resistance phenotypes needs to be further investigated using isogenic strains. Likewise, despite unique polymorphisms have also been detected in some clinical strains for other PBPs, their role in β-lactam resistance, if any, still needs to be experimentally addressed.
Other relevant components of the β-lactam mutational resistome are the porins and RND efflux pumps. Mutational inactivation of OprD is well-known to be the primary carbapenem resistance mechanisms in P. aeruginosa (Lister et al., 2009; Castanheira et al., 2014). OprD inactivation typically results from indels or nonsense mutations, including the Q142X mutation, characteristic of the widespread ST175 high-risk clone (Cabot et al., 2016b). Additionally, some amino acid substitutions have also been recently associated with OprD-driven resistance, particularly in the CF setting (Richardot et al., 2015). Finally, carbapenem resistance may also result from oprD repression caused by mutations in the MexEF-OprN efflux pump regulators (mexS/T) or the ParRS two-component system (Li et al., 2015). Overexpression of MexAB-OprM, caused by mutation of several genes involved in its regulation (mexR, nalC, or nalD) increases MICs of most β-lactams except imipenem, whereas overexpression of MexXY (mexZ, parSR, amgS mutations) is particularly involved in cefepime resistance (Li et al., 2015). Additionally, sequence variations in unique residues are detected in the genes coding for the efflux pump (Del Barrio-Tofiño et al., 2017; López-Causapé et al., 2017); however, their contribution to resistance profiles, if any, still needs to be further explored.
Finally, another potentially relevant mutational β-lactam resistance mechanism is the selection of large (>200 kb) deletions affecting specific parts of the chromosome (Cabot et al., 2016a). Although the basis of the conferred resistance phenotype still needs to be further clarified, these mutants can be recognized by the characteristic brown pigment (pyomelanine) caused by the deletion of one of the affected genes, hmgA, coding for a homogentisate-1,2- dioxygenase. This type of deletion has been documented in both, in vitro evolved β-lactam-resistant mutants and CF isolates (Cabot et al., 2016a; Hocquet et al., 2016). However, the deletion of hmgA is not responsible for the resistance phenotype, which could be linked to the deletion of another of the affected genes, galU. This gene codes for a UDP-glucose pyrophosphorylase involved in the synthesis of the lipopolysaccharide (LPS) core. Indeed, analysis of transposon mutant libraries has shown that inactivation of galU increases ceftazidime and meropenem MICs (Dötsch et al., 2009; Alvarez-Ortega et al., 2010).
Aminoglycoside Mutational Resistome
In the absence of horizontally-acquired aminoglycoside modifying enzymes, resistance to this antibiotic class has been particularly linked to the mutational overexpression of MexXY-OprM. Indeed, mutational overexpression of this pump, mainly caused by mexZ, amgS, or parRS mutations (Table 1), is very frequent among clinical isolates, from both, CF patients and nosocomial infections (Guénard et al., 2014; Prickett et al., 2017). Moreover, recent studies show that the epidemic high-risk clone ST175 overexpresses MexXY due to a specific mutation in mexZ (G195E) (Cabot et al., 2016b). However, recent studies have revealed that the aminoglycoside mutational resistome extends far beyond MexXY overexpression, and that high-level resistance may result from the accumulation of multiple mutations, and the involvement of several novel resistance determinants has been recently documented (El’Garch et al., 2007; Schurek et al., 2008; Feng et al., 2016). Among them is particularly noteworthy fusA1, coding for the elongation factor G. Indeed, specific FusA1 mutations have been associated with aminoglycoside resistance in vitro (Feng et al., 2016; López-Causapé et al., 2018) and among clinical, particularly CF, strains (Chung et al., 2012; Markussen et al., 2014; Greipel et al., 2016; López-Causapé et al., 2017, 2018). Moreover, the implication of fusA1 mutations in aminoglycoside resistance has been recently confirmed through site-directed mutagenesis (Bolard et al., 2017).
Fluoroquinolone Mutational Resistome
The fluoroquinolone mutational resistome generally includes specific missense mutations in DNA gyrase (gyrA and/or gyrB) and topisomerase IV (parC and/or parE) Quinolone Resistance-Determining Regions (QRDRs) (Table 1) (Bruchmann et al., 2013; Kos et al., 2015). High-level fluoroquinolone resistance in P. aeruginosa high-risk clones is nearly universal, and typically involves combinations of mutations in GyrA-T83 and ParC-S87 (Del Barrio-Tofiño et al., 2017). QRDR mutations involved in fluoroquinolone resistance in CF might be more variable (López-Causapé et al., 2017). It is also well-known that the mutational overexpression of efflux pumps modulate fluoroquinolone resistance (Table 1). While the overexpression of MexAB-OprM and MexXY-OprM is globally more frequent among clinical strains, its contribution to clinical fluoroquinolone resistance is likely more modest (Bruchmann et al., 2013). On the other hand, the mutational overexpression of MexEF-OprN or MexCD-OprJ is associated with high-level (clinical) fluoroquinolone resistance, and although their prevalence is considered low except in the CF chronic infection setting, recent data show that it might be higher than expected (Richardot et al., 2015).
Polymixin Mutational Resistome
Whereas the prevalence of polymyxin (colistin and polymyxin B) resistance is still globally low (<5%), it has increased in the last years because of the frequent use of these last-resource antibiotics for the treatment of MDR/XDR nosocomial and CF strains. Polymyxin resistance results most frequently from the modification of the LPS caused by the addition of a 4-amino-4-deoxy-L-arabinose moiety in the lipid A structure (Olaitan et al., 2014; Jeannot et al., 2017). The involved mutations are frequently located in the PmrAB or PhoPQ two-component regulators, which lead to the activation of the arnBCADTEF operon (Barrow and Kwon, 2009). More recent studies have revealed that mutations in the ParRS two-component regulator, not only produce polymyxin resistance due to the activation of the arnBCADTEF operon, but also lead to a MDR phenotype determined by the overprexpression of MexXY and the repression of OprD (Muller et al., 2011). Moreover, two additional two-component regulators, ColRS and CprRS, have been recently found to be involved in polymyxin resistance (Gutu et al., 2013). The analysis of colistin resistance mechanisms among clinical strains is not always straight forward, since the presence of mutations in these two-component regulators is not always associated with clinical colistin resistance, probably denoting partial complementation between the different regulators (Moskowitz et al., 2012; Gutu et al., 2013; López-Causapé et al., 2017). Moreover, recent in vitro evolution assays have revealed the implication of additional mutations in high level colistin resistance, facilitated by the emergence of mutator (mutS deficient) phenotypes (Dößelmann et al., 2017). Particularly noteworthy among them are those occurring in LptD, LpxC, or MigA.
Other Antibiotics
Even if not considered a classical antipseudomonal agent, fosfomycin has emerged in the last decade as a potentially useful antibiotic in urinary tract infections and combined therapy for MDR/XDR P. aeruginosa (Michalopoulos et al., 2011). However, fosfomycin resistance spontaneous mutation rates are high and the mechanism involved is typically the mutational inactivation of glpT, coding for a glycerol-3-phospate permease required for fosfomycin uptake (Castañeda-García et al., 2009; Rodríguez-Rojas et al., 2010). glpT mutations, conferring high-level fosfomycin resistance are also frequently found among MDR/XDR high-risk clones (Del Barrio-Tofiño et al., 2017), and some specific mutations, such as T211P, have been fixed in some widespread lineages as described for ST175 (Cabot et al., 2016b). Another potentially useful antimicrobial for combined therapy against MDR/XDR P. aeruginosa is rifampicin (Cai et al., 2017). However, rifampicin resistance may emerge at high frequency due to the selection of specific missense mutations in rpoB, coding for the beta subunit of the RNA polymerase (Jatsenko et al., 2010). Another example of newer antibiotic families with antipseudomonal activity are the pacidamycins, uridyl peptide antibiotics, targeting translocase I, an essential enzyme in peptidoglycan biosynthesis (Mistry et al., 2013). Emergence of high-level resistance to this antibiotic class has been shown to involve the selection of mutations in the Opp transporter, a binding protein-dependent ABC transporter used for oligopeptide import. Finally, the P. aeruginosa mutational resistome, particularly in the CF setting, may also include resistance to other used antibiotics such as the frequent mutations of domain V of 23S rRNA, conferring macrolide resistance (Mustafa et al., 2017).
Concluding Remarks and Future Perspectives
The analysis of the P. aeruginosa mutational resistome, together with the analysis of the horizontally-acquired resistance determinants, should be useful for establishing the antibiotic resistance genotype, which should correlate with the antibiotic resistance phenotype and as such, it should permit the design of therapeutic strategies and for monitoring the efficacy of administered antibiotic treatments. However, the current applicability of the analysis of the mutational resistome is still limited by the large number of genes involved and the complexity of the resistance phenotypes generated, and, particularly, by the difficulties, in many cases, for differentiating relevant mutations from simple natural polymorphisms. Obviously, the evolution of the mutational resistome is a direct consequence of antimicrobial exposure and as such, it is not surprising that exposure to one antibiotic drives evolution of the mutational resistome for that antibiotic. However, the complexity of the actual resistance profile is further increased by the specificity and interactions among different resistance mechanisms. Indeed, a resistance mutation selected by one antibiotic may have a variable effect among the different agents within the same antibiotic class or family. Likewise, cross resistance (or collateral resistance) implies that exposure to one antibiotic drives also the development of resistance to a different one from the same or other classes. Typically, this is caused by the developed resistance mechanism (such as efflux pump overexpression) affecting simultaneously different antibiotics. Indeed, potential development of cross resistance is a major issue to consider when using antibiotic combinations (Vestergaard et al., 2016). Moreover, cross resistance between antibiotics and antiseptics and other biocides may also occur (Li et al., 2015). Perhaps less obvious is collateral susceptibility, which implies that exposure to one antibiotic increases the susceptibility to a different one (Pál et al., 2015; Imamovic et al., 2017). This might be achieved through two mechanisms. One possible mechanism is that exposure to one antibiotic directly causes increased susceptibility to a different one, for example, mutations in the β-lactamase AmpC increases cephalosporin hydrolysis while reducing that of penicillins or carbapenems (Cabot et al., 2014). The second possibility is that the development of a resistance mechanism impairs the activity of another existing resistance mechanism. An example is competition between the different efflux pumps, since the overexpression of one may impair the expression of another (Mulet et al., 2011). Thus, the evolution of the mutational resistome for a given antibiotic is not only dependent on the exposure to this antibiotic, but it is also conditioned by the simultaneous or even previous exposures to other antibiotics. An illustrative example is provided in a recent in vitro study that demonstrated, for a broad range of antibiotic classes, that the history of exposure and resistance development to a given antibiotic, conditions the dynamics and mechanisms of resistance development when exposed to a second one (Yen and Papin, 2017). In summary, the comprehensive analysis of the mutational resistome of P. aeruginosa in CF and nosocomial infections is expected to become a useful tool for optimizing therapeutic strategies and monitoring the efficacy of administered antibiotic treatments in the near future.
Author Contributions
CL-C and AO wrote the manuscript. CL-C, GC, EdB-T, and AO reviewed the literature.
Funding
The authors were supported by Plan Nacional de I+D+i 2013–2016 and Instituto de Salud Carlos III, Subdirección General de Redes y Centros de Investigación Cooperativa, Ministerio de Economía, Industria y Competitividad, Spanish Network for Research in Infectious Diseases (REIPI RD16/0016) and grant PI15/00088 (PI AO) and co-financed by European Development Regional Fund ERDF “A way to achieve Europe,” Operative program Intelligent Growth 2014–2020. AO was also supported by the European Union through the 11th Call of the Innovative Medicines Initiative (grant COMBACTE-MAGNET).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alvarez-Ortega, C., Wiegand, I., Olivares, J., Hancock, R. E., and Martínez, J. L. (2010). Genetic determinants involved in the susceptibility of Pseudomonas aeruginosa to beta-lactam antibiotics. Antimicrob. Agents Chemother. 54, 4159–4167. doi: 10.1128/AAC.00257-10
Bagge, N., Ciofu, O., Hentzer, M., Campbell, J. I., Givskov, M., and Høiby, N. (2002). Constitutive high expression of chromosomal beta-lactamase in Pseudomonas aeruginosa caused by a new insertion sequence (IS1669) located in ampD. Antimicrob. Agents Chemother. 46, 3406–3411. doi: 10.1128/AAC.46.11.3406-3411.2002
Barrow, K., and Kwon, D. H. (2009). Alterations in two-component regulatory systems of phoPQ and pmrAB are associated with polymyxin B resistance in clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53, 5150–5154. doi: 10.1128/AAC.00893-09
Bassetti, M., Taramasso, L., Giacobbe, D. R., and Pelosi, P. (2012). Management of ventilator-associated pneumonia: epidemiology, diagnosis and antimicrobial therapy. Expert Rev. Anti. Infect. Ther. 10, 585–596. doi: 10.1586/eri.12.36
Berrazeg, M., Jeannot, K., Ntsogo Enguéné, V. Y., Broutin, I., Loeffert, S., Fournier, D., et al. (2015). Mutations in β-Lactamase AmpC increase resistance of Pseudomonas aeruginosa isolates to antipseudomonal cephalosporins. Antimicrob. Agents Chemother. 59, 6248–6255. doi: 10.1128/AAC.00825-15
Bolard, A., Plesiat, P., and Jeannot, K. (2017). Mutations in gene fusA1 as a novel mechanism of aminoglycoside resistance in clinical strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 62, e01835-17. doi: 10.1128/AAC.01835-17
Bruchmann, S., Dötsch, A., Nouri, B., Chaberny, I. F., and Häussler, S. (2013). Quantitative contributions of target alteration and decreased drug accumulation to Pseudomonas aeruginosa fluoroquinolone resistance. Antimicrob. Agents Chemother. 57, 1361–1368. doi: 10.1128/AAC.01581-12
Cabot, G., Bruchmann, S., Mulet, X., Zamorano, L., Moyà, B., Juan, C., et al. (2014). Pseudomonas aeruginosa ceftolozane-tazobactam resistance development requires multiple mutations leading to overexpression and structural modification of AmpC. Antimicrob. Agents Chemother. 58, 3091–3099. doi: 10.1128/AAC.02462-13
Cabot, G., Zamorano, L., Moyà, B., Juan, C., Navas, A., Blázquez, J., et al. (2016a). Evolution of Pseudomonas aeruginosa antimicrobial resistance and fitness under low and high mutation rates. Antimicrob. Agents Chemother. 60, 1767–1778. doi: 10.1128/AAC.02676-15
Cabot, G., López-Causapé, C., Ocampo-Sosa, A. A., Sommer, L. M., Domínguez, M. Á, Zamorano, L., et al. (2016b). Deciphering the resistome of the widespread Pseudomonas aeruginosa Sequence Type 175 international high-risk clone through whole-genome sequencing. Antimicrob. Agents Chemother. 60, 7415–7423.
Cai, Y., Yang, D., Wang, J., and Wang, R. (2017). Activity of colistin alone or in combination with rifampicin or meropenem in a carbapenem-resistant bioluminescent Pseudomonas aeruginosa intraperitoneal murine infection model. J. Antimicrob. Chemother. 73, 456–461. doi: 10.1093/jac/dkx399
Castañeda-García, A., Rodríguez-Rojas, A., Guelfo, J. R., and Blázquez, J. (2009). The glycerol-3-phosphate permease GlpT is the only fosfomycin transporter in Pseudomonas aeruginosa. J. Bacteriol. 191, 6968–6974. doi: 10.1128/JB.00748-09
Castanheira, M., Deshpande, L. M., Costello, A., Davies, T. A., and Jones, R. N. (2014). Epidemiology and carbapenem resistance mechanisms of carbapenem-non-susceptible Pseudomonas aeruginosa collected during 2009-11 in 14 European and Mediterranean countries. J. Antimicrob. Chemother. 69, 1804–1814. doi: 10.1093/jac/dku048
Chen, W., Zhang, Y. M., and Davies, C. (2016). Penicillin-Binding Protein 3 is essential for growth of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 61, e01651-16. doi: 10.1128/AAC.01651-16
Chung, J. C., Becq, J., Fraser, L., Schulz-Trieglaff, O., Bond, N. J., Foweraker, J., et al. (2012). Genomic variation among contemporary Pseudomonas aeruginosa isolates from chronically infected cystic fibrosis patients. J. Bacteriol. 194, 4857–4866. doi: 10.1128/JB.01050-12
D’Costa, V. M., McGrann, K. M., Hughes, D. W., and Wright, G. D. (2006). Sampling the antibiotic resistome. Science 311, 374–377. doi: 10.1126/science.1120800
Del Barrio-Tofiño, E., López-Causapé, C., Cabot, G., Rivera, A., Benito, N., Segura, C., et al. (2017). Genomics and susceptibility profiles of extensively drug-resistant Pseudomonas aeruginosa isolates from Spain. Antimicrob. Agents Chemother. 61, e01589-17. doi: 10.1128/AAC.01589-17
Deplano, A., Denis, O., Poirel, L., Hocquet, D., Nonhoff, C., Byl, B., et al. (2005). Molecular characterization of an epidemic clone of panantibiotic-resistant Pseudomonas aeruginosa. J. Clin. Microbiol. 43, 1198–1204. doi: 10.1128/JCM.43.3.1198-1204.2005
Diaz Caballero, J., Clark, S. T., Coburn, B., Zhang, Y., Wang, P. W., Donaldson, S. L., et al. (2015). Selective sweeps and parallel pathoadaptation drive Pseudomonas aeruginosa evolution in the cystic fibrosis lung. mBio 6, e00981-15. doi: 10.1128/mBio.00981-15
Döring, G., Parameswaran, I. G., and Murphy, T. F. (2011). Differential adaptation of microbial pathogens to airways of patients with cystic fibrosis and chronic obstructive pulmonary disease. FEMS Microbiol. Rev. 35, 124–146. doi: 10.1111/j.1574-6976.2010.00237.x
Dötsch, A., Becker, T., Pommerenke, C., Magnowska, Z., Jänsch, L., and Häussler, S. (2009). Genomewide identification of genetic determinants of antimicrobial drug resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53, 2522–2531. doi: 10.1128/AAC.00035-09
Dößelmann, B., Willmann, M., Steglich, M., Bunk, B., Nübel, U., Peter, S., et al. (2017). Rapid and consistent evolution of colistin resistance in extensively drug-resistant Pseudomonas aeruginosa during morbidostat culture. Antimicrob. Agents Chemother. 61, e00043-17. doi: 10.1128/AAC.00043-17
El’Garch, F., Jeannot, K., Hocquet, D., Llanes-Barakat, C., and Plésiat, P. (2007). Cumulative effects of several nonenzymatic mechanisms on the resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob. Agents Chemother. 51, 1016–1021. doi: 10.1128/AAC.00704-06
Fajardo, A., Martínez-Martín, N., Mercadillo, M., Galán, J. C., Ghysels, B., Matthijs, S., et al. (2008). The neglected intrinsic resistome of bacterial pathogens. PLoS One 3:e1619. doi: 10.1371/journal.pone.0001619
Feng, Y., Jonker, M. J., Moustakas, I., Brul, S., and Ter Kuile, B. H. (2016). Dynamics of mutations during development of resistance by Pseudomonas aeruginosa against five antibiotics. Antimicrob. Agents Chemother. 60, 4229–4236. doi: 10.1128/AAC.00434-16
Fournier, D., Richardot, C., Müller, E., Robert-Nicoud, M., Llanes, C., Plésiat, P., et al. (2013). Complexity of resistance mechanisms to imipenem in intensive care unit strains of Pseudomonas aeruginosa. J. Antimicrob. Chemother. 68, 1772–1780. doi: 10.1093/jac/dkt098
Fraile-Ribot, P. A., Cabot, G., Mulet, X., Periañez, L., Martín-Pena, M. L., Juan, C., et al. (2017a). Mechanisms leading to in vivo ceftolozane/tazobactam resistance development during the treatment of infections caused by MDR Pseudomonas aeruginosa. J. Antimicrob. Chemother. doi: 10.1093/jac/dkx424 [Epub ahead of print].
Fraile-Ribot, P. A., Mulet, X., Cabot, G., Del Barrio-Tofiño, E., Juan, C., Pérez, J. L., et al. (2017b). In vivo emergence of resistance to novel cephalosporin-β-Lactamase inhibitor combinations through the duplication of amino acid D149 from OXA-2 β-Lactamase (OXA-539) in Sequence Type 235 Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 61, e01117-17. doi: 10.1128/AAC.01117-17
Greipel, L., Fischer, S., Klockgether, J., Dorda, M., Mielke, S., Wiehlmann, L., et al. (2016). Molecular epidemiology of mutations in antimicrobial resistance loci of Pseudomonas aeruginosa isolates from airways of Cystic Fibrosis patients. Antimicrob. Agents Chemother. 60, 6726–6734. doi: 10.1128/AAC.00724-16
Guénard, S., Muller, C., Monlezun, L., Benas, P., Broutin, I., Jeannot, K., et al. (2014). Multiple mutations lead to MexXY-OprM-dependent aminoglycoside resistance in clinical strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 58, 221–228. doi: 10.1128/AAC.01252-13
Gutu, A. D., Sgambati, N., Strasbourger, P., Brannon, M. K., Jacobs, M. A., Haugen, E., et al. (2013). Polymyxin resistance of Pseudomonas aeruginosa phoQ mutants is dependent on additional two-component regulatory systems. Antimicrob. Agents Chemother. 57, 2204–2215. doi: 10.1128/AAC.02353-12
Haidar, G., Philips, N. J., Shields, R. K., Snyder, D., Cheng, S., Potoski, B. A., et al. (2017). Ceftolozane-Tazobactam for the treatment of multidrug-resistant Pseudomonas aeruginosa infections: clinical effectiveness and evolution of resistance. Clin. Infect. Dis. 65, 110–120. doi: 10.1093/cid/cix182
Han, S., Zaniewski, R. P., Marr, E. S., Lacey, B. M., Tomaras, A. P., Evdokimov, A., et al. (2010). Structural basis for effectiveness of siderophore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 107, 22002–22007. doi: 10.1073/pnas.1013092107
Hocquet, D., Petitjean, M., Rohmer, L., Valot, B., Kulasekara, H. D., Bedel, E., et al. (2016). Pyomelanin-producing Pseudomonas aeruginosa selected during chronic infections have a large chromosomal deletion which confers resistance to pyocins. Environ. Microbiol. 18, 3482–3493. doi: 10.1111/1462-2920.13336
Imamovic, L., Ellabaan, M. M. H., Dantas Machado, A. M., Citterio, L., Wulff, T., Molin, S., et al. (2017). Drug-driven phenotypic convergence supports rational treatment strategies of chronic infections. Cell 172, 121–134.e14. doi: 10.1016/j.cell.2017.12.012
Jaillard, M., van Belkum, A., Cady, K. C., Creely, D., Shortridge, D., Blanc, B., et al. (2017). Correlation between phenotypic antibiotic susceptibility and the resistome in Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 50, 210–218.
Jatsenko, T., Tover, A., Tegova, R., and Kivisaar, M. (2010). Molecular characterization of Rif(r) mutations in Pseudomonas aeruginosa and Pseudomonas putida. Mutat. Res. 683, 106–114. doi: 10.1016/j.mrfmmm.2009.10.015
Jeannot, K., Bolard, A., and Plésiat, P. (2017). Resistance to polymyxins in Gram-negative organisms. Int. J. Antimicrob. Agents 49, 526–535. doi: 10.1016/j.ijantimicag.2016.11.029
Jorth, P., McLean, K., Ratjen, A., Secor, P. R., Bautista, G. E., Ravishankar, S., et al. (2017). Evolved aztreonam resistance is multifactorial and can produce hypervirulence in Pseudomonas aeruginosa. mBio 8, e00517-17. doi: 10.1128/mBio.00517-17
Juan, C., Maciá, M. D., Gutiérrez, O., Vidal, C., Pérez, J. L., and Oliver, A. (2005). Molecular mechanisms of beta-lactam resistance mediated by AmpC hyperproduction in Pseudomonas aeruginosa clinical strains. Antimicrob. Agents Chemother. 49, 4733–4738. doi: 10.1128/AAC.49.11.4733-4738.2005
Juan, C., Torrens, G., González-Nicolau, M., and Oliver, A. (2017). Diversity and regulation of intrinsic β-lactamases from non-fermenting and other Gram-negative opportunistic pathogens. FEMS Microbiol. Rev. 41, 781–815. doi: 10.1093/femsre/fux043
Juarez, P., Broutin, I., Bordi, C., Plésiat, P., and Llanes, C. (2018). Constitutive activation of MexT by amino acid substitutions results in MexEF-OprN overproduction in clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. doi: 10.1128/AAC.02445-17 [Epub ahead of print].
Juarez, P., Jeannot, K., Plésiat, P., and Llanes, C. (2017). Toxic electrophiles induce expression of the multidrug efflux pump MexEF-OprN in Pseudomonas aeruginosa through a novel transcriptional regulator, CmrA. Antimicrob. Agents Chemother. 61, e00585-17. doi: 10.1128/AAC.00585-17
Kos, V. N., Déraspe, M., McLaughlin, R. E., Whiteaker, J. D., Roy, P. H., Alm, R. A., et al. (2015). The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob. Agents Chemother. 59, 427–436. doi: 10.1128/AAC.03954-14
Lahiri, S. D., Johnstone, M. R., Ross, P. L., McLaughlin, R. E., Olivier, N. B., and Alm, R. A. (2014). Avibactam and class C β-lactamases: mechanism of inhibition, conservation of the binding pocket, and implications for resistance. Antimicrob. Agents Chemother. 58, 5704–5713. doi: 10.1128/AAC.03057-14
Lau, C. H., Krahn, T., Gilmour, C., Mullen, E., and Poole, K. (2015). AmgRS-mediated envelope stress-inducible expression of the mexXY multidrug efflux operon of Pseudomonas aeruginosa. Microbiol. Open 4, 121–135. doi: 10.1002/mbo3.226
Lee, J. Y., and Ko, K. S. (2014). Mutations and expression of PmrAB and PhoPQ related with colistin resistance in Pseudomonas aeruginosa clinical isolates. Diagn. Microbiol. Infect. Dis. 78, 271–276. doi: 10.1016/j.diagmicrobio.2013.11.027
Li, X. Z., Plésiat, P., and Nikaido, H. (2015). The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 28, 337–418. doi: 10.1128/CMR.00117-14
Lister, P. D., Wolter, D. J., and Hanson, N. D. (2009). Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 22, 582–610. doi: 10.1128/CMR.00040-09
López-Causapé, C., Rubio, R., Cabot, G., and Oliver, A. (2018). Evolution of the Pseudomonas aeruginosa aminoglycoside mutational resistome in vitro and in the cystic fibrosis setting. Antimicrob. Agents Chemother. doi: 10.1128/AAC.02583-17 [Epub ahead of print].
López-Causapé, C., Sommer, L. M., Cabot, G., Rubio, R., Ocampo-Sosa, A. A., Johansen, H. K., et al. (2017). Evolution of the Pseudomonas aeruginosa mutational resistome in an international Cystic Fibrosis clone. Sci. Rep. 7:5555. doi: 10.1038/s41598-017-05621-5
Maciá, M. D., Blanquer, D., Togores, B., Sauleda, J., Pérez, J. L., and Oliver, A. (2005). Hypermutation is a key factor in development of multiple-antimicrobial resistance in Pseudomonas aeruginosa strains causing chronic lung infections. Antimicrob. Agents Chemother. 49, 3382–3386. doi: 10.1128/AAC.49.8.3382-3386.2005
Markussen, T., Marvig, R. L., Gómez-Lozano, M., Aanæs, K., Burleigh, A. E., Høiby, N., et al. (2014). Environmental heterogeneity drives within-host diversification and evolution of Pseudomonas aeruginosa. mBio. 5, e01592-14. doi: 10.1128/mBio.01592-14
Michalopoulos, A. S., Livaditis, I. G., and Gougoutas, V. (2011). The revival of fosfomycin. Int. J. Infect. Dis. 15, e732-39. doi: 10.1016/j.ijid.2011.07.007
Mistry, A., Warren, M. S., Cusick, J. K., Karkhoff-Schweizer, R. R., Lomovskaya, O., and Schweizer, H. P. (2013). High-level pacidamycin resistance in Pseudomonas aeruginosa is mediated by an opp oligopeptide permease encoded by the opp-fabI operon. Antimicrob. Agents Chemother. 57, 5565–5571. doi: 10.1128/AAC.01198-13
Moskowitz, S. M., Brannon, M. K., Dasgupta, N., Pier, M., Sgambati, N., Miller, A. K., et al. (2012). PmrB mutations promote polymyxin resistance of Pseudomonas aeruginosa isolated from colistin-treated cystic fibrosis patients. Antimicrob. Agents Chemother. 56, 1019–1030. doi: 10.1128/AAC.05829-11
Moya, B., Dötsch, A., Juan, C., Blázquez, J., Zamorano, L., Haussler, S., et al. (2009). Beta-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog. 5:e1000353. doi: 10.1371/journal.ppat.1000353
Mulet, X., Moyá, B., Juan, C., Macià, M. D., Pérez, J. L., Blázquez, J., et al. (2011). Antagonistic interactions of Pseudomonas aeruginosa antibiotic resistance mechanisms in planktonic but not biofilm growth. Antimicrob. Agents Chemother. 55, 4560–4568. doi: 10.1128/AAC.00519-11
Muller, C., Plésiat, P., and Jeannot, K. (2011). A two-component regulatory system interconnects resistance to polymyxins, aminoglycosides, fluoroquinolones, and β-lactams in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 55, 1211–1221. doi: 10.1128/AAC.01252-10
Mustafa, M. H., Khandekar, S., Tunney, M. M., Elborn, J. S., Kahl, B. C., Denis, O., et al. (2017). Acquired resistance to macrolides in Pseudomonas aeruginosa from cystic fibrosis patients. Eur. Respir. J. 49:1601847. doi: 10.1183/13993003.01847-2016
Olaitan, A. O., Morand, S., and Rolain, J. M. (2014). Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. Front. Microbiol. 5:643. doi: 10.3389/fmicb.2014.00643
Oliver, A., Cantón, R., Campo, P., Baquero, F., and Blázquez, J. (2000). High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288, 1251–1254. doi: 10.1126/science.288.5469.1251
Oliver, A., Mulet, X., López-Causapé, C., and Juan, C. (2015). The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist. Updat. 2, 41–59. doi: 10.1016/j.drup.2015.08.002
Pál, C., Papp, B., and Lázár, V. (2015). Collateral sensitivity of antibiotic-resistant microbes. Trends Microbiol. 23, 401–407. doi: 10.1016/j.tim.2015.02.009
Prickett, M. H., Hauser, A. R., McColley, S. A., Cullina, J., Potter, E., Powers, C., et al. (2017). Aminoglycoside resistance of Pseudomonas aeruginosa in cystic fibrosis results from convergent evolution in the mexZ gene. Thorax 72, 40–47. doi: 10.1136/thoraxjnl-2015-208027
Richardot, C., Plésiat, P., Fournier, D., Monlezun, L., Broutin, I., and Llanes, C. (2015). Carbapenem resistance in cystic fibrosis strains of Pseudomonas aeruginosa as a result of amino acid substitutions in porin OprD. Int. J. Antimicrob. Agents 45, 529–532. doi: 10.1016/j.ijantimicag.2014.12.029
Rodríguez-Rojas, A., Maciá, M. D., Couce, A., Gómez, C., Castañeda-García, A., Oliver, A., et al. (2010). Assessing the emergence of resistance: the absence of biological cost in vivo may compromise fosfomycin treatments for P. aeruginosa infections. PLoS One 5:e10193. doi: 10.1371/journal.pone.0010193
Schurek, K. N., Marr, A. K., Taylor, P. K., Wiegand, I., Semenec, L., Khaira, B. K., et al. (2008). Novel genetic determinants of low-level aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 52, 4213–4219. doi: 10.1128/AAC.00507-08
Skiada, A., Markogiannakis, A., Plachouras, D., and Daikos, G. L. (2011). Adaptive resistance to cationic compounds in Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 37, 187–193. doi: 10.1016/j.ijantimicag.2010.11.019
Suarez, C., Peña, C., Arch, O., Dominguez, M. A., Tubau, F., Juan, C., et al. (2011). A large sustained endemic outbreak of multiresistant Pseudomonas aeruginosa: a new epidemiological scenario for nosocomial acquisition. BMC Infect. Dis. 11:272. doi: 10.1186/1471-2334-11-272
Vestergaard, M., Paulander, W., Marvig, R. L., Clasen, J., Jochumsen, N., Molin, S., et al. (2016). Antibiotic combination therapy can select for broad-spectrum multidrug resistance in Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 47, 48–55. doi: 10.1016/j.ijantimicag.2015.09.014
Vincent, J. L. (2003). Nosocomial infections in adult intensive-care units. Lancet 361, 2068–2077. doi: 10.1016/S0140-6736(03)13644-6
Woodford, N., Turton, J. F., and Livermore, D. M. (2011). Multiresistant Gram-negative bacteria: the role of high-risk clones in the dissemination of antibiotic resistance. FEMS Microbiol. Rev. 35, 736–755. doi: 10.1111/j.1574-6976.2011.00268.x
Keywords: antibiotic resistance, resistome, Pseudomonas aeruginosa, multidrug resistance, evolution, resistance development, mutation
Citation: López-Causapé C, Cabot G, del Barrio-Tofiño E and Oliver A (2018) The Versatile Mutational Resistome of Pseudomonas aeruginosa. Front. Microbiol. 9:685. doi: 10.3389/fmicb.2018.00685
Received: 24 January 2018; Accepted: 23 March 2018;
Published: 06 April 2018.
Edited by:
Gian Maria Rossolini, Università degli Studi di Firenze, ItalyReviewed by:
Daniel Pletzer, The University of British Columbia, CanadaMax Maurin, Université Grenoble Alpes, France
Copyright © 2018 López-Causapé, Cabot, del Barrio-Tofiño and Oliver. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Oliver, antonio.oliver@ssib.es