Cross-talk between mitochondria and proteasome in Parkinson’s disease pathogenesis
- 1 Center for Neuroscience and Cell Biology, University of Coimbra, Coimbra, Portugal
- 2 Faculty of Medicine, University of Coimbra, Coimbra, Portugal
Parkinson’s disease (PD) is the most common progressive neurodegenerative movement disorder, characterized by the selective loss of nigrostriatal dopaminergic neurons, and the presence of intracellular insoluble proteinaceous inclusions, known as Lewy Bodies. Although PD etiopathogenesis remains elusive, the leading hypothesis for the death of specific groups of neurons establishes that mitochondrial dysfunction, alterations in the ubiquitin-proteasomal system (UPS), and oxidative stress are major events that act synergistically causing this devastating disease. In this review we will focus on mitochondrial impairment and its implications on proteasomal function and alpha-synuclein aggregation. We will address the role of mitochondria and proteasome cross-talk in the neuronal loss that leads to PD and discuss how this knowledge might further improve patient therapy.
Parkinson’s Disease Etiopathogenesis
Parkinson’s disease (PD) is a severe and progressive neurodegenerative disorder of the central nervous system (CNS) and is the most common movement disorder. PD is characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) in the ventral midbrain, mostly affecting the nigroestriatal pathway. The consequence of the severe depletion of dopamine (DA) in the striatum is the imbalance of acetylcholine, glutamate and y-aminobutyric acid (GABA) in the subthalamic nucleus, thalamus and cortex, underlying the clinical symptomatology of the disorder (Wichmann and De Long, 2003). The loss of non-dopaminergic neurons, such as norepinephrine-containing neurons in the locus coeruleus, cholinergic neurons in the nucleus basalis of Meynert and serotoninergic neurons in the raphe nuclei have been observed and are also involved in the pathophysiology of PD (Jellinger, 1999).
The clinical features of PD are tremor at rest, rigidity and bradykinesia, often associated to postural instability, gait alterations and dysarthria. These symptoms are mainly explained by deficiency of dopamine in the striatum, whereas minor symptoms such as autonomic dysfunction, depression and cognitive impairment may be consistent with the disease whether or not associated with pathological changes in non-dopaminergic systems. The therapeutic approach to the disorder is symptomatic and palliative. L-dopa is the standard drug, although the disease will progress. The ultimate therapeutic goal, based on the understanding of the etiopathogenesis of the disease has to be restorative and protective.
Even knowing that the main cause of the disease is DAergic cell death, the reason why these cells are progressively dying remains elusive. Genetic, epigenetic and environmental factors, such as head trauma, toxics or drugs have been reported in PD etiopathogenesis (revised by Cardoso et al., 2009). The SNpc is a DA-rich brain region that contains neuromelanin and a high content in iron which have been described to induce oxidative stress being related to the specific neurodegeneration occurring in PD. The decrease in DAergic neurons and the consequent reduction of neuromelanin are responsible for the pale color of PD patients substancia nigra (SN) as compared to age-matched healthy controls and is a neuropathological hallmark of the disorder.
Protein aggregation is also a central feature of PD pathophysiology the presence of Lewy bodies (LBs) being other prominent histopathological hallmark of PD. LBs are eosinophilic intracytoplasmatic aggregates of several proteins such as alpha-synuclein, ubiquitin, synphilin-1, 14-3-3 protein, tubulin and other cytoskeletal proteins (Forno, 1996). LBs are also typical features of other alpha-synucleinopathies such as Dementia with LBs and Multiple system atrophy. However, LBs distribution through the CNS is variable between those disorders and PD. These aggregates may be classified in classical LBs, which are mainly located in brain stem and have a dense hyaline appearance surrounded by a halo, or cortical type LBs, when they are less well defined. The major protein component of both LBs types is alpha-synuclein (Spillantini et al., 1997), whose cellular function remains unknown. Mitochondrial dysfunction, oxidative stress or/and proteasome system impairment, were shown to potentiate alpha-synuclein aggregation in sporadic PD models (revised by Arduino et al., 2010). Despite alpha-synuclein presence in LBs is a histopathological marker for PD, the mechanisms leading to the gradual transition of soluble alpha-synuclein into insoluble LBs or Lewy neuritis are still unknown. In the familial form of PD due to the mutation of parkin gene the classic LBs pathology is not observed. Moreover, toxin-induced parkinsonism is not associated with typical LBs formation, thus suggesting that alpha-synuclein aggregates are not a cause of the disease but probably a chronic mechanism of cell protection against soluble oligomeric alpha-synuclein toxicity.
Evidence exists supporting the notion that oxidative stress and impaired mitochondrial function are involved in the pathogenesis of the disorder. However, the mechanisms responsible for the death of SNpc dopaminergic neurons has not yet been established. In this review we will focus on the role of mitochondria, UPS and oxidative stress as key players in the degeneration of DAergic neurons.
Parkinson’s Disease Forms
The most common type of PD is the sporadic or late-onset (LOPD) form which affects about 1% of individuals older than 65 years (de Lau and Breteler, 2006). This form seems to be related with the aging process and/or the exposure to environmental toxins. However, 5% of PD cases manifesting before 40–50 years, have been referred as familial or early-onset PD (EOPD) (Hatano et al., 2009), with a classical recessive or dominant Mendelian inheritance.
Mutations in some genes are the origin of familial and, in some cases, sporadic PD. In the last decade linkage studies revealed 15 PD-related genetic loci (PARK1-15) (Hatano et al., 2009), and in a recent report a new locus, PARK16, was identified (Satake et al., 2009). Mutations described for these familial forms of PD, include autosomal dominant mutations of alpha-synuclein gene (PARK1) (Polymeropoulos et al., 1997), uchl1 gene (PARK5) and lrrk2 gene (PARK8) (Funayama et al., 2002) or autosomal recessive mutations of parkin gene (PARK2) (Kitada et al., 1998; Mizuno et al., 2008), pink1 gene (PARK6) (Hatano et al., 2004; Valente et al., 2004a), dj-1 gene (PARK7) (Bonifati et al., 2003) and atp13a2 gene (PARK9) (Ramirez et al., 2006). Additionally, in fibroblasts of a single patient, mutations in mitochondrial DNA, nd5 and nd6 genes that codify for two complex I (cx I) subunits (Piccoli et al., 2008) were identified. Despite the overall rarity of the familiar forms of PD, the identification of single genes linked to the disease has yielded crucial insights into possible mechanisms of the PD pathogenesis, giving strong evidences of the involvement of organelles like mitochondria and intracellular degradation pathways as UPS and autophagy in the pathophysiology of PD (revised in Arduino et al., 2010).
Mutations in parkin and uchl1 genes are related with alterations in the UPS pathway. Parkin protein is a 465-aminoacid (a.a.) polypeptide (∼51 kDa) that plays an important role in the UPS as ubiquitin E3 ligase (Shimura et al., 2000). Ubiquitin C-terminal hydrolase L1 (UCHL1) is a 223-a.a. protein which is a component of the UPS, which cleaves the carboxy-terminal peptide bond of polyubiquitine chains, working as a deubiquitinating enzyme (Wilkinson et al., 1989; Larsen et al., 1996, 1998). Mono-ubiquitin recycling by UCHL1 sustains protein degradation. Moreover, both Parkin and UCHL1 were found to be LBs components (Lowe et al., 1990; Shimura et al., 1999; Schlossmacher et al., 2002).
Mutations in pink1 gene are the second most common form of EOPD. This gene encodes a 581 a.a. ubiquitously expressed protein, PINK1 (PTEN induced kinase 1), with serine/threonine kinase activity, found in mitochondria (Hatano et al., 2004; Valente et al., 2004a,b). PINK1 mitochondrial localization is due to a N-terminal mitochondrial-targeting signal (Mills et al., 2008), being one of the most direct evidences of a primary role of mitochondria in PD etiopathogenesis. PINK1 was found in a small group of brain stem LBs as well (Gandhi et al., 2006). Moreover, mitochondria are also indirectly involved in most of other genetic forms of EOPD. Alpha-synuclein mouse model (Martin et al., 2006) and both parkin-knockout mouse and Drosophila models (Greene et al., 2003; Palacino et al., 2004) showed a decreased mitochondrial respiratory activity. Also DJ-1 and LRRK2 proteins were shown to be localized into the mitochondria (Canet-Aviles et al., 2004; West et al., 2005).
Mutations in atp13a2 gene give rise to an autosomal recessive form of early-onset parkinsonism. This gene encodes a predicted lysosomal P-type transmembrane cation transporting ATPase that is localized in the lysosome. Mutant ATP13A2 proteins are retained in the endoplasmic reticulum and degraded by the proteasome (Ramirez et al., 2006). Recently, Gitler et al. (2009) reported a strong genetic interaction between alpha-synuclein and ATP13A2. Moreover, ATP13A2 suppresses alpha-synuclein and manganese toxicity in primary neuron cultures (Gitler et al., 2009).
Analysis of these genes products lead us to conclude that mitochondria and proteasome are deeply involved in the molecular mechanisms of PD pathogenesis. Moreover, in some genetic defects they are involved simultaneously due to the relation of the protein product with both, mitochondria and proteasome, which somehow mean a probable interplay between them.
Parkinson’s Disease, A Disorder of Protein Misfolding
Misfolding diseases are a wide group of devastating disorders characterized by the accumulation of pathological protein aggregates. Despite different origins (sporadic, familial or transmissible), they are sometimes called conformational diseases to emphasize aberrant conformations as the putative cause of deposits that precede or accompany the clinical manifestation of the disease.
Alzheimer’s disease (AD), Prion disorders (PrD), Parkinson’s disease (PD), and Huntington’s disease (HD) are the most typical examples of protein misfolded disorders, which are characterized by protein conformational changes. The misfolded protein process entails protein conformational transitions that go from oligomers, protofibrils or mature aggregates formation (Ciechanover and Brundin, 2003).
The accumulation of those proteins may result from either genetic rare mutations, or post-translational changes mainly due to oxidative/nitrosative stress (further described in Section Neurotoxic Mechanisms), which is more likely to be related to sporadic forms of neurodegenerative diseases (Nakamura and Lipton, 2009).
Even though mutations in alpha-synuclein gene (Ala53Thr, Ala30Pro and Glu46Lys) are very rare causes of hereditary PD, its apparent role as the major structural feature of the LBs, both in EOPD and LOPD, suggests that alpha-synuclein may have a key role in DAergic neuronal cell death.
Firstly identified as synaptic-associated protein in the rat brain (Maroteaux et al., 1988) and highly expressed in pre-synaptic nerve terminals, alpha-synuclein is a 140-a.a. protein whose cellular function remains unknown. Data from the last decade studies indicate that alpha-synuclein has important roles in synaptic vesicle formation and catecholamine metabolism in DAergic neurons (Hatano et al., 2009). Concerning to vesicle formation, Murphy et al. (2000), reported that alpha-synuclein may regulate the size of synaptic vesicles in mature neurons by observing a significant reduction in distal synaptic vesicles pool formation when the expression of alpha-synuclein is suppressed by antisense oligonucleotide technology (Murphy et al., 2000). However, later in 2006, Cooper and colleagues referred that alpha-synuclein blocked endoplasmic reticulum (ER)-to-Golgi vesicular trafficking and induced ER stress followed by cell death (Cooper et al., 2006). In the meanwhile, some studies revealed that alpha-synuclein may modulate signal transduction, cytoskeleton organization and membrane trafficking due to interaction with membrane through lipid rafts (Fortin et al., 2004; Kubo et al., 2005).
Relatively to catecholamine metabolism, it was shown that alpha-synuclein is capable to inhibit DA release in an activity-dependent negative regulator manner (Abeliovich et al., 2000), through exocytosis alterations (Larsen et al., 2006). It is also suggested that alpha-synuclein may modulate the activation of tyrosine hydroxylase (Perez et al., 2002) and down-regulate vesicular monoamine transporter-2 activity (Guo et al., 2008).
A recent study report that alpha-synuclein may also be involved in the regulation of Ca2+ homeostasis specifically increasing Ca2+ entry via L-type Ca2+ channels and, in this way, deregulation promoted by mutant forms can originate neurotoxicity (Hettiarachchi et al., 2009). Moreover, it seems to be clear that alpha-synuclein interacts with tubulin, inhibiting microtubule formation and thus regulating microtubule dynamics in neurons (Zhou et al., 2009). Lee et al. (2006) demonstrated that overexpression of alpha-synuclein caused disruption of microtubule network and impairment of microtubule-dependent trafficking. In the other hand, alpha-synuclein may induce polymerization of purified tubulin into microtubules, a capacity lost by the mutant forms of alpha-synuclein (Alim et al., 2002, 2004). We demonstrated, in cells with chronic mitochondrial deficit (PD cybrids) an increase in free tubulin and in oligomeric alpha-synuclein content (Esteves et al., 2009, 2010a). Additionally, we proved that an increase in free/polymerized tubulin ratio may increase alpha-synuclein oligomerization when a mitochondrial dependent ATP depletion and reactive oxygen species (ROS) generation occurs (Esteves et al., 2009). Interestingly, tubulin can stimulate alpha-synuclein fibrils formation in yeast and it was proved that by inhibiting microtubule dynamics or deleting genes involved in microtubule biogenesis, alpha-synuclein aggregation and toxicity increased (Kim et al., 2008). In this way, microtubule impairment affects alpha-synuclein distribution and toxicity and also may be responsible for mitochondria synaptic depletion, consistent with the “dying back” phenomenon of DAergic neurons degeneration (Figure 1).
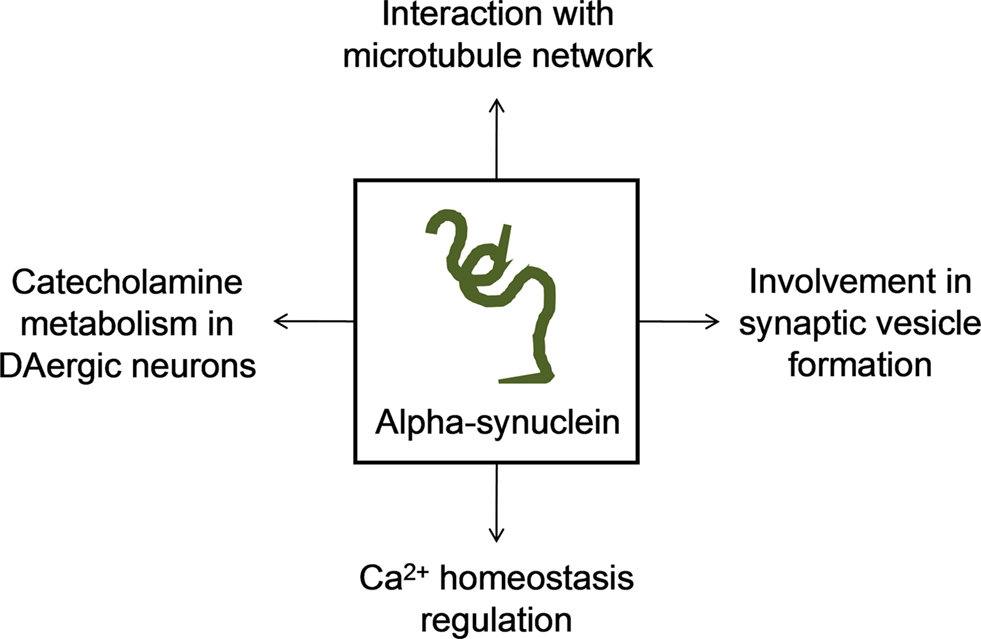
Figure 1. Major proposed functions of alpha-synuclein in the cell.
Alpha-synuclein exists in solution as an unfolded monomer but can easily aggregate and form insoluble antiparallel beta-sheet fibrillar oligomers (Moore et al., 2005). Alpha-synuclein mediated toxicity in PD is thought to be due to soluble intermediate oligomers fraction than to fibrillar deposits, as found in LBs (Outeiro et al., 2008). Toxic-gain of function of mutant protein and over-expression of the human wild-type are both related to PD pathology (Figure 2).
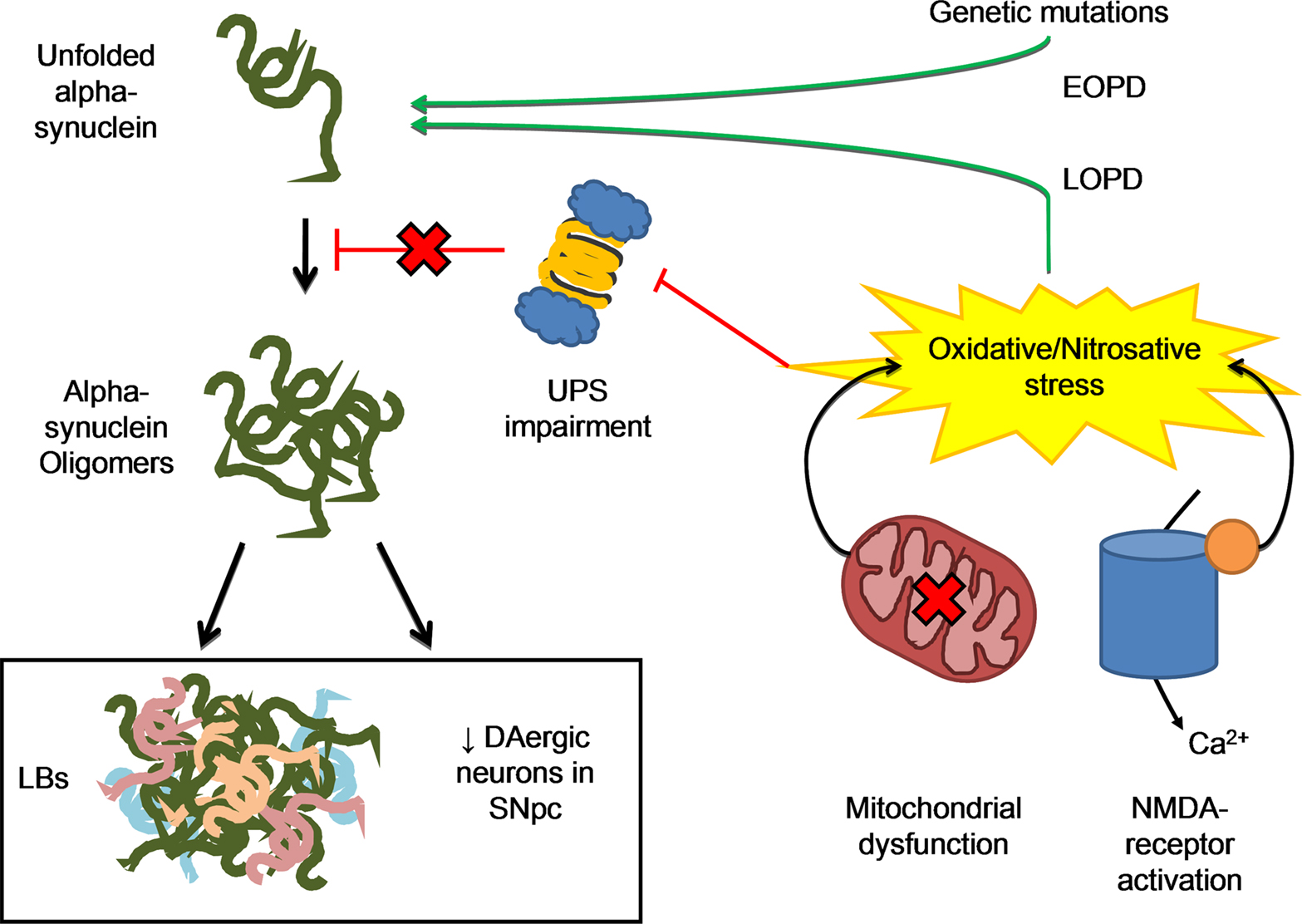
Figure 2. The misfolding process of alpha-synuclein may result from either genetic mutations, responsible for familial forms of the disease, or post-translational changes to the protein mainly due to oxidative/nitrosative stress, which is more likely to be related to sporadic forms of PD. Two main events are related to ROS/RNS formation: (1) excessive Ca2+ influx due to activation of NMDA-type glutamate receptors in nervous system, with activation of neuronal NO synthase as well, as the generation of ROS and (2) Dysfunctional mitochondrial respiration that is one of the major sources of ROS in the cell. Excessive ROS/RNS formation by these two processes may induce UPS impairment, thus resulting in protein aggregation. This may explain the etiology of the two PD neuropathological hallmarks, LBs formation and neuronal death.
Other fact that supports the importance of alpha-synuclein in the pathogenesis of the disease is the decreased sensitivity to MPTP-induced degeneration of DA neurons in alpha-synuclein-deficient mice (Dauer et al., 2002). Inversely, it was shown that MPTP sensitivity is increased in some lines of alpha-synuclein transgenic mice (Nieto et al., 2006; Yu et al., 2008). These results also indicate a probable relationship between mitochondrial dysfunction and alpha-synuclein toxicity, since MPTP is a well known mitochondrial electron transport chain (ETC) cx I inhibitor.
Besides alpha-synuclein accumulation being evident in alpha-synucleinopathies, its degradation pathway is not well defined. Evidences exists that UPS is the primary mechanism for its degradation (Rideout et al., 2001), but it can be inhibited by alpha-synuclein protofibrils (Zhang et al., 2008). Defective proteasome activity is found in transgenic mouse models of alpha-synuclein (Chen et al., 2006). Moreover, Liu et al. (2009) showed that loss of PINK1 function leads to proteasome impairment and alpha-synuclein aggregation.
In this way, mitochondrial dysfunction and UPS impairment seem to be strictly involved and related in formation of alpha-synuclein aggregates, since alpha-synuclein transgenic mice show mitochondrion and UPS loss of function, as shown separately in the studies referred above.
Neurotoxic Mechanisms
Even being this the most unknown field of the disease, there are several evidences that suggest a list of conditions working as neurotoxic mechanisms underlying PD pathogenesis. According to Gallacher and Schapira, oxidative stress and free radical generation, mitochondrial dysfunction, glutamate mediated excitotoxicity, inflammation, oligodendrocyte interaction and neurotrophic factors, UPS impairment, autophagy involvement and apoptosis (Gallagher and Schapira, 2009) are mechanisms involved.
Oxidative stress is considered to compromise the integrity of vulnerable neurons and thus to contribute to neuronal degeneration. The source of the increased oxidative stress observed in PD is unclear but may derive from mitochondrial dysfunction, increased DA metabolism (that can yield excess of hydrogen peroxide and other ROS), increased reactive iron, impairment of antioxidant defenses pathways, and/or due to the highly oxidative intracellular environment within dopaminergic neurons (revised by Cardoso et al., 2009). In PD oxidative stress may be also related to an activation of N-methyl-D-aspartate (NMDA)-type glutamate receptors, with an increased production of RNS (reactive nitrogen species) due to neuronal NO synthase (nNOS) over-activation (Garthwaite et al., 1988; Bredt et al., 1991). A mitochondrial defect generates ROS, specially the superoxide anion (O2-), which rapidly reacts with free radical NO creating peroxinitrite radical (ONOO-) (Beckman et al., 1990; Lipton et al., 1993). Excessive ROS/RNS formation by these two processes may induce UPS impairment and/or misfolding of molecular chaperons, thus resulting in protein aggregation and neuronal damage (Nakamura and Lipton, 2009). This is another point of intersection between mitochondria and UPS functioning, since mitochondria, producing excessive ROS, may adversely affect UPS activity.
In this review we focus those mechanisms we believe to have a central importance in the disease pathogenesis, especially mitochondrial dysfunction and UPS alteration and the correlation between both. At first, it is important to consider mitochondrial dysfunction, because mitochondria are essential organelles for the production of ATP, especially in high energy demand cells such as neurons. Moreover, mitochondria are involved in the regulation of calcium homeostasis, which allows the cell to maintain low levels of cytosolic calcium and avoid neurotoxicity. Mitochondrial respiratory chain cx I in the brain is the major source of endogenous relevant free radicals. Finally, mitochondria also regulates apoptotic cell death. Regarding UPS dysfunction is significant, since proteasome is a major protein degradation cell complex and protein aggregation appears as a main event in PD pathology.
The Role of Mitochondria in PD
As mentioned before, genetic studies highlighted the growing importance of mitochondria role in PD etiopathogenesis, mainly pink1 and parkin genes, whose mutated forms have been linked to EOPD.
A recent report by Liu and colleagues proved that PINK1 exists as a dimer in mitochondrial protein complexes. The protein is composed of a kinase domain, where the mutations are clustered, which is responsible for the enzymatic activity and the formation of the dimer. Moreover, it contains a mitochondrial targeting signal at its N terminal. Although mutations in pink1 do not affect dimerization and localization with mitochondrial complexes, those mutations impair mitochondrial respiration and ATP synthesis in PD cell models. In addition, PINK1 knockdown also contribute to a statistical decrease in oxygen consumption (Liu et al., 2009). It was also observed a significant reduction in mitochondrial respiration in the striatum of PINK1 knockout mice (Gautier et al., 2008) and mitochondrial pathology in pink1 Drosophila mutants (Clark et al., 2006; Park et al., 2006). Thus, and according to Deas and colleagues work, PINK1 is thought to be related with mitochondrial respiration. Moreover, Deas et al. review that PINK1 protects cells against oxidative stress induced apoptosis by interacting with the mitochondrial chaperone tumor necrosis factor receptor-associated protein 1 (TRAP1)/heat shock protein 75 (Hsp75) (Pridgeon et al., 2007). PINK1 is also involved in (1) mitochondria fisson/fusion, functioning as a potential pro-fusion protein in mammals, in (2) mitochondrial trafficking by interacting with Miro and Milton proteins and playing a probable role in anterograde mitochondrial transport, and in (3) mitochondrial calcium homeostasis (Esteves et al., 2010b) by direct regulation of calcium efflux from mitochondria through the mitochondrial sodium/calcium exchanger (Deas et al., 2009).
Mitochondrial dysfunction and increased apoptosis were also shown in Drosophila parkin null mutant or in overexpressed parkin mutation models (Greene et al., 2003). Parkin is a protein that may have a role in maintenance of the outer mitochondrial membrane integrity (Darios et al., 2003). These results were supported by evidences that relate parkin function with mitochondrial dynamics (Riparbelli and Callaini, 2007). Additionally, it seems to be involved in mitochondria trafficking since it was shown that parkin may bind and stabilize microtubules (Yang et al., 2005). Narendra et al. (2008) reported that parkin is recruited to dysfunctional mitochondria and induces their autophagy, what means that degradation of impaired mitochondria in PARK2 EOPD is reduced, contributing to neuronal death.
Stronger evidence of mitochondrial involvement in PD provided by genetic studies, the mitochondrial ETC cx I impairment has been shown to have great relation with PD, since 1989, when Schapira and coworkers first discovered cx I deficiency in SN of postmortem PD brain (Schapira et al., 1989). Additional work confirmed similar results (Schapira et al., 1990a,b; Janetzky et al., 1994; Gu et al., 1998). Moreover, some studies reported a decrease of relevant subunits of cx I in PD human brain (Mizuno et al., 1989; Keeney et al., 2006). Interestingly, MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyrine), a neurotoxin capable to induce PD symptoms in humans (Langston et al.,1983), is a mitochondrial cx I inhibitor. MPTP cross the blood brain barrier and is converted to 1-methyl-4-phenylpyridinium (MPP+) within glial cells and then released to the extracellular space. MPP+ is also concentrated into DAergic neurons via DA transporter. In this way, as cx I deficiency was also found in other PD patients tissues, as platelets and lymphoblasts (Barroso et al., 1993; Schapira, 1994). To address the potential causes of complex I defect, namely if it was due to an environmental toxin or to an alteration of mitochondrial or nuclear DNA, the cytoplasmic hybrid (cybrid) technique, first described by King and Attardi (1989), has been applied. In PD cybrids it was observed a decrease in cx I activity, increase in ROS production and a higher susceptibility to MPP+ (Swerdlow et al., 1996; Cassarino et al., 1997; Esteves et al., 2008). These results indicate that the complex I defect in PD appears to be genetic and arising from mtDNA. Foremost, it was reported the generation of fibrillar and vesicular inclusions in a cybrid model of sporadic PD, which replicates the essential antigenic and structural features of LB, without the need for exogenous protein expression or inhibition of mitochondrial or proteasomal function (Trimmer et al., 2004; Esteves et al., 2010a). Supporting this theory, it is rational that mitochondria in vulnerable PD neurons are under greater stress condition that increases the probability of mtDNA mutation (Soong et al., 1992).
Recently, another incontestable piece of evidence of mitochondrial dysfunction in PD has come from conditional knockout mice, termed “MitoPark” mice, the first animal model showing the slow progressive degeneration of dopamine neurons seen in PD. These mice have a disruption of the gene for mitochondrial transcription factor A (Tfam) in dopaminergic neurons. In addition, this mouse model shows reduced mtDNA expression, reduced respiratory chain function in midbrain dopaminergic neurons which, in turn, leads to a parkinsonian phenotype, with adult onset of slowly progressive impairment of motor function associated to the formation of intraneuronal inclusions and dopamine nerve cell death (Ekstrand et al., 2007). These results are consistent with the involvement of respiratory chain dysfunction in PD pathogenesis. Further, there is evidence of reduced mitochondrial mass and size in mouse SN dopaminergic neurons as compared to non-dopaminergic neurons, suggesting selective vulnerability of dopaminergic neurons as a result of a mitochondrial dysfunction (Liang et al., 2007).
Accordingly, a progressive mitochondrial dysfunction process leads to a loss of ATP, decreased calcium buffering capacity and increased oxidative stress that act synergistically to promote DAergic cell death. Although mitochondrial dysfunction is a major factor, UPS impairment may also be associated as a consequence of mitochondrial dysfunction, or as a causative factor.
The Role of the Ubiquitin Proteasome System in PD
Protein degradation by the UPS consists in a tightly regulated process, starting with target-protein tagging with a polyubiquitine chain by ubiquitin ligases E3 in an ATP dependent manner and ending with degradation by the 26S proteasome, which also requires ATP to assemble 19S and 20S subunits (Goldberg, 2003). The proteasome is a large protease complex that eliminates intracellular misfolded, oxidized or aggregated proteins (Ciechanover and Brundin, 2003). There is proteasome loss of function with aging, reflected on proteasome subunits expression, activity and response to oxidative stress (Bulteau et al., 2000; Keller et al. 2000).
In addition to the existence of EOPD forms caused by mutations in genes that codify proteins of UPS pathway, such as parkin and UCHL1, as previously referred, the colocalization of proteasome subunits in LBs (Ii et al., 1997) and also the presence of ubiquitinated proteins in LB may indicate UPS failure in PD, since ubiquitin is the signal protein for degradation by UPS. Beside UPS impairment being evidently related with some EOPD forms, it was also reported proteasomal dysfunction in the SN in LOPD (McNaught and Jenner, 2001). Tanaka et al. (2001) reported that expression of mutant alpha-synuclein significantly reduces chymotryptic-like, tryptic-like and post-acidic activities of the proteasome. Cell lines expressing wild-type alpha-synuclein also showed a reduction in proteasomal activity but there was no significant difference compared with control cell lines. In the presence of lactacystin, a highly selective proteasome inhibitor, mutant cell lines demonstrated an increased sensitivity to alpha-synuclein toxicity with significantly increased cell death (Tanaka et al., 2001).
Additionally to the relation with mitochondria discussed in the previous section, parkin protein is an E3 ubiquitin-protein ligase. Mutations in the gene of parkin, originate lack of enzymatic activity of this enzyme and consequent misfolded proteins accumulation. Some studies revealed that over-expression of parkin, using viral vectors, may be effective against alpha-synucleinopathy (Lo Bianco et al. 2004; Yamada et al., 2005; Yasuda et al., 2007). Thus, parkin over-expression may be a therapeutic strategy for PD, due to increased UPS activity and misfolded proteins clearance, avoiding aggregates-induced cell death.
PINK1 interacts with several chaperons, like Hsp90 and Cdc37, and loss of this interaction can contribute to PD pathogenesis (Moriwaki et al., 2008). PINK1 also interacts with Parkin which is responsible for PINK1 homeostasis by ubiquitination and posterior degradation via UPS (Deas et al., 2009). PINK1 and parkin also form a functional E3 ligase complex with DJ-1, which promotes degradation of misfolded parkin substrates by UPS, including parkin itself and synphilin-1. PD-pathogenic mutations in genes that code these three proteins are likely to impair E3 ligase activity of the complex, which may explain somehow the PD pathogenesis (Xiong et al., 2009).
Mutations of the gene UCHL1 (PARK5) and alteration of coded protein activity have been found to associate with several neurodegenerative disorders. In addition to a deubiquitination function previously referred, in vitro studies reported that UCHL1 has dimerization-dependent ubiquitin ligase activity (Liu et al., 2002).
Furthermore, McNaught et al. (2004) proposed a progressive PD rat model by systemic administration of proteasome inhibitors. After a latency period of 1–2 weeks rats developed the characteristic symptoms of the disease and also showed, in the postmortem analysis, striatal DA depletion and DAergic cell death in SNpc.
Mitochondrial and Proteasomal Cross-Talk in PD Pathogenesis
After discussing the possible involvement of mitochondria dysfunction and proteasome impairment in both familial and sporadic forms of PD, we will discuss the possible molecular mechanisms that underline mitochondrial and proteasome interplay in the disease course (see Figure 3).
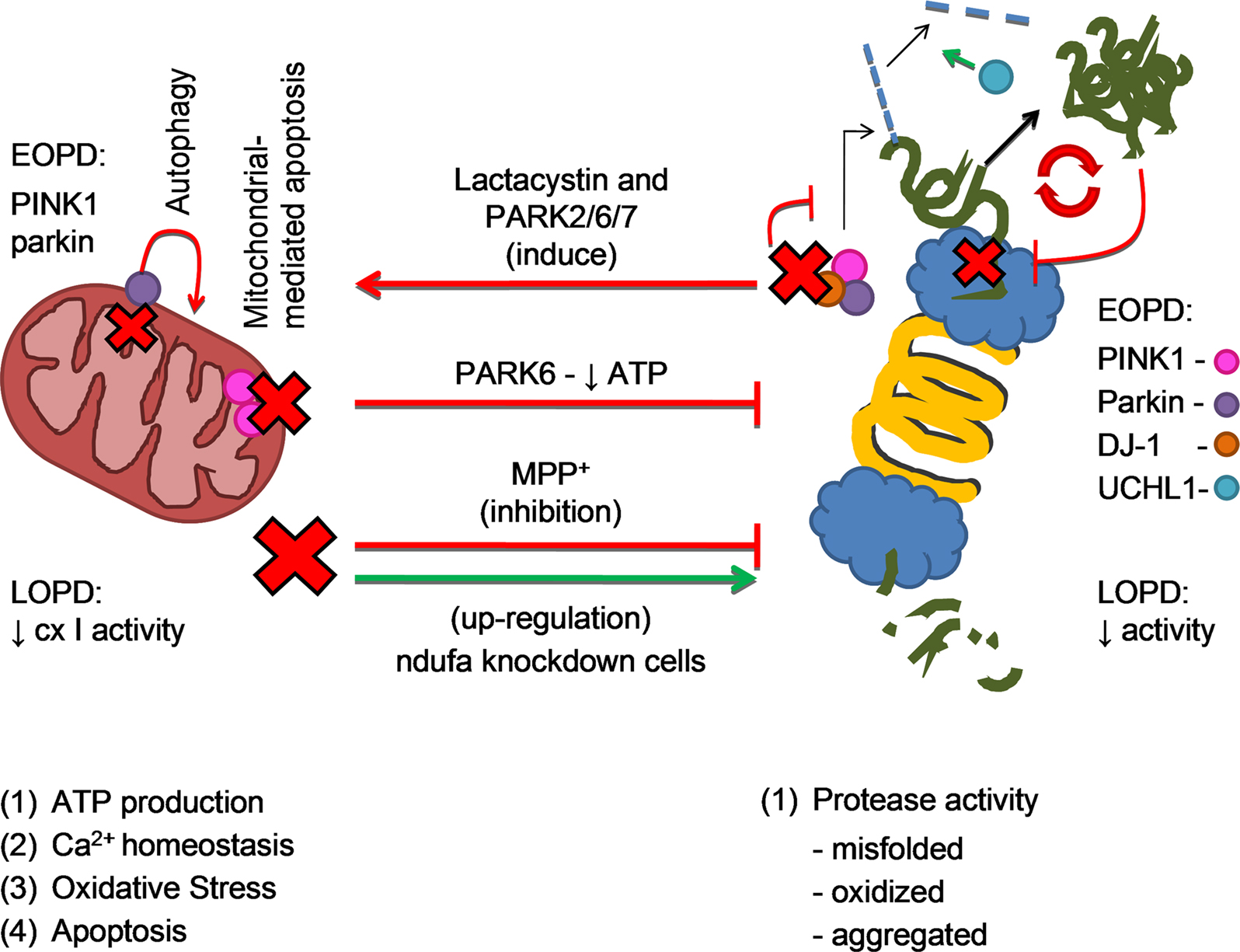
Figure 3. Mitochondrial and proteasomal cross-talk in PD pathogenesis. The four mitochondrial main functions are (1) ATP production (2) regulation of calcium homeostasis (3) oxidative stress and (4) apoptotic cell death mediation. Proteasome is a major protein degradation cell complex. Reduction in mitochondrial cx I activity in LOPD and the acute model of PD (MPP+) inhibit proteasome activity, as it actually happens in LOPD and with aging. Also in PARK6 (PINK1 mutation - EOPD) there is mitochondrial function compromise and therefore proteasome impairment. However, recent and preliminary results shown an up-regulation of proteasomal activity in ndufa knockdown cells. In the opposite way of this two ways crosstalk, lactacystin and/or mutations in genes that code PINK1 (PARK6), parkin (PARK2) and DJ-1 (PARK7) may interfere with the function of the ubiquitin E3 ligase activity complex, inducing alpha-synuclein aggregation and mitochondrial-mediated apoptosis. Accumulation of alpha-synuclein oligomers potentiates proteasome impairment in a vicious cycle. Also described in the figure are parkin function of induce autophagy in dysfunctional mitochondria and UCHL-1 deubiquitinating activity, impaired in PARK2 and PARK5, respectively.
Recent evidence in cell culture systems, where mutant PINK1 or loss of PINK1 was induced, showed a relation between mitochondrial dysfunction and proteasomal impairment (Liu et al., 2009). Liu et al. (2009) demonstrated that proteasomal function is impaired due to a reduction of mitochondrial dependent ATP synthesis. These authors claim that mitochondrial compromise is the primary event followed by proteasome impairment and consequent alpha-synuclein aggregation. Alpha-synuclein aggregates are proved to be able to reduce even more the proteasomal activity, resulting in a vicious cycle.
Moreover, Tanaka et al. (2001) also reported that proteasome inhibition by lactacystin in cells expressing mutant alpha-synuclein increases mitochondria-dependent apoptotic cell death. In other study demonstrating that mitochondria and UPS act synergistically, Shin et al. (2009) showed that only a co-systemic administration of the proteasome inhibitor Z-Ile-Glu(OtBu)-Ala-Leu-aldehyde (PSI) with MPTP in mice, produced a significant reduction in locomotor activity that was correlated with degeneration of the nigrostriatal dopaminergic pathway.
Sun et al. (2009) reported that after proteasome inhibition in dopaminergic neuronal cells, there is a prominent accumulation of polyubiquitinated proteins that are likely to be related with activation of mitochondrial apoptosis and consequent neuronal loss. An increase of polyubiquitinated proteins in mitochondria may be indicative of the potential role of mitochondria as an early key sensor of UPS impairment and accumulation of misfolfed ubiquitinated proteins. In this point of view proteasomal dysfunction seems to appear as the causal disturb, followed by mitochondria participation in the molecular mechanism of the disease.
Moreover, the formation of the ubiquitin E3 ligase activity complex (by PINK1, DJ-1 and parkin) seems to have a neuroprotective role in mitochondria-mediated apoptosis (Darios et al., 2003; Petit et al., 2005; Gorner et al., 2007). Additionally, as referred above, mutation in genes that code these proteins and consequent protein alteration contribute to UPS impairment and at same time mitochondrial dysfunction.
Interestingly, Kikuchi et al. (2003) also demonstrated that proteasomal inhibition in mesencephalic dopaminergic neurons caused mitochondrial dysfunction, reduced glutathione levels, and increased generation of free radicals. Mitochondrial neurotoxins (MPP+, rotenone, 6-OHDA, etc) induce an increase production of ROS and oxidative stress (Sun et al., 2007). This sustained increase could lead to an increase in protein oxidation that overwhelms proteasomal degradative capacity. Moreover, an increase in free radicals production, may induce oxidative modifications of proteasomal subunits that also compromise the proteolytic capacity (for review see Sun et al., 2007). Cheng and coworkers, showed that the antioxidant, puerarin attenuated MPP+-induced proteasomal dysfunction, by reducing the accumulation of ubiquitin-conjugated proteins (Cheng et al., 2009). Thus, oxidative stress may link mitochondrial dysfunction to UPS impairment, whereas the cross-talk between mitochondria and proteasome is likely to be a two ways dead-road inside the cell.
In an effort to clarify the PD pathogenesis we hypothesize that interaction between mitochondrial and proteasome plays a critical role in the DAergic cell death. To support that, some results from our group demonstrated an effective correlation between mitochondrial dysfunction and proteasomal impairment, suggesting that they act synergistically and not only exclusively by themselves.
We reported that MPP+ induced in NT2 human teratocarcinoma a marked increase in ubiquitinylated protein levels, free radicals generation and a decrease in ATP levels. These results indicate that mitochondrial deficits may adversely affect ATP-dependent proteasomal degradation. Accordingly, we also showed a reduction in proteasomal activity (chymotrypsin and post-acidic-like activities) in NT2-MPP+ treated cells after 24 h and in NT2-ρ0 (mitochondrial DNA depleted) cells under basal conditions (Domingues et al., 2008). Our results show that in the absence of a functional mitochondria, due to mitochondrial DNA knockdown, the UPS is down-regulated. In this way, mitochondrial alteration leads to an imbalance in the cellular oxidative status, inducing a proteasomal deregulation, which may exacerbate protein aggregation and consequently degenerative events. Our recent results corroborate the notion that a mitochondrial dysfunction induces an oligomerization of alpha-synuclein (Esteves et al., 2010a). Moreover, it seems that proteasomal dysfunction may potentiate alpha-synuclein oligomerization, which in turn can have an inhibitory effect on the proteasomal activities. Thus, UPS requires a normal mitochondrial activity to work correctly. Interestingly, recent and preliminary results shown an up-regulation of proteasomal activity in ndufa cxI subunit knockdown cells (with a decrease in cxI activity) (Martins Branco et al., unpublished results). These data may indicate an attempt to eliminate alpha-synuclein soluble oligomeric forms. The evident discrepancy between NT2-MPP+ and ndufa knockdown cells may reflect that MPP+ treated cells are an acute toxic model while ndufa knockdown cells represent a chronic mitochondrial dysfunction model whereas an UPS up-regulation reflects a cell rescue phenomenon. Recently, Endo and coworkers showed that a dysfunction of the mitochondrial import system without ATP alterations may up-regulate proteasome activity (Endo et al., 2009). Understand better mitochondrial and UPS interplay may open a new window to PD therapeutics.
Conflict of Interest Statement
The authors declare that this review was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Diogo Martins Branco was supported by Fundação para a Ciência e a Tecnologia, Portugal (BII grant:) and GAPI of Faculdade de Medicina da Universidade de Coimbra, Portugal; This work was supported by PTDC/SAU-NEU/102710/2008, FCT.
References
Abeliovich, A., Schmitz, Y., Fariñas, I., Choi-Lundberg, D., Ho, W. H., Castillo, P. E., Shinsky, N., Verdugo, J. M., Armanini, M., Ryan, A., Hynes, M., Phillips, H., Sulzer, D., and Rosenthal, A. (2000). Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252.
Alim, M. A., Hossain, M. S., Arima, K., Takeda, K., Izumiyama, Y., Nakamura, M., Kaji, H., Shinoda, T., Hisanaga, S., and Ueda, K. (2002). Tubulin seeds alpha-synuclein fibril formation. J. Biol. Chem. 277, 2112–2117.
Alim, M. A., Ma, Q. L., Takeda, K., Aizawa, T., Matsubara, M., Nakamura, M., Asada, A., Saito, T., Kaji, H., Yoshii, M., Hisanaga, S., and Ueda, K. (2004). Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J. Alzheimers Dis. 6, 435–442; discussion 443–439.
Arduino, D. M., Esteves, A. R., Oliveira, C. R., and Cardoso, S. M. (2010). Mitochondrial metabolism modulation: a new therapeutic approach for Parkinson’s disease. CNS Neurol. Disord. Drug Targets 9, 105–119.
Barroso, N., Campos, Y., Huertas, R., Esteban, J., Molina, J. A., Alonso, A., Gutierrez-Rivas, E., and Arenas, J. (1993). Respiratory chain enzyme activities in lymphocytes from untreated patients with Parkinson disease. Clin. Chem. 39, 667–669.
Beckman, J. S., Beckman, T. W., Chen, J., Marshall, P. A., and Freeman, B. A. (1990). Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 87, 1620–1624.
Bonifati, V., Rizzu, P., van Baren, M. J., Schaap, O., Breedveld, G. J., Krieger, E., Dekker, M. C., Squitieri, F., Ibanez, P., Joosse, M., van Dongen, J. W., Vanacore, N., van Swieten, J. C., Brice, A., Meco, G., van Duijn, C. M., Oostra, B. A., and Heutink, P. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259.
Bredt, D. S., Hwang, P. M., Glatt, C. E., Lowenstein, C., Reed, R. R., and Snyder, S. H. (1991). Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature 351, 714–718.
Bulteau, A. L., Petropoulos, I., and Friguet, B. (2000). Age-related alterations of proteasome structure and function in aging epidermis. Exp. Gerontol. 35, 767–777.
Canet-Aviles, R. M., Wilson, M. A., Miller, D. W., Ahmad, R., McLendon, C., Bandyopadhyay, S., Baptista, M. J., Ringe, D., Petsko, G. A., and Cookson, M. R. (2004). The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. U.S.A. 101, 9103–9108.
Cardoso, S. M., Esteves, A. R., Arduíno, D. M., Domingues, A. F., and Oliveira, C. R. (2009). The crucial role of mitochondria in Parkinson’s disease. Recent Res. Devel. Neurosci. 3, 43–84.
Cassarino, D. S., Fall, C. P., Swerdlow, R. H., Smith, T. S., Halvorsen, E. M., Miller, S. W., Parks, J. P., Parker, W. D. Jr., and Bennett, J. P. Jr. (1997). Elevated reactive oxygen species and antioxidant enzyme activities in animal and cellular models of Parkinson’s disease. Biochim. Biophys. Acta 1362, 77–86.
Chen, L., Thiruchelvam, M. J., Madura, K., and Richfield, E. K. (2006). Proteasome dysfunction in aged human alpha-synuclein transgenic mice. Neurobiol. Dis. 23, 120–126.
Cheng, Y. F., Zhu, G. Q., Wang, M., Cheng, H., Zhou, A., Wang, N., Fang, N., Wang, X. C., Xiao, X. Q., Chen, Z. W., and Li, Q. L. (2009). Involvement of ubiquitin proteasome system in protective mechanisms of Puerarin to MPP+-elicited apoptosis. Neurosci. Res. 63, 52–58.
Ciechanover, A., and Brundin, P. (2003). The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40, 427–446.
Clark, I. E., Dodson, M. W., Jiang, C., Cao, J. H., Huh, J. R., Seol, J. H., Yoo, S. J., Hay, B. A., and Guo, M. (2006). Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166.
Cooper, A. A., Gitler, A. D., Cashikar, A., Haynes, C. M., Hill, K. J., Bhullar, B., Liu, K., Xu, K., Strathearn, K. E., Liu, F., Cao, S., Caldwell, K. A., Caldwell, G. A., Marsischky, G., Kolodner, R. D., Labaer, J., Rochet, J. C., Bonini, N. M., and Lindquist, S. (2006). Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313, 324–328.
Darios, F., Corti, O., Lucking, C. B., Hampe, C., Muriel, M. P., Abbas, N., Gu, W. J., Hirsch, E. C., Rooney, T., Ruberg, M., and Brice, A. (2003). Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum. Mol. Genet. 12, 517–526.
Dauer, W., Kholodilov, N., Vila, M., Trillat, A. C., Goodchild, R., Larsen, K. E., Staal, R., Tieu, K., Schmitz, Y., Yuan, C. A., Rocha, M., Jackson-Lewis, V., Hersch, S., Sulzer, D., Przedborski, S., Burke, R., and Hen, R. (2002). Resistance of alphasynuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. U.S.A. 99, 14524–14529.
de Lau, L. M., and Breteler, M. M. (2006). Epidemiology of Parkinson’s disease. Lancet Neurol. 5, 525–535.
Deas, E., Plun-Favreau, H., and Wood, N. W. (2009). PINK1 function in health and disease. EMBO Mol. Med. 1, 152–165.
Domingues, A. F., Arduíno, D. M., Esteves, A. R., Swerdlow, R. H., Oliveira, C. R., and Cardoso, S. M. (2008). Mitochondria and ubiquitin-proteasomal system interplay: relevance to Parkinson’s disease. Free Radic. Biol. Med. 45, 820–825.
Ekstrand, M. I., Terzioglu, M., Galter, D., Zhu, S., Hofstetter, C., Lindqvist, E., Thams, S., Bergstrand, A., Hansson, F. S., Trifunovic, A., Hoffer, B., Cullheim, S., Mohammed, A. H., Olson, L., and Larsson, N. G. (2007). Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. U.S.A. 104, 1325–1330.
Endo, R., Saito, T., Asada, A., Kawahara, H., Ohshima, T., and Hisanaga, S. (2009). Commitment of 1-methyl-4-phenylpyrinidinium ion-induced neuronal cell death by proteasome-mediated degradation of p35 cyclin-dependent kinase 5 activator. J. Biol. Chem. 284, 26029–26039.
Esteves, A. R., Arduino, D. M., Swerdlow, R. H., Oliveira, C. R., and Cardoso, S. M. (2009). Oxidative stress involvement in alpha-synuclein oligomerization in Parkinsons disease cybrids. Antioxid. Redox Signal. 11, 439–448.
Esteves, A., Arduino, D. M., Swerdlow, R. H., Oliveira, C. R., and Cardoso, S. M. (2010a). Microtubule depolymerization potentiates alpha-synuclein oligomerization. Front. Ag. Neurosci. 1:5. doi:10.3389/neuro.24.005.2009.
Esteves, A. R., Arduíno, D. M., Swerdlow, R. H., Oliveira, C. R., and Cardoso, S. M. (2010b). Dysfunctional mitochondria uphold calpain activation: contribution to Parkinson’s disease pathology. Neurobiol. Dis. 37, 723–730.
Esteves, A. R., Domingues, A. F., Ferreira, I. L., Januario, C., Swerdlow, R. H., Oliveira, C. R., and Cardoso, S. M. (2008). Mitochondrial function in Parkinson’s disease cybrids containing an nt2 neuron-like nuclear background. Mitochondrion 8, 219–228.
Forno, L. S. (1996). Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 55, 259–272.
Fortin, D. L., Troyer, M. D., Nakamura, K., Kubo, S. I., Anthony, M. D., and Edwards, R. H. (2004). Lipid rafts mediate the synaptic localization of alpha-synuclein. J. Neurosci. 24, 6715–6723.
Funayama, M., Hasegawa, K., Kowa, H., Saito, M., Tsuji, S., and Obata, F. (2002). A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann. Neurol. 51, 296–301.
Gallagher, D. A., and Schapira, A. H. (2009). Etiopathogenesis and treatment of Parkinson’s disease. Curr. Top Med. Chem. 9, 860–868.
Gandhi, S., Muqit, M. M., Stanyer, L., Healy, D. G., Abou-Sleiman, P. M., Hargreaves, I., Heales, S., Ganguly, M., Parsons, L., Lees, A. J., Latchman, D. S., Holton, J. L., Wood, N. W., and Revesz, T. (2006). PINK1 protein in normal human brain and Parkinson’s disease. Brain 129, 1720–1731.
Garthwaite, J., Charles, S. L., and Chess-Williams, R. (1988). Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 336, 385–388.
Gautier, C. A., Kitada, T., and Shen, J. (2008). Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 105, 11364–11369.
Gitler, A. D., Chesi, A., Geddie, M. L., Strathearn, K. E., Hamamichi, S., Hill, K. J., Caldwell, K. A., Caldwell, G. A., Cooper, A. A., Rochet, J. C., and Lindquist, S. (2009). Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 41, 308–315.
Goldberg, A. L. (2003). Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899.
Gorner, K., Holtorf, E., Waak, J., Pham, T. T., Vogt-Weisenhorn, D. M., Wurst, W., Haass, C., and Kahle, P. J. (2007). Structural determinants of the C-terminal helix-kink-helix motif essential for protein stability and survival promoting activity of DJ-1. J. Biol. Chem. 282, 13680–13691.
Greene, J. C., Whitworth, A. J., Kuo, I., Andrews, L. A., Feany, M. B., and Pallanck, L. J. (2003). Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. U.S.A. 100, 4078–4083.
Gu, M., Cooper, J. M., Taanman, J. W., and Schapira, A. H. (1998). Mitochondrial DNA transmission of the mitochondrial defect in Parkinson’s disease. Ann. Neurol. 44, 177–186.
Guo, J. T., Chen, A. Q., Kong, Q., Zhu, H., Ma, C. M., and Qin, C. (2008). Inhibition of vesicular monoamine transporter-2 activity in alphasynuclein stably transfected SH-SY5Y cells. Cell. Mol. Neurobiol. 28, 35–47.
Hatano, T., Kubo, S., Sato, S., and Hattori, N. (2009). Pathogenesis of familial Parkinson’s disease: new insights based on monogenic forms of Parkinson’s disease. J. Neurochem. 111, 1075–1093.
Hatano, Y., Li, Y., Sato, K., Asakawa, S., Yamamura, Y., Tomiyama, H., Yoshino, H., Asahina, M., Kobayashi, S., Hassin-Baer, S., Lu, C. S., Ng, A. R., Rosales, R. L., Shimizu, N., Toda, T., Mizuno, Y., and Hattori, N. (2004). Novel PINK1 mutations in earlyonset parkinsonism. Ann. Neurol. 56, 424–427.
Hettiarachchi, N. T., Parker, A., Dallas, M. L., Pennington, K., Hung, C. C., Pearson, H. A., Boyle, J. P., Robinson, P., and Peers, C. (2009). alpha-Synuclein modulation of Ca2+ signaling in human neuroblastoma (SH-SY5Y) cells. J. Neurochem. 111, 1192–1201.
Ii, K., Ito, H., Tanaka, K., and Hirano, A. (1997). Immunocytochemical co-localization of the proteasome in ubiquitinated structures in neurodegenerative diseases and the elderly. J. Neuropathol. Exp. Neurol. 56, 125–131.
Janetzky, B., Hauck, S., Youdim, M. B., Riederer, P., Jellinger, K., Pantucek, F., Zochling, R., Boissl, K. W., and Reichmann, H. (1994). Unaltered aconitase activity, but decreased complex I activity in substantia nigra pars compacta of patients with Parkinson’s disease. Neurosci. Lett. 169, 126–128.
Jellinger, K. A. (1999). post mortem studies in Parkinson’s disease-is it possible to detect brain areas for specific symptoms? J. Neural Transm. 56, 1–29.
Keeney, P. M., Xie, J., Capaldi, R. A., and Bennett, J. P. Jr. (2006). Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 26, 5256-5264.
Keller, J. N., Hanni, K. B., and Markesbery, W. R. (2000). Possible involvement of proteasome inhibition in aging: implications for oxidative stress. Mech. Ageing Dev. 113, 61–70.
Kikuchi, S., Shinpo, K., Tsuji, S., Takeuchi, M., and Yamagishi, S., Makita, Z., Niino, M., Yabe, I., Tashiro, K. (2003). Effect of proteasome inhibitor on cultured mesencephalic dopaminergic neurons. Brain Res. 964, 228–236.
Kim, M., Jung, W., Lee, I. H., Bhak, G., Paik, S. R., and Hahn, J. S. (2008). Impairment of microtubule system increases alpha-synuclein aggregation and toxicity. Biochem. Biophys. Res. Commun. 365, 628–635.
King, M. P., and Attardi, G. (1989). Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246, 500–503.
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., Yokochi, M., Mizuno, Y., and Shimizu, N. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608.
Kubo, S. I., Nemani, V. M., Chalkley, R. J., Anthony, M. D., Hattori, N., Mizuno, Y., Edwards, R. H., and Fortin, D. L. (2005). A combinatorial code for the interaction of alpha-synuclein with membranes. J. Biol. Chem. 280, 31664–31672.
Langston, J. W., Ballard, P., Tetrud, J. W., and Irwin, I. (1983). Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980.
Larsen, C. N., Krantz, B. A., and Wilkinson, K. D. (1998). Substrate specificity of deubiquitinating enzymes: ubiquitin C-terminal hydrolases. Biochemistry 37, 3358–3368.
Larsen, C. N., Price, J. S., and Wilkinson, K. D. (1996). Substrate binding and catalysis by ubiquitin C-terminal hydrolases: identification of two active site residues. Biochemistry 35, 6735–6744.
Larsen, K. E., Schmitz, Y., Troyer, M. D., Mosharov, E., Dietrich, P., Quazi, A. Z., Savalle, M., Nemani, V., Chaudhry, F. A., Edwards, R. H., Stefanis, L., and Sulzer, D. (2006). Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci. 26, 11915–11922.
Lee, H. J., Khoshaghideh, F., Lee, S., and Lee, S. J. (2006). Impairment of microtubule-dependent trafficking by overexpression of alpha-synuclein. Eur. J. Neurosci. 24, 3153–3162.
Liang, C. L., Wang, T. T., Luby-Phelps, K., and German, D. C. (2007). Mitochondria mass is low in mouse substantia nigra dopamine neurons: implications for Parkinson’s disease. Exp. Neurol. 203, 370–380.
Lipton, S. A., Choi, Y. B., Pan, Z. H., Lei, S. Z., Chen, H. S., Sucher, N. J., Loscalzo, J., Singel, D. J., and Stamler, J. S. (1993). A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 364, 626– 632.
Liu, W., Vives-Bauza, C., Acín-Peréz-, R., Yamamoto, A., Tan, Y., Li, Y., Magrané, J., Stavarache, M. A., Shaffer, S., Chang, S., Kaplitt, M. G., Huang, X. Y., Beal, M. F., Manfredi, G., and Li, C. (2009). PINK1 defect causes mitochondrial dysfunction, proteasomal deficit and a-synuclein aggregation in cell culture models of Parkinson’s disease. PLoS ONE 4, e4597. doi: 10.1371/journal.pone.0004597.
Liu, Y., Fallon, L., Lashuel, H. A., Liu, Z., and Lansbury, P. T. Jr. (2002). The UCH-L1 gene encodes two opposing enzymatic activities that affect alphasynuclein degradation and Parkinson’s disease susceptibility. Cell 111, 209–218.
Lo Bianco, C., Schneider, B. L., Bauer, M., Sajadi, A., Brice, A., Iwatsubo, T., and Aebischer, P. (2004). Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 17510–17515.
Lowe, J., McDermott, H., Landon, M., Mayer, R. J., and Wilkinson, K. D. (1990). Ubiquitin carboxyl-terminal hydrolase (PGP 9.5) is selectively present in ubiquitinated inclusion bodies characteristic of human neurodegenerative diseases. J. Pathol. 161, 153–160.
Maroteaux, L., Campanelli, J. T., and Scheller, R. H. (1988). Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8, 2804–2815.
Martin, L. J., Pan, Y., Price, A. C., Sterling, W., Copeland, N. G., Jenkins, N. A., Price, D. L., and Lee, M. K. (2006). Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 26, 41–50.
McNaught, K. S., and Jenner, P. (2001). Proteasomal function is impaired in Substantia nigra in Parkinson’s disease. Neurosci. Lett. 297, 191–194.
McNaught, K. S., Perl, D. P., Brownell, A. L., and Olanow, C. W. (2004). Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann. Neurol. 56, 149-162.
Mills, R. D., Sim, C. H., Mok, S. S., Mulhern, T. D., Culvenor, J. G., and Cheng, H. C. (2008). Biochemical aspects of the neuroprotective mechanism of PTEN-induced kinase-1 (PINK1). J. Neurochem. 105, 18–33.
Mizuno, Y., Hattori, N., Kubo, S., Sato, S., Nishioka, K., Hatano, T., Tomiyama, H., Funayama, M., Machida, Y., and Mochizuki, H. (2008). Progress in the pathogenesis and genetics of Parkinson’s disease. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 363, 2215–2227.
Mizuno, Y., Ohta, S., Tanaka, M., Takamiya, S., Suzuki, K., Sato, T., Oya, H., Ozawa, T., and Kagawa, Y. (1989). Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem. Biophys. Res. Commun. 163, 1450–1455.
Moore, D. J., West, A. B., Dawson, V. L., and Dawson, T. M. (2005). Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 28, 57–87.
Moriwaki, Y., Kim, Y. J., Ido, Y., Misawa, H., Kawashima, K., Endo, S., and Takahashi, R. (2008). L347P PINK1 mutant that fails to bind to Hsp90/Cdc37 chaperones is rapidly degraded in a proteasome-dependent manner. Neurosci. Res. 61, 43–48.
Murphy, D. D., Rueter, S. M., Trojanowski, J. Q., and Lee, V. M. (2000). Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 20, 3214–3220.
Nakamura, T., and Lipton, S. A. (2009). Cell death: protein misfolding and neurodegenerative diseases. Apoptosis 14, 455–468.
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803.
Nieto, M., Gil-Bea, F. J., Dalfó, E., Cuadrado, M., Cabodevilla, F., Sánchez, B., Catena, S., Sesma, T., Ribé, E., Ferrer, I., Ramírez, M. J., and Gómez-Isla, T. (2006). Increased sensitivitiy to MPTP in human alpha-synuclein A30P transgenic mice. Neurobiol. Aging 27, 848–856.
Outeiro, T. F., Putcha, P., Tetzlaff, J. E., Spoelgen, R., Koker, M., Carvalho, F., Hyman, B. T., and McLean, P. J. (2008). Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS ONE 3, e1867. doi: 10.1371/journal.pone.0001867.
Palacino, J. J., Sagi, D., Goldberg, M. S., Krauss, S., Motz, C., Wacker, M., Klose, J., and Shen, J. (2004). Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 279, 18614–18622.
Park, J., Yoo, C. I., Sim, C. S., Kim, J. W., Yi, Y., Shin, Y. C., Kim, D. H., and Kim, Y. (2006). A retrospective cohort study of Parkinson’s disease in Korean shipbuilders. Neurotoxicology 27, 445–449.
Perez, R. G., Waymire, J. C., Lin, E., Liu, J. J., Guo, F., and Zigmond, M. J. (2002). A role for alpha-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 22, 3090–3099.
Petit, A., Kawarai, T., Paitel, E., Sanjo, N., Maj, M., Scheid, M., Chen, F., Gu, Y., Hasegawa, H., Salehi-Rad, S., Wang, L., Rogaeva, E., Fraser, P., Robinson, B., St George-Hyslop, P., and Tandon, A. (2005). Wild-type PINK1 prevents basal and induced neuronal apoptosis, a protective effect abrogated by Parkinson disease-related mutations. J. Biol. Chem. 280, 34025–34032.
Piccoli, C., Sardanelli, A., Scrima, R., Ripoli, M., Quarato, G., D’Aprile, A., Bellomo, F., Scacco, S., De Michele, G., Filla, A., Iuso, A., Boffoli, D., Capitanio, N., and Papa, S. (2008). Mitochondrial respiratory dysfunction in familiar parkinsonism associated with PINK1 mutation. Neurochem. Res. 33, 2565–2574.
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., Pike, B., Root, H., Rubenstein, J., Boyer, R., Stenroos, E. S., Chandrasekharappa, S., Athanassiadou, A., Papapetropoulos, T., Johnson, W. G., Lazzarini, A. M., Duvoisin, R. C., Di Iorio, G., Golbe, L. I., and Nussbaum, R. L. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047.
Pridgeon, J. W., Olzmann, J. A., Chin, L. S., and Li, L. (2007). PINK1 Protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 5, e172. doi: 10.1371/journal.pbio.0050172.
Ramirez, A., Heimbach, A., Gründemann, J., Stiller, B., Hampshire, D., Cid, L. P., Goebel, I., Mubaidin, A. F., Wriekat, A. L., Roeper, J., Al-Din, A., Hillmer, A. M., Karsak, M., Liss, B., Woods, C. G., Behrens, M. I., and Kubisch, C. (2006). Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38, 1184–1191.
Rideout, H. J., Larsen, K. E., Sulzer, D., and Stefanis, L. (2001). Proteasomal inhibition leads to formation of ubiquitin/alpha-synuclein-immunoreactive inclusions in PC12 cells. J. Neurochem. 78, 899–908.
Riparbelli, M. G., and Callaini, G. (2007). The Drosophila parkin homologue is required for normal mitochondrial dynamics during spermiogenesis. Dev. Biol. 303, 108–120.
Satake, W., Nakabayashi, Y., Mizuta, I., Hirota, Y., Ito, C., Kubo, M., Kawaguchi, T., Tsunoda, T., Watanabe, M., Takeda, A., Tomiyama, H., Nakashima, K., Hasegawa, K., Obata, F., Yoshikawa, T., Kawakami, H., Sakoda, S., Yamamoto, M., Hattori, N., Murata, M., Nakamura, Y., and Toda, T. (2009). Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 41,1303–1307.
Schapira, A. H. (1994). Evidence for mitochondrial dysfunction in Parkinson’s disease--a critical appraisal. Mov. Disord. 9, 125–138.
Schapira, A. H., Cooper, J. M., Dexter, D., Clark, J. B., Jenner, P., and Marsden, C. D. (1990a). Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 54, 823–827.
Schapira, A. H., Mann, V. M., Cooper, J. M., Dexter, D., Daniel, S. E., Jenner, P., Clark, J. B., and Marsden, C. D. (1990b). Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson’s disease. J. Neurochem. 55, 2142–2145.
Schapira, A. H., Cooper, J. M., Dexter, D., Jenner, P., Clark, J. B., and Marsden, C. D. (1989). Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1, 1269.
Schlossmacher, M. G., Frosch, M. P., Gai, W. P., Medina, M., Sharma, N., Forno, L., Ochiishi, T., Shimura, H., Sharon, R., Hattori, N., Langston, J. W., Mizuno, Y., Hyman, B. T., Selkoe, D. J., and Kosik, K. S. (2002). Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am. J. Pathol. 160, 1655–1667.
Shimura, H., Hattori, N., Kubo, S., Mizuno, Y., Asakawa, S., Minoshima, S., Shimizu, N., Iwai, K., Chiba, T., Tanaka, K., and Suzuki, T. (2000). Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305.
Shimura, H., Hattori, N., Kubo, S., Yoshikawa, M., Kitada, T., Matsumine, H., Asakawa, S., Minoshima, S., Yamamura, Y., Shimizu, N., and Mizuno, Y. (1999). Immunohistochemical and subcellular localization of Parkin protein: absence of protein in autosomal recessive juvenile parkinsonism patients. Ann. Neurol. 45, 668–672.
Shin, M., Jan, C., Jacquard, C., Jaraya, B., Callebert, J., Launay, J. M., Hantraye, P., Remy, P., Palfi, S., and Brouillet, E. (2009). Chronic systemic treatment with a high-dose proteasome inhibitor in mice produces akinesia unrelated to nigrostriatal degeneration. Neurobiol. Aging. [Epub ahead of print].
Soong, N. W., Hinton, D. R., Cortopassi, G., and Arnheim, N. (1992). Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat. Genet. 2, 318–323.
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840.
Sun, F., Kanthasamy, A., Anantharam, V., and Kanthasamy, A. G. (2007). Environmental neurotoxic chemicals-induced ubiquitin proteasome system dysfunction in the pathogenesis and progression of Parkinson’s disease. Pharmacol. Ther. 114, 327–344.
Sun, F., Kanthasamy, A., Anantharam, V., and Kanthasamy, A. G. (2009). Mitochondrial accumulation of polyubiquitinated proteins and differential regulation of apoptosis by polyubiquitination sites Lys-48 and -63. J. Cell. Mol. Med. 13, 1632–1643.
Swerdlow, R. H., Parks, J. K., Miller, S. W., Tuttle, J. B., Trimmer, P. A., Sheehan, J. P., Bennett, J. P. Jr., Davis, R. E., and Parker, W. D. Jr. (1996). Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann. Neurol. 40, 663–671.
Tanaka, Y., Engelender, S., Igarashi, S., Rao, R. K., Wanner, T., Tanzi, R. E., Sawa, A., L Dawson, V., Dawson, T. M., and Ross, C. A. (2001). Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum. Mol. Genet. 10, 919–926.
Trimmer, P. A., Borland, M. K., Keeney, P. M., Bennett, J. P. Jr., and Parker, W. D. Jr. (2004). Parkinson’s disease transgenic mitochondrial cybrids generate Lewy inclusion bodies. J. Neurochem. 88, 800–812.
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., Ali, Z., Del Turco, D., Bentivoglio, A. R., Healy, D. G., Albanese, A., Nussbaum, R., González-Maldonado, R., Deller, T., Salvi, S., Cortelli, P., Gilks, W. P., Latchman, D. S., Harvey, R. J., Dallapiccola, B., Auburger, G., and Wood, N. W. (2004a). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160.
Valente, E. M., Salvi, S., Ialongo, T., Marongiu, R., Elia, A. E., Caputo, V., Romito, L., Albanese, A., Dallapiccola, B., and Bentivoglio, A. R. (2004b). PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann. Neurol. 56, 336–341.
West, A. B., Moore, D. J., Biskup, S., Bugayenko, A., Smith, W. W., Ross, C. A., Dawson, V. L., and Dawson, T. M. (2005). Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. U.S.A. 102, 16842–16847.
Wichmann, T., and De Long, M. R. (2003). Functional neuroanatomy of the basal ganglia in Parkinson’s disease. Adv. Neurol. 91, 9–18.
Wilkinson, K. D., Lee, K. M., Deshpande, S., Duerksen-Hughes, P., Boss, J. M., and Pohl, J. (1989). The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science 246, 670–673.
Xiong, H., Wang, D., Chen, L., Choo, Y. S., Ma, H., Tang, C., Xia, K., Jiang, W., Ronai, Z., Zhuang, X., and Zhang, Z. (2009). Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J. Clin. Invest. 119, 650–660.
Yamada, M., Mizuno, Y., and Mochizuki, H. (2005). Parkin gene therapy for alphasynucleinopathy: a rat model of Parkinson’s disease. Hum. Gene. Ther. 16, 262–270.
Yang, Y., Gehrke, S., Haque, M. E., Imai, Y., Kosek, J., Yang, L., Beal, M. F., Nishimura, I., Wakamatsu, K., Ito, S., Takahashi, R., and Lu, B. (2005). Inactivation of Drosophila DJ-1 leads to impairments of oxidative stress response and phosphatidylinositol 3-kinase/Akt signaling. Proc. Natl. Acad. Sci. U.S.A. 102, 13670–13675.
Yasuda, T., Miyachi, S., Kitagawa, R., Wada, K., Nihira, T., Ren, Y. R., Hirai, Y., Ageyama, N., Terao, K., Shimada, T., Takada, M., Mizuno, Y., and Mochizuki, H. (2007). Neuronal specificity of alpha-synuclein toxicity and effect of Parkin co-expression in primates. Neuroscience 144, 743–753.
Yu, W. H., Matsuoka, Y., Sziraki, I., Hashim, A., Lafrancois, J., Sershen, H., and Duff, K. E. (2008). Increased dopaminergic neuron sensitivity to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in transgenic mice expressing mutant A53T alpha-synuclein. Neurochem. Res. 33, 902–911.
Zhang, N. Y., Tang, Z., and Liu, C. W. (2008). alpha-Synuclein protofibrils inhibit 26 S proteasome-mediated protein degradation: understanding the cytotoxicity of protein protofibrils in neurodegenerative disease pathogenesis. J. Biol. Chem. 18283, 20288–20298.
Zhou, R. M., Huang, Y. X., Li, X. L., Chen, C., Shi, Q., Wang, G. R., Tian, C., Wang, Z. Y., Jing, Y. Y., Gao, C., and Dong, X. P. (2009). Molecular interaction of alpha-synuclein with tubulin influences on the polymerization of microtubule in vitro and structure of microtubule in cells. Mol. Biol. Rep. [Epub ahead of print].
Keywords: Parkinson’s disease, dopamine, alpha-synuclein, mitochondria, proteasome
Citation: Branco DM, Arduino DM, Esteves AR, Silva DFF, Cardoso SM and Oliveira C (2010) Cross-talk between mitochondria and proteasome in Parkinson’s disease pathogenesis. Front. Ag. Neurosci. 2,17:1-10. doi: 10.3389/fnagi.2010.00017
Received: 31 December 2009;
Paper pending published: 18 January 2010;
Accepted: 12 April 2010;
Published online: 21 May 2010.
Edited by:
Paula I. Moreira, University of Coimbra, PortugalReviewed by:
Tiago F. Outeiro, University of Lisbon, PortugalAvi Friedlich, Harvard Medical School, USA
Copyright: © 2010 Branco, Arduino, Esteves, Silva, Cardoso and Oliveira. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Catarina Resende Oliveira, Center for Neuroscience and Cell Biology of Coimbra, University of Coimbra, 3004 Coimbra, Portugal. e-mail: catarina.n.oliveira@gmail.com