 Eleonora Mastrorilli1†
Eleonora Mastrorilli1† Daniele Pietrucci2†
Daniele Pietrucci2† Lisa Barco1*
Lisa Barco1* Serena Ammendola2
Serena Ammendola2 Sara Petrin1
Sara Petrin1 Alessandra Longo1
Alessandra Longo1 Claudio Mantovani3
Claudio Mantovani3 Andrea Battistoni2
Andrea Battistoni2 Antonia Ricci1
Antonia Ricci1 Alessandro Desideri2
Alessandro Desideri2 Carmen Losasso1
Carmen Losasso1- 1Department of Food Safety, National Reference Center for Salmonella, Istituto Zooprofilattico Sperimentale delle Venezie, Legnaro, Italy
- 2Department of Biology, University of Rome Tor Vergata, Rome, Italy
- 3Science Communication Laboratory, Istituto Zooprofilattico Sperimentale delle Venezie, Legnaro, Italy
Over the past decades, Salmonella 4,[5],12:i:- has rapidly emerged and it is isolated with high frequency in the swine food chain. Although many studies have documented the epidemiological success of this serovar, few investigations have tried to explain this phenomenon from a genetic perspective. Here a comparative whole-genome analysis of 50 epidemiologically unrelated S. 4,[5],12:i:-, isolated in Italy from 2010 to 2016 was performed, characterizing them in terms of genetic elements potentially conferring resistance, tolerance and persistence characteristics. Phylogenetic analyses indicated interesting distinctions among the investigated isolates. The most striking genetic trait characterizing the analyzed isolates is the widespread presence of heavy metals tolerance gene cassettes: most of the strains possess genes expected to confer resistance to copper and silver, whereas about half of the isolates also contain the mercury tolerance gene merA. A functional assay showed that these genes might be useful for preventing the toxic effects of metals, thus supporting the hypothesis that they can contribute to the success of S. 4,[5],12:i:- in farming environments. In addition, the analysis of the distribution of type II toxin-antitoxin families indicated that these elements are abundant in this serovar, suggesting that this is another factor that might favor its successful spread.
Introduction
The epidemiology of Salmonella has been characterized by the recurring emergence of strains that periodically pose important threats to human health because of their pathogenicity, their persistence along the food and veterinary chain and their ability to spread widely. The past two decades has seen a rapid worldwide emergence of a new Salmonella serovar, namely monophasic variant of S. Typhimurium, which antigenic formula is 4,[5],12:i:-(Crayford et al., 2014). In spite of the similarity between this new serovar and S. Typhimurium, they show distinct epidemiological, genomic and phenotypic features. From an epidemiological point of view S. 4,[5],12:i:- is strongly associated to the swine food chain (Barco et al., 2015; EFSA, 2017), whereas S. Typhimurium displays a more heterogeneous source distribution being generally associated with cattle food chain, but also with other animal sources (EFSA, 2017). From a genomic point of view S. 4,[5],12:i:- variability is more limited than the S. Typhimurium one (Guerra et al., 2000; Agasan et al., 2002; Dionisi et al., 2009; Soyer et al., 2009; Barco et al., 2015). From a phenotypic point of view the two serovars differ for the lack of expression of the second flagellar phase (Soyer et al., 2009). Moreover, a significant difference has been described among the two serovars in terms of phage type diversity and antimicrobial resistance profiles (Barco et al., 2012).
According to recently published data by the European Food Safety Authority, S. Enteritidis is still the serovar that is more frequently identified as responsible for human and veterinary infections in Europe (EFSA, 2016). However, S. Typhimurium and S. 4,[5],12:i:- are the most common serovars among both human and veterinary isolates in Italy, Belgium, France, Denmark, England, and Galles (Pires et al., 2010; Hopkins et al., 2012). Interestingly, in some of these countries S. 4,[5],12:i:- has overtaken S. Typhimurium and now ranks as the top serovar isolated from animals and foodstuffs. The first S. 4,[5],12:i:- was identified in the late 1980s from poultry in Portugal (Machado and Bernardo, 1990) and between 1998 and 2004 it was the fourth most common serovar in Spain (de la Torre et al., 2003).
This initial clone of S. 4,[5],12:i:-, which was associated with pigs and pork products (de la Torre et al., 2003), was identified as the “Spanish clone” and showed a ASSuT MDR (Ampicillin Streptomycin Sulphonamides Tetracycline Multy Drug Resistances) phenotype with additional resistance to chloramphenicol, gentamicin and trimethoprim mediated by non-conjugative plasmids carrying the spv locus (Guerra et al., 2001; García et al., 2011, 2014; Loftie-Eaton and Rawlings, 2012). Moreover, isolates belonging to the Spanish clone were ascribed to phage type U302, ST-19, and differed from S. Typhimurium LT2 by five major deletions distributed along the chromosome (Garaizar et al., 2002).
A second major clonal line (the “European clone”), spread in several countries across the EU, was characterized by chromosomally encoded resistance to ampicillin, streptomycin, sulphonamides, and tetracyclines (EFSA, 2016) (ASSuT) and lacked the typical S. Typhimurium virulence plasmid (pSLT) (Hauser et al., 2010; Hopkins et al., 2010; Lucarelli et al., 2012; García et al., 2014). This clone was ascribed mainly to phage-types DT193 and DT120 and ST-34 (Bugarel et al., 2012).
S. 4,[5],12:i:- is a monophasic serovar because of the lack of expression of phase-2 flagellar antigen (Hopkins et al., 2012). In Salmonella, the two flagellar phases are expressed alternatively and flagellar phase variation depends on a switch of the transcription of fliC and fljB genes, which encode phase-1 and phase-2 flagellar antigens, respectively (Hopkins et al., 2012). This phenomenon, called “phase variation,” is based on the activity of a DNA invertase, encoded by the hin gene, which enables the reversible inversion of the promoter of the fljAB operon. This genetic element allows transcription of both fljB and fljA, encoding the phase-2 antigen and a negative regulator of the fliC gene, responsible for expression of the phase-1 flagellar antigen, respectively (Barco et al., 2014). Conversely, when the promoter is oriented to inhibit the expression of both fljA and fljB, phase-1 flagellar antigens can be detected because of the lack of repression of fliC (Soyer et al., 2009; Bugarel et al., 2012). The lack of expression of phase-2 flagellar antigen in S. 4,[5],12:i:- is generally associated with the deletion in fljB coding gene; however, to a less frequent extent, other mechanisms can lead to the monophasic status such as mutations and deletions of fljA and hin (Soyer et al., 2009). Furthermore, the invertible promoter that controls the expression of fljB and fliC may become blocked in one position, thereby allowing only the expression of the fliC gene (Zamperini et al., 2007). Several authors (Soyer et al., 2009; Hopkins et al., 2010; Bugarel et al., 2012; Barco et al., 2014) described variants of S. 4,[5],12:i:- that unexpectedly conserved the fljB gene even though are characterized by an inconsistent serological detection of the second flagellar phase. Such isolates have been named “inconsistent” S. Typhimurium or “atypical” monophasic variants of S. Typhimurium (atypical MVST hereafter), and their monophasic status is related to secondary mechanisms different from the absence of the fljB gene. Analysis of the genomic deletions in the phase-2 flagellum locus responsible for the monophasic phenotype of circulating S. 4,[5],12:i:- strains suggests that multiple independent clones may be emerging worldwide. A remarkable amount of genotypic variations that occurred during the expansion of this serovar, including multiple deletion events involving the phase-2 flagellin locus, were recently documented (Petrovska et al., 2016).
Although many studies have documented the ecological success of S. 4,[5],12:i:- serovar, few investigations have been conducted to explain this phenomenon from a genetic perspective. One accredited hypothesis to justify the epidemic spreading of different clones of S. 4,[5],12:i:- is the high occurrence of genes encoding tolerance to toxic concentrations of heavy metals and resistance to antibiotics (Mourão et al., 2016; Petrovska et al., 2016). It is broadly assumed that the spreading of strains showing high resistance to heavy metals has been promoted by the extensive use of feed supplemented with large amounts of copper and zinc to favor animal growth and to limit the use of antimicrobials. This feature is raising considerable concern because, in principle, it may affect the ability of the immune system to control bacterial infections, since macrophages are able to poison intracellular bacteria in the phagosome with large amount of copper and zinc (Neyrolles et al., 2015). Thus, the presence of additional heavy metal resistance genes may further contribute to the natural ability of S. 4,[5],12:i:- to escape the metal-mediated antimicrobial response of human macrophages (Kapetanovic et al., 2016). Moreover, the recent finding that Salmonella motility is modulated by zinc availability (Ammendola et al., 2016) suggests that the ability to face environments characterized by variable metal contents may contribute to the control of flagella expression in monophasic strains.
An additional factor that could be involved in the success of this emergent serovar is the presence of toxin-antitoxin (TA) cassettes. The involvement of these elements in a wide range of biological functions including growth control, defense against phages and biofilm formation may contribute to the success of the isolates harboring these genes in terms of adaptability to changing environments (Di Cesare et al., 2016). Moreover, it has been well established that TA systems, especially type II TA systems, play a central role in persistence (Page and Peti, 2016).
To identify the combination of different factors that could explain the eco-physiological role of this emergent serovar, whole genome sequencing of 50 epidemiologically unrelated S. 4,[5],12:i:- isolates obtained in Italy from animals and foodstuff from 2010 to 2016 was performed. Sequences were evaluated for the presence of genes conferring tolerance to heavy metals and antimicrobials, toxin-antitoxin systems, and specific plasmid replicons. A functional assay was performed to test the impact of heavy metals tolerance genes on bacterial motility in presence of high concentrations of copper. The presented results contribute to clarify the success and diffusion of the S. 4,[5],12:i:- serovar and provide valuable implications in terms of public health.
Materials and Methods
Sample Selection and Collection
Fifty S. enterica serotype 4, [5],12:i:- isolates, also called monophasic variants of Salmonella Typhimurium (MVST), collected by the OIE Reference Laboratory for Salmonellosis (Istituto Zooprofilattico Sperimentale delle Venezie, Legnaro, Italy) and representative of the entire National situation from 2010 to 2016, were selected for this study. Complete information for the 50 investigated isolates is summarized in Figure 1, Supplemental Tables 1, 6. Most of the tested isolates were isolated in 2011 (n = 20) and 2015 (n = 25). Isolates were epidemiological unrelated, 10 of them were identified as atypical MVST, meaning that they phenotypically behave as MVST, even though the fljB gene is conserved (Barco et al., 2014).

Figure 1. Summary infographic of strains. From outer to inner circle: strain name and source of origin; year of isolation (before/after 2011); HMT profile, color coded; TA profile, color coded; AMR profile, color coded (splitted into two circles for readability); replicon profile, color coded (splitted into five circles for readability); phylogenetic tree derived from core genome analysis. Strain clustering is highlighted (red: cluster 1; light-blue: cluster 2; green: cluster 3). Atypical MVST are highlighted with green circles. All 50 the analyzed strains and the two reference genomes (LN999997 and LT2) are included.
The sources of the Salmonella isolates were: 37 from swine, 5 from poultry, 5 from bovine, 1 from humans, 1 from wild birds (heron), and 1 not assigned (Table 1).
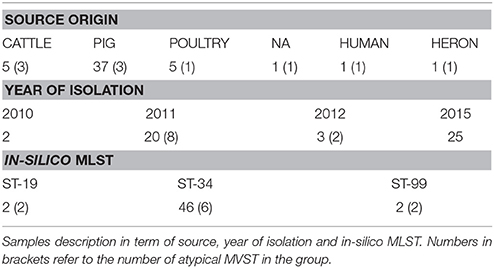
Table 1. Sample description.
All isolates were serotyped according to the White-Kaufmann-Le Minor scheme (Grimont and Weill, 2007) using the traditional slide agglutination method. The S. 4, [5],12:i:- isolates were also phage typed following protocol developed by Anderson et al. (1977); the results were interpreted according to the guidelines established by the International Federation for Enteric Phage Typing (IFEPT, Laboratory of Enteric Pathogens, Health Protection Agency, Colindale, London, UK).
All isolates were supplied with all available information in terms of: country and region of isolation, collection date, and isolation source.
Multilocus Variable-Number Tandem Repeat Analysis
MLVA was performed according to the protocol described by Lindstedt et al. (2007). The size measurements for each locus were estimated using a Genetic Analyzer 3130XL (Applied Biosystems, Life Technologies Corporation, Carlsbad, CA, USA). According to the nomenclature suggested by Larsson et al. (2013), the MLVA results were reported as a string of five numbers representing the variable number of tandem repeats (VNTRs) at the corresponding loci (STTR9–STTR5–STTR6–STTR10pl–STTR3), or as “–” in the case that a PCR product was not obtained for a locus.
Library Preparation
Genomic DNA (gDNA) was extracted using commercial column-based protocols (DNeasy Blood and Tissue kit, Qiagen, and Easy-DNATM gDNA Purification kit, Invitrogen) and purified gDNA was quantified with a Qubit 3.0 Fluorometer (Life Technologies). Libraries for sequencing were prepared using a Nextera XT DNA sample preparation kit (Illumina): briefly, gDNA was subjected to tagmentation, a step in which the DNA is enzymatically cleaved and then tagged with Illumina adapters. Cleaved gDNA was subjected to a second PCR step for indexing with a unique combination of i5 and i7 index primers. PCR products were then purified using Agencourt AMPure XP beads (Beckman Coulter Inc.) and manually normalized to a final molarity of 4 nM using Tris-HCl (10 mM). High-throughput sequencing was performed with MiSeq Reagent kit v3 resulting in 251 bp long paired-ends reads. All the full-length sequences have been deposited in ENA (http://www.ebi.ac.uk/ena; accession number PRJEB21283).
Sequencing and Pre-processing
All the sequencing data were subjected to quality assessments using FastQC v0.11.2 and to length and quality trimming using Trimmomatic v 0.32 (Bolger et al., 2014), according to the following criteria: Illumina adapter removal; removing bases from the head and tail of the sequence if their quality score was below 20; removing sequences if the average quality score in a sliding window of four bases fell below 20; discarding sequences with a final length <100 base pairs.
Bioinformatic Analysis
All trimmed reads belonging to the same sample were assembled using Velvet v1.2.10 (Zerbino and Birney, 2008) and VelvetOptimizer v2.2.4 with options–s 19 (lower hash value)–e 99 (higher hash value)–t 10–f–fastq—shortPaired. Each sample was subject to three assembly runs and the assembly showing the higher n50 was chosen for each sample. All reads were then mapped onto the assembled bacterial genomes to compute the coverage statistics and to verify the quality of the assembly. When a sample showed an apparently large number of assembled contigs other statistics were evaluated, including n50, expected genome size, coverage statistics as well as other information deriving from sample annotation. The sample was considered for subsequent analysis when these metrics fell in a range comparable to the other samples.
All samples were subject to in-silico serotyping using SeqSero (Zhang et al., 2015) and SISTR (Yoshida et al., 2016), starting from both raw reads and assembled data, to confirm the in vitro serotyping.
Identification of Flagellar Genes
Assembled data were searched for flagellar genes (flj genes UniProtKB identifiers: fljA—Phase-1 flagellin repressor: A0A0F7JBE2; fljB—Phase-2 flagellin: Q549S3) using the tblastn algorithm with default parameters to confirm the molecular characterization data available for atypical MVST isolates (Barco et al., 2014).
Identification of Heavy Metal Tolerance Genes
Assembled data were searched for heavy metal tolerance by direct blasting of the nucleotide sequences of eight genes involved in tolerance against copper, tellurite, arsenite, and silver (Mourão et al., 2014) (Supplemental Table 8) using the blastn algorithm with the following options: percentage of identity between query and subject sequence >80%, query coverage >60%, best e-value (which was considered < 1e-15).
Identification of Antibiotic Resistance Genes and Plasmid Replicons
Assembled data were searched for antibiotic resistance genes (ARGs), plasmid replicons and subjected to in-silico multi locus sequence typing using the Bacterial Analysis Pipeline (https://cge.cbs.dtu.dk/services/cge/; Thomsen et al., 2016) with the following options: %ID threshold = 80.00%; selected minimum length = 60% for ResFinder and default parameters for all the other tools. In order to further investigate the presence of the Salmonella virulence plasmid pSLT, associated to the IncFIB(S) replicon in our database, all samples were searched for the spvC marker gene [NCBI, accession number NC_003277.2 (27178.27903, complement)] both by direct blasting with the following options: %ID threshold = 95%; minimum coverage length = 60%, e-value < 1e-15 and by investigation of the annotated genomes. Finally, in order to resolve whether the IncFIB(S) replicon was to be considered indicative of the pSLT (NCBI Reference Sequence accession number: NC_003277.2) or pSLT-BT (GenBank accession number: FN432031.1) plasmid, that share the same replicon, the whole plasmid reference sequences were searched in the genomes.
Identification of Type II Toxin-Antitoxin Families
Assembled data were searched for type II toxin-antitoxin modules by directly blasting the TA database (downloaded from http://202.120.12.135/TADB2/download.html in date 08/29/2016; Shao et al., 2011) using the megablast algorithm with the following options: percentage of identity between query and subject sequence >80%, query coverage >60%, best scoring e-value (which was considered < 1e-15). Database hit were retained if and only if both the toxin and the corresponding antitoxin were contemporarily found in the same region of the assembled genome. Moreover, toxin-antitoxin matches were filtered as follows.
• Removal of multiple matches in the same position. When a multiple match was found, the one deriving from a manually curated reference annotated gene was kept, and the hits with genes with an annotation derived by protein homology were discarded. Matches deriving from a reference annotated by protein homology were kept if and only if there was no corresponding hit with other genes in the database with a manually curated annotation
• Removal of multiple matches in nearby and partially overlapping positions (hits in the same region but with ≤10-bp shift).
Toxin- antitoxin annotation was recovered from the corresponding references (in NCBI or ENA), and the toxin-antitoxin family was extracted; the final profile of the toxin-antitoxin family was determined for each sample by removing the replicate imputed family independently of the position of the hit.
Co-localization of ARGs, HMTG, and TAs on plasmids was investigated. A genomic region of interest was considered to be plasmidic if the same contig was flagged as containing the plasmid replicon sequence using PlasmidFinder.
Phylogenetic Analysis
SNP Based Phylogeny
Assembled data, S. Typhimurium LT2 reference genome (ENA ref AE006468.2) and the most recent reference of S. 4,[5],12:i:- (ENA ref LN999997.1) were subjected to SNP calling and reference-based phylogeny tree building using CSI Phylogeny 1.4 (Kaas et al., 2014) with LN999997.1 as the reference genome and default parameters for SNP filtering and pruning.
Core Genome Phylogeny
Assembled data were subjected to functional annotation using Prokka (Prokka, 2014). To compare our samples with pre-existing genomes references ENA ref AE006468.2 and ENA ref LN999997.1 were also annotated. Annotated data from all strains and the two references were passed to Roary v 3.6.2 (Page et al., 2015) (with options: –e –n –r) to build the pan-genome, core-genome and accessory genome of the strains.
Multiple alignment of core genes was converted from multifasta to phylip format, and then RAxML v 7.2.8 (Stamatakis, 2006) was used to infer the maximum-likelihood core genome phylogeny with the parameters -f a -x 12345 -p 12345 -# autoMRE -m GTRGAMMA. Phylogenetic trees obtained using the SNP-based approach and core genome multi-alignment were both re-rooted on the LT2 reference and compared using TOPD v4.6 (Puigbò et al., 2007) with the split distance (Robinson and Foulds, 1981) and nodal distance (Steel and Penny, 1993), and tested to verify whether the similarity between the two trees was significantly different from random.
Conjugation Assay
The transfer frequency of mcr-1 gene was investigated by conjugation experiments with rifampicin- resistant E. coli J53 as the recipient strain. Donor and recipient strains were grown in LB broth for 24 h at 37°C. A 1:50 dilution was prepared for each strain, and bacterial cells were grown at 37°C to an OD600 0.4. Next, 500 μl of the donor strain was added to 4.5 ml of the recipient strain, and the bacterial suspension was filtered using a 0.22 μm filter laid down on a pre-heated agar tryptose (AT) plate and incubated for 18–24 h at 37°C. The filter was washed with 10 ml of physiological saline and vortexed to completely resuspend the cells. The cellular suspension was centrifuged at 5,000 rpm for 10 min, and the pellet was resuspended in 1 ml of physiological saline after removing the supernatant. Serial dilutions were prepared, and 100 μl was plated in triplicate on Luria Bertani (LB) plates supplemented with colistin (0.7 mg/L) and rifampicin (50 mg/L) to select for transconjugants. The transfer frequencies were calculated as the number of transconjugants obtained per recipient. To screen for the presence of mcr-1 in transconjugant colonies, PCR was performed according to the protocol described by Liu et al. (2016). To determine whether the transconjugants also harbored the hipBA module, a PCR was designed for screening. DNA was extracted by boiling at 99°C for 15 min. PCR primers were as follows: HipA F 5′ GAATTCATGAGCCGGAAACAGCAA 3′; HipA R 5′ AAGCTTTCATTTCTCAGCCAGGC 3′; HipB F 5′ GAATTCATGATCAATAATGATTACCCGTTAAA 3′; HipB R 5′ AAGCTTTTACCAATCCTCCTGTCGT 3′. PCR was performed in a final volume of 50 μl using 1X Buffer Taq Gold, 2 mM MgCl2, 200 μM, or 400 μM dNTPs for HipB and HipA, respectively, 0,4 mM each primer, and 2,5 U Taq Gold (Life Technologies). Thermal cycling consisted of 95°C for 5 min, followed by 30 cycles (95°C for 30 s, 55°C for 30 s, 72°C for 90 s) and a final step at 72°C for 10 min for HipA. For HipB, the annealing temperature was 50°C and the elongation time was 30 s.
Screenings for Swimming Motility
A subset of 15 Salmonella strains were grown in Luria Bertani medium (LB) at 37°C with aeration. Strains were selected for carrying copper tolerance cluster (9 strains) or not (6 strains). In particular, among strains carrying tolerance cluster, IZS-11, IZS-15, IZS-16, DTU2016-1032, DTU2016-1036, DTU2016-1040, and DTU2016-1044 harbored merA, pcoA, pcoD, silA, silE genes, DTU2016-1034 harbored pcoA, pcoD, silA, silE genes, and DTU2016-1031 harbored pcoA, pcoD, silA, silE, terF. Strains IZS-14, IZS-17, 2260-2012, 3443-2011 did not harbor any tolerance genes among those investigated in the present study while strains 4007–2011 and 3528–2011 harbored only merA gene.
Overnight grown inocula were diluted 1:100 in fresh medium and incubated at 37°C with aeration. After 5 h, 4 μl of each strain were dropped on LB motility plates (agar concentration 0,4%), supplemented or not with CuSO4 2 mM. The plates were incubated with the upper side facing up at 37°C overnight. The next day halos of swimming bacteria were screened and their diameters recorded. Copper was prepared as a 0.5 M stock solution by solubilising ultra-pure powder purchased from BDH Laboratory Supplies in ultra-pure water (Ammendola et al., 2016).
Although literature data indicate that copper sulfate is not bactericidal for Salmonella at the concentrations used in this work (Pontel et al., 2007; Petrovska et al., 2016), preliminary assays were carried out to verify possible bactericidal activity of copper sulfate on all the strains under investigation. To this aim, inocula of Salmonella strains, grown overnight in LB medium, were washed once in PBS, resuspended in PBS to a final concentration of 108 cfu/ml and then serially diluted (1:10) in the same buffer. 5 μl of each dilution were spotted onto LB agar plates supplemented or not with CuSO4 2 mM. After an overnight incubation at 37°C the number of colony forming units on both plates was compared to highlight possible copper-induced bacterial killing.
Multivariate Analysis
All the information recovered from previous analyses was summarized in a table to be imported in R (Team, 2008) for statistical analysis. Each category (heavy metal resistance gene profile, antimicrobial resistance profile, toxin-antitoxin system profile, imputed plasmid) was subjected to descriptive analysis, and an extended table in which each categorical variable was replaced with its dummy variable submatrix (i.e., where each gene was flagged as 1 if present in a specific sample, and 0 otherwise) was created for subsequent analysis. Plasmids, HMTGs, AMRs, and TA profiles were tested for co-localization in the same genomic region and for the significance of their association using Fisher's exact test. Multivariate analysis [implemented in the FactoMineR package (Lê et al., 2008)] was used for exploratory data analysis of the extended table, taking into account the grouped structure of the variables. The dummy variables describing AMRs, plasmids, HMTGs, and TAs (1 for presence, 0 for absence) were treated as qualitative variables; metadata variables (collection date, isolation source and region) together with microbiological and molecular variables (i.e., serotype and flagellin gene presence) were treated as supplementary categorical variables. Multiple factor analysis (MFA), data dimensionality and the influence of variable groups on the position of the samples in multidimensional space was investigated; visualization of the related graphs was performed using the package factoextra (Kassambara and Mundt, 2017).
Multilevel multinomial logistic regression [implemented through generalized linear models in the glmnet package (Friedman et al., 2010)] was used to test whether a relationship existed between the profiles of the strains in terms of plasmids, HMTGs, AMRs, TAs, and their cluster position in the phylogenetic trees determined in the previous steps. To investigate each feature weight in predicting sample grouping, only samples belonging to the main clusters identified in the phylogeny analyses were retained. Multinomial logistic regression was subjected to automatic K-fold cross-validation (K = 10) to select the optimal value of the regularization parameter lambda, which determined the overall strength of the lasso penalty. Minimization of misclassification error was set as the criterion for 10-fold cross-validation optimization. The value lambda.min, which is the value of lambda that provides the minimum mean cross-validated error, was selected for the final model.
The gene presence-absence table generated by Roary was imported into R. Each of the annotated gene of the pan-genome was set as 1 in all samples where they could be annotated, and 0 otherwise. Only accessory genomes (i.e., genes that were found in <52 samples) were used as input for hierarchical clustering using the Manhattan distance and Ward agglomeration method to investigate possible sample clustering based on genes shared by only a subset of the cohort. Therefore, predicted genes that were typical of each cluster (i.e., that were present in all strains of the cluster but were absent from all other strains) were investigated for function and GO terms by consulting UniProtKB.
For swimming motility analysis, the strains were divided into two groups according to the presence or absence of the copper resistance cluster. Mean and standard deviation values of motility in LB medium were calculated from replicate samples (n = 3) of each strain. The distribution of the data was tested for normality using the Shapiro-Wilk test, and differences in motility between two groups of samples were assessed using the Mann-Whitney U test.
All methods were carried out in accordance with relevant guidelines and regulations.
Results
Salmonella 4,[5],12:i:- Sample Characterization
Isolates were selected because they belonged to the most common phage-types associated with this serovar (DT193, U311, DT120, U302 DT20A, DT32, DT7, DT7a, RDNC, U302) and displayed different Multilocus variable-number tandem repeat analysis (MLVA) profiles (Supplemental Table 1). The in silico multilocus sequence typing (MLST) profile of the studied strains displayed a scenario in which the majority of the isolates belonged to ST-34 (n = 46; Table 1, Supplemental Table 1).
Bioinformatics Analysis
The raw data showed an average of 968707.94 ± 471051.53 sequenced reads with an average GC% content of 51.6% [range (51–53%)]. Quality trimming removed on average 8.14 ± 3.71% of the strain sequences. The assembled data displayed an average of 171.58 contigs with an average n50 of 242497.7 and an average coverage depth of 68.47 ± 33.72. Complete details are summarized in Supplemental Table 2. Blast research of genes involved in the flagellar structure revealed hits for the fljA and fljB genes on the assembled genomes of all the isolates (n = 10) known to be atypical MVST.
Genomic Regions Encoding Tolerance to Heavy Metals and Resistance to Antimicrobials
The heavy metal tolerance genes found were summarized into 5 HMTG profiles, as reported in Figure 1 and Table 2. Most strains (44/50) possess genes expected to confer resistance to both copper (pcoA-pcoD) and silver (silA-silE), with approximately half of them also exhibiting the mercury tolerance gene merA. In most of the strains, the pcoA, pcoD, silA, and silE genes, when present, were co-localized in the same genomic region (i.e., in the same contig), as reported in Figure 2, with silE preceding silA (5561-bp distance), silA preceding pcoA (4959-bp distance), and pcoA preceding pcoD (1321-bp distance).

Table 2. Sample description by genotype.

Figure 2. Location of pcoA, pcoD, silA, and silE in the identified genomic region. SilP and cusS are also represented in gray, since they were searched after functional annotation highlighted them as conserved in all samples carrying the pcoA-pcoD-silA-silE HMTG profile.
No significant association was found between the presence of this multi-metal tolerance genomic region and the presence of other heavy metal tolerances genes in the same strain. However, Fisher's exact test confirmed a significant association between the presence of the profiles of multi-heavy metal tolerance genes (HMTGs) and the strains isolated after 2011 (p-value < 0.009). In detail, after 2011, more than 97% of the strains carried more than one HMTG, while ~70% showed this pattern before 2011 (Table 3); after 2011 more than 55% showed a HMTG profile containing either merA or terF in addition to the HMTG profile pcoA, pcoD, silA, silE, while only ~17% showed this pattern before 2011.
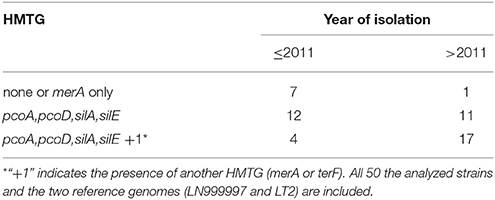
Table 3. Frequency of the heavy metal tolerance profile by year of strain isolation.
Moreover, as reported in Supplemental Table 3. The frequency analysis of annotated genomes revealed that the genomic region containing merA harbored other mercury tolerance genes such as merC (23 of 26 isolates) and merP, merR, and merT (21 of 26 isolates). Additionally, tetracycline resistance genes (tetR, tetC, and tetA) were found in the same region.
The pcoA, pcoD, silA, and silE region contains other copper and silver tolerance genes. In particular, cusS and silP co-occurred in 44 out of 44 strains (Figure 2), and cusR, cusB, cusA, cusF, cusC, and pcoE in 43 out of 44 strains. Furthermore, the arsenic tolerance genes arsD, arsC, arsB co-occurred in the same genomic portion for 42 out of 44 strains, and the cation tolerance genes cutA and sugE were found in the majority of strains (30 ≤ samples ≤ 40; Supplemental Table 3).
The same genomic region containing terF (found only in the strain DTU2016_1031) displayed the toxin-antitoxin cassette hipB-hipA and the tetracycline resistance gene tetA (Supplemental Table 3).
In most of the strains, the pcoA, pcoD, silA, and silE genes, when present, appeared to be co-localized in the same genomic region (i.e., contigs) containing the tetracycline resistance gene tetB. Forty samples showing pcoA, pcoD, silA, silE profile showed also the presence of tetB gene, while four samples showed pcoA, pcoD, silA, silE profile, and absence of tetB gene, respectively. Samples not showing pcoA, pcoD, silA, silE profile showed presence of the tetB gene in four samples, while four samples not showing pcoA, pcoD, silA, silE profile showed absence of the tetB gene. This association was confirmed by Fisher's exact test (p-value = 0.01365).
The acquired antimicrobial resistance gene profiles found in the panel of tested strains are summarized in Table 2. All but five samples showed at least one antimicrobial resistance gene, and most of them showed a complex pattern of multi-resistance. One strain (DTU2016_1031) was found to carry the colistin mcr-1 resistance gene (%ID = 100%, query/HSP = 1,626/1,000).
In most strains, the sulfametoxazol resistance gene sul2 and the streptomycin resistance genes strA and strB, when present, co-localized in the same genomic region (Fisher's exact test with p-value = 6.299e-11), which also contained the plasmid replicon IncQ1. Samples carrying the IncQ1 replicon showed such antimicrobial resistance (AMR) gene profile in 39 samples, while one sample carrying the IncQ1 replicon did not show such AMR gene profile. Samples not carrying the IncQ1 replicon did not show such AMR gene profile in 12 samples, while no sample did not carry the IncQ1 replicon nor such AMR gene profile.
Replicon Profile
The in silico plasmid identification revealed the presence of 22 different plasmid replicon types in the analyzed strains, whose profile are summarized in Table 2. Only two strains (DTU2016_1042 and DTU2016_1043) showed no hit in the plasmid replicon database; all other strains showed from one up to seven different predicted replicon types each, with most of the strains containing from one to three different replicon types. Plasmid replicon IncQ1 was the most common replicon type among the investigated strains being shared by 34 over 50 isolates, followed by replicon ColRNAI that was present in 27 over 50 isolates. All isolates displaying ST-34 lacked the IncFIB(S) replicon, which is a proxy of the Salmonella virulence plasmid pSLT in the used database (Kingsley et al., 2009; Carattoli et al., 2014; Silva et al., 2017), whereas isolates ISZ-14 (ST-19), ISZ-17 (ST-99), and 2260-2012 (ST-99), all classified as atypical MVST, showed a replicon profile containing IncFIB(S). Moreover, when the whole genome of these isolates was matched toward pSLT whole sequence, pSLT was retrieved with 100% of identity and 99% of reference coverage. These results are further supported by the finding that the spvC gene [which is considered to be a marker of the presence of pSLT (Herrero et al., 2009)] was present only in these three isolates.
Genomic Regions Encoding Type II Toxin-Antitoxin Cassettes
The identified type II toxin-antitoxin cassette profiles are summarized in Table 2. All strains shared the putative presence of type II TA cassettes belonging to the rel, fic, vap, hig, phd-doc, and par family, while only a few strains showed additional TA modules belonging to the ccd, let, maz, stborf, vag, or hip family. In particular, TA modules belonging to the ccd family were identified in only five strains (3443-2011, ISZ-14, 2260/2012, ISZ-17, ISZ-1). For two of them (2260-2012, ISZ-17), the ccd TA box appeared to be co-localized on the same genomic region (i.e., the same contig) containing the replicon of the IncFIB(S) plasmid, suggesting that it may be carried by the plasmid. Complete details are summarized in Supplemental Table 9. No significant difference was found in total amount of TA modules (either divided by Type II family or cumulative) between samples isolated before/after 2011.
Interestingly in sample DTU2016_1031, the TA module hipA/hipB co-localized with mcr-1 colistin resistance gene in a genetic region presumably of plasmidic origin, as supported by the concurrent presence of the IncHI2a plasmid replicon and many genes encoding conjugation abilities (e.g., bacterial conjugation TrbI-like protein, plasmid partition protein A, plasmid segregation protein ParM, type-F conjugative transfer system pilin assembly protein). In order to investigate the co-localization we found in the strain DTU2016_1031 of mcr-1 and hipA/hipB genes on a plasmid and its relative transfer frequency, conjugation experiments with E. coli strain J53 were performed. The results demonstrated the plasmidic origin of mcr-1 gene and a transfer frequency of 6 * 10-5 cells per recipient together with the hipA/hipB cassette.
Phylogenetic Analysis
Single Nucleotide polymorphism (SNP) based phylogeny using LN999997.1 as a reference genome, which is a representative multidrug European clone(Petrovska et al., 2016) revealed 2102 high-quality shared SNP positions between each strain and this reference genome. The SNP distance between all strains ranged from 1 to 927 SNPs, generating the cladogram displayed in Figure 3 when S. Typhimurium LT2 (the reference) was used to re-root the tree.

Figure 3. Phylogenetic tree obtained from SNP based phylogeny when LN999997 is used as reference strain; LT2 set as root; green circles highlight atypical MVST. Red: cluster 1; light-blue: cluster 2; green: cluster 3.
As shown in Figure 3, three main clusters were identified. Cluster 1 (highlighted in red) comprises the S. 4,[5],12:i:- reference (ENA ref LN999997.1) and 13 strains, one of which was atypical MVST; the within maximum SNP distance among all strains in the cluster was 65 SNPs. Cluster 2 (highlighted in light blue) comprised 12 strains, four of which were atypical MVST; the within maximum SNP distance among all samples in the cluster was 60 SNPs. Cluster 3 (highlighted in green) comprised 18 strains, none of which was known to be atypical; this cluster included the only strain carrying the mcr-1 resistance gene; the within maximum SNP distance among all samples in the cluster was 73 SNPs. If left unrooted, the tree highlighted another cluster grouping together all samples that were very distantly related from all other samples (847 SNPs), namely, 3443/2011, ISZ-14, ISZ-17, and 2260/2012, which were atypical MVST, and the LT2 reference (data not shown).
Automatic annotation of assembled genomes identified an average of 4664.76 ± 77.86 CDS [range: (4552:5022)], an average of 75.53 ± 4.39 tRNA [range (67:87)] and a mode of three repeated regions per strain (Supplemental Table 2). The pan-genome calculation starting from the annotated files identified a core genome of 3983 genes (genes found in 99% ≤ strains ≤ 100%), 216 soft core genes (95% ≤ strains < 99%), and 593 shell genes (15% ≤ strains < 95%) over a pan-genome of 6992 genes. The multi-FASTA alignment of all the core genes consisted of 3799245 positions, 3799094 of which were determined. The best scoring maximum likelihood tree inferred from bootstrapping is displayed in Figure 4.

Figure 4. Phylogenetic tree obtained from core genome alignment phylogeny; LT2 set as root; green circles highlight atypical MVST. Red: cluster 1; light-blue: cluster 2; green: cluster 3.
Three main clusters were identified with medium-to-high bootstrap value support. These clusters grouped the same strains identified by the SNP-based phylogeny, although sometimes with a different topology. If left unrooted, the cluster containing strains 3443-2011, ISZ-14, 2260-2012, and ISZ-17 and the LT2 reference appeared to be very distantly related to all other strains, in agreement with the SNP cluster assignment. The two phylogenetic trees achieved a split distance value of 0.326530612244898 and a nodal distance of 2.676077, indicating a good level of accordance between the two methods that was significantly different from random chance.
Hierarchical clustering of the Manhattan distance computed over the accessory genome presence/absence (Supplemental Table 4) gene profile is depicted in Figure 5. An average of 645.7115 ± 77.32218 genes per strain was found in the accessory genome, with strain DTU2016_1031 showing the highest number of non-core genes (1,000) and ISZ-4 showing the lowest one (533). Again, strains 3443-2011, ISZ-14, 2260-2012, and 17 clustered with the LT2 reference distantly from all other strains. This cluster was characterized by 1194 accessory genes, 267 of which were common to all five strains. Although with some discrepancies, accessory genome hierarchical clustering showed that the strains tended to group depending on their clustering, as defined by the SNP-based or core genome-based phylogeny. Accessory genes that were typical of each cluster (i.e., that were present in all strains of each cluster, but were absent from all other strains) were annotated with function and GO terms as reported in Supplemental Table 5. In detail, cluster 1 was characterized by 1 predicted gene, cluster 2 by 2 predicted genes, cluster 3 by 15 predicted genes and samples outside clusters by 25 genes that were unique to each of the clusters of strains. Enriched GO terms (associated with ≥1 predicted genes) for each of the clusters were as follows.
1. Outliers—GO molecular function: structural molecule activity [GO:0005198]; GO cellular component: bacterial-type flagellum filament [GO:0009420], extracellular region [GO:0005576], cytoplasm [GO:0005737]; GO biological process: bacterial-type flagellum-dependent cell motility [GO:0071973]
2. Cluster 1—(no enrichment because there is only 1 gene)
3. Cluster 2—GO biological process: DNA binding [GO:0003677], metal ion binding [GO:0046872], transcription factor activity, sequence-specific DNA binding [GO:0003700], transcription, DNA-templated [GO:0006351]
4. Cluster 3—GO molecular function: DNA binding [GO:0003677]

Figure 5. Hierarchical clustering with Manhattan distance and UPGMA over accessory genome presence/absence gene profile. All 50 the analyzed strains and the two reference genomes (LN999997 and LT2) are included.
Multivariate Analysis
Multivariate data analysis of the complete table (Supplemental Table 6) summarizing AMRs, plasmids, HMTGs and TAs as dummy variables together with metadata (collection year, isolation source and geographic area) and molecular/microbiological data (flagellin genes and ST) was performed. MFA was used to incorporate the grouped nature of variables (AMR, HMTG, TA, plasmids). The scree plot obtained from MFA highlights that the first two dimensions explained a good percentage of the total variance (>40%, data not shown), justifying the visualization of the 2D plot results. Six components were retained in the final results to ensure that most of the total original variance was captured.
Figure 6A reports group representations for the first two dimensions. Toxin-antitoxin, heavy metal tolerance and replicon profile appeared to be the most influential factors in determining the positions of the strains in the first dimension, while antibiotic resistance profiles appeared to be the most influential factor in determining the position of the strain in the second dimension. The collection year and other metadata factors appeared to have a minor effect on the sample position in any of the two dimensions.
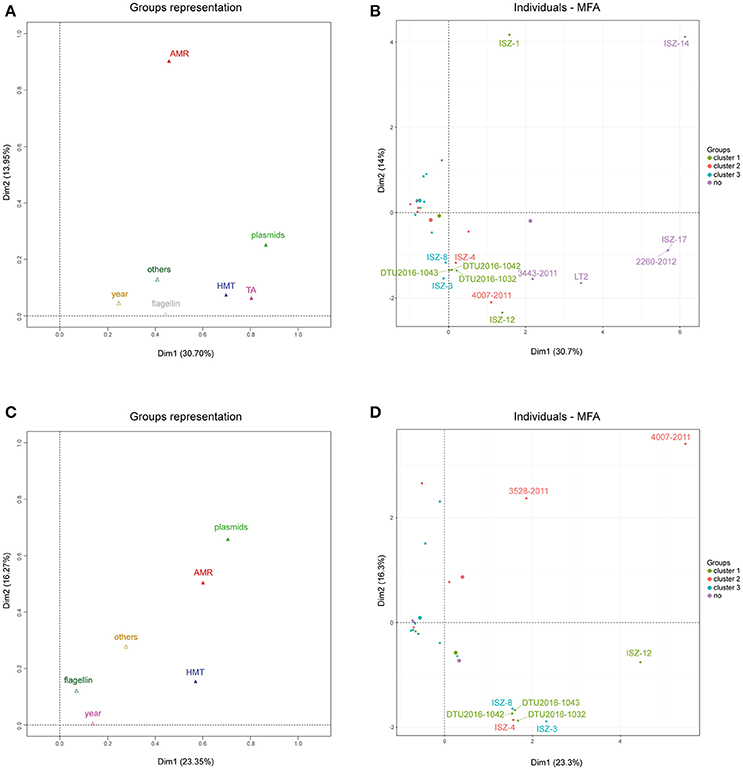
Figure 6. (A) Group representation on the first two dimensions for MFA on the whole dataset. (B) Individuals plot on the first two dimensions for MFA on the whole dataset. Color scales according to sample clustering in the phylogenetic analysis. (C) Group representation on the first two dimensions for MFA on the reduced dataset, after outliers removal. (D) Individuals plot on the first two dimensions for MFA on the reduced dataset, after outliers removal. Color scales according to sample clustering in the phylogenetic analysis. All 50 the analyzed strains and the two reference genomes (LN999997 and LT2) are included.
Figure 6B highlights the contribution of each strain to the construction of each of the first two dimensions and the position of the strain itself in the 2D plane.
The positions of strains 2260-2012, ISZ-17, and ISZ-14 were rightward according to their peculiar combination of TA profiles (rel, fic, vap, hig, phd-doc, rel/par, ccd, maz, ccd/rel, stborf2) and lack of HMTGs; moreover, strain ISZ-14 showed a unique AMR profile, while strains 2260-2012 and ISZ-17 showed no AMR gene. From this analysis, it appeared that almost all the influential strains (ISZ-14, 2260-2012, ISZ-17, LT2, 3443-2011) were actually those identified as outliers in the phylogenetic analysis. Therefore, to investigate finer differences in our cohort of strains, these outliers were excluded, and the analysis was re-run, discarding the variables responsible for their variance (Figures 6C,D) and showing the group representation and individual plots for this analysis.
After excluding the outliers, the percentage of variance captured by the first two axes declined by 7%. Moreover, the type II - TA modules were no longer useful in discriminating between strains (no variance shown in this subset of samples). The heavy metal tolerance profile was still an influential factor in determining the position of the strain in the first dimension, while the antibiotic resistance profile and plasmids appeared to be the influential factor in determining the position of the strain in both the first and second dimension. Strain 4007-2011, which has an extreme position in comparison to all other strains, appeared to be the most important contributor to construction of the axes, showing a unique profile both in terms of AMR (tetB, floR) and in the plasmid replicon profile. Strains ISZ-3, ISZ-4, ISZ-8, DTU2016_1032, DTU2016_1042, and DTU2016_1043 clustered together in the second quadrant. These strains had the following features:
• A diverse profile in terms of plasmid composition;
• No AMR or tetB gene only;
• A common TA profile;
• A HMTG profile with pcoA, pcoD, silA, and silE, with ISZ-3, DTU2016_1042 and DTU2016_1043 also showing merA.
A multinomial logistic regression was applied to investigate important variable for predicting strain clustering. This analysis was restricted to strains belonging to the three main clusters identified in the phylogenetic analysis (44 over 52 total strains), while seven of them were excluded (ISZ-3, ISZ-14, ISZ-15, ISZ-17, LT2, 3443-2011, 2260-2012). K-fold-cross-validation (K = 10) was used to optimize the regularization parameter lambda, resulting in lambda.min = 0.1086056 (value of lambda that provides the minimum mean cross-validated error). The final model, imposing lambda = lambda.min, identified 6 out of 25 variables as significant for determining the cluster label: tetC for AMR genes; ColRNAI, IncQ1, and IncI1 for plasmid replicons; merA for HM tolerance genes. Further details are supplied in Supplemental Table 7.
Motility Test
To determine the effect of the pcoA and pcoD genes on Salmonella motility, 15 strains were tested using an agar motility test in LB medium supplemented or not with CuSO4 2 mM, a concentration of metal that does not display bactericidal activity on Salmonella (Petrovska et al., 2016). The strains were divided into two groups: the first group consisted of samples for which copper resistance genes could not be annotated (n = 6); the second group comprised samples for which copper resistance genes could be annotated on the genome (n = 9). For comparison, we analyzed also the S. Typhimurium reference strain ATCC14028s, which lacks both pcoA and pcoD (Pontel et al., 2007), The halo diameter on LB plates was considered 100%, and the percentage of motility in the presence of copper was calculated for each strain (Figure 7). Strains carrying the copper resistance cluster were significantly less affected by the presence of the metal compared with the strains lacking those genes (Mann-Whitney U test, p-value = 0.0076).

Figure 7. (A) Swimming motility in CuSO4 supplemented plates of Salmonella strains carrying the copper resistance cluster (blue bars) or not (red bars). Each percentage was calculated taking as 100% of motility the halo diameters in plates without CuSO4 supplementation. Statistical difference in motility between the two groups of samples was assessed by the Mann-Withney U test. (B) representative picture showing differences in swimming motility between strains carrying (ISZ-17) or lacking (DTU2016-1040) the copper resistance cluster. At high copper concentration (4 mM) motility is inhibited in both strains. The effect of copper on the motility of the biphasic strain ATCC 14018, which lacks the metal resistance gene cluster, in included for comparison.
Discussion
In this study S. 4,[5],12:i:- was investigated with the aim of exploring genetic signatures possibly associated with its epidemiologic success. Genetic elements coding for tolerance to heavy metals, acquired antimicrobial resistance genes, type II toxin-antitoxin families and plasmid replicon types were studied by analyzing a number of 50 isolates belonging to different sources and collected in Italy from 2010 to 2016.
The results revealed the presence of common genetic features among the investigated isolates that might be involved in the widespread occurrence of such serovar in the swine food chain.
The main genetic trait shared by the investigated strains was represented by the presence of HMTGs encoding efflux systems involved in silver and copper tolerance, such as pcoA, pcoD, silA, and silE, as described by Mourão et al. for European and Spanish circulating clones of S. 4,[5],12:i:- (Mourão et al., 2015).
Tolerance to poisonous metals, mediated by different mechanisms, has been described as being on the rise among many bacterial pathogens (Losasso et al., 2014; Conficoni et al., 2016). This phenomenon has been documented by many authors, with a particular emphasis on pig husbandry (Medardus et al., 2014) and has been attributed to the use of the respective micronutrients in swine feed. Supplements such as copper and zinc, are added in swine and other livestock feed to promote animal growth and to increase feed efficiency. This practice has been rising since the ban of the use of antibiotics as growth promoters in animal feed, which has been effective in the EU since 2006 [Regulation 1831/2003/EC on additives for use in animal nutrition (1831/2003/EC, 2003)]. This use has been posing concerns among the scientific community regarding co-selection for antibiotic resistance in bacteria exposed to heavy metals (particularly copper and zinc but also iron, cobalt, manganese, and arsenic) that are used as growth promoters and for metaphylactic purposes in pig and poultry productions (Baker-Austin et al., 2006; Wales and Davies, 2015).
Unlike biocides (but with commonalities to antibiotic growth promoters), these micronutrients are used at inhibitory rather than lethal concentrations, potentially allowing tolerance phenomena to arise at a higher frequency in the farm environment than following biocide application (Wales and Davies, 2015). However, unlike antibiotic growth promoters, metals are very persistent in the environment (Wales and Davies, 2015). Moreover, due to agricultural practices as well as to other sources such as aquacultural and marine antifouling treatments, heavy metals may accumulate in soil, water, and sediments. Actually it has been estimated that livestock may be the major source of environmental contamination by zinc and copper (Nicholson et al., 2003; Wales and Davies, 2015). In the EU, high inclusion rates are permitted for young piglets which for inorganic zinc compounds may exceed basal requirements by thirty times (Baker-Austin et al., 2006; Norwegian Food Authority, 2014). Supplementation of ruminant feed with micronutrients is much less intensive than for pigs due to their high susceptibility to copper toxicity (Wales and Davies, 2015). The results of the present study indicate that S. 4,[5],12:i:- strains isolated after 2011 displayed a significantly higher prevalence of HMTGs than strains isolated before that date, suggesting that a time-dependent appearance of heavy metal tolerance profiles probably evolved under the selective pressure exerted by the rise in heavy metal concentrations in pig farming environments.
Flagella play an important role in the ability of Salmonella to colonize the gastrointestinal tract (Nicholson et al., 2003; Norwegian Food Authority, 2014; Ammendola et al., 2016) and some studies have indicated that flagellar gene expression is downregulated in presence of chelating agents or in strains that are unable to import zinc (Stecher et al., 2004; Ammendola et al., 2016). Zinc-dependent metabolic pathways are highly susceptible to interference by other transition metals that, when accumulating inside the cell, can compete with zinc for metal-binding sites in proteins (Ammendola et al., 2014). This scenario led us to speculate that Salmonella motility could be decreased in bacteria colonizing the gut of animals fed with copper enriched feed. As a matter of fact, we observed that S. 4,[5],12:i:- motility in agar plates containing 4 mM copper was completely abolished. Lower copper concentrations have variable effects on different Salmonella strains. We found that a 2 mM copper concentration inhibits motility of different S. 4,[5],12:i:- strains lacking the heavy metal resistance gene cluster, as well as that of the biphasic reference strain ATCC14028. A statistical comparison of the motility of S. 4,[5],12:i:- strains containing or lacking HMTGs in plates with 2 mM copper supported the hypothesis that the acquisition of this gene cluster promotes bacterial motility in environments enriched with this metal (Figure 7). Additional studies are required to consolidate this observation and to understand whether this feature may actually favor gut colonization in animals fed with copper-enriched feed. However, beyond the relevance of this specific observation, this experiment indicates that the acquisition of additional heavy metal resistance genes is useful for preventing the toxic effects of metals, thereby suggesting that this is a potential factor contributing to the success of this emerging Salmonella serovar.
The co-localization of silver and copper tolerance genes in the same genomic region, as well as mercury-encoding tolerance genes, suggests the possible acquisition of such determinants via the concerted actions of horizontal transfer of mobile elements. This hypothesis is also supported by the co-localization of genes involved in resistance to tetracycline (tet genes) in the same region harboring HMTGs and is coherent with the observation of Medardus et al. (2014) of a strong association between decreased susceptibility to heavy metals and antimicrobial resistance among Salmonella serovars isolated from swine, swine feed, and barn floors.
Genetic differences between MVST and atypical MVST isolates were detected beyond the well-known and described differences in the flagellin coding genes. Indeed, more than half of the atypical isolates displayed a reduced prevalence of HMTGs.
The frequency of antimicrobial resistance genotypes was high among S. 4,[5],12:i:- isolates with tetra-resistance causing the “ASSuT” phenotype, typical of the European clone, to be the predominant multiple drug resistance profile.
To investigate the potential mobilization of genetic elements, the panel of Salmonella isolates was examined for the presence of plasmid replicons, particularly those belonging to the major Inc family. The results revealed the presence of heterogeneous replicon contents in S. 4,[5],12:i:- isolates. The plasmid replicon IncQ1 was the most common among the isolates. The IncQ1 replicon is identical to the replicon of the broad-host-range RSF1010 plasmid of E. coli, containing the strA, strB, and sul2 resistance genes. This element corresponds to the previously characterized IS26-ΔrepA, repC, sul2, strA, and strB-IS26 transposon, in which part of the IncQ1 replicon has been mobilized together with the streptomycin and sulfonamide resistance genes (Miriagou et al., 2006). This mobile element is widely disseminated on plasmids and bacterial chromosomes (Izumiya et al., 2011) and was previously reported in an international collection of S. Typhimurium isolates originating mostly from humans but also from pigs (Helmuth et al., 1985). Plasmids of the IncQ family are highly mobilizable, stably maintained and transferred among a wide range of bacteria isolated from distinct environments (Smalla et al., 2000), thus suggesting their role in acquiring and transferring genetic material as well as in the concurrent bacterial survival ability in adverse environments. However, the phenotypic and genotypic high diversity of IncQ plasmids has been demonstrated, even for plasmids isolated from the same source (Helmuth et al., 1985; Izumiya et al., 2011). This finding suggests that the replicon may have evolved from a common ancestral plasmid; however, it is unlikely that IncQ plasmids represent a genetic trait for such classification purposes. It remains to be determined whether the resistance genes to sulfamides and streptomycins are located on the chromosomal or plasmidic portion of the isolate genomes, as the former hypothesis was demonstrated by Hendriksen et al. (2015). Since the presence/absence of the Salmonella virulence plasmid pSLT differentiates the European and Spanish clones of MVST circulating in Europe, the results support that the investigated isolates mainly belong to the European clone, being ST-34 and lacking pSLT. Concerning the atypical MVST, pSLT is harbored only by three out of the ten investigated isolates.
The analysis of the investigated strains genomes revealed the spread of type II TA, mainly involved in persistence phenomena. All the type II TA families were identified in the whole isolates panel. Additional genes were found in a small selection of strains. Type II TA modules are involved in bacterial adaptability in response to adverse environmental conditions and contribute to the non-growing phenotype in response to stress, allowing bacteria to self-protect becoming less sensitive to harmful environments (Hayes, 2003). These modules are frequently found in bacterial pathogens, including Salmonella, and are thought to be related to pathogenicity in epidemic bacteria (Georgiades and Raoult, 2011; Lobato-Márquez et al., 2015). Type II TA operons are likely to be acquired by bacteria through horizontal gene transfer and then positively selected due to the benefit given to pathogens in favorable ecological settings (Ramisetty and Santhosh, 2015).
Widespread and virulent Salmonella serovars, such as S. 4,[5],12:i:-, frequently harbor toxin-antitoxin (TA) cassettes (Di Cesare et al., 2016), that are located both in the chromosomal portion of the genome and/or in incompatible plasmids carrying ARGs, promoting persistence phenotypes (Ghafourian et al., 2014) and plasmid stabilization via post-segregation killing (Smalla et al., 2000; Georgiades and Raoult, 2011). S. 4,[5],12:i:- represents one of the three most prevalent serovars (EFSA, 2016), suggesting that the widespread occurrence of many type II TA families among the genome, as demonstrated in the present study, may represent a selective advantage supporting its successful spread.
In addition, the finding that the mcr-1 gene co-localized with the hipA/hipB TA cassette only in the colistin-resistant Salmonella strain, together with the confirmation by the conjugation assay of the plasmidic origin of both mcr-1 and hipA/hipB, suggests a possible active role for type II TA in enhancing plasmid stability and consequently the spread of mcr-1.
Both the SNP-based and the core genome-based phylogenetic analyses of the investigated isolates concordantly indicated a distinction among them based on the above-described genetic traits. Specific genetic signatures might be predicted for each of the identified clusters by accessory genome investigation; remarkably, metal ion binding genes could be identified as distinguishing cluster 2 from the others.
Atypical isolates spread among two of the three identified clusters and represented 100% of the isolates identified outside the clusters, close to the LT2 reference, lacking HMTGs, harboring pSLT and displaying a unique TA profile including additional TA modules (belonging to the maz, ccd/rel family plus stborf) compared to all the other analyzed strains.
Both phylogenetic trees highlighted the presence of a clade including isolates that were closely related the reference genome of S. 4,[5],12:i:- (LN999997), sharing a minimum genomic signature and including the most frequent type II TA families in the investigated panel (rel, fic, vap, hig, phd-doc, rel/par), merA and tet genes. Moreover, the first and second clusters comprehensively collect all strains showing the presence of the mercury tolerance gene merA (either alone or in combination with pcoA, pcoD, silA, and silE), while all the strains considered to be outside any cluster lack all of the searched HMT genes. In addition, multinomial logistic regression supported the significance of a few genetic features, belonging to HMTGs, AMRG and the replicon profile, in discriminating the investigated strains according to the highlighted clusters. The multiple factorial analysis (MFA) obtained by including all the investigated genomic features confirmed that the type II TA families, ARGs, heavy metal tolerance genes and plasmid replicons concurrently explained the variance among the investigated isolates. However, based on the analysis excluding the outlier strains, a different picture was drawn in which the roles of the TA cassettes were lost and the ARGs and replicon types maintained their strength in explaining the observed variance, together with HMTGs, thus confirming the role of such genetic elements in characterizing S. 4,[5],12:i:-.
Additional Information
All the full-length sequences have been deposited in ENA (http://www.ebi.ac.uk/ena; accession number PRJEB21283).
Author Contributions
LB, AR, AB, AD, and CL: Conceptualization; EM, DP, LB, SA, AB, AR, and CL: Methodology; EM and DP; Formal analysis; SP, AL, SA, and CL: Investigation; EM, SP, and AL: Data curation; EM, LB, AB, SA, AD, and CL: Writing—original draft; CM: Visualization; AR: Funding acquisition; AR and CL: Supervision.
Funding
This work was supported by the Italian Ministry of Health (Grant n. RCIZSVe 10/14) and the project Establishing Next Generation sequencing Ability for Genomic analysis in Europe (ENGAGE, GP/EFSA/AFSCO/2015/01/CT1). The conclusions, findings, and opinions expressed in this announcement reflect only the view of the authors and not the official position of the European Food Safety Authority.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Andrea Di Cesare from University of Genova for critical reading of the manuscript and Barbara Di Camillo from University of Padua for methodological support to data analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00715/full#supplementary-material
References
1831/2003/EC, of the E. P. and the C. of the E. U. (2003). Regulation (EC) No 1831/2003 of the European Parliament and of the Council of 22 September 2003 on additives for use in animal nutrition. Off. J. Eur. Union 4.
Agasan, A., Kornblum, J., Williams, G., Pratt, C. C., Fleckenstein, P., Wong, M., et al. (2002). Profile of Salmonella enterica subsp. enterica (subspecies I) serotype 4,5,12:i:- strains causing food-borne infections in New York City. J. Clin. Microbiol. 40, 1924–1929. doi: 10.1128/JCM.40.6.1924-1929.2002
Ammendola, S., Cerasi, M., and Battistoni, A. (2014). Deregulation of transition metals homeostasis is a key feature of cadmium toxicity in Salmonella. Biometals 27, 703–714. doi: 10.1007/s10534-014-9763-2
Ammendola, S., D'Amico, Y., Chirullo, B., Drumo, R., Civardelli, D., Pasquali, P., et al. (2016). Zinc is required to ensure the expression of flagella and the ability to form biofilms in Salmonella enterica sv Typhimurium. Metallomics 8, 1131–1140. doi: 10.1039/c6mt00108d
Anderson, E. S., Ward, L. R., de Saxe, M. J., and De Sa, J. D., (1977). Bacteriophage-typing designations of Salmonella typhimurium. J. Hyg. 78, 297–300. doi: 10.1017/S0022172400056187
Baker-Austin, C., Wright, M. S., Stepanauskas, R., and McArthur, J. V. (2006). Co-selection of antibiotic and metal resistance. Trends Microbiol. 14, 176–182. doi: 10.1016/j.tim.2006.02.006
Barco, L., Barrucci, F., Cortini, E., Ramon, E., Olsen, J. E., Luzzi, I., et al. (2015). Ascertaining the relationship between Salmonella Typhimurium and Salmonella 4,[5],12:i:- by MLVA and inferring the sources of human salmonellosis due to the two serovars in Italy. Front. Microbiol. 6:301. doi: 10.3389/fmicb.2015.00301
Barco, L., Longo, A., Lettini, A. A., Cortini, E., Saccardin, C., Minorello, C., et al. (2014). Molecular characterization of “inconsistent” variants of Salmonella Typhimurium isolated in Italy. Foodborne Pathog. Dis. 11, 497–499. doi: 10.1089/fpd.2013.1714
Barco, L., Mancin, M., Ruffa, M., Saccardin, C., Minorello, C., Zavagnin, P., et al. (2012). Application of the random forest method to analyse epidemiological and phenotypic characteristics of Salmonella 4,[5],12:i:- and Salmonella Typhimurium strains. Zoonoses Public Health 59, 505–512. doi: 10.1111/j.1863-2378.2012.01487.x
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bugarel, M., Vignaud, M. L., Moury, F., Fach, P., and Brisabois, A. (2012). Molecular identification in monophasic and nonmotile variants of Salmonella enterica serovar Typhimurium. Microbiologyopen 1, 481–489. doi: 10.1002/mbo3.39
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Conficoni, D., Losasso, C., Cortini, E., Di Cesare, A., Cibin, V., Giaccone, V., et al. (2016). Resistance to biocides in Listeria monocytogenes collected in meat-processing environments. Front. Microbiol. 7, 1–9. doi: 10.3389/fmicb.2016.01627
Crayford, G., Coombes, J. L., Humphrey, T. J., and Wigley, P. (2014). Monophasic expression of FliC by Salmonella 4,[5],12:I:- DT193 does not alter its pathogenicity during infection of porcine intestinal epithelial cells. Microbiology 160, 2507–2516. doi: 10.1099/mic.0.081349-0
de la Torre, E., Zapata, D., Tello, M., Mejía, W., Frías, N., García Peña, F. J., et al. (2003). Several Salmonella enterica subsp. enterica serotype 4,5,12:i: - Phage types isolated from swine samples originate from serotype typhimurium DT U302. J. Clin. Microbiol. 41, 2395–2400. doi: 10.1128/JCM.41.6.2395-2400.2003
Di Cesare, A., Losasso, C., Barco, L., Eckert, E. M., Conficoni, D., Sarasini, G., et al. (2016). Diverse distribution of toxin-antitoxin II systems in Salmonella enterica serovars. Sci. Rep. 6:28759. doi: 10.1038/srep28759
Dionisi, A. M., Graziani, C., Lucarelli, C., Filetici, E., Villa, L., Owczarek, S., et al. (2009). Molecular characterization of multidrug-resistant strains of Salmonella enterica serotype Typhimurium and monophasic variant (S. 4,[5],12:i:-) isolated from human infections in Italy. Foodborne Pathog. Dis. 6, 711–717. doi: 10.1089/fpd.2008.0240
EFSA (2016). The European Union summary report on trends and sources of zoonoses, zoonotic agents and foodborne outbreaks in 2015. EFSA J. 14:4634. doi: 10.2903/j.efsa.2016.4634
EFSA (2017). The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2016. EFSA J. 15:5077. doi: 10.2903/j.efsa.2017.5077
Friedman, J., Hastie, T., and Tibshirani, R. (2010). Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22. doi: 10.18637/jss.v033.i01
Garaizar, J., Porwollik, S., Echeita, A., Rementeria, A., Herrera, S., Wong, R. M., et al. (2002). DNA microarray-based typing of an atypical monophasic Salmonella enterica serovar. J. Clin. Microbiol. 40, 2074–2078. doi: 10.1128/JCM.40.6.2074-2078.2002
García, P., Guerra, B., Bances, M., Mendoza, M. C., and Rodicio, M. R. (2011). IncA/C plasmids mediate antimicrobial resistance linked to virulence genes in the Spanish clone of the emerging Salmonella enterica serotype 4,[5],12:i:–. J. Antimicrob. Chemother. 66, 543–549. doi: 10.1093/jac/dkq481
García, P., Hopkins, K. L., García, V., Beutlich, J., Mendoza, M. C., Threlfall, J., et al. (2014). Diversity of plasmids encoding virulence and resistance functions in Salmonella enterica subsp. enterica serovar Typhimurium monophasic variant 4,[5],12:i:- strains circulating in Europe. PLoS ONE 9:e89635. doi: 10.1371/journal.pone.0089635
Georgiades, K., and Raoult, D. (2011). Genomes of the most dangerous epidemic bacteria have a virulence repertoire characterized by fewer genes but more toxin-antitoxin modules. PLoS ONE 6:e17962. doi: 10.1371/journal.pone.0017962
Ghafourian, S., Raftari, M., Sadeghifard, N., and Sekawi, Z. (2014). Toxin-antitoxin systems: classification, biological function and application in biotechnology. Curr. Issues Mol. Biol. 16, 9–14.
Grimont, P. A., and Weill, F. X. (2007). Antigenic Formulae of the Salmonella serovars. Paris: WHO collaborating centre for reference and research on Salmonella.
Guerra, B., Laconcha, I., Soto, S. M., González-Hevia, M. A., and Mendoza, M. C. (2000). Molecular characterisation of emergent multiresistant Salmonella enterica serotype [4,5,12:i:-] organisms causing human salmonellosis. FEMS Microbiol. Lett. 190, 341–347. doi: 10.1016/S0378-1097(00)00359-1
Guerra, B., Soto, S. M., Arguelles, J. M., and Mendoza, M. C. (2001). Multidrug resistance is mediated by large plasmids carrying a class 1 integron in the emergent Salmonella enterica serotype [4,5,12:i:]. Antimicrob. Agents Chemother. 45, 1305–1308. doi: 10.1128/AAC.45.4.1305-1308.2001
Hauser, E., Tietze, E., Helmuth, R., Junker, E., Blank, K., Prager, R., et al. (2010). Pork contaminated with Salmonella enterica serovar 4,[5],12:i:-, an emerging health risk for humans. Appl. Environ. Microbiol. 76, 4601–4610. doi: 10.1128/AEM.02991-09
Hayes, F. (2003). Toxins-antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science 301, 1496–1499. doi: 10.1126/science.1088157
Helmuth, R., Stephan, R., Bunge, C., Hoog, B., Steinbeck, A., and Bulling, E. (1985). Epidemiology of virulence-associated plasmids and outer membrane protein patterns within seven common Salmonella serotypes. Infect. Immun. 48, 175–182.
Hendriksen, R. S., Leekitcharoenphon, P., Lukjancenko, O., Lukwesa-Musyani, C., Tambatamba, B., Mwaba, J., et al. (2015). Genomic signature of multidrug-resistant Salmonella enterica serovar Typhi isolates related to a massive outbreak in Zambia between 2010 and 2012. J. Clin. Microbiol. 53, 262–272. doi: 10.1128/JCM.02026-14
Herrero, A., Mendoza, M. C., Threlfall, E. J., and Rodicio, M. R. (2009). Detection of Salmonella enterica serovar Typhimurium with pUO-StVR2-like virulence-resistance hybrid plasmids in the United Kingdom. Eur. J. Clin. Microbiol. Infect. Dis. 28, 1087–1093. doi: 10.1007/s10096-009-0753-1
Hopkins, K. L., de Pinna, E., and Wain, J. (2012). Prevalence of Salmonella enterica serovar 4,[5],12:i:- in England and Wales, 2010. Euro. Surveill. 17:20275. doi: 10.2807/ese.17.37.20275-en
Hopkins, K. L., Kirchner, M., Guerra, B., Granier, S. A., Lucarelli, C., Porrero, M. C., et al. (2010). Multiresistant Salmonella enterica serovar 4,[5],12:i:- in Europe: a new pandemic strain? Euro Surveill. 15:19580. Available online at: http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19580
Izumiya, H., Sekizuka, T., Nakaya, H., Taguchi, M., Oguchi, A., Ichikawa, N., et al. (2011). Whole-genome analysis of Salmonella enterica serovar typhimurium T000240 reveals the acquisition of a genomic island involved in multidrug resistance via IS1 derivatives on the chromosome. Antimicrob. Agents Chemother. 55, 623–630. doi: 10.1128/AAC.01215-10
Kapetanovic, R., Bokil, N. J., Achard, M. E., Ong, C. L., Peters, K. M., Stocks, C. J., et al. (2016). Salmonella employs multiple mechanisms to subvert the TLR-inducible zinc-mediated antimicrobial response of human macrophages. FASEB J. 30, 1901–1912. doi: 10.1096/fj.201500061
Kaas, R. S., Leekitcharoenphon, P., Aarestrup, F. M., and Lund, O. (2014). Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS ONE 9:e104984. doi: 10.1371/journal.pone.0104984
Kingsley, R. A., Msefula, C. L., Thomson, N. R., Kariuki, S., Holt, K. E., Gordon, M. A., et al. (2009). Epidemic multiple drug resistant Salmonella Typhimurium causing invasive disease in sub-Saharan Africa have a distinct genotype. Genome Res. 19, 2279–2287. doi: 10.1101/gr.091017.109
Larsson, J. T., Torpdahl, M., MLVA working group, and Møller Nielsen, E. (2013). Proof-of-concept study for successful inter-laboratory comparison of MLVA results. Euro Surveill. 18:20566. doi: 10.2807/1560-7917.E.S.2013.18.35.20566
Lê, S., Josse, J., and Husson, F. (2008). FactoMineR : an R package for multivariate analysis. J. Stat. Softw. 25, 253–258. doi: 10.18637/jss.v025.i01
Lindstedt, B. A., Torpdahl, M., Nielsen, E. M., Vardund, T., Aas, L., and Kapperud, G. (2007). Harmonization of the multiple-locus variable-number tandem repeat analysis method between Denmark and Norway for typing Salmonella Typhimurium isolates and closer examination of the VNTR loci. J. Appl. Microbiol. 102, 728–735. doi: 10.1111/j.1365-2672.2006.03134.x
Liu, Y. Y., Wang, Y., Walsh, T. R., Yi, L. X., Zhang, R., Spencer, J., et al. (2016). Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect. Dis. 16, 161–168. doi: 10.1016/S1473-3099(15)00424-7
Lobato-Márquez, D., Moreno-Córdoba, I., Figueroa, V., Díaz-Orejas, R., and García-del Portillo, F. (2015). Distinct type I and type II toxin-antitoxin modules control Salmonella lifestyle inside eukaryotic cells. Sci. Rep. 5:9374. doi: 10.1038/srep09374
Loftie-Eaton, W., and Rawlings, D. E. (2012). Diversity, biology and evolution of IncQ-family plasmids. Plasmid 67, 15–34. doi: 10.1016/j.plasmid.2011.10.001
Losasso, C., Belluco, S., Cibin, V., Zavagnin, P., Mičetić, I., Gallocchio, F., et al. (2014). Antibacterial activity of silver nanoparticles: sensitivity of different Salmonella serovars. Front. Microbiol. 5:227. doi: 10.3389/fmicb.2014.00227
Lucarelli, C., Dionisi, A. M., Filetici, E., Owczarek, S., Luzzi, I., and Villa, L. (2012). Nucleotide sequence of the chromosomal region conferring multidrug resistance (R-type ASSuT) in Salmonella Typhimurium and monophasic Salmonella Typhimurium strains. J. Antimicrob. Chemother. 67, 111–114. doi: 10.1093/jac/dkr391
Machado, J., and Bernardo, F. (1990). Prevalence of Salmonella in chicken carcasses in Portugal. J. Appl. Bacteriol. 69, 477–480. doi: 10.1111/j.1365-2672.1990.tb01538.x
Medardus, J. J., Molla, B. Z., Nicol, M., Morrow, W. M., Rajala-Schultz, P. J., Kazwala, R., et al. (2014). In-feed use of heavy metal micronutrients in U.S. Swine production systems and its role in persistence of multidrug-resistant Salmonellae. Appl. Environ. Microbiol. 80, 2317–2325. doi: 10.1128/AEM.04283-13
Miriagou, V., Carattoli, A., and Fanning, S. (2006). Antimicrobial resistance islands: resistance gene clusters in Salmonella chromosome and plasmids. Microbes Infect. 8, 1923–1930. doi: 10.1016/j.micinf.2005.12.027
Mourão, J., Machado, J., Novais, C., Antunes, P., and Peixe, L. (2014). Characterization of the emerging clinically-relevant multidrug-resistant Salmonella enterica serotype 4,[5], 12: i:-(monophasic variant of S. Typhimurium) clones. Eur. J. Clin. Microbiol. Infect. Dis. 33, 2249–2257. doi: 10.1007/s10096-014-2180-1
Mourão, J., Marçal, S., Ramos, P., Campos, J., Machado, J., Peixe, L., et al. (2016). Tolerance to multiple metal stressors in emerging non-typhoidal MDR Salmonella serotypes: a relevant role for copper in anaerobic conditions. J. Antimicrob. Chemother. 71, 2147–2157. doi: 10.1093/jac/dkw120
Mourão, J., Novais, C., Machado, J., Peixe, L., and Antunes, P. (2015). Metal tolerance in emerging clinically relevant multidrug-resistant Salmonella enterica serotype 4,[5],12:i:- clones circulating in Europe. Int. J. Antimicrob. Agents 45, 610–616. doi: 10.1016/j.ijantimicag.2015.01.013
Kassambara, A., and Mundt, F. (2017). factoextra: Extract and Visualize the Results of Multivariate Data Analyses. R package version 1.0.4. Available online at: https://CRAN.R-project.org/package=factoextra
Neyrolles, O., Wolschendorf, F., Mitra, A., and Niederweis, M. (2015). Mycobacteria, metals, and the macrophage. Immunol. Rev. 264, 249–263. doi: 10.1111/imr.12265
Nicholson, F. A., Smith, S. R., Alloway, B. J., Carlton-Smith, C., and Chambers, B. J. (2003). An inventory of heavy metals inputs to agricultural soils in England and Wales. Sci. Total Environ. 311, 205–219. doi: 10.1016/S0048-9697(03)00139-6
Norwegian Food Authority (2014). Zinc and Copper in Pig and Poultry Production—Fate and Effects in the Food Chain and the Environment. Oslo.
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Page, R., and Peti, W. (2016). Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 12, 208–214. doi: 10.1038/nchembio.2044
Petrovska, L., Mather, A. E., Abuoun, M., Branchu, P., Harris, S. R., Connor, T., et al. (2016). Microevolution of monophasic Salmonella typhimurium during epidemic, United Kingdom, 2005–2010. Emerging Infect. Dis. 22, 617–624. doi: 10.3201/eid2204.150531
Pires, S. M., Vigre, H., Makela, P., and Hald, T. (2010). Using outbreak data for source attribution of human salmonellosis and campylobacteriosis in Europe. Foodborne Pathog. Dis. 7, 1351–61. doi: 10.1089/fpd.2010.0564
Pontel, L. B., Audero, M. E., Espariz, M., Checa, S. K., and Soncini, F. C. (2007). GolS controls the response to gold by the hierarchical induction of Salmonella-specific genes that include a CBA efflux-coding operon. Mol. Microbiol. 66, 814–825. doi: 10.1111/j.1365-2958.2007.05963.x
Prokka, S. T. (2014). Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Puigbò, P., Garcia-Vallvé, S., and McInerney, J. O. (2007). TOPD/FMTS: a new software to compare phylogenetic trees. Bioinformatics 23, 1556–1558. doi: 10.1093/bioinformatics/btm135
Ramisetty, B. C., and Santhosh, R. S. (2015). Horizontal gene transfer of chromosomal type II toxin-antitoxin systems of Escherichia coli. FEMS Microbiol. Lett. 363, 1–7. doi: 10.1093/femsle/fnv238
Robinson, D. F., and Foulds, L. R. (1981). Comparison of phylogenetic trees. Math. Biosci. 53, 131–147. doi: 10.1016/0025-5564(81)90043-2
Shao, Y., Harrison, E. M., Bi, D., Tai, C., He, X., Ou, H. Y., et al. (2011). TADB: a web-based resource for Type 2 toxin-antitoxin loci in Bacteria and Archaea. Nucleic Acids Res. 39, D606–D611. doi: 10.1093/nar/gkq908
Silva, C., Puente, J. L., and Calva, E. (2017). Salmonella virulence plasmid: pathogenesis and ecology. Pathog. Dis. doi: 10.1093/femspd/ftx070. [Epub ahead of print].
Smalla, K., Heuer, H., Gotz, A., Niemeyer, D., Krogerrecklenfort, E., and Tietze, E. (2000). Exogenous isolation of antibiotic resistance plasmids from piggery manure slurries reveals a high prevalence and diversity of IncQ-like plasmids. Appl. Environ. Microbiol. 66, 4854–4862. doi: 10.1128/AEM.66.11.4854-4862.2000
Soyer, Y., Switt, A. M., Davis, M. A., Maurer, J., McDonough, P. L., Schoonmaker-Bopp, D. J., et al. (2009). Salmonella enterica serotype 4,5,12:i:-, an emerging Salmonella serotype that represents multiple distinct clones. J. Clin. Microbiol. 47, 3546–3556. doi: 10.1128/JCM.00546-09
Stamatakis, A. (2006). RAxML-VI-HPC: maximum Likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Stecher, B., Hapfelmeier, S., Müller, C., Kremer, M., Stallmach, T., Hardt, W., et al. (2004). Flagella and chemotaxis are required for efficient induction of Salmonella enterica serovar typhimurium colitis in streptomycin-pretreated mice flagella and chemotaxis are required for efficient induction of Salmonella enterica serovar typhimurium colitis. Infect. Immun. 72, 4138–4150. doi: 10.1128/IAI.72.7.4138-4150.2004
Steel, M. A., and Penny, D. (1993). Distributions of tree comparison metrics—some new results. Syst. Biol. 42, 126–141.
Team, D. C. (2008). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Thomsen, M. C., Ahrenfeldt, J., Cisneros, J. L., Jurtz, V., Larsen, M. V., and Hasman, H. (2016). A bacterial analysis platform: an integrated system for analysing bacterial whole genome sequencing data for clinical diagnostics and surveillance. PLoS ONE 11:e0157718. doi: 10.1371/journal.pone.0157718
Wales, A., and Davies, R. (2015). Co-selection of resistance to antibiotics, biocides and heavy metals, and its relevance to foodborne pathogens. Antibiotics 4, 567–604. doi: 10.3390/antibiotics4040567
Yoshida, C. E., Kruczkiewicz, P., Laing, C. R., Lingohr, E. J., Gannon, V. P., Nash, J. H., et al. (2016). The Salmonella in silico typing resource (SISTR): an open web-accessible tool for rapidly typing and subtyping draft Salmonella genome assemblies. PLoS ONE 11:e0147101. doi: 10.1371/journal.pone.0147101
Zamperini, K., Soni, V., Waltman, D., Sanchez, S., Theriault, E. C., Bray, J., et al. (2007). Molecular characterization reveals Salmonella enterica serovar 4,[5],12:i:- from poultry is a variant Typhimurium serovar. Avian Dis. 51, 958–964. doi: 10.1637/7944-021507-REGR.1
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Keywords: Salmonella, tolerance to heavy metals, toxin-antitoxin, antimicrobial resistance, swine foodchain
Citation: Mastrorilli E, Pietrucci D, Barco L, Ammendola S, Petrin S, Longo A, Mantovani C, Battistoni A, Ricci A, Desideri A and Losasso C (2018) A Comparative Genomic Analysis Provides Novel Insights Into the Ecological Success of the Monophasic Salmonella Serovar 4,[5],12:i:-. Front. Microbiol. 9:715. doi: 10.3389/fmicb.2018.00715
Received: 16 February 2018; Accepted: 27 March 2018;
Published: 17 April 2018.
Edited by:
David Rodriguez-Lazaro, University of Burgos, SpainReviewed by:
Masahiro Eguchi, National Institute of Animal Health (NARO), JapanMarcello Trevisani, Università degli Studi di Bologna, Italy
Copyright © 2018 Mastrorilli, Pietrucci, Barco, Ammendola, Petrin, Longo, Mantovani, Battistoni, Ricci, Desideri and Losasso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa Barco, lbarco@izsvenezie.it
†These authors have contributed equally to this work.