Structural and Mechanistic Basis for Extended-Spectrum Drug-Resistance Mutations in Altering the Specificity of TEM, CTX-M, and KPC β-lactamases
 Timothy Palzkill1,2*
Timothy Palzkill1,2*- 1Department of Pharmacology and Chemical Biology, Baylor College of Medicine, Houston, TX, United States
- 2Department of Biochemistry and Molecular Biology, Baylor College of Medicine, Houston, TX, United States
The most common mechanism of resistance to β-lactam antibiotics in Gram-negative bacteria is the production of β-lactamases that hydrolyze the drugs. Class A β-lactamases are serine active-site hydrolases that include the common TEM, CTX-M, and KPC enzymes. The TEM enzymes readily hydrolyze penicillins and older cephalosporins. Oxyimino-cephalosporins, such as cefotaxime and ceftazidime, however, are poor substrates for TEM-1 and were introduced, in part, to circumvent β-lactamase-mediated resistance. Nevertheless, the use of these antibiotics has lead to evolution of numerous variants of TEM with mutations that significantly increase the hydrolysis of the newer cephalosporins. The CTX-M enzymes emerged in the late 1980s and hydrolyze penicillins and older cephalosporins and derive their name from the ability to also hydrolyze cefotaxime. The CTX-M enzymes, however, do not efficiently hydrolyze ceftazidime. Variants of CTX-M enzymes, however, have evolved that exhibit increased hydrolysis of ceftazidime. Finally, the KPC enzyme emerged in the 1990s and is characterized by its broad specificity that includes penicillins, most cephalosporins, and carbapenems. The KPC enzyme, however, does not efficiently hydrolyze ceftazidime. As with the TEM and CTX-M enzymes, variants have recently evolved that extend the spectrum of KPC β-lactamase to include ceftazidime. This review discusses the structural and mechanistic basis for the expanded substrate specificity of each of these enzymes that result from natural mutations that confer oxyimino-cephalosporin resistance. For the TEM enzyme, extended-spectrum mutations act by establishing new interactions with the cephalosporin. These mutations increase the conformational heterogeneity of the active site to create sub-states that better accommodate the larger drugs. The mutations expanding the spectrum of CTX-M enzymes also affect the flexibility and conformation of the active site to accommodate ceftazidime. Although structural data are limited, extended-spectrum mutations in KPC may act by mediating new, direct interactions with substrate and/or altering conformations of the active site. In many cases, mutations that expand the substrate profile of these enzymes simultaneously decrease the thermodynamic stability. This leads to the emergence of additional global suppressor mutations that help correct the stability defects leading to increased protein expression and increased antibiotic resistance.
Introduction
β-lactam antibiotics are the most often-used antimicrobials representing ~65% of antibiotic usage worldwide (Livermore, 2006). The β-lactams act by inhibiting bacterial cell wall biosynthesis. Specifically, they are covalent inhibitors of transpeptidase enzymes, commonly referred to as penicillin-binding proteins (PBPs). These enzymes catalyze a cross-linking reaction of pentapeptides present in the peptidoglycan layer of the cell wall (Lovering et al., 2012).
The most common mechanism of resistance to β-lactam antibiotics is the bacterial production of β-lactamases (Fisher et al., 2005). These enzymes catalyze the hydrolysis of the amide bond present in the β-lactam ring resulting in a product that is an ineffective inhibitor of the PBPs. β-lactamases can be distributed into four classes (A, B, C, and D) based on primary amino acid sequence homology (Ambler, 1980; Fisher et al., 2005). Classes A, C, and D are serine hydrolases that function similarly to classical serine proteases such as chymotrypsin. They catalyze attack of the catalytic serine on the carbonyl carbon of the amide bond to form a covalent acyl-enzyme intermediate that is subsequently hydrolyzed by a water molecule that has been activated by a general base. Class B β-lactamases are zinc metallo-enzymes that contain one or two zinc ions that coordinate a hydroxide ion for direct attack on the carbonyl carbon of the amide and do not proceed through a covalent acyl-enzyme (Palzkill, 2013).
Class A β-lactamases are often encoded on plasmids that can move by conjugation and, as a result, these enzymes are widespread sources of resistance (Bush and Fisher, 2011). Class A β-lactamases are a particular problem for resistance in Gram-negative bacteria including the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumonia, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species; Pendleton et al., 2013). Individual class A β-lactamases display a range of substrate specificities although, as a group, they are commonly known for the efficient hydrolysis of penicillins and early generation cephalosporins. The extensive use of these antibiotics and subsequent spread of class A β-lactamases has led to widespread resistance (Bush and Fisher, 2011). This was countered in the 1980s with the introduction of oxyimino-cephalosporins, which are still good PBP inhibitors but poor substrates for β-lactamases. Mechanism-based inhibitors that target the β-lactamases were also developed to combat resistance (Drawz and Bonomo, 2010). The introduction and subsequent use of both these agents, however, placed selective pressure on bacteria resulting in the evolution of variants of class A enzymes that have gained the ability to hydrolyze oxyimino-cephalosporins or that avoid the action of β-lactamase inhibitors (Petrosino et al., 1998; Gniadkowski, 2008; Salverda et al., 2010).
This review will focus on three groups of class A β-lactamases that have evolved in response to the use of oxyimino-cephalosporins such as cefotaxime and ceftazidime and that are widespread sources of resistance in Gram-negative bacteria (Bonomo, 2017). These groups of enzymes include the TEM, CTX-M, and KPC β-lactamases. Although the evolution of resistance to mechanism-based inhibitors is clearly an important source of resistance, the focus of this review is on the evolution of enzymes with higher activity for catalysis of oxyimino-cephalosporins (Figure 1).
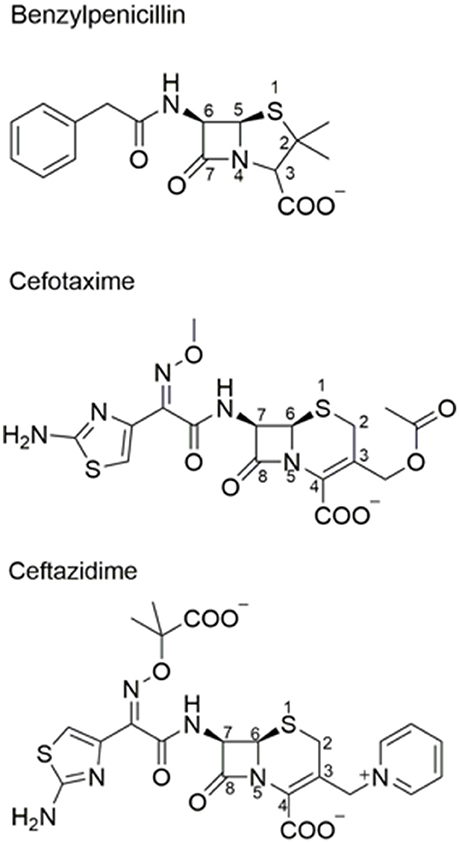
Figure 1. Chemical structures of β-lactam antibiotics. Benzylpenicillin and the oxyimino-cephalosporins cefotaxime and ceftazidime are shown.
Kinetics of β-lactam Hydrolysis by Class A β-lactamases
In order to understand the means by which the TEM, CTX-M, and KPC enzymes and variants inactivate oxyimino-cephalosporins, it is useful to review the kinetics and mechanism by which class A β-lactamases catalyze hydrolysis of β-lactams. Class A β-lactamases are serine hydrolases with a mechanism similar to serine proteases. After formation of the enzyme-substrate complex (ES), the active-site serine attacks and forms a covalent, acyl-enzyme intermediate (EAc). Subsequent catalysis of the hydrolysis of the acyl-enzyme generates the inactive, hydrolyzed β-lactamase product (P) (Hedstrom, 2002; Galleni and Frere, 2007).
Based on this mechanism, the Michaelis-Menten kinetic parameters are described by the following equations.
For β-lactamases, the kinetic parameters kcat, KM, and kcat/KM are composite constants that depend on the rates of multiple steps in the reaction. kcat reflects the magnitude and relationship between the acylation (k2) and deacylation (k3) rate constants (Equation 1). KM is often regarded as an indication of affinity between the substrate and enzyme, i.e., that KM approximates Ks, which is k−1/k1. However, in the β-lactamase mechanism, KM is also dependent on k2 and k3 (Equation 2; Raquet et al., 1994). Assuming k−1>k2, KM approximates substrate affinity (Ks) when the acylation rate (k2) is much slower than the deacylation rate (k3), which is often not the case with β-lactam substrates. When deacylation is rate limiting (k2>k3), the value of KM is lower than Ks and overestimates the affinity of the enzyme for substrate. Amino acid substitutions in β-lactamases that result in changes in KM can be due to changes in Ks, k2 or k3, or a combination thereof. Finally, kcat/KM reflects the rates of steps occurring up to the formation of the acyl-enzyme. The deacylation rate (k3) does not contribute to the value of kcat/KM as seen in equation 3.
Class A β-lactamase Mechanism
Class A β-lactamases utilize a set of conserved amino acid residues in the active site to promote substrate binding, acyl-enzyme formation, and subsequent deacylation to release product. The class A enzymes contain a β and an α/β domain, with the active site situated between these closely connected domains (Herzberg and Moult, 1987; Strynadka et al., 1992). Residues Ser70, Ser130, Asn132, Glu166, Asn170, Lys234, Ser/Thr235, Gly236, and Ala/Ser/Thr237 (Ambler numbering scheme; Ambler et al., 1991) are important contributors to substrate binding and catalysis (Figure 2). The hydroxyl oxygen of Ser70 serves as the nucleophile for attack on the carbonyl carbon of the amide bond (Strynadka et al., 1992; Fisher and Mobashery, 2009). The main chain NH groups of Ser70 and Ala237 act as the oxyanion hole and make hydrogen bonding interactions to stabilize the negative charge that develops on the carbonyl oxygen tetrahedral intermediate during acylation and deacylation (Strynadka et al., 1992). Although the details are controversial, Lys73, Glu166 and a catalytic water are thought to participate in abstracting the proton to activate Ser70 (Herzberg and Moult, 1987; Strynadka et al., 1992; Damblon et al., 1996; Minasov et al., 2002; Meroueh et al., 2005). Ser130 has been proposed to serve as a proton shuttle between Lys73 and the leaving group nitrogen (Strynadka et al., 1992). Asn132 forms a hydrogen bond with the acyl-amide group in the C6/7 side chain of penicillins and cephalosporins for binding and positioning of substrate. Lys234 and Ser/Thr235 make interactions with the C3/C4 substrate carboxylate found on all penicillins, cephalosporins and carbapenems and therefore are important for substrate binding. This interaction may also contribute to stabilization of the acylation and deacylation transition states to facilitate catalysis (Strynadka et al., 1992; Delmas et al., 2010; Fonseca et al., 2012). Glu166 serves as the general base for activating the catalytic water for the deacylation step and amino acid substitutions at this position lead to accumulation of the acyl-enzyme intermediate (Adachi et al., 1991; Delaire et al., 1991; Escobar et al., 1991; Strynadka et al., 1992). Asn170 forms a hydrogen bond to Glu166 and an active site water to help position the residue for activating the water. Both Glu166 and Asn170 are found on an omega loop structure that forms the base of the active site in class A β-lactamases (Figure 2). As discussed below, the omega loop plays an important role in the evolution of increased oxyimino-cephalosporin hydrolysis in class A enzymes. The residues described above are important for catalysis of all β-lactam substrates and mutations at these positions reduce activity. Thus, they form the core residues required for catalysis. Mutations leading to increased catalysis of oxyimino-cephalosporins occur at residues other than these core positions.
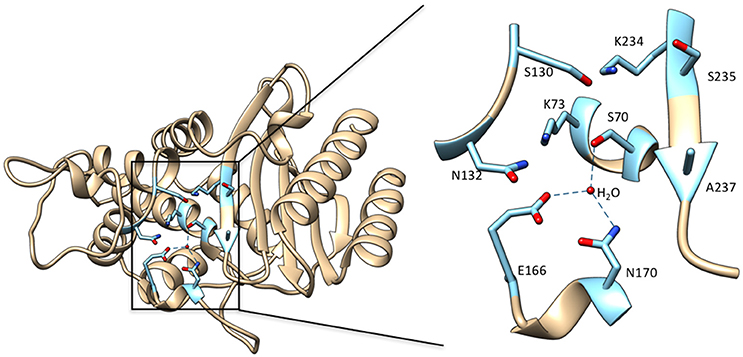
Figure 2. Left: Ribbon diagram of TEM-1 β-lactamase (PDB ID: 1BTL). Key active site residues that are implicated in substrate binding and catalysis and highlighted in cyan. Right: Enlarged view of the boxed region of the active site with the active site residues labeled. The deacylation water molecule is shown as a red sphere with hydrogen bonds from this water to Ser70, Glu166, and Asn170 shown as dotted lines. Hydrogen bonds between residues are not shown.
TEM Extended-Spectrum β-lactamases (ESBLs)
TEM-1 β-lactamase was reported in 1963 in E. coli and Salmonella (Datta and Kontomichalou, 1965). It subsequently spread among Enterobacteriaceae and other Gram-negative pathogens to become a widespread source of β-lactam resistance. TEM-1 efficiently catalyzes the hydrolysis of penicillins and early generation cephalosporins and provides high-level bacterial resistance to these drugs. In part due to the widespread presence of TEM-1, the oxyimino-cephalosporins were introduced in the 1980s (Bush, 2002). The oximino-cephalosporins, such as cefotaxime and ceftazidime, include an oxyimino side chain at the C7 position (Figure 1). The large and inflexible oxyimino side chain is difficult to accommodate into the active site of TEM-1, resulting in slow rates of hydrolysis (Table 1). Ceftazidime differs from cefotaxime by having an even larger side chain that includes a carboxylate group (Figure 1). The increased bulk of ceftazidime is associated with a further reduction in rates of catalysis by TEM-1 compared to cefotaxime (Table 2).
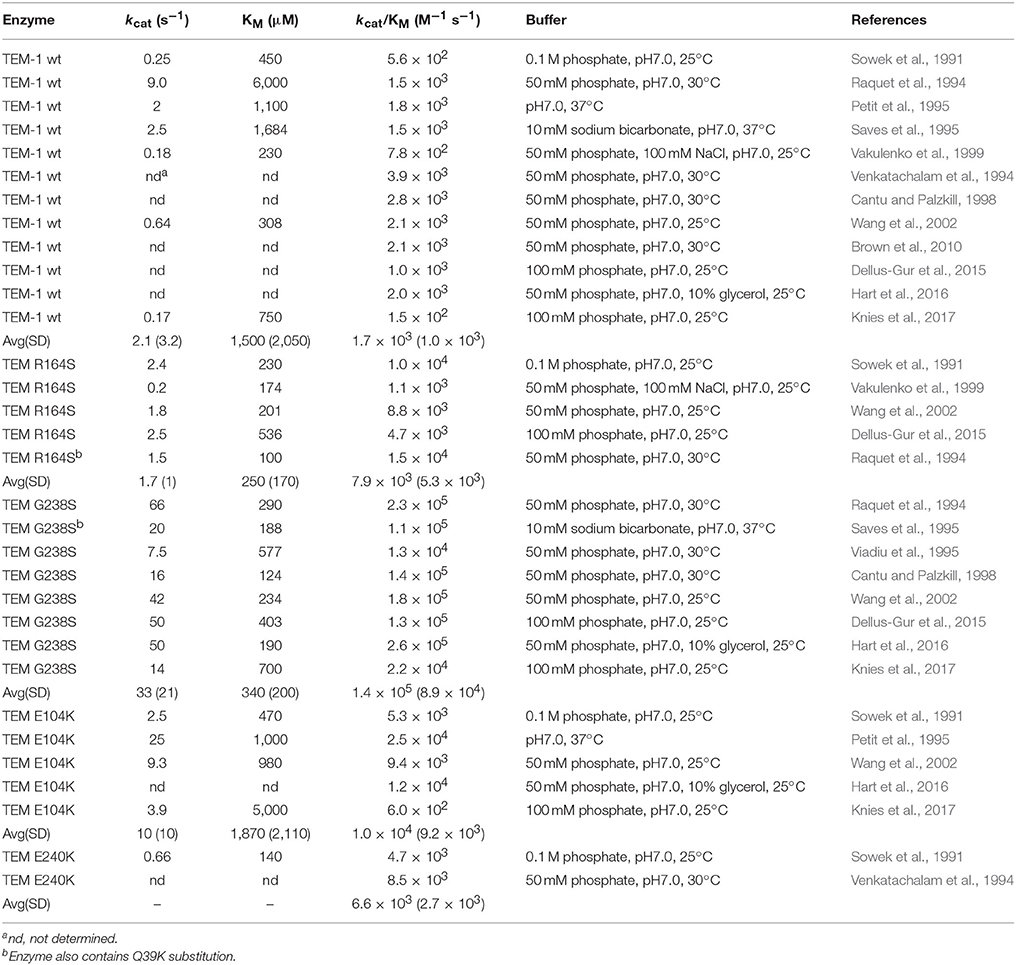
Table 1. Kinetic parameters for cefotaxime hydrolysis by TEM-1 β-lactamase and mutants.
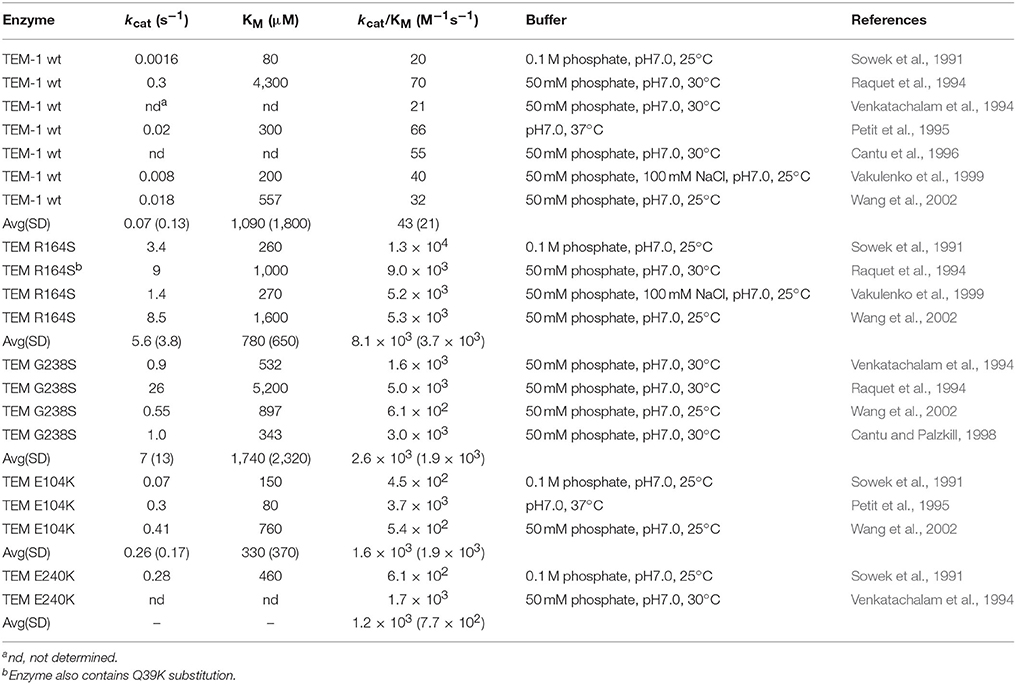
Table 2. Kinetic parameters for ceftazidime hydrolysis by TEM-1 β-lactamase and mutants.
The kinetic parameters for oxyimino-cephalosporin hydrolysis by TEM-1 reveal low kcat and kcat/KM values and high KM values compared with good substrates such as ampicillin or benzylpenicillin. There have been a large number of studies on cefotaxime hydrolysis by TEM-1 and a list of kinetic parameters is compiled in Table 1. The list is likely not comprehensive and there is some variation in assay conditions such as 25°C vs. 30°C but the majority of the listed results were obtained using similar buffer conditions. There is some variation in published values, particularly with regard to KM. KM is generally high and in several studies it was too high to measure accurately (Table 1). The kcat/KM values, however, are relatively consistent and average ~1.7 × 103 M−1s−1 (Table 1). Published kinetic parameters for ceftazidime hydrolysis also exhibit variation in KM but with KM generally high (Table 2). However, kcat/KM values are quite consistent and average ~40 M−1s−1. Therefore, both cefotaxime and ceftazidime are poor substrates for TEM-1 β-lactamase. Ceftazidime, however, is a particularly poor substrate with an ~45-fold lower kcat/KM value than that observed for cefotaxime hydrolysis. Published kcat/KM values for benzylpenicillin hydrolysis are in the range of 107-108 M−1s−1 and therefore cefotaxime and ceftazidime hydrolysis is 104-106-fold less efficient (Christensen et al., 1990). Since kcat/KM reflects the events occurring up to formation of the acyl-enzyme, the low kcat/KM values for cefotaxime and ceftazidime hydrolysis reflect low affinity for substrate binding and/or a slow acylation reaction. Indeed, estimates of Ks for the binding of cefotaxime and ceftazidime to TEM-1 reveal poor affinity with values of 3.8 and 9.9 mM, respectively (Vakulenko et al., 1999). The rate-limiting step for cefotaxime hydrolysis by TEM-1 has been shown to be the acylation step (k2) (Saves et al., 1995). Taken together, these results are consistent with the large, inflexible oxyimino side chain of cefotaxime being poorly accommodated in the active site of TEM-1.
Since the introduction of oxyimino-cephalosporins into clinical practice, variants of TEM-1 with amino acid substitutions that result in increased hydrolysis have been emerging (Petrosino et al., 1998; Salverda et al., 2010; Pimenta et al., 2014). Each unique variant is given a new number and these variants are termed TEM extended spectrum β-lactamases (ESBLs). The number of TEM variants is now greater than 200 (Salverda et al., 2010; Pimenta et al., 2014). The most common substitutions in TEM enzymes displaying enhanced oxyimino-cephalosporin hydrolysis include E104K, R164S/H, M182T, A237T, G238S, and E240K (Figure 3). The effects of these substitutions have been studied extensively using kinetic, biophysical, and structural tools to determine the mechanism by which resistance to oxyimino-cephalosporins is evolving.
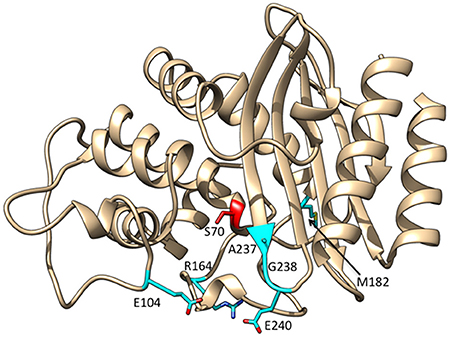
Figure 3. Ribbon diagram of TEM-1 β-lactamase showing the positions at which substitutions commonly occur among enzyme variants with increased catalytic activity for oxyimino-cephalosporins highlighted in cyan. The active site Ser70 nucleophile is highlighted in red.
G238S and R164S Substitutions
The R164S and G238S substitutions are associated with the largest increases in cefotaxime and ceftazidime hydrolysis when introduced into the TEM-1 enzyme. These substitutions are likely the driver substitutions for clinically relevant resistance and are the most often observed in TEM ESBLs. The G238S substitution is predominantly associated with enhanced hydrolysis of cefotaxime and increased resistance. Residue 238 is situated on the β3 strand that forms a side of the active site (Figure 3). A number of studies have shown that the G238S substitution, when introduced into the TEM-1 enzyme, results in an ~80-fold increase in kcat/KM for cefotaxime hydrolysis compared to wild-type TEM-1 and has a value of ~1.4 × 105 M−1s−1 (Table 1). It is somewhat difficult to estimate the changes in kcat and KM relative to TEM-1 as they are often not determined for wild type because of high KM values. However, it is clear that KM for cefotaxime hydrolysis is reduced for G238S compared to TEM-1 as the value is consistently measurable and is in the range of 300 μM while kcat is in the range of 30 s−1 (Table 1). Saves et al. utilized electrospray mass spectroscopy to determine acylation (k2) and deacylation (k3) rates for TEM-1 and the G238S mutant and found acylation is rate-limiting for cefotaxime hydrolysis for TEM-1, as noted above (Saves et al., 1995). Acylation remains rate-limiting for the G238S enzyme but k2 is increased nearly 10-fold (Saves et al., 1995). A significant portion of the increase in kcat/KM observed for the G238S enzyme is thus due to an increase in the acylation rate. Although values for Ks for G238S with cefotaxime have not been determined, the fact that acylation is rate-limiting indicates KM is an approximation of Ks. Since the KM for cefotaxime is reduced for the G238S enzyme compared to TEM-1 (Table 1), this suggests that the increased affinity of the G238S enzyme for cefotaxime also contributes to the increase in kcat/KM.
The G238S substitution in TEM-1 also results in increased ceftazidime hydrolysis, albeit to a lesser extent than that observed for cefotaxime, with a kcat/KM value of ~2.5 × 103 M−1s−1 (Table 2). This is a 60-fold increase over that observed for TEM-1. As with cefotaxime, kcat and KM comparisons to wild-type TEM-1 are difficult. However, the KM for ceftazidime hydrolysis is reduced to a measurable range for G238S with a value of ~1,700 μM while kcat is roughly 7 s−1, although there is high variance in published values (Table 2). A comparison of kcat/KM values for G238S reveals a 60-fold higher catalytic efficiency for cefotaxime vs. ceftazidime hydrolysis.
While the G238S substitution increases the rate of cefotaxime and ceftazidime hydrolysis, it significantly decreases the rate of hydrolysis of penicillins. For example, kcat/KM for ampicillin hydrolysis is reduced 10-fold compared to wild-type TEM-1 while kcat is decreased ~50-fold (Dellus-Gur et al., 2015). Very similar effects were observed for hydrolysis of benzylpenicillin by the G238S enzyme (Cantu and Palzkill, 1998). Because kcat is decreased, the effect of G238S is not simply on binding affinity for penicillins but includes reduced rates of acylation (k2), deacylation (k3), or both. Christensen et al. determined that, for TEM-1, the rates of acylation and deacylation are fast (>1,000 s−1) and neither is rate-limiting for benzylpenicillin hydrolysis (Christensen et al., 1990). Using mass spectroscopy, Saves et al. determined that, for the G238S enzyme, k3 is greatly reduced and is rate-limiting for hydrolysis of benzylpenicillin (Saves et al., 1995). Based on these results, it was suggested the G238S substitution may affect the positioning of the omega loop and alter the action of the Glu166 general base, which is present on the loop, resulting in a decreased deacylation rate (Saves et al., 1995).
Two models have been proposed to explain the mechanism of the G238S substitution. One model involves a hydrogen bond between the side chain hydroxyl of the Ser238 residue and the oxime group of the oxyimino-cephalosporins that would improve affinity of the enzyme for the antibiotics and thereby increase catalytic efficiency (Huletsky et al., 1993). The second model suggests that steric conflicts of the Ser238 side chain with residue Asn170 would lead to movement of the β3 strand or movement of the omega loop thereby causing expansion of the active site to accommodate the larger oxyimino-cephalosporin side chain (Saves et al., 1995; Cantu and Palzkill, 1998). An analysis of kinetic parameters of a series of substitutions at position 238 supported the steric conflict model in that kcat/KM for oxyimino-cephalosporin hydrolysis correlated more closely with side-chain volume than hydrogen bonding potential (Cantu and Palzkill, 1998). In addition, docking and molecular dynamics studies of the G238S mutant with cefotaxime have suggested Ser238 does not hydrogen bond to cefotaxime in the complex (Singh and Dominy, 2012).
The structures of TEM variants from clinical isolates containing multiple substitutions including G238S have been determined. The structure of the TEM-52 enzyme containing the E104K/M182T/G238S substitutions shows the position of the loop region of residues 238–243 is moved by 2.8 Å to widen the active site to accommodate the bulkier side chains of oxyimino-cephalosporins (Figure 4). It was also noted that lysine from the E104K substitution is oriented toward the active site (Orencia et al., 2001). In addition, the structure of the TEM-72 enzyme containing the Q39K, M182T, G238S, and E240K substitutions has been determined (Docquier et al., 2011). Curiously, the TEM-72 enzyme does not show the movement of the 238–243 loop and the active site is not expanded (Figure 4). This could be due to the presence of the E240K substitution adjacent to G238S somehow restraining the conformation. Note, however, that TEM-1 containing the G238S/E240K double mutant has higher catalytic efficiency for cefotaxime hydrolysis than G238S alone, indicating cefotaxime is accommodated into the active site in the simultaneous presence of G238S and E240K (Venkatachalam et al., 1994). Finally, the structure of a TEM G238A mutant has been determined and, similar to TEM-52, shows an expansion of the active site, however, the expansion is due to movement of the omega loop because of steric conflict with the substituted alanine (Wang et al., 2002).
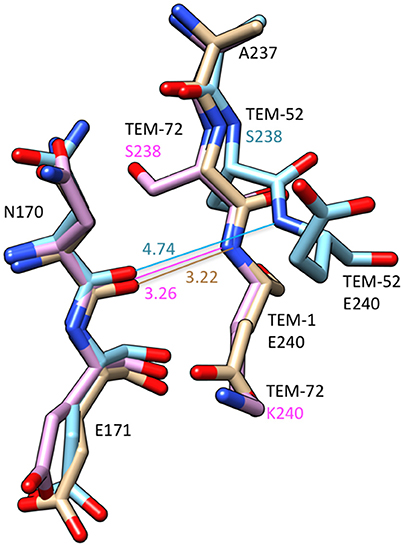
Figure 4. Schematic illustration of a structural comparison of TEM-1 (tan) (PDB ID: 1BTL), TEM-52 (cyan) (1HTZ), and TEM-72 (pink) (3P98). The omega loop residues Asn170-Glu171 and the end of the β3-sheet containing positions 237–240 are shown. The distance between the Asn170 carbonyl oxygen and residue 240 main chain NH are shown. The numbers indicate distances in Å. The distance lines are color coded to match the molecules. The G238S substitution in TEM-52 is associated with a movement of residue 238 and 240 away from Asn170 creating additional space in the active site. Labels for amino acid residues that are substituted relative to wild type are colored according to structure with TEM-52 (cyan), TEM-72 (pink).
The X-ray crystal structure of the G238S mutant has also been determined in a TEM-1 enzyme that also contains stabilizing substitutions that enhance protein expression and crystallization (Dellus-Gur et al., 2015). These substitutions did not influence the catalytic properties of the enzyme compared to G238S without the stabilizing substitutions. The G238S structures were determined under cryogenic conditions and at room temperature (Dellus-Gur et al., 2015). The G238S enzyme exists in two well-defined conformations of the active site loop at the end of the β3-strand containing residue 238. One of these is a more open conformation that could accommodate cefotaxime (Dellus-Gur et al., 2015). Taken together, the structural results on G238S suggest, TEM-72 notwithstanding, the substitution is associated with increased space to accommodate substrate and are broadly consistent with the steric conflict model.
The R164S substitution results in increased hydrolysis of both cefotaxime and ceftazidime, although it is most often associated with TEM ESBLs providing increased resistance to ceftazidime. Several studies reveal the R164S substitution results in a roughly 5-fold increase in kcat/KM for cefotaxime hydrolysis compared to TEM-1 with a value of ~ 8.0 × 103 M−1s−1 (Table 1). Although cefotaxime KM values are often too high to measure for TEM-1, KM is measureable for R164S with values in the range of 250 μM and kcat in the range of 2 s−1 (Table 1). In contrast to the relatively modest increase in cefotaxime hydrolysis mediated by R164S, this substitution is associated with an ~200-fold increase in kcat/KM for ceftazidime hydrolysis compared to TEM-1 with a value of ~8.0 × 103 M−1s−1 compared to 40 M−1s−1 for TEM-1 (Table 2). The increased catalytic efficiency is associated with a consistently measurable KM for R164S with a value of ~800 μM and a kcat of ~6 s−1. The large increase in kcat/KM for ceftazidime hydrolysis by R164S relative to TEM-1 may explain why the substitution is often associated with ESBLs with high ceftazidime resistance. Because kcat/KM is a reflection of rates occurring up to the formation of the acyl-enzyme, the increase in kcat/KM relative to TEM-1 for both cefotaxime and ceftazidime hydrolysis by R164S suggests the substitution is associated with a decrease in Ks (increased affinity) and/or an increase in k2.
As with G238S, the R164S substitution is associated with decreased penicillin hydrolysis. For example, the kcat for ampicillin hydrolysis is decreased 25-fold and kcat/KM is decreased 13-fold for the R164S enzyme relative to wild-type TEM-1 (Dellus-Gur et al., 2015). Similarly, for benzylpenicillin hydrolysis, kcat is decreased 70-fold and kcat/KM is decreased 14-fold (Vakulenko et al., 1999). There are no reported values for Ks, k2 or k3 for penicillin hydrolysis but the 25- and 70-fold reductions in kcat suggests k2 and/or k3 values are significantly lower for penicillin hydrolysis by R164S relative to TEM-1.
Several groups have investigated the structural basis of the change in substrate specificity provided by R164S. Mobashery and colleagues noted that the R164S substitution would create a cavity in the middle of the omega loop and the top of the loop containing Pro167 and Asn170 would collapse to fill the void. This rearrangement creates additional space in the active site (Vakulenko et al., 1999). The structure of TEM-64 (E104K/R164S/M182T), which contains the R164S substitution, has been determined (Wang et al., 2002). It was found the omega loop has an altered conformation where the short helix in the region of residue 170 unwinds and Asn170 moves by 4.5 Å to create a cavity in the active site that could better accommodate the C7β side chain of oxyimino-cephalosporins (Wang et al., 2002; Figure 5). The structure of the R164S mutant has also been determined in a TEM-1 enzyme that, like G238S described above, also contains stabilizing substitutions that enhance protein expression and crystallization without affecting kinetic parameters (Dellus-Gur et al., 2015). Similar to G238S described above, structures were determined at cryogenic conditions and room temperature. The structure of R164S showed the omega loop is conformationally heterogenous (Dellus-Gur et al., 2015). An ensemble of different conformations of the omega loop are present, some of which better accommodate cefotaxime. Binding of oxyimino-cephalosporins is proposed to shift the ensemble toward catalytically active conformations. The structure of a covalently bound boronic-acid analog showed less conformational heterogeneity, supporting this view (Dellus-Gur et al., 2015).
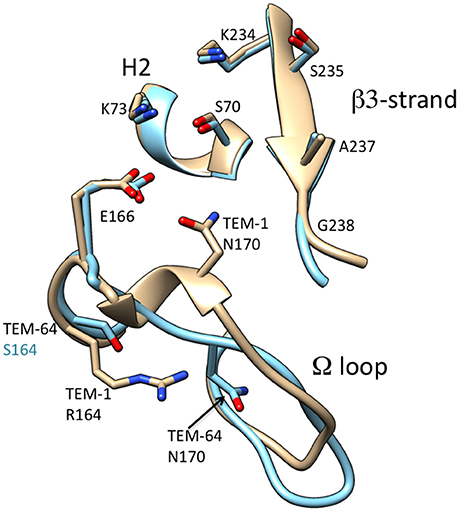
Figure 5. Schematic illustration of the omega loop region as well as the β3-strand and H2 helix of the active site of TEM-1 (tan) (PDB ID: 1BTL) in comparison to that of TEM-64 (cyan) (1JWZ). The omega loop in TEM-64 undergoes a large conformational change moving Asn170 out of the active site. Labels for amino acid residues that are substituted relative to wild type are colored according to structure with TEM-64 (cyan). The boronic acid inhibitor of the TEM-64 structure is omitted for clarity.
E104K and E240K Substitutions
The E104K and E240K substitutions are also associated with increases in the hydrolysis of cefotaxime and ceftazidime. Although these substitutions have been found individually in ESBLs, they are often found in association with the G238S or R164S substitutions. The E104K substitution has been studied by several groups and exhibits an ~6-fold increase in kcat/KM for cefotaxime hydrolysis compared to TEM-1 with a value of 1.0 × 104 M−1s−1(Table 1). KM is consistently measurable for TEM-1 containing the E104K substitution with an average of 1,800 μM, although with high variance in values. kcat is likely also increased relative to TEM-1 and has a value of ~10 s−1 (Table 1). For ceftazidime hydrolysis, the E104K substitution is associated with an ~40-fold increase in kcat/KM compared to TEM-1 (Table 2). The KM value for E104K with ceftazidime is in a measurable range with a value of ~300 μM while kcat is ~0.3 s−1 (Table 2). There are no published results on Ks, k2 or k3 for E104K for these substrates but the increased kcat/KM values suggest minimally that Ks is reduced and/or k2 is increased by the substitution. It has been suggested that electrostatic interactions of E104K could increase binding of oxyimino-cephalosporins. Docking and molecular dynamics, however, suggest the lysine at position 104 does not directly interact with bound cefotaxime, although ceftazidime was not examined (Sowek et al., 1991; Singh and Dominy, 2012).
In contrast to the R164S and G238S substitutions, the E104K substitution has little effect on the hydrolysis of penicillins. The kinetic parameters for benzylpenicillin hydrolysis are very similar to TEM-1 with high kcat, low KM and very high kcat/KM values (Sowek et al., 1991; Hart et al., 2016). Similarly, kinetic parameters are largely unchanged for hydrolysis of 6-furylacrylpenicillanic acid (FAP) (Wang et al., 2002). These results suggest the E104K substitution does not significantly modify active site structures that are necessary for penicillin hydrolysis. Substitutions in TEM-1 that increase oxyimino-cephalosporin hydrolysis therefore need not reduce hydrolysis rates of excellent substrates such as penicillins.
The E240K substitution has been evaluated in multiple publications and exhibits an ~4-fold increase in kcat/KM for cefotaxime hydrolysis compared to TEM-1 with a value of ~6.6 × 103 M−1s−1 (Table 1). The KM for cefotaxime hydrolysis is decreased to a measurable value of ~140 μM while kcat is low and has a value of ~0.5 s−1. With regard to ceftazidime hydrolysis, the E240K substitution results in a roughly 30-fold increase in kcat/KM compared to TEM-1 (Table 2). Similar to the E104K substitution, the E240K substitution results in only modest changes in kinetic parameters for hydrolysis of penicillins such as benzylpenicillin (Sowek et al., 1991). Thus, like E104K, the E240K enzyme is an excellent penicillinase. Finally, a comparison of the effects of E104K vs. E240K on kcat/KM for cefotaxime and ceftazidime reveals that both substitutions result in a ~5-fold increase for cefotaxime but a 30–40-fold increase for ceftazidime relative to TEM-1, indicating these substitutions have the largest impact on ceftazidime hydrolysis.
A237T Substitution
The A237T substitution occurs more rarely than R164S or G238S among TEM ESBLs and when it occurs, it is associated with other substitutions, particularly R164S (www.lahey.org/studies/webt.asp). The A237T substitution was first identified in a selection for mutants of TEM-1 with increased cephalosporin C resistance (Hall and Knowles, 1976). There have been relatively few detailed biochemical studies of the effect of the A237T substitution. Healy et al. showed with purified enzyme that the A237T substitution results in a 2-fold decrease in kcat but a modest (1.3-fold) increase in kcat/KM for hydrolysis of the cephalosporins cephalothin and cephalosporin C (Healy et al., 1989). In addition, they showed the substitution reduces hydrolysis of benzylpenicillin and ampicillin with a 7-fold decrease in kcat and a ~10-fold decrease in kcat/KM. The A237T substitution also decreases the thermal stability of the enzyme by 5°C. Further, Cantu et al. made similar observations with regard to the negative effect of A237T on benzylpenicillin and ampicillin hydrolysis and also showed the substitution causes a modest 1.3-fold increase in kcat/KM for cefotaxime (Cantu et al., 1997). A further suggestion that the A237T substitution increases cefotaxime hydrolysis comes from a study showing that TEM-24 (Q39K/E104K/R164S/A237T/E240K) exhibits a ~10-fold higher Vmax/KM value for cefotaxime hydrolysis relative to TEM-46 (Q39K/E104K/R164S/E240K) (Chanal-Claris et al., 1997). There is no structural information available on a TEM A237T mutant; however, by analogy to structures of the class A enzymes Toho-1, CTX-M-9, and CTX-M-14 in complex with cefotaxime, it is possible that the hydroxyl group of Thr237 makes a hydrogen bond to the C4 carboxylate group to enhance cefotaxime hydrolysis (Shimamura et al., 2002; Delmas et al., 2010; Adamski et al., 2015).
M182T Substitution
The M182T substitution is found in many TEM ESBL enzymes as well as inhibitor resistant enzymes. M182T is found in combination with other amino acid substitutions in ESBLs (www.lahey.org/studies/webt.asp). In contrast to the substitutions described above, residue 182 is not in the vicinity of the active site (Figure 3). The M182T substitution, when introduced by itself into the TEM-1 enzyme, does not alter substrate specificity (Sideraki et al., 2001; Wang et al., 2002). Instead, M182T acts by increasing thermodynamic stability and suppressing aggregation (Sideraki et al., 2001; Wang et al., 2002). A role for the M182T substitution in ESBL evolution was found in a genetic study to identify intragenic second-site suppressor mutations of folding and stability mutants in TEM-1 β-lactamase (Huang and Palzkill, 1997). For this purpose, a destabilizing mutation in the hydrophobic core (L76N) was introduced into TEM-1. This resulted in rapid proteolysis of the enzyme in E. coli and a large reduction in ampicillin resistance. Suppressor mutations (i.e., mutations that restored ampicillin resistance to E. coli containing the TEM-1 L76N gene) were selected after random introduction of point mutations in the TEM gene (Huang and Palzkill, 1997). Using this approach, mutants that reverted the L76N substitution to leucine and isoleucine were discovered; however, the most common suppressor was M182T, which is located 17Å from residue 76 (Huang and Palzkill, 1997). The M182T substitution was shown to restore protein expression levels of TEM-1 L76N in E. coli and, importantly, restored expression of other destabilizing mutations in TEM-1. Based on these results, it was suggested that in natural ESBLs, M182T acts as a suppressor of folding and stability defects associated with substitutions that increase catalysis of extended-spectrum cephalosporins or cause inhibitor resistance (Huang and Palzkill, 1997). It was named a “global suppressor” based on similar properties to intragenic second-site stabilizing mutations that had previously been observed in staphylococcus nuclease that were named global suppressors (Shortle and Lin, 1985).
It was later shown that the TEM-1 L76N substitution results in misfolding leading to aggregation and proteolysis of the enzyme in the periplasmic space of E. coli (Sideraki et al., 2001). The addition of the M182T substitution restored the accumulation of active, folded enzyme in the periplasm and suppressed the formation of aggregates. It was also shown that, in guanidinium hydrochloride denaturation experiments, M182T did not act on the final stability of the L76N enzyme (Sideraki et al., 2001). Subsequently, it has been shown using circular dichroism (CD) measurements at increasing temperatures that M182T does increase the thermodynamic stability of the wild-type TEM-1 enzyme (Wang et al., 2002). In addition, it was shown using the CD assay that TEM mutations that increase oxyimino-cephalosporin hydrolysis such as R164S and G238S, also decrease the thermodynamic stability of the enzyme, i.e., there is a stability-function trade-off (Wang et al., 2002). Previous studies had also shown that R164S and G238S have reduced stability relative to wild-type TEM-1 (Raquet et al., 1995). Adding the M182T substitution to the G238S enzyme results in increased thermodynamic stability suggesting that M182T compensates for TEM ESBL substitutions that trade off protein stability for improved oxyimino-cephalosporin hydrolysis (Wang et al., 2002).
Consistent with the results of Wang et al. (2002), Knies et al. recently showed that M182T increases the thermostability of wild type TEM-1, that the G238S substitution results in lower thermostability of TEM-1, and that the M182T substitution restores stability to the M182T/G238S mutant (Knies et al., 2017). These authors note, however, that the increased thermostability does not correlate with increased cefotaxime MICs. They go on to suggest that the effects of the mutations on other factors may be important, such as folding/misfolding or kinetic stability (Knies et al., 2017). Studies on in vivo (in periplasm) folding kinetics and propensity for misfolded products of TEM ESBL mutants such as G238S and the influence of mutations including M182T on such folding will be of interest to further address the in vivo effect of ESBL substitutions and the role of global suppressor substitutions.
There have been multiple proposals for the structural basis by which M182T increases enzyme thermostability. Farzaneh et al. proposed that the threonine at position 182 results in a new hydrogen bond between residue 182 and the main chain carbonyl group of residue 64 (Farzaneh et al., 1996). This would provide an additional link between the α and αβ domains of β-lactamase and possibly increase stability. Alternatively, it has been shown that Thr182 acts as an N-cap residue for the residue 183-195 α-helix by forming and additional hydrogen bond to Ala185 that would be expected to increase stability (Minasov et al., 2002).
Finally, it is also noteworthy that several groups have now shown that there are, in fact, many substitutions in TEM-1 that increase the stability of the enzyme including V31R, I47V, F60Y, P62S, G78A, V80I, S82H, G92D, R120G, E147G, H153R, M182T, A184V, T188I, L201P, I208M, A224V, E240H, R241H, I247V, T265M, R275Q, R275L, and N276D (Bershtein et al., 2008; Kather et al., 2008; Marciano et al., 2008; Brown et al., 2010; Deng et al., 2012). A number of these substitutions have been observed in TEM ESBL or inhibitor-resistant enzymes from clinical isolates including G92D, R120G, H153R, M182T, A184V, A224V, T265M, R275Q, and N276D (Brown et al., 2010). Therefore, suppression of folding and stability defects is likely achieved by several mutational pathways in natural variants.
Multiple Substitutions and Epistasis in TEM ESBLs
TEM ESBLs from resistant clinical isolates contain 1–5 substitutions and multiply substituted enzymes are the rule with single substitutions being relatively rare (www.lahey.org/studies/webt.asp). Many ESBLs contain a core substitution of either R164S or G238S with additional substitutions such as E104K, M182T, A237T or E240K. Enzymes containing R164S are more often associated with high ceftazidime resistance while those with G238S are associated with high cefotaxime resistance. This is related to the fact, as discussed above, that the R164S substitution gives a large increase in kcat/KM for ceftazidime relative to wild type TEM-1 and G238S gives a large increase in cefotaxime kcat/KM relative to wild type. This categorization is an oversimplification, however, in that R164S does moderately increase cefotaxime hydrolysis and G238S moderately increases ceftazidime hydrolysis relative to wild-type TEM-1.
The addition of substitutions to R164S and G238S mutations results in increased oxyimino-cephalosporin hydrolysis. For example, the addition of E104K to R164S results in a 30-fold increase in kcat/KM for ceftazidime and a 2.4-fold increase for cefotaxime hydrolysis relative to the R164S enzyme (Sowek et al., 1991). Addition of the E240K substitution to R164S results in a 7-fold increase in kcat/KM for ceftazidime but only a modest 1.2-fold increase for cefotaxime hydrolysis (Sowek et al., 1991). Further, the addition of E104K to the G238S enzyme results in a 15-fold increase in kcat/KM for ceftazidime and a 10-fold increase for cefotaxime hydrolysis relative to the G238S enzyme (Wang et al., 2002). The addition of E240K to the G238S enzyme results in a 37-fold increase in kcat/KM for ceftazidime and a 2.6-fold increase for cefotaxime hydrolysis relative to G238S (Venkatachalam et al., 1994). Taken together, these findings show that the E104K and E240K substitutions increase kcat/KM of either R164S or G238S for both ceftazidime and cefotaxime. However, the effect is more pronounced for ceftazidime. It has been noted by Sowek et al., the effect of E104K and E240K on ceftazidime hydrolysis may be due to favorable electrostatic interactions of lysine in either position with the carboxylate group found in the C7β side chain of ceftazidime (Sowek et al., 1991).
R164S or G238S mutations in combination with E104K or E240K generally result in additive effects on catalysis where the combination of two substitutions that each increase hydrolysis leads to even higher levels of hydrolysis in the double mutant. Additive combinations reveal that the two substitutions have independent effects on catalysis (Wells, 1990). However, not all substitutions associated with TEM ESBLs are additive. For example, the combination of E104K and E240K, each of which increases kcat/KM for cefotaxime hydrolysis when introduced individually into the TEM-1 enzyme, results in an E104K/E240K double mutant that hydrolyzes cefotaxime with a kcat/KM at the same level as the E104K substitution alone (Hart et al., 2016). Similarly, the addition of E104K to a R164S/E240K enzyme results in an E104K/R164S/E240K mutant that exhibits the same kcat/KM value for cefotaxime hydrolysis as the R164S/E240K enzyme (Sowek et al., 1991). Therefore, in these examples, the presence of E240K negates the effect of E104K (and vice versa). The lack of additivity means that the two mutations interact, directly or indirectly, and that the interaction has a negative effect on hydrolysis.
Non-additive effects of mutations are termed epistasis (de Visser and Krug, 2014; Dellus-Gur et al., 2015). Epistasis is an important element of evolution because it determines what mutational pathways are accessible to natural selection (de Visser and Krug, 2014; Dellus-Gur et al., 2015). Indeed, these effects have been highlighted for a TEM mutant consisting of a promoter mutation (g4205a) as well as the amino acid substitutions A42G/E104K/M182T/G238S. This variant provides high-level cefotaxime resistance to E. coli (Stemmer, 1994; Orencia et al., 2001). Weinreich et al. showed that out of the 5! = 120 mutational pathways leading from wild type to the final mutant, 102 were selectively inaccessible paths in that some of the individual mutations do not increase cefotaxime resistance on all allelic backgrounds. Because some increase in cefotaxime resistance is required for selection of intermediate mutations, mutational pathways that include a step with no selection advantage will be dead-ends. Thus, negative epistasis excludes some of the possible pathways by which complex mutations may arise (Weinreich et al., 2006).
A well-studied example of epistasis in the TEM system from a structural and functional standpoint is the combination of R164S with G238S. The combination of R164S and G238S, each of which increase resistance toward cefotaxime and ceftazidime, results in a double mutant R164S/G238S with reduced resistance toward each of these (Giakkoupi et al., 2000). The kcat/KM for cefotaxime hydrolysis by the R164S/G238S double mutant is the same as R164S and 20-fold lower than that for G238S, indicating negative epistasis between the substitutions (Dellus-Gur et al., 2015). In each case, the kcat/KM for the double mutant is significantly smaller that the kcat/KM expected if the two mutations were simply additive.
In order to understand the molecular basis of the observed epistasis, the structure of the R164S/G238S double mutant was determined by Dellus-Gur et al. in the context of other stabilizing substitutions that do not impact kinetic parameters as described above for G238S and R164S (Dellus-Gur et al., 2015). As noted above, the G238S substitution induced two dominant conformations of the G238-loop while the R164S substitution induced an ensemble of conformations of the omega loop. The R164S/G238S double mutant exhibited a wider ensemble of conformations than the single mutants (Dellus-Gur et al., 2015). In addition, a non-native interaction between residues 171 and 240 is present which results in a dominant, large change in the position of the catalytically important residue Asn170. It was suggested that the entropic cost of the substrate selecting from the many conformations, in addition to the unfavorable position of Asn170 results in the low kcat for cefotaxime hydrolysis by the double mutant, thereby accounting for the negative epistatic effect of introducing the second mutation (Dellus-Gur et al., 2015). Based on the results, the authors suggest there is a delicate balance between the adaptive benefit of increased structural freedom to allow oxyimino-cephalosporin access to the active site and the cost of diluting the catalytically optimal conformation among many non-productive conformations that decreases catalytic efficiency (Dellus-Gur et al., 2015). This study also provides evidence for the importance of conformational ensembles or sub-states in enzyme action and evolution. The coexistence of multiple sub-states has been referred to as “floppiness” and is recognized as an important component of enzyme catalytic efficiency and evolution (Tokuriki and Tawfik, 2009a; Bar-Even et al., 2015).
Computational Studies of Conformational Heterogeneity of TEM ESBLs
Computational studies also support a role for conformational flexibility in the evolution of altered specificity in the TEM-1 enzyme. It has been shown through docking and molecular dynamics that the G238S and E104K substitutions induce changes in the conformation of the omega loop as well as regions consisting of residues 86–118, 213–229, and 267–271 upon binding cefotaxime (Singh and Dominy, 2012). In addition, a study has demonstrated flexibility in TEM-1 and greater flexibility in ancestral β-lactamases that exhibit broader substrate specificity, thereby linking flexibility with broad specificity (Zou et al., 2015). Further, hidden allosteric sites have been discovered in the TEM enzyme (Horn and Shoichet, 2004; Bowman et al., 2015). These sites are binding pockets that are not present in the crystal structure but become available as the protein structure fluctuates in solution. Such a site was discovered via structure determination of a small molecule inhibitor that was found bound in a pocket 16 Å from the active site that is not apparent in the apoenzyme (Horn and Shoichet, 2004). The conformational changes associated with small molecule binding in the cryptic pocket were communicated to the active site resulting in movement of a key active site residue thereby inhibiting the enzyme (Horn and Shoichet, 2004). Hidden allosteric sites that are sampled among TEM-1 conformations have also been discovered computationally and thiol labeling experiments support their presence in TEM-1 (Bowman and Geissler, 2012; Bowman et al., 2015). Taken together, these studies support a role for conformational heterogeneity in β-lactamases and its potential impact on catalysis via communication with the active site.
Recently, molecular dynamics simulations and Markov State Models (MSMs) were used to examine the mechanism of action of the TEM-1 wild type, E104K, and G238S substitutions on cefotaxime hydrolysis (Hart et al., 2016). The authors showed that docking of cefotaxime into static TEM ESBL structures shows a poor correlation with kcat/KM of the variants, suggesting the static structures do not contain sufficient information to understand the function of the enzymes. The correlation was improved using “Boltzman docking” where MSMs based on molecular dynamics simulations were weighted to the contribution of each state by its equilibrium probability (Hart et al., 2016). The results suggested that consideration of enzyme conformational sub-states rather than a single structure provides more relevant information on TEM ESBL activity against cefotaxime. Next, the authors used MSMs constructed based on molecular dynamics of wild type TEM-1 and the E104K/G238S enzyme that hydrolyzes cefotaxime >1,000-fold faster than wild type and identified states that are more populated by E104K/G238S than by wild type. Interestingly, it was found that the “cefotaximase states” resembled the wild-type structure while the omega loop in the wild type undergoes substantial rearrangements (Hart et al., 2016). This suggests that E104K/G238S hydrolyzes cefotaxime more efficiently than wild type because the omega loop is actually more constrained in a conformation that will accommodate cefotaxime in the double mutant while the wild-type TEM-1 samples many non-productive conformations. Support for this model was obtained by chemical footprinting and the construction of mutations predicted to preferentially occupy active conformational states (Hart et al., 2016). These findings suggest that, contrary to the view that the active site needs to become more open to accommodate cefotaxime, for productive catalysis it needs to be more constrained to limit non-productive sub-states (Hart et al., 2016). Examination of the most populated states of E104K/G238S cefotaximase indicated the serine at position 238 and lysine at position 104 act to pin down the omega loop to limit non-productive conformations (Hart et al., 2016). These results again highlight the importance of conformational sub-states and provide a different view from the idea that the expansion of the TEM-1 active site by ESBL mutations to accommodate oxyimino-cephalosporins is the causative basis of enhanced catalytic efficiency.
CTX-M β-lactamase Variants and Ceftazidime Hydrolysis
CTX-M β-lactamases are class A enzymes that are characterized by the ability to efficiently hydrolyze cefotaxime (Bonnet, 2004). These enzymes have spread globally to become the most widespread ESBLs in Gram-negative bacteria (Cantón et al., 2012). The CTX-M ESBLs are divided into five clusters based on amino acid sequence homology including CTX-M-1, CTX-M-2, CTX-M-8, CTX-M-9, and CTX-M-25 with the names based on the prominent member of each subgroup (D'Andrea et al., 2013). The subgroups differ from one another by >10% amino acid sequence divergence and each subgroup contains a number of variants that differ from one another by <5% sequence divergence (D'Andrea et al., 2013). The CTX-M enzymes are ~35% identical to TEM-1 β-lactamase.
The Toho-1 (CTX-M-44), CTX-M-9, and CTX-M-14 enzymes have been the most intensively studied in terms of structure and mechanism (Shimamura et al., 2002; Chen et al., 2005; Delmas et al., 2010). Toho-1 is in the CTX-M-2 subfamily while CTX-M-9 and −14 are in the CTX-M-9 subfamily and differ only in a V231A substitution in CTX-M-9 relative to CTX-M-14 (Chen et al., 2005; D'Andrea et al., 2013). In addition, CTX-M-9 and CTX-M-14 exhibit similar kinetic parameters for β-lactam substrate hydrolysis (Chen et al., 2005). Therefore, structure and function conclusions from studies of one of these enzymes are likely to apply to the others.
The focus here will be on CTX-M-14 and the role of amino acid substitutions found in variants that increase ceftazidime hydrolysis. In contrast to TEM-1, the CTX-M enzymes are excellent catalysts for the hydrolysis of cefotaxime (Bonnet, 2004). With regard to CTX-M-14, kcat is in the range of 200–400 s−1 and KM ~100 μM while kcat/KM is ~3.0 × 106 M−1s−1 (Chen et al., 2005; Adamski et al., 2015; Patel et al., 2015). Thus, the kcat/KM value for cefotaxime hydrolysis by CTX-M-14 is about 1,500-fold higher than that for TEM-1. Structural studies of CTX-M-14 and CTX-M-9 apoenzymes reveal that, contrary to the expectation that a wider active site would better accommodate cefotaxime, the active site is narrower than that observed for TEM-1 (Chen et al., 2005; Delmas et al., 2010). X-ray structures of CTX-M-14 and CTX-M-9 in the presence of cefotaxime show that the active site expands significantly with cefotaxime bound in the active site (Delmas et al., 2010; Adamski et al., 2015). These structures were solved using an S70G substitution of the nucleophile for acylation in order to trap the unhydrolyzed substrate (Delmas et al., 2010). The expansion is accompanied by the breaking of a hydrogen bond between the main chain carbonyl oxygen of Asn170 and the main chain NH of Asp240 that connects to omega loop the β-3 strand (Delmas et al., 2010) (Figure 6). Note that this is a similar region of the active site as that of TEM-1 where the G238S substitution increases the hydrolysis of oxyimino-cephalosporins and the proposed effect of increasing the size of the active site is similar.
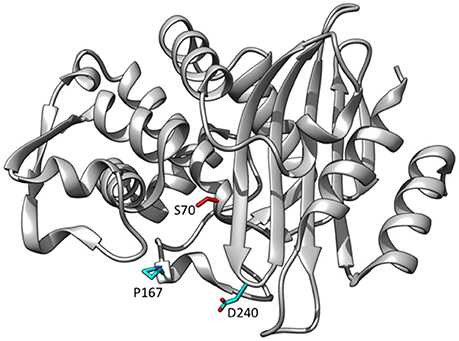
Figure 6. Ribbon diagram of CTX-M-14 β-lactamase showing the positions of residues Pro167 and Asp240 that are substituted in variants able to hydrolyze ceftazidime (cyan). The catalytic residue Ser70 is shown in red. (PDB ID: 1YLT).
Although CTX-M enzymes rapidly hydrolyze cefotaxime, the related oxyimino-cephalosporin ceftazidime is poorly hydrolyzed (Bonnet, 2004). Determinations of kinetic parameters for CTX-M-14 for ceftazidime reveal high KM values of >600 μM, kcat values <5 s−1 and a kcat/KM of 1–5 × 103 M−1s−1 (Chen et al., 2005; Patel et al., 2015). This kcat/KM value is ~1,000-fold less than kcat/KM for cefotaxime hydrolysis by CTX-M-14. Note, however, that this kcat/KM value for ceftazidime is still ~100-fold higher than that observed for TEM-1 indicating CTX-M is a significantly better catalyst for this substrate than TEM-1. The difference in kcat/KM for ceftotaxime vs. ceftazidime by CTX-M enzymes is due to the extra bulk of the C7β side chain of ceftazidime that contains a carboxypropyl group replacing the methyl group found in cefotaxime. This leads to steric clashes in binding ceftazidime in the CTX-M active site (Delmas et al., 2008).
CTX-M D240G Substitution
As noted, the CTX-M enzymes hydrolyze ceftazidime poorly compared to cefotaxime. In the last 15 years, however, variants have emerged that are able to hydrolyze ceftazidime (Bonnet, 2004; Cantón et al., 2012; D'Andrea et al., 2013). In particular, the D240G and P167S substitutions occur individually in multiple CTX-M subgroups where they enhance ceftazidime hydrolysis (Bonnet et al., 2001, 2003; Cartelle et al., 2004; Kimura et al., 2004; Ishii et al., 2007). For example, the D240G substitution has been identified in the CTX-M-1, −2, −9, and −25 subfamilies while the P167S substitution has been identified in variants belonging to the CTX-M-1 and−9 subfamilies (D'Andrea et al., 2013). CTX-M variants containing D240G or P176S from clinical isolates are associated with increased MIC values for ceftazidime and the introduction of the substitutions into CTX-M-14 results in increased ceftazidime MICs for E. coli containing the mutants vs. the wild-type CTX-M-14 (Patel et al., 2015).
The D240G substitution, when introduced into CTX-M-14 or CTX-M-9, results in a 10-fold increase in kcat/KM for ceftazidime hydrolysis (Bonnet et al., 2003; Chen et al., 2005; Patel et al., 2015). Residue 240 is at the end of the β3-strand that flanks the active site and also contains Thr235 and Ser237 that make direct interactions with the C4 carboxylate group of cephalosporins (Delmas et al., 2010; Adamski et al., 2015; Figure 6). The X-ray crystal structure of CTX-M-14 containing the D240G substitution (CTX-M-27) has been determined, and anisotropic B-factor analysis demonstrated increased flexibility of the β3- strand that forms the side of the active site (Chen et al., 2005). This increase in flexibility is thought to expand its substrate profile by allowing access to the bulkier ceftazidime molecule (Chen et al., 2005).
The effect of the D240G substitution was further investigated by determining the structure of CTX-M-9 with the D240G substitution in complex with a glycylboronic acid containing the C7β oximino side chain of ceftazidime which forms a covalent adduct with Ser70 and places the ceftazidime side chain in a similar position as cefotaxime in the Toho-1 E166A-cefotaxime structure (Shimamura et al., 2002; Delmas et al., 2008). A comparison of the positioning of the ceftazidime side chain in the D240G mutant to its position in the wild type CTX-M-9 revealed that the aminothiazole ring was positioned deeper into the active site, more resembling the position of the aminothiazole ring in cefotaxime structures (Delmas et al., 2008). This positioning, in turn, led to more optimal contacts of the NH of the amide group of the ceftazidime side chain with the Ser237 main chain oxygen. Molecular dynamics simulations of ceftazidime acyl-enzyme structures of CTX-M-9 and the D240G mutant based on the ceftazidime-like boronic acid structures also suggested more optimal contacts of Thr235 with the C4 carboxylate of ceftazidime. A new hydrogen bond is observed between the amino group of the aminothiazole ring and the Pro167 backbone oxygen, along with an improved Ser237 main chain O interaction with the amide group (Delmas et al., 2008). Thus, the D240G substitution, via deeper positioning of the aminothiazole ring, appears to improve interactions with ceftazidime with multiple active site groups (Delmas et al., 2008).
CTX-M P167S Substitution
The CTX-M P167S substitution, when introduced into CTX-M-14, results in a 10-fold increase in kcat/KM for ceftazidime hydrolysis, similar to that observed for D240G (Patel et al., 2015). Pro167 resides on the omega loop and the peptide bond between Glu166 and Pro167 is in the cis configuration (Figure 6). The cis peptide bond contributes strongly to the conformation of the omega loop and particularly the positioning of Asn170 (Patel et al., 2017). Pro167 is largely conserved among class A β-lactamases, including TEM-1, and is a cis-proline in these enzymes (Philippon et al., 2016).
Molecular dynamics simulations based on the structure of the Toho-1 (CTX-M-44) enzyme have been performed to assess the effect of the P167S substitution (Kimura et al., 2004). The results suggested that in the P167S enzyme, the aminothiazole ring is displaced to prevent steric clash with Ser167 causing the C4 carboxylate of ceftazidime to hydrogen bond to Ser130 and Ser237 and enhancing hydrolysis (Kimura et al., 2004).
The CTX-M-14 β-lactamase has also been used as a model system to examine the structural changes caused by the P167S substitution that are associated with increased ceftazidime hydrolysis. A number of X-ray structures of CTX-M-14 P167S were solved that enabled an evaluation of the changes in structure between the apoenzyme, the initial enzyme complex with ceftazidime, and the formation of the ceftazidime-acyl enzyme (Patel et al., 2017). The S70G mutation was introduced to P167S to prevent acyl-enzyme formation and capture the structure of the enzyme-substrate complex and the E166A mutation was introduced to P167S to prevent deacylation of the acyl-enzyme to observe the acyl-enzyme structure. It was observed that the P167S apoenzyme closely resembled CTX-M-14 and the peptide bond preceding Ser167 was cis, despite a non-prolyl cis peptide bond being very energetically unfavorable (Jorgensen and Gao, 1988; Patel et al., 2017). The S70G/P167S structure in complex with ceftazidime revealed that the substrate bound with the C8 carbonyl oxygen in the oxyanion hole, as expected for a productive complex. The C4 carboxylate was bound by Thr235 but the C7β side chain was more solvent exposed due to steric constraints with the omega loop (Figure 7). The omega loop retained the conformation observed for the wild-type CTX-M-14 and P167S enzymes, and the peptide bond preceding Ser167 was also cis (Patel et al., 2017; Figure 8). The addition of E166A to the P167S enzyme to create E166A/P167S also showed the omega loop in a closed conformation in the apoenzyme structure, similar to the wild type and P167S apoenzymes (Figure 8). However, the peptide bond preceding Ser167 was trans. The addition of ceftazidime to E166A/P167S to form an acyl-enzyme resulted in a trans peptide bond preceding Ser167 and a large conformational change of the omega loop with Asn170 moving 4.2 Å out of the active site to create a pocket to accommodate the ceftazidime side chain (Patel et al., 2017; Figures 7, 8). This resulted in the aminothiazole ring sinking deeply into the active site in a buried position (Figure 7). These results suggested that both the Ser167 substitution and the presence of acylated ceftazidime are required for the conformational change. Finally, in order to show that the conformational change was due to the P167S substitution and not the E166A substitution that was used to trap the acyl-enzyme, the structure of E166A in complex with ceftazidime was determined. This showed the acyl-ceftazidime in the active site with Pro167 in the cis configuration and the omega loop in the closed conformation indicating the P167S substitution is required for the conformational change (Figures 7, 8). Therefore, the P167S substitution and the presence of acylated ceftazidime were both necessary to drive the structure toward a trans peptide bond at residue 167 and to induce conformational change of the omega loop (Patel et al., 2017).
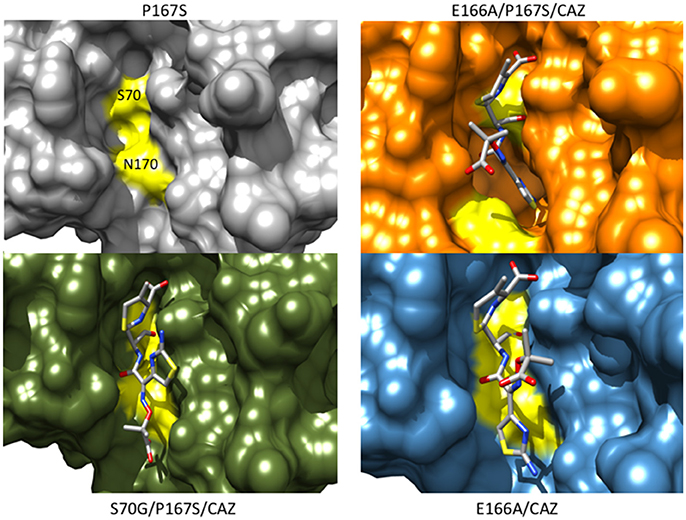
Figure 7. Protein surface representations of the CTX-M-14 P167S (gray) (PDB ID:5TWD), S70G/P167S with ceftazidime (green) (5TWE), E166A/P167S with acylated ceftazidime (orange) (5TW6) and E166A with acylated ceftazidime (blue) (5U53) are shown. The positions of Ser70 and Asn170 on the CTX-M structure are shown in yellow illustrating the movement of the omega loop in the E166A/P167S/CAZ acyl-enzyme separates Ser70 and Asn170 to create space in the active site to accommodate ceftazidime.
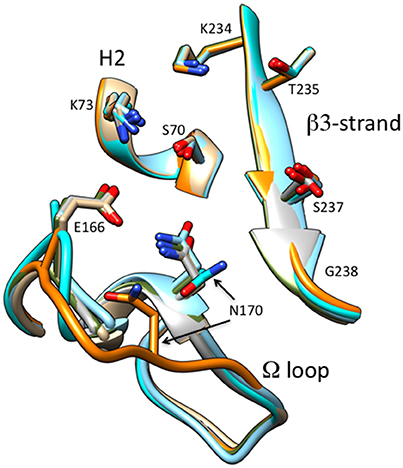
Figure 8. Schematic illustration of the omega loop region as well as the β3-strand and H2 helix of the active site of CTX-M-14 wt (tan) (PDB ID:1YLT) in comparison P167S (gray) (PDB ID:5TWD), S70G/P167S with ceftazidime (green) (5TWE), E166A/P167S with acylated ceftazidime (orange) (5TW6), E166A/P167S apoenzyme (cyan) (5VTH) and E166A with acylated ceftazidime (blue) (5U53). The omega loop in E166A/P167S with acylated ceftazidime undergoes a large conformational change moving Asn170 out of the active site. The ceftazidime molecule is omitted for clarity.
It is interesting to note that the structure of the CTX-M-9 D140G enzyme in complex with the ceftazidime-like boronic acid inhibitor and the CTX-M-14 E166A/P167S enzyme with acylated ceftazidime show the aminothiazole ring sinking to a deeper position in the active site relative to CTX-M-9 or CTX-M-14, respectively (Delmas et al., 2008; Patel et al., 2017). This suggests the P167S and D240G substitutions utilize different paths to a broadly similar end result—altered positioning of the aminothiazole ring and better contacts of substrate with the enzyme. In addition, the TEM-64 (E104K/R164S/M182T) ESBL enzyme with increased activity toward ceftazidime also displays a similar conformational change of the omega loop when complexed to a boronic acid inhibitor (Wang et al., 2002). Finally, a TEM-1 triple mutant (W165Y/E166Y/P167G) selected from a random mutagenesis experiment for increased hydrolysis of ceftazidime displays a large conformational change of the omega loop creating space to accommodate ceftazidime (Stojanoski et al., 2015). These studies suggest that enlargement of the active site via movement of the omega loop, which results in a shift of the Asn170 residue, is an important mechanism by which mutations can lead to hydrolysis of the bulky ceftazidime molecule in class A β-lactamases.
Stabilizing Substitutions in CTX-M Natural Variants
It has been demonstrated that the P167S and D240G substitutions both destabilize the CTX-M enzyme (Chen et al., 2005; Patel et al., 2015). As indicated above, the CTX-M family consists of a large number of variants containing amino acid substitutions (D'Andrea et al., 2013). Many of the substitutions are located outside of the active site and their effect on CTX-M structure and function is largely unknown. The A77V substitution is found in multiple subfamilies including the CTX-M-1, CTX-M-9, and CTX-M-25 groups (Patel et al., 2015). In addition, A77V has been found associated with either the P167S or D240G substitutions in each of these subfamilies. Using the CTX-M-14 model system, addition of the A77V substitution to either a P167S or D240G enzyme results in enhanced steady-state protein expression levels in E. coli relative to the P167S or D240G single mutants (Patel et al., 2015). In addition, the A77V/P167S and A77V/D240G enzymes exhibit increased thermal stability in vitro compared to the P167S and D240G enzymes (Patel et al., 2015). Based on these results, A77V has been suggested to be a global suppressor for CTX-M, analogous to M182T for TEM-1. Given the large number of substitutions among CTX-M variants, it is likely that other global suppressors are present in the family of clinical variants.
KPC β-lactamase Variants and Ceftazidime Hydrolysis
KPC-2 β-lactamase is a class A enzyme that hydrolyzes a broad range of β-lactam antibiotics including pencillins, cephalosporins and carbapenems. This enzyme also efficiently hydrolyzes cefotaxime but only poorly hydrolyzes ceftazidime. The value for kcat/KM for cefotaxime hydrolysis is ~3.0 × 105 M−1s−1 (Yigit et al., 2003; Levitt et al., 2012) while kcat/KM for ceftazidime is ~1.0 × 103 M−1s−1 (Mehta et al., 2015). By comparison, kcat/KM for ceftazidime for KPC-2 is 25-fold higher than that for TEM-1 and similar to the kcat/KM value for CTX-M-14. KPC-2 is an important clinical problem due to its broad substrate profile and wide distribution in enteric bacteria (Nordmann et al., 2009). The problem is compounded by the evolution of KPC variants that with increased ceftazidime hydrolysis rates and resistance (Mehta et al., 2015; Naas et al., 2016). Over 20 variants of KPC have been identified that contain a range of amino acid substitutions (Naas et al., 2016).
A detailed study of the KPC-3 to KPC-11 variants showed that these enzymes exhibit increased kcat/KM values for ceftazidime hydrolysis compared to KPC-2 (Mehta et al., 2015). Amino acid substitutions found among these enzymes include M49I, P104R, P104L, V240G, V240A, and H274Y (Figure 9). The P104R single substitution was found to increase kcat/KM for ceftazidime by 10-fold compared to KPC-2 while P104L exhibited a more modest 2.5-fold increase (Mehta et al., 2015). The V240G substitution increased kcat/KM for ceftazidime by 5-fold and H274Y resulted in a 10-fold increase. Double mutants containing combinations of the single substitutions exhibited further increases in kcat/KM. For example, the V240G/H274Y, P104R/V240G, and P104R/H274Y enzymes displayed 40-, 50-, and 75-fold increases in kcat/KM, respectively, for ceftazidime relative to KPC-2 (Mehta et al., 2015). In contrast, the M49I/H274Y enzyme exhibited similar catalytic efficiency as the H274Y enzyme, suggesting that M49I does not contribute to ceftazidime hydrolysis; however, the M49I substitution alone was not studied. Further, it was shown that the P104R, V240G, and H274Y substitutions act additively rather than cooperatively, i.e., there is no epistasis, when combined into the double mutants. This indicates that the substitutions act independently and do not influence each other's function when present in the double mutants. This would also suggest that the order in which the mutations occur to form the double mutant is not important (Mehta et al., 2015).
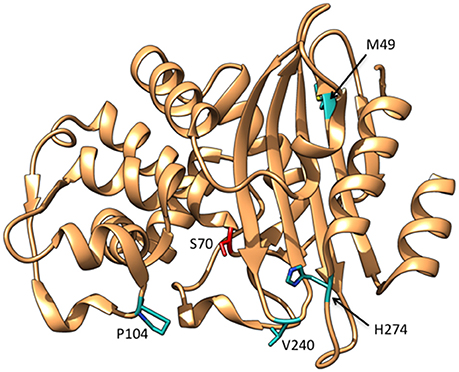
Figure 9. Ribbon diagram of KPC-2 β-lactamase showing the positions of residues Met49, Pro104, Val240, and His274 that are substituted in variants able to hydrolyze ceftazidime (cyan). The catalytic residue Ser70 is shown in red. (PDB ID:2OV5).
There is currently no structural information on the variants so there is limited understanding of their effect on KPC or the mechanism by which they alter specificity. However, molecular modeling of the substitutions onto the KPC-2 structure and computational docking of ceftazidime suggested that the P104R and H274Y substitutions could create new hydrogen bonding interactions with the C7β-oxyimino side chain of ceftazidime (Mehta et al., 2015). Note that the P104R substitution is in the same region of the proteins as the E104K substitution that commonly occurs in TEM ESBL variants. In addition, the V240G substitution is analogous to the CTX-M D240G substitution and could act by a similar mechanism of increasing flexibility of the β3 strand and allowing the aminothiazole ring of ceftazidime to sink deeper into the active site upon binding (Chen et al., 2005; Delmas et al., 2008). Finally, the H274Y substitution appears to be unique to the KPC variants.
As noted above, several substitutions in TEM and CTX-M that increase oxyimino-cephalosporin hydrolysis also decrease stability (Wang et al., 2002; Chen et al., 2005; Patel et al., 2015; Knies et al., 2017). A similar pattern was observed with the substitutions found in the KPC-3 to KPC-11 variants. There was a strong inverse correlation between kcat/KM for ceftazidime hydrolysis and the Tm of the enzymes (Mehta et al., 2015). However, the Tm for the wild type KPC-2 (66°C) is significantly higher than that for TEM or CTX-M and even the most unstable KPC variant is more stable than wild-type TEM-1 (Mehta et al., 2015). Therefore, the high stability of KPC-2 may serve as a buffer to allow the accumulation of substitutions without causing unfolding of the enzyme. This type of stability buffer has been noted previously as a mechanism to enhance protein evolvability (Bloom and Arnold, 2009; Tokuriki and Tawfik, 2009b).
Conclusions
The use of oxyimino-cephalosporins in the clinical setting has led to the evolution of variants of TEM-1, CTX-M, and KPC enzymes that can hydrolyze these drugs. The TEM-1 enzyme does not efficiently hydrolyze either cefotaxime or ceftazidime but there are now many variants that exhibit increased hydrolysis of these drugs. Initial isolates of CTX-M and KPC enzymes hydrolyze cefotaxime but not ceftazidime; however, variants now exist that hydrolyze ceftazidime. For TEM-1, recent studies have pointed to the importance of conformational heterogeneity of variants with ESBL substitutions. Rather than adopting a single, stable conformation, some variants exist in multiple sub-states of conformations that contain active conformers that can accommodate oxyimino-cephalosporins. One study also indicates that sampling of non-productive sub-states underlies the failure of mutant combinations such as R164S/G238S and, in another study, wild-type TEM-1 itself, to hydrolyze oxyimino-cephalosporins. Studies with TEM-1 have also indicated that several ESBL substitutions increase catalytic activity but are associated with a cost in stability. Global suppressor mutations then occur to compensate for the stability loss. It will be of interest to examine the folding, aggregation and stability effects of ESBL mutations and the compensatory effects of suppressors in vivo in the periplasm. The substitutions associated with ceftazidime hydrolysis in CTX-M are different than those observed in TEM, but act through conformational changes in similar regions of the active site as TEM-1. KPC variants are in analogous positions as some TEM and CTX-M substitutions and may act similarly. Conformational heterogeneity and the existence of sub-states have not been examined for CTX-M or KPC variants but may also be an important contributor to the function of these ESBL enzymes.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Funding
β-lactamase research in the author's laboratory is funded by National Institutes of Health grants AI32956 and AI106863.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author wishes to thank Drs. Jin Wang and Jianwei Chen for assistance with preparation of Figure 1. In addition, the author thanks Drs. Hiram Gilbert and B.V.V. Prasad for discussions and comments on the manuscript.
References
Adachi, H., Ohta, T., and Matsuzawa, H. (1991). Site-directed mutants, at position 166, of RTEM-1 beta-lactamase that form a stable acyl-enzyme intermediate with penicillin. J. Biol. Chem. 266, 3186–3191.
Adamski, C. J., Cardenas, A. M., Brown, N. G., Horton, L. B., Sankaran, B., Prasad, B. V., et al. (2015). Molecular basis for the catalytic specificity of the CTX-M extended spectrum β-lactamases. Biochemistry 54, 447–457. doi: 10.1021/bi501195g
Ambler, R. P., Coulson, F. W., Frere, J. M., Ghuysen, J. M., Joris, B., Forsman, M., et al. (1991). A standard numbering scheme for the class A β-lactamases. Biochem. J. 276, 269–272. doi: 10.1042/bj2760269
Ambler, R. P. (1980). The structure of beta-lactamases. Phil. Trans. R. Soc. Lond. B Biol. Sci. 289, 321–331. doi: 10.1098/rstb.1980.0049
Bar-Even, A., Milo, R., Noor, E., and Tawik, D. S. (2015). The moderately efficient enzyme: futile encounters and enzyme floppiness. Biochemistry 54, 4969–4977. doi: 10.1021/acs.biochem.5b00621
Bershtein, S., Goldin, K., and Tawfik, D. S. (2008). Intense neutral drifts yield robust and evolvable consensus proteins. J. Mol. Biol. 379, 1029–1044. doi: 10.1016/j.jmb.2008.04.024
Bloom, J. D., and Arnold, F. H. (2009). In the light of directed evolution: pathways of adaptive protein evolution. Proc. Natl. Acad. Sci. U.S.A. 106(Suppl. 1), 9995–10000. doi: 10.1073/pnas.0901522106
Bonnet, R., Dutour, C., Sampaio, J. L. M., Chanal, C., Sirot, D., Labia, R., et al. (2001). Novel cefotaximase (CTX-M-16) with increased catalytic efficiency due to substitution Asp-240-Gly. Antimicrob. Agents Chemother. 45, 2269–2275. doi: 10.1128/AAC.45.8.2269-2275.2001
Bonnet, R., Recule, C., Baraduc, R., Chanal, C., Sirot, D., De Champs, C., et al. (2003). Effect of D240G substitution in a novel ESBL CTX-M-27. J. Antimicrob. Chemother. 52, 29–35. doi: 10.1093/jac/dkg256
Bonnet, R. (2004). Growing group of extended-spectrum β-lactamases: the CTX-M enzymes. Antimicrob. Agents Chemother. 48, 1–14. doi: 10.1128/AAC.48.1.1-14.2004
Bonomo, R. A. (2017). β-lactamases: a focus on current challenges. Cold Spring Harb. Perspect. Med. 7:a025239. doi: 10.1101/cshperspect.a025239
Bowman, G. R., and Geissler, P. L. (2012). Equilibrium fluctuations of a single folded protein reveal a multitude of potential cryptic allosteric sites. Proc. Natl. Acad. Sci. U.S.A. 109, 11681–11686. doi: 10.1073/pnas.1209309109
Bowman, G. R., Bolin, E. R., Hart, K. M., Maguire, B. C., and Marqusee, S. (2015). Discovery of multiple hidden allosteric sites by combining Markov state models and experiments. Proc. Natl. Acad. Sci. U.S.A. 112, 2734–2739. doi: 10.1073/pnas.1417811112
Brown, N. G., Pennington, J. M., Huang, W., Ayvaz, T., and Palzkill, T. (2010). Multiple global suppressors of protein stability defects facilitate the evolution of extended-spectrum TEM β-lactamases. J. Mol. Biol. 404, 832–846. doi: 10.1016/j.jmb.2010.10.008
Bush, K., and Fisher, J. F. (2011). Epidemiological expansion, structural studies, and clinical challenges of new β-lactamases from Gram-negative bacteria. Ann. Rev. Microbiol. 65, 455–478. doi: 10.1146/annurev-micro-090110-102911
Bush, K. (2002). The impact of β-lactamases on the development of novel antimicrobial agents. Curr. Opin. Invest. Drugs 3, 1284–1290.
Cantón, R., González-Alba, J. M., and Galán, J. C. (2012). CTX-M enzymes: origin and diffusion. Front. Microbiol. 3:110. doi: 10.3389/fmicb.2012.00110
Cantu, C. III., and Palzkill, T. (1998). The role of residue 238 of TEM-1 β-lactamase in the hydrolysis of extended-spectrum antibiotics. J. Biol. Chem. 273, 26603–26609. doi: 10.1074/jbc.273.41.26603
Cantu, C. III., Huang, W., and Palzkill, T. (1996). Selection and characterization of amino acid substitutions at residues 237-240 of TEM-1 β-lactamase with altered substrate specificity for aztreonam and ceftazidime. J. Biol. Chem. 271, 22538–22545. doi: 10.1074/jbc.271.37.22538
Cantu, C. III., Huang, W., and Palzkill, T. (1997). Cephalosporin substrate specificity determinants of TEM-1 β-lactamase. J. Biol. Chem. 272, 29144–29150. doi: 10.1074/jbc.272.46.29144
Cartelle, M., del Mar Tomas, M., Molina, F., Moure, R., Villanueva, R., and Bou, G. (2004). High-level resistance to ceftazidime conferred by a novel enzyme, CTX-M-32, derived from CTX-M-1 through a single Asp240-Gly substitution. Antimicrob. Agents Chemother. 48, 2308–2313. doi: 10.1128/AAC.48.6.2308-2313.2004
Chanal-Claris, C., Sirot, J., Bret, L., Chatron, P., Labia, R., and Sirot, J. (1997). Novel extended-spectrum TEM-type β-lactamase from an Escherichia coli isolate resistant to ceftazidime and susceptible to cephalothin. Antimicrob. Agents Chemother. 41, 715–716.
Chen, Y., Delmas, J., Sirot, J., Shoichet, B., and Bonnet, R. (2005). Atomic resolution structures of CTX-M beta-lactamases: extended spectrum activities from increased mobility and decreased stability. J. Mol. Biol. 348, 349–362. doi: 10.1016/j.jmb.2005.02.010
Christensen, H., Martin, M., and Waley, G. (1990). β-Lactamases as fully efficient enzymes. a. Biochem. J. 266, 853–861.
Damblon, C., Raquet, X., Lian, L. Y., Lamotte-Brasseur, J., Fonze, E., Charlier, P., et al. (1996). The catalytic mechanism of β-lactamases: NMR titration of an active-site lysine residue of the TEM-1 enzyme. Proc. Natl. Acad. Sci. U.S.A. 93, 1747–1752. doi: 10.1073/pnas.93.5.1747
D'Andrea, M. M., Arena, F., Pallecchi, L., and Rossolini, G. M. (2013). CTX-M-type β-lactamases: a succesful story of antibiotic resistance. Int. J. Med. Microbiol. 303, 305–317. doi: 10.1016/j.ijmm.2013.02.008
Datta, N., and Kontomichalou, P. (1965). Penicillinase synthesis controlled by infectious R factors in Enterobacteriacea. Nature 208, 239–241. doi: 10.1038/208239a0
de Visser, J. A., and Krug, J. (2014). Empirical fitness landscapes and the predictability of evolution. Nat. Rev. Genet. 15, 480–490. doi: 10.1038/nrg3744
Delaire, M., Lenfant, F., Labia, R., and Masson, J. M. (1991). Site-directed mutagenesis on TEM-1 β-lactamase: role of Glu166 in catalysis and substrate binding. Protein Eng. 4, 805–810. doi: 10.1093/protein/4.7.805
Dellus-Gur, E., Elias, M., Caselli, E., Prati, F., Salverda, M. L. M., De Visser, J. A. G. M., et al. (2015). Negative epistasis and evolvability in TEM-1 β-lactamase - the thin line between an enzyme's conformational freedom and disorder. J. Mol. Biol. 427, 2396–2409. doi: 10.1016/j.jmb.2015.05.011
Delmas, J., Chen, Y., Prati, F., Robin, F., Shoichet, B. K., and Bonnet, R. (2008). Structure and dynamics of CTX-M enzymes reveal insights into substrate accommodation by extended-spectrum β-lactamases. J. Mol. Biol. 375, 192–201. doi: 10.1016/j.jmb.2007.10.026
Delmas, J., Leyssene, D., Dubois, D., Birck, C., Vazeille, E., Robin, F., et al. (2010). Structural insights into substrate recognition and product expulsion in CTX-M enzymes. J. Mol. Biol. 400, 108–120. doi: 10.1016/j.jmb.2010.04.062
Deng, Z., Huang, W., Bakkalbasi, E., Brown, N. G., Adamski, C. J., Rice, K., et al. (2012). Deep sequencing of systematic combinatorial libraries reveals beta-lactamase sequence constraints at high resolution. J. Mol. Biol. 424, 150–167. doi: 10.1016/j.jmb.2012.09.014
Docquier, J. D., Benvenuti, M., Calderone, V., Rossolini, G. M., and Gangani, S. (2011). Structure of the extended-spectrum β-lactamase TEM-72 inhibited by citrate. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 67, 303–306. doi: 10.1107/S1744309110054680
Drawz, S. M., and Bonomo, R. A. (2010). Three decades of beta-lactamase inhibitors. Clin. Microbiol. Rev. 23, 160–201. doi: 10.1128/CMR.00037-09
Escobar, W. A., Tan, A. K., and Fink, A. L. (1991). Site-directed mutagenesis of β-lactamase leading to accumulation of an acyl-enzyme intermediate. Biochemistry 30, 10783–10787. doi: 10.1021/bi00108a025
Farzaneh, S., Chaibi, E. B., Peduzzi, J., Barthelemy, M., Labia, R., Blazquez, J., et al. (1996). Implication of Ile-69 and Thr-182 residues in kinetic characteristics of IRT-3 (TEM-32) beta-lactamase. Antimicrob. Agents Chemother. 40, 2434–2436.
Fisher, J. F., and Mobashery, S. (2009). Three decades of the class A beta-lactamase acyl-enzyme. Curr. Prot. Pept. Sci. 10, 401–407. doi: 10.2174/138920309789351967
Fisher, J. F., Meroueh, S. O., and Mobashery, S. (2005). Bacterial resistance to beta-lactam antibiotics: compelling opportusism, compelling opportunity. Chem. Rev. 105, 395–424. doi: 10.1021/cr030102i
Fonseca, F., Chudyk, E. I., Van Der Kamp, M. W., Correia, A., Mulholland, A. J., and Spencer, J. (2012). The basis for carbapenem hydrolysis by class A β-lactamases: a combined investigation using crystallography and simulations. J. Amer. Chem. Soc. 134, 18275–18285. doi: 10.1021/ja304460j
Galleni, M., and Frere, J. M. (2007). “Kinetics of β-lactamases and penicillin-binding proteins,” in Enzyme-Mediated Resistance to Antibiotics: Mechanisms, Dissemination, and Prospects for Inhibition, eds R. A. Bonomo and M. E. Tolmasky (Washington, DC: ASM Press), 67–79.
Giakkoupi, P., Tzelepi, E., Tassios, P. T., Legakis, N. J., and Tzouvelekis, L. S. (2000). Detrimental effect of the combination of R164S with G238S in TEM-1 beta-lactamase on the extended-spectrum activity conferred by each single mutation. J. Antimicrob. Chemother. 45, 101–104. doi: 10.1093/jac/45.1.101
Gniadkowski, M. (2008). Evolution of extended-spectrum β-lactamases by mutation. Clin. Microb. Infect. 14, 11–32. doi: 10.1111/j.1469-0691.2007.01854.x
Hall, A., and Knowles, J. R. (1976). Directed selective pressure on a beta-lactamase to analyse molecular changes involved in the development of enzyme function. Nature 264, 803–804. doi: 10.1038/264803a0
Hart, K. M., Ho, C. M., Dutta, S., Gross, M. L., and Bowman, G. R. (2016). Modelling proteins' hidden conformations to predict antibiotic resistance. Nat. Commun. 7:12965. doi: 10.1038/ncomms12965
Healy, W. J., Labgold, M. R., and Richards, J. H. (1989). Substrate preferences in class A β-lactamases: preference for penams vs. cephems. The role of residue 237. Proteins Struct. Func. Genet. 6, 275–283. doi: 10.1002/prot.340060310
Hedstrom, L. (2002). Serine protease mechanism and specificity. Chem. Rev. 102, 4501–4523. doi: 10.1021/cr000033x
Herzberg, O., and Moult, J. (1987). Bacterial resistance to beta-lactam antibiotics: crystal structure of beta-lactamase from Staphylococcus aureus PC1 at 2.5 Å resolution. Science 236, 694–701. doi: 10.1126/science.3107125
Horn, J. R., and Shoichet, B. K. (2004). Allosteric inhibition through core disruption. J. Mol. Biol. 336, 1283–1291. doi: 10.1016/j.jmb.2003.12.068
Huang, W., and Palzkill, T. (1997). A natural polymorphism in β-lactamase is a global suppressor. Proc. Natl. Acad. Sci. U.S.A. 94, 8801–8806. doi: 10.1073/pnas.94.16.8801
Huletsky, A., Knox, J. R., and Levesque, R. C. (1993). Role of Ser-238 and Lys-240 in the hydrolysis of third-generation cephalosporins by SHV-type β-lactamases probed by site-directed mutagenesis and three-dimensional modeling. J. Biol. Chem. 268, 3690–3697.
Ishii, Y., Galleni, M., Ma, L., Frere, J. M., and Yamaguchi, K. (2007). Biochemical characterisation of the CTX-M-14 β-lactamase. Int. J. Antimicrob. Agents 29, 159–164. doi: 10.1016/j.ijantimicag.2006.09.005
Jorgensen, W. L., and Gao, J. (1988). Cis-trans energy difference for the peptide bond in the gas phase and in aqueous solution. J. Amer. Chem. Soc. 110, 4212–4216. doi: 10.1021/ja00221a020
Kather, I., Jakob, R. P., Dobbek, H., and Schmid, F. X. (2008). Increased folding stability of TEM-1 beta-lactamase by in vitro selection. J. Mol. Biol. 383, 238–251. doi: 10.1016/j.jmb.2008.07.082
Kimura, S., Ishiguro, M., Ishii, Y., Alba, J., and Yamaguchi, K. (2004). Role of a mutation at position 167 of CTX-M-19 in ceftazidime hydrolysis. Antimicrob. Agents Chemother. 48, 1454–1460. doi: 10.1128/AAC.48.5.1454-1460.2004
Knies, J. L., Cai, F., and Weinreich, D. M. (2017). Enzyme efficiency but not thermostability drives cefotaxime resistance evolution in TEM-1 β-lactamase. Mol. Biol. Evol. 34, 1040–1054. doi: 10.1093/molbev/msx053
Levitt, P. S., Papp-Wallace, K. M., Taracila, M. A., Hujer, A. M., Winkler, M. L., Smith, K. M., et al. (2012). Exploring the role of a conserved class A residue in the W-loop of KPC-2 β-lactamase. J. Biol. Chem. 287, 31783–31793. doi: 10.1074/jbc.M112.348540
Livermore, D. M. (2006). The β-lactamase threat in enterobacteriaceae, pseudomonas and acinetobacter. Trends Microbiol. 14, 413–420. doi: 10.1016/j.tim.2006.07.008
Lovering, A. L., Safadi, S. S., and Strynadka, N. C. (2012). Structural perspective of peptidoglycan biosynthesis and assembly. Ann. Rev. Biochem. 2012, 451–478. doi: 10.1146/annurev-biochem-061809-112742
Marciano, D. C., Pennington, J. M., Wang, X., Wang, J., Chen, Y., Thomas, V. L., et al. (2008). Genetic and structural characterization of an L201P global suppressor substitution in TEM-1 beta-lactamase. J. Mol. Biol. 384, 151–164. doi: 10.1016/j.jmb.2008.09.009
Mehta, S. C., Rice, K., and Palzkill, T. (2015). Natural variants of the KPC-2 carbapenemase have evolved increased catalytic efficiency for ceftazidime hydrolysis at the cost of enzyme stability. PLoS Pathog. 11:e1004949. doi: 10.1371/journal.ppat.1004949
Meroueh, S. O., Fisher, J. F., Schlegel, H. B., and Mobashery, S. (2005). Ab initio QM/MM study of class A β-lactamase acylation: dual participation of Glu166 and Lys73 in concerted base promotion of Ser70. J. Amer. Chem. Soc. 127, 15397–15407. doi: 10.1021/ja051592u
Minasov, G., Wang, X., and Shoichet, B. K. (2002). An ultrahigh resolution structure of TEM-1 beta-lactamase suggests a role for Glu166 as the general base in acylation. J. Am. Chem. Soc. 124, 5333–5340. doi: 10.1021/ja0259640
Naas, T., Dortet, L., and Iorga, B. I. (2016). Structural and functional aspects of class A carbapenemases. Curr. Drug Targets 17, 1006–1028. doi: 10.2174/1389450117666160310144501
Nordmann, P., Cuzon, G., and Naas, T. (2009). The real threat of Klebsiella pneumoniae carbapenemase-producing bacteria. Lancet Infect. Dis. 9, 228–236. doi: 10.1016/S1473-3099(09)70054-4
Orencia, M. C., Yoon, J. S., Ness, J. E., Stemmer, W. P. C., and Stevens, R. C. (2001). Predicting the emergence of antibiotic resistance by directed evolution and structural analysis. Nat. Struc. Biol. 8, 238–242. doi: 10.1038/84981
Palzkill, T. (2013). Metallo-β-lactamase structure and function. Ann. N.Y. Acad. Sci. 1277, 91–104. doi: 10.1111/j.1749-6632.2012.06796.x
Patel, M. P., Fryszcyn, B. G., and Palzkill, T. (2015). Characterization of the global stabilizing substitution A77V and its role in the evolution of CTX-M β-lactamases. Antimicrob. Agents Chemother. 59, 6741–6748. doi: 10.1128/AAC.00618-15
Patel, M. P., Hu, L., Stojanoski, V., Sankaran, B., Prasad, B. V., and Palzkill, T. (2017). The drug-resistant variant P167S expands the substrate profile of CTX-M β-lactamases for oxyimino-cephalosporin antibiotics by enlarging the active site upon acylation. Biochemistry 56, 3443–3453. doi: 10.1021/acs.biochem.7b00176
Pendleton, J. N., Gorman, S. P., and Gilmore, B. F. (2013). Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti. Infect. Ther. 11, 297–308. doi: 10.1586/eri.13.12
Petit, A., Maveyraud, L., Lenfant, F., Samama, J. P., Labia, R., and Masson, J. M. (1995). Multiple substitutions at position 104 of ß-lactamase TEM-1: assessing the role of this residue in substrate specificity. Biochem. J. 305, 33–40. doi: 10.1042/bj3050033
Petrosino, J., Cantu Iii, C., and Palzkill, T. (1998). β-lactamases: protein evolution in real time. Trends Microbiol. 6, 323–327. doi: 10.1016/S0966-842X(98)01317-1
Philippon, A., Slama, P., Deny, P., and Labia, R. (2016). A structure-based classification of class A β-lactamases, a broadly diverse family of enzymes. Clin. Microb. Rev. 29, 29–57. doi: 10.1128/CMR.00019-15
Pimenta, A. C., Fernandes, R., and Moreira, I. S. (2014). Evolution of drug resistance: insight on TEM-1 β-lactamases structure and activity and β-lactam antibiotics. Mini Rev. Med. Chem. 14, 111–122. doi: 10.2174/1389557514666140123145809
Raquet, X., Lamotte-Brasseur, J., Fonze, E., Goussard, S., Courvalin, P., and Frere, J. M. (1994). TEM beta-lactamase mutants hydrolysing third-generation cephalosporins-A kinetic and molecular modelling analysis. J. Mol. Biol. 244, 625–639. doi: 10.1006/jmbi.1994.1756
Raquet, X., Vanhove, M., Lamotte-Brasseur, J., Goussard, S., Courvalin, P., and Frere, J. M. (1995). Stability of TEM beta-lactamase mutants hydrolyzing third generation cephalosporins. Proteins 23, 63–72. doi: 10.1002/prot.340230108
Salverda, M. L. M., De Visser, J. A. G. M., and Barlow, M. (2010). Natural evolution of TEM-1 β-lactamase: experimental reconstruction and clinical relevance. FEMS Microbiol. Rev. 2010, 1–22. doi: 10.1111/j.1574-6976.2010.00222.x
Saves, I., Burlet-Schiltz, O., Maveyraud, L., Samama, J. P., Prome, J. C., and Masson, J. M. (1995). Mass spectral kinetic study of acylation and deacylation during hydrolysis of penicillins and ceftotaxime by β-lactamase TEM-1 and the G238S mutant. Biochemistry 34, 11660–11667. doi: 10.1021/bi00037a003
Shimamura, T., Ibuka, A., Fushinobu, S., Wakagi, T., Ishiguro, M., Ishii, Y., et al. (2002). Acyl-intermediate structures of the extended-spectrum class A β-lactamase, Toho-1, in complex with cefotaxime, cephalothin, and benzylpenicillin. J. Biol. Chem. 277, 46601–46608. doi: 10.1074/jbc.M207884200
Shortle, D., and Lin, B. (1985). Genetic analysis of Staphylococcal nuclease: identification of three intragenic “global suppressors” of nuclease minus mutations. Genetics 110, 539–555.
Sideraki, V., Huang, W., Palzkill, T., and Gilbert, H. F. (2001). A secondary drug resistance mutation of TEM-1 beta-lactamase that suppresses misfolding and aggregation. Proc. Natl. Acad. Sci. U.S.A. 98, 283–288. doi: 10.1073/pnas.011454198
Singh, M. K., and Dominy, B. N. (2012). The evolution of cefotaximase activity in the TEM-1 β-lactamase. J. Mol. Biol. 415, 205–220. doi: 10.1016/j.jmb.2011.10.041
Sowek, J. A., Singer, S. B., Ohringer, S., Malley, M. F., Dougherty, T. J., Gougoutas, J. Z., et al. (1991). Substitution of lysine at position 104 or 240 of TEM-1pTZ18R b-lactamase enhances the effect of serine-164 substitution on hydrolysis or affinity for cephalosporins and the monobactam aztreonam. Biochemistry 30, 3179–3188. doi: 10.1021/bi00227a004
Stemmer, W. (1994). Rapid evolution of a protein in vitro by DNA shuffling. Nature 370, 389–391. doi: 10.1038/370389a0
Stojanoski, V., Chow, D. C., Hu, L., Sankaran, B., Gilbert, H. F., Prasad, B. V., et al. (2015). A triple mutant in the Ω-loop of TEM-1 β-lactamase changes the substrate profile via a large conformational change and an altered general base for catalysis. J. Biol. Chem. 290, 10382–10394. doi: 10.1074/jbc.M114.633438
Strynadka, N. C. J., Adachi, H., Jensen, S. E., Johns, K., Sielecki, A., Betzel, C., et al. (1992). Molecular structure of the acyl-enzyme intermediate in β-lactam hydrolysis at 1.7 Å resolution. Nature 359, 700–705. doi: 10.1038/359700a0
Tokuriki, N., and Tawfik, D. S. (2009a). Protein dynamism and evolvability. Science 324, 203–207. doi: 10.1126/science.1169375
Tokuriki, N., and Tawfik, D. S. (2009b). Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 19, 596–604. doi: 10.1016/j.sbi.2009.08.003
Vakulenko, S. B., Taibi-Tronche, P., Toth, M., Massova, I., Lerner, S. A., and Mobashery, S. (1999). Effects on substrate profile by mutational substitutions at positions 164 and 179 of the class A TEMpUC19 β-lactamase from Escherichia coli. J. Biol. Chem. 274, 23052–23060. doi: 10.1074/jbc.274.33.23052
Venkatachalam, K. V., Huang, W., Larocco, M., and Palzkill, T. (1994). Characterization of TEM-1 ß-lactamase mutants from positions 238 to 241 with increased catalytic efficiency for ceftazidime. J. Biol. Chem. 269, 23444–23450.
Viadiu, H., Osuna, J., Fink, A. L., and Soberon, X. (1995). A new TEM β-lactamase double mutant with broadened specificity reveals substrate-dependent functional interactions. J. Biol. Chem. 270, 781–787. doi: 10.1074/jbc.270.2.781
Wang, X., Minasov, G., and Shoichet, B. K. (2002). Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J. Mol. Biol. 320, 85–95. doi: 10.1016/S0022-2836(02)00400-X
Weinreich, D. M., Delaney, N. F., Depristo, M. A., and Hartl, D. L. (2006). Darwinian evolution can follow only very few mutational paths to fitter proteins. Science 312, 111–114. doi: 10.1126/science.1123539
Wells, J. A. (1990). Additivity of mutational effects in proteins. Biochemistry 29, 8509–8517. doi: 10.1021/bi00489a001
Yigit, H., Queenan, A. M., Rasheed, J. K., Biddle, J. W., Domenech-Sanchez, A., Alberti, S., et al. (2003). Carbapenem-resistant strain of Klebsiella oxytoca harboring carbapenem-hydrolyzing β-lactamase KPC-2. Antimicrob. Agents Chemother. 47, 3881–3889. doi: 10.1128/AAC.47.12.3881-3889.2003
Keywords: β-lactamase, β-lactam antibiotics, protein evolution, antibiotic resistance, enzyme structure, enzyme mechanism
Citation: Palzkill T (2018) Structural and Mechanistic Basis for Extended-Spectrum Drug-Resistance Mutations in Altering the Specificity of TEM, CTX-M, and KPC β-lactamases. Front. Mol. Biosci. 5:16. doi: 10.3389/fmolb.2018.00016
Received: 27 December 2017; Accepted: 08 February 2018;
Published: 23 February 2018.
Edited by:
Christopher Davies, Medical University of South Carolina, United StatesReviewed by:
Focco Van Den Akker, Case Western Reserve University, United StatesSergei Vakulenko, University of Notre Dame, United States
Copyright © 2018 Palzkill. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timothy Palzkill, timothyp@bcm.edu