Drug-induced psychosis: how to avoid star gazing in schizophrenia research by looking at more obvious sources of light
- Division of Psychological Medicine, Institute of Psychiatry, King’s College London, London, UK
The prevalent view today is that schizophrenia is a syndrome rather than a specific disease. Liability to schizophrenia is highly heritable. It appears that multiple genetic and environmental factors operate together to push individuals over a threshold into expressing the characteristic clinical picture. One environmental factor which has been curiously neglected is the evidence that certain drugs can induce schizophrenia-like psychosis. In the last 60 years, improved understanding of the relationship between drug abuse and psychosis has contributed substantially to our modern view of the disorder suggesting that liability to psychosis in general, and to schizophrenia in particular, is distributed trough the general population in a similar continuous way to liability to medical disorders such as hypertension and diabetes. In this review we examine the main hypotheses resulting from the link observed between the most common psychotomimetic drugs (lysergic acid diethylamide, amphetamines, cannabis, phencyclidine) and schizophrenia.
The prevalent view among researchers today is that schizophrenia is a syndrome rather than a specific disease. Liability to this syndrome is highly heritable but the fact that concordance between monozygotic twins is less than 50% suggests an important role for environmental influences (Tandon et al., 2008; MacDonald and Schulz, 2009). It appears that multiple genetic and environmental factors operate together to push individuals over a threshold into expressing the characteristic clinical picture (Tsuang et al., 2004; Di Forti et al., 2007a).
One environmental factor which has been curiously neglected is the evidence that certain drugs can contribute to the onset of schizophrenia-like psychosis. Since the mid-twentieth century, more knowledge has accumulated concerning this than any other causal factor. Here, therefore, we examine the evidence that acute administration of drugs such as lysergic acid diethylamide (LSD), amphetamines, phencyclidine (PCP), and cannabis can induce psychotic symptoms, and consider the implications for our understanding of schizophrenia.
However, before we discuss these issues it is appropriate to remind ourselves of a few definitions. The clinical definition of a psychotic disorder restricts the term to those illnesses with delusions and/or prominent hallucinations, with the hallucinations occurring in the absence of insight into their pathological nature (DSM-IV). A drug with psychotomimetic actions is one which induces temporary states of altered perception (McGraw-Hill, 1993), often mimicking the symptoms of psychosis, including delusions, and/or hallucinations (“psychoto-” from psychosis, and “-mimetic” from the Greek μιμητιℵ?ς “in imitation of”). Thus, experimental studies examine those effects of drugs that resemble the positive symptoms (e.g., hallucinations, delusions, paranoia, and disorganized thinking) and negative symptoms (affective flattening, anhedonia, amotivation, attentional impairment, and cognitive impairment) of schizophrenia (D’Souza et al., 2004).
LSD and the Serotonin Hypothesis
The first drug to attract major attention as a possible model of psychosis was LSD. LSD was synthesized in 1938 by Hofmann (1980), but its hallucinogenic properties were not discovered till Hofmann accidentally spilt some on his skin 5 years later, when he recalled later “I happened to experience its unforeseeable effects in my own body – or rather, in my own mind,” seeing uninterrupted streams of “fantastic pictures, extraordinary shapes with intense kaleidoscopic play of colors”.
In the 1960s, the use of LSD and other recreational drugs spread among young people across the Western world. Timothy Leary, an advocate of psychedelic drugs, was fired from Harvard University, where he was a lecturer from 1959 to 1963, and founded the League for Spiritual Discovery (L.S.D). His phrase “Turn on, tune in, drop out!” became an anthem of the hippie movement. More serious individuals such as the famous author, Aldous Huxley also advocated the use of LSD “to make this trivial world sublime,” and even took the drug on his deathbed.
Neuropharmacologists began to ask whether schizophrenia might be caused by chemical alterations similar to those produced by LSD. Shaw and Woolley (1956) hypothesized that LSD might act on 5-HT receptors, and later Anden et al. (1968) suggested a direct agonist effect at 5-HT receptors in the CNS. The idea that the psychotic state induced by LSD resulted from actions at serotonin receptors led to the suggestion that LSD provides a “model psychosis” and to the serotonin hypothesis of schizophrenia.
One consequence was a great deal of interest in other hallucinogens such as N,N-dimethyltryptamine (DMT; Manske, 1931), an endogenous substance suspected of being synthesized by the pineal gland (Rodriguez, 2007). Szara (1956) gave DMT to healthy volunteers and reported hallucinations, distortions of perception and body image, speech disturbances, and euphoria. The idea arose that schizophrenia might be caused by endogenous hallucinogens, and differences in levels of DMT in urine and blood between schizophrenic patients and controls were reported (Franzen and Gross, 1965; Gillin et al., 1976). Narasimhachari et al. (1972) gave an MAO inhibitor (tranylcypromine) and cysteine to both groups and found an exacerbation of symptoms and an increase in urinary DMT only in schizophrenics. In 1979 one of us (RMM), studying the excretion of DMT and its precursor N-methyltryptamine (NMT), found a greater DMT excretion in cases of schizophrenia compared to controls (Murray et al., 1979); the reasons for these differences were never established (Strassman, 1991).
Glennon et al. (1983) proposed the 5-HT2 receptor subtypes as the specific target of hallucinogenic drugs such as LSD. Subsequent studies in rodents supported this (Nichols, 2004), and investigations in healthy volunteers provided evidence of 5-HT2A receptor mediation in producing psychotomimetic effects in humans (Vollenweider et al., 1998; Carter et al., 2007). The serotonin hypothesis received further support from the finding of 5-HT2-receptor abnormalities in post-mortem schizophrenic brains (Mita et al., 1986; Gurevich and Joyce, 1997), but in vivo studies of 5-HT2-receptor abnormalities reported conflicting results. Recently, Rasmussen et al. (2010) assessed 30 antipsychotic-naïve schizophrenic patients and matched healthy controls, finding that the patients had lower serotonin 2A binding in the frontal cortex. Positron emission tomography (PET) scanning in humans showed that hallucinogens as psilocybin produced a marked activation of the pre-frontal cortex caused by serotonergic (Arora and Meltzer, 1991; Joyce et al., 1993; Vollenweider et al., 1997). The evidence that clozapine is particularly efficacious in the care of treatment resistant schizophrenia provoked further interest because of the 5-HT2 receptor antagonism induced by this drug and other atypical antipsychotics (Lieberman et al., 1998; Meltzer, 1999; Tamminga and Holcomb, 2005).
Breakey et al. (1974) who investigated the relationship between LSD use and the age of onset of schizophrenia commented “On the basis of these results it seems plausible to conclude that the taking of drugs might play some precipitating role in the onset of schizophrenia, bringing this disorder on more quickly.” This statement has been amply confirmed. Vardy and Kay (1983), comparing patients hospitalized for LSD psychosis with first-break schizophrenics, suggested that LSD psychosis was a drug-induced schizophreniform reaction; moreover they concluded that there was a greater psychotic response to LSD in persons with genetic predisposition to schizophrenia.
Amphetamine, Crystal Meth, and the Dopamine Theory
As seen in Figure 1, the heyday of research into LSD and other hallucinogens was in the 1960s. However, new laws restricted researchers’ ability to study these drugs, and it became clear that LSD produced a psychosis with more visual and fewer auditory aberrations than is typical of schizophrenia. Interest switched to amphetamines.
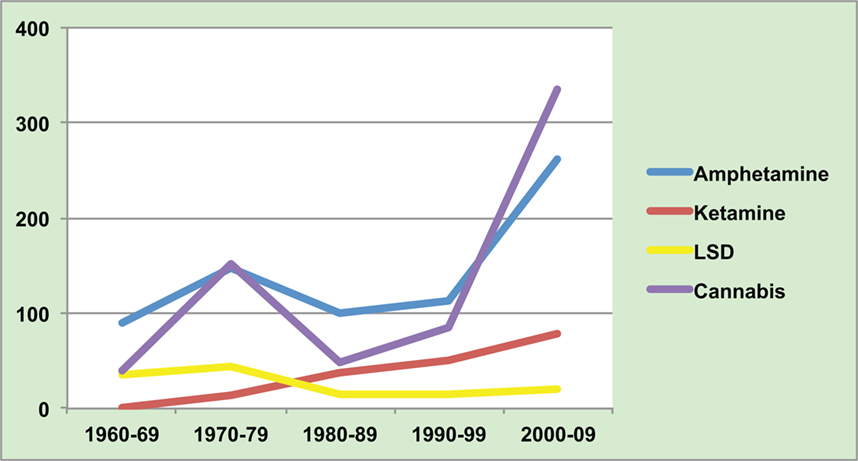
Figure 1. Number of published papers (Pub Med) on illicit drugs and psychosis.
The similarity of amphetamine psychosis to schizophrenia was first clearly described in the 1950s by Tatetsu et al. (1956) and was substantiated by a similar report in Britain from Connell (1958) Subsequently, Angrist et al. (1974) found that amphetamine administered experimentally produced a picture similar to paranoid psychosis in healthy individuals and exacerbated psychotic symptoms in approximately one-third of schizophrenic patients (Lieberman et al., 1987); moreover neuroleptic drugs blocked these psychotogenic effects of amphetamines (Espelin and Done, 1968). Amphetamine was found to stimulate dopamine outflow, while, in contrast, antipsychotics were found to block dopamine receptors in the brain (Carlsson and Lindqvist, 1963; Van Rossum, 1967). Subsequently SPET and PET studies demonstrated that acute administration of amphetamine induced greater striatal dopamine release in first-episode schizophrenia patients compared to healthy controls (Laruelle et al., 1996; Breier et al., 1997). Together these observations provided the basis for the dopamine hypothesis of schizophrenia (Snyder, 1972).
Since the 1990s there has been a marked increase in use of methamphetamine (sometimes termed crystal meths) particularly in South-East Asia [United Nations Office on Drugs Crime (UNODC), 2009], producing a rapid rise in methamphetamine psychosis in the same countries (Chen et al., 2003). In 2003 Chen and colleagues recruited 445 amphetamine users from a psychiatric hospital and a detention center in Taiwan. One hundred sixty-three had methamphetamine psychosis which presented a picture almost identical to that of paranoid schizophrenia. In most cases the symptoms slowly disappeared following cessation of the drug but in a proportion they did not remit readily, if ever (Grelotti et al., 2010). Chen and colleagues subsequently addressed the question of why some methamphetamine users became psychotic while others appeared to be able to continue their use without major problems. Not surprisingly, earlier and greater use of methamphetamine was associated with increased risk of psychosis. More interestingly, methamphetamine users with a pre-existing psychosis-prone personality were more likely to develop frank psychosis as were those with a greater familial risk. Psychosis-prone personality and family history were also associated with a more prolonged psychotic episode (Chen et al., 2003).
Recently researchers have looked for genes that may confer a vulnerability to psychosis subsequent to methamphetamine use. Bousman et al. (2009) reviewed 38 genetic association studies, and reported four genes associated with psychosis: DTNBP1, which codes for dystrobrevin-binding protein 1, OPRM1, which codes for the mu-opioid receptor, SNCA, implicated in the modulation of dopamine transmission, and a single haplotype of SOD2, which encodes a mitochondrial protein important in cellular defense against oxidative damage. It remains to be seen how robust these associations are.
Generally speaking, amphetamine, and methamphetamine do not produce a psychosis after initial use but after repeated use. An appropriate theory has to explain this and also why abstinent methamphetamine abusers can be precipitated back into a psychosis not only by further methamphetamine abuse by also by social stress. One possibility is dopamine sensitization (Ljeberman et al., 1990; Glenthøj and Hemmingsen, 1997). In rodents, repeated administration of amphetamine leads to reversed tolerance and an increased neurochemical and behavioral reaction to each dose. Boileau and colleagues tested whether this might happen also in humans. They administered dextroamphetamine by mouth on days 1, 3, and 5 to 10 healthy volunteers, and measured the effect on striatal dopamine release before exposure, then the day of first exposure, then 2 weeks later after the third dose, using the PET/[11C] raclopride technique. Each dose of amphetamine caused greater dopamine release in the ventral striatum together with greater behavioral responses. Indeed, 1 year later there was a greater psychomotor response and greater increase dopamine release compared to the initial dose, in the ventral striatum, progressively extending to the dorsal caudate and putamen (Boileau et al., 2006). Such findings have led to the “dopamine sensitization” hypothesis of schizophrenia which postulates that a sensitized dopamine system is responsible for the genesis of psychotic symptoms (Peleg-Raibstein et al., 2009).
Prolonged methamphetamine abuse may produce permanent damage to neurons. Thus, a PET study showed a reduction in dopamine transporter density in methamphetamine users, which seemed to be related to the duration of methamphetamine use, and to the chronic psychotic state in methamphetamine users (Iyo et al., 2004).
Angel Dust Provokes the Glutamate Theory
The “dopamine theory” of schizophrenia has dominated attempts to explain the positive symptoms seen in schizophrenic patients (Kapur and Mamo, 2003) but is less able to account for negative symptoms. Drugs acting on the glutamate system have been proposed to model these. Kotz and Merkel (1926) discovered the compound, 1-piperidinocyclohexanecarbonitrile, and thus laid the groundwork for the preparation of PCP which was developed as an intravenous general anesthetic in the 1940s by Parke, Davis & Co. Unfortunately, up to 50% of adult patients given PCP developed agitation and hallucinations (Greifenstein et al., 1958; Johnstone et al., 1958; Collins et al., 1960), so the focus of investigation of PCP shifted from its possible use as an anesthetic to its capacity to produce model psychoses (Luby et al., 1962). Acute administration of PCP to healthy volunteers produced severe disturbances in body image, affect and thinking with impressive similarity to “certain primary symptoms of the schizophrenic process” (Luby et al., 1959); as well as severe impairments of attention, motor function, and proprioceptive acuity (Rosenbaum et al., 1959). Although PCP was removed from clinical use in 1965, it became popular as a recreational drug as “angel dust.”
Subsequently, ketamine, another glutamate antagonist was introduced as an anesthetic, and later reports of ketamine induced psychosis started to appear. In 1983 Anis and colleagues showed that ketamine and phencyclidine selectively reduce responses of central neurons acting at the N-methyl-D-aspartate (NMDA) receptor (Anis et al., 1983; Seeman and Tallerico, 2005). The observation that PCP and ketamine are NMDA antagonists together with the findings of lower levels of glutamate (by about 50%) in cerebrospinal fluid samples from schizophrenic patients compared to controls gave rise to the “glutamate hypothesis of schizophrenia” (Kim et al., 1980). Although, this latter finding was not consistently replicated, the idea of hypoglutamatergic transmission in schizophrenia has continued to attract great interest as has the effects of NMDA antagonists (Tamminga and Holcomb, 2005). A dissenting view comes from Pomerol and colleagues who gave ketamine to 10 healthy volunteers. The commonest feature was referential thinking of a delusional nature, together with a range of perceptual abnormalities perhaps best described as dissociative. However, it did not induce hallucinations, and the authors were doubtful about its ability to cause thought disorder; furthermore, although negative-like symptoms resulted they could not exclude the possibility that this was simply due to its anesthetic effects (Pomarol-Clotet et al., 2006).
According to the glutamate deficiency theory, drugs which enhance glutamate neurotransmission should improve psychotic symptoms. Trials of glutamate potentiation using glycine and related compounds have not reported any antipsychotic effects (Deakin and Simpson, 1997) but in 2007 Lilly developed LY2140023 which is a pro-drug of LY404039, a selective agonist for metabotropic glutamate 2/3 (mGlu2/3) receptors. A randomized, three-armed, double-blind, placebo-controlled study showed that schizophrenic patients treated with LY2140023 improved to a similar extent to those treated with olanzapine, both showing greater improvement on the positive and negative syndrome scale (PANSS) than placebo (Patil et al., 2007). The precise mechanism by which this drug acts on schizophrenia is not yet fully understood. However, mGluR2/3 agonists have been shown to inhibit dopamine release leading to the suggestion that dopamine abnormalities in psychosis could be secondary to a primary hypofunction in glutamate neurotransmission (Snyder and Murphy, 2008). Seeman and Tallerico (2005) set out to test this last hypothesis, and found that phencyclidine and ketamine act on the high affinity state of dopamine D2 receptors (dissociation constants of 3 and 55 nM, respectively) suggesting a direct or indirect dopamine-mimetic action of phencyclidine.
Cannabis: The Sacred and Ubiquitous HERB
Cannabis is the world’s favorite illicit recreational drug. Its use grew in many countries in the latter part of the twentieth century and now more than 160 million people take it every day. Within Western Europe there have been marked variations in its use (Figure 2) with some countries showing a recent decline in the drug’s popularity, e.g., UK, while in others its use continues to grow, e.g., Italy [European Monitoring Centre for Drugs, and Drug Addiction Annual Report, 2009].
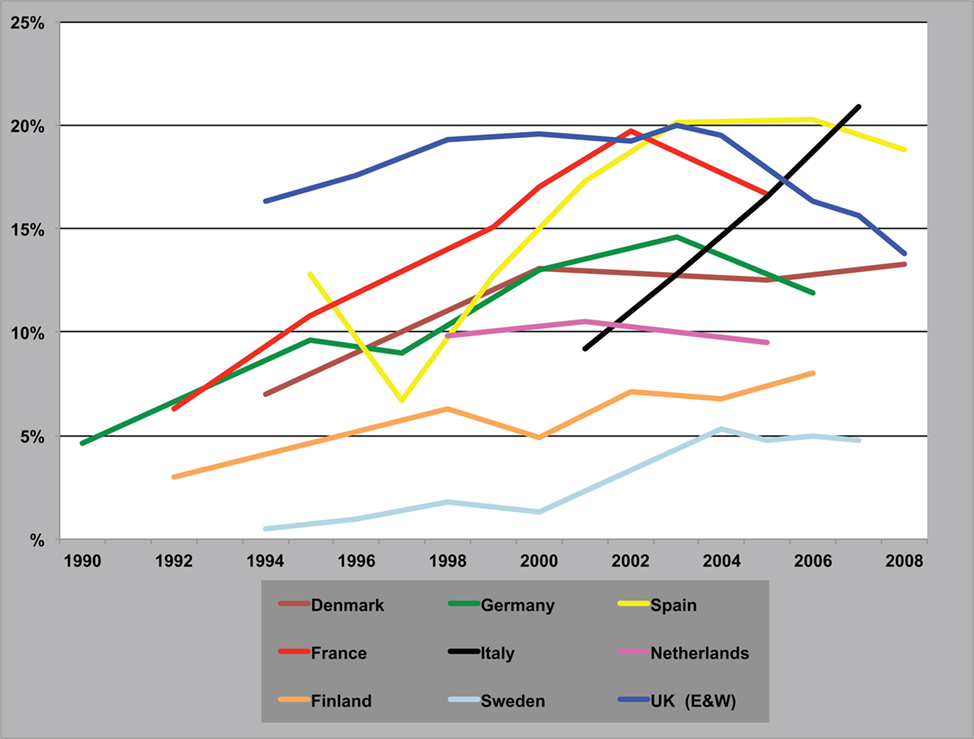
Figure 2. Trends in the prevalence of “cannabis use in the last year” among young adults. Annual Report, 2009.
Cannabis has been linked to the development of psychotic symptoms since time immemorial. The world’s oldest pharmacopeia, the Pen-Ts’ao Chin, traditionally attributed to the Chinese Emperor Shen Nung (2737 a.c.), suggests cannabis use as a remedy for rheumatic pain, intestinal constipation, disorders of the female reproductive system, and malaria, but adds that “if taken in excess will produce visions of devils … over a long term, it makes one communicate with spirits and lightens one’s body…” (Zuardi, 2006). In the mid-nineteenth century the French psychiatrist Moreau de Tour noted the phenomenological similarities between drug-induced states and psychosis. Indeed, he proposed the use of hashish to study madness a full century before the LSD model of psychosis (Beringer, 1927; Hofmann, 1980).
The first convincing evidence that cannabis might be a risk factor for psychosis came from the Swedish Conscripts Study on in which 45,570 inductees into the military were followed-up for 15 years. This study reported a risk for schizophrenia 2.4 times higher among those who had used cannabis by 18 years than among non-users. Moreover, there was a dose–response relationship in that risk for schizophrenia rose to 6.0 times in heavy cannabis smokers (use of cannabis more than 50 times at initial interview; Andreasson et al., 1987). After 15 years of silence, other epidemiological studies on cannabis use and schizophrenia started to be published (Arseneault et al., 2002; Van Os et al., 2002; Zammit et al., 2002; Fergusson et al., 2003). As summarized in Table 1, today consistent evidence supports an association between heavy cannabis use and the risk of psychotic symptoms and illness (Green et al., 2005; Henquet et al., 2005; Fergusson et al., 2006; Barnett et al., 2007; Di Forti et al., 2007b; Moore et al., 2007; Mc Grath et al., 2010) even if the strength and nature of that association is still an object of discussion (D’Souza et al., 2004).
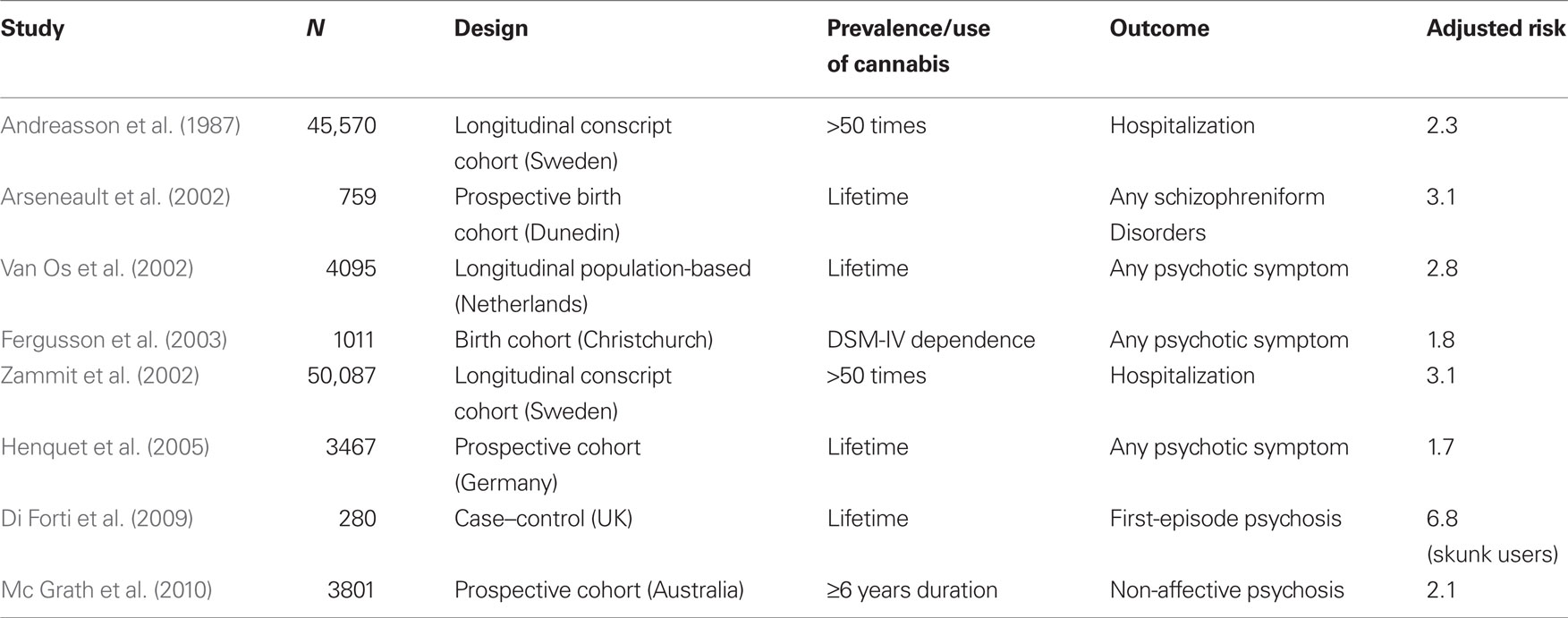
Table 1. Studies suggesting an association between cannabis use and psychosis.
Recent meta-analyses reported an increase of the risk between 1.4 and 1.9 times in people using cannabis that might account for 8–14% of cases of schizophrenia in different countries (Moore et al., 2007). Furthermore, Di Forti et al. (2009) showed a positive association between the potency and frequency of cannabis use in increasing the risk of psychotic illness.
Delta-9-tetrahydrocannabinol (THC) is the main psychoactive ingredient in cannabis; when given experimentally in sufficient dosage, it can produce transient psychotic symptoms and impaired memory via stimulation of CB1 receptors, targets for the endogenous cannabinoid [endocannabinoid (eCB)] transmitters (Matsuda et al., 1990; Freund et al., 2003; Zuurman et al., 2010). Several studies have shown that either pure THC or synthetic CB1 agonists elicit psychotic symptoms in a proportion of healthy volunteers (Leweke et al., 1999a,b; D’Souza et al., 2004; Morrison et al., 2009; Zuurman et al., 2009). CB1 receptors are widely distributed throughout the central nervous system, particularly in the hippocampus, amygdala, cerebellum, basal ganglia, and pre-frontal cortex (Herkenham et al., 1991). They are primarily expressed on glutamate and GABA-ergic terminals (Freund et al., 2003; Chevaleyre et al., 2006), and upon stimulation by cannabinoids, act to reduce pre-synaptic glutamate or GABA release (Chevaleyre et al., 2006). The glutamate and GABA inputs to the band of dopamine neurons in the ventral midbrain express CB1 receptors and numerous animal studies have shown that exogenous cannabinoids alter the balance of excitation and inhibition reaching dopamine cells (Lupica and Riegel, 2005; Matyas et al., 2008). Although a net inhibition of dopamine cells has been described (Melis et al., 2004), by far the most commonly documented effect is an increase in firing with attendant elevations of dopamine release at downstream forebrain territories (Lupica and Riegel, 2005).
One testable hypothesis which arises is whether the psychotomimetic properties of THC in humans are mediated via excess dopamine release. Bossong et al. (2009) used a dopamine D2/D3 receptor tracer (raclopride) and positron emission tomography, to examine striatal synaptic dopamine release. The tracer binding was significantly reduced in the ventral striatum and the precommissural dorsal putamen after inhalation of THC compared to placebo, implying an increased release of endogenous dopamine in these regions. However, the increase is modest compared to that obtained with amphetamine and cocaine, and two other studies have failed to find such clear-cut results (Stokes et al., 2009). For example, another PET study by Stokes et al. (2009) found only a non-significant increase in dopamine release in the striatum following oral THC. Later Stokes et al. (2010) re-analyzed their initial data and showed significant decreases in cortical [11C]-raclopride binding potential after administration of THC, suggesting dopamine release in the cortex. Our own group observed clear psychotic phenomena (including thought-echo and passivity) in a group of 10 healthy volunteers given IV THC during SPET scanning but there were no significant differences in DA release between THC and placebo sessions, nor were positive symptoms and DA release related (Barkus et al., 2010). Furthermore, D’Souza et al. (2005, 2008) showed that D2 blockers did not offer “protection” against acute THC-psychosis, either in healthy controls or in stable schizophrenic patients. However, the validity of this interpretation needs to be investigated and whether the psychotomimetic properties of THC arise by way of downstream dopaminergic events is currently unclear.
Indeed, the reverse may be true. One of the downstream effects of stimulating D2 receptors in the striatum is the synthesis and release of endocannabinoid transmitters (Giuffrida et al., 1999; Centonze et al., 2004; Kreitzer and Malenka, 2005; Yin and Lovinger, 2006; D’Souza et al., 2008; Stokes et al., 2010) at the dendritic spines of medium spiny neurons (MSNs; Uchigashima et al., 2007). In turn, eCBs act in a so-called retrograde fashion to inhibit neighboring GABA and glutamate terminals (Gerdeman et al., 2002; Robbe et al., 2002; Freiman et al., 2006; Adermark et al., 2009). There is accumulating evidence that a number of key physiological processes within the striatum depend upon D2 → eCB → CB1 signaling and the subsequent modification of fast amino-acid based transmission (Calabresi et al., 2007; Kreitzer and Malenka, 2008). Such processes are believed to be vital for the selection and initiation of psychomotor behaviors and for associative learning, or in other words, the normal functions of the striatum in health (Graybiel, 2005; Yin and Knowlton, 2006; Houk et al., 2007; Kreitzer and Malenka, 2007). One question which arises is whether the pro-psychotic effects of not just THC, but stimulant drugs as well involve the striatal D2 → eCB → CB1 trans-synaptic pathway. If this were the case then it might explain why D2 blockade has little effect on THC-psychosis because THC “enters” the pathway at a point which is distal to D2 receptors. In contrast pharmacological manipulations at CB1 would be predicted to impact upon stimulant (D2 initiated), psychosis, since CB1 is downstream of D2. Several lines of evidence suggest that this might be the case.
It is clear that stimulant sensitization or adaptations to repeated drug administration as opposed to single drug exposures, are important for the emergence of psychosis. One of the outcomes of repeated stimulant administration is a functional up-regulation of striatal CB1 receptors (Centonze et al., 2007). Furthermore, several recent studies have shown that either CB1 knockout or blockade of CB1 receptors by potent CB1 antagonists impairs stimulant sensitization (Corbille et al., 2007; Thiemann et al., 2008a,b). In a particularly elegant design it was demonstrated that microinjections of a potent CB1 antagonist directly into the ventral striatum reduced the expression of behavioral sensitization to methamphetamine (Chiang and Chen, 2007). Findings with the first generation CB1 blocker rimonabant in sensitization paradigms have been inconsistent (Lesscher et al., 2005; Thiemann et al., 2008b), but rimonabant, as opposed to newer CB1 antagonists, only partially blocks the effects of THC in humans (Huestis et al., 2001; Zuurman et al., 2010) which might explain why an early trial of rimonabant in schizophrenia failed (Meltzer et al., 2004). Rimonabant is also ineffective in animal models of cocaine reward, in contrast to CB1 antagonists such as AM251 (Gardiner, personal communication).
Another cannabinoid manipulation has shown more promise. The molecule cannabidiol (CBD), a constituent of free-growing, natural Cannabis sativa (but largely eliminated from high potency cannabis preparations such as skunk; Potter et al., 2008) appears to have antipsychotic properties in humans (Leweke et al., 2000; Zuardi et al., 2009). At low doses, the receptor pharmacology of CBD involves pharmacological antagonism of CB1 agonists (despite low affinity for the orthosteric site of CB1 receptors) and inhibition of adenosine re-uptake. CBD also inhibits the degradation of the endocannabinoid molecule anandamide and acts at a “third” cannabis receptor termed GPR55 (Pertwee, 2007). CBD displays efficacy against stimulant (Zuardi et al., 1991; Moreira and Guimaraes, 2005) and NMDA models of psychosis in animals (Long et al., 2006), and against L-DOPA (Zuardi et al., 2009), ketamine (Bosi et al., 2003), and THC elicited psychosis in humans (Zuardi et al., 1982; Bhattacharyya et al., 2009). A single head-to-head trial of CBD vs amisulpride in 42 schizophrenic patients found equivalent efficacy (Leweke, 2007), but further data, particularly over the longer term, is required before we can definitely conclude that these antipsychotic effects are real.
Overall, the above suggests a model in which pharmacological manipulations to neurochemical systems controlling traffic through striatal circuits can be either pro- or antipsychotic. The most notable feature is the consistency of the pharmacology with which facilitating the D2 → eCB → CB1 axis appears to be pro-psychotic.
Conclusion
We have seen that a great deal of knowledge has accumulated concerning drug-induced psychoses over the last 50 years. However, the picture presenting in the clinic, as opposed to that seen in the experimental setting, has not been widely acknowledged as telling us much about schizophrenia itself. Perhaps the main reason for this is that when drug-induced psychoses were first identified, schizophrenia was considered a discrete disease with a likely single genetic cause. Consequently, drug-induced psychoses, even those phenomenologically very similar to schizophrenia, were seen as phenocopies rather than examples of the schizophrenia syndrome consequent upon the operation of a particular cause. However, now that schizophrenia is generally viewed as at the extreme end of a continuum of psychosis where the combined effect of genetic and environmental factors pushes the individual over a threshold into the expression of the disorder. In the light of this new knowledge it is time to re-appraise the drug-induced psychoses.
Certainly the major pharmacological theories of schizophrenia have their origins in the effects of drugs of abuse; in chronological order, the effects of LSD initiated the serotonergic model; amphetamines the dopamine hypothesis, PCP, and ketamine the glutamatergic model. Most recently the effects of cannabis have provoked interest in the role of endocannabinoids. None of these models account for the complete picture of schizophrenia, but this is not to be expected if we regard the condition as one with multiple causes and consequent heterogeneity of the clinical picture. Rather the various drugs mimic different aspects of the disorder. Stimulants and THC are particularly likely to induce paranoia beliefs (Javitt and Zukin, 1990), whereas LSD is more closely associated with visual illusions/hallucinations (Smith et al., 2009). The non-competitive NMDA antagonists PCP and ketamine appear to induce negative symptoms, and oneroid states (characterized by perceptual illusions, perplexity, and delusional thinking in the context of clouding of consciousness; Gonzalez-Maeso and Sealfon, 2009). What is common between the different classes of drug is the promotion of a fundamental change in the subject’s experience of reality, whether acutely, during drug intoxication (in the case of LSD, ketamine, and THC) or as a result of an adaptive process secondary to repeated use (stimulants and THC).
How then is it possible to discriminate between drugs which distort the experience of reality in a way which closely corresponds to aspects of schizophrenia from those that do not? One approach is to postulate that if a drug-model pushes the CNS toward psychosis AND if it is also “true” that a diametrically opposing pharmacological manipulation pulls a schizophrenic psychosis back into reality, then the drug-model in question constitutes a reasonable model of schizophrenic-like psychosis. Some examples may illuminate this idea. Agonists at the 5HT receptor transform reality in a fundamental way; yet 5HT2 antagonists have no efficacy against schizophrenic psychosis (Zuardi, 2006). Similarly, NMDA channel blockers distort the experience of reality, yet, despite much effort and theoretical support, drugs which enhance NMDA channel opening have not yet been proven to be effective in schizophrenia.
In contrast, opposing pharmacological manipulations at D2 receptors can elicit clear bi-directional responses at the psychological level. Drugs which drive D2-mediated signaling to excess have pro-psychotic properties AND D2 blockers are effective against schizophrenic psychosis. On this basis, one can infer that, at a neurochemical level, the DA model psychosis is more akin to reality-distortion in schizophrenia than are either the serotonergic or glutamatergic models.
In recent years it has been suggested that excess dopamine synthesis and release in the striatum is the final, common arbiter of positive psychotic symptoms (Broome et al., 2005; Di Forti et al., 2007a; Murray et al., 2008) probably through a pathological dysfunction of the “reward” pathway (Strassman, 1984). Do all of the drugs which induce psychotic symptoms do so via their effects on the dopamine system? Certainly, as we have seen, an argument can be made that they all impact on the dopamine system either directly or indirectly. However, it is not clear that this effect accounts for their propensity to induce the characteristic positive symptoms of schizophrenia. Another caveat is that the dopamine model can probably no longer be considered in isolation from the emerging endocannabinoid model. Earlier we utilized the relatively simple concept of a D2 → eCB → CB1 axis to describe the known physiological relations of the two neurochemical systems within the striatum (Kreitzer and Malenka, 2008). Notably, opposing pharmacological manipulations at CB1 receptors can also elicit bi-directional responses, either pro- or antipsychotic. Moreover, CB1 manipulations appear to be effective against dopamine D2 mediated psychoses.
Finally, not all addictive drugs which impact on the dopamine system appear to be able to induce psychotic symptoms. For example, neither cigaret smoking, nor use of opiates induce psychotic symptoms. One possible reason is that the fact that nicotine and opiate receptors (and responses) show a rapid de-sensitization following exposure to their respective agonists. This illustrates the general theme of this paper; determining the neurochemical differences between those drugs whose effects do and which do not mimic particular aspects of schizophrenia is worthy of investigation in humans as well as in animals.
Conflict of Interest Statement:
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adermark, L., Talani, G., and Lovinger, D. M. (2009). Endocannabinoid-dependent plasticity at GABAergic and glutamatergic synapses in the striatum is regulated by synaptic activity. Eur. J. Neurosci. 29, 32–41.
Anden, N. E., Corrodi, H., Fuxe, K., and Hokfelt, T. (1968). Evidence for a central 5-hydroxytryptamine receptor stimulation by lysergic acid diethylamide. Br. J. Pharmacol. 34, 1–7.
Andreasson, S., Allebeck, P., Engstrom, A., and Rydberg, U. (1987). Cannabis and Schizophrenia: a longitudinal study of Swedish conscripts. Lancet 26, 1483–1486.
Angrist, B., Sathananthan, G., Wilk, S., and Gershon, S. (1974). Amphetamine psychosis: behavioural and biochemical aspects. J. Psychiatr. Res. 11, 13–23.
Anis, N. A., Berry, S. C., Burton, N. R., and Lodge, D. (1983). The dissociative anesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methyl-D-aspartate. Br. J. Pharmacol. 79, 565–575.
Arora, R. C., and Meltzer, H. Y. (1991). Serotonin 2 (5-HT2) receptor binding in the frontal cortex of schizophrenic patients. J. Neural Transm. 85, 19–29.
Arseneault, L., Cannon, M., Poulton, R., Murray, R., Caspi, A., and Moffitt, T. E. (2002). Cannabis use in adolescence and risk for adult psychosis: longitudinal prospective study. BMJ 325, 1212–1213.
Barkus, E., Morrison, P. D., Vuletic, D., Dickson, J. C., Ell, P. J., Pilowsky, L. S., Brenneisen, R., Holt, D. W., Powell, J., Kapur, S., and Murray, R. M. (2010). Does intravenous Δ9-tetrahydrocannabinol increase dopamine release? A SPET study. J. Psychopharmacol. doi: 10.1177/0269881110382465. [Epub ahead of print].
Barnett, J. H., Werners, U., Secher, S. M., Hill, K. E, Brazil, R., Masson, K., Pernet, D. E., Kirkbride, J. B., Murray, G. K., Bullmore, E. T., and Jones, P. B. (2007). Substance use in a population-based clinic sample of people with first-episode psychosis. Br. J. Psychiatry 190, 515–520.
Beringer, K. (1927). Der Meskalinrausch. Seine Geschichte und Erscheinungsweise. Berlin: Julius Springer.
Bhattacharyya, S., Morrison, P. D., Fusar-Poli, P., Martin-Santos, R., Borgwardt, S., Winton-Brown, T., Nosarti, C., O’ Carroll, C. M., Seal, M., Allen, P., Mehta, M. A., Stone, J. M., Tunstall, N., Giampietro, V., Kapur, S., Murray, R. M., Zuardi, A. W., Crippa, J. A., Atakan, Z., and McGuire, P. K. (2009). Opposite effects of delta-9-tetrahydrocannabinol and cannabidiol on human brain function and psychopathology. Neuropsychopharmacology 35, 764–774.
Boileau, I., Dagher, A., Leyton, M., Gunn, R. N., Baker, G. B., Diksic, M., and Benkelfat, C. (2006). Modeling sensitization to stimulants in humans. An [11C] Raclopride/Positron Emission Tomography Study in healthy me. Arch. Gen. Psychiatry 63, 1386–1395.
Bosi, D., Hallak, J., Dursun, S., Deakin, J., and Zuardi, A. (2003). Effects of cannabidiol on (s)-ketamine-induced psychopathology in healthy volunteers. J. Psychopharmacol. 17, A55.
Bossong, M. G., van Berckel, B. N., Boellaard, R., Zuurman, L., Schuit, R. C., Windhorst, A. D., van Gerven, J. M., Ramsey, N. F., Lammertsma, A. A., and Kahn, R. S. (2009). Delta 9-tetrahydrocannabinol induces dopamine release in the human striatum. Neuropsychopharmacology 34, 759–766.
Bousman, C. A., Glatt, S. J., Everall, I. P., and Tsuang, M. T. (2009). Genetic association studies of methamphetamine use disorders: a systematic review and synthesis. Am. J. Med. Genet. 150B, 1025–1049.
Breakey, W. R., Goodell, H., Lorenz, P. C., and McHugh, P. R. (1974). Hallucinogenic drugs as precipitants of schizophrenia. Psychol. Med. 4, 255–261.
Breier, A., Su, T. P., Saunders, R., Carson, R. E., Kolachana, B. S., de Bartolomeis, A., Weinberger, D. R., Weisenfeld, N., Malhotra, A. K., Eckelman, W. C., and Pickar, D. (1997). Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc. Natl. Acad. Sci. U.S.A. 94, 2569–2574.
Broome, M. R., Woolley, J. B., Tabraham, P., Johns, L. C., Bramon, E., Murray, G. K., Pariante, C., McGuire, P. K., and Murray, R. M. (2005). What causes the onset of psychosis? Schizophr. Res. 79, 23–34.
Calabresi, P., Picconi, B., Tozzi, A., and Di Filippo, M. (2007). Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 30, 211–219.
Carlsson, A., and Lindqvist, M. (1963). Effect of chlorpromazine or haloperidol on formation of 3-methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol. Toxicol. 20, 140–144.
Carter, O. L., Hasler, F., Pettigrew, J. D., Wallis, G. M., Liu, G. B., and Vollenweider, F. X. (2007). Psilocybin links binocular rivalry switch rate to attention and subjective arousal levels in humans. Psychopharmacology 195, 415–424.
Centonze, D., Battista, N., Rossi, S., Mercuri, N. B., Finazzi-Agro, A., Bernardi, G., Calabresi, P., and Maccarrone, M. (2004). A critical interaction between dopamine D2 receptors and endocannabinoids mediates the effects of cocaine on striatal GABAergic transmission. Neuropsychopharmacology 29, 1488–1497.
Centonze, D., Rossi, S., De Chiara, V., Prosperetti, C., Battista, N., Bernardi, G., Mercuri, N. B., Usiello, A., and Maccarrone, M. (2007). Chronic cocaine sensitizes striatal GABAergic synapses to the stimulation of cannabinoid CB1 receptors. Eur. J. Neurosci. 25, 1631–1640.
Chen, C. K., Lin, S. K., Sham, P. C., Ball, D., Loh, E. W., Hsiao, C. C., Chiang, Y. L., Ree, S. C., Lee, C. H., and Murray, R. M. (2003). Pre-morbid characteristics and co-morbidity of methamphetamine users with and without psychosis. Psychol. Med. 33, 1407–1414.
Chevaleyre, V., Takahashi, K. A., and Castillo, P. E. (2006). Endocannabinoid-mediated synaptic plasticity in the CNS. Annu. Rev. Neurosci. 29, 37–76.
Chiang, Y. C., and Chen, J. C. (2007). The role of the cannabinoid type 1 receptor and down-stream cAMP/DARPP-32 signal in the nucleus accumbens of methamphetamine-sensitized rats. J. Neurochem. 103, 2505–2517.
Collins, V. J, Gorospe, C. A., and Rovenstine, E. A. (1960). Intravenous nonbarbiturate, non narcotic analgesics: preliminary studies. I. Cyclohexylamines. Anesth. Analg. 39, 303–306.
Connell, P. H. (1958). Amphetamine Psychosis. Maudsley Monographs. No. 5. London: Oxford University Press.
Corbille, A. G., Valjent, E., Marsicano, G., Ledent, C., Lutz, B., Herve, D., and Girault, J. (2007). Role of cannabinoid type 1 receptors in locomotor activity and striatal signaling in response to psychostimulants. J. Neurosci. 27, 6937–6947.
Deakin, J. F., and Simpson, D. C. (1997). A two-process theory of schizophrenia: evidence from studies in post-mortem brain. J. Psych. Res. 31, 277–295.
Di Forti, M., Lappin, J. M., and Murray, R. M. (2007a). Risk factors for schizophrenia. All roads lead to dopamine. Eur. Neuropsychopharmacol. 17, S101–S107.
Di Forti, M., Morrison, P. D., Butt, A., and Murray, R. M. (2007b). Cannabis use and psychiatric and cognitive disorders: the chicken or the egg? Curr. Opin. Psychiatry 20, 228–234.
Di Forti, M., Morgan, C., Dazzan, P., Pariante, C., Mondelli, V., Reis Marques, T., Handley, R., Aas, M., Luzi, S., Russo, M., Paparelli, A., Butt, A., Stilo, S. A., Wiffen, B., Powell, J., and Murray, R. M. (2009). Use of high potency cannabis is associated with a greater risk of psychosis. Br. J. Psychiatry 195, 488–491.
D’Souza, D. C., Abi-Saab, W. M., Madonick, S., Forselius-Bielen, K., Doersch, A., Braley, G., Gueorguieva, R., Cooper, T. B., and Krystal, J. H. (2005). Delta-9-tetrahydrocannabinol effects in schizophrenia: implications for cognition, psychosis, and addiction. Biol. Psychiatry 57, 594–608.
D’Souza, D. C., Braley, G., Blaise, R., Vendetti, M., Oliver, S., Pittman, B., Ranganathan, M., Bhakta, S., Zimolo, Z., Cooper, T., and Perry, E. (2008). Effects of haloperidol on the behavioral, subjective, cognitive, motor, and neuroendocrine effects of Delta-9-tetrahydrocannabinol in humans. Psychopharmacology 198, 587–603.
D’Souza, D. C., Perry, E., MacDougall, L., Ammerman, Y., Cooper, T., Yu-Te, W., Braley, G., Gueorguieva, R., and Harrison, K. J. (2004). The psychotomimetic effects of intravenous delta-9-tetrahydrocannabinol in healthy individuals: implications for psychosis. Neuropsychopharmacology 29, 1558–1572.
Espelin, D. E., and Done, A. K. (1968). Amphetamine poisoning: effectiveness of chiorpromazine. N. Engl. J. Med. 278, 1361–1365.
European Monitoring Centre for Drugs, and Drug Addiction Annual Report. (2009). The State of the Drugs Problem in Europe. Luxembourg: Publications Office of the European Union 2009, 99.
Fergusson, D. M., Horwood, L. J., and Swain-Campbell, N. R. (2003). Cannabis dependence and psychotic symptoms in young people. Psychol. Med. 33, 15–21.
Fergusson, D. M., Poulton, R., Smith, P. F., and Boden, J. M. (2006). Cannabis and psychosis. BMJ 332, 172–175.
Franzen, F., and Gross, H. (1965). Tryptamine, N,N-dimethyltryptamine, N,N-dimethyl-5-hydroxytryptamine and 5-methoxytryptamine in human blood and urine. Nature 206, 1052.
Freiman, I., Anton, A., Monyer, H., Urbanski, M. J., and Szabo, B. (2006). Analysis of the effects of cannabinoids on identified synaptic connections in the caudate-putamen by paired recordings in transgenic mice. J. Physiol. 575, 789–806.
Freund, T. F., Katona, I., and Piomelli, D. (2003). Role of endogenous cannabinoids in synaptic signaling. Physiol. Rev. 83, 1017–1066.
Gerdeman, G. L., Ronesi, J., and Lovinger, D. M. (2002). Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat. Neurosci. 5, 446–451.
Gillin, J. C., Kaplan, J., Stillman, R., and Wyall, R. J. (1976). The psychedelic model of schizophrenia: the case of N,N-dimethyltryptamine. Am. J. Psychiatry 133, 203–208.
Giuffrida, A., Parsons, L. H., Kerr, T. M., Rodriguez de Fonseca, F., Navarro, M., and Piomelli, D. (1999). Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat. Neurosci. 2, 358–363.
Glennon, R. A., Rosecrans, J. A., and Young, R. (1983). Drug-induced discrimination: a description of the paradigm and a review of its specific application to the study of hallucinogenic agents. Med. Res. Rev. 3, 289–340.
Glenthøj, B. Y., and Hemmingsen, R. (1997). Dopaminergic sensitization: implications for the pathogenesis of schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 21, 22–46.
Gonzalez-Maeso, J., and Sealfon, S. (2009). Psychedelics and schizophrenia. Trends Neurosci. 32, 225–232.
Graybiel, A. M. (2005). The basal ganglia: learning new tricks and loving it. Curr. Opin. Neurobiol. 15, 638–644.
Green, B., Young, R., and Kavanagh, D. (2005). Cannabis use and misuse prevalence among people with psychosis. Br. J. Psychiatry 187, 306–313.
Greifenstein, F. E., Yoskitake, J., DeVault, M., and Gajewski, J. E. (1958). A study of 1-aryl-cyclohexylamine for anesthesia. Anesth. Analg. 37, 283–294.
Grelotti, D. G., Kanayama, G., and Pope, H. G. (2010). Remission of persistent methamphetamine-induced psychosis after electroconvulsive therapy. Am. J. Psychiatry 167, 17–23.
Gurevich, E. V., and Joyce, J. N. (1997). Alterations in the cortical serotonergic system in schizophrenia: a postmortem study. Biol. Psychiatry 42, 529–545.
Henquet, C., Murray, R. M., Linszen, D., and van Os, J. (2005). The environment and schizophrenia: the role of cannabis use. Schizophr. Bull. 31, 608–612.
Herkenham, M., Lynn, A. B., Johnson, M. R., Melvin, L. S., de Costa, B. R., and Rice, K. C. (1991). Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J. Neurosci. 11, 563–583.
Houk, J. C., Bastianen, C., Fansler, D., Fishbach, A., Fraser, D., Reber, P. J., Roy, S. A., and Simo, L. S. (2007). Action selection and refinement in subcortical loops through basal ganglia and cerebellum. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 362, 1573–1583.
Huestis, M. A., Gorelick, D. A., Heishman, S. J., Preston, K. L., Nelson, R. A., Moolchan, E. T., and Frank, R. A. (2001). Blockade of effects of smoked marijuana by the CB1-selective cannabinoid receptor antagonist SR141716. Arch. Gen. Psychiatry 58, 322–328.
Iyo, M., Sekine, Y., and Mori, N. (2004). Neuromechanism of developing methamphetamine psychosis: a neuroimaging study. Ann. N. Y. Acad. Sci. 1025, 288–329.
Javitt, D. C., and Zukin, S. R. (1990). Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 148, 1301–1308.
Johnstone, M., Evans, V., and Baigel, S. (1958). Sernyl (Cl-395) in clinical anesthesia. Br. J. Anaesth. 31, 433–439.
Joyce, J. N., Shane, A., Lexow, N., Winokur, A., Casanova, M., and Kleinmann, J. E. (1993). Serotonin uptake sites and serotonin receptors are altered in the limbic system of schizophrenics. Neuropsychopharmacology 8, 315–336.
Kapur, D., and Mamo, D. (2003). Half a century of antipsychotics and still a central role for opamine D2 receptors. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 1981–1990.
Kim, J. S., Kornhuber, H. H., Schmid-Burgk, W., and Holzmuller, B. (1980). Low cerebrospinal fluid glutamate in schizophrenic patients and a new hypothesis on schizophrenia. Neurosci. Lett. 20, 379–382.
Kreitzer, A. C., and Malenka, R. C. (2005). Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J. Neurosci. 25, 10537–10545.
Kreitzer, A. C., and Malenka, R. C. (2007). Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature 445, 643–647.
Kreitzer, A. C., and Malenka, R. C. (2008). Striatal plasticity and basal ganglia circuit function. Neuron 60, 543–554.
Laruelle, M., Abi-Dargham, A., van Dyck, C. H, Gil, R., D’Souza, C. D., Erdos, J., McCance, E., Rosenblatt, W., Fingado, C., Zoghbi, S. S., Baldwin, R. M., Seibyl, J. P., Krystal, J. H., Charney, D. S., and Innis, R. B. (1996). Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc. Natl. Acad. Sci. U.S.A. 93, 9235–9240.
Lesscher, H. M., Hoogveld, E., Burbach, J. P., van Ree, J. M., and Gerrits, M. A. (2005). Endogenous cannabinoids are not involved in cocaine reinforcement and development of cocaine-induced behavioural sensitization. Eur. Neuropsychopharmacol. 15, 31–37.
Leweke, F. M., Giuffrida, A., Wurster, U., Emrich, H. M., and Piomelli, D. (1999a). Elevated endogenous cannabinoids in schizophrenia. Neuroreport 10, 1665–1669.
Leweke, F. M., Schneider, U., Thies, M., Münte, T. F., and Emrich, H. M. (1999b). Effects of synthetic D9-tetrahydrocannabinol on binocular depth inversion of natural and artificial objects in man. Psychopharmacology 142, 230–235.
Leweke, F. M., Schneider, U., Radwan, M., Schmidt, E., and Emrich, H. M. (2000). Different effects of nabilone and cannabidiol on binocular depth inversion in man. Pharmacol. Biochem. Behav. 66, 175–181.
Lieberman, J. A., Kane, J. M., and Alvir, J. (1987). Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacology 91, 415–433.
Lieberman, J. A, Mailman, R. B., Duncan, G., Sikich, L., Chakos, M., Nichols D. E., and Kraus, J. E. (1998). Serotonergic basis of antipsychotic drug effects in schizophrenia. Biol. Psychiatry 44, 1099–1117.
Ljeberman, J. A., Klnon, B. J., and Loebel, A. D. (1990). Dopaminergic mechanisms in idiopathic and drug-induced psychoses. Schizoph. Bull. 16, 97–110.
Long, L. E., Malone, D. T., and Taylor, D. A. (2006). Cannabidiol reverses MK-801-induced disruption of prepulse inhibition in mice. Neuropsychopharmacology 31, 795–803.
Luby, E. D., Cohen, B. D., Rosenbaum, F., Gottlieb, J., and Kelley, R. (1959). Study of a new schizophrenomimetic drug, sernyl. Arch. Neurol. Psychiatry 81, 363–369.
Luby, E. D., Gottlieb, J. S., Cohen, B. D., Rosenbaum, G., and Domino, E. F. (1962). Model psychoses and schizophrenia. Am. J. Psychiatry 119, 61–67.
Lupica, C. R., and Riegel, A. C. (2005). Endocannabinoid release from midbrain dopamine neurons: a potential substrate for cannabinoid receptor antagonist treatment of addiction. Neuropharmacology 48, 1105–1116.
MacDonald, A. W., and Schulz, S. C. (2009). What we know: findings that every theory of schizophrenia should explain. Schizophr. Bull. 35, 493–508.
Manske, R. (1931). A synthesis of the methyltryptamines and some derivatives. Can. J. Res. 5, 592–600.
Matsuda, L. A., Lolait, S. J., Brownstein, M. J., Young, A. C., and Bonner, T. I. (1990). Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346, 561–564.
Matyas, F., Urban, G. M., Watanabe, M., Mackie, K., Zimmer, A., Freund, T. F., and Katona, I. (2008). Identification of the sites of 2-arachidonoylglycerol synthesis and action imply retrograde endocannabinoid signaling at both GABAergic and glutamatergic synapses in the ventral tegmental area. Neuropharmacology 54, 95–107.
Mc Grath, J., Welham, J., Scott, J., Varghese, D., Degenhardt, L., Hayatbakhsh, M. R., Alati, R., Williams, G. M., Bor, W., and Najman, J. M. (2010). Association between cannabis use and psychosis-related outcomes using sibling pair analysis in a cohort of young adult. Arch. Gen. Psychiatry 67, 440–447.
McGraw-Hill. (1993). Encyclopedia of Science and Technology, 5th Edn. New York: The McGraw-Hill Companies Inc.
Melis, M., Perra, S., Muntoni, A. L., Pillolla, G., Lutz, B., Marsicano, G., Di Marzo, V., Gessa, G. L., and Pistis, M. (2004). Prefrontal cortex stimulation induces 2-arachidonoyl-glycerol-mediated suppression of excitation in dopamine neurons. J. Neurosci. 24 , 10707–10715.
Meltzer, H. Y. (1999). The role of serotonin in antipsychotic drug action. Neuropsychopharmacology 21, 106s–115s.
Meltzer, H. Y., Arvanitis, L., Bauer, D., and Rein, W. (2004). Placebo-controlled evaluation of four novel compounds for the treatment of schizophrenia and schizoaffective disorder. Am. J. Psychiatry 161, 975–984.
Mita, T., Hanada, S., Nishino, N., Kuno, T., Nakai, H., Yamdori, T., Mizoi, Y., and Tanaka, C. (1986). Decreased serotonin S2 and increased dopamine D2 receptors in chronic schizophrenics. Biol. Psychiatry 21, 1407–1414.
Moore, T. H. M., Zammit, S., Lingford-Hughes, A., Barnes, T. R. E., Jones, P. B., Burke, M., and Lewis, G. (2007). Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet 370, 319–328.
Moreira, F. A., and Guimaraes, F. S. (2005). Cannabidiol inhibits the hyperlocomotion induced by psychotomimetic drugs in mice. Eur. J. Pharmacol. 512, 199–205.
Morrison, P. D., Zois, V., McKeown, D. A., Lee, T. D., Holt, D. W., Powell, J. F., Kapur, S., and Murray, R. M. (2009). The acute effects of synthetic intravenous Delta9-tetrahydrocannabinol on psychosis, mood and cognitive functioning. Psychol. Med. 39 , 1607–1616.
Murray, R. M., Lappin, J., and Di Forti, M. (2008). Schizophrenia: from developmental deviance to dopamine dysregulation. Eur. Neuropsychopharmacol. 18, S129–S134.
Murray, R. M., Oon, M. C. H., Rodnight, R., Birley, J. L. T., and Smith, A. (1979). Increased excretion of dimethyltryptamine and certain features of psychosis. A possible association. Arch. Gen. Psychiatry 36, 644–664.
Narasimhachari, N., Avalos, J., Fujimori, M., and Himwich, H. E. (1972). Studies of drugfree schizophrenics and controls. Biol. Psychiatry 5, 311–318.
Patil, S. T., Zhang, L., Martenyi, F., Lowe, S. L., Jackson, K. A., Andreev, B. V., Avedisova, A. S., Bardenstein, L. M., Gurovich, I. Y., Morozova, M. A., Mosolov, S. N., Neznanov, N. G., Reznik, A. M., Smulevich, A. B., Tochilov, V. A., Johnson, B. G., Monn, J. A., and Schoepp, D. D. (2007). Activation of mGluR2/3 receptors as a new approach to treat schizophrenia: a randomized phase 2 clinical trial. Nat. Med. 13, 1102–1107.
Peleg-Raibstein, D., Yee, B. J., and Hauser, J. (2009). The amphetamine sensitization model of schizophrenia: relevance beyond psychotic symptoms? Psychopharmacology 206, 603–621.
Pertwee, R. G. (2007). The diverse CB(1) and CB(2) receptor pharmacology of three plant cannabinoids: delta(9)- tetrahydrocannabinol, cannabidiol and Delta(9)-tetrahydrocannabivarin. Br. J. Pharmacol. 153, 199–215.
Pomarol-Clotet, E., Honey, G. D., Murray, G. K., Corlett, P. R., Abasalom, A. R., Lee, M., McKenna, P. J., Bullmore, E. T., and Fletcher, P. C. (2006). Psychological effects of ketamine in healthy volunteers. Br. J. Psychiatry 189, 173–179.
Potter, D. J., Clark, P., and Brown, M. B. (2008). Potency of delta 9-THC and other cannabinoids in cannabis in England in 2005: implications for psychoactivity and pharmacology. J. Forensic Sci. 53, 90–94.
Rasmussen, H., Erritzoe, D., Andersen, R., Ebdrup, B. H., Aggernaes, B., Oranje, B., Kalbitzer, J., Madsen, J., Pinborg, L. H., Baare’, W., Svarer, C., Lublin, H., Knudsen, G. M., and Glenthoj, B. (2010). Decreased frontal serotonin2A receptor binding in antipsychotic-naive patients with first-episode schizophrenia. Arch. Gen. Psychiatry 67, 9–16.
Robbe, D., Kopf, M., Remaury, A., Bockaert, J., and Manzoni, O. J. (2002). Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc. Natl. Acad. Sci. U.S.A. 99, 8384–8388.
Rodriguez, M. A. (2007). A methodology for studying various interpretations of the N,N-dimethyltryptamine-induced alternate reality. Nat. Neurosci. 21, 67–84.
Rosenbaum, G., Cohen, B. D., Luby, E. D., Gottlieb, J. S., and Yelen, D. (1959). Cornparisons of Sernyl with other drugs. Arch. Gen. Psychiatry 1, 651–656.
Seeman, P., and Tallerico, T. (2005). Dopamine receptor contribution to the action of PCP, LSD and ketamine psychotomimetics. Mol. Psychiatry 10, 877–883.
Shaw, E., and Woolley, D. W. (1956). Some serotonin-like activities of lysergic acid diethylamide. Science 124, 121.
Smith, M. J., Thirthalli, J., Abdallah, A. B., Murray, R. M., and Cottler, L. B. (2009). Prevalence of psychotic symptoms in substance users: a comparison across substances. Compr. Psychiatry 50, 245–250.
Snyder, E. M., and Murphy, M. R. (2008). Schizophrenia therapy: beyond atypical antipsychotics. Nat. Rev. 7, 471–472.
Snyder, S. H. (1972). Catecholamines in the brain as mediators of amphetamine psychosis. Arch. Gen. Psychiatry 27, 169–179.
Stokes, P. R., Mehta, M. A., Curran, H. V., Breen, G., and Grasby, P. M. (2009). Can recreational doses of THC produce significant dopamine release in the human striatum? Neuroimage 48, 186–190.
Stokes, P. R. A., Egerton, A., Watson, B., Reid, A., Breen, G., Lingford-Hughes, A., Nutt, D. J., and Mehta, M. A. (2010). Significant decreases in frontal and temporal [11C]-raclopride binding after THC challenge. Neuroimage 52, 1521–1527.
Strassman, R. J. (1984). Adverse reaction to psychedelic drugs. A review of the literature. J. Nerv. Ment. Dis. 172, 577–595.
Strassman, R. J. (1991). Human hallucinogenic drug research in the United States: a present-day case history and review of the process. J. Psychoactive Drug 23, 29–38.
Szara, S. (1956). Dimethyltryptamine: its metabolism in man: the relation of its psychotic effect to serotonin metabolism. Experientia 12, 441–442.
Tamminga, C. A., and Holcomb, H. H. (2005). Phenotype of schizophrenia: a review and formulation. Mol. Psychiatry 10, 27–39.
Tandon, R., Keshavan, M. S., and Nasrallah, H. A. (2008). Schizophrenia, “Just the Facts” What we know in 2008. Epidemiology and etiology. Schizophr. Res. 102, 1–18.
Thiemann, G., Di Marzo, V., Molleman, A., and Hasenohrl, R. U. (2008a). The CB(1) cannabinoid receptor antagonist AM251 attenuates amphetamine-induced behavioural sensitization while causing monoamine changes in nucleus accumbens and hippocampus. Pharmacol. Biochem. Behav. 89, 384–391.
Thiemann, G., van der Stelt, M., Petrosino, S., Molleman, A., Di Marzo, V., and Hasenohrl, R. U. (2008b). The role of the CB1 cannabinoid receptor and its endogenous ligands, anandamide and 2-arachidonoylglycerol, in amphetamine-induced behavioural sensitization. Behav. Brain Res. 187, 289–296.
Tsuang, M. T., Bar, J. L., Stone, W. S., and Faraone, S. V. (2004). Gene–environment interactions in mental disorders. World Psychiatry 3, 73–83.
Uchigashima, M., Narushima, M., Fukaya, M., Katona, I., Kano, M., and Watanabe, M. (2007). Subcellular arrangement of molecules for 2-arachidonoyl-glycerol-mediated retrograde signaling and its physiological contribution to synaptic modulation in the striatum. J. Neurosci. 27, 3663–3676.
United Nations Office on Drugs, and Crime (UNODC). (2009). “Patterns and trends of amphetamine-type stimulants and other drugs in East and South-East Asia (and neighbouring regions),” in Annual Report. Vienna: Vienna International Centre.
Van Os, J., Bak, M., Hanssen, M., Bij, R. V., de Graaf, R., and Verdoux, H. (2002). Cannabis use and psychosis: a longitudinal population-based study. Am. J. Epidemiol. 156 , 319–327.
Van Rossum, J. M. (1967). “The significance of dopamine-receptor blockade for the action of neuroleptic drugs,” in Neuropsychopharmacology, Proceedings 5th Collegium Internationale Neuropsychopharmacologicum, eds H. Brill, J. Cole, P. Deniker, H. Hippius, and P. B. Bradley (Amsterdam: Excerpta Medica Foundation), 321–329.
Vardy, M. M., and Kay, S. R. (1983). LSD psychosis or LSD-induced schizophrenia? A multimethod inquiry. Arch. Gen. Psychiatry 40, 877–883.
Vollenweider, F. X., Leenders, K. L., Scharfetter, C., Maguire, P., Stadelmann, O., and Angst, J. (1997). Positron emission tomography and fluorodeoxyglucose studies of metabolic hyperfrontality and psychopathology in the psilocybin model of psychosis. Neuropsychopharmacology 16, 357–372.
Vollenweider, F. X., Vollenweider-Scherpenhuyzen, M. F. I., Bäbler, A., Vogel, H., and Hell, D. (1998). Psilocybin induces schizophrenia-like psychosis in humans via a serotonin-2 agonist action. Neuroreport 9, 3897–3902.
Yin, H. H., and Knowlton, B. J. (2006). The role of the basal ganglia in habit formation. Nat. Rev. 7, 464–476.
Yin, H. H., and Lovinger, D. M. (2006). Frequency-specific and D2 receptor-mediated inhibition of glutamate release by retrograde endocannabinoid signaling. Proc. Natl. Acad. Sci. U.S.A. 103, 8251–8256.
Zammit, L. S., Allebeck, P., Andreasson, S., Lundberg, I., and Lewis, G. (2002). Self reported cannabis use as a risk factor for schizophrenia in Swedish conscripts of 1969: historical cohort study. BMJ 325, 1199–1212.
Zuardi, A., Crippa, J., Hallak, J., Pinto, J., Chagas, M., Rodrigues, G., Dursun, S. M., and Tumas, V. (2009). Cannabidiol for the treatment of psychosis in Parkinson’s disease. J. Psychopharmacol. 23, 979–983.
Zuardi, A. W. (2006). History of cannabis as a medicine: a review. Rev. Bras. Psiquiatr. 28, 153–157.
Zuardi, A. W., Rodrigues, J. A., and Cunha, J. M. (1991). Effects of cannabidiol in animal models predictive of antipsychotic activity. Psychopharmacology 104, 260–264.
Zuardi, A. W, Shirakawa, I., Finkelfarb, E., and Karniol, I. G. (1982). Action of cannabidiol on the anxiety and other effects produced by delta 9-THC in normal subjects. Psychopharmacology 76, 245–250.
Zuurman, L., Passier, P. C., de Kam, M. L., Kleijn, H. J., Cohen, A. F., and van Gerven, J. M. (2009). Pharmacodynamic and pharmacokinetic effects of the intravenous CB1 receptor agonist Org 26828 in healthy male volunteers. J. Psychopharmacol. 24, 1689–1696.
Keywords: LSD, schizophrenia, amphetamines, psychosis, cannabis, PCP
Citation: Paparelli A, Di Forti M, Morrison PD and Murray RM (2011) Drug-induced psychosis: how to avoid star gazing in schizophrenia research by looking at more obvious sources of light. Front. Behav. Neurosci. 5:1. doi: 10.3389/fnbeh.2011.00001
Received: 01 March 2010;
Paper pending published: 19 October 2010;
Accepted: 02 January 2011;
Published online: 17 January 2011.
Edited by:
Andreas Meyer-Lindenberg, Central Institute of Mental Health, GermanyReviewed by:
Andreas Meyer-Lindenberg, Central Institute of Mental Health, GermanyJoshua W. Buckholtz, Vanderbilt University, USA
Markus Leweke, Central Institute of Mental Health, Germany
Copyright: © 2011 Paparelli, Di Forti, Morrison and Murray. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Alessandra Paparelli, Institute of Psychiatry, King’s College London, De Crespigny Park, London SE5 8AF, UK. e-mail: alessandra.a.paparelli@kcl.ac.uk