Interaction between therapeutic interventions for Alzheimer’s disease and physiological Aβ clearance mechanisms
 Christopher D. Morrone
Christopher D. Morrone Mingzhe Liu
Mingzhe Liu Sandra E. Black
Sandra E. Black JoAnne McLaurin
JoAnne McLaurin- 1Biological Sciences, Sunnybrook Research Institute, Toronto, ON, Canada
- 2Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, ON, Canada
- 3Canadian Partnership for Stroke Recovery, Sunnybrook Research Institute, Toronto, ON, Canada
- 4Department of Medicine (Neurology), University of Toronto, Toronto, ON, Canada
- 5Hurvitz Brain Sciences Research Program, Sunnybrook Research Institute, Sunnybrook Health Sciences Centre and University of Toronto, Toronto, ON, Canada
Most therapeutic agents are designed to target a molecule or pathway without consideration of the mechanisms involved in the physiological turnover or removal of that target. In light of this and in particular for Alzheimer’s disease, a number of therapeutic interventions are presently being developed/investigated which target the amyloid-β peptide (Aβ). However, the literature has not adequately considered which Aβ physiological clearance pathways are necessary and sufficient for the effective action of these therapeutics. In this review, we evaluate the therapeutic strategies targeting Aβ presently in clinical development, discuss the possible interaction of these treatments with pathways that under normal physiological conditions are responsible for the turnover of Aβ and highlight possible caveats. We consider immunization strategies primarily reliant on a peripheral sink mechanism of action, small molecules that are reliant on entry into the CNS and thus degradation pathways within the brain, as well as lifestyle interventions that affect vascular, parenchymal and peripheral degradation pathways. We propose that effective development of Alzheimer’s disease therapeutic strategies targeting Aβ peptide will require consideration of the age- and disease-specific changes to endogenous Aβ clearance mechanisms in order to elicit maximal efficacy.
Introduction
Alzheimer’s disease (AD) is characterized in part by the accumulation of the amyloid-beta peptide (Aβ) within the brain parenchyma leading to cellular injury and ultimately death, as well as along blood vessels resulting in vascular dysfunction (Querfurth and LaFerla, 2010). It is suggested that the imbalance between Aβ production and clearance in aging drives Alzheimer’s disease progression in late onset Alzheimer’s disease (Hardy and Selkoe, 2002). In light of this, many clinical trials have been initiated over the last 20 years that have targeted removal or inhibition of Aβ production with very limited to no success.1 The failure of these trials has been attributed to targeting Aβ too late in the disease process, after the now recognized prodromal phase of Aβ accumulation in the absence of clinical symptoms (Sperling et al., 2011). Although these treatment strategies show high success rates in rodent models with amyloid precursor protein (APP) overexpression, a major confound is the lack of physiological deficits from aging and long term amyloid burden in these models, which are present in AD patients. Subsequently, endogenous Aβ degradation is diminished more prominently in AD than in disease models, a fact which is often overlooked in the development of treatments. This is a major contributing factor to the lack of clinical efficacy of Aβ specific therapies (Tanzi et al., 2004). We present three classes of AD treatments, and discuss how they rely on, and interact with, three physiological pathways of Aβ clearance. Firstly, we examine Aβ immunization strategies, and their dependence on the peripheral sink. Then we discuss the effectiveness of small molecule therapies targeting APP cleavage in the context of reduced Aβ degradation in the brain parenchyma. Finally, we present an array of lifestyle interventions in the clinic for AD, and discuss how these preserve vascular health, and how this may ultimately enhance interstitial fluid (ISF) and soluble Aβ drainage mechanisms. We survey therapeutic strategies that are presently in clinical development and describe the Aβ degradation pathways that are necessary for drug efficacy while highlighting potential disease-specific confounds that may contribute to previously failed trials.
Immunization Strategies
Currently, there are both passive and active immunization strategies being developed for Alzheimer’s disease. These antibodies target various components of the AD pathology, such as Aβ and tau, but we will focus on those targeting Aβ (Table 1).
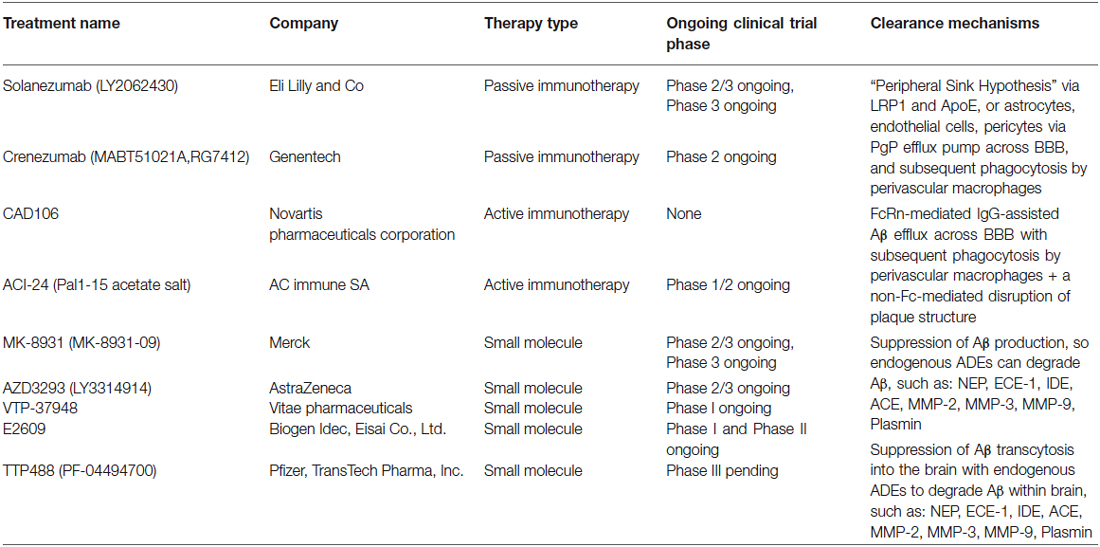
Table 1. Therapeutic interventions presently under investigation for Alzheimer’s disease.
The most prominent area of immunization research has largely been focused on the development of antibodies against Aβ. One passive immunization antibody currently undergoing clinical trials is solanezumab [LY2064230]. It is the humanized IgG1 analog of the murine m266.2 antibody, an anti-Aβ monoclonal antibody (Samadi and Sultzer, 2011). In vitro studies have shown solanezumab to have a strong affinity (Kd of 10−12) towards the middle region of Aβ13-28 and thus acts primarily via a peripheral sink mechanism, although other physiological buffers and endogenous Aβ binding proteins may occur (DeMattos et al., 2001). The “peripheral sink” hypothesis is based on the premise that antibodies minimally transverse the blood-brain barrier (BBB) and thus clearance of Aβ from the brain relies on antibodies binding to Aβ within the bloodstream. Antibodies directed towards Aβ shift the balance of Aβ from the brain and the surrounding vasculature, leading to an efflux of Aβ into the periphery (DeMattos et al., 2001). However, in AD, this “peripheral sink” may be compromised, as Aβ efflux mechanisms may be less efficient or countered by influx of Aβ transcytosis into the brain (Kurz and Perneczky, 2011). Through rodent studies, it was shown that m266 treatment rapidly increased plasma Aβ40/42, which directly correlated with the amount of brain Aβ burden pre-treatment. These data support the idea that m266 acted as a “peripheral sink” to directly facilitate the efflux of Aβ from the brain (DeMattos et al., 2002). Another advantage of solanezumab is the selective binding to soluble Aβ, which greatly decreased the incidence of vasogenic edema and microhaemorrhages that were associated with earlier antibodies (Racke et al., 2005), called Amyloid Associated Imaging Abnormalities in human trials (Sperling et al., 2011). Phase 2 trials conducted in 52 mild-to-moderate AD patients in a double blind, placebo-controlled manner demonstrated a dose-dependent increase in plasma Aβ40 and Aβ42 as well as an increase in unbound Aβ42 in the CSF. These results suggested that solanezumab bound soluble Aβ, which thereby disrupted the equilibrium between soluble and insoluble Aβ within the CNS, resulting in reduction in the deposited Aβ burden (Farlow et al., 2012). Two Phase 3 trials, EXPEDITION-1 and -2 conducted in patients with mild-to-moderate AD did not show statistically significant improvement in cognition or activities of daily living as measured by the Alzheimer’s Disease Assessment Scales, (ADASCog 11 and 12) or the Alzheimer’s Disease Centers Scale for Activities of Daily Living (ADCS-ADL); however, there were significant differences in secondary outcome measures (Doody et al., 2014). Currently, there is a third Phase 3 trial in 2,100 mild AD patients who have elevated levels of Aβ plaques (EXPEDITION-3, ClinicalTrials.gov identifier: NCT01900665), a Phase 2/3 trial to test solanezumab in carriers of the APP, presenilin-1 and presenilin-2 Alzheimer’s gene mutations (DIAN Study, ClinicalTrials.gov identifier: NCT01760005), and a Phase 2 study in seniors deemed to be at high risk for AD who have amyloid positive PET scans (A4 Study, ClinicalTrials.gov identifier: NCT02008357).
Early studies, using passive immunization in AD patients, were often associated with microhemorrhages and vasogenic edema (Racke et al., 2005). In an attempt to avoid these side-effects, a humanized anti-Aβ monoclonal antibody with an IgG4 backbone called crenezumab (MABT5102A) was created (Adolfsson et al., 2012). Crenezumab is similar to solanezumab in that both target the midsection of Aβ; however, the IgG4 isotype reduces the risk of Fcγ receptor-mediated overactivation of microglial cells potentially leading to deleterious proinflammatory responses, while maintaining effective Aβ phagocytosis and clearance (Adolfsson et al., 2012). Furthermore, crenezumab recognizes both soluble Aβ oligomers and multiple Aβ aggregates that are present in AD brains (Adolfsson et al., 2012). In vitro experiments have shown that crenezumab both neutralized and protected neurons against toxic Aβ oligomers (Adolfsson et al., 2012). Phase 1 trials have shown to be extremely promising, as a dose-dependent increase in total plasma Aβ levels was observed to serum crenezumab concentrations demonstrating significant target interactions (Adolfsson et al., 2012). As well, no trial participants developed vasogenic edema, demonstrating the safety of high dose treatment (Adolfsson et al., 2012). Currently, there are several Phase 2 trials with crenezumab being tested as a method of prevention for carriers of the PSEN1 E280A mutations, (Alzheimer’s Prevention Initiative, ClinicalTrials.gov identifier: NCT01998841), on brain amyloid burden in mild to moderate AD (BLAZE Study, ClinicalTrials.gov identifier: NCT01397578) and a long-term safety extension study (ClinicalTrials.gov identifier: NCT01723826).
There has also been some success in terms of active Aβ immunotherapies for treatment of AD. Active Aβ immunotherapies are potentially more cost-effective and long-lasting compared to passive Aβ immunotherapies, which require recurring antibody infusions. Studies have shown an increase in cross-reactive, potentially protective Aβ autoantibodies as a result of active immunotherapy treatment in vervets, which are much lower in AD patients when compared to healthy, age-matched individuals (Weksler et al., 2002; Britschgi et al., 2009). However, active Aβ immunotherapies often require the use of a strong adjuvant for antibody production that may be detrimental to elderly patients who already exhibit an above average proinflammatory cytokine levels (Michaud et al., 2013b).
CAD106 is one such active immunization strategy that is currently in clinical trials. CAD106 was designed with multiple Aβ1-6 coupled to a virus-like Qβ particle, to avoid activation of inflammatory T cells (Wiessner et al., 2011). In APP transgenic mouse studies, CAD106 administration generated Aβ-specific antibodies without activation of Aβ-specific T cells (Wiessner et al., 2011). In prevention trials, CAD106 immunization significantly reduced plaque formation in APP24 mice, however in treatment trials of advanced plaque formation, efficacy was reduced. CAD106 treatment had no effect on levels of vascular Aβ and proinflammatory cytokines, and did not increase microhemorrhages. CAD106 treatment in rhesus monkeys, which share similar Aβ sequences to humans, showed a dose dependent increase in antibody production and Phase 1 trials in humans have found similar results (Wiessner et al., 2011; Winblad et al., 2012). Phase 1 trials showed that CAD106 did not have any adverse effects related to the treatment and a significant portion of the patients treated (67% in cohort 1 and 82% in cohort 2) developed Aβ antibody response that met the responder threshold (Winblad et al., 2012). Phase 2 trials were designed to establish antibody responses and tolerability to various doses, different regions of injection, and different doses of adjuvant. Partial results from Phase 2 findings have reported that long-term exposure to high amounts of CAD106 did not have any additional safety findings (Graf et al., 2014; ClinicalTrials.gov identifier: NCT01097096).
ACI-24 is another active immunization method under developed (Nicolau et al., 2002). ACI-24 is an Aβ1-15 peptide that is bound to liposomal surfaces through two palmitoylated lysine residues, forming a tandem at each end of the peptide (Muhs et al., 2007). The Aβ1-15 sequences were chosen as it retained the B cell epitope of Aβ, but lacked the T cell activation epitope (Monsonego et al., 2001, 2003). ACI-24 treatment restored memory deficits in mice, and produced mainly isotopes IgG, IgG2b, and IgG3, whereby the first two IgGs are related to noninflammatory Th2 response and the latter is a T cell-independent IgG subclass (Gavin et al., 1998; Muhs et al., 2007). Double transgenic mice treated with ACI-24 showed an improvement in memory function which correlated with an increase in IgG antibodies (Sigurdsson et al., 2004; Muhs et al., 2007). Furthermore, treatment resulted in significant reductions in both insoluble Aβ40 and Aβ42, as well as soluble Aβ42, with slight decreases in soluble Aβ40. This effect was observed without additional microglial activation, astrogliosis, or proinflammatory cytokine production (Muhs et al., 2007). Currently, there is a Phase 1/2 trial to examine the safety, tolerability, immunogenicity and efficacy of ACI-24 in mild-to-moderate AD patients (EudraCT Number: 2008-006257-40).
Efflux Pathways for Clearance of Aβ
Since only 0.1% of all peripherally administered or in vivo generated antibodies cross the BBB, then the efflux of Aβ from the brain and subsequent degradation pathways in the periphery will be required for effective passive or active immunization strategies targeting Aβ (Banks et al., 2002; Morgan, 2011). The major efflux pathway for Aβ across the BBB is via the low density lipoprotein receptor-related protein -1 (LRP-1; Kanekiyo and Bu, 2014). LRP-1 is a large multi-functional receptor that regulates endocytosis of multiple ligands directly or indirectly through interaction with other ligands, such as Apolipoprotein E (ApoE), α2-macroglobulin, or other receptor associated proteins many of which have been implicated in AD pathogenesis (Liu et al., 2013; Kanekiyo and Bu, 2014). LRP-1 is abundantly expressed on neurons, glia and vascular cells within the brain and thus, is ideally located as a mechanism for Aβ clearance. For the present argument, LRP-1 is expressed on the microvascular including capillaries, venules and arterioles (Sagare et al., 2013). Furthermore, in AD patients and a mouse model of cerebrovascular amyloid angiopathy (CAA), LRP-1 staining is greatly reduced on vessels and is co-localized to amyloid plaques (Shibata et al., 2000; Deanne et al., 2004; Donahue et al., 2006). LRP-1 expression is also reduced in an age-dependent manner on microvasculature adding further to the potential deficits in Aβ efflux from the brain (Johanson et al., 2006). As mentioned, LRP-1 transports other ligands such as ApoE, the major lipid/protein chaperone in the brain which is known to bind directly to Aβ. There are 3 ApoE isoforms, ApoEε4 representing a risk factor for AD and having the lowest affinity for Aβ, ApoEε2 which represents a protective factor with a high binding affinity, and ApoEε3 which has an affinity between the two (Liu et al., 2013). Thus the differential affinity of the ApoE isoforms for Aβ binding could have further deleterious implications for the clearance of Aβ.
Studies on aging and AD have suggested that there is an increased permeability of the BBB with age and AD progression, as a result of oxidative stress and vascular changes, suggesting that passive diffusion of Aβ across the BBB might also contribute to Aβ efflux (Kalaria and Hedera, 1995; Marques et al., 2013). Under these conditions, a role for perivascular macrophages, pericyte or endothelial cell uptake, and degradation of Aβ after diffusion may also contribute (Verbeek et al., 2000; Hawkes and McLaurin, 2009). However, it has been proposed that macrophage function decreases with aging and pericytes are extremely sensitive to Aβ-induced toxicity, therefore the balance between these processes must be considered (Sengillo et al., 2013).
Lastly, the receptor for advanced glycation end-products (RAGE) plays an important role in AD by contributing to Aβ-induced neuronal dysfunction, microglial activation, and a key role in Aβ transcytosis into the brain (Yan et al., 1996, 2012; Deane et al., 2003; Origlia et al., 2008, 2010). In AD, Aβ can act as a ligand for RAGE and subsequently stimulate the upregulation of RAGE via a positive feed-back mechanism (Bierhaus et al., 2005). With RAGE-induced influx of Aβ into the brain, RAGE activity may negate the already compromised effects of Aβ efflux pathways, resulting in no change to the overall brain Aβ levels (Kurz and Perneczky, 2011). An oral inhibitor of RAGE, TPP488 (PF-04494700) has been shown to block these interactions. In vitro studies showed that TPP488 inhibited soluble RAGE binding to RAGE ligands, but more importantly in this context, to Aβ42.2 Phase 2 trials in people with mild-to-moderate AD showed that TPP488 was well tolerated in subjects that received either a low 10 mg or a high 20 mg dose (Sabbagh et al., 2011; ClinicalTrials.gov identifiers: NCT00566397, NCT00141661). However, results were inconclusive with respect to plasma Aβ levels and inflammatory markers, and there were no significant differences in cognitive and functional measures (Sabbagh et al., 2011). Recently, a Phase 3 trial of TPP488 in mild-to-moderate AD patients starting in 2014 was announced. The combination use of promoting Aβ clearance from the brain and blocking re-entry may provide a more powerful treatment then either strategy alone.
Small Molecule Inhibitors
Currently, small-molecule inhibitors are being developed to inhibit various processes involved in Aβ plaque formation in AD. One class of inhibitors that are presently under examination by various studies are β-site APP Cleaving Enzyme (BACE) inhibitors. As β-secretase plays a major role in the production of Aβ peptides from APP, BACE inhibitors may reduce the amount of Aβ that is produced, allowing for endogenous Aβ clearance mechanisms to function more effectively. However, for these small-molecule inhibitors to work, they must cross both the BBB and enter the appropriate compartments within neurons (Vassar et al., 1999; Gabathuler, 2010). Therefore, in addition to target efficacy, BACE, these therapeutics must be developed with the appropriate molecular weight and charge to overcome these challenges (Pardridge, 2007). Specific BACE inhibitors are discussed below and summarized in Table 1.
MK8931, a small molecule inhibitor of BACE 1 and BACE2, has shown in Phase 1 trials that single doses of up to 500 mg were well tolerated and met with reductions in CSF Aβ of up to 92% in healthy individuals (Forman et al., 2012). Furthermore, MK8931 has shown to have a relatively long half-life, 20 h, which is ideal for single daily dosing paradigms (Forman et al., 2012; Stone et al., 2013). Currently, Phase 2/3 trials are examining the safety and efficacy of MK-8931 at daily dosages of 12 and 40 mg, as well as long-term treatment effects on ADAS-cog and ADCS-ADL scores (EPOCH, ClinicalTrials.gov identifier: NCT01739348 and APECS, NCT01953601).
AZD3293 (LY3314814) is an oral, brain permeable BACE 1 inhibitor, currently being developed by AstraZeneca and Eli Lilly (Haeberlein et al., 2013). Mouse and guinea pig studies have shown AZD3293 treatment resulted in a dose- and time-dependent reduction in the amount of Aβ40/42 and soluble β-APP in the brain, CSF and plasma (Haeberlein et al., 2013). Results from quantitative analyses of soluble α-APP and β-APP in healthy volunteers showed that AZD3293 treatment resulted in a dose-dependent decrease in the amount of soluble β-APP in the CSF, while soluble α-APP showed a similar broadly dose-dependent increase (Höglund et al., 2014). Phase 1 trials of AZD 3293 have recently been completed. A Phase 2/3 trial is currently recruiting, and will examine the safety and efficacy of the AZD 3293 over 2 years of treatment in early AD (ClinicalTrials.gov identifier: NCT02245737). Clinical Dementia Rating-Sum of Boxes (CDR-SOB) will be used as the primary outcome measure, with ADAS-COG and ADCS-ADL as secondary outcome measures, as well as other imaging and clinical markers.
Another oral BACE inhibitor, VTP-37948, currently in development by Vitae Pharmaceuticals, has shown good brain penetrance and reduced CSF Aβ levels by up to 80% in preclinical studies.3 At present, VTP-37948 is in Phase 1 trials with healthy individuals to examine safety and tolerability, as well as the pharmacokinetics and pharmacodynamics of the drug.
E2609 is a BACE1 inhibitor that is being developed by Eisai Ltd. It has been shown to significantly inhibit Aβ40/42 production in both the CSF and plasma of cynomolgus monkeys after oral dosing (Lucas et al., 2012). Partial results from Phase 1 studies have shown E2609 was well tolerated across all dosage treatments (up to 800 mg), and had a prolonged effect in reducing plasma Aβ40/42 after a single dosage in healthy individuals (Lai et al., 2012). Currently, trials have been completed on patients with MCI and those with evidence of Aβ pathology, and a dose-finding Phase 2 trial is underway in patients with MCI and mild AD (ClinicalTrials.gov identifier: NCT02322021). They also examined the safety and pharmacology of the E2609 across Japanese and Caucasian populations.
Further studies on BACE inhibitors must be conducted to determine efficacy in target engagement as well as potential off-target deleterious side-effects. Post-mortem analyses of AD brains demonstrated increased BBB permeability in brain regions, such as the hippocampus, which may aid BACE inhibitors to reach their targets (Montagne et al., 2015). However, >90% of AD patients exhibit CAA, amyloid deposition within the vasculature of the central nervous system, resulting in hypoperfusion as well as a physical barrier to influx of BACE inhibitors (Revesz et al., 2002; Thal et al., 2008). BACE cleaves many substrates, that play important roles in the nervous system, and thus inhibition of BACE may also lead to deleterious effects throughout the body. One example is neuregulin-1, a peptide that is essential for heart and nervous system development as well as the maintenance of muscle spindles (Britsch, 2007). Other BACE1 substrates, seizure-protein 6, L1, CHL1 and contactin-2, are important neural cell adhesion molecules that are crucial for guidance and maintaining neural circuits (Kuhn et al., 2012; Zhou et al., 2012). Furthermore, BACE1 KO mice demonstrate problems in axon targeting, although this may represent a developmental issue, re-programming of neural circuits and adult neurogenesis (Rajapaksha et al., 2011). Therefore, the consequences of total BACE inhibition on the critical function of these other substrates need to be examined further, as the effects of these BACE inhibitors on Aβ offers a promising therapeutic route. An alternate and possibly less prone to deleterious side-effects may be the development of modulators of BACE activity rather than inhibitors.
Intraparenchymal Degradation Pathways
In order for small molecule therapies to be effective in AD, they not only need to exhibit high CNS bioavailability but also utilize endogenous parenchymal Aβ catabolism pathways. In regards to the BACE 1 inhibitors described above, these inhibitors will decrease new Aβ production and thus potentially prevent further neuronal and vascular damage. However, at time of treatment most AD patients will have a pre-existing Aβ load within the CNS that may require catabolism for full recovery. The endogenous parenchymal catabolic pathways for Aβ are regulated by a number of degrading enzymes in the extracellular space, secreted chaperones that monitor proteostasis, as well as glial cell uptake and degradation by lysosomal, autophagic and proteosomal pathways (Guénette, 2003; Tanzi et al., 2004; Lai and McLaurin, 2012; Wyatt et al., 2012).
Enzymatic degradation of Aβ peptides is accomplished by a number of enzymes including, but not limited to, neprilysin (NEP), insulin degrading enzyme (IDE), angiotensin converting enzyme (ACE) and various matrix metalloproteinases (MMPs). NEP is the most extensively studied, and has been shown to degrade both Aβ40 and Aβ42 in vitro and in vivo (Iwata et al., 2000, 2001). Furthermore, recent studies in aged mice and AD patients have shown decreased levels of NEP in the hippocampus and temporal gyrus, regions with a high amyloid load in AD (Yasojima et al., 2001; Iwata et al., 2002). However, the degradation of Aβ is complicated and cannot be accomplished by a single enzyme, as Kms and Aβ aggregation state will play a role. IDE has also been shown to have Aβ degrading activity, as IDE deficient mice have increased brain Aβ levels and over-expression in an AD mouse model decreases Aβ levels (Farris et al., 2003; Leissring et al., 2003). Furthermore, insulin competes with Aβ for IDE degradation and thus in Diabetes Mellitus, Aβ levels are increased within the CNS (Qiu and Folstein, 2006). The role of MMPs, ACE, endothelin-converting enzyme and others all contribute to Aβ catabolism however the precise role for each enzyme is not fully elucidated.
A recent hypothesis suggests that a family of secreted chaperones, which exist in the extracellular space, patrol the brain for misfolded proteins and aid in clearance (Wyatt et al., 2012). The chaperones relating to Aβ clearance that have been identified are clusterin (also referred to as ApoJ), α2-macroglobulin and ApoE. As mentioned above, all three chaperones are co-receptors for LRP-1 and thus aid in Aβ catabolism via uptake by neurons, glia or vascular cells.
In AD, astrocytes and microglia are the immune effectors of the CNS and thus play a role in injury resolution via limiting effects of toxic Aβ species (Guénette, 2003). Astrocytes become activated and surround amyloid plaques in an attempt to limit the damage to surrounding neuropil (Akiyama et al., 2000). Examination of human AD brain demonstrated the presence of N-truncated Aβ within astrocytes and more specifically Aβ was detected in lysosomal granules of astrocytes, thus suggesting phagocytosis and degradation (Funato et al., 1998; Thal et al., 1999; Nagele et al., 2003). In support of the pathological findings, adult mouse astrocytes have been shown to degrade Aβ deposits in brain sections, and phagocytose extracellular Aβ (Wyss-Coray et al., 2003; Koistinaho et al., 2004; Mandrekar et al., 2009). Although astrocyte uptake and degradation is less efficient then resident microglial cells, this pathway may contribute to Aβ clearance under treatment strategies.
Microglial cells are the resident phagocytes of the CNS and play a significant role in Aβ clearance (reviewed in Lai and McLaurin, 2012). Although microglial cells in vitro readily phagocytose and degrade soluble and fibrillary Aβ, there is some controversy regarding efficacy under the pathological conditions present in AD brains (Lee and Landreth, 2010). Similar to astrocytes, microglia surround amyloid plaques, and electron microscopy studies have suggested the intracellular presence of Aβ in endosomal compartments (Frackowiak et al., 1992). Furthermore, Aβ can be detected within lysosomal compartments of non-plaque associated microglial cells after treatment with an anti-aggregant compound, 1-fluoro-scyllo-inositol (Hawkes et al., 2012). Thus, although some literature suggests that accumulation of Aβ and amyloid plaques may be the result of immuno-incompetent microglia in AD, the presence of small molecules therapies, such as immunotherapy, curcumin and scyllo-inositol, that boost phagocytosis support a role for microglia in therapeutic interventions (Wilcock et al., 2004; McLaurin et al., 2006; Yanagisawa et al., 2010).
Lifestyle Interventions
In recent years, AD research has begun to focus on changes that can be made in daily living and activity which potentially decrease risk and delay symptomatic expression of AD. During the International Conference on Nutrition and the Brain a compilation of the presented data led to the identification of 7 changes to be integrated into daily living, six involving diet and the seventh recommending exercise (Barnard et al., 2014). Exercise and diet alterations have been associated with improvements in symptomatic and pathophysiological AD outcomes in both animal models and humans (Luchsinger et al., 2002; Stranahan et al., 2012; Hawkes et al., 2015; Lim et al., 2015; Lin et al., 2015). Regular, controlled diet and exercise are likely protective against AD pathogenicity through the maintenance of cardiovascular and cerebrovascular health. As mentioned above, the vasculature plays a crucial role in the clearance of Aβ across the BBB because of receptors (i.e., LRP-1, RAGE) expressed on the plasma membranes of capillary, arteriole, and small venule cells (Deane et al., 2003; Sagare et al., 2013). It has recently become apparent that regular and healthy pulsation of blood vessels promote a convective bulk flow of the brain’s parenchymal ISF, which acts to clear toxic solutes such as Aβ (Carare et al., 2008; Weller et al., 2008; Iliff et al., 2012). Lifestyle interventions, including exercise, diet and sleep, may have direct beneficial effects on Aβ clearance, as well as act indirectly through enhancing vascular health.
Exercise
There are >20 ongoing and recently completed clinical trials assessing the efficacy of different exercise interventions on AD symptoms and pathology.4 Thoroughly detailing these trials is beyond the scope of this review, however a few points will be noted. Firstly, exercise has had beneficial effects on many cognitive assessments (de Andrade et al., 2013; Winchester et al., 2013; Okonkwo et al., 2014). Secondly, AD interventional benefits are seen in a multitude of exercises ranging from walking to high-intensity aerobic physical activity (Venturelli et al., 2011; Nascimento et al., 2012; Vidoni et al., 2012; Hoffmann et al., 2013; Suttanon et al., 2013; Winchester et al., 2013; Arcoverde et al., 2014; Okonkwo et al., 2014). Finally, regular exercise over years is correlated with significantly slowed Aβ deposition over time, perhaps in part due to enhanced Aβ clearance (Okonkwo et al., 2014).
The beneficial effects of aerobic exercise on brain health have been well studied in recent years, with a focus on enhanced neurogenesis, increased levels of neurotrophic factors such as brain-derived neurotrophic factor (BDNF), and reduced risk for AD (Voss et al., 2013). Lin et al. (2015) demonstrated that exercise is associated with improved Aβ clearance mechanisms by upregulation of LRP-1 protein levels whereas RAGE was unchanged (Lin et al., 2015). Interestingly, one study in the Tg2576 AD mouse model demonstrated reduced soluble Aβ, but unchanged total Aβ, as a result of exercise (Nichol et al., 2008). This further suggests that Aβ clearance mechanisms specifically are upregulated with exercise. BDNF signaling in the brainstem of mice has recently been shown to increase the excitability of parasympathetic cholinergic neurons which, by way of the vagal nerve, act to lower resting heart rate (Wan et al., 2014). This provides molecular evidence for a mechanism by which aerobic exercise can reinforce vascular health (Wan et al., 2014; Mattson, 2015). Since the structurally and functionally impaired cerebrovasculature in AD dampens blood vessel-dependent Aβ clearance pathways, physical activities which can upregulate BDNF signaling and provide enhanced amyloid removal to the periphery, would be beneficial in slowing disease progression (Dorr et al., 2012; Lai et al., 2015).
Diet
Healthy diet maintenance is important in the modulation of AD symptoms and pathology, assisting endogenous mechanisms including neurogenesis, antioxidant protection, and Aβ clearance (Aliev et al., 2013; Maruszak et al., 2014). Type 2 Diabetes Mellitus (T2DM) is an acquired disease caused by hyperglycaemia and potentially insulin resistance. The main risk factors for the incidence and prevalence of T2DM are obesity, primarily from high fat diets, and age (Barbagallo and Dominguez, 2014; Centers for Disease Control and Prevention, 2014). Studies have shown that T2DM patients are at an increased risk of developing dementia, and conversely, 80% of AD patients have T2DM (Janson et al., 2004; Biessels et al., 2006). In a recent study by Hawkes et al. (2015) mice that were subject to a high fat diet during gestation and early life exhibited deficits in perivascular clearance of Aβ, an effect which was exacerbated when the high fat diet was lifelong. Also, vascular deposits of Aβ in aged human cases of hyperlipidemia were significantly greater when compared to aged-matched people with normal lipid levels post-mortem (Hawkes et al., 2015).
Dietary plans have been well researched in AD. The main three are the Mediterranean, ketogenic, and caloric restriction diets (Aliev et al., 2013; Maruszak et al., 2014). The Mediterranean diet involves replacement of meat products, especially red meat, with plant-based alternatives, and primarily olive and fish oils for additional fats (Yannakoulia et al., 2015). Clinical studies on the effect of this diet on cognition in MCI/AD are inconsistent, with some showing benefits and others not (Olsson et al., 2015; Yannakoulia et al., 2015). Contrary to the Mediterranean diet, ketogenic and caloric restriction diets involve a significant reduction in food intake (Maruszak et al., 2014; Paoli et al., 2014). The ketogenic diet aims to create a state of fasting within the body (Paoli et al., 2014). This reduces metabolic induced stresses, including damage from reactive oxidative species and pathogenic mitochondrial biogenesis (Paoli et al., 2014). Ketogenic diets may also decrease the production of advanced glycation end products, which accumulate on Aβ plaques, potentially assisting in one of the aforementioned clearance cascades by decreasing reuptake of Aβ by RAGE (Deane et al., 2003; Srikanth et al., 2011; Paoli et al., 2014). Caloric restriction on the other hand, is achieved by a moderate decrease in overall intake of calories (Maruszak et al., 2014). Caloric restriction has been associated with a reduced risk of AD and memory improvements in the elderly, and has been a beneficial intervention in mouse models of AD (Lee et al., 2000, 2002; Luchsinger et al., 2002; Gustafson et al., 2003; Wu et al., 2008; Witte et al., 2009). This diet has also been demonstrated to decrease Aβ pathology through enhancing non-amyloidogenic APP cleavage by α-secretase and increasing clearance of Aβ through upregulated IDE levels (Farris et al., 2004; Wang et al., 2005; Tang and Chua, 2008). There are two current clinical trials on the effectiveness of caloric restriction in MCI/AD.
Sleep
Sleep and circadian rhythm disturbances are common in aging and AD patients (Floyd et al., 2000; Cipriani et al., 2014; Zelinski et al., 2014). Circadian rhythms are controlled by the suprachiasmatic nucleus (SCN) in the hypothalamus, which acts like a biological clock in its management of many physiological functions (Reppert and Weaver, 2001; Coogan et al., 2013; Videnovic et al., 2014; Zelinski et al., 2014). Mouse models of AD exhibit circadian rhythm alterations suggesting a link to Aβ (Sterniczuk et al., 2010; Baño Otalora et al., 2012). Even self-reported disruptions in sleep indicate a 33% increased risk of dementia and a 51% increased risk of AD (Benedict et al., 2014). Higher brain amyloid burden and lower CSF Aβ levels were observed in sleep deprived and narcoleptic patients without AD (Spira et al., 2013; Liguori et al., 2014). AD animal models in which sleep is deprived show similar trends of increased amyloid load, as well as increased memory dysfunction (Kang et al., 2009; Rothman et al., 2013; Di Meco et al., 2014).
A study in cognitively normal middle-aged men measured CSF biomarkers of AD by an intrathecal catheter (Ooms et al., 2014; AWAKE study, ClinicalTrials.gov identifier: NCT01194713). A 6% decrease in Aβ42 levels were observed following an unrestricted sleep, however no change was observed in participants who remained awake (Ooms et al., 2014). The benefits of sleep on soluble Aβ levels in the brain may have been underestimated due to measurement of only spinal CSF (Ooms et al., 2014). There is also enhanced clearance by the glymphatic system during sleep, leading to additional Aβ efflux into the blood and cervical lymph nodes (Szentistványi et al., 1984; Iliff et al., 2012; Xie et al., 2013). Using in vivo two-photon microscopy in sleeping and in anesthetized mice, Xie et al. (2013) measured a 60% increase in the brain parenchyma, associated with an increase in the rate of exchange of CSF-ISF. These results suggest that regular sleep may serve to aid in the clearance of toxic solutes, such as Aβ, from the extracellular space in the brain, and normalization of sleep patterns may serve as beneficial lifestyle intervention in the treatment of AD (Xie et al., 2013).
There are 7 active clinical trials for sleep interventions in AD, two of which are directly concerned with sleep apnea. Sleep apnea causes pauses or disruptions in breathing during sleep, and is correlated with cognitive dysfunction in AD patients (Janssens et al., 2000; National Heart, Lung, and Blood Institute, 2012). It is potentially associated with AD through mechanisms such as cellular oxidative stress, hypoxia, and sleep disturbances (Pan and Kastin, 2014). The two current clinical trials for sleep apnea and AD are assessing the effectiveness of a continuous positive airway pressure (CPAP) device on measurements including cognition, quality of daily living and CSF levels of Aβ42 (AZAP, ClinicalTrials.gov identifier: NCT01400542 and SNAP, ClinicalTrials.gov identifier: NCT01962779). The use of CPAP has effectively improved sleeping conditions in patients with mild-to-moderate AD, an effect that was maintained for 3 weeks (Cooke et al., 2009).
Sleep apnea leads to impaired vascular health, with increased arterial stiffness and blood pressure, decreased cerebral blood flow, and thickened carotid intima-media (Daulatzai, 2012; Ciccone et al., 2014). The tunica intima and tunica media are the innermost perivascular layers along cerebral arteries, and the tunica media is the area within which the majority of ISF flows on its way out of the brain during perivascular clearance (Carare et al., 2008; Weller et al., 2008, 2010). Therefore if sleep disturbances are thickening this area, the rate at which bulk flow of ISF travels would theoretically be decreased. This, in conjunction with the impaired arterial health in sleep apnea, would potentially diminish the perivascular clearance of Aβ, leading to increased CAA and amyloid in the brain parenchyma (Daulatzai, 2012). Nighttime wakefulness also leads to a decrease in glymphatic Aβ clearance because of the inability for parenchymal expansion, which normally speeds the rate of CSF-ISF exchange during sleep (Xie et al., 2013). Both glymphatic and perivascular clearance mechanisms are further diminished by the impaired vasculature present in sleep disorders due to irregular blood vessel pulsations (Carare et al., 2008; Iliff et al., 2013b).
Vascular Health
Hypertension, increased body mass index, abnormal glucose regulation, hyperlipidemia, and hypercholesterolemia are all vascular related risk factors for AD and cognitive decline (Kivipelto and Solomon, 2008; Reynolds et al., 2010; Tolppanen et al., 2012; Liu et al., 2014; Deckers et al., 2015). AD patients analyzed for the relationships between cardiovascular risk factors and cognition showed lower scores on the MMSE and ADAS-COG in hypertensive patients compared to those with normal blood pressure, and lower scores on the MMSE and Clinical Dementia Rating in patients with hyperlipidemia compared to those without (Lobanova and Qureshi, 2014). Such findings suggest vascular injury may play a role in cognitive dysfunction in AD. Vasoactive drugs therefore might have the potential to treat aspects of AD, including deficits in cognition. Two current vasoactive drugs in development for AD are nilvadipine and cilostazol (Ikeda, 1999; Nimmrich and Eckert, 2013). Nilvadipine is a dihydropyridine that blocks calcium channels and prevents cognitive decline in patients with MCI (Hanyu et al., 2007; Nimmrich and Eckert, 2013). Although intervention with Nilvadipine leads to decreased hypertension, its main effect on cognition is postulated to be through neuroprotection, potentially by decreased calcium-mediated excitotoxicity of neurons (Takakura et al., 1992; Hanyu et al., 2007; Nimmrich and Eckert, 2013). A Phase 3 placebo-controlled trial of nilvadipine in mild-moderate AD is underway (NILVAD, ClinicalTrials.gov identifier: NCT02017340).
Cilostazol has multiple beneficial effects on the vasculature, antiplatelet activity, and is an inhibitor of the cAMP and cGMP regulator phosphodiesterase type 3 (PDE3; Ikeda, 1999; Saito and Ihara, 2014). Studies of cilostazol in human dementia/AD patients showed some benefit on slowing cognitive decline and increasing cerebral perfusion (Sakurai et al., 2013; Ihara et al., 2014). Patients with mild dementia, but not moderate to severe dementia, maintained higher MMSE scores when treated with cilostazol in conjunction with the acetylcholinesterase inhibitor donepezil, compared to donepezil alone (Ihara et al., 2014; CASID study, ClinicalTrials.gov identifier: NCT01409564). Slower decline in ADAS-COG scoring was also seen in another study, along with increased regional cerebral blood flow to the right anterior cingulate lobe (Sakurai et al., 2013). Cilostazol is theorized to slow AD progression by promoting perivascular clearance of ISF, which contains soluble Aβ, in part due to it vasodilation and regulation of blood vessel pulsations (Carare et al., 2008; Han et al., 2013; Saito and Ihara, 2014). Additionally, PDE3 expression is increased in arterial cells with Aβ deposition, primarily in smooth muscle cells (Maki et al., 2014). Smooth muscle cells are present within the tunica media of the perivascular space along leptomeningeal arteries, a pathway through which ISF, including soluble Aβ, is cleared from the brain (Weller, 2005; Kwee and Kwee, 2007; Carare et al., 2008; Weller et al., 2008). In a mouse model of CAA, cilostazol protected against vascular and cognitive deficits, and decreased Aβ deposits, potentially because of enhanced perivascular clearance (Maki et al., 2014). This suggests beneficial effects of cilostazol in AD on perivascular clearance of ISF, and on a vascular protective signaling cascade involving inhibition of PDE3 and increased cAMP and cGMP activity.
Perivascular Clearance
There are two proposed mechanisms by which drainage of the brain’s extracellular fluid, also referred to as ISF, occur (Carare et al., 2008; Iliff et al., 2012). The first involves a bulk flow of ISF and solutes within arterial perivascular spaces, across the BBB, where they enter cervical lymph nodes (Szentistványi et al., 1984; Carare et al., 2008). Perivascular spaces, or Virchow-Robin spaces, are small areas around blood vessel walls, including the smooth muscle cells which line arteries, that are in continuity with the subarachnoid space from the point where blood vessels penetrate into the brain parenchyma (Weller, 2005; Kwee and Kwee, 2007; Weller et al., 2008). The pia mater sheaths the perivascular space along leptomeningeal arteries until they branch off into smaller arterioles and capillaries (Weller, 2005; Kwee and Kwee, 2007; Carare et al., 2008). According to one theory, flow of ISF occurs along the basement membranes of capillaries and arterioles until it converges upon leptomeningeal arteries, where the fluid continues to drain in the perivascular tunica media, the space between the arterial smooth muscle cells, and in the tunica adventitia, the basement membranes of the smooth muscle cells (Carare et al., 2008; Weller et al., 2008, 2010). There is further convergence with leptomeningeal and major cerebral arteries before the ISF drains completely out of the brain into the cervical lymph nodes (Szentistványi et al., 1984; Weller et al., 2010). This theory implies that flow of ISF occurs retrogradely only along capillaries, arterioles, and arteries, not along venules or veins (Figure 1; Carare et al., 2008; Arbel-Ornath et al., 2013).
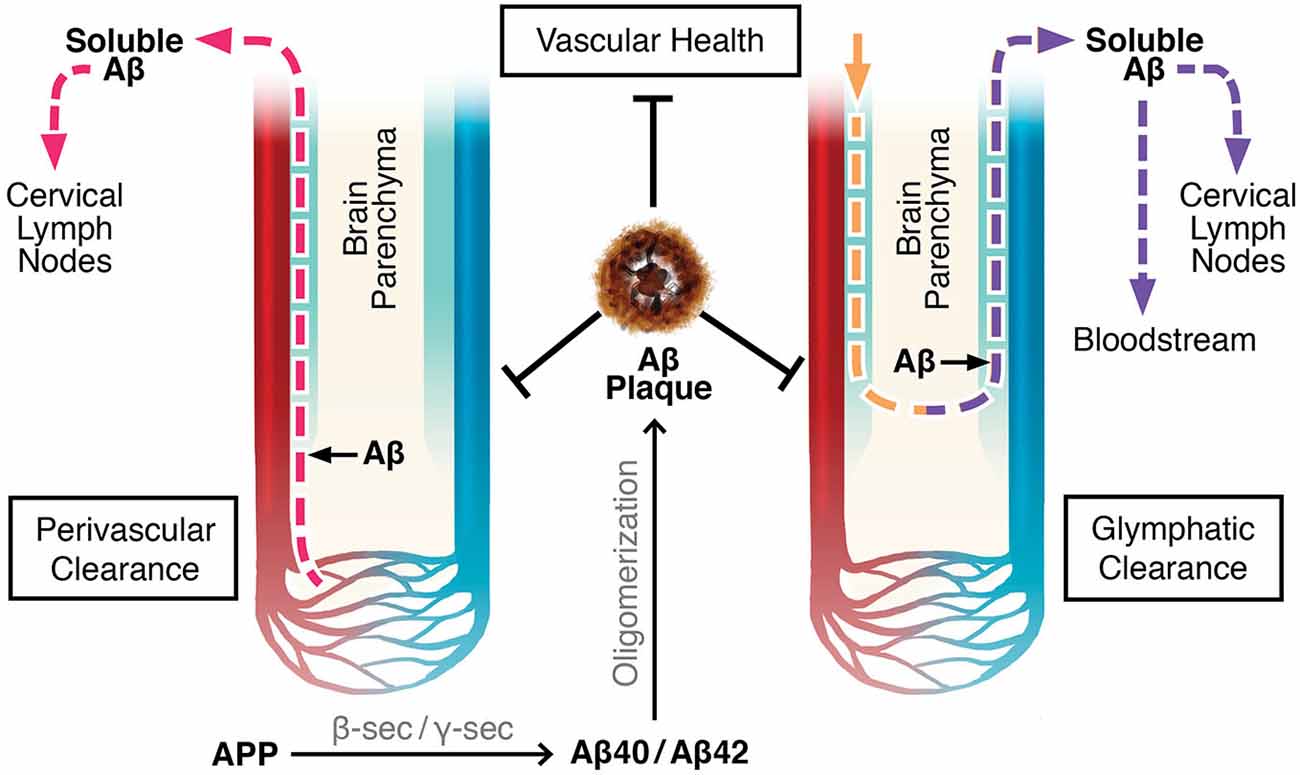
Figure 1. Perivascular and glymphatic drainage of brain interstitial fluid (ISF). These mechanisms drain brain ISF and parenchymal solutes (e.g., Aβ) to the periphery. Perivascular clearance involves ISF efflux along capillaries, arterioles, and arteries within the perivascular space. Glymphatics involve CSF influx and CSF/ISF efflux within the perivascular space along arteries and veins, respectively. Color Legend, red: arteries/arterioles/capillaries; blue: veins/venules; light blue: perivascular space; pink dotted line: ISF efflux; yellow dotted line: CSF influx; purple dotted line: CSF/ISF efflux.
Evidence for perivascular clearance has been obtained by experiments involving radio- or fluorescent labeled tracers. Furthermore, multi-photon imaging and mathematical models have provided strong support (Szentistványi et al., 1984; Schley et al., 2006; Carare et al., 2008; Wang and Olbricht, 2011; Arbel-Ornath et al., 2013). ISF drainage was theorized to be by a convective bulk flow mechanism by assessing the clearance of multiple radiolabeled compounds of differing molecular weights. These compounds cleared at the same rate, confirming that ISF did not drain by diffusion, in which case the smaller molecules would have moved faster (Cserr et al., 1981; Abbott, 2004; Carare et al., 2008). Using multi-photon imaging through a surgically dissected cranial window in mice, Arbel-Ornath et al. (2013) observed active perivascular clearance of ISF in vivo. Consistent with previous conclusions, the fluorescent tracer flowed out of the brain along capillaries and arteries but not veins (Arbel-Ornath et al., 2013). Perivascular clearance is reliant on proper vasculature flow, as it is non-existent after cardiac arrest and is diminished with decreased perfusion (Carare et al., 2008; Arbel-Ornath et al., 2013). Theoretical models propose that the pulsating motion of blood flowing into the brain acts to push ISF in the opposing direction, thereby explaining why the draining of ISF requires functioning vasculature, and why fluid is not cleared along veins (Schley et al., 2006; Carare et al., 2008; Wang and Olbricht, 2011).
Although Aβ self-aggregates into fibers and forms plaques, there is still a significant portion of soluble species of Aβ that is released to the extracellular space (Selkoe, 2001). Since convective bulk flow of ISF drains solutes, perivascular drainage represents one of the major Aβ clearance mechanisms and could be impaired in AD (Carare et al., 2008). The APP/PS1 AD mouse model showed a 60% increased retention of fluorescent tracer, compared to wild type mice (Arbel-Ornath et al., 2013). The presence of Aβ plaques on arteries cause increased tortuosity and decreased calibre which in turn disrupts and lengthens blood flow transit times (Dorr et al., 2012). This irregularity in the vasculature is likely interrupting the pulsating motion which drives perivascular clearance. Also, it was found that fluorescent conjugated tracers and Aβ40 when injected are present where plaques would form in AD (Hawkes et al., 2013, 2014). The hippocampus of non-transgenic mice showed an aging-related decrease in perivascular clearance of injected Aβ40, suggesting the importance of this clearance mechanism to AD pathogenesis (Hawkes et al., 2013). Together, these conclusions support a positive feedback loop for CAA and AD, where impaired perivascular ISF drainage leads to Aβ deposition along the arteries, which causes weakened vasculature functioning, thereby allowing increased levels of Aβ to remain in the brain parenchyma, more specifically in the hippocampus (Carare et al., 2008; Hawkes et al., 2013).
Glymphatics
The glymphatic system is another ISF drainage system recently proposed by Maiken Nedergaard’s group (Iliff et al., 2012; Iliff and Nedergaard, 2013). Glymphatics involve the para-arterial, unidirectional flow of CSF from the cisterna magna of the subarachnoid space, along penetrating leptomeningeal arteries, towards the pituitary and pineal recesses of the 3rd ventricle, and into the brain within the perivascular (Virchow-Robin) space (Iliff et al., 2012, 2013a). CSF enters the brain parenchyma and mixes with ISF in a process reliant on aquaporin-4 (AQP4) channels present on the perivascular endfeet of astrocytes. The extracellular fluid composed of CSF and ISF is proposed to exit the parenchyma by drainage out of the brain within the venous perivascular space of large calibre veins only, where it then flows into the cervical lymph nodes, or absorbs into the blood across arachnoid villi on dural sinuses (Figure 1; Szentistványi et al., 1984; Iliff et al., 2012). Similar to perivascular clearance, glymphatic flow is driven by the pulsation of blood vessels; however, the former is by bidirectional flow along the vasculature, and the latter is unidirectional (Hadaczek et al., 2006; Schley et al., 2006; Wang and Olbricht, 2011; Iliff et al., 2013b).
The glymphatic system was observed in vivo in mice after an intrathecal injected contrast agent was followed by magnetic resonance imaging, and through a closed cranial window with two-photon laser scanning microscopy (Iliff et al., 2012, 2013a). These results were reproduced using clinically-relevant levels of intrathecal injections in rats and mice (Yang et al., 2013). In confirmation that arterial pulsations drive the unidirectional flow of CSF into the parenchyma, unilateral ligation of the mouse carotid artery decreased blood flow and pulsality as well as flow of the injected tracers. Subsequently, treatment with dobutamine to increase blood flow and pulsality, increased tracer movement (Iliff et al., 2013b).
Similar to perivascular clearance, soluble Aβ is cleared from the brain parenchyma by glymphatic drainage (Carare et al., 2008; Iliff et al., 2012). Interestingly, the rates of CSF-ISF exchange significantly decrease with age, suggesting an age-related decline in the glymphatic clearance of toxic solutes, such as Aβ (Kress et al., 2014). Unlike perivascular clearance, the physics of the overall drainage system are affected by molecular size, because larger tracers were slower to enter the parenchyma following subarachnoid injection, suggesting a reliance on AQP4 channels for CSF-ISF exchange (Iliff et al., 2012). In healthy conditions, the expression of AQP4 is highly polarized to astrocytic endfeet (Iliff and Nedergaard, 2013). However, with neuroinflammation as well as with age, this expression profile changes, with decreased AQP4 at the perivascular endfeet and increased levels in the soma. This may explain the impairment in glymphatic flow with age, suggesting another mechanism by which Aβ clearance may be diminished in AD (Iliff and Nedergaard, 2013; Kress et al., 2014). Additionally, both the perivascular and the glymphatic clearance systems are impaired following an ischaemic stroke in mice (Arbel-Ornath et al., 2013; Gaberel et al., 2014). Because of a close clinical relationship with stroke, this provides further evidence to why drainage of Aβ may be impaired in AD (Viswanathan et al., 2009).
In addition, late-onset AD is thought to relate to impaired cerebral clearance of Aβ (Mawuenyega et al., 2010). This is in contrast to overproduction which leads to amyloid accumulation from mutations affecting APP processing (e.g., mutations in APP or presenilin complex), resulting in early-onset Autosomal Dominant AD or trisomy 21, which predisposes to early-onset dementia in Down’s syndrome. In this context, collagenosis of the deep penetrating venular system could be predicted to affect the glymphatic clearance of amyloid and other toxic proteins along the perivascular venular pathways. Collagenosis causes wall thickening and stenosis of the deep venular system and correlates with confluent periventricular white matter hyperintensities (pvWMH), which is seen in elderly controls and Alzheimer’s patients on Magnetic Resonance images (Moody et al., 1995; Black et al., 2009). PvWMH are associated with hypertension and other vascular risk factors, older age and possible genetic factors. Recent work from our group suggests the correlation of pvWMH is strongest with significant stenosis of the medium to large venules. We hypothesize that this leads to BBB leakage and perivenous edema, which further disrupts clearance mechanisms for amyloid and other toxins along the perivascular pathways (Black et al., 2009; Gao et al., 2012). It is of note that pvWMH are also associated with CAA and visible as microbleeds, which would be consistent with disruption of the perivascular pathway, thereby exacerbating deposition of periarteriole Aβ (Pettersen et al., 2008).
CAA and AD associated arterial damage, as described above, would decrease the functioning of both perivascular and glymphatic drainage by impairing arterial pulsations (Carare et al., 2008; Dorr et al., 2012; Iliff et al., 2013b). Despite little Aβ deposition, the structural and functional integrity of venules and veins also deteriorate in AD, which may lead to further impairments in amyloid clearance (Revesz et al., 2003; Weller et al., 2009; Lai et al., 2015). The presence of atherosclerosis is common in patients with AD, causing further irregularities in vascularization including the thickening of the perivascular space, which likely decreases vascular associated Aβ clearance (Ross, 1993; Frink, 2002; Roher et al., 2003). One of the major hallmarks of perivascular clearance is that soluble Aβ deposits along capillaries and arteries as ISF drains (Hawkes et al., 2013, 2014). Since para-venous drainage out of the brain is proposed to occur in the glymphatic system, it is surprising that Aβ deposits are not abundantly seen along venules and veins (Revesz et al., 2003; Weller et al., 2009; Iliff et al., 2012). There are a couple of explanations for this observation. First, it is possible that the lack of smooth muscle cells on veins provide less substrates for Aβ to deposit on Weller et al. (2008). Secondly, peripheral monocytes have been reported to enter the lumen of veins, but not arteries, and clear soluble Aβ (Michaud et al., 2013a).
Healthy lifestyle interventions play an important role in the maintenance of the preceding Aβ clearance mechanisms. As discussed above, consistent exercise, diet and sleep contribute to proper structural and functional integrity of the vasculature, maintaining regular blood vessel pulsations, which are the driving force behind both the perivascular and glymphatic clearance mechanisms (Carare et al., 2008; Iliff et al., 2013b). Together these conclusions stress the importance of these lifestyle interventions in the maintenance of vasculature health, and subsequently in delaying the progression of AD.
Conclusion and Future Perspectives
In this review article, we have summarized three different therapeutic approaches that rely on various combinations of physiological Aβ clearance mechanisms. We have highlighted some caveats to these approaches and attempted to highlight mechanisms that might function in synergy to remove toxic Aβ from the CNS as a disease-modifying therapy. The function of the Aβ clearance mechanisms, efflux at the BBB, catabolism within the CNS or drainage via either the perivascular drainage pathway or glymphatics, must be considered when designing new therapeutics and interpreting results from ongoing clinical trials, including the potential exacerbation of clearance mechanisms from small vessel arteriolar, capillary and venular disease. We propose that clinical trial failure may not be the sole result of an ineffective drug candidate or wrong patient population but may represent a lack of endogenous clearance mechanisms needed to support drug effects.
Although all cases of AD are characterized pathologically in the same manner, the cause of disease and co-morbidities that contribute to disease progression vary extensively between patients. In light of this, multi-modal therapeutic approaches may need to be considered, with an eye on personalized medicine to account for the variability in presentation of AD patients, including reliable quantification of subtypes of small vessel disease as well as patterns of gray and white matter atrophy, which is overlooked by many currently popular automatic pipelines. Combination therapies are already in practice for various diseases and thus may also be necessary for AD. Furthermore, the treatment strategies may need to vary depending on disease state or age of the individual at presentation, with the goal of treating the disease or delaying the onset.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was supported by Canadian Institutes of Health Research Grants (JM: FRN37857;102467), Cryptic Rite Charitable Foundation (JM).
Footnotes
- ^ www.clinicaltrials.gov
- ^ http://www.ttpharma.com
- ^ http://www.alzforum.org
- ^ www.clinicaltrials.gov
References
Abbott, N. J. (2004). Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem. Internat. 45, 545–552. doi: 10.1016/j.neuint.2003.11.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Adolfsson, O., Pihlgren, M., Toni, N., Varisco, Y., Buccarello, A. L., Antoniello, K., et al. (2012). An effector-reduced anti-β-amyloid (Aβ) antibody with unique Aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J. Neurosci. 32, 9677–9689. doi: 10.1523/JNEUROSCI.4742-11.2012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G. M., et al. (2000). Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421. doi: 10.1016/S0197-4580(00)00124-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aliev, G., Ashraf, G. M., Kaminsky, Y. G., Sheikh, I. A., Sudakov, S. K., Yakhno, N. N., et al. (2013). Implication of the nutritional and nonnutritional factors in the context of preservation of cognitive performance in patients with dementia/depression and Alzheimer disease. Am. J. Alzheimers Dis. Other Demen. 28, 660–670. doi: 10.1177/1533317513504614
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Arbel-Ornath, M., Hudry, E., Eikermann-Haerter, K., Hou, S., Gregory, J. L., Zhao, L., et al. (2013). Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer’s disease mouse models. Acta Neuropathol. 126, 353–364. doi: 10.1007/s00401-013-1145-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Arcoverde, C., Deslandes, A., Moraes, H., Almeida, C., Araujo, N. B., Vasques, P. E., et al. (2014). Treadmill training as an augmentation treatment for Alzheimer’s disease: a pilot randomized controlled study. Arq. Neuropsiquiatr. 72, 190–196. doi: 10.1590/0004-282x20130231
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Banks, W. A., Terrell, B., Farr, S. A., Robinson, S. M., Nonaka, N., and Morley, J. E. (2002). Passage of amyloid beta protein antibody across the blood brain barrier in a mouse model of Alzheimer’s disease. Peptides 23, 2223–2226. doi: 10.1016/S0196-9781(02)00261-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baño Otalora, B., Popovic, N., Gambini, J., Popovic, M., Viña, J., Bonet-Costa, V., et al. (2012). Circadian system functionality, hippocampal oxidative stress and spatial memory in the APPswe/PS1dE9 transgenic model of Alzheimer disease: effects of melatonin or ramelteon. Chronobiol. Internat. 29, 822–834. doi: 10.3109/07420528.2012.699119
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barbagallo, M., and Dominguez, L. J. (2014). Type 2 diabetes mellitus and Alzheimer’s disease. World J. Diabetes 5, 889–893. doi: 10.4239/wjd.v5.i6.889
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barnard, N. D., Bush, A. I., Ceccarelli, A., Cooper, J., de Jager, C. A., Erickson, K. I., et al. (2014). Dietary and lifestyle guidelines for the prevention of Alzheimer’s disease. Neurobiol. Aging 35(Suppl. 2), S74–S78. doi: 10.1016/j.neurobiolaging.2014.03.033
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Benedict, C., Byberg, L., Cedernaes, J., Hogenkamp, P. S., Giedratis, V., Kilander, L., et al. (2014). Self-reported sleep disturbance is associated with Alzheimer’s disease risk in men. Alzheimers Dement. doi: 10.1016/j.jalz.2014.08.104. [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bierhaus, A., Humpert, P. M., Morcos, M., Wendt, T., Chavakis, T., Arnold, B., et al. (2005). Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. (Berl). 83, 876–886. doi: 10.1007/s00109-005-0688-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Biessels, G. J., Staekenborg, S., Brunner, E., Brayne, C., and Scheltens, P. (2006). Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 5, 64–74. doi: 10.1016/s1474-4422(05)70284-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Black, S. E., Gao, F., and Bilbao, J. (2009). Understanding white matter disease: imaging-pathological correlations in vascular cognitive impairment. Stroke 40(Suppl. 3), S48–S52. doi: 10.1161/strokeaha.108.537704
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Britsch, S. (2007). The Neuregulin-I/ErbB Signaling System in Development and Disease (Vol. 190). Berlin: Springer Science and Business Media.
Britschgi, M., Olin, C. E., Johns, H. T., Takeda-Uchimura, Y., LeMieux, M. C., Rufibach, K., et al. (2009). Neuroprotective natural antibodies to assemblies of amyloidogenic peptides decrease with normal aging and advancing Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 106, 12145–12150. doi: 10.1073/pnas.0904866106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Carare, R. O., Bernardes-Silva, M., Newman, T. A., Page, A. M., Nicoll, J. A. R., Perry, V. H., et al. (2008). Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol. Appl. Neurobiol. 34, 131–144. doi: 10.1111/j.1365-2990.2007.00926.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Centers for Disease Control and Prevention. (2014). Estimates of Diabetes and Its Burden in the United States. National Diabetes Statistics Report. Atlanta, GA: US Department of Health and Human Services. Available online at: http://www.cdc.gov/diabetes/pubs/statsreport14.htm
Ciccone, M. M., Scicchitano, P., Zito, A., Cortese, F., Boninfante, B., Falcone, V. A., et al. (2014). Correlation between inflammatory markers of atherosclerosis and carotid intima-media thickness in obstructive sleep Apnea. Molecules 19, 1651–1662. doi: 10.3390/molecules19021651
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cipriani, G., Lucetti, C., Danti, S., and Nuti, A. (2014). Sleep disturbances and dementia. Psychogeriatrics 15, 65–74. doi: 10.1111/psyg.12069
Coogan, A. N., Schutová, B., Husung, S., Furczyk, K., Baune, B. T., Kropp, P., et al. (2013). The circadian system in Alzheimer’s disease: disturbances, mechanisms and opportunities. Biol. Psychiatry 74, 333–339. doi: 10.1016/j.biopsych.2012.11.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cooke, J. R., Ancoli-Israel, S., Liu, L., Loredo, J. S., Natarajan, L., Palmer, B. S., et al. (2009). Continuous positive airway pressure deepens sleep in patients with Alzheimer’s disease and obstructive sleep apnea. Sleep Med. 10, 1101–1106. doi: 10.1016/j.sleep.2008.12.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cserr, H. F., Cooper, D. N., Suri, P. K., and Patlak, C. S. (1981). Efflux of radiolabeled polyethylene glycols and albumin from rat brain. Am. J. Physiol. 240, F319–F328.
Daulatzai, M. A. (2012). Quintessential risk factors: their role in promoting cognitive dysfunction and Alzheimer’s disease. Neurochem. Res. 37, 2627–2658. doi: 10.1007/s11064-012-0854-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
de Andrade, L. P., Gobbi, L. T. B., Coelho, F. G. M., Christofoletti, G., Costa, J. L. R., and Stella, F. (2013). Benefits of multimodal exercise intervention for postural control and frontal cognitive functions in individuals with Alzheimer’s disease: a controlled trial. J. Am. Geriatr. Soc. 61, 1919–1926. doi: 10.1111/jgs.12531
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Deane, R., Du Yan, S., Submamaryan, R. K., LaRue, B., Jovanovic, S., Hogg, E., et al. (2003). RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 9, 907–913. doi: 10.1038/nm890
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Deanne, R., Wu, Z., Sagare, A., Davis, J., Du Yan, S., Hamm, K., et al. (2004). LRP/amyloid-beta peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 45, 333–344. doi: 10.1016/j.neuron.2004.07.017
Deckers, K., van Boxtel, M. P. J., Schiepers, O. J. G., de Vugt, M., Muñoz Sánchez, J. L., Anstey, K. J., et al. (2015). Target risk factors for dementia prevention: a systematic review and Delphi consensus study on the evidence from observational studies. Int. J. Geriatr. Psychiatry 30, 234–246. doi: 10.1002/gps.4245
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
DeMattos, R. B., Bales, K. R., Cummins, D. J., Dodart, J. C., Paul, S. M., and Holtzman, D. M. (2001). Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 98, 8850–8855. doi: 10.1073/pnas.151261398
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
DeMattos, R. B., Bales, K. R., Cummins, D. J., Paul, S. M., and Holtzman, D. M. (2002). Brain to plasma amyloid-β efflux: a measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science 295, 2264–2267. doi: 10.1126/science.1067568
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Di Meco, A., Joshi, Y. B., and Praticò, D. (2014). Sleep deprivation impairs memory, tau metabolism and synaptic integrity of a mouse model of Alzheimer’s disease with plaques and tangles. Neurobiol. Aging 35, 1813–1820. doi: 10.1016/j.neurobiolaging.2014.02.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Donahue, J. E., Flaherty, S. L., Johanson, C. E., Duncan, J. A. 3rd, Silverberg, G. D., Miller, M. C., et al. (2006). RAGE, LRP-1 and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 112, 405–415. doi: 10.1007/s00401-006-0115-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Doody, R. S., Thomas, R. G., Farlow, M., Iwatsubo, T., Vellas, B., Joffe, S., et al. (2014). Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 311–321. doi: 10.1056/NEJMoa1312889
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dorr, A., Sahota, B., Chinta, L. V., Brown, M. E., Lai, A. Y., Ma, K., et al. (2012). Amyloid-β-dependent compromise of microvascular structure and function in a model of Alzheimer’s disease. Brain 135(Pt. 10), 3039–3050. doi: 10.1093/brain/aws243
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Farlow, M., Arnold, S. E., van Dyck, C. H., Aisen, P. S., Snider, B. J., Porsteinsson, A. P., et al. (2012). Safety and biomarker effects of solanezumab in patients with Alzheimer’s disease. Alzheimers Dement. 8, 261–271. doi: 10.1016/j.jalz.2011.09.224
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Farris, W., Mansourian, S., Chang, Y., Lindsley, L., Eckman, E. A., Frosch, M. P., et al. (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid-beta-protein and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. U S A 100, 4162–4167. doi: 10.1073/pnas.0230450100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Farris, W., Mansourian, S., Leissring, M. A., Eckman, E. A., Bertram, L., Eckman, C. B., et al. (2004). Partial loss-of-function mutations in insulin-degrading enzyme that induce diabetes also impair degradation of amyloid β-protein. Am. J. Pathol. 164, 1425–1434. doi: 10.1016/s0002-9440(10)63229-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Floyd, J. A., Medler, S. M., Ager, J. W., and Janisse, J. J. (2000). Age-related changes in initiation and maintenance of sleep: a meta-analysis. Res. Nurs. Health 23, 106–117. doi: 10.1002/(sici)1098-240x(200004)23:2<106::aid-nur3>3.0.co;2-a
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Forman, M., Tseng, J., Palcza, J., Leempoels, J., Ramael, S., Krishna, G., et al. (2012). “The novel BACE inhibitor MK-8931 dramatically lowers CSF Aβ peptides in healthy subjects: results from a rising single dose study (PL02. 004),” in 64th American Academy of Neurology Annual Meeting (New Orleans: Neurology).
Frackowiak, J., Wisniewski, H. M., Wegiel, J., Merz, G. S., Iqbal, K., and Wang, K. C. (1992). Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol. 84, 225–233. doi: 10.1007/bf00227813
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Frink, R. J. (2002). Inflammatory Atherosclerosis: Characteristics of the Injurious Agent. Sacramento (CA): Heart Research Foundation.
Funato, H., Yoshimura, M., Yamazaki, T., Saido, T. C., Ito, Y., Yokofijita, J., et al. (1998). Astrocytes containing amyloid-b protein (Ab) positive granules are associated with Ab40-positive diffuse plaques in the aged human brain. Am. J. Pathol. 152, 983–992.
Gabathuler, R. (2010). Approaches to transport therapeutic drugs across the blood-brain barrier to treat brain diseases. Neurobiol. Dis. 37, 48–57. doi: 10.1016/j.nbd.2009.07.028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gaberel, T., Gakuba, C., Goulay, R., Martinez De Lizarrondo, S., Hanouz, J.-L., Emery, E., et al. (2014). Impaired glymphatic perfusion after strokes revealed by contrast-enhanced MRI: a new target for fibrinolysis? Stroke 45, 3092–3096. doi: 10.1161/strokeaha.114.006617
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gao, F., Noor, R., Keith, J., Nadeau, Y., Kiss, A., and Black, S. (2012). Relationship between collagenosis of the deep medullary veins and periventricular white matter hyperintensities on magnetic resonance imaging in Alzheimer’s disease: does one size fit all? Alzheimers Dement. 8(Suppl. 4), P297. doi: 10.1016/j.jalz.2012.05.2146
Gavin, A. L., Barnes, N., Dijstelbloem, H. M., and Hogarth, P. M. (1998). Cutting edge: identification of the mouse IgG3 receptor: implications for antibody effector function at the interface between innate and adaptive immunity. J. Immunol. 160, 20–23.
Graf, A., Riviere, M. E., Caputo, A., Farlow, M. R., Marotta, G., Sanchez-Valle, R., et al. (2014). Active Aβ immunotherapy cad106 phase ii dose-adjuvant finding study: safety and CNS biomarkers. Alzheimers Dement. 10, P274. doi: 10.1016/j.jalz.2014.04.448
Guénette, S. Y. (2003). Astrocytes: a cellular player in Aβ clearance and degradation. Trends Mol. Med. 9, 279–280. doi: 10.1016/s1471-4914(03)00112-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gustafson, D., Rothenberg, E., Blennow, K., Steen, B., and Skoog, I. (2003). An 18-year follow-up of overweight and risk of Alzheimer disease. Arch. Intern. Med. 163, 1524–1528. doi: 10.1001/archinte.163.13.1524
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hadaczek, P., Yamashita, Y., Mirek, H., Tamas, L., Bohn, M. C., Noble, C., et al. (2006). The “perivascular pump” driven by arterial pulsation is a powerful mechanism for the distribution of therapeutic molecules within the brain. Mol. Ther. 14, 69–78. doi: 10.1016/j.ymthe.2006.02.018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Haeberlein, S. B., Cebers, G., Höglund, K., Salter, H., Eketjäll, S., Bogstedt, A., et al. (2013). AZD3293, a potent and selective orally active, brain-permeable BACE1 inhibitor. Alzheimers Dement. J. Alzheimers Assoc. 9:P813. doi: 10.1016/j.jalz.2013.05.1689
Han, S. W., Lee, S.-S., Kim, S. H., Lee, J. H., Kim, G. S., Kim, O.-J., et al. (2013). Effect of cilostazol in acute lacunar infarction based on pulsatility index of transcranial Doppler (ECLIPse): a multicenter, randomized, double-blind, placebo-controlled trial. Eur. Neurol. 69, 33–40. doi: 10.1159/000338247
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hanyu, H., Hirao, K., Shimizu, S., Sato, T., Kiuchi, A., and Iwamoto, T. (2007). Nilvadipine prevents cognitive decline of patients with mild cognitive impairment. Int. J. Geriatr. Psychiatry 22, 1264–1266. doi: 10.1002/gps.1851
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hardy, J., and Selkoe, D. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hawkes, C. A., Deng, L.-H., Fenili, D., Nitz, M., and McLaurin, J. (2012). In vivo uptake of β-amyloid by non-plaque associated microglia. Curr. Alzheimer Res. 9, 890–901. doi: 10.2174/156720512803251084
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hawkes, C. A., Gatherer, M., Sharp, M. M., Dorr, A., Yuen, H. M., Kalaria, R., et al. (2013). Regional differences in the morphological and functional effects of aging on cerebral basement membranes and perivascular drainage of amyloid-β from the mouse brain. Aging Cell 12, 224–236. doi: 10.1111/acel.12045
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hawkes, C. A., Gentleman, S. M., Nicoll, J. A., and Carare, R. O. (2015). Prenatal high-fat diet alters the cerebrovasculature and clearance of β-amyloid in adult offspring. J. Pathol. 235, 619–631. doi: 10.1002/path.4468
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hawkes, C. A., Jayakody, N., Johnston, D. A., Bechmann, I., and Carare, R. O. (2014). Failure of perivascular drainage of β-amyloid in cerebral amyloid angiopathy. Brain Pathol. 24, 396–403. doi: 10.1111/bpa.12159
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hawkes, C. A., and McLaurin, J. (2009). Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. U S A 106, 1261–1266. doi: 10.1073/pnas.0805453106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hoffmann, K., Frederiksen, K. S., Sobol, N. A., Beyer, N., Vogel, A., Simonsen, A. H., et al. (2013). Preserving cognition, quality of life, physical health and functional ability in Alzheimer’s disease: the effect of physical exercise (ADEX trial): rationale and design. Neuroepidemiology 41, 198–207. doi: 10.1159/000354632
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Höglund, K., Salter, H., Zetterberg, H., Andreason, U., Olsson, T., Alexander, R., et al. (2014). Monitoring the soluble amyloid precursor protein alpha (sAPPα) and beta (sAPPβ) fragments in plasma and CSF from healthy individuals treated with BACE inhibitor AZD3293 in a multiple ascending dose study: pharmacokinetic and pharmacodynamic correlate. Alzheimers Dement. J. Alzheimers Assoc. 10:P447. doi: 10.1016/j.jalz.2014.05.605
Ihara, M., Nishino, M., Taguchi, A., Yamamoto, Y., Hattori, Y., Saito, S., et al. (2014). Cilostazol add-on therapy in patients with mild dementia receiving donepezil: a retrospective study. PLoS One 9:e89516. doi: 10.1371/journal.pone.0089516
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ikeda, Y. (1999). Antiplatelet therapy using cilostazol, a specific PDE3 inhibitor. Thromb. Haemost. 82, 435–438.
Iliff, J. J., Lee, H., Yu, M., Feng, T., Logan, J., Nedergaard, M., et al. (2013a). Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J. Clin. Invest. 123, 1299–1309. doi: 10.1172/JCI67677
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iliff, J. J., and Nedergaard, M. (2013). Is there a cerebral lymphatic system? Stroke 44, S93–S95. doi: 10.1161/STROKEAHA.112.678698
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iliff, J. J., Wang, M., Liao, Y., Plog, B. A., Peng, W., Gundersen, G. A., et al. (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 4:147ra111. doi: 10.1126/scitranslmed.3003748
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iliff, J. J., Wang, M., Zeppenfeld, D. M., Venkataraman, A., Plog, B. A., Liao, Y., et al. (2013b). Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J. Neurosci. 33, 18190–18199. doi: 10.1523/JNEUROSCI.1592-13.2013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iwata, N., Takaki, Y., Fukami, S., Tsubuki, S., and Saido, T. C. (2002). Region-specific reduction of Aβ degrading endopeptidase, neprilysin, in mouse hippocampus upon aging. J. Neurosci. Res. 70, 493–500. doi: 10.1002/jnr.10390
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iwata, N., Tsubuki, S., Takaki, Y., Shirotani, K., Lu, B., Gerard, N. P., et al. (2001). Metabolic regulation of brain Aα by neprilysin. Science 292, 1550–1552. doi: 10.1126/science.1059946
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iwata, N., Tsubuki, S., Takaki, Y., Watanabe, K., Sekiguchi, M., Hosoki, E., et al. (2000). Identification of the major Abeta 1–42 degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat. Med. 6, 143–150. doi: 10.1038/72237
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Janson, J., Laedtke, T., Parisi, J. E., Brien, P. O., Petersen, R. C., and Butler, P. C. (2004). Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 53, 474–481. doi: 10.2337/diabetes.53.2.474
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Janssens, J. P., Pautex, S., Hilleret, H., and Michel, J. P. (2000). Sleep disordered breathing in the elderly. Aging (Milano) 12, 417–429.
Johanson, C., Flaherty, S., Messier, A., Duncan, J. I., and Silverberg, G. (2006). Expression of the beta-amyloid transporter, LRP-1, in aging choroid plexus: implications for the CSF-brain system in NPH and Alzheimer’s disease. Cerebrospinal Fluid Res. 3:S29. doi: 10.1186/1743-8454-3-S1-S29
Kalaria, R. N., and Hedera, P. (1995). Differential degeneration of the cerebral microvasculature in Alzheimer’s disease. Neuroreport 6, 477–480. doi: 10.1097/00001756-199502000-00018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kanekiyo, T., and Bu, G. (2014). The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front. Aging Neurosci. 6:93. doi: 10.3389/fnagi.2014.00093
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kang, J.-E., Lim, M. M., Bateman, R. J., Lee, J. L., Smyth, L. P., Cirrito, J. R., et al. (2009). Amyloid-β dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007. doi: 10.1126/science.1180962
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kivipelto, M., and Solomon, A. (2008). Alzheimer’s disease - the ways of prevention. J. Nutr. Health Aging 12, 89S–94S. doi: 10.1007/bf02982595
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Koistinaho, M., Lin, S., Wu, X., Esterman, M., Koger, D., Hanson, J., et al. (2004). Apolipoprotein E promotes astrocytes colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 10, 719–726. doi: 10.1038/nm1058
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kress, B. T., Iliff, J. J., Xia, M., Wang, M., Wei, H., Zeppenfeld, D., et al. (2014). Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 76, 845–861. doi: 10.1002/ana.24271
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kuhn, P. H., Koroniak, K., Hogl, S., Colombo, A., Zeitschel, U., Willem, M., et al. (2012). Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 31, 3157–3168. doi: 10.1038/emboj.2012.173
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kurz, A., and Perneczky, R. (2011). Amyloid clearance as a treatment target against Alzheimer’s disease. J. Alzheimers Dis. 24, 61–73. doi: 10.3233/JAD-2011-102139
Kwee, R. M., and Kwee, T. C. (2007). Virchow-Robin spaces at MR imaging. Radiographics 27, 1071–1086. doi: 10.1148/rg.274065722
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lai, R., Albala, B., Kaplow, J. M., Aluri, J., Yen, M., and Satlin, A. (2012). First-in-human study of E2609, a novel BACE1 inhibitor, demonstrates prolonged reductions in plasma beta-amyloid levels after single dosing. Alzheimers Dement. 8:P96. doi: 10.1016/j.jalz.2012.05.237
Lai, A. Y., Dorr, A., Thomason, L. A. M., Koletar, M. M., Sled, J. G., Stefanovic, B., et al. (2015). Venular degeneration leads to vascular dysfunction in a transgenic model of Alzheimer’s disease. Brain 138, 1046–1058. doi: 10.1093/brain/awv023
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lai, A. Y., and McLaurin, J. (2012). Clearance of amyloid-b by microglia and macrophages: the issue of what, when and where. Future Neurol. 7, 165–176. doi: 10.2217/fnl.12.6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, J., Duan, W., Long, J. M., Ingram, D. K., and Mattson, M. P. (2000). Dietary restriction increases the number of newly generated neural cells and induces BDNF expression, in the dentate gyrus of rats. J. Mol. Neurosci. 15, 99–108. doi: 10.1385/jmn:15:2:99
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, J., Duan, W., and Mattson, M. P. (2002). Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J. Neurochem. 82, 1367–1375. doi: 10.1046/j.1471-4159.2002.01085.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, C. Y., and Landreth, G. E. (2010). The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 117, 949–960. doi: 10.1007/s00702-010-0433-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Leissring, M. A., Farris, W., Chang, Y., Walsh, D. M., Wu, X., Sun, X., et al. (2003). Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology and premature death. Neuron 40, 1087–1093. doi: 10.1016/s0896-6273(03)00787-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liguori, C., Placidi, F., Albanese, M., Nuccetelli, M., Izzi, F., Marciani, M. G., et al. (2014). CSF beta-amyloid levels are altered in narcolepsy: a link with the inflammatory hypothesis? J. Sleep Res. 23, 420–424. doi: 10.1111/jsr.12130
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lim, Y. Y., Pietrzak, R. H., Bourgeat, P., Ames, D., Ellis, K. A., Rembach, A., et al. (2015). Relationships between performance on the cogstate brief battery, neurodegeneration and Aβ accumulation in cognitively normal older adults and adults with MCI. Arch. Clin. Neuropsychol. 30, 49–58. doi: 10.1093/arclin/acu068
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lin, T.-W., Shih, Y.-H., Chen, S.-J., Lien, C.-H., Chang, C.-Y., Huang, T.-Y., et al. (2015). Running exercise delays neurodegeneration in amygdala and hippocampus of Alzheimer’s disease (APP/PS1) transgenic mice. Neurobiol. Learn. Mem. 118, 189–197. doi: 10.1016/j.nlm.2014.12.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, C.-C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. doi: 10.1038/nrneurol.2012.263
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, G., Yao, L., Liu, J., Jiang, Y., Ma, G., Chen, Z., et al. (2014). Cardiovascular disease contributes to Alzheimer’s disease: evidence from large-scale genome-wide association studies. Neurobiol. Aging 35, 786–792. doi: 10.1016/j.neurobiolaging.2013.10.084
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lobanova, I., and Qureshi, A. I. (2014). The association between cardiovascular risk factors and progressive hippocampus volume loss in persons with Alzheimer’s disease. J. Vasc. Interv. Neurol. 7, 52–55.
Lucas, F., Fukushima, T., and Nozaki, Y. (2012). Novel BACE1 inhibitor, E2609, lowers Aβ levels in the cerebrospinal fluid and plasma in nonhuman primates. Alzheimers Dement. 8:P224. doi: 10.1016/j.jalz.2012.05.2022
Luchsinger, J. A., Tang, M. X., Shea, S., and Mayeux, R. (2002). Caloric intake and the risk of Alzheimer disease. Arch. Neurol. 59, 1258–1263. doi: 10.1001/archneur.59.8.1258
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maki, T., Okamoto, Y., Carare, R. O., Hase, Y., Hattori, Y., Hawkes, C. A., et al. (2014). Phosphodiesterase III inhibitor promotes drainage of cerebrovascular β-amyloid. Ann. Clin. Transl. Neurol. 1, 519–533. doi: 10.1002/acn3.79
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mandrekar, S., Jiang, Q., Lee, C. Y., Koenigsknecht-Talboo, J., Holtzman, D., and Landreth, G. E. (2009). Microglia mediate the clearance of soluble Aβ through fluid phase macropinocytosis. J. Neurosci. 29, 4252–4262. doi: 10.1523/JNEUROSCI.5572-08.2009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marques, F., Sousa, J. C., Sousa, N., and Palha, J. A. (2013). Blood-brain-barriers in aging and Alzheimer’s disease. Mol. Neurosci. 8:38. doi: 10.1186/1750-1326-8-38
Maruszak, A., Pilarski, A., Murphy, T., Branch, N., and Thuret, S. (2014). Hippocampal neurogenesis in Alzheimer’s disease: is there a role for dietary modulation? J. Alzheimers Dis. 38, 11–38. doi: 10.3233/JAD-131004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mattson, M. P. (2015). Lifelong brain health is a lifelong challenge: from evolutionary principles to empirical evidence. Ageing Res. Rev. 20, 37–45. doi: 10.1016/j.arr.2014.12.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mawuenyega, K. G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., et al. (2010). Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330:1774. doi: 10.1126/science.1197623
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McLaurin, J., Kierstead, M. E., Brown, M. E., Hawkes, C. A., Lambermon, M. H. L., Phinney, A. L., et al. (2006). Cyclohexanehexol-based inhibitors of Ab-aggregation prevent and reverse Alzheimer-like features in a transgenic model of Alzheimer disease. Nat. Med. 12, 801–808. doi: 10.1038/nm1423
Michaud, M., Balardy, L., Moulis, G., Gaudin, C., Peyrot, C., Vellas, B., et al. (2013b). Proinflammatory cytokines, aging and age-related diseases. J. Am. Med. Dir. Assoc. 14, 877–882. doi: 10.1016/j.jamda.2013.05.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Michaud, J.-P., Bellavance, M.-A., Préfontaine, P., and Rivest, S. (2013a). Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell Rep. 5, 646–653. doi: 10.1016/j.celrep.2013.10.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Monsonego, A., Maron, R., Zota, V., Selkoe, D. J., and Weiner, H. L. (2001). Immune hyporesponsiveness to amyloid β-peptide in amyloid precursor protein transgenic mice: implications for the pathogenesis and treatment of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 98, 10273–10278. doi: 10.1073/pnas.191118298
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Monsonego, A., Zota, V., Karni, A., Krieger, J. I., Bar-Or, A., Bitan, G., et al. (2003). Increased T cell reactivity to amyloid β protein in older humans and patients with Alzheimer disease. J. Clin. Invest. 112, 415–422. doi: 10.1172/jci18104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Montagne, A., Barnes, S. R., Sweeney, M. D., Halliday, M. R., Sagare, A. P., Zhao, Z., et al. (2015). Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302. doi: 10.1016/j.neuron.2014.12.032
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Moody, D. M., Brown, W. R., Challa, V. R., and Anderson, R. L. (1995). Periventricular venous collagenosis: association with leukoaraiosis. Radiology 194, 469–476. doi: 10.1148/radiology.194.2.7824728
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Morgan, D. (2011). Immunotherapy for Alzheimer’s disease. J. Intern. Med. 269, 54–63. doi: 10.1111/j.1365-2796.2010.02315.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Muhs, A., Hickman, D. T., Pihlgren, M., Chuard, N., Giriens, V., Meerschman, C., et al. (2007). Liposomal vaccines with conformation-specific amyloid peptide antigens define immune response and efficacy in APP transgenic mice. Proc. Natl. Acad. Sci. U S A 104, 9810–9815. doi: 10.1073/pnas.0703137104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nagele, R. G., D’Andrea, M. R., Lee, H., Venkataraman, V., and Wang, H. Y. (2003). Astrocytes accumulate Abeta 42 and give rise to astrocytic amyloid plaques in Alzheimer’s disease brains. Brain Res. 971, 197–209. doi: 10.1016/s0006-8993(03)02361-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nascimento, C. M. C., Teixeira, C. V. L., Gobbi, L. T. B., Gobbi, S., and Stella, F. (2012). A controlled clinical trial on the effects of exercise on neuropsychiatric disorders and instrumental activities in women with Alzheimer’s disease. Rev. Bras. Fisioter. 16, 197–204. doi: 10.1590/s1413-35552012005000017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
National Heart, Lung, and Blood Institute (2012). What is Sleep Apnea? US Department of Health and Human Services. Available online at: http://www.nhlbi.nih.gov/health/health-topics/topics/sleepapnea
Nichol, K. E., Poon, W. W., Parachikova, A. I., Cribbs, D. H., Glabe, C. G., and Cotman, C. W. (2008). Exercise alters the immune profile in Tg2576 Alzheimer mice toward a response coincident with improved cognitive performance and decreased amyloid. J. Neuroinflammation 5:13. doi: 10.1186/1742-2094-5-13
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nicolau, C., Greferath, R., Balaban, T. S., Lazarte, J. E., and Hopkins, R. J. (2002). A liposome-based therapeutic vaccine against β-amyloid plaques on the pancreas of transgenic NORBA mice. Proc. Natl. Acad. Sci. U S A 99, 2332–2337. doi: 10.1073/pnas.022627199
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nimmrich, V., and Eckert, A. (2013). Calcium channel blockers and dementia. Br. J. Pharmacol. 169, 1203–1210. doi: 10.1111/bph.12240
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Okonkwo, O. C., Schultz, S. A., Oh, J. M., Larson, J., Edwards, D., Cook, D., et al. (2014). Physical activity attenuates age-related biomarker alterations in preclinical AD. Neurology 83, 1753–1760. doi: 10.1212/WNL.0000000000000964
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Olsson, E., Karlström, B., Kilander, L., Byberg, L., Cederholm, T., and Sjögren, P. (2015). Dietary patterns and cognitive dysfunction in a 12-year follow-up study of 70 year old men. J. Alzheimers Dis. 43, 109–119. doi: 10.3233/JAD-140867
Ooms, S., Overeem, S., Besse, K., Rikkert, M. O., Verbeek, M., and Claassen, J. A. H. R. (2014). Effect of 1 night of total sleep deprivation on cerebrospinal fluid β-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol. 71, 971–977. doi: 10.1001/jamaneurol.2014.1173
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Origlia, N., Bonadonna, C., Rosellini, A., Leznik, E., Arancio, O., Yan, S. S., et al. (2010). Microglial receptor for advanced glycation end product-dependent signal pathway drives β-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex. J. Neurosci. 30, 11414–11425. doi: 10.1523/jneurosci.2127-10.2010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Origlia, N., Righi, M., Capsoni, S., Cattaneo, A., Fang, F., Stern, D. M., et al. (2008). Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-β-mediated cortical synaptic dysfunction. J. Neurosci. 28, 3521–3530. doi: 10.1523/jneurosci.0204-08.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pan, W., and Kastin, A. J. (2014). Can sleep apnea cause Alzheimer’s disease? Neurosci. Biobehav. Rev. 47, 656–669. doi: 10.1016/j.neubiorev.2014.10.019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Paoli, A., Bianco, A., Damiani, E., and Bosco, G. (2014). Ketogenic diet in neuromuscular and neurodegenerative diseases. Biomed. Res. Int. 2014:474296. doi: 10.1155/2014/474296
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pardridge, W. M. (2007). Blood-brain barrier delivery. Drug Discov. Today 12, 54–61. doi: 10.1016/j.drudis.2006.10.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pettersen, J. A., Sathiyamoorthy, G., Gao, F. Q., Szilagyi, G., Nadkarni, N. K., St George-Hyslop, P., et al. (2008). Microbleed topography, leukoaraiosis and cognition in probable Alzheimer disease from the Sunnybrook dementia study. Arch. Neurol. 65, 790–795. doi: 10.1001/archneur.65.6.790
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Qiu, W. Q., and Folstein, M. F. (2006). Insulin, insulin degrading enzyme and amyloid-beta peptide in Alzheimer’s disease; review and hypothesis. Neurobiol. Aging 27, 190–198. doi: 10.1016/j.neurobiolaging.2005.01.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Querfurth, H. W., and LaFerla, F. M. (2010). Alzheimer’s disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Racke, M. M., Boone, L. I., Hepburn, D. L., Parsadainian, M., Bryan, M. T., Ness, D. K., et al. (2005). Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid β. J. Neurosci. 25, 629–636. doi: 10.1523/jneurosci.4337-04.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rajapaksha, T. W., Eimer, W. A., Bozza, T. C., and Vassar, R. (2011). The Alzheimer ’s β-secretase enzyme BACE1 is required for accurate axon guidance of olfactory sensory neurons and normal glomerulus formation in the olfactory bulb. Mol. Neurodegener. 6:88. doi: 10.1186/1750-1326-6-88
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Reppert, S. M., and Weaver, D. R. (2001). Molecular analysis of mammalian circadian rhythms. Annu. Rev. Physiol. 63, 647–676. doi: 10.1146/annurev.physiol.63.1.647
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Revesz, T., Ghiso, J., Lashley, T., Plant, G., Rostagno, A., Frangione, B., et al. (2003). Cerebral amyloid angiopathies: a pathologic, biochemical and genetic view. J. Neuropathol. Exp. Neurol. 62, 885–898.
Revesz, T., Holton, J. L., Lashley, T., Plant, G., Rostagno, A., Ghiso, J., et al. (2002). Sporadic and familial cerebral amyloid angiopathies. Brain Pathol. 12, 343–357. doi: 10.1111/j.1750-3639.2002.tb00449.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Reynolds, C. A., Gatz, M., Prince, J. A., Berg, S., and Pedersen, N. L. (2010). Serum lipid levels and cognitive change in late life. J. Am. Geriatr. Soc. 58, 501–509. doi: 10.1111/j.1532-5415.2010.02739.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roher, A. E., Esh, C., Kokjohn, T. A., Kalback, W., Luehrs, D. C., Seward, J. D., et al. (2003). Circle of willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler. Thromb. Vasc. Biol. 23, 2055–2062. doi: 10.1161/01.atv.0000095973.42032.44
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ross, R. (1993). The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 362, 801–809. doi: 10.1038/362801a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rothman, S. M., Herdener, N., Frankola, K. A., Mughal, M. R., and Mattson, M. P. (2013). Chronic mild sleep restriction accentuates contextual memory impairments and accumulations of cortical Aβ and pTau in a mouse model of Alzheimer’s disease. Brain Res. 1529, 200–208. doi: 10.1016/j.brainres.2013.07.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sabbagh, M. N., Agro, A., Bell, J., Aisen, P. S., Schweizer, E., and Galasko, D. (2011). PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer’s disease. Alzheimer Dis. Assoc. Disord. 25, 206–212. doi: 10.1097/wad.0b013e318204b550
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sagare, A. P., Bell, R. D., and Zlokovic, B. V. (2013). Neurovascular defects and faulty amyloid-b vascular clearance in Alzheimer’s disease. J. Alzheimers Dis. 33, S87–S100. doi: 10.3233/JAD-2012-129037
Saito, S., and Ihara, M. (2014). New therapeutic approaches for Alzheimer’s disease and cerebral amyloid angiopathy. Front. Aging Neurosci. 6:290. doi: 10.3389/fnagi.2014.00290
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sakurai, H., Hanyu, H., Sato, T., Kume, K., Hirao, K., Kanetaka, H., et al. (2013). Effects of cilostazol on cognition and regional cerebral blood flow in patients with Alzheimer’s disease and cerebrovascular disease: a pilot study. Geriatr. Gerontol. Int. 13, 90–97. doi: 10.1111/j.1447-0594.2012.00866.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Samadi, H., and Sultzer, D. (2011). Solanezumab for Alzheimer’s disease. Expert Opin. Biol. Ther. 11, 787–798. doi: 10.1517/14712598.2011.578573
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schley, D., Carare-Nnadi, R., Please, C. P., Perry, V. H., and Weller, R. O. (2006). Mechanisms to explain the reverse perivascular transport of solutes out of the brain. J. Theor. Biol. 238, 962–974. doi: 10.1016/j.jtbi.2005.07.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sengillo, J. D., Winkler, E. A., Walker, C. T., Sullivan, J. S., Johnson, M., and Zlokovic, B. V. (2013). Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 23, 303–310. doi: 10.1111/bpa.12004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shibata, M., Yamada, S., Kumar, S. R., Calero, M., Bading, J., Frangione, B., et al. (2000). Clearance of Alzheimer’s amyloid-β(1–40) peptide from brain by LDL receptor-related protein-1 at the blood brain barrier. J. Clin. Invest. 106, 1489–1499. doi: 10.1172/jci10498
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sigurdsson, E. M., Knudsen, E., Asuni, A., Fitzer-Attas, C., Sage, D., Quartermain, D., et al. (2004). An attenuated immune response is sufficient to enhance cognition in an Alzheimer’s disease mouse model immunized with amyloid-β derivatives. J. Neurosci. 24, 6277–6282. doi: 10.1523/jneurosci.1344-04.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sperling, R. A., Aisen, P. S., Beckett, L. A., Bennett, D. A., Craft, S., Fagan, A. M., et al. (2011). Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 280–292. doi: 10.1016/j.jalz.2011.03.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Spira, A. P., Gamaldo, A. A., An, Y., Wu, M. N., Simonsick, E. M., Bilgel, M., et al. (2013). Self-reported sleep and β-amyloid deposition in community-dwelling older adults. JAMA Neurol. 70, 1537–1543. doi: 10.1001/jamaneurol.2013.4258
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Srikanth, V., Maczurek, A., Phan, T., Steele, M., Westcott, B., Juskiw, D., et al. (2011). Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 32, 763–777. doi: 10.1016/j.neurobiolaging.2009.04.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sterniczuk, R., Dyck, R. H., Laferla, F. M., and Antle, M. C. (2010). Characterization of the 3xTg-AD mouse model of Alzheimer’s disease: part 1. Circadian changes. Brain Res. 1348, 139–148. doi: 10.1016/j.brainres.2010.05.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stone, J., Kleijn, H. J., Dockendorf, M., Ma, L., Palcza, J., Tseng, J., et al. (2013). Consistency of Bace inhibitor-mediated brain amyloid production inhibition by MK-8931 in Alzheimer’s disease patients and healthy young adults. Alzheimers Dement. J. Alzheimers Assoc. 9, P690–P691. doi: 10.1016/j.jalz.2013.04.362
Stranahan, A. M., Martin, B., and Maudsley, S. (2012). Anti-inflammatory effects of physical activity in relationship to improved cognitive status in humans and mouse models of Alzheimers disease. Curr. Alzheimer Res. 9, 86–92. doi: 10.2174/156720512799015019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Suttanon, P., Hill, K. D., Said, C. M., Williams, S. B., Byrne, K. N., LoGiudice, D., et al. (2013). Feasibility, safety and preliminary evidence of the effectiveness of a home-based exercise programme for older people with Alzheimer’s disease: a pilot randomized controlled trial. Clin. Rehabil. 27, 427–438. doi: 10.1177/0269215512460877
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Szentistványi, I., Patlak, C. S., Elliss, R. A., and Cserr, H. F. (1984). Drainage of interstitial fluid from different regions of rat brain. Am. J. Physiol. 246, F835–F844.
Takakura, S., Sogabe, K., Satoh, H., Mori, J., Fujiwara, T., Totsuka, Z., et al. (1992). Nilvadipine as a neuroprotective calcium entry blocker in a rat model of global cerebral ischemia. A comparative study with nicardipine hydrochloride. Neurosci. Lett. 141, 199–202. doi: 10.1016/0304-3940(92)90894-d
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, B. L., and Chua, C. E. L. (2008). SIRT1 and neuronal diseases. Mol. Aspects Med. 29, 187–200. doi: 10.1016/j.mam.2007.02.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tanzi, R. E., Moir, R. D., and Wagner, S. L. (2004). Clearance of Alzheimer’s Aβ peptide: the many roads to perdition. Neuron 43, 605–608. doi: 10.1016/s0896-6273(04)00533-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thal, D. R., Griffin, W. S. T., de Vos, R. A., and Ghebremedhin, E. (2008). Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathol. 115, 599–609. doi: 10.1007/s00401-008-0366-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thal, D. R., Sassing, I., Schultz, C., Haas, C., Braak, E., and Braak, H. (1999). Fleecy amyloid deposits in the internal layers of the human entorhinal cortex are comprised of N-terminal truncated fragments of Aβ. J. Neuropathol. Exp. Neurol. 58, 210–216. doi: 10.1097/00005072-199902000-00010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tolppanen, A.-M., Solomon, A., Soininen, H., and Kivipelto, M. (2012). Midlife vascular risk factors and Alzheimer’s disease: evidence from epidemiological studies. J. Alzheimers Dis. 32, 531–540. doi: 10.3233/JAD-2012-120802
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., et al. (1999). β-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. doi: 10.1126/science.286.5440.735
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Venturelli, M., Scarsini, R., and Schena, F. (2011). Six-month walking program changes cognitive and ADL performance in patients with Alzheimer. Am. J. Alzheimers Dis. Other Demen. 26, 381–388. doi: 10.1177/1533317511418956
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Verbeek, M. M., Van Nostrand, W. E., Otte-Höller, I., Wesseling, P., and De Waal, R. M. (2000). Amyloid-β-induced degeneration of human brain pericytes is dependent on the apolipoprotein E genotype. Ann. N Y Acad. Sci. 903, 187–199. doi: 10.1111/j.1749-6632.2000.tb06368.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Videnovic, A., Lazar, A. S., Barker, R. A., and Overeem, S. (2014). ‘The clocks that time us’-circadian rhythms in neurodegenerative disorders. Nat. Rev. Neurol. 10, 683–693. doi: 10.1038/nrneurol.2014.206
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vidoni, E. D., Van Sciver, A., Johnson, D. K., He, J., Honea, R., Haines, B., et al. (2012). A community-based approach to trials of aerobic exercise in aging and Alzheimer’s disease. Contemp. Clin. Trials 33, 1105–1116. doi: 10.1016/j.cct.2012.08.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Viswanathan, A., Rocca, W. A., and Tzourio, C. (2009). Vascular risk factors and dementia: how to move forward? Neurology 72, 368–374. doi: 10.1212/01.wnl.0000341271.90478.8e
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Voss, M. W., Vivar, C., Kramer, A. F., and van Praag, H. (2013). Bridging animal and human models of exercise-induced brain plasticity. Trends Cogn. Sci. 17, 525–544. doi: 10.1016/j.tics.2013.08.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wan, R., Weigand, L. A., Bateman, R., Griffioen, K., Mendelowitz, D., and Mattson, M. P. (2014). Evidence that BDNF regulates heart rate by a mechanism involving increased brainstem parasympathetic neuron excitability. J. Neurochem. 129, 573–580. doi: 10.1111/jnc.12656
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, J., Ho, L., Qin, W., Rocher, A. B., Seror, I., Humala, N., et al. (2005). Caloric restriction attenuates beta-amyloid neuropathology in a mouse model of Alzheimer’s disease. FASEB J. 19, 659–661. doi: 10.1096/fj.04-3182fje
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, P., and Olbricht, W. L. (2011). Fluid mechanics in the perivascular space. J. Theor. Biol. 274, 52–57. doi: 10.1016/j.jtbi.2011.01.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weksler, M. E., Relkin, N., Turkenich, R., LaRusse, S., Zhou, L., and Szabo, P. (2002). Patients with Alzheimer disease have lower levels of serum anti-amyloid peptide antibodies than healthy elderly individuals. Exp. Gerontology 37, 943–948. doi: 10.1016/s0531-5565(02)00029-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weller, R. O. (2005). Microscopic morphology and histology of the human meninges. Morphologie 89, 22–34. doi: 10.1016/s1286-0115(05)83235-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weller, R. O., Boche, D., and Nicoll, J. A. R. (2009). Microvasculature changes and cerebral amyloid angiopathy in Alzheimer’s disease and their potential impact on therapy. Acta Neuropathol. 118, 87–102. doi: 10.1007/s00401-009-0498-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weller, R. O., Galea, I., Carare, R. O., and Minagar, A. (2010). Pathophysiology of the lymphatic drainage of the central nervous system: implications for pathogenesis and therapy of multiple sclerosis. Pathophysiology 17, 295–306. doi: 10.1016/j.pathophys.2009.10.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weller, R. O., Subash, M., Preston, S. D., Mazanti, I., and Carare, R. O. (2008). Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 18, 253–266. doi: 10.1111/j.1750-3639.2008.00133.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wiessner, C., Wiederhold, K. H., Tissot, A. C., Frey, P., Danner, S., Jacobson, L. H., et al. (2011). The second-generation active Aβ immunotherapy CAD106 reduces amyloid accumulation in APP transgenic mice while minimizing potential side effects. J. Neurosci. 31, 9323–9331. doi: 10.1523/JNEUROSCI.0293-11.2011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wilcock, D. M., Rojiani, A., Rosenthal, A., Levkowitz, G., Subbarao, S., Alamed, J., et al. (2004). Passive amyloid immunotherapy clears amyloid and transiently activated microglia in a transgenic mouse model of amyloid deposition. J. Neurosci. 24, 6144–6151. doi: 10.1523/jneurosci.1090-04.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Winblad, B., Andreasen, N., Minthon, L., Floesser, A., Imbert, G., Dumortier, T., et al. (2012). Safety, tolerability and antibody response of active Aβ immunotherapy with CAD106 in patients and with Alzheimer’s disease: randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 11, 597–604. doi: 10.1016/s1474-4422(12)70140-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Winchester, J., Dick, M. B., Gillen, D., Reed, B., Miller, B., Tinklenberg, J., et al. (2013). Walking stabilizes cognitive functioning in Alzheimer’s disease (AD) across one year. Arch. Gerontol. Geriatr. 56, 96–103. doi: 10.1016/j.archger.2012.06.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Witte, A. V., Fobker, M., Gellner, R., Knecht, S., and Flöel, A. (2009). Caloric restriction improves memory in elderly humans. Proc. Natl. Acad. Sci. U S A 106, 1255–1260. doi: 10.1073/pnas.0808587106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, P., Shen, Q., Dong, S., Xu, Z., Tsien, J. Z., and Hu, Y. (2008). Calorie restriction ameliorates neurodegenerative phenotypes in forebrain-specific presenilin-1 and presenilin-2 double knockout mice. Neurobiol. Aging 29, 1502–1511. doi: 10.1016/j.neurobiolaging.2007.03.028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wyatt, A. R., Yerbury, J. J., Dabbs, R. A., and Wilson, M. R. (2012). Roles of extracellular chaperones in amyloidosis. J. Mol. Biol. 421, 499–516. doi: 10.1016/j.jmb.2012.01.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wyss-Coray, T., Loike, J. D., Brionne, T. C., Lu, E., Anakov, R., Yan, F., et al. (2003). Adult mouse astrocytes degrade amyloid-b in vitro and in vivo. Nat. Med. 9, 453–457. doi: 10.1038/nm838
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xie, L., Kang, H., Xu, Q., Chen, M. J., Liao, Y., Thiyagarajan, M., et al. (2013). Sleep drives metabolite clearance from the adult brain. Science 342, 373–377. doi: 10.1126/science.1241224
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yan, S. D., Chen, X., Fu, J., Chen, M., Zhu, H., Roher, A., et al. (1996). RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 382, 685–691. doi: 10.1038/382685a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yan, S. S., Chen, D., Yan, S., Guo, L., and Chen, J. X. (2012). RAGE is a key cellular target for Aβ-induced perturbation in Alzheimer’s disease. Front. Biosci. (Schol. Ed.) 4, 240–250. doi: 10.2741/S265
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yanagisawa, D., Shirai, N., Amatsubo, T., Taguchi, H., Hirao, K., Urushitani, M., et al. (2010). Relationship between tautomeric stuctures of curcumin derivatives and their Abeta-binding activities in the context of therapies for Alzheimer’s disease. Biomaterials 31, 4179–4185. doi: 10.1016/j.biomaterials.2010.01.142
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, L., Kress, B. T., Weber, H. J., Thiyagarajan, M., Wang, B., Deane, R., et al. (2013). Evaluating glymphatic pathway function utilizing clinically relevant intrathecal infusion of CSF tracer. J. Transl. Med. 11:107. doi: 10.1186/1479-5876-11-107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yannakoulia, M., Kontogianni, M., and Scarmeas, N. (2015). Cognitive health and Mediterranean: just diet or lifestyle pattern? Ageing Res. Rev. 20, 74–78. doi: 10.1016/j.arr.2014.10.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yasojima, K., Akiyama, H., McGeer, E. G., and McGeer, P. L. (2001). Reduced neprilysin in high plaque areas of Alzheimer’s disease brain: a possible relationship to deficient degradation of beta-amyloid peptide. Neurosci. Lett. 297, 97–100. doi: 10.1016/s0304-3940(00)01675-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zelinski, E. L., Deibel, S. H., and McDonald, R. J. (2014). The trouble with circadian clock dysfunction: multiple deleterious effects on the brain and body. Neurosci. Biobehav. Rev. 40, 80–101. doi: 10.1016/j.neubiorev.2014.01.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhou, L., Barão, S., Laga, M., Bockstael, K., Borgers, M., Gijsen, H., et al. (2012). The neural cell adhesion molecules L1 and CHL1 are cleaved by BACE1 protease in vivo. J. Biol. Chem. 287, 25927–25940. doi: 10.1074/jbc.M112.377465
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: Alzheimer’s disease, amyloid-β peptide, clearance, vaccine, therapeutics, lifestyle factors
Citation: Morrone CD, Liu M, Black SE and McLaurin J (2015) Interaction between therapeutic interventions for Alzheimer’s disease and physiological Aβ clearance mechanisms. Front. Aging Neurosci. 7:64. doi: 10.3389/fnagi.2015.00064
Received: 25 January 2015; Accepted: 13 April 2015;
Published online: 05 May 2015.
Edited by:
George E. Barreto, Pontificia Universidad Javeriana, ColombiaReviewed by:
Emmanuel Planel, Centre Hospitalier de l’Université Laval, CanadaMicha Wilhelmus, VU University Medical Center, Netherlands
Copyright © 2015 Morrone, Liu, Black and McLaurin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: JoAnne McLaurin, Biological Sciences, Sunnybrook Research Institute, 2075 Bayview Ave., Room S113, Toronto, ON M4N 3M5, Canada, jmclaurin@sri.utoronto.ca