Differential regulation of collapsin response mediator protein 2 (CRMP2) phosphorylation by GSK3ß and CDK5 following traumatic brain injury
 Sarah M. Wilson1
Sarah M. Wilson1  Ki Duk Park
Ki Duk Park Rajesh Khanna
Rajesh Khanna- 1Paul and Carole Stark Neurosciences Research Institute, Indiana University School of Medicine, Indianapolis, IN, USA
- 2Center for Neuro-Medicine, Brain Science Institute, Korea Institute of Science and Technology, Seoul, Korea
- 3Department of Pharmacology, College of Medicine, University of Arizona, Tucson, AZ, USA
Aberrant ion channel function has been heralded as a main underlying mechanism driving epilepsy and its symptoms. However, it has become increasingly clear that treatment strategies targeting voltage-gated sodium or calcium channels merely mask the symptoms of epilepsy without providing disease-modifying benefits. Ion channel function is likely only one important cog in a highly complex machine. Gross morphological changes, such as reactive sprouting and outgrowth, may also play a role in epileptogenesis. Mechanisms responsible for these changes are not well-understood. Here we investigate the potential involvement of the neurite outgrowth-promoting molecule collapsin response mediator protein 2 (CRMP2). CRMP2 activity, in this respect, is regulated by phosphorylation state, where phosphorylation by a variety of kinases, including glycogen synthase kinase 3 β (GSK3β) renders it inactive. Phosphorylation (inactivation) of CRMP2 was decreased at two distinct phases following traumatic brain injury (TBI). While reduced CRMP2 phosphorylation during the early phase was attributed to the inactivation of GSK3β, the sustained decrease in CRMP2 phosphorylation in the late phase appeared to be independent of GSK3β activity. Instead, the reduction in GSK3β-phosphorylated CRMP2 was attributed to a loss of priming by cyclin-dependent kinase 5 (CDK5), which allows for subsequent phosphorylation by GSK3β. Based on the observation that the proportion of active CRMP2 is increased for up to 4 weeks following TBI, it was hypothesized that it may drive neurite outgrowth, and therefore, circuit reorganization during this time. Therefore, a novel small-molecule tool was used to target CRMP2 in an attempt to determine its importance in mossy fiber sprouting following TBI. In this report, we demonstrate novel differential regulation of CRMP2 phosphorylation by GSK3β and CDK5 following TBI.
Introduction
Precise regulation of voltage- and ligand-gated ion channels is essential for proper function of both the peripheral and central nervous systems. As perturbations to these tightly-controlled systems can result in diverse neuropathologies, regulators of ion channel function have become prime targets for therapeutic intervention. We have previously demonstrated that the intracellular phosphoprotein collapsin response mediator protein 2 (CRMP2) is a positive regulator of both ligand- and voltage-gated calcium channels (Brittain et al., 2009, 2012a) and can be targeted as such to provide therapeutic relief (Brittain et al., 2011a,b; Wilson et al., 2012a). While phosphorylation of CRMP2 increases its interaction with the N-type voltage-gated calcium channel (Brittain et al., 2012b), we recently discovered a novel posttranslational modification (SUMOylation) that negatively impacts CRMP2's ability to enhance calcium influx (Ju et al., 2013). Intriguingly, through SUMOylation, a previously unidentified link between CRMP2 and trafficking of voltage-gated sodium channels was unearthed (Dustrude et al., 2013). CRMP2 is therefore in the unique position of potentially possessing the ability to impact voltage-gated calcium and sodium channels, as well as the ligand-gated N-methyl-D-aspartate (NMDA) receptor. A common pathology to which all three of these channel types are thought to contribute is epilepsy (for review see Ghasemi and Schachter, 2011; Oliva et al., 2012; Siwek et al., 2012).
Nearly 2.3 million people in the United States alone are burdened by epilepsy (CDC, 2012), a neurological condition classified by spontaneously recurring seizures (Goddard et al., 1969), with an estimated 150,000 more diagnosed each year (Hirtz et al., 2007; England et al., 2012). It is estimated that nearly half of epilepsy cases are classified as complex partial seizures, the majority of which originate from foci within the temporal lobe (Hauser and Kurland, 1975; Manford et al., 1992a,b; Larner, 1995; Panayiotopoulos, 2005). In many patients, temporal lobe epilepsy (TLE) is initiated by a traumatic event such as traumatic brain injury (TBI), febrile seizures, status epilepticus (SE), tumors, stroke, or infection (Kharatishvili and Pitkanen, 2010; Yang et al., 2010a; O'Dell et al., 2012). These events are often followed by an asymptomatic latency period lasting upwards of 10 years prior to the development of spontaneous recurring seizures (de Lanerolle et al., 2003; Sharma et al., 2007; Yang et al., 2010a). That these arguably diverse insults can lead to a similar phenotype suggests the possibility of shared epileptogenic mechanisms. The majority of antiepileptic drugs (AEDs) available today target the voltage-gated sodium channel. One of these, Lacosamide (Vimpat®) (R-N-benzyl 2-acetamido-3-methoxypropionamide) ((R)-LCM), does so through a unique mechanism. Instead of affecting current density or steady-state gating kinetics, (R)-LCM selectively enhances sodium channel slow inactivation (Errington et al., 2008). Another characteristic that sets (R)-LCM apart from other AEDs is its ability to target CRMP2 (Beyreuther et al., 2007; Park et al., 2009). Notably, expression levels of CRMP2 have been shown to alter the ability of (R)-LCM to impact sodium channel function (Wang et al., 2010).
Given CRMP2's remarkable ability to regulate ion channel function, it can be at times difficult to consider its many other functions, particularly those for which it was first identified (i.e., neurite outgrowth and guidance) (Goshima et al., 1995). Importantly, of the myriad of CRMP2 functions, it is the ability to promote neurite outgrowth that is impacted by (R)-LCM, not those associated with ion channel function (Wang and Khanna, 2011; Wilson et al., 2012b). CRMP2 promotes neurite outgrowth by two distinct mechanisms: (Brittain et al., 2009) binding and transporting tubulin dimers from the soma to distal projections (Fukata et al., 2002; Kimura et al., 2005) and (Brittain et al., 2012a) stabilizing the growing end of the microtubule by promoting the inherent GTPase activity of tubulin (Chae et al., 2009), the latter of which is impaired by (R)-LCM (Wilson et al., 2012b). Aberrant growth and reorganization of neuronal circuits, specifically that of the dentate mossy fibers within the hippocampus, is commonly observed in post-mortem tissue samples from TLE patients and in animal models of the disease (for review see Koyama and Ikegaya, 2004; Sutula, 2004). Notably, CRMP2 was recently suggested to be involved in mossy fiber sprouting in the SE model of TLE (Lee et al., 2012). Under normal conditions, mossy fibers project from the granule cell layer of the dentate gyrus into the CA3 region of the hippocampus where they form synapses with pyramidal cells (Andersen et al., 1969). During TLE, mossy fibers are observed to innervate the inner molecular layer where they synapse onto the dendrites of other dentate granule cells, leading to the formation of recurrent excitatory circuits (Blaabjerg and Zimmer, 2007). To date, the molecular mechanisms contributing to mossy fiber sprouting are relatively unknown. Recent focus has centered on the involvement of growth-factor cascades, particularly that of the tropomycin-related kinase receptor B (TrkB). Activation of the TrkB receptor eventually leads to the phosphorylation and inactivation of glycogen-synthase kinase β (GSK3β) by protein kinase B (Akt) (Cross et al., 1995; Alessi et al., 1996; Bhave et al., 1999). Inactivation of GSK3β has been observed following insults commonly associated with TLE, such as TBI (Shapira et al., 2007; Dash et al., 2011; Zhao et al., 2012), hypoxia-ischemia (Sasaki et al., 2001; Endo et al., 2006; Xiong et al., 2012), and SE (Lee et al., 2012).
The ability of CRMP2 to promote neurite outgrowth is governed by its phosphorylation state. Phosphorylation by a variety of kinases, including GSK3β, renders CRMP2 inactive (Arimura et al., 2000, 2005; Brown et al., 2004; Cole et al., 2004, 2006; Uchida et al., 2005, 2009; Yoshimura et al., 2005; Hou et al., 2009). Specifically, GSK3β phosphorylates CRMP2 at threonines 509, 514, and serine 518, thereby reducing its affinity for tubulin (Yoshimura et al., 2005). As inactivation of GSK3β is observed following TLE-related insults, it is possible that these insults may also lead to a decrease in GSK3β-phosphorylated (inactive) CRMP2, thereby promoting outgrowth. Indeed we have recently demonstrated that targeting CRMP2 with (R)-LCM can prevent the increased excitatory connectivity of layer V pyramidal neurons in the neocortical isolation (undercut) model of posttraumatic epileptogenesis (Wilson et al., 2012b). Therefore, if loss of CRMP2 phosphorylation is a contributing factor in mossy fiber sprouting, it may be possible to prevent this reorganization through the use of (R)-LCM and its derivatives. In this report we determine if GSK3β phosphorylation of CRMP2 is altered at various stages following a TLE-related insult, as well as, the impact of GSK3β inactivation on CRMP2 function. Ultimately, the role of CRMP2 in mossy fiber sprouting is investigated by selectively targeting CRMP2 function in vivo.
Lacosamide was originally discovered to be stereoselective, as much higher concentrations of the (S)- configuration are required to halt epileptiform activity both in vitro and in vivo compared to the (R)- configuration (Andurkar et al., 1999; LeTiran et al., 2001; Lees et al., 2006). In this report we employ the use of (S)-LCM, which retains the ability to target CRMP2-mediated neurite outgrowth without impacting sodium channel function, to determine if GSK3β phosphorylation of CRMP2 is altered at various stages following a TLE-related insult. Ultimately, the role of CRMP2 in mossy fiber sprouting is investigated by selectively targeting CRMP2 function in vivo.
Materials and Methods
Materials
All reagents were purchased from Sigma (St. Louis, MO, USA) unless otherwise indicated. (S)-LCM was provided by the laboratory of Dr. Ki Duk Park, Center for Neuro-Medicine, Brain Science Institute, Korea Institute of Science and Technology. A 100 mM solution was made up in dimethylsulfoxide (DMSO) and stored in small aliquots at −20°C. The final concentration of 200 μM was chosen as it phenocopied the effect of CRMP2 siRNA knockdown (data not shown).
Cortical Neuron Culture
Rat cortical neuron cultures were prepared from cortices dissected from embryonic day 19 (E19) rats as described (Goslin and Banker, 1989), with some modifications. Briefly, cortices were dissected out of E19 rats, and cells were dissociated enzymatically and mechanically (trituration through Pasteur pipette) in a Papain solution (12 U/ml; Worthington) containing Leibovitz's L-15 medium (Invitrogen), 0.42 mg/ml cysteine (Sigma), 250 U/ml DNase 1 (type IV; Sigma), 25 mM NaHCO3, penicillin (50 U/ml)/streptomycin (50 μg/ml), 1 mM sodium pyruvate, and 1 mg/ml glucose (Invitrogen). After dissociation, the cells were gently washed by sequential centrifugation in Neurobasal medium containing either 2 mg/ml or 20 mg/ml BSA and Pen/Strep, glucose, pyruvate, and DNase1 (as above) and then plated on poly-D-lysine-coated coverslips or 96-well plates at ~400 cells per mm2. Growth media (1 ml/well or 100 μl/well for 12- and 96-well plates, respecitvely) consisted of Neurobasal medium continaing 2% NuSerum, 2% NS21 (Chen et al., 2008), supplemented with penicillin/streptomycin (100 U/ml; 50 μg/ml), 0.1 mM L-Glutamine and 0.4 mML-glutamax (Invitrogen). 5-fluoro-2′-deoxyuridine (1.5 μg/mL) (Sigma) was added 48 h after plating to reduce the number of non-neuronal cells. After 4 days in culture and 2× each week thereon, half of the growth medium was replaced with medium without 5-fluoro-2′-deoxyuridine.
Neurite Outgrowth
Primary cortical neurons plated on 96-well culture plates were transfected via lipofectamine 2000 (Invitrogen) with EGFP at 4 DIV 48 h before imaging with the ImageXpress Micro (Molecular Devices). Immediately prior to imaging, media was exchanged with sterile phosphate buffered saline (PBS). The overexpression of EGFP allowed for visualization of a small percentage of neurons while maintaining optimal cell densities required for survival. EGFP fluorescence was imaged at 4× magnification. To enable laser-based autofocus, laser offset was determined via z-stack. Optimum exposure time was also determined to prevent saturation.
Analysis of neurite outgrowth was completed using a neurite outgrowth analysis protocol within the MetaXpress software (Molecular Devices). Cell soma and processes are detected by defining separate size and fluorescence intensity threshold parameters. Cells were excluded if they were determined not to be neurons based on morphology, if processes extended beyond the image field, or if no processes were longer than 50 μm. The following parameters are recorded and summarized into a final “total outgrowth” parameter: number of processes, number of branches, mean process length, and maximum process length.
Whole-Cell Patch-Clamp Recordings
Whole-cell voltage recordings were performed at RT on primary cultured cortical neurons (7 DIV) using an EPC 10 Amplifier (HEKA Electronics). Electrodes were pulled from thin-walled borosilicate glass capillaries (Warner Instruments) with a P-97 electrode puller (Sutter Instrument) such that the final electrode resistances were 2–3 MΩ when filled with internal solutions. The internal solution for recording Na+ currents contained (in mM): 110 CsCl, 5 MgSO4, 10 EGTA, 4 ATP Na2, and 25 HEPES (pH 7.2, 290–310 mOsmo/L). For recording Na+ currents, the external solution contained (in mM): 100 NaCl, 10 tetraethylammonium chloride (TEA-Cl), 1 CaCl2, 1 CdCl2., 1 MgCl2, 10 D-glucose, 4 4-AP, 0.1 NiCl2, 10 HEPES (pH 7.3, 310–315 mOsm/L). Whole-cell capacitance and series resistance (70–80%) were compensated with the amplifier. Cells were considered only when the seal resistance was more than 1 GΩ and the series resistance was less than 10 MΩ. Linear leak currents were digitally subtracted by P/4.
Immunoblot Assay and Quantification
Protein samples were boiled in Laemmli sample buffer for 5 min and fractionated on 4–15% separating SDS polyacrylamide gels. Apparent molecular weights were determined using broad range standards (Fisher). Following electrophoresis, proteins were transferred to PVDF membranes (Invitrogen) for immunoblotting. Membranes were occasionally stained with ponceau (BioRad) to monitor transfer efficiency. Following transfer, membranes were blocked for 1 h in 5% skim milk powder +0.05% BSA in TBST at RT. Primary antibody incubations [goat anti-rabbit GSK3β and GSK3β pSer9 (Millipore), goat anti-rabbit CRMP2 (Sigma), donkey anti-sheep CRMP2 pThr509/514 (Kinasource), goat-anti-rabbit CRMP2 pSer522 (ECM Biosciences), goat anti-rabbit CDK5 (Cell Signaling), or goat anti-mouse βIII-tubulin (Promega)] were either 2 h at RT or overnight at 4°C. Membranes were extensively washed in TBST and incubated in secondary antibody [goat anti-rabbit, goat anti-mouse, or donkey anti-sheep IgG horseradish peroxidase (HRP)] (G Biosciences) or (goat anti-rabbit, goat anti-mouse IgG dylight 650 or 800 conjugated) (Pierce) (1:15,000). Membranes incubated with HRP-conjugated secondary antibodies were washed extensively in TBST prior to probing with Enhanced Chemiluminescence Western blotting substrate (Fisher) before exposure to photographic film. Blots were exposed for a range of durations to ensure the generation of a print in which the film is not saturated. Membranes incubated with dylight-conjugated secondary antibodies were washed extensively in TBST prior to imaging with the LI-COR Odyssey imaging system. Both images obtained from film and LI-COR were digitized and quantified using Un-Scan-It gel V6.1 scanning software (Silk Scientific Inc., Orem), limiting our analysis to the linear range. Immunoblot images were digitized and analyzed using UnscanIt software, limiting analysis to the digital range. Briefly, densitometric values were obtained for each band and normalized to a loading control (tubulin) for each respective sample. In place of a single background measurement, background values for each band were obtained from an directly adjacent area to avoid issues of background discrepancy. To better allow for direct comparisons, measurements for CDK5- and GSK3β-phosphorylated CRMP2 were compared to values for total CRMP2 and Tubulin from the same immunoblot following a stripping and reprobing procedure. The same method was also applied for comparing phosphorylated-GSK3 to total GSK3 and tubulin. While this method provides a direct comparison between levels of each protein within a single, loaded sample, the stripping procedure does decrease the esthetic quality of the immunoblot.
Traumatic Brain Injury
All procedures involving animals were approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine and were carried out according to NIH guidelines and regulations. Animals were doubly-housed and maintained in a 12 h light/12 h dark cycle environment with access to foor and water ad libitum. Adult male Sprague–Dawley rats (275–300 g) were subjected to controlled cortical impact (CCI) injury. Rats were anesthetized with a ketamine/xylazine mixture (80 and 5 mg/kg, respectively) and placed in a stereotaxic frame prior to TBI. Using sterile procedures, the skin was retracted, and a ~4 mm craniotomy was performed ~3 mm lateral to midline and 3 mm posterior to the bregma suture. The skullcap was removed without disruption of the dura. The impacting tip was angled on a medial-lateral plane so that it was perpendicular to the exposed cortical surface. The deformation impact depth was set a 1.5 mm, and the piston velocity was controlled at 3.0 m/s. Following impact, the exposed tissue was covered with bone wax (Henry Schein) and the midline incision was sutured with 5.0 monofilament (Ethicon). Following surgery, animals received a bolus of sterile saline and post-operative analgesic Buprenorphine (0.5 mg/kg). During all surgical procedures and recovery, the core body temperature of the animals were maintained at 36–37°C. Sham animals received the same craniotomy and post-operative care.
In vivo Administration of (S)-Lacosamide ((S)-LCM)
To provide continuous infusion, (S)-LCM was delivered via an implanted osmotic mini-pump (Alzet). To compensate for animal growth over the 4-week treatment period, the animals were weighed prior to surgery and the amount of (S)-LCM was adjusted to account for the expected weight gain and an infusion rate of 2.5 μl/h to allow for administration of an average of 5 mg/kg per day of (S)-LCM or <0.01% DMSO for vehicle. Immediately following CCI surgery, sterile mini-pumps were subcutaneously implanted. An incision was made on the back, between the shoulder blades. A small pocket was created by carefully separating skin from muscle near the incision site. The mini-pump was placed into the pocket and the incision was closed with 5.0 monofilament.
Tissue Processing
For immunoblots: at 24 h or 4 weeks post-TBI, animals were sacrificed and transcardially perfused with 0.1 M phosphate buffer. For perfusion, an incision is made in the left ventricle to allow insertion of a needle attached to a peristaltic pump through the ventricle and into the ascending aorta. The needle was clamped into position and a second incision was made in the right atrium for drainage. Following perfusion, brains were extracted and hippocampi ipsilateral and contralateral to the injury site were dissected, frozen in liquid nitrogen, and stored at −80°C. Prior to immunoblot assay, tissue was thawed and homogenized using a sonicator.
For TIMM staining: at 4 weeks following TBI, animals were sacrificed and transcardially perfused (as previously described) with a sodium sulfide perfusate solution (150 mM NaS2, 8.1 mM Na2HPO4, 1.9 mM NaH2PO4), followed by 4% paraformaldehyde. Following perfusion, brains were extracted and placed in 4% paraformaldehyde for 24 h at 4°C. Brains were then transferred to 0.1 M phosphate buffer + 30% sucrose for 48 h at 4°C. Tissue was embedded into Optimal Cutting Temperature (OCT) compound (Tissue-Tek) on dry ice. Coronal slices (35 μm thickness) were made on a cryostat (Leica). Slices were mounted onto gelatin-coated microscope slides and stored at −20°C.
TIMM Staining
Tissue sections were allowed to thawed and processed for TIMM staining with the RAPID TIMM Stain Kit (FD Neurotechnologies). Tissue sections were washed in 0.1 M phosphate buffer 3 times, 3 min each and transferred to the TIMM solution (132 mM Citric Acid, 79.5 mM Sodium Citrate, 153.6 mM Hydroquinone, 5 mM AgNO3, 30% Gum Arabic), where they were rocked gently in the dark for 45–60 min at 30°C. Sections were then rinsed in ddH2O for 3 min in the dark, followed by gently washing them in running water for 30 min to remove excess stain. Sections were dehydrated in 50, 75, and 95% ethanol for 3 min each. Sections were incubated in absolute ethanol 3 times, 3 min each and then cleared in xylene (Fisher) 3 times, 3 min each. Coverslips were added using a resinous mounting medium (Aquamount) (Fisher).
Both low and higher magnification images were obtained using a light microscope (Nikon 90i) and scored by three observers blinded to the conditions, based on the scale originally established by Cavazos et al. (1991). Briefly, the scoring system ranks TIMM staining on a scale of 0–5, with 0 being the absence of TIMM granules within the supragranular region and 5 indicating the existence of a dense band of TIMM granules within the supragranular region, extending into the inner molecular layer. To avoid issues of variance among animals, scores were compared from contralateral and ipsilateral hippocampi from the same animal to yield the difference in TIMM scoring (Ispilateral—Contralateral).
Data Analysis
All data points are shown as mean ± s.e.m. Statistical differences between control and experimental conditions were determined by using ANOVA with a Dunnett's or Tukey's post-hoc test or a Student's t-test when comparing only two conditions. Values of p < 0.05 were judged to be statistically significant.
Results
In regards to its outgrowth-promoting function, the activity of CRMP2 is regulated by its phosphorylation state. In the unphosphorylated form, CRMP2 is considered active and thereby growth-promoting; however, upon phosphorylation by a variety of kinases, most notably GSK3β, CRMP2 is rendered inactive (Arimura et al., 2000, 2005; Brown et al., 2004; Cole et al., 2004, 2006; Uchida et al., 2005, 2009; Yoshimura et al., 2005; Hou et al., 2009). Tonically active under naïve conditions, GSK3β is inactivated following insults commonly associated with TLE such as TBI (Shapira et al., 2007; Dash et al., 2011; Zhao et al., 2012), hypoxia-ischemia (Sasaki et al., 2001; Endo et al., 2006; Xiong et al., 2012), and SE (Lee et al., 2012). This inactivation of GSK3β may lead to an overall decrease in the level of phosphorylated (inactive) CRMP2, thereby promoting neurite outgrowth.
GSK3β Phosphorylation of CRMP2 Under Naïve Conditions
In order for inactivation of GSK3β to impact CRMP2 function, a proportion of CRMP2 must be phosphorylated by GSK3β under normal conditions. Additionally, changes in CRMP2 phosphorylation by GSK3β should occur on a relatively fast time-scale. To determine the extent of GSK3β phosphorylation of CRMP2, primary cultured cortical neurons were exposed to the GSK3β inhibitor Lithium Chloride (LiCl) (10 mM) or the protein phosphatase inhibitor okadaic acid (200 nM) for 18–24 h (Figure 1A). Western blot analysis was performed with an antibody specific to GSK3β-mediated phosphorylation of CRMP2 at Thr509 and Thr514. Importantly, CRMP2 appears to be phosphorylated by GSK3β under naïve/control conditions (Figures 1B,C). Prevention of dephosphorylation by okadaic acid increased CRMP2 phosphorylation by ~2-fold (12.1 ± 1.3) compared to control (6.6 ± 0.6), while LiCl-mediated inhibition of GSK3β resulted in an ~90% loss of CRMP2 phosphorylation (0.6 ± 0.3) (p < 0.05) (Figures 1B,C). Levels of total CRMP2 protein remained unchanged (control: 0.31 ± 0.03; okadaic acid: 0.41 ± 0.05; and LiCl: 0.37 ± 0.03) (p > 0.05) (Figure 1D). This data suggests that a proportion of CRMP2 is phosphorylated by GSK3β under normal conditions and loss of GSK3β activity dramatically dramatically results in a near-complete loss of phosphorylated CRMP2 within 18–24 h. GSK3β phosphorylation of CRMP2 appears to be dynamically regulated, as evidenced by active dephosphorylation under control conditions.
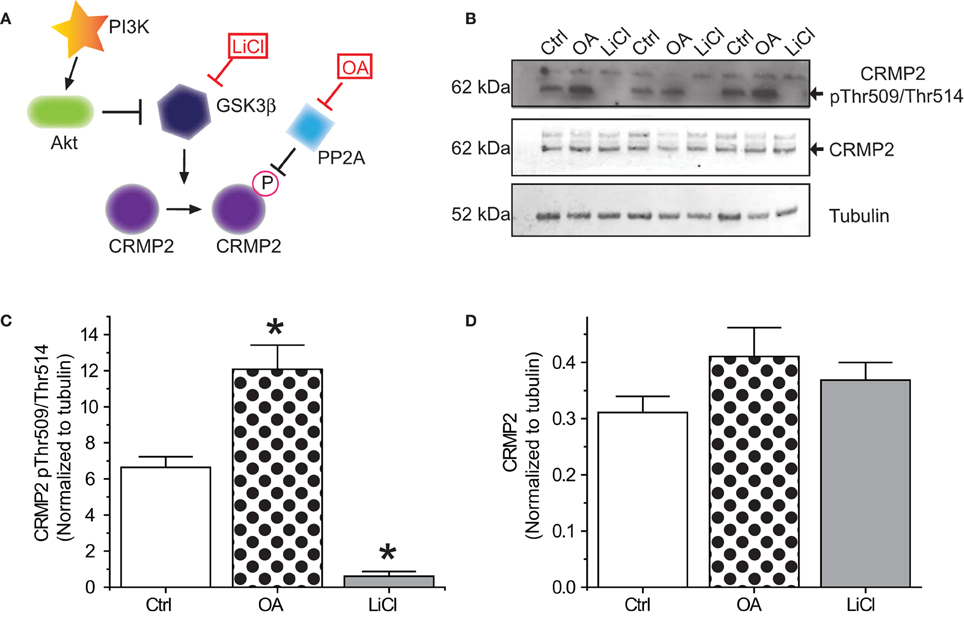
Figure 1. Phosphorylation of CRMP2 by GSKβ. (A) Signaling cascade involved in changes in phosphorylation of CRMP2 by GSK3β. Inactivation of GSK3β occurs following activation of Akt or exogenous exposures to lithium chloride (LiCl). Dephosphorylation of CRMP2 by PP2A is prevented by exposure to okadaic acid (OA). (B) Levels of GSK3β-phosphorylated CRMP2 and total CRMP2 following 24 h treatment with LiCl (10 mM) or OA (200 nM). (C,D) Summary of western blot analysis of GSK3β-phosphorylated CRMP2 and total CRMP2 levels in cortical neurons ± LiCl or OA (*p < 0.05 vs. Ctrl, One-Way ANOVA, Dunnet's post-hoc analysis) (values represent mean ± s.e.m.) (n = 3).
GSK3β Inhibition Increases Neurite Outgrowth via CRMP2
In regards to its ability to promote neurite outgrowth, phosphorylation by GSK3β effectively inactivates CRMP2 by reducing its affinity for tubulin (Yoshimura et al., 2005). Therefore, inactivation of GSK3β should be growth promoting. To determine if the LiCl-induced loss of GSK3β-phosphorylated CRMP2 directly alters CRMP2-mediated neurite outgrowth within the same timeframe, EGFP-transfected cortical neurons were exposed to lithium chloride for 18–24 h to determine the effect of GSK3β inhibition on neurite outgrowth (Figure 2A). Immediately following exposure, neurons were imaged using the ImageXpress Micro system and neurite outgrowth was determined via the MetaXpress software system. As expected, inhibition of GSK3β increased total outgrowth (154.5 ± 5.9) compared to controls (99.8 ± 4.0) (p < 0.05) (Figures 2B–D). We had previously demonstrated that (S)-LCM, the inactive enantiomer of (R)-LCM, retains the ability to target CRMP2 function and can therefore be used in place of knockdown strategies to determine the role of CRMP2 in a particular process (Wilson and Khanna, unpublished). To ensure that the LiCl-induced increase in outgrowth was in fact due to changes in CRMP2 activity, the experiment was repeated in the presence of (S)-LCM (200 μM). As (S)-LCM alone decreases outgrowth, it was included in both LiCl-treated and control conditions. Without (S)-LCM, LiCl increased neurite outgrowth by 54.9 ± 5.9% (Figure 2D). In contrast, in the presence of (S)-LCM, LiCl increased neurite outgrowth by 15.3 ± 5.1% (p < 0.05) (Figures 2D,E).
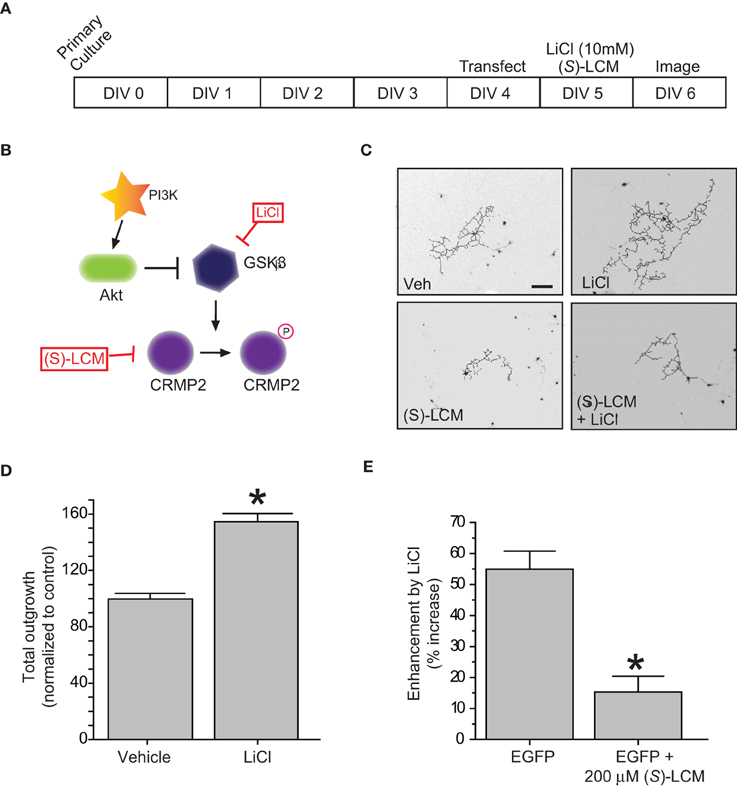
Figure 2. Inactivation of GSK3β enhances neurite outgrowth in a CRMP2-dependent manner. (A) Experimental timeline. Cortical neurons were transfected with EGFP at 4 DIV and exposed to vehicle (<0.01% DMSO), LiCl (10 mM), (S)-LCM (200 μM), or LiCl + (S)-LCM for 24 h starting at 5 DIV and imaged at 6 DIV. (B) GSK3β signaling cascade. (C) Representative tracings of neurons transfected with EGFP and exposed to LiCl, (S)-LCM, or both. (Scale bar = 150 μm). (D) Total outgrowth of neurons exposed to LiCl for 24 h (*p < 0.05, student's t-test) (values represent mean ± s.e.m.). (E) Enhancement of outgrowth by LiCl under conditions of (S)-LCM treatment (*p < 0.05 vs. EGFP, One-Way ANOVA, Dunnet's post-hoc analysis) (values represent mean ± s.e.m.) (n = 92–150 cells from 8 separate culture wells).
(S)-LCM does not Target Voltage-Gated Sodium Channels
It is essential to verify that (S)-LCM is unable to impact voltage-gated sodium channels in this system. Therefore, sodium channel slow inactivation of cultured cortical neurons was determined by holding cells at −70 mV, conditioning to potentials ranging from −100 to +20 mV (in +10 mV increments) for 5 s, moved to a hyperpolarizing pulse of −120 mV for 150 ms to allow fast-inactivated channels to recover, and a single depolarizing pulse to 0 mV was applied for 15 ms to determine the fraction of channels available (Figure 3A). The addition of 200 μM (S)-LCM did not alter the onset or extent of slow inactivation (Figures 3B–D). We next determined the effect of (S)-LCM on fast inactivation and steady-state activation. For fast inactivation, cells were held at −80 mV, conditioned to potentials ranging from −120 to −10 mV (in +10 mV increments) for 500 ms and the fraction of available current was determined by a 20 ms test pulse at 0 mV (Figure 3E). For steady-state activation, cells were held at −80 mV and current was measured at potentials ranging from −70 to +80 mV (in +10 mV increments) for 500 ms (Figure 3E). Neither fast inactivation nor steady-state inactivation was effected by (S)-LCM (Figure 3F).
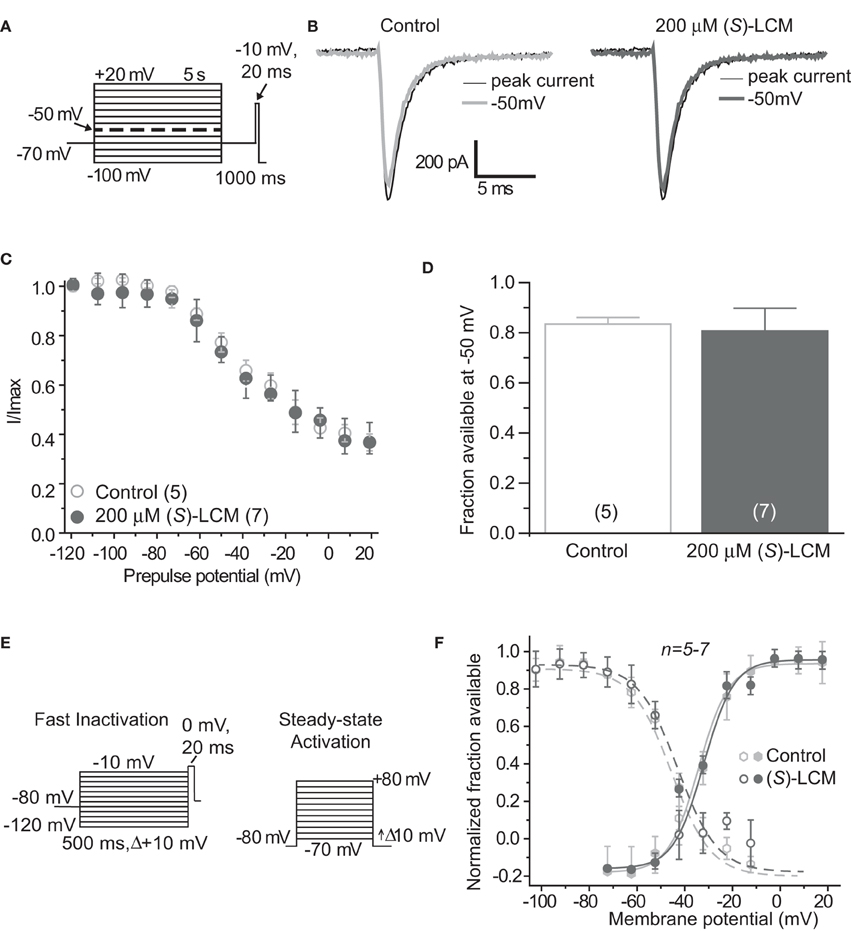
Figure 3. (S)-Lacosamide ((S)-LCM) does not affect biophysical properties of voltage-gated sodium currents in cortical neurons. (A) Voltage protocol for slow inactivation. (B) Representative current traces from cortical neurons in the absence (control, 0.1% DMSO) or presence of 200 μM (S)-LCM. The thin black and thick gray traces represent the currents evoked at −100 and −50 mV, respectively (also highlighted in the voltage protocol as a dashed thick line). (C) Summary of steady-state slow activation curves for neurons treated with DMSO (control) or 200 μM (S)-LCM. No drug-induced slow inactivation was evident at voltages more depolarizing that −80 mV in neurons treated with (S)-LCM. (D) Summary of the fraction of current available at −50 mV for neurons treated with DMSO (control) or 200 μM (S)-LCM (p > 0.05, Student's t-test). (E) Voltage protocol for fast inactivation (left) and activation (right). (F) Representative Boltzmann fits for steady-state fast inactivation and activation for neurons treated with 0.1% DMSO (control) and or 200 μM (S)-LCM are shown. Values for V1/2, the voltage of half-maximal inactivation and activation and the slope factors (k) were derived from Boltzmann distribution fits to the individual recordings and averaged to determine the mean (±s.e.m.) voltage dependence of steady-state inactivation and activation, respectively. There were no differences between control and drug for any of the parameters tested (p > 0.05, One-Way ANOVA). Data are from 5 to 7 cells per condition.
Loss of CRMP2 Phosphorylation Following TBI
The development of TLE following TBI, accounts for 20% of symptomatic epilepsy (Agrawal et al., 2006). Evidence of increased Akt activation within the hippocampus as well as other regions has been observed following TBI (Zhang et al., 2006; Zhao et al., 2012). Corresponding to changes in Akt activity, levels of phosphorylated (inactive) GSK3β are also increased following TBI (Shapira et al., 2007; Dash et al., 2011; Zhao et al., 2012). To determine if changes in CRMP2 phosphorylation could be the driving force behind the increased neurite outgrowth in the hippocampus following TLE-related insults, hippocampal tissue was collected at both early (24 h) and late (4 weeks) phases following TBI in adult male rats. Consistent with previous reports, levels of Ser9-phosphorylated (inactivated) GSK3β were increased in the early phase following TBI (1.31 ± 0.08) compared to sham controls (1.00 ± 0.05) (p < 0.05) (Figures 4A,B). Importantly, total expression of GSK3β remained unchanged [(1.00 ± 0.11) vs. (0.81 ± 0.12)] (p > 0.05) (Figures 4A,B). The increase in GSK3β phosphorylation appeared to be transient, as levels did not differ at 4 weeks following TBI (0.97 ± 0.12) compared to sham controls (1.00 ± 0.06) (p > 0.05) (Figures 4C,D).
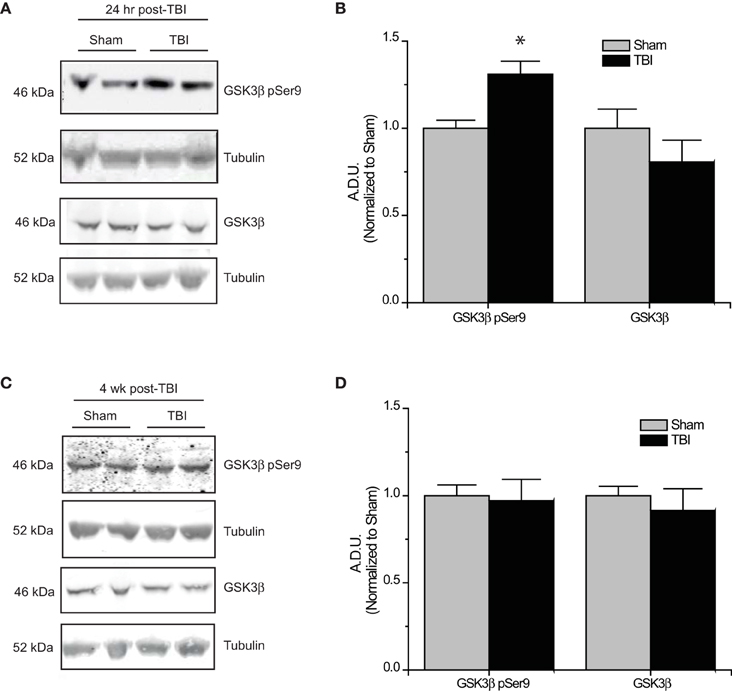
Figure 4. Changes in GSK3β phosphorylation following TBI. (A) Western blots of phosphorylated and total GSK3β from hippocampal tissue 24 h following TBI. (B) Summary of GSK3β pSer9 and total GSK3β levels 24 h following TBI. [Data is depicted as arbitrary densitometric units (A.D.U.)]. (C) Western blots of phosphorylated and total GSK3β from hippocampal tissue 4 weeks following TBI. (D) Summary of GSK3β pSer9 and total GSK3β levels 4 weeks following TBI (*p < 0.05 vs. sham, Student's t-test) (values represent mean ± s.e.m.) (n = 4–5).
Subsequent to the observed inactivation of GSK3β, levels of GSK3β-phosphorylated CRMP2 were reduced in the early phase following TBI (0.52 ± 0.07) compared to sham controls (1.00 ± 0.13) (p < 0.05) (Figures 5A,B). No change in total CRMP2 expression was observed [(1.10 ± 0.12) vs. (1.00 ± 0.04)] (p > 0.05) (Figures 5A,B). Despite the observed transience of GSK3β inactivation, levels of GSK3β-phosphorylated CRMP2 remained reduced in the late phase following TBI (0.62 ± 0.08) compared to sham controls (1.00 ± 0.09) (p < 0.05) (Figures 5C,D). These results suggest that there is an increased level of active (unphosphorylated) CRMP2 in both the early and late phases following TBI. Interestingly, while the decrease in CRMP2 phosphorylation at 24 h post-TBI is directly correlated with an inactivation of GSK3β, the sustained decrease observed at 4 weeks post-TBI appears to be independent of changes in GSK3β activity.
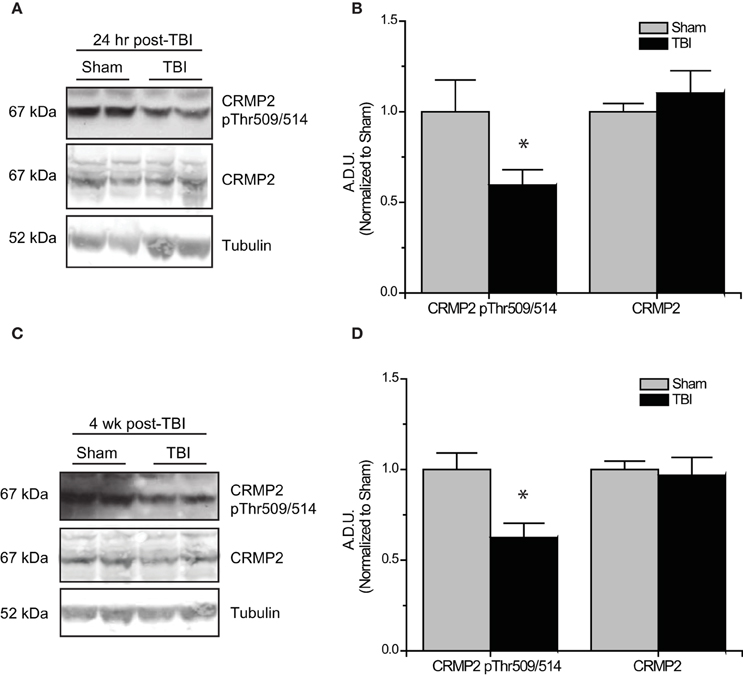
Figure 5. Changes in CRMP2 phosphorylation by GSK3β following TBI. (A) Western blots of GSK3β-phosphorylated and total CRMP2 from hippocampal tissue 24 h following TBI. (B) Summary of CRMP2 pThr509/514 and total CRMP2 levels 24 h following TBI (raw data represents protein of interest normalized to tubulin and further normalized to sham to allow for easy comparison) [data is represented as arbitrary densitometric units (A.D.U)]. (C) Western blots of GSK3β-phosphorylated and total CRMP2 from hippocampal tissue 4 weeks following TBI. (D) Summary of CRMP2 pThr509/514 and total CRMP2 levels 4 weeks following TBI. Data was normalized to sham conditions for ease of comparison. (*p < 0.05 vs. sham, Student's t-test) (values represent mean ± s.e.m. from n = 4–5).
“Primed” CRMP2 is Decreased in the Late, but not Early Phase Following TBI
Phosphorylation of GSK3β is not the only avenue through which phosphorylation of its substrates is regulated. GSK3β substrate recognition can be complex, as there is no strict consensus motif, often requiring prior phosphorylation (priming) at a serine slightly c-terminal to the GSK3β site(s) (DePaoli-Roach, 1984; Fiol et al., 1988). This type of hierarchical phosphorylation allows for complex regulation at multiple levels. In the case of CRMP2, it must first be phosphorylated at serine 522 by the serine/threonine kinase cyclin-dependent protein kinase 5 (CDK5) in order to be phosphorylated by GSK3β (Yoshimura et al., 2005; Cole et al., 2006) (Figure 6A). Sequential phosphorylation of CRMP2 by CDK5 and GSK3β has been demonstrated for many facets of CRMP2 function, most importantly, neurite outgrowth and growth cone collapse (Brown et al., 2004; Uchida et al., 2005). As decreased levels of GSK3β-phosphorylated CRMP2 were observed in the late phase following TBI that were not secondary to changes in GSK3β expression or activity, it is possible that the changes in levels of GSK3β-phosphorylated CRMP2 may be attributed to a decrease in phosphorylation by CDK5. Therefore, levels of CDK5-phosphorylated CRMP2 were determined from hippocampal tissue collected at early (24 h) and late (4 weeks) time points following TBI. Notably, CDK5 phosphorylation of CRMP2 at 24 h following TBI did not differ from sham controls [(1.05 ± 0.07) vs. (1.00 ± 0.12)] (p > 0.05) (Figures 6B,C). However, at 4 weeks following injury, levels of CDK5-phosphorylated CRMP2 were decreased (0.69 ± 0.07) compared to sham controls (1.00 ± 0.03) (p < 0.05) (Figures 6B,C). These results suggest that CRMP2 is differentially regulated during early and late phases following injury. While a loss of GSK3β activity accounts for decreases in CRMP2 phosphorylation immediately following injury, the same phenotype during later phases is attributed to a loss of priming by CDK5.
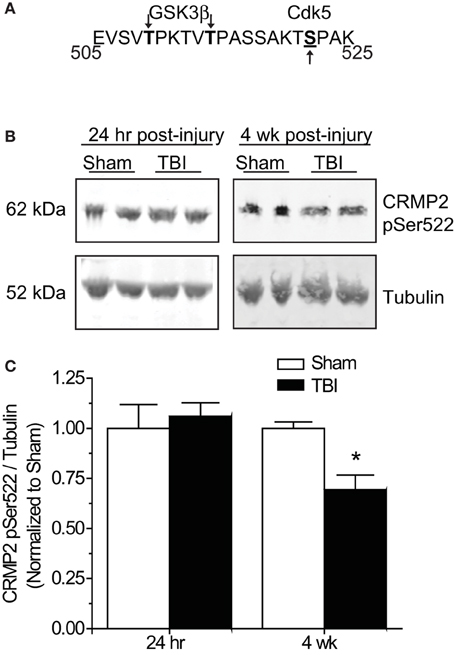
Figure 6. Changes in CRMP2 phosphorylation by CDK5 following TBI. (A) Schematic of GSK3β and Cdk5 phosphorylation sites within the rat CRMP2 sequence. Numbers represent amino acid residues within the CRMP2 sequence. (B) Western blots of CDK5-phosphorylared CRMP2 from hippocampal tissue 24 h and 4 weeks following TBI. (C) Summary of CRMP2 pSer522 levels at both 24 h and 4 weeks following TBI. For ease of comparison, data was normalized to the sham conditions. Levels of CRMP2 pSer522 were decreased at 4 weeks, but not 24 h following TBI. (*p < 0.05, student's t-test) (values represent mean ± s.e.m. from n = 4–5).
Effects of Targeting CRMP2 in vivo on Mossy Fiber Sprouting
As changes in CRMP2 phosphorylation, and presumably activity, are observed within the hippocampus at both early and late time points following TBI, CRMP2 may be involved in both the induction and maintenance of mossy fiber sprouting following injury. To determine the importance of CRMP2 in this phenomenon, osmotic minipumps containing (S)-LCM (140 mg/kg) were implanted (subcutaneously) immediately following TBI surgery in adult male rats. This method allowed for continuous delivery of ~5 mg/kg (S)-LCM per day (~0.21 mg/kg per hour) over the course of 4 weeks (Figure 7A). The extent of aberrant mossy fiber sprouting is easily identified due to the high amount of chelatable zinc within mossy fibers which can be visualized via a silver-sulfide staining method (TIMM staining) (Timm, 1958; Zimmer, 1973). Therefore, at the cessation of treatment, bilateral hippocampal tissue was obtained and processed for TIMM staining, to reveal the extent of mossy fiber sprouting within the inner molecular layer of the hippocampus.
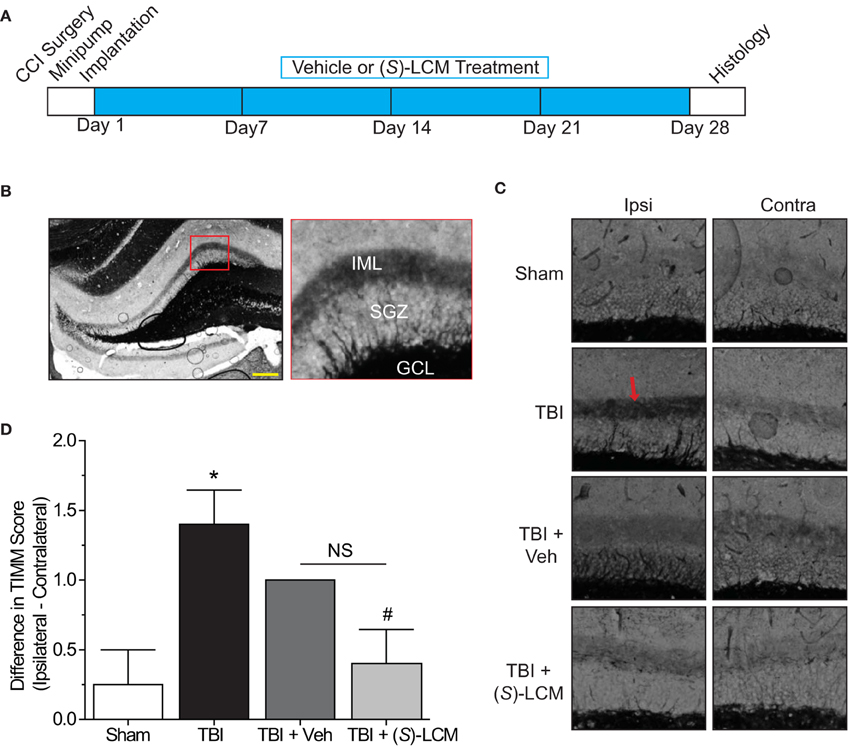
Figure 7. Effects of targeting CRMP2 in vivo on mossy fiber sprouting. (A) Timeline of experimental design. Animals received either controlled cortical impact or sham (craniotomy) surgery. Immediately following surgery, animals were implanted with osmotic mini-pumps containing either vehicle or (S)-LCM to be continuously infused at <0.01% DMSO and ~5 mg/kg per day. Following 4 weeks of treatment, tissue samples were prepared for histology. (B) Representative low-magnification image of a TIMM-stained coronal section. Red box depicts region quantified for the extent of mossy fiber sprouting into the inner molecular layer. (C) Representative 10×-magnification images of ipsilateral and contralateral TIMM-stained hippocampi. TBI led to a dense laminar band of TIMM reactivity within the supragranular zone extending to the inner molecular layer (red arrow). (D) Summary of TIMM scores from animals exposed to sham or TBI surgery ± vehicle or (S)-LCM. To minimize the impact of variance among animals, data is represented as the difference in TIMM score between ipsilateral and contralateral hippocampi from the same animal. (S)-LCM treatment prevented the TBI-induced increase in mossy fiber sprouting, however did not differ from vehicle. (*p < 0.05 vs. sham; #p < 0.05 vs. TBI; One-Way ANOVA, Tukey's post-hoc analysis) (values represent mean ± s.e.m. from n = 4–5). Scale bar = 250 μm.
As expected, TBI led to increased TIMM differences (1.40 ± 0.25) compared to sham controls (0.25 ± 0.25) (p < 0.05) (Figures 7B–D). Importantly, differences in TIMM scores did not differ between sham (0.25 ± 0.25) and naïve animals (0.00 ± 0.32) (p > 0.05). Intriguingly, (S)-LCM treatment prevented the TBI-induced increase in TIMM differences (0.40 ± 0.25) compared to animals receiving TBI alone (1.40 ± 0.25) (p < 0.05) (Figures 7B–D). However, the changes in TIMM scores following TBI did not differ between animals receiving (S)-LCM (0.40 ± 0.25) and vehicle (~0.01%DMSO) (1.00 ± 0.00) (p > 0.05) (Figures 7C–D). Therefore, it cannot definitively be concluded that CRMP2 is necessary for mossy fiber sprouting following TBI.
Discussion
In order for changes in GSK3β activity to impact CRMP2 function, a balance of GSK3β-phosphorylated and unphosphorylated CRMP2 must be present. Importantly, GSK3β phosphorylation of CRMP2 appears to be dynamically regulated in naïve neurons, as inhibition of GSK3β led to an almost complete loss of phosphorylation within 24 h. Additionally, the increase in phosphorylation following okadaic acid exposure provides evidence for active dephosphorylation. Therefore, changes in GSK3β activity can directly impact CRMP2 function. Indeed, inhibition of GSK3β led to increases in neurite outgrowth in a CRMP2-dependent manner. Given that inactivation of GSK3β has previously been demonstrated following TBI, decreased phosphorylation of CRMP2 may account for the changes in neurite elongation and branching observed within the hippocampus. Our findings indicate that TBI leads to decreased GSK3β phosphorylation of CRMP2 at both 24 h and 4 weeks post-injury. As mossy fiber sprouting is considered a progressive process, the maintained loss of phosphorylation throughout later phases following injury is an important finding. In contrast to early phases following TBI, the loss of GSK3β phosphorylation at 4 weeks post-injury is likely not attributed to a prolonged inactivation of GSK3β. In fact, previous reports suggest that levels of Akt-phosphorylated GSK3β return to baseline within 14 days (Dash et al., 2011). These findings suggest that while CRMP2 may play an integral role in promoting neurite outgrowth both immediately following injury as well as in later phases, the mechanisms underlying the increase in CRMP2 activity during these phases may differ. Indeed, our results revealed that the decrease in GSK3β-phosphorylated CRMP2 during later phases following injury was in fact attributed to a decrease in priming by CDK5.
Mossy fiber sprouting in TLE can likely be divided into 2 distinct phases: the induction phase, during which sprouting and outgrowth are attributed directly to the precipitating insult such as TBI, hypoxia-ischemia, or SE, and the maintenance phase (Sutula, 2004; Pitkänen and Lukasiuk, 2009). Therefore, early events following injury, such as inactivation of GSK3β, can be attributed to injury-induced mechanisms (i.e., activation of pro-survival signaling pathways). The latter phase involves processes secondary to the original insult such as hyperexcitability and network synchronization. Interestingly, we have previously demonstrated that CDK5 priming of CRMP2 is decreased in response to prolonged neuronal activity and that the loss of priming is directly translated into a reduction in GSK3β-phosphorylated CRMP2 (Wilson and Khanna, unpublished). As TBI can lead to progressive hyperexcitability (Yang et al., 2010b), the sustained loss of CRMP2 phosphorylation is potentially due to progressive changes in neuronal function which are secondary to the precipitating injury, such as activity-driven changes in CDK5 function.
Overall, phosphorylation of CRMP2 appears to be differentially regulated through both induction (early) and maintenance (late) phases following TBI. As such, targeting CRMP2-mediated neurite outgrowth throughout these stages may be sufficient to attenuate the progression of mossy fiber sprouting. Indeed, the extent of mossy fiber sprouting in animals that had received continuous administration of (S)-LCM following TBI was markedly decreased compared to untreated animals. However, the trending effect of vehicle administration is a confounding factor that prevents a definitive conclusion from being drawn. Changes in hippocampal cell death likely do not account for the observed changes in TIMM staining, as dentate granule cell survival is typically not impacted by moderate TBI (Lowenstein et al., 1992; Grady et al., 2003). Furthermore, (S)-LCM is unlikely to affect hippocampal cell death as a recent study demonstrated that the parent compound (R)-LCM does not alter neuronal survival following TBI (Pitkanen et al., 2014). The lack of significant separation between (S)-LCM- and vehicle-treated groups may be a result of the nature of administration, despite the previous success observed with infusion of (R)-LCM in a separate study (Licko et al., 2013). While continuous subcutaneous infusion was considered to be preferable over daily intraperitoneal injections, it is possible that inflammation at the implantation site may have been a factor.
Although the exact role of CRMP2 in mossy fiber sprouting has not yet been determined, it is possible that the loss of GSK3β phosphorylation immediately following injury contributes to the induction of mossy fiber sprouting while the loss of priming by CDK5 in later phases contributes to the maintenance of mossy fiber sprouting. It is of great interest that these mechanistically distinct events culminate in a similar end-point: an increase in the amount of active CRMP2 (Figure 8). The reduction in mossy fiber sprouting in (S)-LCM-treated animals suggests, at the very least, that CRMP2 may be one factor involved in initiation and progression of mossy fiber sprouting.
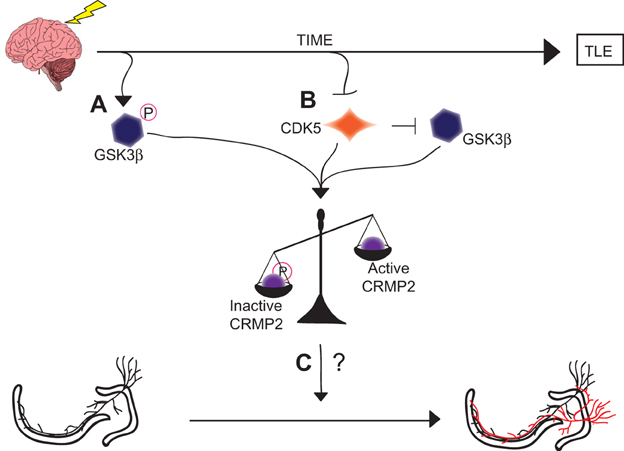
Figure 8. Graphical summary of findings. (A) GSK3β is phosphorylated and thereby inactivated in the early phases following injury. This inactivation leads to decreased amounts of phosphorylated (inactive) CRMP2. (B) CDK5 phosphorylation of CRMP2 is decreased in the later phases following injury. This decrease in phosphorylation also indirectly reduces levels of GSK3β-phosphorylated CRMP2 through a loss of priming, resulting in an overall increase in the proportion of active CRMP2. (C) Despite the sustained increase in unphosphorylated (active) CRMP2 in the hippocampus, the role of CRMP2 in aberrant mossy fiber sprouting following injury remains unclear, as denoted by the “?.”
Authors and Contributors
Participated in Research Design—Sarah M. Wilson, Rajesh Khanna. Conducted Experiments—Sarah M. Wilson, Seul Ki Yeon, Xiao-Fang Yang. Performed Data Analysis—Sarah M. Wilson, Seul Ki Yeon, Ki Duk Park, Xiao-Fang Yang. Wrote the Manuscript—Sarah M. Wilson, Rajesh Khanna.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Weina Ju for help with some of the immunoblots, and members of the Stark Neurosciences Research Institute for helpful comments and discussions. We express many thanks to Dr. Fletcher White and Matthew Ripsch for their generous guidance in experimental design, as well as, the use of their equipment. This work was supported, in part, by a grant a National Scientist Development from the American Heart Association (SDG5280023 to Rajesh Khanna) and a grant from the Showalter foundation (to Rajesh Khanna). Sarah M. Wilson was partially funded by a Stark Fellowship and a Larry Kays Medical Neuroscience award.
References
Agrawal, A., Timothy, J., Pandit, L., and Manju, M. (2006). Post-traumatic epilepsy: an overview. Clin. Neurol. Neurosurg. 108, 433–439. doi: 10.1016/j.clineuro.2005.09.001
Alessi, D. R., Andjelkovic, M., Caudwell, B., Cron, P., Morrice, N., Cohen, P., et al. (1996). Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 15, 6541–6551.
Andersen, P., Bliss, T. V., Lomo, T., Olsen, L. I., and Skrede, K. K. (1969). Lamellar organization of hippocampal excitatory pathways. Acta Physiol. Scand. 76, 4a–5a.
Andurkar, S. V., Stables, J. P., and Kohn, H. (1999). The anticonvulsant activities of N-benzyl 3-methoxypropionamides. Bioorg. Med. Chem. 7, 2381–2389. doi: 10.1016/S0968-0896(99)00186-8
Arimura, N., Inagaki, N., Chihara, K., Menager, C., Nakamura, N., Amano, M., et al. (2000). Phosphorylation of collapsin response mediator protein-2 by Rho-kinase. Evidence for two separate signaling pathways for growth cone collapse. J. Biol. Chem. 275, 23973–23980. doi: 10.1074/jbc.M001032200
Arimura, N., Menager, C., Kawano, Y., Yoshimura, T., Kawabata, S., Hattori, A., et al. (2005). Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Mol. Cell Biol. 25, 9973–9984. doi: 10.1128/MCB.25.22.9973-9984.2005
Beyreuther, B. K., Freitag, J., Heers, C., Krebsfanger, N., Scharfenecker, U., and Stohr, T. (2007). Lacosamide: a review of preclinical properties. CNS Drug Rev. 13, 21–42. doi: 10.1111/j.1527-3458.2007.00001.x
Bhave, S. V., Ghoda, L., and Hoffman, P. L. (1999). Brain-derived neurotrophic factor mediates the anti-apoptotic effect of NMDA in cerebellar granule neurons: signal transduction cascades and site of ethanol action. J. Neurosci. 19, 3277–3286.
Blaabjerg, M., and Zimmer, J. (2007). The dentate mossy fibers: structural organization, development and plasticity. Prog. Brain Res. 163, 85–107. doi: 10.1016/S0079-6123(07)63005-2
Brittain, J. M., Chen, L., Wilson, S. M., Brustovetsky, T., Gao, X., Ashpole, N. M., et al. (2011b). Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2). J. Biol. Chem. 286, 37778–37792. doi: 10.1074/jbc.M111.255455
Brittain, J. M., Duarte, D. B., Wilson, S. M., Zhu, W., Ballard, C., Johnson, P. L., et al. (2011a). Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca(2+) channel complex. Nat. Med. 17, 822–829. doi: 10.1038/nm.2345
Brittain, J. M., Pan, R., You, H., Brustovetsky, T., Brustovetsky, N., Zamponi, G. W., et al. (2012a). Disruption of NMDAR-CRMP-2 signaling protects against focal cerebral ischemic damage in the rat middle cerebral artery occlusion model. Channels (Austin) 6, 52–59. doi: 10.4161/chan.18919
Brittain, J. M., Piekarz, A. D., Wang, Y., Kondo, T., Cummins, T. R., and Khanna, R. (2009). An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J. Biol. Chem. 284, 31375–31390. doi: 10.1074/jbc.M109.009951
Brittain, J. M., Wang, Y., Eruvwetere, O., and Khanna, R. (2012b). Cdk5-mediated phosphorylation of CRMP-2 enhances its interaction with CaV2.2. FEBS Lett. 586, 3813–3818. doi: 10.1016/j.febslet.2012.09.022
Brown, M., Jacobs, T., Eickholt, B., Ferrari, G., Teo, M., Monfries, C., et al. (2004). Alpha2-chimaerin, cyclin-dependent Kinase 5/p35, and its target collapsin response mediator protein-2 are essential components in semaphorin 3A-induced growth-cone collapse. J. Neurosci. 24, 8994–9004. doi: 10.1523/JNEUROSCI.3184-04.2004
Cavazos, J. E., Golarai, G., and Sutula, T. P. (1991). Mossy fiber synaptic reorganization induced by kindling: time course of development, progression, and permanence. J. Neurosci. 11, 2795–2803.
CDC, CfDCaP. (2012). “Epilepsy in adults and access to care - United States, 2010,” in MMWR, Vol. 61 (Washington, DC), 909–913.
Chae, Y. C., Lee, S., Heo, K., Ha, S. H., Jung, Y., Kim, J. H., et al. (2009). Collapsin response mediator protein-2 regulates neurite formation by modulating tubulin GTPase activity. Cell. Signal. 21, 1818–1826. doi: 10.1016/j.cellsig.2009.07.017
Chen, Y., Stevens, B., Chang, J., Milbrandt, J., Barres, B. A., and Hell, J. W. (2008). NS21: re-defined and modified supplement B27 for neuronal cultures. J. Neurosci. Methods 171, 239–247. doi: 10.1016/j.jneumeth.2008.03.013
Cole, A. R., Causeret, F., Yadirgi, G., Hastie, C. J., McLauchlan, H., McManus, E. J., et al. (2006). Distinct priming kinases contribute to differential regulation of collapsin response mediator proteins by glycogen synthase kinase-3 in vivo. J. Biol. Chem. 281, 16591–16598. doi: 10.1074/jbc.M513344200
Cole, A. R., Knebel, A., Morrice, N. A., Robertson, L. A., Irving, A. J., Connolly, C. N., et al. (2004). GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J. Biol. Chem. 279, 50176–50180. doi: 10.1074/jbc.C400412200
Cross, D. A., Alessi, D. R., Cohen, P., Andjelkovich, M., and Hemmings, B. A. (1995). Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789. doi: 10.1038/378785a0
Dash, P. K., Johnson, D., Clark, J., Orsi, S. A., Zhang, M., Zhao, J., et al. (2011). Involvement of the glycogen synthase kinase-3 signaling pathway in TBI pathology and neurocognitive outcome. PLoS ONE 6:e24648. doi: 10.1371/journal.pone.0024648
de Lanerolle, N. C., Kim, J. H., Williamson, A., Spencer, S. S., Zaveri, H. P., Eid, T., et al. (2003). A retrospective analysis of hippocampal pathology in human temporal lobe epilepsy: evidence for distinctive patient subcategories. Epilepsia 44, 677–687. doi: 10.1046/j.1528-1157.2003.32701.x
DePaoli-Roach, A. A. (1984). Synergistic phosphorylation and activation of ATP-Mg-dependent phosphoprotein phosphatase by F A/GSK-3 and casein kinase II (PC0.7). J. Biol. Chem. 259, 12144–12152.
Dustrude, E. T., Wilson, S. M., Ju, W., Xiao, Y., and Khanna, R. (2013). CRMP2 protein SUMOylation modulates NaV1.7 channel trafficking. J. Biol. Chem. 288, 24316–24331. doi: 10.1074/jbc.M113.474924
Endo, H., Nito, C., Kamada, H., Nishi, T., and Chan, P. H. (2006). Activation of the Akt//GSK3[beta] signaling pathway mediates survival of vulnerable hippocampal neurons after transient global cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 26, 1479–1489. doi: 10.1038/sj.jcbfm.9600303
England, M. J., Liverman, C. T., Schultz, A. M., and Strawbridge, L. M. (2012). Epilepsy across the spectrum: promoting health and understanding. A summary of the institute of medicine report. Epilepsy Behav. 25, 266–276. doi: 10.1016/j.yebeh.2012.06.016
Errington, A. C., Stohr, T., Heers, C., and Lees, G. (2008). The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol. Pharmacol. 73, 157–169. doi: 10.1124/mol.107.039867
Fiol, C. J., Haseman, J. H., Wang, Y. H., Roach, P. J., Roeske, R. W., Kowalczuk, M., et al. (1988). Phosphoserine as a recognition determinant for glycogen synthase kinase-3: phosphorylation of a synthetic peptide based on the G-component of protein phosphatase-1. Arch. Biochem. Biophys. 267, 797–802. doi: 10.1016/0003-9861(88)90089-6
Fukata, Y., Itoh, T. J., Kimura, T., Menager, C., Nishimura, T., Shiromizu, T., et al. (2002). CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat. Cell Biol. 4, 583–591. doi: 10.1038/ncb825
Ghasemi, M., and Schachter, S. C. (2011). The NMDA receptor complex as a therapeutic target in epilepsy: a review. Epilepsy Behav. 22, 617–640. doi: 10.1016/j.yebeh.2011.07.024
Goddard, G. V., McIntyre, D. C., and Leech, C. K. (1969). A permanent change in brain function resulting from daily electrical stimulation. Exp. Neurol. 25, 295–330. doi: 10.1016/0014-4886(69)90128-9
Goshima, Y., Nakamura, F., Strittmatter, P., and Strittmatter, S. M. (1995). Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature 376, 509–514. doi: 10.1038/376509a0
Goslin, K., and Banker, G. (1989). Experimental observations on the development of polarity by hippocampal neurons in culture. J. Cell Biol. 108, 1507–1516. doi: 10.1083/jcb.108.4.1507
Grady, M. S., Charleston, J. S., Maris, D., Witgen, B. M., and Lifshitz, J. (2003). Neuronal and glial cell number in the hippocampus after experimental traumatic brain injury: analysis by stereological estimation. J. Neurotrauma 20, 929–941. doi: 10.1089/089771503770195786
Hauser, W. A., and Kurland, L. T. (1975). The epidemiology of epilepsy in Rochester, Minnesota, 1935 through 1967. Epilepsia 16, 1–66. doi: 10.1111/j.1528-1157.1975.tb04721.x
Hirtz, D., Thurman, D. J., Gwinn-Hardy, K., Mohamed, M., Chaudhuri, A. R., and Zalutsky, R. (2007). How common are the “common” neurologic disorders? Neurology 68, 326–337. doi: 10.1212/01.wnl.0000252807.38124.a3
Hou, S. T., Jiang, S. X., Aylsworth, A., Ferguson, G., Slinn, J., Hu, H., et al. (2009). CaMKII phosphorylates collapsin response mediator protein 2 and modulates axonal damage during glutamate excitotoxicity. J. Neurochem. 111, 870–881. doi: 10.1111/j.1471-4159.2009.06375.x
Ju, W., Li, Q., Wilson, S. M., Brittain, J. M., Meroueh, L., and Khanna, R. (2013). SUMOylation alters CRMP2 regulation of calcium influx in sensory neurons. Channels 7, 153–159. doi: 10.4161/chan.24224
Kharatishvili, I., and Pitkanen, A. (2010). Posttraumatic epilepsy. Curr. Opin. Neurol. 23, 183–188. doi: 10.1097/WCO.0b013e32833749e4
Kimura, T., Watanabe, H., Iwamatsu, A., and Kaibuchi, K. (2005). Tubulin and CRMP-2 complex is transported via Kinesin-1. J. Neurochem. 93, 1371–1382. doi: 10.1111/j.1471-4159.2005.03063.x
Koyama, R., and Ikegaya, Y. (2004). Mossy fiber sprouting as a potential therapeutic target for epilepsy. Curr. Neurovasc. Res. 1, 3–10. doi: 10.2174/1567202043480242
Larner, A. J. (1995). Axonal sprouting and synaptogenesis in temporal lobe epilepsy: possible pathogenetic and therapeutic roles of neurite growth inhibitory factors. Seizure 4, 249–258. doi: 10.1016/S1059-1311(95)80001-8
Lee, C.-Y., Jaw, T., Tseng, H.-C., Chen, I. C., and Liou, H.-H. (2012). Lovastatin modulates glycogen synthase kinase-3β pathway and inhibits mossy fiber sprouting after pilocarpine-induced status epilepticus. PLoS ONE 7:e38789. doi: 10.1371/journal.pone.0038789
Lees, G., Stohr, T., and Errington, A. C. (2006). Stereoselective effects of the novel anticonvulsant lacosamide against 4-AP induced epileptiform activity in rat visual cortex in vitro. Neuropharmacology 50, 98–110. doi: 10.1016/j.neuropharm.2005.08.016
LeTiran, A., Stables, J. P., and Kohn, H. (2001). Functionalized amino acid anticonvulsants: synthesis and pharmacological evaluation of conformationally restricted analogues. Bioorg. Med. Chem. 9, 2693–2708. doi: 10.1016/S0968-0896(01)00204-8
Licko, T., Seeger, N., Zellinger, C., Russmann, V., Matagne, A., and Potschka, H. (2013). Lacosamide treatment following status epilepticus attenuates neuronal cell loss and alterations in hippocampal neurogenesis in a rat electrical status epilepticus model. Epilepsia 54, 1176–1185. doi: 10.1111/epi.12196
Lowenstein, D. H., Thomas, M. J., Smith, D. H., and McIntosh, T. K. (1992). Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J. Neurosci. 12, 4846–4853.
Manford, M., Hart, Y. M., Sander, J. W., and Shorvon, S. D. (1992a). National General Practice Study of Epilepsy (NGPSE): partial seizure patterns in a general population. Neurology 42, 1911–1917. doi: 10.1212/WNL.42.10.1911
Manford, M., Hart, Y. M., Sander, J. W., and Shorvon, S. D. (1992b). The national general practice study of epilepsy. the syndromic classification of the international league against epilepsy applied to epilepsy in a general population. Arch. Neurol. 49, 801–808. doi: 10.1001/archneur.1992.00530320025008
O'Dell, C. M., Das, A., Wallace, G. T., Ray, S. K., and Banik, N. L. (2012). Understanding the basic mechanisms underlying seizures in mesial temporal lobe epilepsy and possible therapeutic targets: a review. J. Neurosci. Res. 90, 913–924. doi: 10.1002/jnr.22829
Oliva, M., Berkovic, S. F., and Petrou, S. (2012). Sodium channels and the neurobiology of epilepsy. Epilepsia 53, 1849–1859. doi: 10.1111/j.1528-1167.2012.03631.x
Panayiotopoulos, C. P. (2005). The Epilepsies: Seizures, Syndromes and Management. Oxfordshire: Bladon Medical Publishing, an imprint of Springer Science+Business Media.
Park, K. D., Morieux, P., Salome, C., Cotten, S. W., Reamtong, O., Eyers, C., et al. (2009). Lacosamide isothiocyanate-based agents: novel agents to target and identify lacosamide receptors. J. Med. Chem. 52, 6897–6911. doi: 10.1021/jm9012054
Pitkanen, A., Immonen, R., Ndode-Ekane, X., Grohn, O., Stohr, T., and Nissinen, J. (2014). Effect of lacosamide on structural damage and functional recovery after traumatic brain injury in rats. Epilepsy Res. 108, 653–665. doi: 10.1016/j.eplepsyres.2014.02.001
Pitkänen, A., and Lukasiuk, K. (2009). Molecular and cellular basis of epileptogenesis in symptomatic epilepsy. Epilepsy Behav. 14(1 Suppl. 1), 16–25. doi: 10.1016/j.yebeh.2008.09.023
Sasaki, C., Hayashi, T., Zhang, W. R., Warita, H., Manabe, Y., Sakai, K., et al. (2001). Different expression of glycogen synthase kinase-3beta between young and old rat brains after transient middle cerebral artery occlusion. Neurol. Res. 23, 588–592. doi: 10.1179/016164101101199054
Shapira, M., Licht, A., Milman, A., Pick, C. G., Shohami, E., and Eldar-Finkelman, H. (2007). Role of glycogen synthase kinase-3beta in early depressive behavior induced by mild traumatic brain injury. Mol. Cell. Neurosci. 34, 571–577. doi: 10.1016/j.mcn.2006.12.006
Sharma, A. K., Reams, R. Y., Jordan, W. H., Miller, M. A., Thacker, H. L., and Snyder, P. W. (2007). Mesial temporal lobe epilepsy: pathogenesis, induced rodent models and lesions. Toxicol. Pathol. 35, 984–999. doi: 10.1080/01926230701748305
Siwek, M., Henseler, C., Broich, K., Papazoglou, A., and Weiergraber, M. (2012). Voltage-gated Ca(2+) channel mediated Ca(2+) influx in epileptogenesis. Adv. Exp. Med. Biol. 740, 1219–1247. doi: 10.1007/978-94-007-2888-2_55
Sutula, T. P. (2004). Mechanisms of epilepsy progression: current theories and perspectives from neuroplasticity in adulthood and development. Epilepsy Res. 60, 161–171. doi: 10.1016/j.eplepsyres.2004.07.001
Timm, F. (1958). Zur Histochemie der Schwermetalle Das Sulfid-Silberverfahren. Dtsch. Z. Ges. Gerichtl. Med. 46, 706–711.
Uchida, Y., Ohshima, T., Sasaki, Y., Suzuki, H., Yanai, S., Yamashita, N., et al. (2005). Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer's disease. Genes Cells 10, 165–179. doi: 10.1111/j.1365-2443.2005.00827.x
Uchida, Y., Ohshima, T., Yamashita, N., Ogawara, M., Sasaki, Y., Nakamura, F., et al. (2009). Semaphorin3A signaling mediated by Fyn-dependent tyrosine phosphorylation of collapsin response mediator protein 2 at tyrosine 32. J. Biol. Chem. 284, 27393–27401. doi: 10.1074/jbc.M109.000240
Wang, Y., Brittain, J. M., Jarecki, B. W., Park, K. D., Wilson, S. M., Wang, B., et al. (2010). In silico docking and electrophysiological characterization of lacosamide binding sites on collapsin response mediator protein 2 (CRMP-2) identifies a pocket important in modulating sodium channel slow inactivation. J. Biol. Chem. 285, 25296–25307. doi: 10.1074/jbc.M110.128801
Wang, Y., and Khanna, R. (2011). Voltage-gated calcium channels are not affected by the novel anti-epileptic drug lacosamide. Transl. Neurosci. 2, 13–22. doi: 10.2478/s13380-011-0002-9
Wilson, S. M., Schmutzler, B. S., Brittain, J. M., Dustrude, E. T., Ripsch, M. S., Pellman, J. J., et al. (2012a). Inhibition of transmitter release and attenuation of aids therapy-induced and tibial nerve injury-related painful peripheral neuropathy by novel synthetic Ca2+ channel peptides. J. Biol. Chem. 287, 35065–35077. doi: 10.1074/jbc.M112.378695
Wilson, S. M., Xiong, W., Wang, Y., Ping, X., Head, J. D., Brittain, J. M., et al. (2012b). Prevention of posttraumatic axon sprouting by blocking collapsin response mediator protein 2-mediated neurite outgrowth and tubulin polymerization. Neuroscience 210, 451–466. doi: 10.1016/j.neuroscience.2012.02.038
Xiong, T., Tang, J., Zhao, J., Chen, H., Zhao, F., Li, J., et al. (2012). Involvement of the Akt/GSK-3β/CRMP-2 pathway in axonal injury after hypoxic–ischemic brain damage in neonatal rat. Neuroscience 216, 123–132. doi: 10.1016/j.neuroscience.2012.04.052
Yang, L., Afroz, S., Michelson, H. B., Goodman, J. H., Valsamis, H. A., and Ling, D. S. (2010b). Spontaneous epileptiform activity in rat neocortex after controlled cortical impact injury. J. Neurotrauma 27, 1541–1548. doi: 10.1089/neu.2009.1244
Yang, T., Zhou, D., and Stefan, H. (2010a). Why mesial temporal lobe epilepsy with hippocampal sclerosis is progressive: uncontrolled inflammation drives disease progression? J. Neurol. Sci. 296, 1–6. doi: 10.1016/j.jns.2010.06.002
Yoshimura, T., Kawano, Y., Arimura, N., Kawabata, S., Kikuchi, A., and Kaibuchi, K. (2005). GSK-3[beta] Regulates Phosphorylation of CRMP-2 and Neuronal Polarity. Cell 120, 137–149. doi: 10.1016/j.cell.2004.11.012
Zhang, X., Chen, Y., Ikonomovic, M. D., Nathaniel, P. D., Kochanek, P. M., Marion, D. W., et al. (2006). Increased phosphorylation of protein kinase B and related substrates after traumatic brain injury in humans and rats. J. Cereb. Blood Flow Metab. 26, 915–926. doi: 10.1038/sj.jcbfm.9600238
Zhao, S., Fu, J., Liu, X., Wang, T., Zhang, J., and Zhao, Y. (2012). Activation of Akt/GSK-3beta/beta-catenin signaling pathway is involved in survival of neurons after traumatic brain injury in rats. Neurol. Res. 34, 400–407. doi: 10.1179/1743132812Y.0000000025
Keywords: CRMP2, GSK3β, CDK5, phosphorylation, mossy fiber sprouting, TIMM staining, epileptogenesis, (S)-Lacosamide
Citation: Wilson SM, Ki Yeon S, Yang X-F, Park KD and Khanna R (2014) Differential regulation of collapsin response mediator protein 2 (CRMP2) phosphorylation by GSK3ß and CDK5 following traumatic brain injury. Front. Cell. Neurosci. 8:135. doi: 10.3389/fncel.2014.00135
Received: 05 March 2014; Accepted: 29 April 2014;
Published online: 28 May 2014.
Edited by:
Christophe Altier, University of Calgary, CanadaReviewed by:
Adam R. Cole, Garvan Institute for Medical Research, AustraliaHailan Yao, Roskamp Institute, USA
Copyright © 2014 Wilson, Ki Yeon, Yang, Park and Khanna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajesh Khanna, Department of Pharmacology, College of Medicine, University of Arizona, Life Sciences North, Rm 650, PO Box 245050, 1501 North Campbell Drive, Tucson, AZ 85724, USA e-mail: rkhanna@email.arizona.edu