 Jidong Zhang
Jidong Zhang Pengmian Feng
Pengmian Feng Hao Lin
Hao Lin Wei Chen
Wei Chen- 1Department of Immunology, Zunyi Medical College, Zunyi, China
- 2Hebei Province Key Laboratory of Occupational Health and Safety for Coal Industry, School of Public Health, North China University of Science and Technology, Tangshan, China
- 3Key Laboratory for Neuro-Information of Ministry of Education, Center for Informational Biology, School of Life Science and Technology, University of Electronic Science and Technology of China, Chengdu, China
- 4Department of Physics, Center for Genomics and Computational Biology, School of Sciences, North China University of Science and Technology, Tangshan, China
N6-methyladenosine (m6A) plays important roles in a branch of biological and physiological processes. Accurate identification of m6A sites is especially helpful for understanding their biological functions. Since the wet-lab techniques are still expensive and time-consuming, it's urgent to develop computational methods to identify m6A sites from primary RNA sequences. Although there are some computational methods for identifying m6A sites, no methods whatsoever are available for detecting m6A sites in microbial genomes. In this study, we developed a computational method for identifying m6A sites in Escherichia coli genome. The accuracies obtained by the proposed method are >90% in both 10-fold cross-validation test and independent dataset test, indicating that the proposed method holds the high potential to become a useful tool for the identification of m6A sites in microbial genomes.
Introduction
At present, ~150 kinds of RNA modifications have been found in different RNA species (Boccaletto et al., 2018), which not only enrich the genetic information, but also play critical roles in a variety of biological processes as mentioned in a recent review (Roundtree et al., 2017). Among these modifications, the N6-methyladenosine (m6A) is the most abundant posttranscriptional modification and has been found in the three domains of life. m6A has been found to participate in various biological activities, such as mRNA splicing (Nilsen, 2014), mRNA translation (Wang et al., 2015), mRNA maturation (Hoernes et al., 2016), stem cell proliferation (Bertero et al., 2018), and even a series of diseases (Zhang et al., 2016; Cui et al., 2017; Li et al., 2017).
In order to reveal its biological functions, different kinds of high-throughput sequencing techniques have been proposed to map the locations of m6A on genome wide (Dominissini et al., 2013; Linder et al., 2015; Wan et al., 2015; Hong et al., 2018). Although these techniques promoted the research progress on understanding the biological functions and the identification of RNA modifications, they are still labor-intensive and cost-ineffective. In addition, the resolution of detecting m6A sites for most techniques is still not satisfactory. Therefore, it's necessary to develop novel methods to detect m6A sites.
Giving the credit to the experimental data yielded by these high-throughput sequencing techniques as reported in a recent work (Chen X. et al., 2017), some machine learning based computational methods have been proposed to identify m6A sites (Chen et al., 2015a,b, 2016a, 2017b,c; Zhou et al., 2016). Although these methods are really good complements to experimental methods for detecting m6A sites, to the best of our knowledge, so far there is no computational tool available for detecting m6A sites in microbial genomes.
Stimulated by the successful applications of machine learning methods in computational genomics and proteomics (Chen et al., 2012; Feng et al., 2013; Cao et al., 2016, 2017a,b; Hu et al., 2018), in the present work, we presented a support vector machine (SVM) based method for identifying m6A sites in the Escherichia coli (E. coli) genome. By encoding the RNA sequences using nucleotide chemical property and accumulated nucleotide frequency, the proposed method obtained promising performances in 10-fold cross validation test. Moreover, we also validated the method on the independent dataset and obtained satisfactory results.
Materials and Methods
Benchmark Dataset
The m6A site containing sequences of E. coli genome were obtained from the RMBase database (Xuan et al., 2018). All the sequences are 41 bp long with the m6A site in the center. To overcome redundancy and reduce the homology bias, sequences with more than 80% sequence similarity were removed by using the CD-HIT program (Fu et al., 2012). After such a screening procedure, 2,055 m6A site containing sequences were retained and regarded as positive samples.
The negative samples (non-m6A site containing sequences) were obtained by choosing the 41-bp long sequences with the central adenosine that was not experimentally confirmed occurring methylation on its 6th nitrogen. By doing so, we could obtain a large number of negative samples. After removing sequences with identify >80%, the number of negative samples are still dramatically larger than that of positive samples. To balance out the numbers between positive and negative samples in model training, we randomly picked out the same number of negative samples and repeated this process 10 times. Therefore, 10 negative subsets were obtained, and each of them includes 2,055 non-m6A site containing sequences. The positive and negative samples thus obtained are provided in Supplementary Material.
Sequence Encoding Scheme
Inspired by recent studies (Chen et al., 2016b,c,d, 2017a,d; Feng et al., 2017), in order to transfer the RNA sequences into discrete vectors that can be recognized and handled by machine learning methods, we encoded RNA sequences using nucleotide chemical properties and accumulated nucleotide frequency. Their brief descriptions are as following.
The four nucleotides, namely, adenine (A), guanine (G), cytosine (C), and uracil (U) can be classified into three different groups according to their physicochemical properties, i.e., ring structures, secondary structures, and chemical functionality (Chen et al., 2016b,c,d, 2017a,d; Feng et al., 2017). Therefore, based on the different physicochemical properties, the four coordinates (1, 1, 1), (0, 0, 1), (1, 0, 0), and (0, 1, 0) were used to represent the four bases (A, C, G, and U) of RNA, respectively.
In order to include nucleotide composition surrounding the modification site as well, the accumulated nucleotide frequency of any nucleotide nj at position i was also used to represent RNA sequences and was defined as
where |Ni| is the length of the sliding substring concerned, l denotes each of the site locations counted in the substring, qϵ{A, C, G, U}.
By integrating both nucleotide physicochemical properties and accumulated nucleotide frequency, an L nt long RNA sequence could be represented a 4L-dimensional vector (Chen et al., 2016b,c,d, 2017a,d; Feng et al., 2017).
Support Vector Machine
As an efficient supervised machine learning algorithm, SVM has been widely used in the realm of bioinformatics (Cao et al., 2014; Li et al., 2017; Wang et al., 2017b; Zhang et al., 2017). Its basic idea is to transform the input data into a high dimensional feature space and then determine the optimal separating hyperplane.
In the current study, the implementation of SVM was performed by using the LibSVM package 3.18, available at http://www.csie.ntu.edu.tw/~cjlin/libsvm/. The radial basis kernel function (RBF) was used to obtain the classification hyperplane. The grid search method was applied to optimize its regularization parameter C and kernel parameter γ.
Evaluation Metrics
The performance was evaluated by using the following four metrics, namely sensitivity (Sn), specificity (Sp), Accuracy (Acc), and the Mathew's correlation coefficient (MCC), which can be expressed as
where TP, TN, FP, and FN represent true positive, true negative, false positive, and false negative, respectively.
To further evaluate the performance of the current method more objectively, inspired by recent works (Wang et al., 2017a), the ROC (receiver operating characteristic) curve was also plotted. Its vertical coordinate indicates the true positive rate (sensitivity) and the horizontal coordinate indicates the false positive rate (1-specificity). The area under the ROC curve (auROC) is an indicator of the performance quality of a binary classifier, i.e., the value 0.5 of auROC is equivalent to random prediction while the value 1 of auROC represents a perfect one.
Results and Discussions
Performance for m6A Site Identification
In statistical prediction, independent dataset test, K-fold cross-validation test and jackknife test are often used to derive the metric values for a predictor (Chou, 2011). In order to saving computational time, the 10-fold cross-validation test was used to examine the performance of the proposed method. In 10-fold cross-validation test, the samples in the dataset are randomly partitioned into 10 equal sized sub-datasets. Of the 10 sub-datasets, a single sub-dataset is retained as the validation data for testing the model, and the remaining 9 sub-datasets are used as training data. The process is then repeated 10 times, with each of the 10 sub-datasets used exactly once as the validation data.
By encoding RNA sequences using nucleotide chemical property and accumulated nucleotide frequency, each sample in the dataset was represented by a (4 × 41) = 164-dimensional vector and used as the input of SVM. The 10-fold cross-validation test results for identifying m6A sites in E. coli were listed in Table 1. In addition, to demonstrate that whether its accuracy is sensitive to the selection of negative data, the method was also tested on the other nine negative datasets, respectively. Their predictive results of the 10-fold cross-validation were also provided in Table 1.
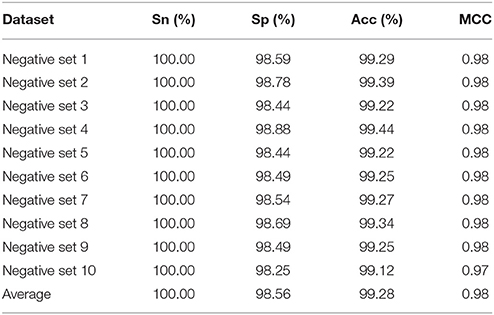
Table 1. The 10-fold cross validation predictive results by using different negative datasets for identifying m6A sites in E. coli.
As indicated in Table 1, we found that the predictive accuracy is not affected by the selection of negative data. In addition, the 10 ROC curves obtained based on the 10 different negative datasets were also plotted in Figure 1. It was found that their auROCs are all higher than 0.98. These results demonstrate the reliability and robustness of the model developed in this study.
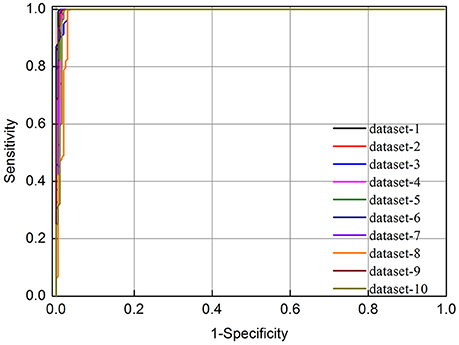
Figure 1. The ROC curves of 10-fold cross validation test for identifying m6A sites in E. coli based on different negative datasets. The vertical coordinate is the true positive rate (Sn) while horizontal coordinate is the false positive rate (1-Sp).
Comparison With Other Methods
In order to demonstrate the effectiveness of nucleotide chemical property and accumulated nucleotide frequency for identifying m6A sites in E. coli, we compared the performance of the proposed method with that of the method based on other commonly used RNA sequence features. Chen et al. have proposed the pseudo nucleotide composition (PseKNC) to represent RNA sequences (Chen et al., 2014a,b), in which both the local and global sequence order information w included. Since it has been proposed in 2014, PseKNC have been used in in many branches of computational genomics (Guo et al., 2014; Lin et al., 2014, 2017). Therefore, we employed the SVM to perform the comparisons between the model based on nucleotide chemical property and accumulated nucleotide frequency features and that based on the PseKNC features (Chen et al., 2015a). The 10-fold cross-validation test results were listed in Table 2.
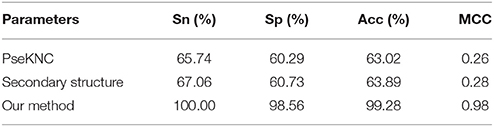
Table 2. Comparison of different parameters for identifying m6A sites in E. coli.
As indicated in a recent study (Schwartz et al., 2013), the m6A modification is also affected by RNA secondary structures. Therefore, we performed the prediction of m6A sites by using RNA secondary structure. To this end, all the sequences in the benchmark dataset were encoded by using their second structures. The details about the encoding scheme based on secondary structures can be found in a recent work (Xue et al., 2005). By doing so, each RNA sequence is converted to a 32 dimensional vector (Xue et al., 2005) and used as the input feature of SVM. Its 10-fold cross-validation test results were also listed in Table 2.
As shown in Table 2, the predictive performance of the method based on nucleotide chemical property and accumulated nucleotide frequency is dramatically higher than that based on PseKNC and RNA secondary structure.
Validation on Independent Dataset
The proposed method trained based on the benchmark dataset from the E. coli genome was further used to identify the m6A sites in the P. aeruginosa genome. For this purpose, we firstly collected the 5,814 experimentally confirmed m6A sites from the RMBase to form an independent dataset, which is given in Supporting Information S2. Of the 5,814 m6A sites in the P. aeruginosa, 5,809 were correctly identified, indicating that the proposed method is really quite promising for identifying m6A sites in microbial genomes.
Conclusion
In this study, we present a computational method to identify m6A sites in the E. coli genome by encoding the RNA sequences using nucleotide chemical property and accumulated nucleotide frequency. The results obtained based on the benchmark dataset and independent dataset demonstrate that the proposed method is powerful and promising in discovering m6A sites. We hope that the proposed method will be helpful for the future research on m6A sites in microbial genomes.
Since user-friendly and publicly accessible web-servers (Feng et al., 2018)and databases (Liang et al., 2017) represent the direction of developing new prediction method, we will make efforts in our future work to provide a web-server for the method presented in this paper.
Author Contributions
HL and WC: conceived and designed the experiments; JZ and PF: performed the experiments; HL and WC: wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Nature Science Foundation of China (Nos. 31771471, 61772119), Program for the Top Young Innovative Talents of Higher Learning Institutions of Hebei Province (No. BJ2014028), the Outstanding Youth Foundation of North China University of Science and Technology (No. JP201502), and the Fundamental Research Funds for the Central Universities of China (Nos. ZYGX2015Z006, ZYGX2016J125, ZYGX2016J118), Natural Science Foundation of Guizhou Province (QKH-2016-1167); The Scientific and Technological Innovation Project for Oversea Students of Guizhou province (QR-2016-20); High School Science and Technology Talent Support Project of Guizhou Province (QJH-KY-2016-079).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00955/full#supplementary-material
References
Bertero, A., Brown, S., Madrigal, P., Osnato, A., Ortmann, D., Yiangou, L., et al. (2018). The SMAD2/3 interactome reveals that TGFβ controls m(6)A mRNA methylation in pluripotency. Nature 555, 256–259. doi: 10.1038/nature25784
Boccaletto, P., Machnicka, M. A., Purta, E., Piatkowski, P., Baginski, B., Wirecki, T. K., et al. (2018). MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46, D303–D307. doi: 10.1093/nar/gkx1030
Cao, R., Adhikari, B., Bhattacharya, D., Sun, M., Hou, J., and Cheng, J. (2017a). QAcon: single model quality assessment using protein structural and contact information with machine learning techniques. Bioinformatics 33, 586–588. doi: 10.1093/bioinformatics/btw694
Cao, R., Bhattacharya, D., Hou, J., and Cheng, J. (2016). DeepQA: improving the estimation of single protein model quality with deep belief networks. BMC Bioinformatics 17:495. doi: 10.1186/s12859-016-1405-y
Cao, R., Freitas, C., Chan, L., Sun, M., Jiang, H., and Chen, Z. (2017b). ProLanGO: protein function prediction using neural machine translation based on a recurrent neural network. Molecules 22:E1732. doi: 10.3390/molecules22101732
Cao, R., Wang, Z., Wang, Y., and Cheng, J. (2014). SMOQ: a tool for predicting the absolute residue-specific quality of a single protein model with support vector machines. BMC Bioinformatics 15:120. doi: 10.1186/1471-2105-15-120
Chen, W., Feng, P., Ding, H., and Lin, H. (2016a). Identifying N (6)-methyladenosine sites in the Arabidopsis thaliana transcriptome. Mol. Genet. Genomics 291, 2225–2229. doi: 10.1007/s00438-016-1243-7
Chen, W., Feng, P., Ding, H., Lin, H., and Chou, K.-C. (2015a). iRNA-methyl: identifying N-6-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem. 490, 26–33. doi: 10.1016/j.ab.2015.08.021
Chen, W., Feng, P., Tang, H., Ding, H., and Lin, H. (2016b). Identifying 2′-O-methylationation sites by integrating nucleotide chemical properties and nucleotide compositions. Genomics 107, 255–258. doi: 10.1016/j.ygeno.2016.05.003
Chen, W., Feng, P., Tang, H., Ding, H., and Lin, H. (2016c). RAMPred: identifying the N-1-methyladenosine sites in eukaryotic transcriptomes. Sci. Rep. 6:31080. doi: 10.1038/srep31080
Chen, W., Feng, P., Yang, H., Ding, H., Lin, H., and Chou, K.-C. (2017a). iRNA-AI: identifying the adenosine to inosine editing sites in RNA sequences. Oncotarget 8, 4208–4217. doi: 10.18632/oncotarget.13758
Chen, W., Lei, T.-Y., Jin, D.-C., Lin, H., and Chou, K.-C. (2014a). PseKNC: A flexible web server for generating pseudo K-tuple nucleotide composition. Anal. Biochem. 456, 53–60. doi: 10.1016/j.ab.2014.04.001
Chen, W., Lin, H., Feng, P. M., Ding, C., Zuo, Y. C., and Chou, K. C. (2012). iNuc-PhysChem: a sequence-based predictor for identifying nucleosomes via physicochemical properties. PLoS ONE 7:e47843. doi: 10.1371/journal.pone.0047843
Chen, W., Tang, H., and Lin, H. (2017b). MethyRNA: a web server for identification of N-6-methyladenosine sites. J. Biomol. Struct. Dyn. 35, 683–687. doi: 10.1080/07391102.2016.1157761
Chen, W., Tang, H., Ye, J., Lin, H., and Chou, K.-C. (2016d). iRNA-PseU: Identifying RNA pseudouridine sites. Mol. Ther. Nucleic Acids 5, 155–163. doi: 10.1038/mtna.2016.37
Chen, W., Tran, H., Liang, Z., Lin, H., and Zhang, L. (2015b). Identification and analysis of the N-6-methyladenosine in the Saccharomyces cerevisiae transcriptome. Sci. Rep. 5:13859. doi: 10.1038/srep13859
Chen, W., Xing, P., and Zou, Q. (2017c). Detecting N6-methyladenosine sites from RNA transcriptomes using ensemble support vector machines. Sci. Rep. 7:40242. doi: 10.1038/srep40242
Chen, W., Yang, H., Feng, P., Ding, H., and Lin, H. (2017d). iDNA4mC: identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 33, 3518–3523. doi: 10.1093/bioinformatics/btx479
Chen, W., Zhang, X., Brooker, J., Lin, H., Zhang, L., and Chou, K.-C. (2014b). PseKNC-General: a cross-platform package for generating various modes of pseudo nucleotide compositions. Bioinformatics 31, 119–120. doi: 10.1093/bioinformatics/btu602
Chen, X., Sun, Y. Z., Liu, H., Zhang, L., Li, J. Q., and Meng, J. (2017). RNA methylation and diseases: experimental results, databases, web servers and computational models. Brief Bioinform. doi: 10.1093/bib/bbx142. [Epub ahead of print].
Chou, K. C. (2011). Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 273, 236–247. doi: 10.1016/j.jtbi.2010.12.024
Cui, Q., Shi, H., Ye, P., Li, L., Qu, Q., Sun, G., et al. (2017). m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 18, 2622–2634. doi: 10.1016/j.celrep.2017.02.059
Dominissini, D., Moshitch-Moshkovitz, S., Salmon-Divon, M., Amariglio, N., and Rechavi, G. (2013). Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nat. Protoc. 8, 176–189. doi: 10.1038/nprot.2012.148
Feng, P., Ding, H., Yang, H., Chen, W., Lin, H., and Chou, K.-C. (2017). iRNA-PseColl: identifying the occurrence sites of different RNA modifications by incorporating collective effects of nucleotides into PseKNC. Mol. Ther. Nucleic Acids 7, 155–163. doi: 10.1016/j.omtn.2017.03.006
Feng, P. M., Chen, W., Lin, H., and Chou, K. C. (2013). iHSP-PseRAAAC: identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem. 442, 118–125. doi: 10.1016/j.ab.2013.05.024
Feng, P., Yang, H., Ding, H., Lin, H., Chen, W., and Chou, K. C. (2018). iDNA6mA-PseKNC: Identifying DNA N(6)-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics doi: 10.1016/j.ygeno.2018.01.005. [Epub ahead of print].
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Guo, S.-H., Deng, E.-Z., Xu, L.-Q., Ding, H., Lin, H., Chen, W., et al. (2014). iNuc-PseKNC: a sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics 30, 1522–1529. doi: 10.1093/bioinformatics/btu083
Hoernes, T. P., Huttenhofer, A., and Erlacher, M. D. (2016). mRNA modifications: dynamic regulators of gene expression? RNA Biol. 13, 760–765. doi: 10.1080/15476286.2016.1203504
Hong, T., Yuan, Y., Chen, Z., Xi, K., Wang, T., Xie, Y., et al. (2018). Precise antibody-independent m6A identification via 4SedTTP-involved and FTO-assisted strategy at single-nucleotide resolution. J. Am. Chem. Soc. doi: 10.1021/jacs.7b13633. [Epub ahead of print].
Hu, H., Zhang, L., Ai, H., Zhang, H., Fan, Y., Zhao, Q., et al. (2018). HLPI-Ensemble: Prediction of human lncRNA-protein interactions based on ensemble strategy. RNA Biol. doi: 10.1080/15476286.2018.1457935. [Epub ahead of print].
Li, Z., Weng, H., Su, R., Weng, X., Zuo, Z., Li, C., et al. (2017). FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell 31, 127–141. doi: 10.1016/j.ccell.2016.11.017
Liang, Z. Y., Lai, H. Y., Yang, H., Zhang, C. J., Yang, H., Wei, H. H., et al. (2017). Pro54DB: a database for experimentally verified sigma-54 promoters. Bioinformatics 33, 467–469. doi: 10.1093/bioinformatics/btw630
Lin, H., Deng, E.-Z., Ding, H., Chen, W., and Chou, K.-C. (2014). iPro54-PseKNC: a sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res. 42, 12961–12972. doi: 10.1093/nar/gku1019
Lin, H., Liang, Z.-Y., Tang, H., and Chen, W. (2017). Identifying sigma70 promoters with novel pseudo nucleotide composition. IEEE/ACM Trans. Comput. Biol. Bioinform. doi: 10.1109/TCBB.2017.2666141. [Epub ahead of print].
Linder, B., Grozhik, A. V., Olarerin-George, A. O., Meydan, C., Mason, C. E., and Jaffrey, S. R. (2015). Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772. doi: 10.1038/nmeth.3453
Nilsen, T. W. (2014). Molecular biology. Internal mRNA methylation finally finds functions. Science 343, 1207–1208. doi: 10.1126/science.1249340
Roundtree, I. A., Evans, M. E., Pan, T., and He, C. (2017). Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200. doi: 10.1016/j.cell.2017.05.045
Schwartz, S., Agarwala, S. D., Mumbach, M. R., Jovanovic, M., Mertins, P., Shishkin, A., et al. (2013). High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell 155, 1409–1421. doi: 10.1016/j.cell.2013.10.047
Wan, Y., Tang, K., Zhang, D., Xie, S., Zhu, X., Wang, Z., et al. (2015). Transcriptome-wide high-throughput deep m(6)A-seq reveals unique differential m(6)A methylation patterns between three organs in Arabidopsis thaliana. Genome Biol. 16:272. doi: 10.1186/s13059-015-0839-2
Wang, F., Huang, Z. A., Chen, X., Zhu, Z., Wen, Z., Zhao, J., et al. (2017a). LRLSHMDA: Laplacian Regularized Least Squares for Human Microbe-Disease Association prediction. Sci. Rep. 7:7601. doi: 10.1038/s41598-017-08127-2
Wang, X., Zhao, B. S., Roundtree, I. A., Lu, Z., Han, D., Ma, H., et al. (2015). N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399. doi: 10.1016/j.cell.2015.05.014
Wang, Y., You, Z., Li, X., Chen, X., Jiang, T., and Zhang, J. (2017b). PCVMZM: using the probabilistic classification vector machines model combined with a zernike moments descriptor to predict protein-protein interactions from protein sequences. Int. J. Mol. Sci. 18:1029. doi: 10.3390/ijms18051029
Xuan, J. J., Sun, W. J., Lin, P. H., Zhou, K. R., Liu, S., Zheng, L. L., et al. (2018). RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 46, D327–D334. doi: 10.1093/nar/gkx934
Xue, C., Li, F., He, T., Liu, G. P., Li, Y., and Zhang, X. (2005). Classification of real and pseudo microRNA precursors using local structure-sequence features and support vector machine. BMC Bioinformatics 6:310. doi: 10.1186/1471-2105-6-310
Zhang, C., Samanta, D., Lu, H., Bullen, J. W., Zhang, H., Chen, I., et al. (2016). Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. U.S.A. 113, E2047–E2056. doi: 10.1073/pnas.1602883113
Zhang, L., Ai, H., Chen, W., Yin, Z., Hu, H., Zhu, J., et al. (2017). CarcinoPred-EL: novel models for predicting the carcinogenicity of chemicals using molecular fingerprints and ensemble learning methods. Sci. Rep. 7:2118. doi: 10.1038/s41598-017-02365-0
Keywords: N6-methyladenosine, machine learning method, nucleotide physicochemical properties, microbial genome, pseudo nucleotide composition
Citation: Zhang J, Feng P, Lin H and Chen W (2018) Identifying RNA N6-Methyladenosine Sites in Escherichia coli Genome. Front. Microbiol. 9:955. doi: 10.3389/fmicb.2018.00955
Received: 13 March 2018; Accepted: 24 April 2018;
Published: 14 May 2018.
Edited by:
Hongsheng Liu, Liaoning University, ChinaReviewed by:
Yongqiang Xing, Inner Mongolia University of Science and Technology, ChinaRenzhi Cao, Pacific Lutheran University, United States
Copyright © 2018 Zhang, Feng, Lin and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Lin, hlin@uestc.edu.cn
Wei Chen, chenweiimu@gmail.com