Deciphering the Astrocyte Reaction in Alzheimer’s Disease
 Beatriz G. Perez-Nievas
Beatriz G. Perez-Nievas Alberto Serrano-Pozo
Alberto Serrano-Pozo- 1Institute of Psychiatry, King’s College London, London, United Kingdom
- 2Alzheimer’s Research Unit, MassGeneral Institute for Neurodegenerative Diseases (MIND), Department of Neurology, Massachusetts General Hospital, Boston, MA, United States
Reactive astrocytes were identified as a component of senile amyloid plaques in the cortex of Alzheimer’s disease (AD) patients several decades ago. However, their role in AD pathophysiology has remained elusive ever since, in part owing to the extrapolation of the literature from primary astrocyte cultures and acute brain injury models to a chronic neurodegenerative scenario. Recent accumulating evidence supports the idea that reactive astrocytes in AD acquire neurotoxic properties, likely due to both a gain of toxic function and a loss of their neurotrophic effects. However, the diversity and complexity of this glial cell is only beginning to be unveiled, anticipating that astrocyte reaction might be heterogeneous as well. Herein we review the evidence from mouse models of AD and human neuropathological studies and attempt to decipher the main conundrums that astrocytes pose to our understanding of AD development and progression. We discuss the morphological features that characterize astrocyte reaction in the AD brain, the consequences of astrocyte reaction for both astrocyte biology and AD pathological hallmarks, and the molecular pathways that have been implicated in this reaction.
Introduction
The term “glia” was first coined by Virchow to refer to the non-neuronal cells that form the “glue” of the brain (Virchow, 1858). Different types of glial cells were distinguished in the early years, including astroglia (van Lenhossék, 1895), microglia (del Río-Hortega and Penfield, 1892) and oligodendroglia (del Río-Hortega, 1921).
Astroglia or astrocytes were named after their stellate shape under the microscope. Soon after the development of appropriate staining methods, it became apparent that both acute (i.e., traumatic brain or spinal cord injury, stroke) and chronic (epilepsy, neurodegenerative diseases) insults to the central nervous system (CNS) are associated with a dramatic change in astrocyte morphology. In these conditions astrocytes appear hypertrophic and overexpress two intermediate-filament proteins of their cytoskeleton: glial fibrillar acidic protein (GFAP) and vimentin. These two characteristics qualify the astrocytes as “reactive”, as opposed to “resting” non-reactive astrocytes, which are not truly quiescent but exert many of the functions listed below. We prefer the terms astrocytic “reaction” or “response” over “astrocytosis” or “astrogliosis”, because the suffix –osis implies a pathological state of astrocytes or astroglia, which at present is not well characterized.
In this review article, we will first summarize the current knowledge about the physiological roles of astrocytes. A review of the morphological and molecular basis of astrocyte reaction in Alzheimer’s disease (AD) will follow next, with special emphasis in its consequences for AD pathophysiology. We will highlight the main ongoing scientific controversies and the remaining areas of uncertainty.
Astrocytes in the Healthy Brain
Morphological and Molecular Heterogeneity of Astrocytes
Traditionally astrocytes have been classified in protoplasmic and fibrous. Protoplasmic astrocytes are cortical astrocytes, with no or minimal GFAP immunoreactivity in normal conditions, and with a “bushy” appearance owing to the profuse ramification of their processes in fine prolongations or leaflets, which reach the pre-and post-synaptic elements of the neurons (Figure 1A). By contrast, fibrous astrocytes are GFAP-immunoreactive astrocytes found in the white matter along the myelinated axons (Figure 1B). Research interest has focused by far on protoplasmic cortical astrocytes, in detriment of fibrous white matter astrocytes. Subpial interlaminar astrocytes are a third kind of astrocyte (Figure 1C). These are exclusively found in the most superficial layer of the cortex of primates and are characteristic for the flat shape of their soma and their long perpendicular processes towards the layers III and IV of the cortex (Colombo et al., 2000; Oberheim et al., 2009).
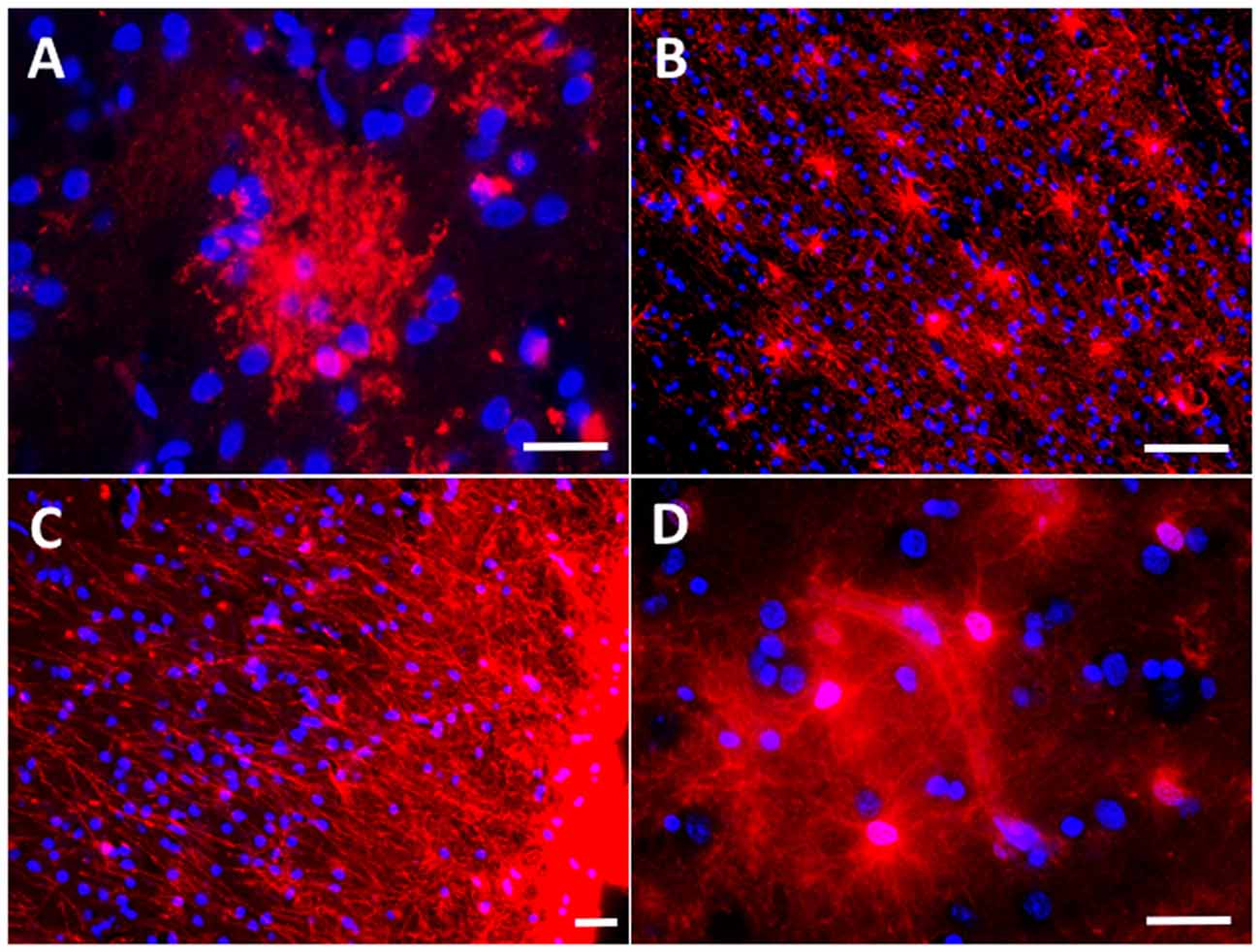
Figure 1. Astrocyte morphological features in the normal brain. (A) Protoplasmic astrocyte in layer II of the occipital neocortex with its typical bushy appearance depicted with immunohistochemistry for the glutamate transporter GLT-1/EAAT2. (B) Fibrous astrocytes in the white matter of the temporal lobe are rich in glial fibrillary acidic protein (GFAP). (C) Subpial interlaminar astrocytes in the frontal association neocortex of a healthy control individual form a palisade of processes which extend towards deep layers perpendicularly to the cortical surface and are GFAP-immunoreactive. (D) Perivascular astrocytes with their vascular endfeet wrapping a capillary vessel, here shown with immunohistochemistry for aldehyde dehydrogenase 1 L1 (constitutively present in all astrocytes). Scale bars: 10 μm in (A,C,D); 20 μm in (B).
However, the morphological, functional and molecular heterogeneity of astrocytes is much broader and just starting to emerge (Sosunov et al., 2014; Chai et al., 2017; John Lin et al., 2017). For example, using the extracellular matrix receptor CD44 as pan-astrocytic marker, cortical and hippocampal human astrocytes have been recently classified in CD44+ astrocytes with and without long processes. CD44+ astrocytes with long processes are distinct from protoplasmic astrocytes in that they are located in the subpial layer (interlaminar astrocytes), deep cortical layers and hippocampus, and express high levels of GFAP and S100β and low levels of glutamine synthetase and glutamate transporters. CD44+ astrocytes without long processes are variable in shape and number and display a mixed phenotype between protoplasmic and fibrous astrocytes (Sosunov et al., 2014). A regional network-specific specialization of astrocytes has recently been unveiled. For example, mouse striatal and hippocampal astrocytes differ in some morphological aspects (higher astrocyte-neuron ratio in striatal, shorter astrocyte-synapse distance in hippocampal), electrophysiological properties (calcium spontaneous and evoked responses in hippocampal astrocytes), and transcriptomic and proteomic signature (Chai et al., 2017). Up to five distinct subpopulations of astrocytes have been distinguished in the mouse CNS, with variable proportions depending on the CNS region, each characterized by a characteristic transcriptomic program, and a different migration, proliferative and synaptogenic potential during development (John Lin et al., 2017).
Physiological Roles of Astrocytes
A prolific research in the last two decades has expanded the role of astrocytes from a mere structural function to critical functions in CNS development, modulation of synaptic activity and glutamate homeostasis, blood-brain barrier (BBB) formation and neurovascular coupling, and inflammatory response. The basis of this astrocyte functional specialization remains largely unraveled, but it is plausible that specific subpopulations of astrocytes with distinct morphology and molecular equipment carry out different functions in different neural circuits (Chai et al., 2017).
Neurodevelopment
The refinement of the neural circuits during CNS development requires axon growth and synapse formation as well as pruning of redundant unnecessary synapses and axons. Recently, both microglia and astrocytes have been implicated in these processes. Astrocytes have been shown to promote the formation of excitatory synapses during CNS development (Allen et al., 2012) and to engulf and eliminate both excitatory and inhibitory synapses during prenatal development, and also in the adult brain (Chung et al., 2013).
Synaptic Function
Approximately 60% of excitatory synapses in the CA1 region of the rat hippocampus are tripartite, that is, have an astrocyte leaflet next to the presynaptic bouton and the postsynaptic dendritic spine (Ventura and Harris, 1999). Both cortical and hippocampal astrocytes are distributed in essentially non-overlapping domains (so called “islands”) with very little inter-digitation between their fine processes, a phenomenon termed “tiling” (Bushong et al., 2002; Halassa et al., 2007). In the mouse cortex, each astrocyte wraps an average of four neurons and up to 600 dendrites from different neurons (Halassa et al., 2007). The intimate relationship between astrocytes and neurons and the ratio astrocyte-neuron enable astrocytes to coordinate synaptic networks.
Astrocytes can modulate synaptic transmission and plasticity mainly by the re-uptake of glutamate from the synaptic cleft through their membrane glutamate transporters GLT-1 (also called excitatory amino acid transporter 2 or EAAT2; Figure 1A) and GLAST (also called excitatory amino acid transporter 1 or EAAT1). GLT-1 is more abundant than GLAST. Of note, the degree of astrocytic coverage of synapses is thought to be changing and this dynamic process can also impact the concentration and time course of glutamate at the synaptic cleft. If the coverage is reduced or there is a down-regulation of glutamate transporters, the resulting increased glutamate concentration for a prolonged time can cause either its spillover and activation of extra-synaptic neuronal NMDA receptors, leading to neuronal excitotoxicity, or its binding to presynaptic neuronal metabotropic glutamate receptors (mGluRs class III) leading to inhibition of glutamate release from the presynaptic neuron (Oliet et al., 2001; Figure 2A).
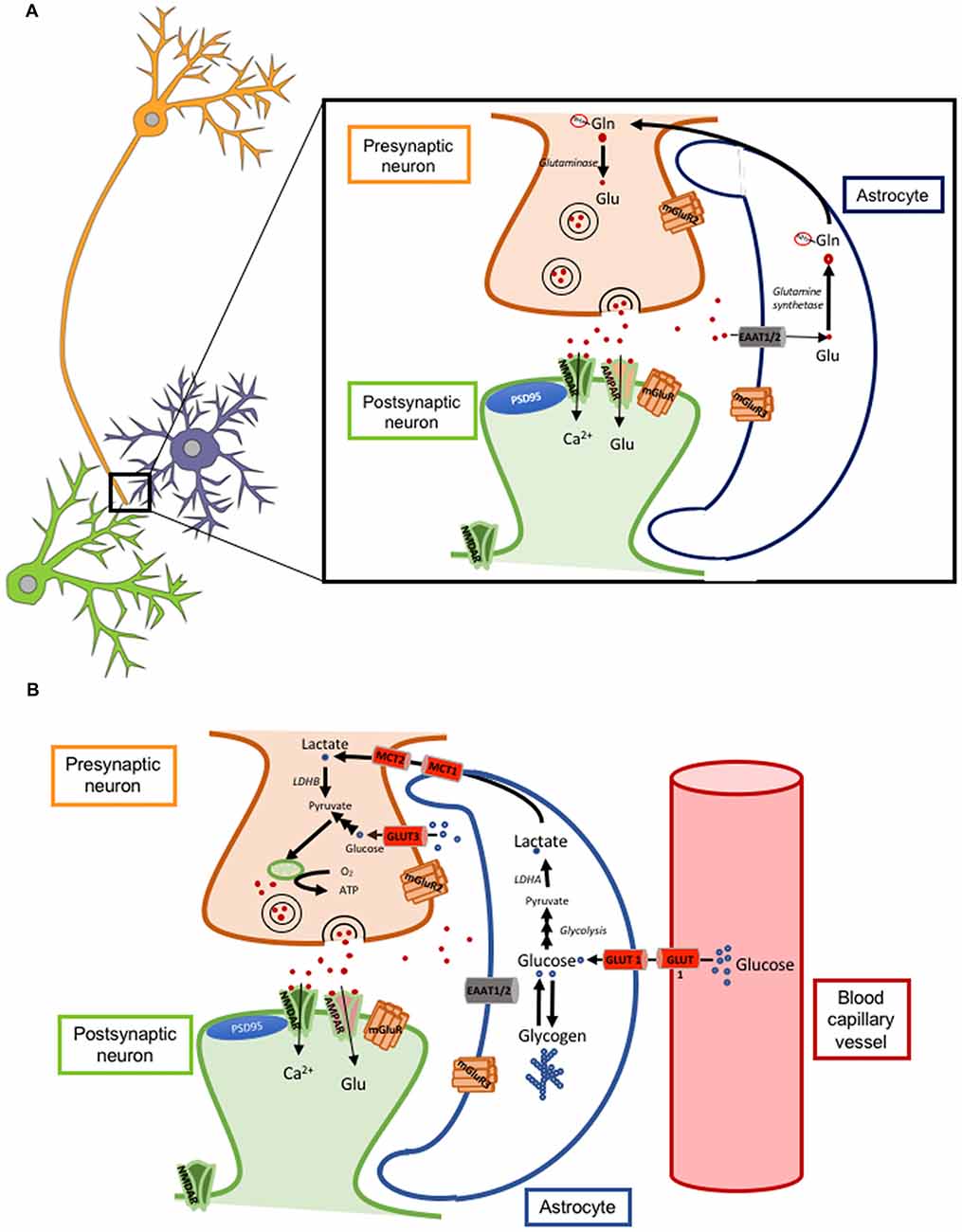
Figure 2. Astrocyte-neuron glutamate-glutamine cycle and lactate shuttle. (A) Cartoon illustrating a tripartite excitatory synapse with the glutamate-glutamine cycle. (B) Cartoon illustrating a tripartite excitatory synapse and a glioneurovascular unit with the lactate shuttle. Abbreviations: AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; ATP, adenosine triphosphate; EAAT, excitatory amino acid transporter; Gln-NH3, glutamine; Glu, glutamate; GLUT, glucose transporter; LDH, lactate dehydrogenase; MCT, monocarboxylate transporter; mGluR, metabotropic glutamate receptor; NMDAR, N-methyl-D-aspartate receptor.
In addition, several groups have proposed that astrocytes can release small molecules that act as modulators of synaptic activity, a phenomenon they termed gliotransmission. The main purported gliotransmitters are adenosine triphosphate (ATP) and D-serine. During long term potentiation (LTP), astrocytes would release ATP to the synaptic cleft, which would be then rapidly hydrolyzed to adenosine by extracellular ectonucleotidases. Adenosine would bind A1 adenosine receptors in neurons to suppress excitatory transmission in neighboring non-stimulated pathways, a function named heterosynaptic depression (Pascual et al., 2005). On the other hand, astrocytes would synthesize and release D-serine, which would bind the glycine site in the NMDA receptor of post-synaptic neuron, a binding that is necessary for the opening of the NMDA receptor gate upon glutamate binding and for LTP (Henneberger et al., 2010). Extracellular D-serine is at least as abundant as glycine but an up to three times more potent ligand of the glycine site. To produce D-serine, astrocytes would uptake L-serine from the extracellular space and convert it with the cytosolic enzyme serine racemase using pyridoxal 5’-phosphate as cofactor, whereas D-serine degradation is carried out by the also cytosolic enzyme D-amino acid oxidase.
Recent studies, however, have challenged the concept of gliotransmission and a heated debate is currently ongoing (Fiacco and McCarthy, 2018; Savtchouk and Volterra, 2018). The gliotransmission body of evidence was largely built upon observations of in vitro studies using primary astrocyte cultures as well as in vivo studies using a dominant-negative SNARE (dnSNARE) mouse model under the GFAP promoter to suppress synaptic-like vesicle release specifically in astrocytes (Pascual et al., 2005). The observation that this promoter is leaky to neurons, that is, the dnSNARE transgene was largely expressed by neurons rather than astrocytes, has essentially invalidated many of the above reports (Fujita et al., 2014; Sloan and Barres, 2014). Similarly, the notion that D-serine is produced and released by astrocytes has recently been challenged (Wolosker et al., 2016). Using BAC-transgenic mice expressing enhanced green fluorescent protein (eGFP) under the serine racemase promoter (Srr), Ehmsen et al. (2013) elegantly showed that serine racemase is expressed almost exclusively in neurons. In any case, D-serine levels have been reported to be unchanged in soluble cortical extracts from AD subjects compared to aged controls (Chouinard et al., 1993; Nagata et al., 1995).
Neurovascular Unit
The astrocytic endfeet are a structural part of the BBB together with the endothelial cells, basement membrane and pericytes (Figure 1D). Together with pericytes (Hall et al., 2014), astrocytes are thought to help coordinate blood flow with neuronal activity, a concept known as neurovascular coupling (Mishra et al., 2016). In addition, astrocytes control water flux between the brain and the bloodstream through surface water channels called aquaporins (AQ), especially AQ1 (also expressed in choroid plexus) and AQ4 (also expressed in ependymal cells). Moreover, astrocytes have recently been involved in the paravascular clearance of toxic solutes through the expression of AQ4 in the astrocyte perivascular endfeet. The discovery of this mechanism was possible by tracking small fluorescent tracers injected in the subarachnoid space of living mice with multiphoton microscopy through a craniotomy. This novel drainage system, proposed by Nedeegaard and termed glymphatic pathway, posits that the arterial pulse wave within the brain determines the rapid movement of cerebrospinal fluid (CSF) from the subarachnoid cortical space into the paravascular space of the penetrating arteries, and from them to the capillary beds and the interstitial fluid (ISF), where an exchange of toxic solutes would take place. Efflux of these solutes would then occur though the paravenous spaces. Sleep, anesthesia, exercise, body posture—supine and specially lateral positions—facilitate this glymphatic transport, whereas sleep deprivation and prone position reduce its rate (Xie et al., 2013; Lee et al., 2015; He et al., 2017; von Holstein-Rathlou et al., 2018). This clearance system has been reported to largely rely on the expression of AQ4 in the astrocyte perivascular endfeet, because it is severely impaired in AQ4 knock-out mice (Iliff et al., 2012, 2013). The implications of these findings in AD pathophysiology will be discussed in detail below.
Energy Metabolism
Glucose is the main source of energy for the brain. Glucose utilization has been traditionally correlated with neuronal activity and, since the mid 1980s, the radiotracer 18-fluorodeoxyglucose ([18F]-FDG) has been used for PET imaging of neuronal activity and for the diagnosis of AD, where there is a typical symmetric bilateral temporo-parietal hypometabolism. However, a recent study with micro-PET in rats has elegantly demonstrated that astrocytes also actively uptake glucose and contribute, at least to some extent, to the brain [18F]-FDG PET signal (Zimmer et al., 2017). Of note, the uptake of glucose by astrocytes is coupled with the uptake of glutamate, because blocking the glutamate transporter GLT-1 prevents glucose utilization and aerobic glycolysis in astrocytes (Zimmer et al., 2017). Astrocytes store the glucose in the form of glycogen and contribute to fuel neurons by supplying lactate in certain situations such as hypoglycemia and ischemia, a phenomenon often referred to as lactate shuttle (Mächler et al., 2016; Figure 2B). Of note, lactate has been reported to be necessary for LTP and memory formation by supplying energy to the neurons (Suzuki et al., 2011).
Morphological Features of Astrocyte Reaction in Alzheimer’s Disease
Reactive Astrocytes Associate With Alzheimer’s Pathological Hallmarks
AD is the most common neurodegenerative disease and the most common cause of dementia. Pathologically, AD is defined by the presence of two core lesions: amyloid plaques and neurofibrillary tangles (NFTs). Amyloid plaques are extracellular deposits of the amyloid β peptide (Aβ), which is a normal by-product resulting from the sequential cleavage of the transmembrane protein amyloid β precursor protein (AβPP) by the aspartyl-proteases β- and γ-secretases. NFTs are intracellular inclusions of the microtubule associated protein tau, which in the AD brain is aberrantly hyperphosphorylated and misfolded. Besides the development of these lesions, AD is associated with the disappearance of synapses, dendritic branches and neurons (Serrano-Pozo et al., 2011a). The first descriptions of the existence of prominent cortical astrocytic and microglial reactions in the AD brain date from the late 1980s (Beach and McGeer, 1988; Beach et al., 1989; Itagaki et al., 1989), but amyloid plaques, NFTs, and their effects on neurons and synapses have monopolized researchers’ interest for a long time, and the role of non-neuronal cells such as microglia and astrocytes is only recently gaining scientific momentum.
While reactive astrocytes are not exclusive of AD, the relationship between reactive astrocytes and AD pathological hallmarks, particularly amyloid plaques, is the most intimate in any neurodegenerative disease. In fact, senile amyloid plaques are defined among other features by the presence of a cluster of reactive astrocytes that penetrate and embrace amyloid deposits with their processes, fragmenting and isolating plaques from the surrounding neuropil (Itagaki et al., 1989; Figures 3A,B), and reactive astrocytes follow the laminar distribution of amyloid plaques in the association cortex (Beach and McGeer, 1988). Postmortem quantitative neuropathological studies have shown that the number of reactive astrocytes in the vicinity of amyloid plaques increases as the disease advances (Pike et al., 1995; Vehmas et al., 2003) and is independent of plaque size and apolipoprotein E (APOE) genotype (Figures 3C–F).
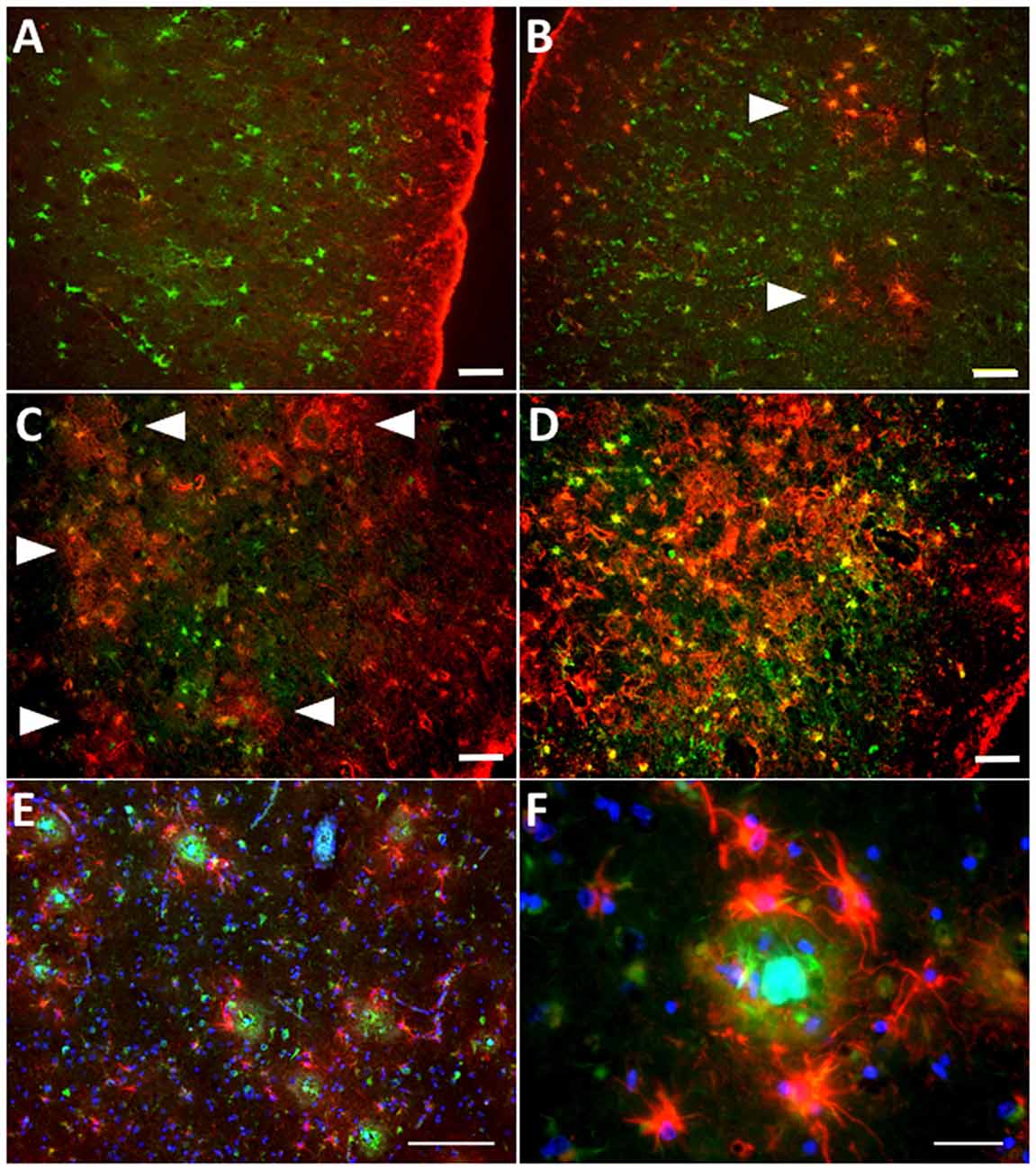
Figure 3. Astrocyte reaction to amyloid plaques in the Alzheimer’s brain. (A–D) Double fluorescent immunohistochemistry for GFAP (red) and the enzyme glutamine synthetase (green) showing the progression of astrocyte reaction (defined by GFAP immunoreactivity) in the temporal association neocortex (Brodmann’s area 38) of a healthy control subject without Alzheimer’s disease (AD) changes (A), another healthy control individual with sparse neuritic plaques (B), and two patients with an AD diagnosis (C,D). Arrowheads point to increasingly numerous clusters of GFAP-immunoreactive astrocytes, which become confluent in advanced stages of the disease. (E) Clusters of GFAP-immunoreactive astrocytes (red) surrounding dense-core amyloid plaques (Thioflavine-S positive, green) in the temporal association neocortex (Brodmann’s area 38) of an AD patient. (F) GFAP-immunoreactive astrocytes penetrate and surround dense-core amyloid plaques with their processes. Scale bars: 50 μm in (A–D), 100 μm in (E), and 10 μm in (F).
The association between reactive astrocytes and NFTs—the other core pathological lesion of the disease—has received much less attention, not only in AD but also in all the other tauopathies such as Pick’s disease, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease (AGD), or chronic traumatic encephalopathy (CTE). However, immunohistochemical and electron microscopy studies have shown that reactive astrocytes can also penetrate with their processes the extracellular “ghost” NFTs present in the midst of the neuropil in advanced AD (Ikeda et al., 1992a,b; Figures 4A–D). Therefore, these end-stage NFTs can exhibit both tau and GFAP immunoreactivities (Probst et al., 1982; Irwin et al., 2012). Postmortem quantitative neuropathological studies have shown that this spatial association between reactive astrocytes and NFTs also parallels the progression of the disease (Simpson et al., 2010).
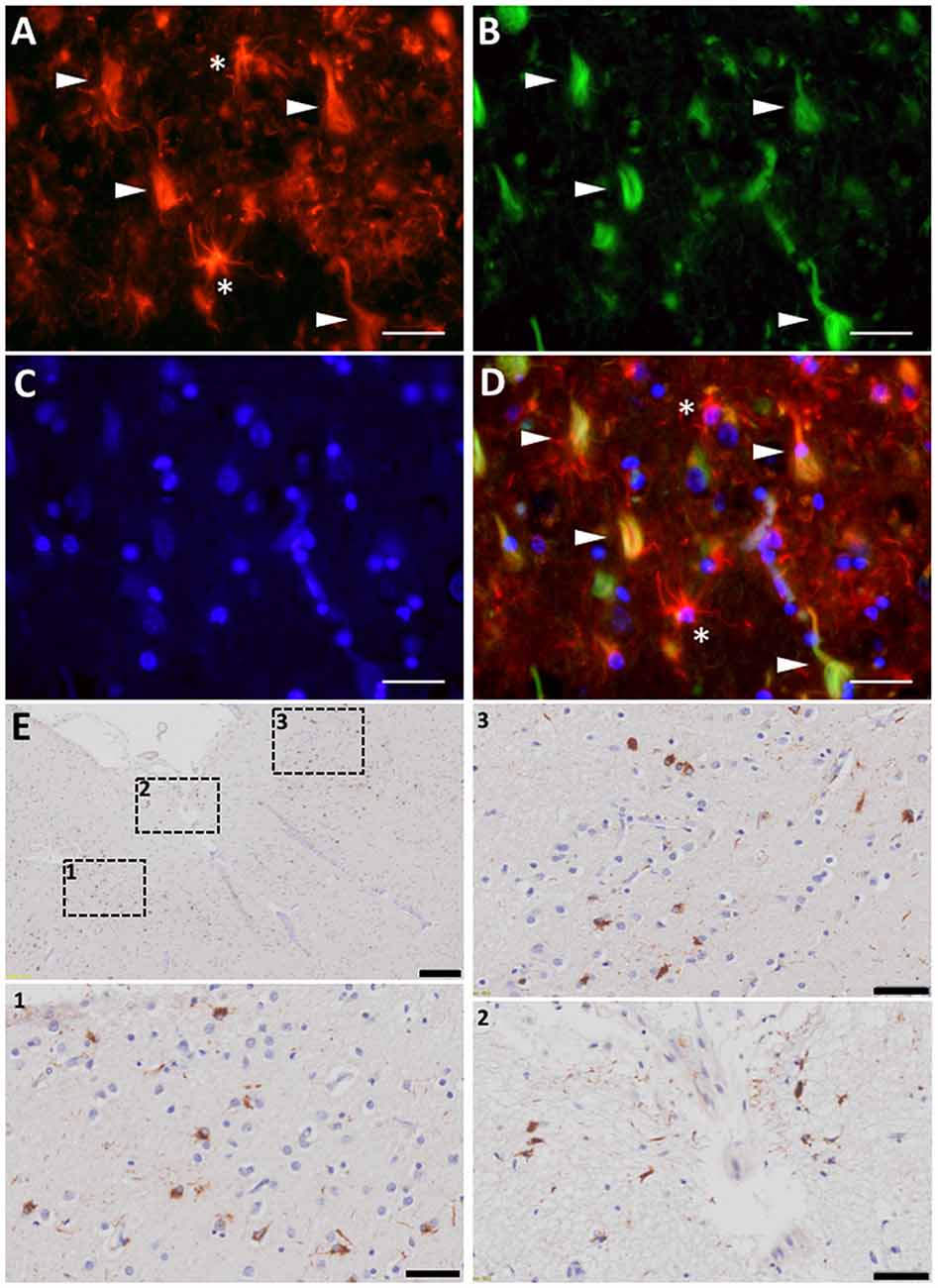
Figure 4. Astrocyte reaction to neurofibrillary tangles (NFTs) in the Alzheimer’s brain. (A–D) Fluorescent immunohistochemical staining for GFAP (A, red) with Thioflavine-S (B, green) and DAPI (C, blue) staining in the temporal association neocortex (Brodmann’s Area 38) of an individual with a diagnosis of AD. Reactive (GFAP+) astrocytes surround and penetrate extracellular “ghost” NFTs, so that these late-stage extracellular NFTs become GFAP-immunoreactive (D, merge). (E) Peroxidase-DAB immunohistochemical staining for total tau showing thorn-shaped tau-immunoreactive astrocytes in the subpial (inset 2) and mid layers (insets 1 and 3) of the frontal association cortex (Brodmann’s areas 8/9) of a subject with a diagnosis with AD. Scale bars: 10 μm in (A–D), 200 μm in (E), and 50 μm in insets 1–3.
Migration or Just Reorientation of Processes?
When astrocytes respond to an experimental acute injury and become reactive, they occupy the same neuropil volume and do not lose their even and non-overlapping distribution within the neuropil despite the hypertrophy and increased number of their main processes (Wilhelmsson et al., 2006). Whether reactive astrocytes migrate towards amyloid plaques or just reorient their processes towards them has not been investigated in detail. This distinction is important because the physiologic distribution of astrocytes in non-overlapping domains described above is thought to be crucial for their homeostatic and synaptic functions.
Several studies provide indirect evidence that astrocytes direct their processes but do not substantially move their cell bodies towards the plaques. The observation that the number of reactive astrocytes does not correlate with plaque size argues against their chemotactic migration towards plaques and in favor of a reaction of local “resting” or homeostatic astrocytes. A more recent statistical physics-based spatial analysis of astrocytes imaged in vivo in the cortex of APPswe/PS1dE9 and wild-type littermates confirmed that plaques in these transgenic mice do not alter the spatial distribution of astrocytes much (Galea et al., 2015). By contrast, homeostatic microglial cells are known to be highly dynamic and motile surveillants of the brain (Nimmerjahn et al., 2005), activated microglial cells have been shown to migrate towards dense-core amyloid plaques in mice in vivo (Meyer-Luehmann et al., 2008), and the number of activated microglial cells in the vicinity of plaques does correlate with plaque size, supporting a chemotactic effect (Serrano-Pozo et al., 2013b).
The reorientation of astrocyte processes towards amyloid plaques is dependent on the intermediate filaments of the cytoskeleton because depletion of GFAP and vimentin from astrocytes in a mouse model of AD causes a failure of astrocytes to penetrate within plaques and a decrease in astrocyte-plaque overlap (Kraft et al., 2013; Kamphuis et al., 2015). This phenomenon is so prominent that the palisade of long interlaminar astroglial processes, characteristic of the supragranular layers of the isocortex of primates, is severely disrupted or even virtually absent in AD and Down’s syndrome patients with AD neuropathological changes (Colombo et al., 2000, 2002, 2005). Importantly, these interlaminar astrocytes are likely major contributors to the microcolumnar functional organization of neurons in the isocortex (perpendicular to the pial surface), which is disrupted in AD, with thinner microcolumns and wider spaces between them (Buldyrev et al., 2000).
The up-regulation of GFAP in reactive astrocytes can be so dramatic in advanced AD that the GFAP protein can deposit in the astrocyte primary processes forming eosinophilic elongated structures called Rosenthal fibers (Wegiel and Wisniewski, 1994). These Rosenthal fibers are very much like those that define Alexander’s disease, a rare, usually childhood-onset, progressive leukodystrophy that causes spastic quadriparesis, seizures and intellectual decline. Interestingly, Alexander’s disease is due to mutations in the GFAP gene that result in decreased solubility and subsequent deposition of the GFAP protein within the cytoplasm of the white matter fibrous astrocytes (Hsiao et al., 2005; Hagemann et al., 2006). The insolubility of GFAP in advanced AD and Alexander’s disease could have deleterious effects in astrocyte biology by both overwhelming their protein degradation systems, autophagy (Tang et al., 2008) and proteasome (Tang et al., 2010; Orre et al., 2013), and restricting their range of migration and process motion.
Proliferation or Just Phenotypic Change?
Because reactive astrocytes are conventionally identified by their GFAP immunoreactivity and GFAP expression in non-reactive (resting) astrocytes is often below the detection level of immunohistochemistry, it is not uncommon to misinterpret the enhanced GFAP immunoreactivity seen in the AD brain as evidence of astrocyte proliferation. A few studies have found increased expression of cell division markers (i.e., cyclins, PCNA, Ki67) within astrocytes in the brain of AD subjects (Nagy et al., 1997; Wharton et al., 2005).
Despite these studies, there is more substantial and stronger evidence against proliferation of astrocytes or glial progenitors in AD. Experiments injecting BrdU in plaque-bearing mice have definitively demonstrated that only microglia exhibits significant proliferation, particularly in the proximity of plaques (Bondolfi et al., 2002; Kamphuis et al., 2012; Luccarini et al., 2012; Sirko et al., 2013). The same conclusion was achieved by a recent human postmortem study using the proliferative marker PCNA, Aβ and Iba1 or GFAP triple immunohistochemistry (Marlatt et al., 2014). Similar numbers of GFAP-positive astrocytes were found in wild-type and 3xTg AD mice hippocampus, where >80% astrocytes are GFAP-immunoreactive (Olabarria et al., 2010). Last, the number of cortical astrocytes was found to be similar between healthy and AD brains in two stereology-based quantitative human postmortem studies: one using morphological identification of astrocytes in Nissl-stained sections (Pelvig et al., 2003), and another using double fluorescent immunohistochemistry with GFAP and aldehyde dehydrogenase 1 isoform L1 (ALDH1L1) or glutamine synthetase as pan-astrocytic constitutive markers (Serrano-Pozo et al., 2013a).
Immortal or Senescent and Mortal?
Cell death is the other side of the coin with respect to cell proliferation. The finding of a similar number of astrocytes in AD and normal brains and the lack of correlation between total astrocyte number and AD progression could have two interpretations: (1) there is no significant astrocyte proliferation or death in either the normal or the AD brain; and (2) there is significant proliferation and death but both rates are similar. Several postmortem studies using terminal transferase-mediated dUTP-biotin nick end labeling (TUNEL) and caspase 3 activated immunohistochemistry (Smale et al., 1995; Li et al., 1997; Kobayashi et al., 2004) have reported apoptotic astrocytes, but other authors have not found astrocyte apoptosis (Sugaya et al., 1997). These studies are likely confounded by the cause of death and the potential pro-apoptotic effects of the agonal period immediately before death. The finding of increased CSF levels of astrocytic markers in AD patients with respect to healthy control subjects, including GFAP (Jesse et al., 2009; Ishiki et al., 2016) and glutamine synthetase (Gunnersen and Haley, 1992; Tumani et al., 1999), argues in favor of some astrocyte cell death.
Another possibility is that astrocytes reach a senescent state in the AD brain. Morphologically, senescent-looking astrocytes have been characterized in the hippocampus and entorhinal cortex of 3xTg AD mice as astrocytes that are located far from amyloid plaques (>50 μm), with atrophied cell somas, and simplified processes (Olabarria et al., 2010; Yeh et al., 2011). However, other authors have reported that the size of astrocyte somas far from amyloid plaques (>60 μm) in an APP/PS1 transgenic mouse model is comparable to that of wild-type mice (Brawek et al., 2018). Another more dramatic morphological change that may resemble astrocyte senescence is the so-called clasmatodendrosis. Clasmatodendritic astrocytes are characteristic for perikaryal swelling and accumulation of lysosomes and vacuoles, condensed chromatin leading to pyknotic nuclei, and fragmented or beaded processes. Recently, astrocytes expressing green-fluorescent protein (GFP) under the GFAP promoter have been reported to be sensitive to clasmatodendrosis in a plaque-bearing AD mouse model (Daschil and Humpel, 2016). However, it should be noted that this phenomenon has been described mostly in the fibrous astrocytes from periventricular and deep white matter lesions of individuals with vascular dementia and/or AD, rather than in cortical astrocytes. Moreover, the development of clasmatodendrosis has been attributed to ischemia, hypoxia, local disruption of the local BBB and acidification of the local environment, rather than to the toxicity of AD pathological hallmarks (Tomimoto et al., 1997; Sahlas et al., 2002; Chen et al., 2016). p16INK4a and matrix metalloproteinase 1 (MMP1) have been proposed as molecular markers defining senescent astrocytes (Bhat et al., 2012).
Functional Consequences of Astrocyte Reaction in Alzheimer’s Disease
As with the morphological aspects, considerable controversy surrounds the topic of the functional consequences of astrocyte reaction in AD. Conceptually, AD-associated astrocyte reaction could entail a loss of the normal functions of the astrocyte (i.e., modulation of synaptic function, BBB integrity and function), a gain of a new toxic function (i.e., inflammation), or both.
Plaque Formation or Plaque Clearance?
Whether reactive astrocytes contribute to plaque formation and maturation or to Aβ clearance and plaque growth restriction is still debated. Multiple immunohistochemical and immunoelectron microscopy postmortem studies on the AD brain have revealed that astrocytes and, to a lesser extent, microglia, contain granules of non-fibrillar Aβ, presumably engulfed from extracellular diffuse amyloid deposits (Akiyama et al., 1996, 1999; Funato et al., 1998; Yamaguchi et al., 1998; Thal et al., 2000; Oide et al., 2006). However, this observation has been also interpreted as evidence for an active participation of reactive astrocytes in plaque formation (Nagele et al., 2003), rather than clearance. A feed-forward mechanism has been proposed by which reactive astrocytes could up-regulate BACE1 and promote the amyloidogenic processing of AβPP in response to noxious stimuli such as inflammation (Zhao et al., 2011) or ischemia (Hartlage-Rübsamen et al., 2003). It is noteworthy that Aβ-containing astrocytes have been found preferentially near fleecy N-terminally truncated Aβ deposits in the human entorhinal cortex (Thal et al., 2000) and that astrocytes have been implicated in the generation and secretion of N-terminally truncated Aβ in a BACE1-independent manner (Oberstein et al., 2015).
On the other hand, ex vivo and in vivo experiments applying adult mouse or human astrocytes on the brain of plaque-bearing mice have confirmed that astrocytes uptake and clear Aβ, with diffuse deposits over larger fibrillar aggregates being preferentially removed (Koistinaho et al., 2004; Pihlaja et al., 2008, 2011; Nielsen et al., 2009, 2010), These experimental studies are in agreement with a postmortem immunohistochemical study that has confirmed the presence of oligomeric protofibrillar forms of Aβ within reactive astrocytes using conformation-specific antibodies (Lasagna-Reeves and Kayed, 2011). Two main mechanisms have been proposed: secretion of Aβ-degrading enzymes and phagocytosis (uptake and lysosomal degradation).
The phagocytosis of Aβ by astrocytes requires the participation of APOE because APOE null astrocytes are not efficient at amyloid plaque removal (Koistinaho et al., 2004). Using intracortical microinjection of fluorescently-labeled Aβ and in vivo multiphoton microscopy through a craniotomy in 5xFAD mice—characterized by a fast amyloid plaque deposition, it has been shown that the phagocytic potential of astrocytes declines with aging (Iram et al., 2016). Interestingly, in these mice the complement factor C1q can facilitate Aβ phagocytosis by astrocytes (Iram et al., 2016). Enhancing lysosomal biogenesis in astrocytes through viral delivery of the transcription factor EB (TFEB) results in increased Aβ phagocytosis and degradation in APP/PS1 mice (Xiao et al., 2014).
Postmortem immunohistochemical studies have revealed that astrocytes express some of the known Aβ-degrading enzymes. For example, endothelin-converting enzyme-2 (ECE-2) is up-regulated in AD astrocytes, although also in neurons and some microglial cells (Palmer et al., 2009). MMP3 is present in plaques and in white matter fibrous astrocytes in the AD brain (Yoshiyama et al., 2000; but see also, Baig et al., 2008). Astrocyte deficiency of lipoprotein-related protein 1 (LRP1), an apoE receptor, reduces the secretion of Aβ-degrading enzymes such as matrix metalloproteases (MMPs) 6 and 9 and insulin degrading enzyme (IDE) and accelerates amyloid plaque deposition (Liu et al., 2017). Of note, IDE expression has been shown to increase in reactive astrocytes around plaques in the Tg2576 AD mouse model (Leal et al., 2006).
It should be noted that both amyloid plaque burden and plaque size distribution remain relatively stable throughout the disease progression, indicating that there is no significant net plaque growth and clearance, that is, the clearance mechanisms might just enable to neutralize growth but not to effectively eliminate plaques (Hyman et al., 1995; Serrano-Pozo et al., 2011b, 2012). Although depletion of GFAP and vimentin from astrocytes led to a failure of penetration into amyloid plaques in a mouse model of AD, it is uncertain whether plaque growth and amyloid deposition subsequently accelerate because the two existing studies showed conflicting results (Kraft et al., 2013; Kamphuis et al., 2015). Therefore, although there is no proportionality between plaque size and number of surrounding reactive astrocytes (Serrano-Pozo et al., 2013b), it is plausible that reactive astrocytes form an effective physical barrier around the plaques and limit their growth.
Interestingly, besides Aβ, astrocytes can accumulate other proteins associated with neurodegeneration such as tau and α-synuclein. The presence of tau-immunoreactive “tufted” astrocytes pathologically defines PSP, whereas the finding of tau-immunoreactive “astrocytic plaques” defines the pathological diagnosis of CBD. An aging-related tau astrogliopathy (ARTAG) has recently been described with “thorn-shaped” and “granular fuzzy” tau-positive astrocytes in the medial temporal lobe and other brain regions (Kovacs et al., 2016; Liu et al., 2016). These thorn-shaped astrocytes have also been identified in the brain of individuals with AD (Lace et al., 2012; López-González et al., 2013; Figure 4E). In fact, classic electron microscopy studies in the 1990s already described the presence of paired helical filaments of tau in astrocytes in AD (Ikeda et al., 1992a; Yamazaki et al., 1995; Arima et al., 1998). Of note, although NFTs in AD are composed of similar amounts of three repeat (3R) and four repeat (4R)-tau, astrocyte tau immunoreactivity in AD, ARTAG, and the classic tauopathies PSP and CBD is predominantly 4R-tau, not 3R. Whether astrocytes have a predilection for up-taking 4R-tau over 3R-tau, or whether they degrade 3R-tau more readily than 4R-tau, remains unknown. Similarly to tau, α-synuclein accumulation has been found within astrocytes in the basal forebrain of subjects with sporadic Parkinson’s disease (PD; Braak et al., 2007). Although it is possible that astrocytes express low levels of these genes and up-regulate their expression in these neurodegenerative diseases, it is more plausible that they take up misfolded forms of these proteins from the ISF, since both tau and α-synuclein are thought to be released by neurons at the synapse and to propagate from neuron to neuron trans-synaptically (Luk et al., 2012; de Calignon et al., 2012). Whether and how astrocyte reaction impacts the prion-like propagation of these proteins, and the consequences of these astrocyte proteinopathies for the astrocyte biology remain to be investigated.
Neuroprotection or Neurotoxicity?
A simplistic view of reactive astrocytes as neuroprotective in AD alludes to the formation of a scar-like physical barrier between the amyloid plaques and the surrounding neuropil. Reactive astrocytes around amyloid plaques may, thus, limit the collateral damage from diffusible soluble Aβ oligomeric species by isolating the reservoirs of these especially neurotoxic Aβ species. For example, in a quantitative neuropathological study from the population-based Medical Research Council-Cognitive Function and Aging Study (MRC-CFAS), the number of diffuse and compact plaques lacking astrocyte reaction in layer VI of the cingulate cortex was independently associated with worse cognition (lower MMSE score; Mathur et al., 2015). The attenuation of astrocyte reaction by depletion of GFAP and vimentin led to a multiplication of plaque-associated dystrophic neurites, also suggesting a net neuroprotective effect of reactive astrocytes in AD (Kraft et al., 2013). On the other hand, reactive astrocytes around neuritic plaques have recently been shown to engulf plaque-associated dystrophic neurites of APPPS1 mice and AD patients and this phagocytic function has been deemed neuroprotective (Gomez-Arboledas et al., 2018). These observations contrast with other studies supporting the idea that astrocyte reaction contributes to neurodegeneration in dementia due to AD. Astrocytes isolated from aged 5xFAD offer much less neurotrophic support when co-cultured with neonatal neurons as compared with astrocytes from wildtype mice (Iram et al., 2016). Postmortem studies comparing subjects with dementia due to AD and cognitively intact subjects with high levels of amyloid plaques and NFTs (so called “high pathology control”, “asymptomatic AD”, or “mismatch AD” cases) have shown that the latter group lacks the prominent microglial and astrocyte responses typical of the AD brain, and have lower or normal levels of inflammatory cytokines and preserved neuron number and synaptic density (Lue et al., 1996; Perez-Nievas et al., 2013).
An unbiased way to approach this question is to study the transcriptomic profile of astrocytes from the AD brain compared to the normal aging brain. It should be noted that a substantial region-dependent shift in the astrocyte transcriptome has been reported with aging in both mouse and human normal brains (Soreq et al., 2017; Boisvert et al., 2018). Specifically, aged murine astrocytes down-regulate cholesterol synthesis and up-regulate synaptic elimination and immune pathways, whereas homeostatic and glutamate neurotransmission genes do not appear to change much with aging (Boisvert et al., 2018). Several human postmortem studies using laser capture microdissection (LCM) of astrocytes in brains from AD patients and non-demented healthy controls have investigated the transcriptomic changes that occur in AD astrocytes. Simpson et al. (2011) used GFAP to identify and capture astrocytes by LCM and compared the transcriptomic profile between subjects with an advanced Braak stage of NFTs (V–VI) and subjects with low Braak stages (I–II). They observed a dysregulation of genes associated with the actin cytoskeleton, proliferation, apoptosis, and ubiquitin-mediated proteolysis at low Braak stages, that contrasted with an altered regulation of intracellular signaling pathways, including insulin, phosphatidylinositol 3-kinase (PI3K)/Akt, and mitogen-activated protein kinase (MAPK) pathways at high levels of AD pathology. In another more recent LCM-based transcriptomic study using ALDH1L1 rather than GFAP as astrocytic marker, the authors found an up-regulation of genes encoding both astrocytic immune response and mitochondrial machinery in the posterior cingulate gyrus—an area of abundant and early Aβ deposition—of AD patients compared to healthy subjects (Sekar et al., 2015). The advantage of using ALDH1L1 instead of GFAP is that non-reactive “resting” GFAP-negative astrocytes are better represented in the healthy control group, allowing for the detection of more subtle differences in gene expression between health and disease.
Although meritorious, the results of human postmortem transcriptomic studies should be taken with caution because they can be affected by several confounders including the effects on gene expression of the cause of death and the agonal period prior to death (i.e., hypoxia, ischemia, sepsis), the postmortem interval, and the common presence of mixed pathologies such as Lewy bodies and cerebrovascular disease. Moreover, it is not easy to dissect the effect of amyloid plaques and NFTs on astrocyte gene expression. By contrast, transcriptomic studies in transgenic mice are devoid of these confounders. Orre et al. (2014) performed a transcriptional analysis on acutely isolated astrocytes from the cortex of aged controls and APPswe/PS1dE9 AD mice using GLT-1 as astrocytic marker for fluorescently-assisted cell sorting (FACS). These mice develop amyloid plaques similar to human AD plaques, but not NFTs. In this transgenic AD mouse model, astrocytes exhibited a proinflammatory immune phenotype and a reduced expression of neuronal support genes and genes involved in neuronal communication. Based on the distinct transcriptomic profiles observed in mouse models of acute brain injury, such as the lipopolysaccharide (LPS) model of neuroinflammation and the stroke model of middle cerebral artery occlusion, Barres proposed a classification of astrocytes in neurotoxic (A1) and neuroprotective (A2; Zamanian et al., 2012). These authors recently showed that, in vitro, A1 astrocytes lose their ability to promote neuronal survival, neurite outgrowth, synapse formation and phagocytosis, leading to neuron and oligodendrocyte death, and postulated that A1 astrocytes can be identified by the expression of complement fraction 3 (C3) and are abundant in many human neurological diseases such as AD, amyotrophic lateral sclerosis, Huntington’s disease, and multiple sclerosis (Liddelow et al., 2017). Following this classification, an age-dependent neurotoxic transcriptomic signature (A1) has recently been reported in astrocytes isolated from a tauopathy mouse model under the human APOEε4 knock-in (KI) background, as compared with the APOEε3 KI background (Shi et al., 2017).
Thus, taken together, astrocyte transcriptomic studies from human AD brain and AD mouse models support the idea that astrocyte reaction in AD involves a gain of neurotoxic function and loss of neuroprotective function of astrocytes.
Effects on Synaptic Function
There is growing evidence indicating that astrocyte reaction impairs the normal function of astrocytes as modulators of neuronal synaptic transmission. Most studies have focused on glutamatergic transmission and have attributed reactive astrocytes a crucial participation in glutamate-mediated neuronal excitotoxicity.
Using in vivo multiphoton microscopy through a craniotomy and the glutamate-sensitive probe iGluSnFR delivered with an intracortical injection of an adeno-associated viral vector, Hefendehl et al. (2016) have recently reported that the microenvironment surrounding plaques in an APPPS1 mouse model has chronically elevated glutamate concentrations, and that neurons in the immediate vicinity of plaques do not appropriately respond to stimuli, such as hindlimb pinch in the somatosensory cortex or visual stimuli in the visual cortex, as judged by the in vivo imaging analysis of calcium transients and glutamate concentration. This increased peri-plaque glutamate concentration correlated with a reduced expression of GLT-1/EAAT2 in the reactive (GFAP+) astrocytes surrounding plaques, and was partially corrected by the intravenous administration of the antibiotic ceftriaxone, which is known to up-regulate the expression of GLT1-/EAAT2 by astrocytes (Hefendehl et al., 2016). Of note, soluble Aβ oligomers reduce the expression of the glutamate transporter GLT-1/EAAT2 in astrocyte cultures through a mechanism involving calcineurin (CN)/nuclear factor of activated T cells (NFAT) pathway (Abdul et al., 2009) and oxidative stress (Scimemi et al., 2013). This reduction of GLT-1/EAAT2 expression by astrocytes parallels the progression of AD pathology in the human brain (Simpson et al., 2010). Recently, it has been proposed that preservation of GLT-1/EAAT2 expression in GFAP+ reactive astrocytes could be a mechanism of resilience against AD neuropathological changes (Kobayashi et al., 2018). Other authors have shown a decreased solubility of GLT-1/EAAT2 in the brain of patients with AD, which could potentially impair the re-uptake of synaptic glutamate (Woltjer et al., 2010).
A second proposed mechanism for astrocyte-mediated neurotoxicity is the downregulation of glutamine synthetase expression level or activity, leading to a subsequent reduction in the astrocyte capacity to detoxify neuronal glutamate to glutamine. Indeed, acute viral-induced astrocyte reaction can induce neuronal glutamate excitotoxicity via down-regulation of the astrocyte cytosolic enzyme glutamine synthetase (Ortinski et al., 2010) and glutamine synthetase levels have been reported to be reduced in the 3xTg mouse prefrontal cortex (Olabarria et al., 2011; Kulijewicz-Nawrot et al., 2013). However, whether this is also the case in the human AD brain remains unclear (Le Prince et al., 1995; Tumani et al., 1999; Serrano-Pozo et al., 2013a). Notwithstanding these conflicting reports, glutamine synthetase catalytic activity has been shown to be sensitive to oxidation and may be impaired by oxidative damage in AD (Smith et al., 1991; Hensley et al., 1994, 1995). Perhaps as compensatory mechanism against glutamate excitotoxicity, glutamine synthetase can be up-regulated in groups of cortical pyramidal neurons in AD brains, although not selectively in the vicinity of plaques (Robinson, 2000; Serrano-Pozo et al., 2013a). The common denominator of all these mechanisms is thought to be an increase in extracellular glutamate levels leading to excessive activation of extra-synaptic NMDA receptors and, thereby, neuronal excitotoxicity (Li et al., 2011). This common mechanism could explain the finding of calcium overload (Kuchibhotla et al., 2008) and hyperactive neurons (Busche et al., 2008, 2012) found in the proximity of plaques using multiphoton calcium imaging in live APP/PS1 mice, although the latter finding was attributed to a decreased gabaergic inhibition instead of an excess of glutamate.
A third alternative novel mechanism proposed for astrocyte-mediated neurotoxicity is an enhancement of gabaergic tone, rather than excessive glutamatergic transmission or reduced gabaergic inhibition. Reactive astrocytes from plaque-bearing AD mouse models have been shown to induce neuronal tonic inhibition via increase in GABA release to the synaptic cleft (Mitew et al., 2013; Jo et al., 2014; Wu et al., 2014). A recent detailed histological study has described a transient increase in GABA immunoreactivity of astrocytes in middle aged (but not young or very old) APP/PS1 transgenic mice, specifically in reactive astrocytes surrounding dense-core plaques (Brawek et al., 2018). This mechanism could explain the co-existence of an abnormally high proportion of silent neurons in multiphoton calcium imaging studies on living APP/PS1 mice (Busche et al., 2008, 2012).
Effects on Blood Brain Barrier Integrity and Function
Besides forming amyloid plaques in the brain parenchyma, Aβ can deposit in the wall of small cortical and leptomeningeal vessels and capillaries, a condition called cerebral amyloid angiopathy (CAA). CAA can occur without concomitant AD pathology, but more commonly is present in up to 90% of individuals with a postmortem diagnosis of AD, usually with a mild degree (Serrano-Pozo et al., 2011a). In addition to the possibility of causing lobar intracerebral and focal subarachnoid hemorrhages due to vessel rupture, CAA can cause brain hypoperfusion and subsequent ischemia, and independently contributes to AD-related cognitive decline (Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study, 2001; Pfeifer et al., 2002; Arvanitakis et al., 2011; Serrano-Pozo et al., 2013c). Moreover, an increased CSF/plasma albumin ratio, indicating increased BBB permeability, has been repeatedly shown in AD and other dementias (Skoog et al., 1998; Bowman et al., 2007; Skillbäck et al., 2017).
Since astrocytes are part of the BBB, the contribution of astrocyte reaction to BBB disruption and amyloid plaque and CAA accumulation are gaining increasing research interest. Does astrocyte reaction affect BBB integrity in AD and CAA? Indeed, a number of astrocyte abnormalities have been described in the neurovascular unit of mouse models of AD and CAA and human AD brains including: (1) swelling and detachment of astrocyte endfeet; (2) altered secretion of proteins that are part of the basement membrane or the extracellular matrix; (3) reduction of both endothelial and astrocytic glucose transporter 1 (Glut1); (4) reduced astrocytic expression of monocarboxylate transporter 1 (MCT1) resulting in a decreased lactate release; and (5) loss of potassium and water channels (AQ; Wilcock et al., 2009; Hawkes et al., 2011; Merlini et al., 2011). Furthermore, astrocyte-dependent cerebral vasoreactivity was reported to be impaired in amyloid-laden vessels in a mouse model of CAA (Kimbrough et al., 2015).
In addition, it is possible that astrocyte reaction impairs the clearance of Aβ at the BBB. Weller et al. (2008) proposed that Aβ is cleared from the ISF through a perivascular drainage from capillaries to cortical and leptomeningeal arteries through the basement membranes. They proposed that the suction effect after the arterial pulse wave drives this perivascular drainage and that the stiffening of the arterial walls with aging and vascular risk factors could attenuate this arterial pulsatility and favor Aβ accumulation in capillaries and arteries in the form of CAA and in the brain parenchyma in the form of amyloid plaques (Weller et al., 2008; Hawkes et al., 2011, 2014; Arbel-Ornath et al., 2013). In 2012, Nedergaard redefined this hypothesis by proposing that toxic solutes including Aβ are cleared from the ISF through a paravascular drainage system that involves astrocytes and would serve as the brain lymphatic system, hence the term glymphatic (Iliff et al., 2012, 2013). Specifically, the glymphatic system hypothesis postulates that there is an inflow of CSF from the subarachnoid space to the paravascular space of penetrating arteries, between the basement membrane of the endothelium and the smooth muscle cell layer; then, low molecular weight solutes including Aβ pass to the ISF through the astrocyte end-feet of the BBB and efflux from the brain parenchyma through the paravenous space by a convective bulk-flow driven by the arterial pulse wave and the arterio-venous pressure gradient, rather than simple diffusion. Noteworthy, sleep has been shown to potentiate this transport system (Xie et al., 2013), and sleep deprivation has been shown to enhance Aβ deposition (Kang et al., 2009). This glymphatic drainage system would be largely dependent on the expression of AQ4 in the astrocyte perivascular endfeet, because genetic deletion of AQ4 severely impairs this glymphatic system, disrupts the paravascular flow of Aβ (Iliff et al., 2012), and leads to an increased amyloid burden in the form of both plaques and CAA, and further cognitive impairment in APP/PS1 mice (Xu et al., 2015). In addition, aging has been associated with a decreased perivascular localization of AQ4 and, thus, impaired paravascular clearance of endogenous Aβ in wild-type mice (Kress et al., 2014).
However, these studies have been recently disputed. Using computational models and similar multiphoton microscopy experiments in wild-type and AQ4 null living mice and rats, the Verkman lab has refuted both the hypothesis that arterial pulse-driven convective bulk flow rather than simple diffusion is responsible for the vast part of solute movement through the brain parenchyma, as well as the involvement of AQ4 in solute transport in the rodent brain (Jin et al., 2016; Smith et al., 2017). Another recent study applied computational modeling to 3D-reconstruction of electron microscopy images of the neuropil and concluded than diffusion rather than bulk-flow accounts for the transport of interstitial solutes (Holter et al., 2017). Other computational modeling studies have tried to explain cerebral metabolite clearance by proposing that there is a network of astrocytes connected through AQ4 channels that serve as sites of low resistance to bulk flow, and that the arterial pulsation determines a fast para-arterial transport through dispersion (the combined effect of local mixing and diffusion in the para-arterial space), rather than bulk flow (Asgari et al., 2015, 2016). Studies on AQ in the human AD brain have also yielded conflicting results. Aquaporin 1 (AQ1) immunoreactivity has been shown to increase in close proximity to amyloid plaques and CAA in some studies (Misawa et al., 2008; Hoshi et al., 2012), whereas other authors found no difference in AQ1 immunoreactivity between AD and healthy control subjects, reported AQ1 to be expressed primarily in the white matter rather than the cortical astrocytes, and observed a decrease in expression with aging (Moftakhar et al., 2010). Furthermore, in another study, the researchers only found significantly increased levels of AQ1 by Western blot at early stages of the disease (Braak II; Pérez et al., 2007). With regards to aquaporin 4 (AQ4), no significant differences in protein levels were found between AD and healthy control subjects by Western blot (Pérez et al., 2007), but an enhanced immunoreactivity associated with amyloid plaques and CAA has been reported by other authors (Moftakhar et al., 2010; Hoshi et al., 2012), suggesting the possibility of a redistribution of AQ4 within astrocytes in AD. More recently, in a population-based clinic-pathological study, Zeppenfeld et al. (2017) reported an increased global AQ4 immunoreactivity in AD patients compared to aged and young healthy controls, associated with a loss of perivascular localization that correlated inversely with an increased plaque burden and a higher Braak NFT stage, which seemingly reconciles the human data with the mouse data above.
Signaling Pathways Involved in Astrocyte Reaction in Alzheimer’s Disease
To date, in vivo and postmortem evidence has implicated four main signaling pathways in the astrocyte reaction in AD: the Janus kinase (JAK)/STAT3, the calcium/CN/NFAT, the NFκB and the MAPK pathways. Figure 5 depicts these molecular cascades that transmit information from extracellular signals to target genes in the nucleus. Other pathways probably involved in astrocyte reaction but with less in vivo supporting evidence are calpain and caspase activation.
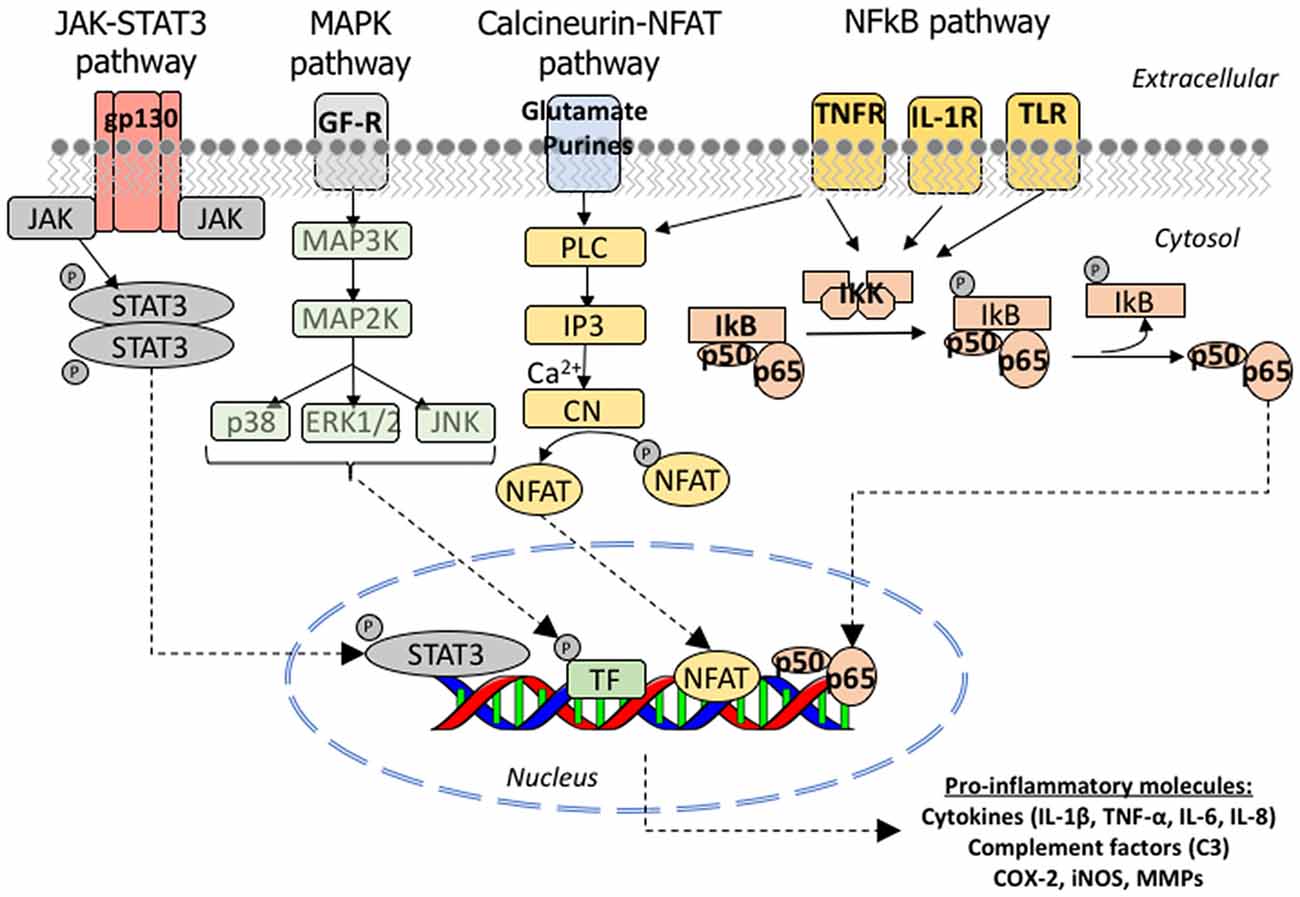
Figure 5. Molecular signaling pathways involved in astrocyte reaction in AD. Cartoon illustrating the JAK/STAT3, MAPK, CN/NFAT and NFκB signaling cascades. JAK/STAT3 pathway: the JAKs (JAK 1, 2, 3 and Tyk2) are tyrosine-kinases associated with the intracellular domain of cytokine receptors. The binding of a cytokine ligand to its receptor triggers a dimerization or oligomerization of the associated JAK and this leads to its transphosphorylation and activation. The activated JAK then phosphorylates key tyrosine residues in the cytokine receptor, which leads to the recruitment of STAT (STAT1, 2, 3, 4, 5A, 5B or 6) and a single tyrosine phosphorylation at the Y of its C-terminus. Once phosphorylated, STAT forms homo or heterodimers and is imported to the nucleus, where it complexes with other transcription factors, notably NFkB, to bind its target genes. MAPK pathway: the p38 mitogen-activating protein kinase (p38 MAPK) is a family of serine-threonine kinases of which p38α is the most studied due to its ubiquitous expression at high levels. p38α phosphorylates multiple substrates including transcription factors, DNA and RNA binding proteins, other serine-threonine kinases (including GSK3β), and cell cycle and pro-apoptotic proteins. Calcineurin/NFAT pathway: the phosphatase function of CN is activated upon the binding of calcium and calmodulin to its regulatory subunit. Once activated, CN removes several phosphate residues from the N-terminus of the transcription factors NFATc (nuclear factor of activated T cells) proteins, named NFATc1 to 4, and exposes their nuclear localization motif, leading to their rapid entry into the nucleus. NFATc targets are largely genes encoding cytokines, growth factors and their receptors, and cell adhesion proteins, as well as many microRNAs. NFκB pathway: NFκB is a heterodimeric transcription factor composed of the subunits p65/RelA and p50, which belong to the Rel family of proteins. It is normally located in the cytoplasm in an inactive form due to its binding to the inhibitor IκB, which masks its nuclear localization signal located in the p65 subunit. Activation of IKK complex by certain extracellular signals can dissociate NFκB and IκB by promoting the phosphorylation of the IκB-α, which is a necessary step for IκB-α ubiquitination and subsequent degradation by the proteasome. Once free from IκB, NFκB can translocate to the nucleus and bind to the 10 base pair consensus-sequence GGGACTTTCC GGGRNNYYCC in the promoter of its target genes (NFκB response elements). Abbreviations: CN, calcineurin; COX, cyclo-oxygenase; ERK, extracellular signal-regulated kinases; IL, interleukin; IL1-R, interleukin 1 receptor; iNOS, inducible nitric oxide synthetase; IP3, inositol-3-phosphate; JAK, Janus kinase; MAPK, mitogen-activated protein kinase; MMPs, matrix metalloproteases; NFAT, nuclear factor of activated T cells; PL3, phospholipase 3; STAT, signal transducer and activator of transcription; TLR, toll-like receptor; TNF-α, tumor necrosis factor α; TNF-R, tumor necrosis factor receptor.
JAK/STAT3 Pathway
The JAK-STAT (signal transducer and activator of transcription) is a signaling pathway activated by extracellular cytokines (Mertens and Darnell, 2007). The levels of STAT1 protein were shown to be increased in both the cytosolic and particulated fractions from the cortex of AD patients compared to healthy subjects (Kitamura et al., 1997). Recently, STAT3 immunoreactivity was shown to be increased in the nucleus of GFAP- and vimentin-immunoreactive astrocytes in the APPswe/PS1dE9 and 3xTg mouse models of AD. Lentiviral expression of suppressor of cytokine signaling protein 3 (SOCS3)—a negative regulator of this pathway, which is upregulated by STAT and switches off the activity of JAK—specifically in astrocytes led not only to a decrease in nuclear STAT3 immunoreactivity but also to a reduction of GFAP immunoreactivity, implicating the JAK/STAT3 pathway in astrocytic reaction (Ben Haim et al., 2015).
Calcium/Calcineurin/NFAT Pathway
The calcium/CN/NFAT pathway links cytosolic calcium levels to gene expression (Crabtree and Schreiber, 2009). CN has been reported to play a central role in AD pathophysiology both in neuronal and astrocytic phenotypes. In neurons, CN excessive activation has deleterious consequences, both functional and morphological. Soluble Aβ oligomers bind to NMDA receptors and increase cytosolic calcium, leading to CN activation and enhanced long term depression (LTD), which contributes to memory impairment. The morphological substrate of CN overactivation is the triad of synaptic loss, dystrophic neurites, and neuron loss (Wu et al., 2010). Inhibiting CN/NFAT pathway in APP/PS1 mice, either by treatment with the CN inhibitor immunosuppressant drug tacrolimus (also called FK-506; Rozkalne et al., 2011) or through the viral-mediated neuronal expression of the specific inhibitor peptide VIVIT, restored this phenotype (Hudry et al., 2012).
Soluble Aβ oligomers also determine a cytosolic calcium overload and CN activation in astrocytes in vitro, which leads to an overexpression of GFAP and, subsequently, a reactive phenotype characterized by the release of inflammatory cytokines and the down-regulation of the glutamate transporter GLT-1/EAAT2 (Norris et al., 2005; Abdul et al., 2009). In vivo studies also support CN overactivation in astrocytes. Resting calcium level and frequency and synchrony of calcium transients are increased in reactive astrocytes from APP/PS1 mice (Kuchibhotla et al., 2009; Delekate et al., 2014). CN immunoreactivity is increased in hippocampal astrocytes of aged wild-type mice and APP/PS1 mice (Norris et al., 2005). A high activity proteolytic fragment of CN with 45–48 KDa molecular weight range is highly expressed by astrocytes surrounding plaques in human postmortem brain sections (Pleiss et al., 2016). In the human AD hippocampus, CN-Aα and NFAT3 have been shown to increasingly translocate to the astrocyte nucleus as the disease advances, whereas NFAT1 has been shown to translocate to the nucleus mainly in the stage of mild cognitive impairment (Abdul et al., 2009). Inhibition of CN/NFAT pathway in hippocampal astrocytes through the selective viral-mediated expression of VIVIT in astrocytes improved cognitive and synaptic function, reduced glial activation, lowered Aβ levels, increased GLT-1 expression, and reduced glutamate-mediated neuronal excitotoxicity in plaque-bearing AD mice (Furman et al., 2012; Sompol et al., 2017).
NFκB Pathway
The NFκB pathway is activated by membrane receptor signals from Toll-like receptors, TNFα receptor, T-cell receptor and B-cell receptor. Among NFκB target genes are cytokines (Il-1, IL-6, TNFα, COX-2), complement proteins (i.e., C3), major histocompatibility complex (MHC) class 1 and 2, β-2 microglobulin and APOE (Hayden et al., 2006). Of note, complement fraction C3 has been proposed as a specific marker of a subtype of reactive GFAP-positive astrocytes that are neurotoxic (so-called A1 astrocytes; Liddelow et al., 2017).
Among the stimuli that can trigger this pathway are Aβ1–40 and Aβ1–42, S100β protein and reactive oxygen species (ROS). In vitro experiments in primary culture of rat cortical astrocytes have shown that Aβ at nM or μM concentrations can activate NFκB leading to an upregulation of pro-inflammatory cytokines IL-1 and IL-6 (Bales et al., 1998) and inducible nitric oxide synthetase (iNOS; Akama et al., 1998). The astrocytic secreted S100β protein can also up-regulate iNOS in cultured astrocytes in an autocrine fashion through a NFκB-dependent mechanism (Lam et al., 2001).
Importantly, suppression of NFκB in the APPswe/PS1dE9 ameliorated astrocytic reaction. However, intriguingly, the level of the NFκB inhibitor IκB-α was normal in 3xTg AD mice compared to wild-type littermates, arguing against NFκB activation in this mouse model (Ben Haim et al., 2015). The levels of NFκB p65 protein were shown to be increased in the cortex from AD patients compared to healthy subjects (Kitamura et al., 1997). However, neuropathological studies in AD brains addressing the cell type responsible for this increase have revealed that NFκB immunoreactivity is increased in the cytoplasm and nucleus of neurons, especially those surrounding amyloid plaques or bearing a NFT (Terai et al., 1996; Kaltschmidt et al., 1997; Ferrer et al., 1998). According to these studies the astrocytic NFκB immunoreactivity is either undetectable (Ferrer et al., 1998), only present in the cytoplasm of cortical layer I astrocytes and to a similar extent than in control brains (Terai et al., 1996), or only present in reactive astrocytes around diffuse plaques (Kaltschmidt et al., 1997).
MAPK Pathway
The mitogen-activating protein kinase (MAPK) pathway is represented by three main families of kinases, all of which are activated by extracellular signals: the p38 MAPK, the c-Jun kinase (JNK), and the extracellular signal-regulated kinases (ERK 1 and 2).
The p38 MAPK together with Jun kinase (JNK) are known as the stress kinases. p38α is activated by very diverse extracellular stimuli including UV light, heat and osmotic shock, oxidative stress, cytokines, chemokines, hormones and growth factors and is thought to have a key role in the cell response to many extracellular threats, leaning the fate of the cell towards survival vs. apoptosis. Inhibition of the p38 MAPK pathway in astrocytes has anti-inflammatory effects in astrocytes in vitro (Da Silva et al., 1997; Bhat et al., 2012).
The ERKs are essentially activated by trophic factors and are thought to play an important role in synaptic plasticity. Importantly, a postmortem immunohistochemical study revealed that this pathway is upregulated in the AD brain specifically in GFAP+ reactive astrocytes, but only in the white matter and only at early AD stages corresponding to mild dementia. In moderate and advanced AD dementia, the astrocytic expression was equivalent to non-demented control individuals and predominated in cortical pyramidal neurons, their axons, and plaque-associated dystrophic neurites (Webster et al., 2006). The authors speculated with a protective role of this signaling pathway in astrocytes against early neuronal and synaptic damage.
Apolipoprotein E Genotype and Astrocyte Reaction
The APOEε4 allele remains the strongest genetic risk factor for the development of AD. Compared to the most common genotype in the general population ε3/ε3, carrying one copy of the ε4 allele increases the risk of developing AD ~2–3 times, whereas homozygous (ε4/ε4) individuals have a ~8–12-fold higher risk. Moreover, the ε4 allele anticipates the onset of AD in a dose-dependent manner (Corder et al., 1993). By contrast, the ε2 allele is protective against the development of AD (Corder et al., 1994; Serrano-Pozo et al., 2015). The main normal function of the APOE is the transport of cholesterol within the brain in the form of high-density lipoprotein (HDL) particles. However, apoE4 has been shown to favor Aβ accumulation by promoting its aggregation in the form of amyloid plaques (Hyman et al., 1995) and soluble Aβ oligomers (Hashimoto et al., 2012), and by reducing Aβ clearance (Castellano et al., 2011). Remarkably, although large clinico-pathological studies have established that the APOEε4 allele does not increase either the burden or the Braak stages of NFTs independently of amyloid plaques (Serrano-Pozo et al., 2015; Farfel et al., 2016), apoE4 has recently been shown to promote tau pathology and neurodegeneration in a mouse model of tauopathy (Shi et al., 2017).
Because, together with microglia, astrocytes are the main source of apoE within the brain, whether the APOE genotype has any influence on the astrocyte reaction found in the AD brain is a research topic of growing interest. Using stereology-based quantitative methods in postmortem human AD brain specimens, APOEε4 carriers and non-carriers did not differ significantly in either the number of total, resting (GFAP-negative), or reactive (GFAP-positive) astrocytes (Serrano-Pozo et al., 2013a), the progression of astrocyte reaction along the clinical course of the disease (Serrano-Pozo et al., 2011b), or the proximity of the association between reactive astrocytes and plaques (Serrano-Pozo et al., 2016), but see also (Mathur et al., 2015). However, it is still possible that reactive astrocytes behave differently in different APOE genetic backgrounds. For example, APOEε4 KI mice show increased numbers of reactive astrocytes and activated microglia, increased levels of pro-inflammatory cytokines, and decreased levels of synaptic markers after administration of LPS, compared with APOEε3 and APOEε2 KI mice, suggesting that APOEε4 astrocytes may be more susceptible to react to pro-inflammatory stimuli (Zhu et al., 2012). Indeed, E4FAD and P301S/E4 mice (corresponding to the 5xFAD mouse model of brain β-amiloidosis and the P301S tau transgenic mouse of tauopathy under a human APOEε4 KI background, respectively) have a hyperactivated microglia and increased levels of proinflammatory cytokines as compared to the APOEε3 and APOEε2 KI double transgenic mice (Rodriguez et al., 2014; Shi et al., 2017). On the other hand, human inducible pluripotent stem cell (hiPSC)-derived astrocytes from APOEε4/ε4 human subjects exhibit a loss of neurotrophic function with respect to neurons and synapses in vitro (Zhao et al., 2017). In another study, APOEε4 astrocytes showed decreased potential to phagocytose synapses, whereas this ability was enhanced in APOEε2 astrocytes compared to APOEε3. However, APOEε4 KI mice had an increased proportion of C1q-tagged synapses (and APOEε2 KI mice a decreased proportion) compared to APOEε3 KI mice, which would make them vulnerable to elimination by microglia (Chung et al., 2016).
Cross-Talk Between Astrocytes and Microglia
Although astrocytes and microglia share the consideration of glial cells, microglial cells are thought to derive from mesenchymal cells of the yolk sac. Microglia are the innate immune cells of the brain, likewise macrophages in other organs. As such, they are highly dynamic cells that continuously survey the brain tissue in normal conditions, migrate to areas of injury (Nimmerjahn et al., 2005), and phagocytose virus, bacteria and neuronal debris (Fuhrmann et al., 2010). There are multiple lines of evidence supporting the existence of a cross-talk between astrocytes and microglia in AD. First, like astrocytes, activated microglia decorate dense-core amyloid (senile) plaques (Itagaki et al., 1989; Serrano-Pozo et al., 2013b, 2016), where they establish an intimate relationship with astrocytes (Bouvier et al., 2016). Second, there is a strong correlation between the number of activated microglial cells and that of reactive astrocytes, and both parallel disease progression (Serrano-Pozo et al., 2011b). Third, the attenuation of astrocyte reaction around plaques observed in APP/PS1: GFAP/Vimentin double knock out mice is associated with increased numbers of activated microglia around plaques (Kraft et al., 2013). Fourth, the “paracrine” secretory function of microglial cells can change astrocyte phenotype and vice versa. Inflammatory cytokines secreted by microglia (i.e., TNFα, IL-1, C1q) can transform neuroprotective resting astrocytes (A2) into neurotoxic (A1). Conversely, astrocytes can release C3 complement fraction via NFκB in response to oligomeric Aβ and C3 can in turn activate microglia through its C3R receptor (Lian et al., 2016). Fifth, microglial processes can be present in excitatory synapses together with astrocyte processes and pre- and post-synaptic neuronal elements, forming a “quadripartite” synapse. Activated microglia can cause direct synaptotoxicity through the secretion of C1q (Hong et al., 2016). Sixth, astrocytes appear to influence the degree of microglial reactivity in mouse models of brain β-amyloidosis (Rodriguez et al., 2014) and tauopathy (Shi et al., 2017) in an apoE isoform-dependent fashion (E4 > E3 > E2). Thus, it is likely that reactive astrocytes and activated microglia do not “play solo”, but act in a concerted fashion.
Biomarkers of Astrocyte Reaction
Since astrocyte reaction appears to be linked to the neurodegenerative process in AD, it follows that imaging and CSF biomarkers of astrocyte reaction could be useful to improve the accuracy of the clinical diagnosis of AD dementia and monitor and predict the progression of the disease.
With regards to CSF biomarkers, initially a candidate-based approach led researchers to measure the levels of classic astrocyte markers in CSF such as GFAP, S100β, and glutamine synthetase. Compared to healthy controls, GFAP levels have been found to be higher in the CSF of AD, dementia with Lewy bodies (DLB), frontotemporal lobar degeneration (FTLD) and Creutzdfeldt-Jakob disease (CJD) patients, whereas S100β was found to be elevated in CJD, but not in AD. Reports on elevated CSF levels of glutamine synthetase in AD are conflicting (Gunnersen and Haley, 1992; Tumani et al., 1999; Timmer et al., 2015). However, it is a “non-a priori hypothesis” proteomic approach that has rendered the most promising CSF biomarker of astrocyte reaction to date: chitinase 3 protein-like 1 (also called YKL-40). This CSF biomarker has been shown to predict progression from normal cognition to MCI and from MCI to AD dementia (Craig-Schapiro et al., 2010). Subsequent studies confirmed a correlation between CSF YKL-40 levels and biomarkers of neurodegeneration, such as CSF total and phospho-tau levels and cortical thickness, at the earliest stages of AD (Antonell et al., 2014; Alcolea et al., 2015). However, elevated CSF YKL-40 levels are not specific of AD; the highest levels appear to occur in sporadic CJD patients, followed by AD and tauopathies (PSP, CBD and Pick’s disease), whereas patients with vascular dementia and with Parkinson disease dementia or DLB have been reported to have normal levels (Llorens et al., 2017). Importantly, YKL-40 is expressed by GFAP-immunoreactive astrocytes near plaques and CAA-laden vessels (Craig-Schapiro et al., 2010; Llorens et al., 2017), but also in white matter fibrous astrocytes and in random cortical protoplasmic and perivascular astrocytes (Llorens et al., 2017). A recent postmortem quantitative neuropathological study in AD and other tauopathies has described that YKL-40 is only expressed by a subset of GFAP-immunoreactive astrocytes and that there is a positive correlation between YKL-40 and tau immunoreactivities, in agreement with the correlations found in CSF (Querol-Vilaseca et al., 2017).
The development of PET radiotracers specific for astrocyte reaction has proven to be more challenging than for activated microglia. While PET radioligands of the translocator protein 18 kDa (TSPO), also called peripheral benzodiazepine receptor (PBR), have been used to depict microglial activation in vivo for almost two decades (Cagnin et al., 2001), no such established radiotracer exists for reactive astrocytes. However, it should be noted that TSPO is not only up-regulated in activated microglial cells but also in reactive astrocytes, and that it is also expressed by endothelial and vascular smooth muscle cells (Cosenza-Nashat et al., 2009). Recently, [11C]-deuterium-L-deprenyl ([11C]-DED), a modified inhibitor of the monoamino oxidase B (MAO-B) enzyme, has been proposed as a PET imaging biomarker of astrocyte reaction (Carter et al., 2012; Schöll et al., 2015; Rodriguez-Vieitez et al., 2016). Indeed, MAO-B was shown to be up-regulated in GFAP-immunoreactive astrocytes many years ago (Nakamura et al., 1990; Jossan et al., 1991). However, the specificity of this radiotracer requires further validation because MAO-B is also expressed by neurons and the contribution of each cell type to the radiotracer uptake remains to be clarified.
The results of astrocyte-specific transcriptomic studies in the human AD brain and AD mouse models will inform the development of new CSF and PET imaging biomarkers specific of astrocyte reaction (Zamanian et al., 2012; Orre et al., 2014; Liddelow et al., 2017).
Conclusion
In summary, recent evidence directly implicates astrocytes in the pathophysiology of AD and supports the idea that astrocyte reaction against amyloid plaques and NFTs leads to a loss of their neurotrophic potential and a gain of neurotoxic properties. Large gaps of knowledge remain regarding the molecular pathways involved in this reaction and its functional consequences for both astrocytes themselves, and neurons and their synapses. Future research should also address the implication of each AD pathological hallmark in astrocyte reaction, and further characterize the cross-talk between astrocytes and microglia and the influence of the APOE genotype. Technological advances will help answer some of these questions, including some new in vitro tools (human induced pluripotent stem cells (hiPSCs)-derived astrocytes, 3D-cultures, brain organoids, protocols of isolation and purification of astrocytes from the adult mouse and human brains), recently developed unbiased and high-throughput molecular biology techniques (i.e., single-cell RNAseq), new mouse models and gene delivery approaches for astrocyte-specific manipulations, and more specific CSF biomarkers and PET radiotracers for in vivo studies of astrocyte reaction in the human brain.
Author Contributions
AS-P and BGP-N designed this review outline, performed literature review, prepared the figures and wrote the manuscript.
Funding
This work was supported by Alzheimer’s Research UK (ARUK-RF2014-2) to BGP-N, National Institute of Neurological Disorders and Stroke (NINDS R25NS065743) to AS-P and Alzheimer’s Association (AACF-17-524184) to AS-P.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdul, H. M., Sama, M. A., Furman, J. L., Mathis, D. M., Beckett, T. L., Weidner, A. M., et al. (2009). Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J. Neurosci. 29, 12957–12969. doi: 10.1523/JNEUROSCI.1064-09.2009
Akama, K. T., Albanese, C., Pestell, R. G., and Van Eldik, L. J. (1998). Amyloid β-peptide stimulates nitric oxide production in astrocytes through an NFκB-dependent mechanism. Proc. Natl. Acad. Sci. U S A 95, 5795–5800. doi: 10.1073/pnas.95.10.5795
Akiyama, H., Mori, H., Saido, T., Kondo, H., Ikeda, K., and McGeer, P. L. (1999). Occurrence of the diffuse amyloid β-protein (Aβ) deposits with numerous Aβ-containing glial cells in the cerebral cortex of patients with Alzheimer’s disease. Glia 25, 324–331. doi: 10.1002/(sici)1098-1136(19990215)25:4<324::aid-glia2>3.0.co;2-5
Akiyama, H., Schwab, C., Kondo, H., Mori, H., Kametani, F., Ikeda, K., et al. (1996). Granules in glial cells of patients with Alzheimer’s disease are immunopositive for C-terminal sequences of β-amyloid protein. Neurosci. Lett. 206, 169–172. doi: 10.1016/s0304-3940(96)12474-5
Alcolea, D., Vilaplana, E., Pegueroles, J., Montal, V., Sánchez-Juan, P., González-Suárez, A., et al. (2015). Relationship between cortical thickness and cerebrospinal fluid YKL-40 in predementia stages of Alzheimer’s disease. Neurobiol. Aging 36, 2018–2023. doi: 10.1016/j.neurobiolaging.2015.03.001
Allen, N. J., Bennett, M. L., Foo, L. C., Wang, G. X., Chakraborty, C., Smith, S. J., et al. (2012). Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486, 410–414. doi: 10.1038/nature11059
Antonell, A., Mansilla, A., Rami, L., Lladó, A., Iranzo, A., Olives, J., et al. (2014). Cerebrospinal fluid level of YKL-40 protein in preclinical and prodromal Alzheimer’s disease. J. Alzheimers Dis. 42, 901–908. doi: 10.3233/JAD-140624
Arbel-Ornath, M., Hudry, E., Eikermann-Haerter, K., Hou, S., Gregory, J. L., Zhao, L., et al. (2013). Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer’s disease mouse models. Acta Neuropathol. 126, 353–364. doi: 10.1007/s00401-013-1145-2
Arima, K., Izumiyama, Y., Nakamura, M., Nakayama, H., Kimura, M., Ando, S., et al. (1998). Argyrophilic tau-positive twisted and non-twisted tubules in astrocytic processes in brains of Alzheimer-type dementia: an electron microscopical study. Acta Neuropathol. 95, 28–39. doi: 10.1007/s004010050762
Arvanitakis, Z., Leurgans, S. E., Wang, Z., Wilson, R. S., Bennett, D. A., and Schneider, J. A. (2011). Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann. Neurol. 69, 320–327. doi: 10.1002/ana.22112
Asgari, M., de Zélicourt, D., and Kurtcuoglu, V. (2015). How astrocyte networks may contribute to cerebral metabolite clearance. Sci. Rep. 5:15024. doi: 10.1038/srep15024
Asgari, M., de Zélicourt, D., and Kurtcuoglu, V. (2016). Glymphatic solute transport does not require bulk flow. Sci. Rep. 6:38635. doi: 10.1038/srep38635
Baig, S., Kehoe, P. G., and Love, S. (2008). MMP-2, -3 and -9 levels and activity are not related to Aβ load in the frontal cortex in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 34, 205–215. doi: 10.1111/j.1365-2990.2007.00897.x
Bales, K. R., Du, Y., Dodel, R. C., Yan, G. M., Hamilton-Byrd, E., and Paul, S. M. (1998). The NF-κB/Rel family of proteins mediates Aβ-induced neurotoxicity and glial activation. Mol. Brain Res. 57, 63–72. doi: 10.1016/s0169-328x(98)00066-7
Beach, T. G., and McGeer, E. G. (1988). Lamina-specific arrangement of astrocytic gliosis and senile plaques in Alzheimer’s disease visual cortex. Brain Res. 463, 357–361. doi: 10.1016/0006-8993(88)90410-6
Beach, T. G., Walker, R., and McGeer, E. G. (1989). Patterns of gliosis in Alzheimer’s disease and aging cerebrum. Glia 2, 420–436. doi: 10.1002/glia.440020605
Ben Haim, L., Ceyzériat, K., Carrillo-de Sauvage, M. A., Aubry, F., Auregan, G., Guillermier, M., et al. (2015). The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J. Neurosci. 35, 2817–2829. doi: 10.1523/JNEUROSCI.3516-14.2015
Bhat, R., Crowe, E. P., Bitto, A., Moh, M., Katsetos, C. D., Garcia, F. U., et al. (2012). Astrocyte senescence as a component of Alzheimer’s disease. PLoS One 7:e45069. doi: 10.1371/journal.pone.0045069
Boisvert, M. M., Erikson, G. A., Shokhirev, M. N., and Allen, N. J. (2018). The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. 22, 269–285. doi: 10.1016/j.celrep.2017.12.039
Bondolfi, L., Calhoun, M., Ermini, F., Kuhn, H. G., Wiederhold, K.-H., Walker, L., et al. (2002). Amyloid-associated neuron loss and gliogenesis in the neocortex of amyloid precursor protein transgenic mice. J. Neurosci. 22, 515–522. doi: 10.1523/JNEUROSCI.22-02-00515.2002
Bouvier, D. S., Jones, E. V., Quesseveur, G., Davoli, M. A., A Ferreira, T., Quirion, R., et al. (2016). High resolution dissection of reactive glial nets in Alzheimer’s disease. Sci. Rep. 6:24544. doi: 10.1038/srep24544
Bowman, G. L., Kaye, J. A., Moore, M., Waichunas, D., Carlson, N. E., and Quinn, J. F. (2007). Blood-brain barrier impairment in Alzheimer disease: stability and functional significance. Neurology 68, 1809–1814. doi: 10.1212/01.WNL.0000262031.18018.1a
Braak, H., Sastre, M., and Del Tredici, K. (2007). Development of α-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 114, 231–241. doi: 10.1007/s00401-007-0244-3
Brawek, B., Chesters, R., Klement, D., Müller, J., Lerdkrai, C., Hermes, M., et al. (2018). A bell-shaped dependence between amyloidosis and GABA accumulation in astrocytes in a mouse model of Alzheimer’s disease. Neurobiol. Aging 61, 187–197. doi: 10.1016/j.neurobiolaging.2017.09.028
Buldyrev, S. V., Cruz, L., Gomez-Isla, T., Gomez-Tortosa, E., Havlin, S., Le, R., et al. (2000). Description of microcolumnar ensembles in association cortex and their disruption in Alzheimer and Lewy body dementias. Proc. Natl. Acad. Sci. U S A 97, 5039–5043. doi: 10.1073/pnas.060009897
Busche, M. A., Chen, X., Henning, H. A., Reichwald, J., Staufenbiel, M., Sakmann, B., et al. (2012). Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 109, 8740–8745. doi: 10.1073/pnas.1206171109
Busche, M. A., Eichhoff, G., Adelsberger, H., Abramowski, D., Wiederhold, K.-H., Haass, C., et al. (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321, 1686–1689. doi: 10.1126/science.1162844
Bushong, E. A., Martone, M. E., Jones, Y. Z., and Ellisman, M. H. (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 22, 183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002
Cagnin, A., Brooks, D. J., Kennedy, A. M., Gunn, R. N., Myers, R., Turkheimer, F. E., et al. (2001). In vivo measurement of activated microglia in dementia. Lancet 358, 461–467. doi: 10.1016/s0140-6736(01)05625-2
Carter, S. F., Schöll, M., Almkvist, O., Wall, A., Engler, H., Långström, B., et al. (2012). Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J. Nucl. Med. 53, 37–46. doi: 10.2967/jnumed.110.087031
Castellano, J. M., Kim, J., Stewart, F. R., Jiang, H., DeMattos, R. B., Patterson, B. W., et al. (2011). Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 3:89ra57. doi: 10.1126/scitranslmed.3002156
Chai, H., Diaz-Castro, B., Shigetomi, E., Monte, E., Octeau, J. C., Yu, X., et al. (2017). Neural circuit-specialized astrocytes: transcriptomic, proteomic, morphological, and functional evidence. Neuron 95, 531.e9–549.e9. doi: 10.1016/j.neuron.2017.06.029
Chen, A., Akinyemi, R. O., Hase, Y., Firbank, M. J., Ndung’u, M. N., Foster, V., et al. (2016). Frontal white matter hyperintensities, clasmatodendrosis and gliovascular abnormalities in ageing and post-stroke dementia. Brain 139, 242–258. doi: 10.1093/brain/awv328
Chouinard, M. L., Gaitan, D., and Wood, P. L. (1993). Presence of the N-methyl-D-aspartate-associated glycine receptor agonist, D-serine, in human temporal cortex: comparison of normal, Parkinson, and Alzheimer tissues. J. Neurochem. 61, 1561–1564. doi: 10.1111/j.1471-4159.1993.tb13657.x
Chung, W.-S., Clarke, L. E., Wang, G. X., Stafford, B. K., Sher, A., Chakraborty, C., et al. (2013). Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400. doi: 10.1038/nature12776
Chung, W.-S., Verghese, P. B., Chakraborty, C., Joung, J., Hyman, B. T., Ulrich, J. D., et al. (2016). Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc. Natl. Acad. Sci. U S A 113, 10186–10191. doi: 10.1073/pnas.1609896113
Colombo, J. A., Fuchs, E., Härtig, W., Marotte, L. R., and Puissant, V. (2000). “Rodent-like” and “primate-like” types of astroglial architecture in the adult cerebral cortex of mammals: a comparative study. Anat. Embryol. 201, 111–120. doi: 10.1007/pl00008231
Colombo, J. A., Quinn, B., and Puissant, V. (2002). Disruption of astroglial interlaminar processes in Alzheimer’s disease. Brain Res. Bull. 58, 235–242. doi: 10.1016/s0361-9230(02)00785-2
Colombo, J. A., Reisin, H. D., Jones, M., and Bentham, C. (2005). Development of interlaminar astroglial processes in the cerebral cortex of control and Down’s syndrome human cases. Exp. Neurol. 193, 207–217. doi: 10.1016/j.expneurol.2004.11.024
Corder, E. H., Saunders, A. M., Risch, N. J., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., et al. (1994). Protective effect of apolipoprotein E type 2 allele for late onset alzheimer disease. Nat. Genet. 7, 180–184. doi: 10.1038/ng0694-180
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Cosenza-Nashat, M., Zhao, M.-L., Suh, H.-S., Morgan, J., Natividad, R., Morgello, S., et al. (2009). Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol. 35, 306–328. doi: 10.1111/j.1365-2990.2008.01006.x
Crabtree, G. R., and Schreiber, S. L. (2009). SnapShot: Ca2+-calcineurin-NFAT signaling. Cell 138:210, 210.e1. doi: 10.1016/j.cell.2009.06.026
Craig-Schapiro, R., Perrin, R. J., Roe, C. M., Xiong, C., Carter, D., Cairns, N. J., et al. (2010). YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol. Psychiatry 68, 903–912. doi: 10.1016/j.biopsych.2010.08.025
Da Silva, J., Pierrat, B., Mary, J. L., and Lesslauer, W. (1997). Blockade of p38 mitogen-activated protein kinase pathway inhibits inducible nitric-oxide synthase expression in mouse astrocytes. J. Biol. Chem. 272, 28373–28380. doi: 10.1074/jbc.272.45.28373
Daschil, N., and Humpel, C. (2016). Green-fluorescent protein+ astrocytes attach to β-amyloid plaques in an Alzheimer mouse model and are sensitive for clasmatodendrosis. Front. Aging Neurosci. 8:75. doi: 10.3389/fnagi.2016.00075
Delekate, A., Fuchtemeier, M., Schumacher, T., Ulbrich, C., Foddis, M., and Petzold, G. C. (2014). Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 5:5422. doi: 10.1038/ncomms6422
de Calignon, A., Polydoro, M., Suárez-Calvet, M., William, C., Adamowicz, D. H., Kopeikina, K. J., et al. (2012). Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73, 685–697. doi: 10.1016/j.neuron.2011.11.033
del Río-Hortega, P. (1921). Estudios sobre la neuroglía: la glía de escasas radiaciones (oligodendroglía). Trab. Lab Histol. Patol. 1–15, 1–43.
del Río-Hortega, P., and Penfield, W. (1892). Cerebral cicatrix: the reaction of neuroglia and microglia to brain wounds. Bull. Johns Hopkins Hosp. 41, 278–303.
Ehmsen, J. T., Ma, T. M., Sason, H., Rosenberg, D., Ogo, T., Furuya, S., et al. (2013). D-serine in glia and neurons derives from 3-phosphoglycerate dehydrogenase. J. Neurosci. 33, 12464–12469. doi: 10.1523/JNEUROSCI.4914-12.2013
Farfel, J. M., Yu, L., De Jager, P. L., Schneider, J. A., and Bennett, D. A. (2016). Association of APOE with tau-tangle pathology with and without β-amyloid. Neurobiol. Aging 37, 19–25. doi: 10.1016/j.neurobiolaging.2015.09.011
Ferrer, I., Martí, E., López, E., and Tortosa, A. (1998). NF-kB immunoreactivity is observed in association with β A4 diffuse plaques in patients with Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 24, 271–277. doi: 10.1046/j.1365-2990.1998.00116.x
Fiacco, T. A., and McCarthy, K. D. (2018). Multiple lines of evidence indicate that gliotransmission does not occur under physiological conditions. J. Neurosci. 38, 3–13. doi: 10.1523/JNEUROSCI.0016-17.2017
Fuhrmann, M., Bittner, T., Jung, C. K. E., Burgold, S., Page, R. M., Mitteregger, G., et al. (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 13, 411–413. doi: 10.1038/nn.2511
Fujita, T., Chen, M. J., Li, B., Smith, N. A., Peng, W., Sun, W., et al. (2014). Neuronal transgene expression in dominant-negative SNARE mice. J. Neurosci. 34, 16594–16604. doi: 10.1523/JNEUROSCI.2585-14.2014
Funato, H., Yoshimura, M., Yamazaki, T., Saido, T. C., Ito, Y., Yokofujita, J., et al. (1998). Astrocytes containing amyloid β-protein (Aβ)-positive granules are associated with Aβ40-positive diffuse plaques in the aged human brain. Am. J. Pathol. 152, 983–992.
Furman, J. L., Sama, D. M., Gant, J. C., Beckett, T. L., Murphy, M. P., Bachstetter, A. D., et al. (2012). Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J. Neurosci. 32, 16129–16140. doi: 10.1523/JNEUROSCI.2323-12.2012
Galea, E., Morrison, W., Hudry, E., Arbel-Ornath, M., Bacskai, B. J., Gómez-Isla, T., et al. (2015). Topological analyses in APP/PS1 mice reveal that astrocytes do not migrate to amyloid-β plaques. Proc. Natl. Acad. Sci. U S A 112, 15556–15561. doi: 10.1073/pnas.1516779112
Gomez-Arboledas, A., Davila, J. C., Sanchez-Mejias, E., Navarro, V., Nuñez-Diaz, C., Sanchez-Varo, R., et al. (2018). Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia 66, 637–653. doi: 10.1002/glia.23270
Gunnersen, D., and Haley, B. (1992). Detection of glutamine synthetase in the cerebrospinal fluid of Alzheimer diseased patients: a potential diagnostic biochemical marker. Proc. Natl. Acad. Sci. U S A 89, 11949–11953. doi: 10.1073/pnas.89.24.11949
Hagemann, T. L., Connor, J. X., and Messing, A. (2006). Alexander disease-associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and a white matter stress response. J. Neurosci. 26, 11162–11173. doi: 10.1523/JNEUROSCI.3260-06.2006
Halassa, M. M., Fellin, T., Takano, H., Dong, J.-H., and Haydon, P. G. (2007). Synaptic islands defined by the territory of a single astrocyte. J. Neurosci. 27, 6473–6477. doi: 10.1523/JNEUROSCI.1419-07.2007
Hall, C. N., Reynell, C., Gesslein, B., Hamilton, N. B., Mishra, A., Sutherland, B. A., et al. (2014). Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508, 55–60. doi: 10.1038/nature13165
Hartlage-Rübsamen, M., Zeitschel, U., Apelt, J., Gärtner, U., Franke, H., Stahl, T., et al. (2003). Astrocytic expression of the Alzheimer’s disease β-secretase (BACE1) is stimulus-dependent. Glia 41, 169–179. doi: 10.1002/glia.10178
Hashimoto, T., Serrano-Pozo, A., Hori, Y., Adams, K. W., Takeda, S., Banerji, A. O., et al. (2012). Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J. Neurosci. 32, 15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012
Hawkes, C. A., Härtig, W., Kacza, J., Schliebs, R., Weller, R. O., Nicoll, J. A., et al. (2011). Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 121, 431–443. doi: 10.1007/s00401-011-0801-7
Hawkes, C. A., Jayakody, N., Johnston, D. A., Bechmann, I., and Carare, R. O. (2014). Failure of perivascular drainage of β-amyloid in cerebral amyloid angiopathy. Brain Pathol. 24, 396–403. doi: 10.1111/bpa.12159
Hayden, M. S., West, A. P., and Ghosh, S. (2006). SnapShot: NF-κB signaling pathways. Cell 127, 1286–1287. doi: 10.1016/j.cell.2006.12.005
He, X.-F., Liu, D.-X., Zhang, Q., Liang, F.-Y., Dai, G.-Y., Zeng, J.-S., et al. (2017). Voluntary exercise promotes glymphatic clearance of amyloid β and reduces the activation of astrocytes and microglia in aged mice. Front. Mol. Neurosci. 10:144. doi: 10.3389/fnmol.2017.00144
Hefendehl, J. K., LeDue, J., Ko, R. W. Y., Mahler, J., Murphy, T. H., and MacVicar, B. A. (2016). Mapping synaptic glutamate transporter dysfunction in vivo to regions surrounding Aβ plaques by iGluSnFR two-photon imaging. Nat. Commun. 7:13441. doi: 10.1038/ncomms13441
Henneberger, C., Papouin, T., Oliet, S. H. R., and Rusakov, D. A. (2010). Long-term potentiation depends on release of D-serine from astrocytes. Nature 463, 232–236. doi: 10.1038/nature08673
Hensley, K., Carney, J. M., Mattson, M. P., Aksenova, M., Harris, M., Wu, J. F., et al. (1994). A model for β-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc. Natl. Acad. Sci. U S A 91, 3270–3274. doi: 10.1073/pnas.91.8.3270
Hensley, K., Hall, N., Subramaniam, R., Cole, P., Harris, M., Aksenov, M., et al. (1995). Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J. Neurochem. 65, 2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x
Holter, K. E., Kehlet, B., Devor, A., Sejnowski, T. J., Dale, A. M., Omholt, S. W., et al. (2017). Interstitial solute transport in 3D reconstructed neuropil occurs by diffusion rather than bulk flow. Proc. Natl. Acad. Sci. U S A 114, 9894–9899. doi: 10.1073/pnas.1706942114
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hoshi, A., Yamamoto, T., Shimizu, K., Ugawa, Y., Nishizawa, M., Takahashi, H., et al. (2012). Characteristics of aquaporin expression surrounding senile plaques and cerebral amyloid angiopathy in Alzheimer disease. J. Neuropathol. Exp. Neurol. 71, 750–759. doi: 10.1097/NEN.0b013e3182632566
Hsiao, V. C., Tian, R., Long, H., Der Perng, M., Brenner, M., Quinlan, R. A., et al. (2005). Alexander-disease mutation of GFAP causes filament disorganization and decreased solubility of GFAP. J. Cell Sci. 118, 2057–2065. doi: 10.1242/jcs.02339
Hudry, E., Wu, H.-Y., Arbel-Ornath, M., Hashimoto, T., Matsouaka, R., Fan, Z., et al. (2012). Inhibition of the NFAT pathway alleviates amyloid β neurotoxicity in a mouse model of Alzheimer’s disease. J. Neurosci. 32, 3176–3192. doi: 10.1523/JNEUROSCI.6439-11.2012
Hyman, B. T., West, H. L., Rebeck, G. W., Buldyrev, S. V., Mantegna, R. N., Ukleja, M., et al. (1995). Quantitative analysis of senile plaques in Alzheimer disease: observation of log-normal size distribution and molecular epidemiology of differences associated with apolipoprotein E genotype and trisomy 21 (Down syndrome). Proc. Natl. Acad. Sci. U S A 92, 3586–3590. doi: 10.1073/pnas.92.8.3586
Ikeda, K., Haga, C., Akiyama, H., Kase, K., and Iritani, S. (1992a). Coexistence of paired helical filaments and glial filaments in astrocytic processes within ghost tangles. Neurosci. Lett. 148, 126–128. doi: 10.1016/0304-3940(92)90820-w
Ikeda, K., Haga, C., Oyanagi, S., Iritani, S., and Kosaka, K. (1992b). Ultrastructural and immunohistochemical study of degenerate neurite-bearing ghost tangles. J. Neurol. 239, 191–194. doi: 10.1007/bf00839138
Iliff, J. J., Wang, M., Liao, Y., Plogg, B. A., Peng, W., Gundersen, G. A., et al. (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 4:147ra111. doi: 10.1126/scitranslmed.3003748
Iliff, J. J., Wang, M., Zeppenfeld, D. M., Venkataraman, A., Plog, B. A., Liao, Y., et al. (2013). Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J. Neurosci. 33, 18190–18199. doi: 10.1523/JNEUROSCI.1592-13.2013
Iram, T., Trudler, D., Kain, D., Kanner, S., Galron, R., Vassar, R., et al. (2016). Astrocytes from old Alzheimer’s disease mice are impaired in Aβ uptake and in neuroprotection. Neurobiol. Dis. 96, 84–94. doi: 10.1016/j.nbd.2016.08.001
Irwin, D. J., Cohen, T. J., Grossman, M., Arnold, S. E., Xie, S. X., Lee, V. M.-Y., et al. (2012). Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain J. Neurol. 135, 807–818. doi: 10.1093/brain/aws013
Ishiki, A., Kamada, M., Kawamura, Y., Terao, C., Shimoda, F., Tomita, N., et al. (2016). Glial fibrillar acidic protein in the cerebrospinal fluid of Alzheimer’s disease, dementia with Lewy bodies, and frontotemporal lobar degeneration. J. Neurochem. 136, 258–261. doi: 10.1111/jnc.13399
Itagaki, S., McGeer, P. L., Akiyama, H., Zhu, S., and Selkoe, D. (1989). Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol. 24, 173–182. doi: 10.1016/0165-5728(89)90115-x
Jesse, S., Steinacker, P., Cepek, L., von Arnim, C. A. F., Tumani, H., Lehnert, S., et al. (2009). Glial fibrillary acidic protein and protein S-100B: different concentration pattern of glial proteins in cerebrospinal fluid of patients with Alzheimer’s disease and Creutzfeldt-Jakob disease. J. Alzheimers Dis. 17, 541–551. doi: 10.3233/JAD-2009-1075
Jin, B.-J., Smith, A. J., and Verkman, A. S. (2016). Spatial model of convective solute transport in brain extracellular space does not support a “glymphatic” mechanism. J. Gen. Physiol. 148, 489–501. doi: 10.1085/jgp.201611684
Jo, S., Yarishkin, O., Hwang, Y. J., Chun, Y. E., Park, M., Woo, D. H., et al. (2014). GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 20, 886–896. doi: 10.1038/nm.3639
John Lin, C.-C., Yu, K., Hatcher, A., Huang, T.-W., Lee, H. K., Carlson, J., et al. (2017). Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 20, 396–405. doi: 10.1038/nn.4493
Jossan, S. S., Gillberg, P. G., Gottfries, C. G., Karlsson, I., and Oreland, L. (1991). Monoamine oxidase B in brains from patients with Alzheimer’s disease: a biochemical and autoradiographical study. Neuroscience 45, 1–12. doi: 10.1016/0306-4522(91)90098-9
Kaltschmidt, B., Uherek, M., Volk, B., Baeuerle, P. A., and Kaltschmidt, C. (1997). Transcription factor NF-κB is activated in primary neurons by amyloid β peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc. Natl. Acad. Sci. U S A 94, 2642–2647. doi: 10.1073/pnas.94.6.2642
Kamphuis, W., Kooijman, L., Orre, M., Stassen, O., Pekny, M., and Hol, E. M. (2015). GFAP and vimentin deficiency alters gene expression in astrocytes and microglia in wild-type mice and changes the transcriptional response of reactive glia in mouse model for Alzheimer’s disease. Glia 63, 1036–1056. doi: 10.1002/glia.22800
Kamphuis, W., Orre, M., Kooijman, L., Dahmen, M., and Hol, E. M. (2012). Differential cell proliferation in the cortex of the APPswePS1dE9 Alzheimer’s disease mouse model. Glia 60, 615–629. doi: 10.1002/glia.22295
Kang, J.-E., Lim, M. M., Bateman, R. J., Lee, J. J., Smyth, L. P., Cirrito, J. R., et al. (2009). Amyloid-β dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007. doi: 10.1126/science.1180962
Kimbrough, I. F., Robel, S., Roberson, E. D., and Sontheimer, H. (2015). Vascular amyloidosis impairs the gliovascular unit in a mouse model of Alzheimer’s disease. Brain 138, 3716–3733. doi: 10.1093/brain/awv327
Kitamura, Y., Shimohama, S., Ota, T., Matsuoka, Y., Nomura, Y., and Taniguchi, T. (1997). Alteration of transcription factors NF-κB and STAT1 in Alzheimer’s disease brains. Neurosci. Lett. 237, 17–20. doi: 10.1016/s0304-3940(97)00797-0
Kobayashi, E., Nakano, M., Kubota, K., Himuro, N., Mizoguchi, S., Chikenji, T., et al. (2018). Activated forms of astrocytes with higher GLT-1 expression are associated with cognitive normal subjects with Alzheimer pathology in human brain. Sci. Rep. 8:1712. doi: 10.1038/s41598-018-19442-7
Kobayashi, K., Hayashi, M., Nakano, H., Shimazaki, M., Sugimori, K., and Koshino, Y. (2004). Correlation between astrocyte apoptosis and Alzheimer changes in gray matter lesions in Alzheimer’s disease. J. Alzheimers Dis. 6, 623–632; discussion 673–681. doi: 10.3233/jad-2004-6606
Koistinaho, M., Lin, S., Wu, X., Esterman, M., Koger, D., Hanson, J., et al. (2004). Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat. Med. 10, 719–726. doi: 10.1038/nm1058
Kovacs, G. G., Ferrer, I., Grinberg, L. T., Alafuzoff, I., Attems, J., Budka, H., et al. (2016). Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol. 131, 87–102. doi: 10.1007/s00401-015-1509-x
Kraft, A. W., Hu, X., Yoon, H., Yan, P., Xiao, Q., Wang, Y., et al. (2013). Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 27, 187–198. doi: 10.1096/fj.12-208660
Kress, B. T., Iliff, J. J., Xia, M., Wang, M., Wei, H. S., Zeppenfeld, D., et al. (2014). Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 76, 845–861. doi: 10.1002/ana.24271
Kuchibhotla, K. V., Goldman, S. T., Lattarulo, C. R., Wu, H.-Y., Hyman, B. T., and Bacskai, B. J. (2008). Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59, 214–225. doi: 10.1016/j.neuron.2008.06.008
Kuchibhotla, K. V., Lattarulo, C. R., Hyman, B. T., and Bacskai, B. J. (2009). Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323, 1211–1215. doi: 10.1126/science.1169096
Kulijewicz-Nawrot, M., Syková, E., Chvátal, A., Verkhratsky, A., and Rodríguez, J. J. (2013). Astrocytes and glutamate homoeostasis in Alzheimer’s disease: a decrease in glutamine synthetase, but not in glutamate transporter-1, in the prefrontal cortex. ASN Neuro 5, 273–282. doi: 10.1042/an20130017
Lace, G., Ince, P. G., Brayne, C., Savva, G. M., Matthews, F. E., de Silva, R., et al. (2012). Mesial temporal astrocyte tau pathology in the MRC-CFAS ageing brain cohort. Dement. Geriatr. Cogn. Disord. 34, 15–24. doi: 10.1159/000341581
Lam, A. G., Koppal, T., Akama, K. T., Guo, L., Craft, J. M., Samy, B., et al. (2001). Mechanism of glial activation by S100β: involvement of the transcription factor NFκB. Neurobiol. Aging 22, 765–772. doi: 10.1016/s0197-4580(01)00233-0
Lasagna-Reeves, C. A., and Kayed, R. (2011). Astrocytes contain amyloid-β annular protofibrils in Alzheimer’s disease brains. FEBS Lett. 585, 3052–3057. doi: 10.1016/j.febslet.2011.08.027
Le Prince, G., Delaere, P., Fages, C., Lefrançois, T., Touret, M., Salanon, M., et al. (1995). Glutamine synthetase (GS) expression is reduced in senile dementia of the Alzheimer type. Neurochem. Res. 20, 859–862. doi: 10.1007/bf00969698
Leal, M. C., Dorfman, V. B., Gamba, A. F., Frangione, B., Wisniewski, T., Castaño, E. M., et al. (2006). Plaque-associated overexpression of insulin-degrading enzyme in the cerebral cortex of aged transgenic tg2576 mice with Alzheimer pathology. J. Neuropathol. Exp. Neurol. 65, 976–987. doi: 10.1097/01.jnen.0000235853.70092.ba
Lee, H., Xie, L., Yu, M., Kang, H., Feng, T., Deane, R., et al. (2015). The effect of body posture on brain glymphatic transport. J. Neurosci. 35, 11034–11044. doi: 10.1523/JNEUROSCI.1625-15.2015
Li, W. P., Chan, W. Y., Lai, H. W., and Yew, D. T. (1997). Terminal dUTP nick end labeling (TUNEL) positive cells in the different regions of the brain in normal aging and Alzheimer patients. J. Mol. Neurosci. 8, 75–82. doi: 10.1007/bf02736774
Li, S., Jin, M., Koeglsperger, T., Shepardson, N. E., Shankar, G. M., and Selkoe, D. J. (2011). Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 31, 6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011
Lian, H., Litvinchuk, A., Chiang, A. C.-A., Aithmitti, N., Jankowsky, J. L., and Zheng, H. (2016). Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J. Neurosci. 36, 577–589. doi: 10.1523/jneurosci.2117-15.2016
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Liu, A. K. L., Goldfinger, M. H., Questari, H. E., Pearce, R. K. B., and Gentleman, S. M. (2016). ARTAG in the basal forebrain: widening the constellation of astrocytic tau pathology. Acta Neuropathol. Commun. 4:59. doi: 10.1186/s40478-016-0330-7
Liu, C.-C., Hu, J., Zhao, N., Wang, J., Wang, N., Cirrito, J. R., et al. (2017). Astrocytic LRP1 mediates brain Aβ clearance and impacts amyloid deposition. J. Neurosci. 37, 4023–4031. doi: 10.1523/jneurosci.3442-16.2017
Llorens, F., Thüne, K., Tahir, W., Kanata, E., Diaz-Lucena, D., Xanthopoulos, K., et al. (2017). YKL-40 in the brain and cerebrospinal fluid of neurodegenerative dementias. Mol. Neurodegener. 12:83. doi: 10.1186/s13024-017-0226-4
López-González, I., Carmona, M., Blanco, R., Luna-Muñoz, J., Martínez-Mandonado, A., Mena, R., et al. (2013). Characterization of thorn-shaped astrocytes in white matter of temporal lobe in Alzheimer’s disease brains. Brain Pathol. 23, 144–153. doi: 10.1111/j.1750-3639.2012.00627.x
Luccarini, I., Grossi, C., Traini, C., Fiorentini, A., Ed Dami, T., and Casamenti, F. (2012). Aβ plaque-associated glial reaction as a determinant of apoptotic neuronal death and cortical gliogenesis: a study in APP mutant mice. Neurosci. Lett. 506, 94–99. doi: 10.1016/j.neulet.2011.10.056
Lue, L. F., Brachova, L., Civin, W. H., and Rogers, J. (1996). Inflammation, Aβ deposition, and neurofibrillary tangle formation as correlates of Alzheimer’s disease neurodegeneration. J. Neuropathol. Exp. Neurol. 55, 1083–1088. doi: 10.1097/00005072-199655100-00008
Luk, K. C., Kehm, V., Carroll, J., Zhang, B., O’Brien, P., Trojanowski, J. Q., et al. (2012). Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. doi: 10.1126/science.1227157
Mächler, P., Wyss, M. T., Elsayed, M., Stobart, J., Gutierrez, R., von Faber-Castell, A., et al. (2016). In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab. 23, 94–102. doi: 10.1016/j.cmet.2015.10.010
Marlatt, M. W., Bauer, J., Aronica, E., van Haastert, E. S., Hoozemans, J. J. M., Joels, M., et al. (2014). Proliferation in the Alzheimer hippocampus is due to microglia, not astroglia and occurs at sites of amyloid deposition. Neural Plast. 2014:693851. doi: 10.1155/2014/693851
Mathur, R., Ince, P. G., Minett, T., Garwood, C. J., Shaw, P. J., Matthews, F. E., et al. (2015). A reduced astrocyte response to β-amyloid plaques in the ageing brain associates with cognitive impairment. PLoS One 10:e0118463. doi: 10.1371/journal.pone.0118463
Merlini, M., Meyer, E. P., Ulmann-Schuler, A., and Nitsch, R. M. (2011). Vascular β-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAβ mice. Acta Neuropathol. 122, 293–311. doi: 10.1007/s00401-011-0834-y
Mertens, C., and Darnell, J. E. (2007). SnapShot: JAK-STAT signaling. Cell 131:612. doi: 10.1016/j.cell.2007.10.033
Meyer-Luehmann, M., Spires-Jones, T. L., Prada, C., Garcia-Alloza, M., de Calignon, A., Rozkalne, A., et al. (2008). Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nature 451, 720–724. doi: 10.1038/nature06616
Misawa, T., Arima, K., Mizusawa, H., and Satoh, J. (2008). Close association of water channel AQP1 with amyloid-β deposition in Alzheimer disease brains. Acta Neuropathol. 116, 247–260. doi: 10.1007/s00401-008-0387-x
Mishra, A., Reynolds, J. P., Chen, Y., Gourine, A. V., Rusakov, D. A., and Attwell, D. (2016). Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci. 19, 1619–1627. doi: 10.1038/nn.4428
Mitew, S., Kirkcaldie, M. T. K., Dickson, T. C., and Vickers, J. C. (2013). Altered synapses and gliotransmission in Alzheimer’s disease and AD model mice. Neurobiol. Aging 34, 2341–2351. doi: 10.1016/j.neurobiolaging.2013.04.010
Moftakhar, P., Lynch, M. D., Pomakian, J. L., and Vinters, H. V. (2010). Aquaporin expression in the brains of patients with or without cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 69, 1201–1209. doi: 10.1097/nen.0b013e3181fd252c
Nagata, Y., Borghi, M., Fisher, G. H., and D’Aniello, A. (1995). Free D-serine concentration in normal and Alzheimer human brain. Brain Res. Bull. 38, 181–183. doi: 10.1016/0361-9230(95)00087-u
Nagele, R. G., D’Andrea, M. R., Lee, H., Venkataraman, V., and Wang, H.-Y. (2003). Astrocytes accumulate Aβ42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 971, 197–209. doi: 10.1016/s0006-8993(03)02361-8
Nagy, Z., Esiri, M. M., and Smith, A. D. (1997). Expression of cell division markers in the hippocampus in Alzheimer’s disease and other neurodegenerative conditions. Acta Neuropathol. 93, 294–300. doi: 10.1007/s004010050617
Nakamura, S., Kawamata, T., Akiguchi, I., Kameyama, M., Nakamura, N., and Kimura, H. (1990). Expression of monoamine oxidase B activity in astrocytes of senile plaques. Acta Neuropathol. 80, 419–425. doi: 10.1007/bf00307697
Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study. (2001). Pathological correlates of late-onset dementia in a multicentre, community-based population in England Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet 357, 169–175. doi: 10.1016/s0140-6736(00)03589-3
Nielsen, H. M., Mulder, S. D., Beliën, J. A. M., Musters, R. J. P., Eikelenboom, P., and Veerhuis, R. (2010). Astrocytic Aβ1–42 uptake is determined by Aβ-aggregation state and the presence of amyloid-associated proteins. Glia 58, 1235–1246. doi: 10.1002/glia.21004
Nielsen, H. M., Veerhuis, R., Holmqvist, B., and Janciauskiene, S. (2009). Binding and uptake of A β1–42 by primary human astrocytes in vitro. Glia 57, 978–988. doi: 10.1002/glia.20822
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Norris, C. M., Kadish, I., Blalock, E. M., Chen, K.-C., Thibault, V., Porter, N. M., et al. (2005). Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. J. Neurosci. 25, 4649–4658. doi: 10.1523/jneurosci.0365-05.2005
Oberheim, N. A., Takano, T., Han, X., He, W., Lin, J. H. C., Wang, F., et al. (2009). Uniquely hominid features of adult human astrocytes. J. Neurosci. 29, 3276–3287. doi: 10.1523/jneurosci.4707-08.2009
Oberstein, T. J., Spitzer, P., Klafki, H.-W., Linning, P., Neff, F., Knölker, H.-J., et al. (2015). Astrocytes and microglia but not neurons preferentially generate N-terminally truncated Aβ peptides. Neurobiol. Dis. 73, 24–35. doi: 10.1016/j.nbd.2014.08.031
Oide, T., Kinoshita, T., and Arima, K. (2006). Regression stage senile plaques in the natural course of Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 32, 539–556. doi: 10.1111/j.1365-2990.2006.00767.x
Olabarria, M., Noristani, H. N., Verkhratsky, A., and Rodríguez, J. J. (2010). Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58, 831–838. doi: 10.1002/glia.20967
Olabarria, M., Noristani, H. N., Verkhratsky, A., and Rodríguez, J. J. (2011). Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: mechanism for deficient glutamatergic transmission? Mol. Neurodegener. 6:55. doi: 10.1186/1750-1326-6-55
Oliet, S. H., Piet, R., and Poulain, D. A. (2001). Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science 292, 923–926. doi: 10.1126/science.1059162
Orre, M., Kamphuis, W., Dooves, S., Kooijman, L., Chan, E. T., Kirk, C. J., et al. (2013). Reactive glia show increased immunoproteasome activity in Alzheimer’s disease. Brain 136, 1415–1431. doi: 10.1093/brain/awt083
Orre, M., Kamphuis, W., Osborn, L. M., Jansen, A. H. P., Kooijman, L., Bossers, K., et al. (2014). Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 35, 2746–2760. doi: 10.1016/j.neurobiolaging.2014.06.004
Ortinski, P. I., Dong, J., Mungenast, A., Yue, C., Takano, H., Watson, D. J., et al. (2010). Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat. Neurosci. 13, 584–591. doi: 10.1038/nn.2535
Palmer, J. C., Baig, S., Kehoe, P. G., and Love, S. (2009). Endothelin-converting enzyme-2 is increased in Alzheimer’s disease and up-regulated by Aβ. Am. J. Pathol. 175, 262–270. doi: 10.2353/ajpath.2009.081054
Pascual, O., Casper, K. B., Kubera, C., Zhang, J., Revilla-Sanchez, R., Sul, J.-Y., et al. (2005). Astrocytic purinergic signaling coordinates synaptic networks. Science 310, 113–116. doi: 10.1126/science.1116916
Pelvig, D. P., Pakkenberg, H., Regeur, L., Oster, S., and Pakkenberg, B. (2003). Neocortical glial cell numbers in Alzheimer’s disease. A stereological study. Dement. Geriatr. Cogn. Disord. 16, 212–219. doi: 10.1159/000072805
Pérez, E., Barrachina, M., Rodríguez, A., Torrejón-Escribano, B., Boada, M., Hernández, I., et al. (2007). Aquaporin expression in the cerebral cortex is increased at early stages of Alzheimer disease. Brain Res. 1128, 164–174. doi: 10.1016/j.brainres.2006.09.109
Perez-Nievas, B. G., Stein, T. D., Tai, H.-C., Dols-Icardo, O., Scotton, T. C., Barroeta-Espar, I., et al. (2013). Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 136, 2510–2526. doi: 10.1093/brain/awt171
Pfeifer, L. A., White, L. R., Ross, G. W., Petrovitch, H., and Launer, L. J. (2002). Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology 58, 1629–1634. doi: 10.1212/wnl.58.11.1629
Pihlaja, R., Koistinaho, J., Kauppinen, R., Sandholm, J., Tanila, H., and Koistinaho, M. (2011). Multiple cellular and molecular mechanisms are involved in human Aβ clearance by transplanted adult astrocytes. Glia 59, 1643–1657. doi: 10.1002/glia.21212
Pihlaja, R., Koistinaho, J., Malm, T., Sikkilä, H., Vainio, S., and Koistinaho, M. (2008). Transplanted astrocytes internalize deposited β-amyloid peptides in a transgenic mouse model of Alzheimer’s disease. Glia 56, 154–163. doi: 10.1002/glia.20599
Pike, C. J., Cummings, B. J., and Cotman, C. W. (1995). Early association of reactive astrocytes with senile plaques in Alzheimer’s disease. Exp. Neurol. 132, 172–179. doi: 10.1016/0014-4886(95)90022-5
Pleiss, M. M., Sompol, P., Kraner, S. D., Abdul, H. M., Furman, J. L., Guttmann, R. P., et al. (2016). Calcineurin proteolysis in astrocytes: implications for impaired synaptic function. Biochim. Biophys. Acta 1862, 1521–1532. doi: 10.1016/j.bbadis.2016.05.007
Probst, A., Ulrich, J., and Heitz, P. U. (1982). Senile dementia of Alzheimer type: astroglial reaction to extracellular neurofibrillary tangles in the hippocampus. An immunocytochemical and electron-microscopic study. Acta Neuropathol. 57, 75–79. doi: 10.1007/bf00688880
Querol-Vilaseca, M., Colom-Cadena, M., Pegueroles, J., San Martín-Paniello, C., Clarimon, J., Belbin, O., et al. (2017). YKL-40 (Chitinase 3-like I) is expressed in a subset of astrocytes in Alzheimer’s disease and other tauopathies. J. Neuroinflammation 14:118. doi: 10.1186/s12974-017-0893-7
Robinson, S. R. (2000). Neuronal expression of glutamine synthetase in Alzheimer’s disease indicates a profound impairment of metabolic interactions with astrocytes. Neurochem. Int. 36, 471–482. doi: 10.1016/s0197-0186(99)00150-3
Rodriguez, G. A., Tai, L. M., LaDu, M. J., and Rebeck, G. W. (2014). Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition. J. Neuroinflammation 11:111. doi: 10.1186/1742-2094-11-111
Rodriguez-Vieitez, E., Saint-Aubert, L., Carter, S. F., Almkvist, O., Farid, K., Schöll, M., et al. (2016). Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer’s disease. Brain 139, 922–936. doi: 10.1093/brain/awv404
Rozkalne, A., Hyman, B. T., and Spires-Jones, T. L. (2011). Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model mice. Neurobiol. Dis. 41, 650–654. doi: 10.1016/j.nbd.2010.11.014
Sahlas, D. J., Bilbao, J. M., Swartz, R. H., and Black, S. E. (2002). Clasmatodendrosis correlating with periventricular hyperintensity in mixed dementia. Ann. Neurol. 52, 378–381. doi: 10.1002/ana.10310
Savtchouk, I., and Volterra, A. (2018). Gliotransmission: beyond black-and-white. J. Neurosci. 38, 14–25. doi: 10.1523/jneurosci.0017-17.2017
Schöll, M., Carter, S. F., Westman, E., Rodriguez-Vieitez, E., Almkvist, O., Thordardottir, S., et al. (2015). Early astrocytosis in autosomal dominant Alzheimer’s disease measured in vivo by multi-tracer positron emission tomography. Sci. Rep. 5:16404. doi: 10.1038/srep16404
Scimemi, A., Meabon, J. S., Woltjer, R. L., Sullivan, J. M., Diamond, J. S., and Cook, D. G. (2013). Amyloid-β1–42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J. Neurosci. 33, 5312–5318. doi: 10.1523/JNEUROSCI.5274-12.2013
Sekar, S., McDonald, J., Cuyugan, L., Aldrich, J., Kurdoglu, A., Adkins, J., et al. (2015). Alzheimer’s disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. Neurobiol. Aging 36, 583–591. doi: 10.1016/j.neurobiolaging.2014.09.027
Serrano-Pozo, A., Betensky, R. A., Frosch, M. P., and Hyman, B. T. (2016). Plaque-associated local toxicity increases over the clinical course of Alzheimer disease. Am. J. Pathol. 186, 375–384. doi: 10.1016/j.ajpath.2015.10.010
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011a). Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Serrano-Pozo, A., Mielke, M. L., Gómez-Isla, T., Betensky, R. A., Growdon, J. H., Frosch, M. P., et al. (2011b). Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol. 179, 1373–1384. doi: 10.1016/j.ajpath.2011.05.047
Serrano-Pozo, A., Gómez-Isla, T., Growdon, J. H., Frosch, M. P., and Hyman, B. T. (2013a). A phenotypic change but not proliferation underlies glial responses in Alzheimer disease. Am. J. Pathol. 182, 2332–2344. doi: 10.1016/j.ajpath.2013.02.031
Serrano-Pozo, A., Muzikansky, A., Gómez-Isla, T., Growdon, J. H., Betensky, R. A., Frosch, M. P., et al. (2013b). Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J. Neuropathol. Exp. Neurol. 72, 462–471. doi: 10.1097/nen.0b013e3182933788
Serrano-Pozo, A., Qian, J., Monsell, S. E., Frosch, M. P., Betensky, R. A., and Hyman, B. T. (2013c). Examination of the clinicopathologic continuum of Alzheimer disease in the autopsy cohort of the National Alzheimer Coordinating Center. J. Neuropathol. Exp. Neurol. 72, 1182–1192. doi: 10.1097/nen.0000000000000016
Serrano-Pozo, A., Mielke, M. L., Muzitansky, A., Gómez-Isla, T., Growdon, J. H., Bacskai, B. J., et al. (2012). Stable size distribution of amyloid plaques over the course of Alzheimer disease. J. Neuropathol. Exp. Neurol. 71, 694–701. doi: 10.1097/nen.0b013e31825e77de
Serrano-Pozo, A., Qian, J., Monsell, S. E., Betensky, R. A., and Hyman, B. T. (2015). APOEε2 is associated with milder clinical and pathological Alzheimer disease. Ann. Neurol. 77, 917–929. doi: 10.1002/ana.24369
Shi, Y., Yamada, K., Liddelow, S. A., Smith, S. T., Zhao, L., Luo, W., et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. doi: 10.1038/nature24016
Simpson, J. E., Ince, P. G., Lace, G., Forster, G., Shaw, P. J., Matthews, F., et al. (2010). Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol. Aging 31, 578–590. doi: 10.1016/j.neurobiolaging.2008.05.015
Simpson, J. E., Ince, P. G., Shaw, P. J., Heath, P. R., Raman, R., Garwood, C. J., et al. (2011). Microarray analysis of the astrocyte transcriptome in the aging brain: relationship to Alzheimer’s pathology and APOE genotype. Neurobiol. Aging 32, 1795–1807. doi: 10.1016/j.neurobiolaging.2011.04.013
Sirko, S., Behrendt, G., Johansson, P. A., Tripathi, P., Costa, M., Bek, S., et al. (2013). Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog. Cell Stem Cell 12, 426–439. doi: 10.1016/j.stem.2013.04.007
Skillbäck, T., Delsing, L., Synnergren, J., Mattsson, N., Janelidze, S., Nägga, K., et al. (2017). CSF/serum albumin ratio in dementias: a cross-sectional study on 1861 patients. Neurobiol. Aging 59, 1–9. doi: 10.1016/j.neurobiolaging.2017.06.028
Skoog, I., Wallin, A., Fredman, P., Hesse, C., Aevarsson, O., Karlsson, I., et al. (1998). A population study on blood-brain barrier function in 85-year-olds: relation to Alzheimer’s disease and vascular dementia. Neurology 50, 966–971. doi: 10.1212/wnl.50.4.966
Sloan, S. A., and Barres, B. A. (2014). Looks can be deceiving: reconsidering the evidence for gliotransmission. Neuron 84, 1112–1115. doi: 10.1016/j.neuron.2014.12.003
Smale, G., Nichols, N. R., Brady, D. R., Finch, C. E., and Horton, W. E. (1995). Evidence for apoptotic cell death in Alzheimer’s disease. Exp. Neurol. 133, 225–230. doi: 10.1006/exnr.1995.1025
Smith, C. D., Carney, J. M., Starke-Reed, P. E., Oliver, C. N., Stadtman, E. R., Floyd, R. A., et al. (1991). Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc. Natl. Acad. Sci. U S A 88, 10540–10543. doi: 10.1073/pnas.88.23.10540
Smith, A. J., Yao, X., Dix, J. A., Jin, B.-J., and Verkman, A. S. (2017). Test of the “glymphatic” hypothesis demonstrates diffusive and aquaporin-4-independent solute transport in rodent brain parenchyma. Elife 6:e27679. doi: 10.7554/eLife.27679
Sompol, P., Furman, J. L., Pleiss, M. M., Kraner, S. D., Artiushin, I. A., Batten, S. R., et al. (2017). Calcineurin/NFAT signaling in activated astrocytes drives network hyperexcitability in Aβ-bearing mice. J. Neurosci. 37, 6132–6148. doi: 10.1523/jneurosci.0877-17.2017
Soreq, L., UK Brain Expression Consortium, North American Brain Expression Consortium, Rose, J., Soreq, E., Hardy, J., et al. (2017). Major shifts in glial regional identity are a transcriptional hallmark of human brain aging. Cell Rep. 18, 557–570. doi: 10.1016/j.celrep.2016.12.011
Sosunov, A. A., Wu, X., Tsankova, N. M., Guilfoyle, E., McKhann, G. M., and Goldman, J. E. (2014). Phenotypic heterogeneity and plasticity of isocortical and hippocampal astrocytes in the human brain. J. Neurosci. 34, 2285–2298. doi: 10.1523/jneurosci.4037-13.2014
Sugaya, K., Reeves, M., and McKinney, M. (1997). Topographic associations between DNA fragmentation and Alzheimer’s disease neuropathology in the hippocampus. Neurochem. Int. 31, 275–281. doi: 10.1016/s0197-0186(96)00158-1
Suzuki, A., Stern, S. A., Bozdagi, O., Huntley, G. W., Walker, R. H., Magistretti, P. J., et al. (2011). Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823. doi: 10.1016/j.cell.2011.02.018
Tang, G., Perng, M. D., Wilk, S., Quinlan, R., and Goldman, J. E. (2010). Oligomers of mutant glial fibrillary acidic protein (GFAP) Inhibit the proteasome system in alexander disease astrocytes and the small heat shock protein αB-crystallin reverses the inhibition. J. Biol. Chem. 285, 10527–10537. doi: 10.1074/jbc.m109.067975
Tang, G., Yue, Z., Talloczy, Z., Hagemann, T., Cho, W., Messing, A., et al. (2008). Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum. Mol. Genet. 17, 1540–1555. doi: 10.1093/hmg/ddn042
Terai, K., Matsuo, A., and McGeer, P. L. (1996). Enhancement of immunoreactivity for NF-κB in the hippocampal formation and cerebral cortex of Alzheimer’s disease. Brain Res. 735, 159–168. doi: 10.1016/s0006-8993(96)00310-1
Thal, D. R., Schultz, C., Dehghani, F., Yamaguchi, H., Braak, H., and Braak, E. (2000). Amyloid β-protein (Aβ)-containing astrocytes are located preferentially near N-terminal-truncated Aβ deposits in the human entorhinal cortex. Acta Neuropathol. 100, 608–617. doi: 10.1007/s004010000242
Timmer, N. M., Herbert, M. K., Claassen, J. A. H. R., Kuiperij, H. B., and Verbeek, M. M. (2015). Total glutamine synthetase levels in cerebrospinal fluid of Alzheimer’s disease patients are unchanged. Neurobiol. Aging 36, 1271–1273. doi: 10.1016/j.neurobiolaging.2014.12.010
Tomimoto, H., Akiguchi, I., Wakita, H., Suenaga, T., Nakamura, S., and Kimura, J. (1997). Regressive changes of astroglia in white matter lesions in cerebrovascular disease and Alzheimer’s disease patients. Acta Neuropathol. 94, 146–152. doi: 10.1007/s004010050686
Tumani, H., Shen, G., Peter, J. B., and Brück, W. (1999). Glutamine synthetase in cerebrospinal fluid, serum and brain: a diagnostic marker for Alzheimer disease? Arch. Neurol. 56, 1241–1246. doi: 10.1001/archneur.56.10.1241
van Lenhossék, M. (1895). “Die Stützzellen des Rückenmarkes,” in Der Feinere Bau Des Nervensystems Im Lichte Neuester Forschungen, ed. B. H. Kornfeld (Berlin: Fischer’s Medicine), 176–247.
Vehmas, A. K., Kawas, C. H., Stewart, W. F., and Troncoso, J. C. (2003). Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol. Aging 24, 321–331. doi: 10.1016/s0197-4580(02)00090-8
Ventura, R., and Harris, K. M. (1999). Three-dimensional relationships between hippocampal synapses and astrocytes. J. Neurosci. 19, 6897–6906.
Virchow, R. (1858). “Rückenmark und Gehirn,” in Die Cellularpathologie, ed. R. Virchow (Berlin: Verlag von August Hirschwald), 239–254.
von Holstein-Rathlou, S., Petersen, N. C., and Nedergaard, M. (2018). Voluntary running enhances glymphatic influx in awake behaving, young mice. Neurosci. Lett. 662, 253–258. doi: 10.1016/j.neulet.2017.10.035
Webster, B., Hansen, L., Adame, A., Crews, L., Torrance, M., Thal, L., et al. (2006). Astroglial activation of extracellular-regulated kinase in early stages of Alzheimer disease. J. Neuropathol. Exp. Neurol. 65, 142–151. doi: 10.1097/01.jnen.0000199599.63204.6f
Wegiel, J., and Wisniewski, H. M. (1994). Rosenthal fibers, eosinophilic inclusions and anchorage densities with desmosome-like structures in astrocytes in Alzheimer’s disease. Acta Neuropathol. 87, 355–361. doi: 10.1007/s004010050098
Weller, R. O., Subash, M., Preston, S. D., Mazanti, I., and Carare, R. O. (2008). Perivascular drainage of amyloid-β peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 18, 253–266. doi: 10.1111/j.1750-3639.2008.00133.x
Wharton, S. B., Williams, G. H., Stoeber, K., Gelsthorpe, C. H., Baxter, L., Johnson, A. L., et al. (2005). Expression of Ki67, PCNA and the chromosome replication licensing protein Mcm2 in glial cells of the ageing human hippocampus increases with the burden of Alzheimer-type pathology. Neurosci. Lett. 383, 33–38. doi: 10.1016/j.neulet.2005.04.019
Wilcock, D. M., Vitek, M. P., and Colton, C. A. (2009). Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience 159, 1055–1069. doi: 10.1016/j.neuroscience.2009.01.023
Wilhelmsson, U., Bushong, E. A., Price, D. L., Smarr, B. L., Phung, V., Terada, M., et al. (2006). Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc. Natl. Acad. Sci. U S A 103, 17513–17518. doi: 10.1073/pnas.0602841103
Wolosker, H., Balu, D. T., and Coyle, J. T. (2016). The rise and fall of the d-serine-mediated gliotransmission hypothesis. Trends Neurosci. 39, 712–721. doi: 10.1016/j.tins.2016.09.007
Woltjer, R. L., Duerson, K., Fullmer, J. M., Mookherjee, P., Ryan, A. M., Montine, T. J., et al. (2010). Aberrant detergent-insoluble excitatory amino acid transporter 2 accumulates in Alzheimer disease. J. Neuropathol. Exp. Neurol. 69, 667–676. doi: 10.1097/NEN.0b013e3181e24adb
Wu, Z., Guo, Z., Gearing, M., and Chen, G. (2014). Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzhiemer’s disease model. Nat. Commun. 5:4159. doi: 10.1038/ncomms5159
Wu, H.-Y., Hudry, E., Hashimoto, T., Kuchibhotla, K., Rozkalne, A., Fan, Z., et al. (2010). Amyloid β induces the morphological neurodegenerative triad of spine loss, dendritic simplification and neuritic dystrophies through calcineurin activation. J. Neurosci. 30, 2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010
Xiao, Q., Yan, P., Ma, X., Liu, H., Perez, R., Zhu, A., et al. (2014). Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. J. Neurosci. 34, 9607–9620. doi: 10.1523/JNEUROSCI.3788-13.2014
Xie, L., Kang, H., Xu, Q., Chen, M. J., Liao, Y., Thiyagarajan, M., et al. (2013). Sleep drives metabolite clearance from the adult brain. Science 342, 373–377. doi: 10.1126/science.1241224
Xu, Z., Xiao, N., Chen, Y., Huang, H., Marshall, C., Gao, J., et al. (2015). Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Aβ accumulation and memory deficits. Mol. Neurodegener. 10:58. doi: 10.1186/s13024-015-0056-1
Yamaguchi, H., Sugihara, S., Ogawa, A., Saido, T. C., and Ihara, Y. (1998). Diffuse plaques associated with astroglial amyloid β protein, possibly showing a disappearing stage of senile plaques. Acta Neuropathol. 95, 217–222. doi: 10.1007/s004010050790
Yamazaki, M., Nakano, I., Imazu, O., and Terashi, A. (1995). Paired helical filaments and straight tubules in astrocytes: an electron microscopic study in dementia of the Alzheimer type. Acta Neuropathol. 90, 31–36. doi: 10.1007/bf00294456
Yeh, C.-Y., Vadhwana, B., Verkhratsky, A., and Rodríguez, J. J. (2011). Early astrocytic atrophy in the entorhinal cortex of a triple transgenic animal model of Alzheimer’s disease. ASN Neuro 3, 271–279. doi: 10.1042/AN20110025
Yoshiyama, Y., Asahina, M., and Hattori, T. (2000). Selective distribution of matrix metalloproteinase-3 (MMP-3) in Alzheimer’s disease brain. Acta Neuropathol. 99, 91–95. doi: 10.1007/pl00007428
Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G., et al. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012
Zeppenfeld, D. M., Simon, M., Haswell, J. D., D’Abreo, D., Murchison, C., Quinn, J. F., et al. (2017). Association of perivascular localization of aquaporin-4 with cognition and Alzheimer disease in aging brains. JAMA Neurol. 74, 91–99. doi: 10.1001/jamaneurol.2016.4370
Zhao, J., Davis, M. D., Atagi, Y., Shinohara, M., Graff-Radford, N. R., Younkin, S. G., et al. (2017). APOEε4/ε4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum. Mol. Genet. 26, 2690–2700. doi: 10.1093/hmg/ddx155
Zhao, J., O’Connor, T., and Vassar, R. (2011). The contribution of activated astrocytes to Aβ production: implications for Alzheimer’s disease pathogenesis. J. Neuroinflammation 8:150. doi: 10.1186/1742-2094-8-150
Zhu, Y., Nwabuisi-Heath, E., Dumanis, S. B., Tai, L. M., Yu, C., Rebeck, G. W., et al. (2012). APOE genotype alters glial activation and loss of synaptic markers in mice. Glia 60, 559–569. doi: 10.1002/glia.22289
Keywords: Alzheimer’s disease, amyloid plaques, astrocytes, glia, microglia, neurofibrillary tangles
Citation: Perez-Nievas BG and Serrano-Pozo A (2018) Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front. Aging Neurosci. 10:114. doi: 10.3389/fnagi.2018.00114
Received: 03 February 2018; Accepted: 03 April 2018;
Published: 25 April 2018.
Edited by:
Nibaldo C. Inestrosa, Pontificia Universidad Católica de Chile, ChileReviewed by:
Valentina Echeverria Moran, Bay Pines VA Healthcare System (VHA), United StatesAlberto Lleo, Hospital Sant Pau, Spain
Copyright © 2018 Perez-Nievas and Serrano-Pozo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alberto Serrano-Pozo, aserrano1@mgh.harvard.edu