Aging Microglia—Phenotypes, Functions and Implications for Age-Related Neurodegenerative Diseases
 Björn Spittau1,2*
Björn Spittau1,2*- 1Department of Molecular Embryology, Faculty of Medicine, Institute for Anatomy and Cell Biology, University of Freiburg, Freiburg, Germany
- 2Institute of Anatomy, University of Rostock, Rostock, Germany
Aging of the central nervous system (CNS) is one of the major risk factors for the development of neurodegenerative pathologies such as Parkinson’s disease (PD) and Alzheimer’s disease (AD). The molecular mechanisms underlying the onset of AD and especially PD are not well understood. However, neuroinflammatory responses mediated by microglia as the resident immune cells of the CNS have been reported for both diseases. The unique nature and developmental origin of microglia causing microglial self-renewal and telomere shortening led to the hypothesis that these CNS-specific innate immune cells become senescent. Age-dependent and senescence-driven impairments of microglia functions and responses have been suggested to play essential roles during onset and progression of neurodegenerative diseases. This review article summarizes the current knowledge of microglia phenotypes and functions in the aging CNS and further discusses the implications of these age-dependent microglia changes for the development and progression of AD and PD as the most common neurodegenerative diseases.
Introduction
Microglia are the resident immune cells of the central nervous system (CNS) and are involved in a multifaceted range of physiological as well as pathophysiological functions (Prinz and Priller, 2014). Recent scientific efforts have resulted in the elucidation of the developmental origin, the molecular signature and the versatile functions of these macrophage-like glia cells and gained insight into their nature. Especially, the developmental origin of microglia and the associated phenomenon that these CNS-specific immune cells underlie a self-propelling regeneration causing telomere shortening has resulted in the hypothesis that microglia age, develop age-dependent cellular dystrophy and become senescent (Streit, 2006). Microglial senescence has been linked to functional changes that are high likely to contribute to an age-dependent increase of microglia-mediated neuroinflammatory responses, which are believed to further threaten aged neurons and, thus, drive the progression of age-related neurodegenerative pathologies, such as Alzheimer’s disease (AD) and Parkinson’s disease (PD). This review article summarizes the current knowledge of functional and phenotypic properties of aged microglia and highlights the contribution of aged microglia to the development and progression of AD and PD.
Microglia Origin
The origin of microglia has long been debated and initial studies suggested that microglia differ substantially from perivascular and meningeal macrophages, which were described to derive from blood-borne monocytes and are constantly replenished (Hickey and Kimura, 1988; Bechmann et al., 2001). Recent reports have demonstrated that both, CNS-specific macrophages and microglia do not develop from circulating monocytes. CNS macrophages including perivascular macrophages, meningeal macrophages and to a lesser extent choroid plexus macrophages arise from hematopoietic precursors during embryonic development in a PU.1-dependent manner and further establish stable cell populations that do not experience turnover and replacement from bone-marrow-derived cells (Goldmann et al., 2016). The developmental origin of microglia has been revealed by elegant fate-mapping experiments, which clearly demonstrated that PU.1 and Irf8-dependent pathways are crucial for microglial embryonic development (Kierdorf et al., 2013). Moreover, this development from yolk sac progenitors is independent of IL-34. However, microglia migration towards and population of the CNS parenchyma relies on neuronal expression and secretion of IL-34, which serves as the most potent homing factor for microglia precursors (Greter et al., 2012). In order to sense IL-34, the microglial expression of colony stimulating factor-1 receptor (CSF1R) is required. Interestingly, the presence of CSF1 itself is not necessary for proper microglia development and colonization of the CNS (Ginhoux et al., 2010). The development of sophisticated methods to immediately isolate microglia and to further analyze their respective gene expression patterns have resulted in the observation that microglia display a unique microglia transcriptome demonstrating that microglia are distinct from other macrophage populations (Beutner et al., 2013). The establishment of this microglia-specific gene expression profile in mice is restricted to the first postnatal weeks and includes distinct microglia genes such as Olfml3, Tmem119, Hexb and Fcrls, which indicate microglia maturation (Bennett et al., 2016). Butovsky et al. (2014) have demonstrated that this maturation is dependent on the presence of Transforming growth factor beta 1 (TGFβ1). Using an elegant transgenic mice approach to overcome the lethal inflammatory phenotype of TGFβ1 mutant mice, this seminal study demonstrated that lack of TGFβ1 in the CNS results in decreased microglia numbers and loss of the microglia-specific gene expression signature. However, the molecular mechanisms of microglia maturation are likely to be more complicated and may not entirely rely on CNS-specific mediators. Recently, the contribution of gut microbiota has been demonstrated to regulate microglia maturation and functions in the CNS (Erny et al., 2015).
Upon establishment of the blood-brain-barrier (BBB), microglia undergo a constant self-renewal in order to maintain their population and it has been shown that no progenitor recruitment from blood circulation is contributing to this maintenance of microglia numbers throughout the lifetime (Ajami et al., 2007). Next to proliferation, microglial apoptosis contributes to maintaining the turnover of microglia in the adult CNS (Askew et al., 2017). A challenging central question is whether microglial self-renewal is the result of asymmetric cell division of microglial precursors in the CNS. Using selective CSF1R inhibitors, Elmore et al. (2014) were able to show that microglia could be effectively depleted (90% depletion) from the CNS. Surprisingly, a complete repopulation was observed and was mediated through a proliferation of nestin-positive precursors, which finally differentiated into microglia (Elmore et al., 2014). These results suggest that the microglia turnover in the adult CNS is mediated from a yet unknown precursor cell population or even from a not yet located stem cell niche. It will be of utmost interest to further characterize the endogenous microglia replenishment in the healthy and diseased CNS.
Microglia Activation
The paradigm that microglia retain a resting state under non-pathological conditions and only react to endogenous and exogenous inflammatory stimuli has been critically evaluated by Nimmerjahn et al. (2005). Using 2-photon imaging, the authors have shown that microglia are constantly surveying their microenvironment in order to rapidly react and migrate towards impairments such as neuron death, BBB leakage or extracellular ATP accumulation (Nimmerjahn et al., 2005). This observation led to the conclusion that microglia might have important functions under physiological conditions. Recent reports have contributed to broaden our understanding that microglia capacities in the CNS include several essential features such as synaptic pruning (Stevens et al., 2007; Paolicelli et al., 2011; Hoshiko et al., 2012; Schafer et al., 2012), influencing functions of activated and/or over-activated neurons (Panatier and Robitaille, 2012), supporting cortical neuron survival (Ueno et al., 2013), shaping axonal projections (Pont-Lezica et al., 2014; Squarzoni et al., 2014), synapse formation during learning in the adult CNS (Parkhurst et al., 2013) as well as maintenance of synaptic functions in the mature retina (Wang et al., 2016). Although many of the abovementioned functions are mediated during the embryonic and postnatal development of the CNS, microglia support neuronal circuits and, thus, important neurological CNS functions throughout adulthood and aging.
Under pathological conditions microglia rapidly change their morphology and adopt activation states in order to adequately react to the activation-causing stimuli. Microglia proliferation has been reported after traumatic CNS injuries (Streit et al., 1999) and degenerative pathologies including optic nerve lesion (Wohl et al., 2010), AD (Kamphuis et al., 2012), prion disease (Gómez-Nicola et al., 2013), PD (Machado et al., 2016) or ischemia (Li et al., 2013).
Based on the nature of distinct microglia stimuli, pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) have been categorized and are detected by microglial receptors including Toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-like receptors (RLRs), AIM2-like receptors (ALRs) as well as C-type lectins (Kigerl et al., 2014). In analogy to macrophages, which display remarkable plasticities in response to distinct environmental triggers (Mosser and Edwards, 2008), microglia activation has been subdivided into M1-like and M2-like activation states (Prinz and Priller, 2014). Although M1- and M2-like microglia polarization can be sufficiently induced in vitro by using Th1 cytokines such as IFNγ (Zhou et al., 2015) and Th2 cytokines such as IL4 (Zhou et al., 2012), these distinct activation patterns do not seem to be applicable in vivo (Ransohoff, 2016). Moreover, macrophage activation patterns classified as M1 and M2 further needs thorough revision due to increasing knowledge about distinct macrophage stimuli (Martinez and Gordon, 2014). With regard to their unique molecular signatures, resident microglia have to be distinguished from infiltrating monocytes/macrophages in terms of gene expression patterns, surface receptors and functions, respectively. Especially under pathological conditions microglia and macrophages display distinct functional properties. Infiltrating monocytes are able to contribute to the resident microglia pool during experimental autoimmune encephalomyelitis (EAE) and have been demonstrated to trigger EAE progression in this context (Ajami et al., 2011). Moreover, T-cell mediated macrophage activation seems to be essential for inflammatory demyelination processes observed during EAE (Yamasaki et al., 2014). Similar observations for monocyte/macrophage contribution to neuroinflammation and subsequent neurodegeneration has been reported for generalized seizures an animal model for epilepsy (Varvel et al., 2016). Of note, inflammatory M1 macrophages entering the aged brain due to BBB impairments have been demonstrated to negatively regulate synaptic functions and LTP formation in the hippocampus (Costello et al., 2016).
Recently identified microglia-specific genes such as Tmem119 (Bennett et al., 2016) or Fcrls (Butovsky et al., 2014) and/or sophisticated microglia isolation protocols (de Haas et al., 2007) can be used in order to identify region-specific microglial differences. Differences in microglial surface receptor expressions (de Haas et al., 2008) and gene expression patterns (Doorn et al., 2015) have been reported and suggest the existence of different functional distinct microglia subpopulations in the CNS. This microglial diversity may be responsible for regionally localized homeostatic functions and might further underlie region-specific sensitivities to microglial dysregulation and involvement in age-related neurodegenerative processes (Grabert et al., 2016).
Aging Microglia: Priming, Functions and Phenotypes
In healthy rodents, microglia make up 5%–12% of all CNS-specific cells. However, the distribution is diverse and some brain areas display higher densities of microglia (Lawson et al., 1990). Interestingly, the nigrostriatal system including substantia nigra (SN) and the caudate putamen (CPu) show significantly higher microglia densities than adjacent brain regions (Sharaf et al., 2013). Similar observations have been made in humans where microglia represent 0.5%–16.6% of all cells in the brain parenchyma. Moreover, the numbers of microglia in the white matter are higher compared to gray matter (Mittelbronn et al., 2001). Several studies have tried to address the age-related changes in microglia numbers in different species with diverse results. Whereas no obvious changes in Iba1+ microglia numbers have been observed in the aged rat hippocampus (VanGuilder et al., 2011), reductions in microglia numbers were detected in the aged nigrostriatal system and cerebral cortex (Sharaf et al., 2013). However, in aged rhesus monkeys (25–35 years) the numbers of microglia increased and the cells displayed heterogeneous intracellular inclusions indicative of increased phagocytosis and reduced capacity to digest engulfed particles (Peters et al., 1991). Dystrophic changes in microglia have been detected in aged individuals and were much more prevalent in older subjects (68-year-old) than in the younger ones (38-year-old; Streit et al., 2004). Interestingly, it has been described that dystrophic or senescent microglia might undergo age-dependent degeneration and are believed to lose their neuroprotective functions, thereby, contributing to the age-dependent onset of sporadic AD (sAD; Streit et al., 2009). Altered microglia morphology and reduced arborization have been reported in the human brain during aging and in AD patients (Davies et al., 2016). It remains unclear whether the observed morphological changes are signs of microglia degeneration and a recent study suggests that the reported microglia dystrophy might reflect age-related cytoskeleton alterations (Tischer et al., 2016). Interestingly, direct Tau uptake by microglia has been reported (Bolós et al., 2016) and is enhanced by anti-tau antibodies in an Fc-receptor-dependent manner (Luo et al., 2015). Moreover, hyperphosphorylated Tau was detectable in aged common marmoset brains and was present in dystrophic microglia suggesting that clearance of pathological protein aggregates might foster microglia dystrophy (Rodriguez-Callejas et al., 2016).
Due to the unique nature of microglia and especially due to their self-renewal capacity, a telomere shortening was hypothesized during aging of microglia. Whereas cells with high proliferative potential, such as stem cells or cancer cells have increased telomerase activity in order to maintain telomere length, somatic cells with limited replication potential possess less telomerase activity (Satyanarayana et al., 2004; Blasco, 2007). Indeed, telomere shortening has been reported in cultured microglia (Flanary and Streit, 2004) and was associated with dementia in human AD brain samples (Flanary et al., 2007). However, it seems that mouse microglia do not develop dystrophy and telomere shortening under normal conditions due to the limited lifespan of mice. Transgenic approaches, such as the Ercc1 mutant mice, a DNA repair-deficient mouse model that displays features of accelerated aging in multiple tissues including the CNS have contributed to the understanding how aging might affect microglia functions. In Ercc1 mutant mice, microglia display hallmark features of priming and increased responses to systemic lipopolysaccharide (LPS) exposure as reflected by cytokine expression and phagocytosis (Raj et al., 2014). Mice lacking the telomerase RNA component (TERC) are characterized by an accelerated aging phenotype associated with enhanced microglia activation in response to LPS and a subsequent immune cell infiltration upon BBB dysregulation (Raj et al., 2015). These results indicate that microglial aging result in functional impairments and increased microglia activation, which is probably involved in the onset and/or progression of neurodegeneration.
Age-dependent microglia activation has been described in aged rodents (Perry et al., 1993), rats (Ogura et al., 1994), humans (Streit and Sparks, 1997) as well as non-human primates (Sheffield and Berman, 1998). Hallmarks of age-dependent microglia activation are increased expression of MHCII (Henry et al., 2009; VanGuilder et al., 2011), CD68 (Wong et al., 2005; Griffin et al., 2006) as well as increased levels of TLRs (Letiembre et al., 2007). Aged microglia have been characterized by the presence of lipofuscin inclusions, reduced processes complexity and increased expression pro-inflammatory (TNFα, IL1β, IL6) and anti-inflammatory (IL10, TGFβ1) cytokines. After LPS challenge aged microglia exhibit increased expression of TNFα, IL1β, IL6 and IL10 (Sierra et al., 2007). Furthermore, increased expression of macrophage inflammatory protein (MIP)1α, MIP1β and RANTES in different brain regions of aged mice have been detected (Felzien et al., 2001). In general, the reactivity of microglia upon stimulation seems to be increased during aging, which is further reflected by enhanced microglia activation in aged mice after injection of activating cytokines IL1β and IL12 to the hippocampus (Lee et al., 2013). This phenomenon has been described as microglia priming and TLR2, TLR3 and TLR4 seem to be essential to prime microglia but not astrocytes for ATP-dependent interleukin-1β release (Facci et al., 2014). Microglia priming induces a highly conserved transcriptional signature with aging- and disease-specific aspects (Holtman et al., 2015), which is dependent on High mobility group box 1 (HMGB1). HMGB1 mediates the neuroinflammatory priming in the aged CNS and inhibition of HMGB1 functions appears to desensitize aged microglia to an immune challenge, thus preventing exaggerated behavioral and neuroinflammatory responses following microglia stimulation (Fonken et al., 2016).
Expression levels of the complement genes, C3 and complement factor B (CFB), both of which being previously associated with age-related macular degeneration (AMD), increased during aging suggesting that senescent retinal microglia may contribute to complement dysregulation during disease pathogenesis and progression (Ma et al., 2013). Although these studies suggest an inflammatory microglia phenotype in aged mice, RNA sequencing revealed that aged microglia display decreased expression of genes associated with endogenous ligand recognition and upregulated genes associated with microbe recognition and host defense. Most interestingly, aged microglia presented increased expression of genes related to neuroprotection and neurorestoration (Hickman et al., 2013).
Among the functional impairments of aged microglia, a reduced capacity to engulf amyloid-β fibrils (Floden and Combs, 2011) and reduced chemotaxis, process motility and migration towards laser-induced injury and extracellular ATP (Damani et al., 2011) have been described. Moreover, cultured microglia from aged mice show a stronger reaction upon ATP-triggered activation, which is characterized by increased nitric oxide (NO) and TNFα release (Lai et al., 2013). Recently, it has been reported that myelin pieces are gradually released from aging myelin sheaths and are subsequently cleared by microglia. This myelin fragmentation increased with age and led to the formation of insoluble, lipofuscin-like lysosomal inclusions in microglia contributing to microglial senescence and immune dysfunctions in aging mice (Safaiyan et al., 2016). Age-dependent microglia dysfunctions might be further enhanced by loss of endogenous regulation of microglia functions and activation states. TGFβ1 has been demonstrated to promote quiescence of microglia in vitro (Spittau et al., 2013) and in vivo (Butovsky et al., 2014) and is a promising candidate to regulate microglia activation states. Moreover, TGFβ1 inhibits IFNγ-induced microglia activation and degeneration of midbrain dopaminergic (mDA) neurons (Zhou et al., 2015) and induces microglia-mediated engulfment of apoptotic cells via induction of microglial Mfge8 expression (Spittau et al., 2015). Age-dependent impairment of TGFβ1 signaling was lately described to reduce the protective functions of microglia promoting cytotoxic activation and potentiating microglia-mediated neurodegeneration (Tichauer et al., 2014).
Although most of the abovementioned studies have been performed in rodents, recent studies indicate that microglia priming and age-dependent microglia activation is also detectable in the human CNS. In vivo imaging using (R)-[(11)C]PK11195 and positron emission tomography revealed that activated microglia appear in several cortical and subcortical areas during healthy aging (Schuitemaker et al., 2012). Further, glial-specific genes shift their regional expression patterns during aging and especially microglia-specific genes globally display increased expression during aging (Soreq et al., 2017). Taken together, priming of microglia and age-related changes in microglia functions and activations are likely to be involved in the development and progression of neurodegenerative diseases such as AD and PD.
Aging and AD and PD
The most frequent neurodegenerative diseases are AD and PD, both of which sharing a common risk factor namely aging. AD is characterized by progressive cognitive impairments and behavioral disturbances. Neuropathological hallmarks such as neuron death, dystrophic neurites, synapse loss, amyloid plaques and neurofibrillary tangles are accompanied by reactive gliosis (Serrano-Pozo et al., 2011). Basically, AD can be categorized into two distinct subtypes: sAD and familial AD (fAD). Whereas sAD represent a polygenic disorder, fAD is linked to mutations and polymorphisms in genes such as Amyloid precursor protein (APP), Presenilin 1 (PSEN1) or Presenilin 2 (PSEN2) and account for 5%–10% of all AD cases (Bagyinszky et al., 2014). PD is characterized by the progressive loss of mDA neurons in the SN pars compacta and the subsequent reduction in dopamine levels in the basal ganglia, which results in classical movement disturbances such as akinesia, rigidity and tremor. Similar to AD, PD cases can be divided into sporadic PD (sPD) and familial PD (fPD), the latter of which represents approximately 20%–25% of all PD cases. A common hallmark of sPD and fPD is the presence of intracellular inclusions termed Lewy bodies (Goedert, 2001; Jellinger, 2001). α-Synuclein (αSyn) has been identified as a major component of Lewy bodies in sporadic and familial cases and is believed to be the central player in PD etiology (Spillantini et al., 1998). Activated microglia have been described in both pathologies (McGeer et al., 1988) and microglia-mediated neuroinflammatory responses are believed to contribute to disease onset, severity and progression of AD (Heneka et al., 2015) and PD (Block et al., 2007).
In AD mouse models, microglia seem to build up a barrier that prevents neurotoxic effects of proto fibrillar Amyloid-β (Aβ) hotspots around amyloid plaques (Condello et al., 2015). Furthermore, microglia phagocytose Aβ in order to promote clearance of Aβ plaques. Aged microglia display reduced Aβ phagocytosis (Floden and Combs, 2011) and recently it has been demonstrated that Aβ engulfment by aged microglia results in Aβ redistribution rather than in a biophysical degradation of the protein aggregates (Njie et al., 2012). Interestingly, young microglia have been recently demonstrated to restore the amyloid clearance of aged microglia in an elegant ex vivo organotypic brain slice co-culturing approach (Daria et al., 2017). In transgenic APP23 mice, exacerbated age-dependent microglia activation and disturbances in microglial cytoskeletal regulations have been described which are high likely to contribute to further neurodegeneration (Janssen et al., 2016). Next to the deteriorated response of aged and primed microglia, complement factor C3 secreted from reactive astrocytes has been reported to interact with the microglial C3a receptor (C3aR) thereby mediating Aβ pathology and neuroinflammation in AD mouse models (Lian et al., 2016). However, lack of C3aR in APP transgenic mice results in decreased, rather than increased, Aβ deposition and C3aR-deficient microglia are more effective in degrading extracellular Aβ (Czirr et al., 2017). Although increased microglia responses as well as impairments of microglia-mediated clearance of Aβ seem to promote the progression of AD, the role of microglia contribution to AD pathology remains unclear. Interestingly, the ablation of microglia in APP transgenic mouse strains crossed with CD11b-HSVTK mice, in which nearly complete depletion of microglia was achieved after ganciclovir application, neither Aβ plaque formation nor amyloid-associated neuron dystrophy was depended on the presence of microglia (Grathwohl et al., 2009). Further studies will be essential in order to broaden our understanding of how microglia contribute to disease onset and progression and a special focus should be given on the role of aged microglia to elucidate the impact of aging on microglial functions in AD.
In PD, marked microglia reactions in the human SN have been observed, which are associated with extraneuronal neuromelanin deposits (Beach et al., 2007). Furthermore, human neuromelanin is able to induce microglia-mediated neuroinflammation and neurodegeneration in rats (Zecca et al., 2008) indicating that the release of neuromelanin from degenerating mDA neurons is a potent trigger for microglia activation. In rodents, toxin-based models, such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) are employed to understand the contribution of microglia to onset and progression of mDA neuron degeneration (Machado et al., 2016). Although the contribution of microglia-mediated neuroinflammation has been clearly demonstrated (Block et al., 2007; Machado et al., 2016), the impact of aging is often ignored in these studies. Interestingly, microglia priming increases the response to the herbicide paraquat and, thus resulting in increased mDA neurodegeneration (Purisai et al., 2007) and aged monkeys display stronger and persistent microglia reactivity after MPTP application indicating microglial involvement in age-dependent degeneration of mDA neurons (Kanaan et al., 2008). It is high likely that age-dependent microglia priming involves epigenetic modifications as reported by Tang et al. (2014). The authors demonstrated that histone H3K27me3 demethylase Jumonji domain containing 3 (Jmjd3) is essential for M2-like microglia activation. Inhibition of Jmjd3 resulted in exacerbation of MPTP-induced mDA degeneration and aged mice displayed reduced Jmjd3 expression and increased H3K27me3 suggesting an age-dependent switch in microglia activation phenotypes (Tang et al., 2014). The effect of aging on the severity of MPTP-induced neurodegeneration is further supported by a study using Senescence-accelerated mouse prone 8 (SAMP8), a mouse line with early onset of senility which presents increased microglia activation and increased neurodegeneration after MPTP intoxication (Liu et al., 2010). Next to toxin-based models for PD, αSyn transgenic mice are often used to understand how αSyn aggregates contribute to neuroinflammation and neurodegeneration in PD. Non-aggregated αSyn is able to trigger TLR-mediated immune responses of microglia, a phenomenon that might contribute to the onset of sporadic and/or familial αSyn-related PD forms (Roodveldt et al., 2013). Notably, telomere shortening has been shown to accelerate αSyn pathology, which is linked to limited microglia function in the brainstem (Scheffold et al., 2016). This observation might be explained by age-dependent microglia deficits. Indeed, isolated microglia from adult mice display phagocytosis impairment of free and exosome-associated αSyn oligomers associated with enhanced TNFα secretion (Bliederhaeuser et al., 2016). Taken together, the contribution of aged microglia to the progressive nature of PD is most likely and the fact that the nigrostriatal system displays a high density of microglia (Sharaf et al., 2013) further supports the hypothesis that microglia are involved in PD pathogenesis. However, the molecular and functional changes of aged microglia are only partially understood and their contribution to neurodegeneration and neuroinflammation in aged individuals need to be further addressed in future studies.
Conclusion
Aging has been clearly demonstrated to affect microglia functions and activation states in vitro and in vivo and it further appears that the unique nature of microglia contributes to their age-dependent functional impairment. Moreover, the onset, severity and progression of neurodegenerative diseases such as AD and PD are influenced by aging and aging-associated changes in microglia functions (Figure 1). However, it is not clear whether aged microglia are responsible for the exacerbation of neurodegeneration in aged individuals or whether aged neurons itself are more prone to degenerative cues. Furthermore, the contribution of systemic age-dependent changes, such as obesity and metabolic diseases like diabetes, are likely to affect microglia and neuroinflammatory responses. Overall, the effect of aging on microglia needs to be further analyzed in order to better understand the molecular mechanisms underlying age-related changes in microglia phenotypes and functions.
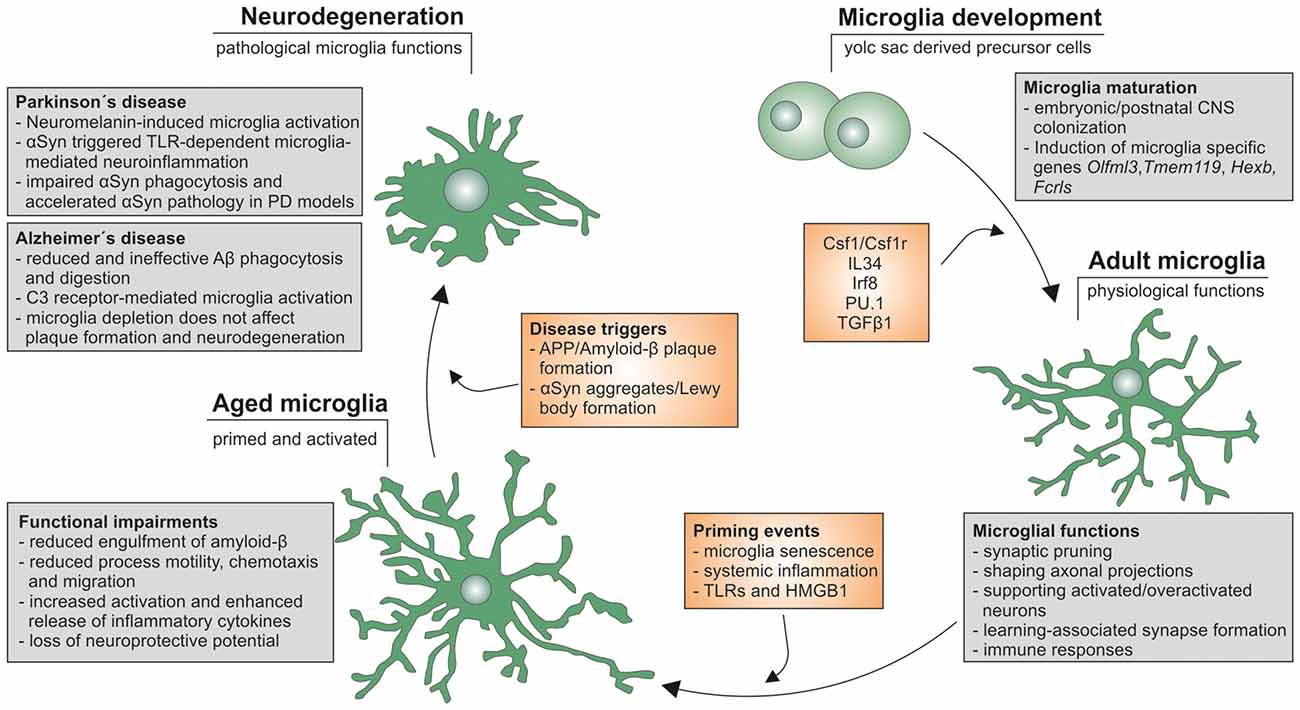
Figure 1. Microglia life cycle. Schematic summary of microglial development, maturation as well as aging and the functional changes influencing onset, severity and progression of neurodegeneration in Alzheimer’s disease (AD) and Parkinson’s disease (PD).
Author Contributions
BS wrote the manuscript.
Funding
This work was funded by grants from the Deutsche Forschungsgemeinschaft (DFG, SP 1555/2-1).
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ajami, B., Bennett, J. L., Krieger, C., Tetzlaff, W., and Rossi, F. M. V. (2007). Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 10, 1538–1543. doi: 10.1038/nn2014
Ajami, B., Bennett, J. L., Krieger, C., McNagny, K. M., and Rossi, F. M. V. (2011). Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 14, 1142–1149. doi: 10.1038/nn.2887
Askew, K., Li, K., Olmos-Alonso, A., Garcia-Moreno, F., Liang, Y., Richardson, P., et al. (2017). Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep. 18, 391–405. doi: 10.1016/j.celrep.2016.12.041
Bagyinszky, E., Youn, Y. C., An, S. S. A., and Kim, S. (2014). The genetics of Alzheimer’s disease. Clin. Interv. Aging 9, 535–551. doi: 10.2147/CIA.S51571
Beach, T. G., Sue, L. I., Walker, D. G., Lue, L. F., Connor, D. J., Caviness, J. N., et al. (2007). Marked microglial reaction in normal aging human substantia nigra: correlation with extraneuronal neuromelanin pigment deposits. Acta Neuropathol. 114, 419–424. doi: 10.1007/s00401-007-0250-5
Bechmann, I., Priller, J., Kovac, A., Böntert, M., Wehner, T., Klett, F. F., et al. (2001). Immune surveillance of mouse brain perivascular spaces by blood-borne macrophages. Eur. J. Neurosci. 14, 1651–1658. doi: 10.1046/j.0953-816x.2001.01793.x
Bennett, M. L., Bennett, F. C., Liddelow, S. A., Ajami, B., Zamanian, J. L., Fernhoff, N. B., et al. (2016). New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. U S A 113, E1738–E1746. doi: 10.1073/pnas.1525528113
Beutner, C., Linnartz-Gerlach, B., Schmidt, S. V., Beyer, M., Mallmann, M. R., Staratschek-Jox, A., et al. (2013). Unique transcriptome signature of mouse microglia. Glia 61, 1429–1442. doi: 10.1002/glia.22524
Blasco, M. A. (2007). Telomere length, stem cells and aging. Nat. Chem. Biol. 3, 640–649. doi: 10.1038/nchembio.2007.38
Bliederhaeuser, C., Grozdanov, V., Speidel, A., Zondler, L., Ruf, W. P., Bayer, H., et al. (2016). Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol. 131, 379–391. doi: 10.1007/s00401-015-1504-2
Block, M. L., Zecca, L., and Hong, J.-S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69. doi: 10.1038/nrn2038
Bolós, M., Llorens-Martín, M., Jurado-Arjona, J., Hernández, F., Rábano, A., and Avila, J. (2016). Direct evidence of internalization of tau by microglia in vitro and in vivo. J. Alzheimers Dis. 50, 77–87. doi: 10.3233/JAD-150704
Butovsky, O., Jedrychowski, M. P., Moore, C. S., Cialic, R., Lanser, A. J., Gabriely, G., et al. (2014). Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143. doi: 10.1038/nn.3599
Condello, C., Yuan, P., Schain, A., and Grutzendler, J. (2015). Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 6:6176. doi: 10.1038/ncomms7176
Costello, D. A., Keenan, K., McManus, R. M., Falvey, A., and Lynch, M. A. (2016). The age-related neuroinflammatory environment promotes macrophage activation, which negatively impacts synaptic function. Neurobiol. Aging 43, 140–148. doi: 10.1016/j.neurobiolaging.2016.04.001
Czirr, E., Castello, N. A., Mosher, K. I., Castellano, J. M., Hinkson, I. V., Lucin, K. M., et al. (2017). Microglial complement receptor 3 regulates brain Aβ levels through secreted proteolytic activity. J. Exp. Med. 214, 1081–1092. doi: 10.1084/jem.20162011
Damani, M. R., Zhao, L., Fontainhas, A. M., Amaral, J., Fariss, R. N., and Wong, W. T. (2011). Age-related alterations in the dynamic behavior of microglia. Aging Cell 10, 263–276. doi: 10.1111/j.1474-9726.2010.00660.x
Daria, A., Colombo, A., Llovera, G., Hampel, H., Willem, M., Liesz, A., et al. (2017). Young microglia restore amyloid plaque clearance of aged microglia. EMBO J. 35, 583–603. doi: 10.15252/embj.201694591
Davies, D. S., Ma, J., Jegathees, T., and Goldsbury, C. (2016). Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer’s disease. Brain Pathol. doi: 10.1111/bpa.12456 [Epub ahead of print].
de Haas, A. H., Boddeke, H. W. G. M., and Biber, K. (2008). Region-specific expression of immunoregulatory proteins on microglia in the healthy CNS. Glia 56, 888–894. doi: 10.1002/glia.20663
de Haas, A. H., Boddeke, H. W. G. M., Brouwer, N., and Biber, K. (2007). Optimized isolation enables ex vivo analysis of microglia from various central nervous system regions. Glia 55, 1374–1384. doi: 10.1002/glia.20554
Doorn, K. J., Brevé, J. J. P., Drukarch, B., Boddeke, H. W., Huitinga, I., Lucassen, P. J., et al. (2015). Brain region-specific gene expression profiles in freshly isolated rat microglia. Front. Cell. Neurosci. 9:84. doi: 10.3389/fncel.2015.00084
Elmore, M. R. P., Najafi, A. R., Koike, M. A., Dagher, N. N., Spangenberg, E. E., Rice, R. A., et al. (2014). Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82, 380–397. doi: 10.1016/j.neuron.2014.02.040
Erny, D., Hrabě de Angelis, A. L., Jaitin, D., Wieghofer, P., Staszewski, O., David, E., et al. (2015). Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 18, 965–977. doi: 10.1038/nn.4030
Facci, L., Barbierato, M., Marinelli, C., Argentini, C., Skaper, S. D., and Giusti, P. (2014). Toll-like receptors 2, -3 and -4 prime microglia but not astrocytes across central nervous system regions for ATP-dependent interleukin-1β release. Sci. Rep. 4:6824. doi: 10.1038/srep06824
Felzien, L. K., McDonald, J. T., Gleason, S. M., Berman, N. E., and Klein, R. M. (2001). Increased chemokine gene expression during aging in the murine brain. Brain Res. 890, 137–146. doi: 10.1016/s0006-8993(00)03090-0
Flanary, B. E., Sammons, N. W., Nguyen, C., Walker, D., and Streit, W. J. (2007). Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res. 10, 61–74. doi: 10.1089/rej.2006.9096
Flanary, B. E., and Streit, W. J. (2004). Progressive telomere shortening occurs in cultured rat microglia, but not astrocytes. Glia 45, 75–88. doi: 10.1002/glia.10301
Floden, A. M., and Combs, C. K. (2011). Microglia demonstrate age-dependent interaction with amyloid-β fibrils. J. Alzheimers Dis. 25, 279–293. doi: 10.3233/JAD-2011-101014
Fonken, L. K., Frank, M. G., Kitt, M. M., D’Angelo, H. M., Norden, D. M., Weber, M. D., et al. (2016). The alarmin HMGB1 mediates age-induced neuroinflammatory priming. J. Neurosci. 36, 7946–7956. doi: 10.1523/JNEUROSCI.1161-16.2016
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Goedert, M. (2001). Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2, 492–501. doi: 10.1038/35081564
Goldmann, T., Wieghofer, P., Jordão, M. J. C., Prutek, F., Hagemeyer, N., Frenzel, K., et al. (2016). Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 17, 797–805. doi: 10.1038/ni.3423
Gómez-Nicola, D., Fransen, N. L., Suzzi, S., and Perry, V. H. (2013). Regulation of microglial proliferation during chronic neurodegeneration. J. Neurosci. 33, 2481–2493. doi: 10.1523/JNEUROSCI.4440-12.2013
Grabert, K., Michoel, T., Karavolos, M. H., Clohisey, S., Baillie, J. K., Stevens, M. P., et al. (2016). Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 19, 504–516. doi: 10.1038/nn.4222
Grathwohl, S. A., Kälin, R. E., Bolmont, T., Prokop, S., Winkelmann, G., Kaeser, S. A., et al. (2009). Formation and maintenance of Alzheimer’s disease β-amyloid plaques in the absence of microglia. Nat. Neurosci. 12, 1361–1363. doi: 10.1038/nn.2432
Greter, M., Lelios, I., Pelczar, P., Hoeffel, G., Price, J., Leboeuf, M., et al. (2012). Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity 37, 1050–1060. doi: 10.1016/j.immuni.2012.11.001
Griffin, R., Nally, R., Nolan, Y., McCartney, Y., Linden, J., and Lynch, M. A. (2006). The age-related attenuation in long-term potentiation is associated with microglial activation. J. Neurochem. 99, 1263–1272. doi: 10.1111/j.1471-4159.2006.04165.x
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Henry, C. J., Huang, Y., Wynne, A. M., and Godbout, J. P. (2009). Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain Behav. Immun. 23, 309–317. doi: 10.1016/j.bbi.2008.09.002
Hickey, W. F., and Kimura, H. (1988). Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science 239, 290–292. doi: 10.1126/science.3276004
Hickman, S. E., Kingery, N. D., Ohsumi, T. K., Borowsky, M. L., Wang, L.-C., Means, T. K., et al. (2013). The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905. doi: 10.1038/nn.3554
Holtman, I. R., Raj, D. D., Miller, J. A., Schaafsma, W., Yin, Z., Brouwer, N., et al. (2015). Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol. Commun. 3:31. doi: 10.1186/s40478-015-0203-5
Hoshiko, M., Arnoux, I., Avignone, E., Yamamoto, N., and Audinat, E. (2012). Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J. Neurosci. 32, 15106–15111. doi: 10.1523/JNEUROSCI.1167-12.2012
Janssen, L., Dubbelaar, M. L., Holtman, I. R., de Boer-Bergsma, J., Eggen, B. J. L., Boddeke, H. W. G. M., et al. (2016). Aging, microglia and cytoskeletal regulation are key factors in the pathological evolution of the APP23 mouse model for Alzheimer’s disease. Biochim. Biophys. Acta 1863, 395–405. doi: 10.1016/j.bbadis.2016.11.014
Kamphuis, W., Orre, M., Kooijman, L., Dahmen, M., and Hol, E. M. (2012). Differential cell proliferation in the cortex of the APPswePS1dE9 Alzheimer’s disease mouse model. Glia 60, 615–629. doi: 10.1002/glia.22295
Kanaan, N. M., Kordower, J. H., and Collier, T. J. (2008). Age and region-specific responses of microglia, but not astrocytes, suggest a role in selective vulnerability of dopamine neurons after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure in monkeys. Glia 56, 1199–1214. doi: 10.1002/glia.20690
Kierdorf, K., Erny, D., Goldmann, T., Sander, V., Schulz, C., Perdiguero, E. G., et al. (2013). Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 16, 273–280. doi: 10.1038/nn.3318
Kigerl, K. A., de Rivero Vaccari, J. P., Dietrich, W. D., Popovich, P. G., and Keane, R. W. (2014). Pattern recognition receptors and central nervous system repair. Exp. Neurol. 258, 5–16. doi: 10.1016/j.expneurol.2014.01.001
Lai, A. Y., Dibal, C. D., Armitage, G. A., Winship, I. R., and Todd, K. G. (2013). Distinct activation profiles in microglia of different ages: a systematic study in isolated embryonic to aged microglial cultures. Neuroscience 254, 185–195. doi: 10.1016/j.neuroscience.2013.09.010
Lawson, L. J., Perry, V. H., Dri, P., and Gordon, S. (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39, 151–170. doi: 10.1016/0306-4522(90)90229-w
Lee, D. C., Ruiz, C. R., Lebson, L., Selenica, M.-L. B., Rizer, J., Hunt, J. B. Jr., et al. (2013). Aging enhances classical activation but mitigates alternative activation in the central nervous system. Neurobiol. Aging 34, 1610–1620. doi: 10.1016/j.neurobiolaging.2012.12.014
Letiembre, M., Hao, W., Liu, Y., Walter, S., Mihaljevic, I., Rivest, S., et al. (2007). Innate immune receptor expression in normal brain aging. Neuroscience 146, 248–254. doi: 10.1016/j.neuroscience.2007.01.004
Li, T., Pang, S., Yu, Y., Wu, X., Guo, J., and Zhang, S. (2013). Proliferation of parenchymal microglia is the main source of microgliosis after ischaemic stroke. Brain 136, 3578–3588. doi: 10.1093/brain/awt287
Lian, H., Litvinchuk, A., Chiang, A. C.-A., Aithmitti, N., Jankowsky, J. L., and Zheng, H. (2016). Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J. Neurosci. 36, 577–589. doi: 10.1523/JNEUROSCI.2117-15.2016
Liu, J., Wang, M.-W., Gu, P., Ma, Q.-Y., Wang, Y.-Y., Geng, Y., et al. (2010). Microglial activation and age-related dopaminergic neurodegeneration in MPTP-treated SAMP8 mice. Brain Res. 1345, 213–220. doi: 10.1016/j.brainres.2010.05.043
Luo, W., Liu, W., Hu, X., Hanna, M., Caravaca, A., and Paul, S. M. (2015). Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci. Rep. 5:11161. doi: 10.1038/srep11161
Ma, W., Cojocaru, R., Gotoh, N., Gieser, L., Villasmil, R., Cogliati, T., et al. (2013). Gene expression changes in aging retinal microglia: relationship to microglial support functions and regulation of activation. Neurobiol. Aging 34, 2310–2321. doi: 10.1016/j.neurobiolaging.2013.03.022
Machado, V., Zöller, T., Attaai, A., and Spittau, B. (2016). Microglia-mediated neuroinflammation and neurotrophic factor-induced protection in the MPTP mouse model of Parkinson’s disease-lessons from transgenic mice. Int. J. Mol. Sci. 17:E151. doi: 10.3390/ijms17020151
Martinez, F. O., and Gordon, S. (2014). The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6:13. doi: 10.12703/P6-13
McGeer, P. L., Itagaki, S., Boyes, B. E., and McGeer, E. G. (1988). Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1291. doi: 10.1212/WNL.38.8.1285
Mittelbronn, M., Dietz, K., Schluesener, H. J., and Meyermann, R. (2001). Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 101, 249–255.
Mosser, D. M., and Edwards, J. P. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969. doi: 10.1038/nri2448
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Njie, E. G., Boelen, E., Stassen, F. R., Steinbusch, H. W. M., Borchelt, D. R., and Streit, W. J. (2012). Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol. Aging 33, 195.e1–195.e12. doi: 10.1016/j.neurobiolaging.2010.05.008
Ogura, K., Ogawa, M., and Yoshida, M. (1994). Effects of ageing on microglia in the normal rat brain: immunohistochemical observations. Neuroreport 5, 1224–1226.doi: 10.1097/00001756-199406020-00016
Panatier, A., and Robitaille, R. (2012). The soothing touch: microglial contact influences neuronal excitability. Dev. Cell 23, 1125–1126. doi: 10.1016/j.devcel.2012.11.015
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R. III., Lafaille, J. J., et al. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi: 10.1016/j.cell.2013.11.030
Perry, V. H., Matyszak, M. K., and Fearn, S. (1993). Altered antigen expression of microglia in the aged rodent CNS. Glia 7, 60–67. doi: 10.1002/glia.440070111
Peters, A., Josephson, K., and Vincent, S. L. (1991). Effects of aging on the neuroglial cells and pericytes within area 17 of the rhesus monkey cerebral cortex. Anat. Rec. 229, 384–398. doi: 10.1002/ar.1092290311
Pont-Lezica, L., Beumer, W., Colasse, S., Drexhage, H., Versnel, M., and Bessis, A. (2014). Microglia shape corpus callosum axon tract fasciculation: functional impact of prenatal inflammation. Eur. J. Neurosci. 39, 1551–1557. doi: 10.1111/ejn.12508
Prinz, M., and Priller, J. (2014). Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci. 15, 300–312. doi: 10.1038/nrn3722
Purisai, M. G., McCormack, A. L., Cumine, S., Li, J., Isla, M. Z., and Di Monte, D. A. (2007). Microglial activation as a priming event leading to paraquat-induced dopaminergic cell degeneration. Neurobiol. Dis. 25, 392–400. doi: 10.1016/j.nbd.2006.10.008
Raj, D. D. A., Jaarsma, D., Holtman, I. R., Olah, M., Ferreira, F. M., Schaafsma, W., et al. (2014). Priming of microglia in a DNA-repair deficient model of accelerated aging. Neurobiol. Aging 35, 2147–2160. doi: 10.1016/j.neurobiolaging.2014.03.025
Raj, D. D. A., Moser, J., van der Pol, S. M. A., van Os, R. P., Holtman, I. R., Brouwer, N., et al. (2015). Enhanced microglial pro-inflammatory response to lipopolysaccharide correlates with brain infiltration and blood-brain barrier dysregulation in a mouse model of telomere shortening. Aging Cell 14, 1003–1013. doi: 10.1111/acel.12370
Ransohoff, R. M. (2016). A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci. 19, 987–991. doi: 10.1038/nn.4338
Rodriguez-Callejas, J. D., Fuchs, E., and Perez-Cruz, C. (2016). Evidence of tau hyperphosphorylation and dystrophic microglia in the common marmoset. Front. Aging Neurosci. 8:315. doi: 10.3389/fnagi.2016.00315
Roodveldt, C., Labrador-Garrido, A., Gonzalez-Rey, E., Lachaud, C. C., Guilliams, T., Fernandez-Montesinos, R., et al. (2013). Preconditioning of microglia by α-synuclein strongly affects the response induced by toll-like receptor (TLR) stimulation. PLoS One 8:e79160. doi: 10.1371/journal.pone.0079160
Safaiyan, S., Kannaiyan, N., Snaidero, N., Brioschi, S., Biber, K., Yona, S., et al. (2016). Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 19, 995–998. doi: 10.1038/nn.4325
Satyanarayana, A., Manns, M. P., and Rudolph, K. L. (2004). Telomeres and telomerase: a dual role in hepatocarcinogenesis. Hepatology 40, 276–283. doi: 10.1002/hep.20308
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Scheffold, A., Holtman, I. R., Dieni, S., Brouwer, N., Katz, S.-F., Jebaraj, B. M. C., et al. (2016). Telomere shortening leads to an acceleration of synucleinopathy and impaired microglia response in a genetic mouse model. Acta Neuropathol. Commun. 4:87. doi: 10.1186/s40478-016-0364-x
Schuitemaker, A., van der Doef, T. F., Boellaard, R., van der Flier, W. M., Yaqub, M., Windhorst, A. D., et al. (2012). Microglial activation in healthy aging. Neurobiol. Aging 33, 1067–1072. doi: 10.1016/j.neurobiolaging.2010.09.016
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Sharaf, A., Krieglstein, K., and Spittau, B. (2013). Distribution of microglia in the postnatal murine nigrostriatal system. Cell Tissue Res. 351, 373–382. doi: 10.1007/s00441-012-1537-y
Sheffield, L. G., and Berman, N. E. (1998). Microglial expression of MHC class II increases in normal aging of nonhuman primates. Neurobiol. Aging 19, 47–55. doi: 10.1016/s0197-4580(97)00168-1
Sierra, A., Gottfried-Blackmore, A. C., McEwen, B. S., and Bulloch, K. (2007). Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 55, 412–424. doi: 10.1002/glia.20468
Soreq, L., UK Brain Expression Consortium, North American Brain Expression Consortium, Rose, J., Soreq, E., Hardy, J., et al. (2017). Major shifts in glial regional identity are a transcriptional hallmark of human brain aging. Cell Rep. 18, 557–570. doi: 10.1016/j.celrep.2016.12.011
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998). α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. U S A 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
Spittau, B., Rilka, J., Steinfath, E., Zöller, T., and Krieglstein, K. (2015). TGFβ1 increases microglia-mediated engulfment of apoptotic cells via upregulation of the milk fat globule-EGF factor 8. Glia 63, 142–153. doi: 10.1002/glia.22740
Spittau, B., Wullkopf, L., Zhou, X., Rilka, J., Pfeifer, D., and Krieglstein, K. (2013). Endogenous transforming growth factor-beta promotes quiescence of primary microglia in vitro. Glia 61, 287–300. doi: 10.1002/glia.22435
Squarzoni, P., Oller, G., Hoeffel, G., Pont-Lezica, L., Rostaing, P., Low, D., et al. (2014). Microglia modulate wiring of the embryonic forebrain. Cell Rep. 8, 1271–1279. doi: 10.1016/j.celrep.2014.07.042
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. doi: 10.1016/j.cell.2007.10.036
Streit, W. J. (2006). Microglial senescence: does the brain’s immune system have an expiration date? Trends Neurosci. 29, 506–510. doi: 10.1016/j.tins.2006.07.001
Streit, W. J., Braak, H., Xue, Q.-S., and Bechmann, I. (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 118, 475–485. doi: 10.1007/s00401-009-0556-6
Streit, W. J., Sammons, N. W., Kuhns, A. J., and Sparks, D. L. (2004). Dystrophic microglia in the aging human brain. Glia 45, 208–212. doi: 10.1002/glia.10319
Streit, W. J., and Sparks, D. L. (1997). Activation of microglia in the brains of humans with heart disease and hypercholesterolemic rabbits. J. Mol. Med. 75, 130–138. doi: 10.1007/s001090050097
Streit, W. J., Walter, S. A., and Pennell, N. A. (1999). Reactive microgliosis. Prog. Neurobiol. 57, 563–581. doi: 10.1016/s0301-0082(98)00069-0
Tang, Y., Li, T., Li, J., Yang, J., Liu, H., Zhang, X. J., et al. (2014). Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson’s disease. Cell Death Differ. 21, 369–380. doi: 10.1038/cdd.2013.159
Tichauer, J. E., Flores, B., Soler, B., Eugenín-von Bernhardi, L., Ramírez, G., and von Bernhardi, R. (2014). Age-dependent changes on TGFβ1 Smad3 pathway modify the pattern of microglial cell activation. Brain Behav. Immun. 37, 187–196. doi: 10.1016/j.bbi.2013.12.018
Tischer, J., Krueger, M., Mueller, W., Staszewski, O., Prinz, M., Streit, W. J., et al. (2016). Inhomogeneous distribution of Iba-1 characterizes microglial pathology in Alzheimer’s disease. Glia 64, 1562–1572. doi: 10.1002/glia.23024
Ueno, M., Fujita, Y., Tanaka, T., Nakamura, Y., Kikuta, J., Ishii, M., et al. (2013). Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 16, 543–551. doi: 10.1038/nn.3358
VanGuilder, H. D., Bixler, G. V., Brucklacher, R. M., Farley, J. A., Yan, H., Warrington, J. P., et al. (2011). Concurrent hippocampal induction of MHC II pathway components and glial activation with advanced aging is not correlated with cognitive impairment. J. Neuroinflammation 8:138. doi: 10.1186/1742-2094-8-138
Varvel, N. H., Neher, J. J., Bosch, A., Wang, W., Ransohoff, R. M., Miller, R. J., et al. (2016). Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc. Natl. Acad. Sci. U S A 113, E5665–E5674. doi: 10.1073/pnas.1604263113
Wang, X., Zhao, L., Zhang, J., Fariss, R. N., Ma, W., Kretschmer, F., et al. (2016). Requirement for microglia for the maintenance of synaptic function and integrity in the mature retina. J. Neurosci. 36, 2827–2842. doi: 10.1523/JNEUROSCI.3575-15.2016
Wohl, S. G., Schmeer, C. W., Witte, O. W., and Isenmann, S. (2010). Proliferative response of microglia and macrophages in the adult mouse eye after optic nerve lesion. Invest. Ophthalmol. Vis. Sci. 51, 2686–2696. doi: 10.1167/iovs.09-4537
Wong, A. M., Patel, N. V., Patel, N. K., Wei, M., Morgan, T. E., de Beer, M. C., et al. (2005). Macrosialin increases during normal brain aging are attenuated by caloric restriction. Neurosci. Lett. 390, 76–80. doi: 10.1016/j.neulet.2005.07.058
Yamasaki, R., Lu, H., Butovsky, O., Ohno, N., Rietsch, A. M., Cialic, R., et al. (2014). Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 211, 1533–1549. doi: 10.1084/jem.20132477
Zecca, L., Wilms, H., Geick, S., Claasen, J.-H., Brandenburg, L.-O., Holzknecht, C., et al. (2008). Human neuromelanin induces neuroinflammation and neurodegeneration in the rat substantia nigra: implications for Parkinson’s disease. Acta Neuropathol. 116, 47–55. doi: 10.1007/s00401-008-0361-7
Zhou, X., Spittau, B., and Krieglstein, K. (2012). TGFβ signalling plays an important role in IL4-induced alternative activation of microglia. J. Neuroinflammation 9:210. doi: 10.1186/1742-2094-9-210
Keywords: aging, microglia, neurodegeneration, Alzheimer’s disease (AD), Parkinson’s disease (PD)
Citation: Spittau B (2017) Aging Microglia—Phenotypes, Functions and Implications for Age-Related Neurodegenerative Diseases. Front. Aging Neurosci. 9:194. doi: 10.3389/fnagi.2017.00194
Received: 20 March 2017; Accepted: 30 May 2017;
Published: 14 June 2017.
Edited by:
Yu Tang, University of Texas Southwestern Medical Center, United StatesReviewed by:
Wai T. Wong, National Eye Institute (NIH), United StatesClaudia Perez-Cruz, Centro de Investigaciones y Estudios Avanzados CINVESTAV, Mexico
Copyright © 2017 Spittau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Björn Spittau, bjoern.spittau@anat.uni-freiburg.de