Molecular regulation of auditory hair cell death and approaches to protect sensory receptor cells and/or stimulate repair following acoustic trauma
 Christine T. Dinh
Christine T. Dinh Stefania Goncalves
Stefania Goncalves Esperanza Bas
Esperanza Bas Thomas R. Van De Water
Thomas R. Van De Water Azel Zine
Azel Zine- 1University of Miami Ear Institute, University of Miami Miller School of Medicine, Miami, FL, USA
- 2Integrative and Adaptive Neurosciences, Aix-Marseille Université, CNRS, UMR 7260, Marseille, France
- 3Faculty of Pharmacy, Biophysics Department, University of Montpellier, Montpellier, France
Loss of auditory sensory hair cells (HCs) is the most common cause of hearing loss. This review addresses the signaling pathways that are involved in the programmed and necrotic cell death of auditory HCs that occur in response to ototoxic and traumatic stressor events. The roles of inflammatory processes, oxidative stress, mitochondrial damage, cell death receptors, members of the mitogen-activated protein kinase (MAPK) signal pathway and pro- and anti-cell death members of the Bcl-2 family are explored. The molecular interaction of these signal pathways that initiates the loss of auditory HCs following acoustic trauma is covered and possible therapeutic interventions that may protect these sensory HCs from loss via apoptotic or non-apoptotic cell death are explored.
Introduction
Auditory hair cells (HCs) are important for the conversion of acoustic sound energy into electrical impulses that travel to the auditory centers of the brain for hearing. Sensorineural hearing loss (SNHL) is a form of hearing impairment that occurs most commonly from damagedHCs within the cochlea; it is a prevalent disability, affecting one in five people and more than 48 million Americans (Lin et al., 2011). There are numerous causes of acquired SNHL. Some of these etiologies include viral infections, platinum-based chemotherapeutic agents, aminoglycoside antibiotics, acoustic trauma, labyrinthine concussion, cochlear hypoxia, radiation exposure, cochlear implant electrode insertion trauma, and meningitis (Kuhn et al., 2011). Genetic mutations can also cause structural or physiologic abnormalities within the cochlea and impair cochlear homeostasis. These insults to the inner ear can promote cell death of auditory HCs and hearing loss.
Several signaling cascades are activated following an insult to the cochlea; these pathways can be pro-inflammatory, pro-death, and even pro-survival. The signaling cascades that occur on a cellular and a molecular level are highly complex and in many ways entwine; there is significant cross communication between pathways and it is the culmination of all of these activities that tilt the pendulum of cell survival and cell death in one direction or the other. Although there has been great progress in understanding the pathways of cell death in auditory HCs, several pro-death concepts are still speculative and extrapolated from studies of other cell types. As research in necrobiology of the inner ear progresses, these pathways specific to auditory HC death will be more stream-lined and refined. This review will discuss general concepts and pathways in apoptosis and necrosis that contribute to the understanding of key events in acoustic trauma. Several drug therapies take advantage of key events that occur following acoustic injury by targeting, promoting, or inhibiting different pathways that favor cell survival.
Cell Death
Auditory HCs undergo cell death through apoptosis and necrosis, in response to different insults to the inner ear. Apoptosis is an organized form of programmed cell death that is characterized by blebbing of the cell membrane, shrinkage of the cell body, fragmentation of the nucleus, condensation of the chromatin, and cleavage of chromosomal DNA. The cell is then divided into numerous fragments called apoptotic bodies that are disposed by phagocytosis (Figure 1). Particular to HCs undergoing apoptotic cell death, mitochondria swell, stereocilia are disrupted and lost, cuticular plates shrink, and chromatin condensation occurs in the nucleus, prior to disruption of junctional complexes and extrusion of auditory HCs (Hirose et al., 2004). Necrosis, originally thought to be an uncontrolled process, is another form of cell death that appears to be highly regulated. During necrotic cell death, the cell volume increases, the organelles swell, and the plasma membrane ruptures. There is spillage of intracellular contents in the extracellular space and postlytic DNA fragmentation occurs (Figure 1). Necrosis of auditory HCs can occur in concert with mechanisms of apoptosis. Most of the molecular and cellular mechanisms responsible for apoptosis and necrosis in auditory HCs originate in studies of other cell types. A review of general concepts in apoptosis and necrosis and stress signaling is offered below.
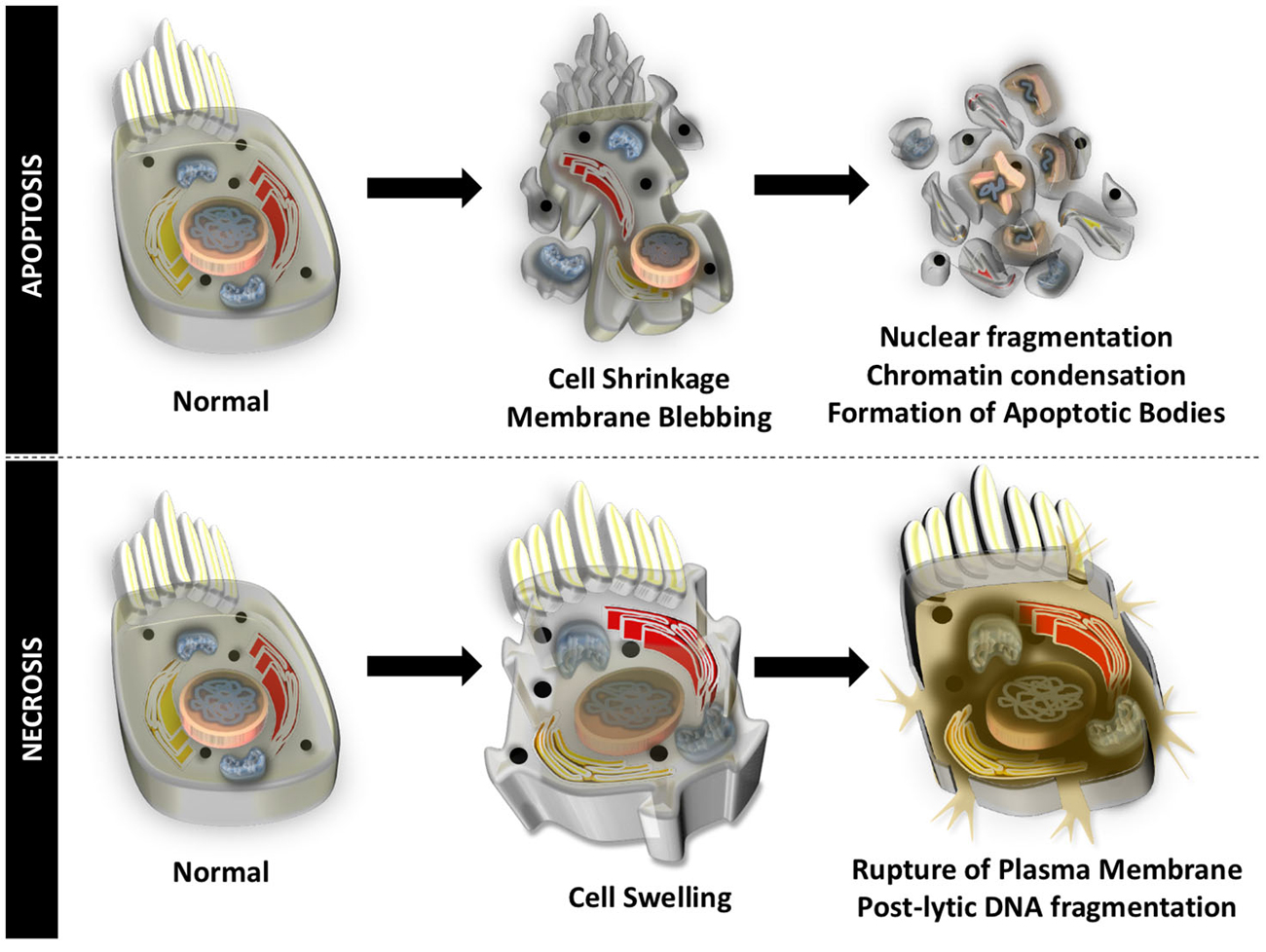
Figure 1. Apoptotic and necrotic cell death. Apoptosis is an organized form of programmed cell death that is characterized by shrinkage of the cell body, blebbing of the cell membrane, chromatin condensation, fragmentation of the nucleus, cleavage of chromosomal DNA, and division into numerous cell fragments (apoptotic bodies).Necrosis occurs when the volume of the cell increases, organelles swell, the plasma membrane ruptures, intracellular contents spill into the extracellular matrix, and there is postlytic DNA fragmentation.
Apoptosis
Apoptosis can occur through two different signaling cascades, called the intrinsic [mitochondrial death] and extrinsic [death receptor, DR] pathways. These two particular pathways can interweave and can promote synergism in cell death in many contexts (Roy and Nicholson, 2000).
Intrinsic Pathway
In the intrinsic pathway, a stress signal initiates a disturbance between the pro- and anti-apoptotic proteins of the Bcl-2 family that promote supramolecular openings or activation of mega-channels in the outer membrane of the mitochondria (Marzo et al., 1998; Kuwana et al., 2002). In particular, Bcl-2-like protein 4 (Bax) activation is a key regulator of this phenomenon. This results in the release of pro-death proteins from the intermembrane space of the mitochondria into the cytosol. These regulatory mitochondrial proteins that are liberated into the cytoplasm can activate both caspase-dependent and caspase-independent cell death pathways. These proteins include cytochrome c (cyt c), endonuclease G (EndoG), apoptosis inducing factor (AIF), second mitochondria-derived activator of caspases/direct inhibitor of apoptosis protein binding protein with low pI (Smac/DIABLO), and mammalian homolog of bacterial high temperature requirement protein A2 (Omi/HtrA2; Figure 2).
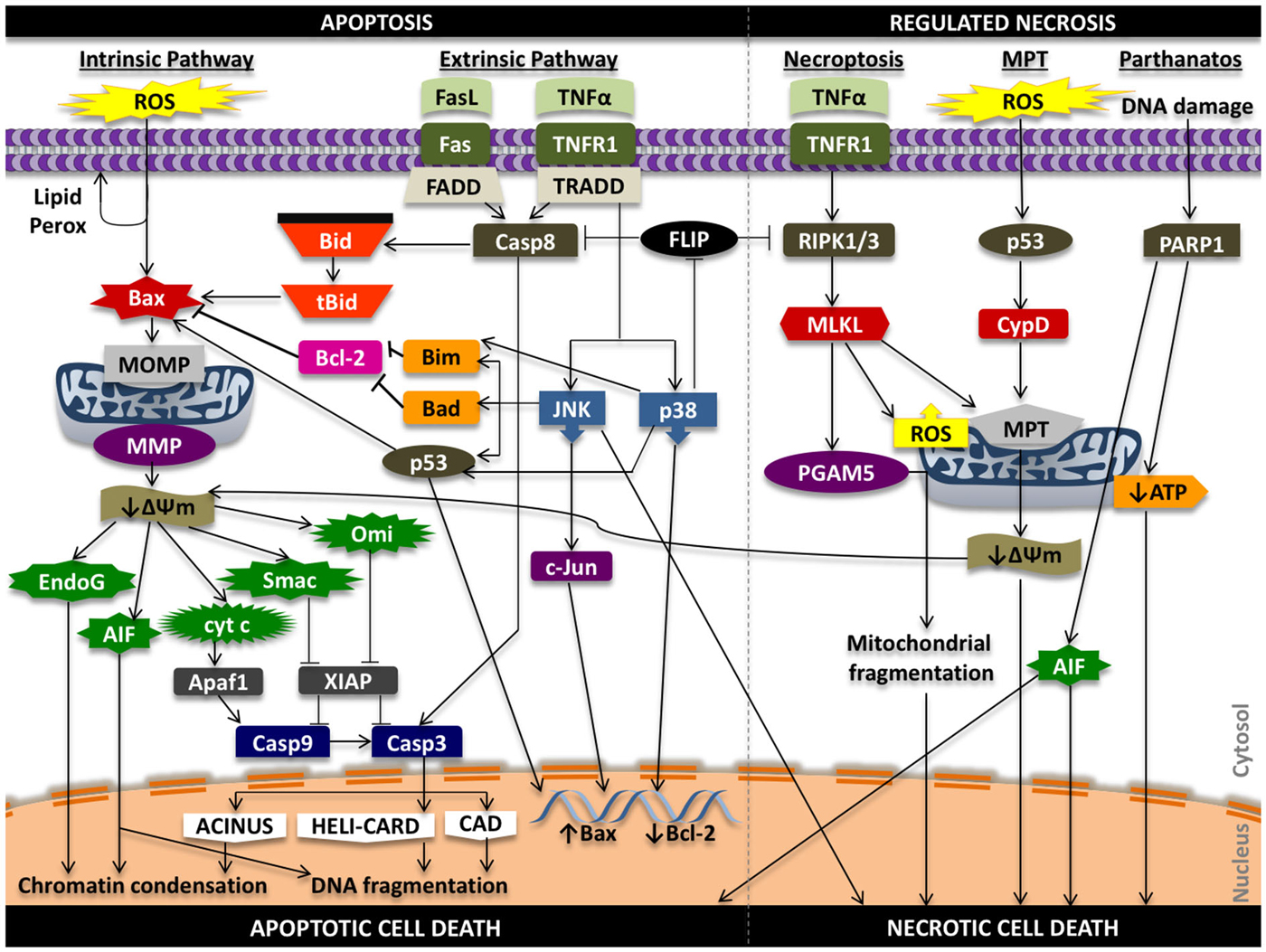
Figure 2. Signaling in apoptosis and regulated necrosis. ROS can promote lipid peroxidation of the phosopholipid membrane, proteolytic degradation, and mitochondrial and cellular DNA damage that can propagate the apoptotic effects of oxidative stress. Oxidative stress can activate pro-apoptotic Bax protein that initiates damage to the outer mitochondrial membrane (i.e., pore formation) that results in release of pro-death proteins from the mitochondria into the cytosol that act in caspase-independent and caspase-dependent mechanisms to induce chromatin condensation, DNA fragmentation and intrinsic apoptotic cell death. Ligands such as Fas ligand (FasL) and tumor necrosis factor alpha (TNFα) can promote extrinsic programmed cell death through caspase-8 activation and downstream caspase-3 activity, while also triggering intrinsic apoptosis through truncation of Bid (tBid). Tumor necrosis factor receptor type 1 (TNFR1) signaling can induce phosphorylation and activation of p38 and JNK that can lead to Bax-mediated mitochondrial cell death and phosphorylation of transcription factors that increase pro-death and reduce pro-survival gene expression. Activation of TNFR1 can also initiate a RIPK1 and RIPK3-dependent form of regulated necrosis (necroptosis) through regulation of PGAM5, ROS, and the mitochondrial permeability transition (MPT). MPT-mediated regulated necrotic cell death can be triggered following oxidative stress; downstream signaling results in activation of cyclophilin D (CypD), increased permeability of the inner mitochondrial membrane, and mitochondrial swelling and rupture that promotes necrosis. Alterations in the transmembrane potential of the mitochondria can also induce intrinsic cell death. Lastly, hyper-activation of PARP1 from persistent DNA damage that can inhibit mitochondrial ATP synthesis and promote mitochondrial release of AIF to initiate necrotic form of cell death. AIF, in this situation, can promote both apoptosis and necrosis. ΔΨm, mitochondrial membrane potential; Casp3, caspase-3; Casp8, caspase-8; Casp9, caspase-9; MMP, mitochondrial membrane permeability; MOMP, mitochondrial outer membrane permeability.
When released into the cytoplasm, cyt c binds with apoptotic protease-activating factor-1 (Apaf-1) and recruits pro-caspase-9 to form an apoptosome (Cain et al., 2000). This apoptosome can induce caspase-3 dependent apoptosis (Bratton et al., 2001). Subsequently, caspase-3 initiates apoptosis by promoting DNA fragmentation through caspase-activated DNase (CAD), chromatin condensation via apoptotic chromatin condensation inducer in the nucleus (ACINUS), and acceleration of DNA degradation through cleavage of cytosolic helicase with an N-terminal caspase-recruitment domain (HELI-CARD; Liu et al., 1997; Enari et al., 1998; Sahara et al., 1999; Kovacsovics et al., 2002).
EndoG is a mitochondrion-specific nuclease that translocates into the nucleus and works in concert with exonucleases and DNAse I to ensure efficient nucleosomal fragmentation of DNA, independent of caspase activation (Li et al., 2001; Widlak et al., 2001). Similar to EndoG, AIF is also a caspase-independent death effector; once released into the cytosol, AIF migrates into the nucleus to stimulate chromatin condensation and large scale DNA fragmentation (Lorenzo et al., 1999; Daugas et al., 2000).
Smac and Omi/HtrA2 are similar because both promote caspase-dependent apoptosis by binding and inhibiting X-linked inhibitor of apoptosis protein (XIAP). XIAP is a cytosolic protein that has three baculoviral inhibitory repeat (BIR) domains—BIR1 and BIR2 specifically bind and inhibit caspase-3 and -7, while BIR3 is a specific inhibitor of caspase-9 (Deveraux et al., 1999). Smac functions by neutralizing the caspase-inhibiting properties of XIAP, thereby promoting caspase-3, -7, and -9 activations (Chai et al., 2000, 2001). Similar to Smac, Omi/HtrA2 competes with caspase -3, -7, and -9 for XIAP binding and therefore promotes caspase-dependent cell death (Suzuki et al., 2001; Hegde et al., 2002). However, Omi/HtrA2 is a ubiquitously expressed mitochondrial serine protease that can also promote apoptosis through caspase-independent activity through alternate mechanisms that rely on its serine protease properties (Li et al., 2002).
Extrinsic Death Pathway
The extrinsic cell death pathway is intricate and involves several molecular interactions that occur in succession: (1) binding of a death ligand to its complementary receptor; (2) recruitment of adaptor molecules such as FAS-associated death domain protein (FADD) and tumor necrosis factor receptor type 1-associated death domain protein (TRADD); (3) binding, dimerizing, and activation of initiator caspase-8 and -10; and (4) formation of a death-inducing signaling complex (DISC; Itoh and Nagata, 1993; Tartaglia et al., 1993; Chinnaiyan et al., 1995; Hsu et al., 1995; Nagata, 1999; Fischer et al., 2006). DISC is a multi-protein complex that subsequently cleaves and promotes executioner caspase-3 and -7 activities to promote programmed cell death (Figure 2).
The most well recognized and studied death ligands include TNFα and FasL or CD95L. Their complementary receptors are TNFR1, also known as p55 or CD120a and Fas receptor (FasR, also referred to as CD95 or apoptosis antigen 1, APO-1), respectively (Itoh and Nagata, 1993; Tartaglia et al., 1993). Other DRs that have been described include TNF-like receptor apoptosis mediating protein (TRAMP, also called DR3, APO-3), TNF-related apoptosis inducing ligand receptors-1 (TRAIL-R1 or DR4) and -2 (TRAIL-R2, also named DR5 and APO-2), and DR6 (Bodmer et al., 1997; Guicciardi and Gores, 2009).
Initiators caspase-8 and caspase-10 can cleave and activate effector caspase-3 to initiate programmed cell death (Ng et al., 1999; Wang et al., 2001; Fischer et al., 2006). Caspase-8 can also promote effector caspase-7 activity. In addition, both caspase-8 and caspase-10 can cleave Bcl-2 homology 3 interacting domain death agonist (BID) into truncated BID (tBID) that triggers mitochondrial cell death pathways mediated by Bax and Bcl-2 homologous antagonist killer (Bak) activation (Chandler et al., 1998; Li et al., 1998; Luo et al., 1998; Korsmeyer et al., 2000; Kandasamy et al., 2003; Milhas et al., 2005). Bax and Bak are pro-death proteins that belong to the Bcl-2 family of proteins that can stimulate mitochondrial release of pro-apoptotic proteins such as cyt c and Smac. There are likely other levels of modulation between the intrinsic and extrinsic pathways of apoptosis that have not yet been discovered.
Necrosis
Mechanisms of regulated necrosis include necroptosis and MPT. Parthanatos, ferroptosis and pyroptosis have also been described as mechanisms of non-apoptotic cell death; however, they may not represent distinct forms of necrosis (Galluzzi et al., 2012; Vanden Berghe et al., 2014; Figure 2).
Necroptosis
Receptor interacting protein kinases (RIPK) are important regulators of cell survival and cell death. Necroptosis refers to a RIPK3-dependent molecular cascade of events that promotes regulated necrotic cell death (Galluzzi et al., 2012). Necroptosis and apoptosis share similar upstream signaling elements and regulator molecules such as Fas-associated death domain-like interleukin-1β-converting enzyme-like inhibitory proteins (FLIP) and cellular inhibitors of apoptosis proteins 1 and 2 (cIAP1 and cIAP2; McComb et al., 2012; Silke and Strasser, 2013). When caspase-8 is inhibited by genetic manipulation or pharmacologic therapies, RIPK3 is recruited to RIPK1 to form a necrosis-inducing complex (necrosome) that phosphorylates pseudokinase mixed lineage kinase domain-like protein (MLKL) and engages in downstream activities that lead to RIPK1/RIPK3 dependent necroptosis (He et al., 2009; Zhang et al., 2009; Vandenabeele et al., 2010). The events that occur downstream to promote necroptosis are controversial, convoluted, and poorly characterized, but may include production of mitochondrial reactive oxygen species (ROS), activation of mitochondrial phosphatase, i.e., PGAM5, and induction of MPT (Wang et al., 2012a,b; Marshall and Baines, 2014). PGAM5 is a mitochondrial protein phosphatase that can activate dynamin related protein 1 (Drp1) and its GTPase activity, which can stimulate fragmentation of mitochondria and execution of necrosis (Wang et al., 2012a,b).
Mitochondrial Permeability Transition
The MPT refers to an abrupt increase in the permeability of the inner mitochondrial membrane to small solutes that lead to osmotic influx of water into the mitochondrial matrix, mitochondrial swelling and rupture of the outer mitochondrial membrane (Tsujimoto and Shimizu, 2007). Although it is not well characterized, cyclophilin D (CypD; also known as peptidylprolyl isomerase F) is believed to be crucial in the formation of the permeability transition pore complex associated with MPT-dependent necrosis (Li et al., 2004; Baines et al., 2005; Nakagawa et al., 2005). Interestingly, the shift in the transmembrane potential in MPT can also initiate intrinsic cell death by halting the bioenergetics and redox functions of the mitochondria and initiating release of pro-apoptotic mitochondrial proteins into the cytosol (Marchetti et al., 1996; Scarlett and Murphy, 1997; Brenner and Grimm, 2006; Kroemer et al., 2007).
Other Forms of Non-Apoptotic Cell Death
Parthanatos is a poly(ADP-ribose) polymerase 1 (PARP1) dependent form of non-apoptotic death. PARP1 is recruited to sites of DNA damage where it is able to catalyze ADP ribosylation of various proteins and histones and generate negative charges by overconsumption of nicotinamide adenine dinucleotide (NAD) during the ADP ribosylation process. Ultimately, other factors important for DNA repair are recruited. However, when PARP1 becomes hyper-activated in situations of persistent DNA damage, there is a depletion of NAD and inhibition of mitochondrial ATP synthesis, which is essential in ATP-dependent apoptosis pathways (Yu et al., 2006; Wang et al., 2011). Overactivation of PARP-1 can lead to unregulated synthesis of poly (ADP-ribose) (PAR) that can bind to and initiate release of AIF into the cytoplasm. PARP-1 mediated AIF expression can promote DNA condensation and widespread cell death that is independent of caspases and distinct from apoptosis (Andrabi et al., 2008). There is controversy whether parthanatos represents a form of regulated necrosis or is a distinct entity.
Ferroptosis refers to an iron-dependent form of non-apoptotic cell death that is morphologically, biochemically, and genetically different from both apoptosis and necrosis (Dixon et al., 2012). Pyroptosis refers to caspase-1 dependent cell death that exhibits a spectrum of morphotypes that range from necrosis to purely apoptosis (Kepp et al., 2010). Caspase-1 is activated through a supramolecular complex called a pyroptosome or inflammasome and is thought to cause pore formation in the plasma membrane that leads to osmotic cell lysis (necrosis) and activation of caspase-7 (apoptosis) (Fink and Cookson, 2006; Fernandes-Alnemri et al., 2007; Lamkanfi et al., 2008). The elements behind regulated necrosis are still unclear; however, research in this topic has become more and more prevalent especially in the context of cancer research.
Auditory Hair Cell Death
Depending on the cochlear insult, various elements of the apoptotic and necrotic cell death pathway are activated (Op de Beeck et al., 2011; Figure 1). In acoustic trauma, loss of auditory HCs occur through direct mechanical injury and activation of apoptosis and necrosis related pathways (Saunders et al., 1985; Pirvola et al., 2000; Yang et al., 2004). Intense noise exposure can displace the tympanic membrane, vibrate the ossicles, and create large displacements of the basilar membrane that can sheer and injure stereocilia, auditory HCs, and supporting cells of the organ of Corti important for hearing. Downstream stress signaling from acoustic trauma are activated, including expression of free radicals and pro-inflammatory factors that trigger both apoptotic and necrotic cell death (discussed in subsequent sections). Following intense noise exposure Bax activation promotes mitochondrial release of pro-apoptotic factors (i.e., cyt c, AIF, and EndoG), caspase-3 activation, and intrinsic cell death (Nicotera et al., 2003; Yamashita et al., 2004a,b; Han et al., 2006; Wang et al., 2007a,b). Acoustic trauma also stimulates TNFR1 and Fas DR signaling that activates downstream pathway leading to caspase-8 activity and extrinsic apoptotic cell death (Nicotera et al., 2003; Fujioka et al., 2006; Jamesdaniel et al., 2011). Although the molecular mechanisms are still unclear, necrosis of outer HCs occur early following acoustic trauma and persist weeks after acoustic trauma (Yang et al., 2004; Hu et al., 2006). Lastly, intense noise exposure can also promote loss of auditory HCs through HC extrusion (Cotanche et al., 1997).
Although there are several drugs that can injure auditory HCs, the most commonly encountered ototoxic drugs are aminoglycosides and cisplatin. In brief, aminoglycoside antibiotics, such as gentamicin, amikacin, kanamycin, and neomycin, can initiate apoptotic cell death in auditory HCs and vestibular HCs. In particular, the outer HCs of the basal turn are the most vulnerable to aminoglycoside ototoxicity. The most predominant form of cell death is apoptosis, however necrotic features are also seen following exposure to aminoglycosides (Jiang et al., 2006). Aminoglycosides can induce mitochondrial apoptotic cell death and DNA fragmentation through oxidative stress, Bax activation, mitochondrial release of cyt c and caspase-3 activity (Mangiardi et al., 2004; Matsui et al., 2004; Coffin et al., 2013). Evidence for the role of caspase-8 and extrinsic apoptosis is aminoglycoside ototoxicity is not strong (Tabuchi et al., 2007; Ding et al., 2010).
Cisplatin is a platinum-based chemotherapeutic drug used to treat cancer and can promote ototoxicity, nephrotoxicity, and even neurotoxicity. Cisplatin predominantly affects the outer HCs of the basal turn of the cochlea; spiral ganglion neurons are affected by this drug. Similar to aminoglycosides, cisplatin stimulates free radical production in the cochlea that leads to Bax activation, mitochondrial release of cyt c, caspase activation, and intrinsic apoptotic cell death (Rybak et al., 2007). Unfortunately, regulated necrosis and potential pathways associated with this form of cell death are still unclear in the field of cell biology. The contribution of pathways described in regulated necrosis is still speculative in cochlear injury. Progress in necrobiology research can improve our understanding of necrosis in auditory HCs.
Stress Signaling in Auditory Hair Cell Death
There are several signaling pathways that are involved in apoptosis and necrosis of auditory HCs, including (1) expression of extracellular pro-inflammatory cytokines such as tumor necrosis factor alpha (TNFα) and recruited of neutrophils and macrophages to the cochlea; and (2) generation of oxidative stress in the form of ROS and reactive nitrogen species (RNS) such as superoxide (), peroxynitrite (ONOO−), and hydroxyl () radicals (Figure 2). These stress signals can activate intrinsic and extrinsic apoptotic cell death of auditory HCs as well as initiate molecular mechanisms for necrosis. Although signaling mechanisms discussed are extrapolated from studies investigating the effects of various cochlear insults on apoptosis and necrosis in auditory HCs and studies of other cell types, many of these pathways are relevant and expressed following acoustic trauma. Studies pertaining to stress signaling in the cochlea are bolded in subsequent sections.
Inflammatory Cytokines and Chemokines
Several cytokines and chemokines are upregulated in the cochlea following an inner ear insult. TNFα is one of the most well studied and possibly most important cytokines as it possesses pro-inflammatory properties and can also initiated cell death of auditory HCs. TNFα is expressed within the cochlea during acoustic trauma, cisplatin ototoxicity, aminoglycoside exposure, electrode insertion trauma, autoimmune inner ear disease, as well as various other traumas to the inner ear (Ichimiya et al., 2000; Satoh et al., 2002, 2003; Aminpour et al., 2005; Zou et al., 2005; Fujioka et al., 2006; Van Wijk et al., 2006; So et al., 2007; Bas et al., 2009, 2012a,b).
Following a cochlear insult, the stria vascularis and spiral ligament express and release TNFα, which promotes leukocyte migration through the spiral modiolar vein and tributaries (Keithley et al., 2008). TNFα also stimulates expression of other cytokines, chemokines, and adhesion molecules such as interleukin (IL)-1β, IL-6, monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-2 (MIP-2), soluble intercellular adhesion molecule-1 (siCAM-1), vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and vascular endothelial growth factor (VEGF; Yoshida et al., 1999; Ichimiya et al., 2000, 2003; Maeda et al., 2005). These inflammatory mediators can promote migration and adhesion of inflammatory cells such as neutrophils, monocytes, macrophages, lymphocytes, eosinophils, basophils, and even natural killer cells. (Taub et al., 1995; Proost et al., 1996; Mutsaers et al., 1997; Deshmane et al., 2009). These inflammatory cells are themselves a major source of cytokines and chemokines, creating a positive feedback loop that propagates and intensifies the inflammatory process.
In particular, expression of TNFα can promote neutrophil migration, adhesion, and generation of superoxide free radicals in the cochlea (Tsujimoto et al., 1986; Figari et al., 1987). Neutrophils, one type of polymorphonuclear white blood cell, are among the first immune cells to arrive in response to inflammation. They relesase a number of pro-inflammatory cytokines that stimulate migration of other immune cells, such as monocytes (Mutsaers et al., 1997). In response to local cytokines, monocytes will differentiate into M1 and M2 macrophages. M1 macrophages are primarily involved in clearance of injured cells; they can secrete inflammatory factors, stimulate inducible nitrous oxide synthetase (iNOS) activity, and generate oxidative stress in the form of ROS and RNS that can diffuse into surrounding tissue. M2 macrophages are known to release several growth factors from the transforming growth factor-beta (TGF-β) family of proteins, which are important for initiation of a fibroproliferative response, wound healing, and tissue repair (Mahdavian Delavary et al., 2011).
Not only is TNFα a leukocyte attractant; extracellular TNFα can bind to tumor necrosis factor receptor 1 (TNFR1) on the cell surface of auditory HCs and initiate a signaling cascade that can lead to cell death (Dinh et al., 2008a,b, 2011, 2013; Haake et al., 2009; Figure 2). Adaptor protein TRADD, receptor interacting protein (RIP), and FADD are recruited along with caspase-8 to form a DISC that cleaves and activates caspase-3 and -7 and triggers extrinsic apoptosis. TNFα-mediated Bax activation and intrinsic programmed cell death also occurs, likely through truncation of Bid.
The binding of TNFα to TNFR1 can also promote formation of another signaling complex consisting of TNFR1, TRADD, RIP, and TNF receptor-associated factor 2 (TRAF2) that promotes mitogen-activated protein kinase (MAPK) and nuclear factor kappa B (NFkB) signaling (Shen and Pervaiz, 2006). NFkB is a transcription factor that can mediate cellular inflammation, proliferation, and apoptosis. When activated, NFkB can translocate into the nucleus of an affected cell and activate transcription of several genes that are pro-inflammatory, pro-survival and pro-death, depending on the cell type (Pahl, 1999). TNFα initiates NFkB signaling in an attempt to block cell death by up regulation of pro-survival genes Bcl-2 and Bcl-xL in auditory HCs (Haake et al., 2009; Dinh et al., 2011).
High levels of IL-1beta (β) have also been detected in OC and cochlea following various inner ear challenges such as exposure to gentamicin, electrode insertion trauma, and autoimmune responses mediated by inner ear tissues (Pathak et al., 2011; Bas et al., 2012a,b). This cytokine can promote formation of a pyroptosome or inflammasome that mediates caspase-1 dependent cell death via pyroptosis (Brough and Rothwell, 2007). As discussed earlier, caspase-1 activation can promote apoptosis by inducing caspase-7 activity or necrosis through pore formation in the cell membrane and cell lysis through an osmotic imbalance (Fink and Cookson, 2006; Fernandes-Alnemri et al., 2007; Lamkanfi et al., 2008). In summary, TNFα expression in the cochlea can promote a robust inflammatory response and activation of downstream pro-apoptotic cell signaling that can promote cell death of auditory HCs. Blocking TNFα expression and associated signaling cascades may mitigate the inflammatory and pro-death responses in the cochlea. In particular, glucocorticoids have been shown to reduce inflammatory cell trafficking as well as inhibit TNFα-mediated downstream pathways of cell death (Dinh et al., 2008a,b; Haake et al., 2009). Steroids can also activate NFkB signaling that promotes cell survival pathways following TNFα-induced death of auditory HCs (Dinh et al., 2011). Inhibition of Bax expression by short interfering RNA can also abrogate TNFα-mediated HC loss (Dinh et al., 2013).
MAPK Signaling
In response to TNFα, auditory HCs can initiate MAPK signaling pathways. There are several distinct groups of MAPKs; however, the most extensively studied MAPKs are extracellular signal-regulated kinases (ERK) 1 and 2 (ERK1/2), c-Jun N-terminal kinases (JNK 1, 2, and 3), and p38 kinases (Roux and Blenis, 2004). Growth factors are the primary initiators of ERK1/2 activation, while JNK and p38 activities are stimulated by a number of physical and chemical stressors such as oxidative stress, UV irradiation, hypoxia, and various inflammatory cytokines including TNFα (Chen et al., 2001; Kyriakis and Avruch, 2001). There are three tiers of protein kinases that comprise each family of MAPKs: (1) MAPK; (2) MAPK kinase (MAPKK); and (3) MAPKK kinase (MAPKKK; Figure 3). MAPKKK phosphorylates and activates MAPKK, which then phosphorylates and activates MAPK to then phosphorylate target substrates (Keshet and Seger, 2010).
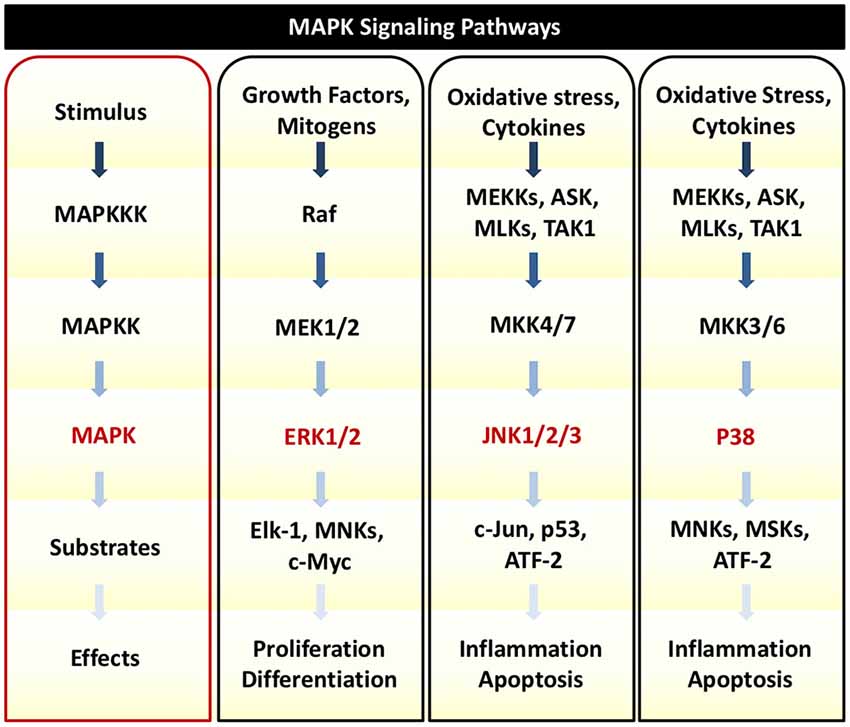
Figure 3. MAPK Signaling. There are three tiers of protein kinases that comprise each family of MAPKs: (1) MAPK; (2) MAPK kinase (MAPKK); and (3) MAPKK kinase (MAPKKK). Following a specific stimulus, MAPKKK phosphorylates and activates MAPKK, which then phosphorylates and activates MAPK to then phosphorylate target substrates that can regulate cellular proliferation, survival, inflammation, and cell death. The three main classes of MAPK include ERK1/2, JNK1/2/3, and p38. Oxidative stress and cytokines such as TNFα can trigger JNK and p38 to phosphorylate downstream transcription factors that initiate pro-death and pro-inflammatory signaling and gene transcription.
While ERK1/2 signaling has been implicated in cell proliferation and survival, TNFα activation of JNK and p38 signaling cascades promotes downstream events associated with cell death (Wajant et al., 2003). P38 is a mediator of apoptosis and it does so through several transcriptional and posttranscriptional molecular mechanisms. P38 activation can promote Bax mRNA and protein expression while phosphorylating and deactivating pro-survival Bcl-2 protein, thereby stimulating Bax-mediated mitochondrial apoptosis and cyt c release (Porras et al., 2004; Capano and Crompton, 2006; Markou et al., 2009; Owens et al., 2009). Pro-death factors such as Bcl-2-like protein 11 (Bim, a pro-apoptotic regulator), FasL, and FasR have also been positively regulated following p38 MAPK activation to stress stimuli (Hsu et al., 1999; Stephanou et al., 2001; Porras et al., 2004; Cai et al., 2006). Through inhibition of cellular FLIP (c-FLIP) in the DISC of the extrinsic apoptotic pathway, p38 may also trigger Fas-mediated caspase-8 dependent programmed cell death (Tourian et al., 2004). There is some evidence that p38 may also promote apoptosis by blocking ERK1/2 signaling cascades associated with cell survival (Porras et al., 2004). Blocking p38 MAPK activity has been associated with protection against noise trauma, aminoglycoside, cisplatin, radiation and TNFα-induced ototoxicity (Wei et al., 2005; Tabuchi et al., 2010; Bas et al., 2012a,b; Park et al., 2012; Wang et al., 2012a,b; Maeda et al., 2013; Kim et al., 2014; Kurioka et al., 2014; Shin et al., 2014).
JNK has three isoforms: JNK1, JNK2, and JNK3. JNK1 and JNK2 are expressed ubiquitously while JNK3 is primarily localized to cardiac and neuronal tissues. In response to an extracellular stimulus, JNK signaling can promote apoptosis through two distinct mechanisms: one targeting the nucleus and the other targeting the mitochondria. JNK can translocate to the nucleus and phosphorylate c-Jun and other transcription factors such as p53, subsequently promoting expression of pro-apoptotic genes (such as TNFα, FasL, Bak, Bim, and Bax) and blocking transcription of anti-apoptotic genes (such as Bcl-2) (Faris et al., 1998; Budhram-Mahadeo et al., 1999; Eichhorst et al., 2000; Fan et al., 2001; Whitfield et al., 2001; Liu and Lin, 2005; Oleinik et al., 2007; Dhanasekaran and Reddy, 2008; Amaral et al., 2010). The JNK-mediated upregulation of pro-inflammatory and pro-apoptotic proteins and downregulation of pro-survival proteins can provide the necessary substrates for fueling pro-death cellular events.
In addition, activated JNK can phosphorylate a number of proteins that are intimately involved in mitochondrial cell death. By phosphorylating the pro-death proteins Bim and Bad, activated JNK can neutralize Bcl-2 and Bcl-xL protein inhibition while promoting Bax activation of intrinsic apoptotic cell death (Ottilie et al., 1997; Puthalakath et al., 1999; Donovan et al., 2002; Lei et al., 2002; Lei and Davis, 2003; Wang et al., 2007a,b). Furthermore, phosphorylated JNK can indirectly trigger pro-apoptotic Bax-mediated mitochondrial death signaling through truncation of Bid. The constellation of these events results in release of cyt c, Smac, and other mitochondrial pro-death proteins into the cytoplasm that promote caspase-dependent and -independent mechanisms of programmed cell death (Tournier et al., 2000; Madesh et al., 2002; Deng et al., 2003; Dhanasekaran and Reddy, 2008).
Although JNK activity is primarily linked to apoptotic cell death, prolonged JNK activation is thought to divert TNFα-induced cell death from apoptosis to necrosis. Elevated levels of ROS can promote prolonged JNK activation, while prolonged JNK signaling can lead to increased mitochondrial production of ROS, creating a positive feedback loop that favors TNFα-induced necrotic cell death (Sakon et al., 2003; Wajant et al., 2003; Ventura et al., 2004).
Similar to p38, JNK signaling is implicated in noise, aminoglycoside, cisplatin, radiation and TNFα-initiated loss of auditory HCs and with a resultant hearing loss. The insertion of an electrode array of a cochlea implant into the cochlea also exhibits effects of JNK and downstream molecular events. Direct inhibition of JNK by either CEP-1347 or D-JNKI-1 (aka AM-111) have demonstrated protection against HC and hearing losses following aminoglycoside, acoustic, and electrode insertion injury to the cochlea (Ylikoski et al., 2002; Wang et al., 2003, 2007a,b; Eshraghi et al., 2006, 2010; Jiang et al., 2006; Suckfuell et al., 2007). Indirect modulation of JNK activity by various drug therapies have been associated with HC and hearing protection (Nakamagoe et al., 2010; Pyun et al., 2011; Shin et al., 2014).
Oxidative Stress
ROS are free radicals containing oxygen. They are very reactive and in high amounts can damage cells. Extracellular ROS can be generated by activated phagocytes such as neutrophils, monocytes, and macrophages, and play a vital role in host defense against pathogens (Evans and Halliwell, 1999). Intracellular ROS are primarily produced by the cell’s mitochondria (Turrens, 2003). Oxygen-derived free radicals can induce apoptosis and necrosis by peroxidation of a cell’s phospholipid membranes, proteolytic degradation, and DNA damage in mitochondria and in the nucleus (Beckman and Ames, 1997; Kohen and Nyska, 2002).
Oxygen (O2) contains two unpaired electrons. Superoxide anion () is the product of one electron reduction of O2 and is the precursor for several ROS. can react with nitric oxide and produce RNS, such as peroxynitrite, that are also detrimental to cell viability (Beckman and Koppenol, 1996).
The main enzymatic source of is NAD phosphate (NADPH) oxidases, which are located on the cellular membrane of phagocytes. NADPH oxidases can transfer electrons from NADPH to O2 to produce (Dworakowski et al., 2006). The mitochondrial electron transport chain (ETC) is the predominant non-enzymatic supplier of . The ETC involves several redox enzymes that act in sequence to couple electron transfer to proton translocation across the mitochondrial membrane, creating a transmembrane electrochemical proton gradient. This gradient is crucial for driving ATP synthesis and cellular events that depend on ATP as an energy source. The ETC can leak electrons to O2, partially reducing this molecule to (Turrens, 2003). Overproduction of ROS can promote cell death following various stress signals.
Classically, ROS is not associated with the DR (death receptor)-mediated apoptosis; however there is some recent evidence that oxidative stress can turn on extrinsic cell death signaling. This again demonstrates the many levels of communication that occur both upstream and downstream in events of programmed cell death. ROS can activate apoptosis signal-regulating kinase-1 (ASK-1), which is a MAPKKK that can phosphorylate and activate mediators of the JNK and p38 pathways of extrinsic programmed cell death (Nagai et al., 2007). ROS may also promote JNK-mediated extrinsic apoptosis through oxidation and inhibition of MAPK phosphatases that normally suppress JNK activity (Kamata et al., 2005).
Intrinsic cell death can also be affected by oxidative stress. High levels of ROS can initiate nuclear accumulation of FOXO3 (O subclass of the forkhead family of transcription factors) that can trigger up regulation of genes important for cell death such as Bim and Bcl-6 (Hagenbuchner et al., 2012; Sinha et al., 2013). FOXO3 can promote Bim translocation to the mitochondria and knockdown of anti-apoptotic protein Bcl-xL, which can modulate Bax-mediated mitochondrial cell death (Sedlak et al., 1995; Khawaja et al., 2008). It is hypothesized that FOXO3 can regulate the expression and activation of various pro-apoptotic and pro-survival proteins of the Bcl-2 family that can lead to mitochondrial stress, transient opening of the MPT pore, collapse of the mitochondrial membrane potential, release of mitochondrial pro-death proteins into the cytoplasm, and transient increases in ROS from the ETC. This downstream mitochondrial production of ROS can promote a positive feedback loop that converges into apoptosis (Sinha et al., 2013). In this positive response, overproduction of ROS can then promote more oxidation of lipids, proteins, and nucleic acids, disruption of mitochondrial integrity and induction of apoptosis and possibly necrosis. In addition, these free radicals can also oxidize cardiolipin, which results in tBID binding of the voltage-dependent anion channel (VDAC) of the MPT pore and downstream events associated with intrinsic apoptosis (Kagan et al., 2005).
Cochlear tissues produce high levels of ROS in response to a variety of challenges, such as gentamicin, radiation, cisplatin, electrode insertion trauma, and noise exposure (Henderson et al., 2006; Rybak et al., 2007; Choung et al., 2009; Low et al., 2009; Bas et al., 2012a,b). Cells can counteract the effects of oxidative stress through an antioxidant defense system that comprises free radical scavengers and antioxidant enzymes such as reduced glutathione (GSH), superoxide dismutases, and catalase. Insufficient levels of antioxidants such as GSH and vitamins A, C, and E are also associated with oxidative stress. It is the imbalance produced by ROS and antioxidant activity that can help determine the fate of an affected cell. Antioxidants and free-radical scavengers that have demonstrated protection against cochlear injury include iron chelators, manipulation of dietary glutathione, D-Methionine, lipoic acid, N-acetylcysteine, alpha-tocopherol, and m40403 (a superoxide dismutase mimetic), resveratrol, nitro-L-arginine methyl ester (L-NAME) and co-enzyme (Evans and Halliwell, 1999; Ohlemiller et al., 1999, 2000; Huang et al., 2000; Teranishi et al., 2001; Seidman et al., 2003; Nicotera et al., 2004; Samson et al., 2008; Fetoni et al., 2009; Rewerska et al., 2013). Dexamethasone is a synthetic steroid that has demonstrated beneficial effects against several inner ear disorders; dexamethasone can block the expression of ROS as well as reduce the extracellular inflammatory response and block extrinsic cell death signaling through activation of NFkB (Bas et al., 2012a).
Acoustic Trauma and Auditory HC Death
Although a number of stressors can initiate pro-inflammatory and pro-cell death signaling within the cochlea, the most studied insult to the inner ear is acoustic trauma. Acoustic trauma initiates a sequence of events within the cochlea that can culminate in apoptosis and regulated necrosis of auditory HCs (Figure 4).
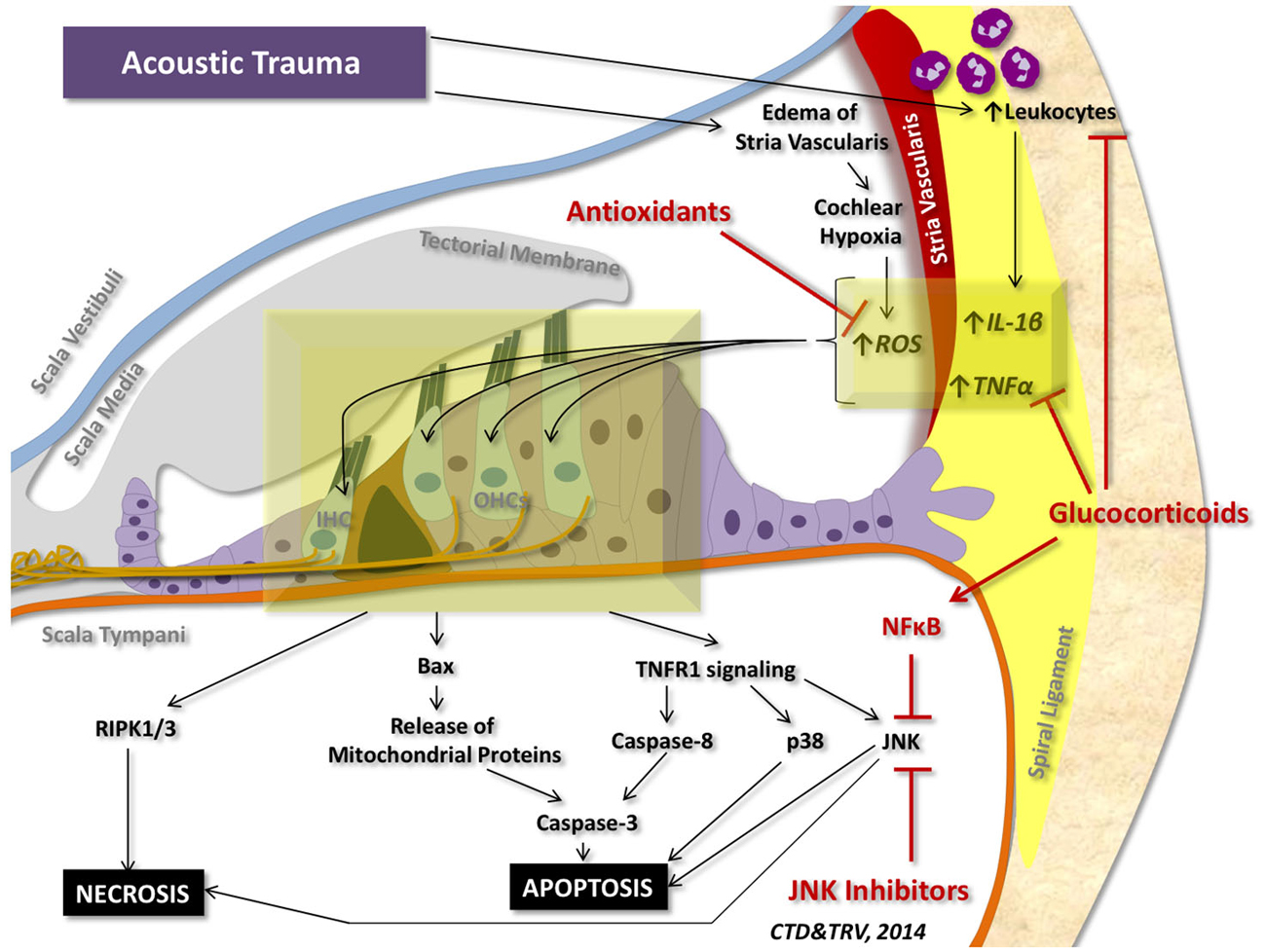
Figure 4. Cell death signaling following acoustic trauma. Acoustic trauma can promote edema of the stria vascularis, resulting in cochlear hypoxia, formation of ROS, and oxidative stress. Noise trauma can also stimulate the spiral ligament to express cytokines (such as TNFα and IL-1β) and chemokines that initiate migration of leukocytes to the cochlea; leukocytes will then release a number of other pro-inflammatory factors and free radicals that propagate the inflammatory process. Oxidative stress and pro-inflammatory cytokines can travel to the inner hair cells (IHC) and outer hair cells (OHCs) of the organ of Corti and induce intrinsic and extrinsic apoptotic signaling cascades. Prolonged activation of JNK and induction of RIPK1/3 activity can promote necrotic cell death. Glucocorticoids have demonstrated protection against acoustic trauma, in part by reducing leukocyte migration, decreasing expression of pro-inflammatory factors, and activating NFκB inhibition of JNK signaling. Antioxidants and free radical scavengers can neutralize ROS and downstream effects of oxidative stress. Inhibitors of JNK can also mitigate noise-induced loss of auditory HCs by preventing downstream signaling cascades that lead to apoptosis and necrosis.
Loud noise exposures produce large displacements of the tympanic membrane and propagate waves of mechanical energy into the inner ear. These intense pulses can produce rapid displacement of cochlear fluid within the inner ear, causing shearing force damage to the OC and basilar membrane, injuring auditory HCs, and altering the endocochlear potential (EP). Acoustic trauma can also initiate inflammation and edema of the stria vascularis and compromise the blood supply to the cochlea (Smith et al., 1985). Since there is minimal redundancy in cochlear blood flow, even transient reductions in cochlear blood flow can result in hypoxia and injury to auditory HCs. Injuries to the stria vascularis and spiral ligament occur following noise trauma, damaging type II and type IV fibrocytes that are important for maintenance of theEP) and lead to permanent hearing loss (Hirose and Liberman, 2003).
Acoustic trauma can lead to ROS release by the marginal cells of the stria vascularis as a result of reductions in cochlear blood flow and cochlear hypoxia (Yamane et al., 1995; Ohlemiller et al., 1999). It is controversial how hypoxia results in oxidative stress; however, it may be related to an increase in oxygen demand by auditory HCs followed by increased aerobic respiration by mitochondria to produce ATP and production of free radical byproducts via the ETC (Yamane et al., 1995; Taylor, 2008). The lateral wall structures of the cochlea also express cytokines (such as TNFα and IL-1β) and chemokines following acoustic trauma, which stimulate migration of leukocytes into the cochlea via the spiral modiolar vein (Keithley et al., 2008). These inflammatory cells will in turn release more cytokines and chemokines as well as produce ROS and RNS that will propagate the inflammatory process in the inner ear.
When TNFα binds to its complementary receptor TNFR1, TRADD and FADD are recruited along with caspase-8. Caspase-8 can activate executioner caspase-3 to promote extrinsic cell death through DNA fragmentation and chromatin condensation via CAD, ACINUS, and HELI-CARD or it can cleave BID into tBID, which can activate Bax-mediated intrinsic, mitochondrial cell death (Liu et al., 1997; Enari et al., 1998; Li et al., 1998; Kovacsovics et al., 2002; Nicotera et al., 2003; Wang et al., 2003; Murai et al., 2008; Jamesdaniel et al., 2011; Infante et al., 2012). Furthermore, loud noise exposure can upregulate TNFα-mediated p38 and JNK signaling in the sensory epithelium of the inner ear that promotes transcription of pro-death genes and Bax-mediated mitochondrial release of apoptotic proteins such as cyt c (Wang et al., 2007a,b; Dinh et al., 2008a,b; Infante et al., 2012; Jamesdaniel et al., 2011). Fas-mediated apoptosis also occurs following acoustic trauma (Jamesdaniel et al., 2011).
Oxidative stress from noise exposure can also initiate intrinsic apoptotic cell death in auditory HCs, resulting in mitochondrial release of cyt c into the cytoplasm, generation of apoptosomes, and activation of caspase-3 dependent cell death. AIF and EndoG are also released into the cytosol, translocate into the nucleus, and can initiate chromatin condensation and DNA fragmentation of auditory HCs (Nicotera et al., 2003; Yamashita et al., 2004a,b; Han et al., 2006; Henderson et al., 2006).
In addition to apoptosis, acoustic trauma can result in regulated necrosis through RIPK3/RIPK1 activation in rats, that was reversed with necrosis inhibitor necrostatin-1 (Zheng et al., 2014).
Acoustic trauma can also promote swelling and rupture of dendritic terminals of cochlear nerve afferent fibers (Spoendlin, 1971). Intense noise exposure can trigger inner HCs to release significant amounts of glutamate into the synapses with type I fibers of the cochlear nerve. The glutamate overwhelms the postsynaptic glutamate receptors and results in ion influx into dendritic terminals of the cochlear nerve that leads to loss of auditory nerve fibers (Pujol et al., 1990; Kujawa and Liberman, 2009).
Furthermore, intense noise exposure can increase intracellular calcium in auditory HCs and activate calcium-dependent calpains—cysteine proteases that promote proteolysis and breakdown of cytoskeletal and membrane proteins, kinases, phosphatases, and transcription factors (Wang et al., 1999). Subsequently, calpain cleaves and activates calcineurin that promotes Bad-mediated mitochondrial release of apoptotic factors that lead to intrinsic cell death (Le Prell et al., 2007).
Otoprotective drugs can target different levels in apoptosis and necrosis signaling pathways following acoustic trauma (Figure 4). Glucocorticoids can reduce extracellular inflammatory cell trafficking, reduce the level of TNFα expression in spiral ligament fibrocytes, and bind to its glucocorticoid receptors in auditory HCs to activate pro-survival NFkB signaling and inhibit JNK signaling. Additionally, they can reduce oxidative stress, and regulate pro-death and pro-survival gene transcription (Maeda et al., 2005; Tahera et al., 2006; Dinh et al., 2008a,b, 2011; Bas et al., 2012b). Regulation of MAPK signaling can also protect against noise-induced hearing loss by reducing p38 and JNK MAPKs (Tabuchi et al., 2010). In particular, direct and indirect inhibitors of JNK can block JNK phosphorylation of c-Jun and p53 and suppress downstream activation of intrinsic and extrinsic forms of apoptosis and potentially necrosis following acoustic trauma (Sakon et al., 2003; Wang et al., 2003, 2007a,b; Ventura et al., 2004; Coleman et al., 2007; Suckfuell et al., 2007). Lastly, antioxidants and free-radical scavengers can neutralize ROS that is responsible for lipid peroxidation of cell membranes, protein degradation, DNA injury and other mechanisms related to oxidative stress induced apoptosis and necrosis of noise-injured auditory HCs (Seidman et al., 2003; Nicotera et al., 2004; Nordang and Anniko, 2005; Samson et al., 2008; Fetoni et al., 2009; Rewerska et al., 2013).
Conclusion
Numerous diverse insults to the inner ear can cause auditory HC damage and hearing loss. The evolutionarily conserved apoptotic and necrotic cell death signaling that occurs in auditory HCs is shared among many ototoxic and traumatic stressor events. The most well studied molecular mechanisms behind cell death in auditory HCs involve TNFα signaling, JNK and p38 activation and the effect of high levels of oxidative stress. Although their effects on intrinsic and extrinsic pathways of apoptosis have been studied extensively, there are likely many levels of cross communication between signaling cascades that are still undiscovered. Research in this area is becoming more prevalent, as well as research into mechanisms of regulated necrosis in auditory HCs. A number of otoprotective drug therapies target different levels along this pro-inflammatory pro-death signaling cascade to block downstream events that lead to cell death and promote auditory HC viability and hearing protection.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Amaral, J. D., Xavier, J. M., Steer, C. J., and Rodrigues, C. M. (2010). The role of p53 in apoptosis. Discov. Med. 9, 145–152.
Aminpour, S., Tinling, S. P., and Brodie, H. A. (2005). Role of tumor necrosis factor-alpha in sensorineural hearing loss after bacterial meningitis. Otol. Neurotol. 26, 602–609. doi: 10.1097/01.mao.0000178121.28365.0d
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Andrabi, S. A., Dawson, T. M., and Dawson, V. L. (2008). Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann. N Y Acad. Sci. 1147, 233–241. doi: 10.1196/annals.1427.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baines, C. P., Kaiser, R. A., Purcell, N. H., Blair, N. S., Osinska, H., Hambleton, M. A., et al. (2005). Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662. doi: 10.1038/nature03434
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bas, E., Gupta, C., and Van De Water, T. R. (2012a). A novel organ of corti explant model for the study of cochlear implantation trauma. Anat. Rec. (Hoboken) 295, 1944–1956. doi: 10.1002/ar.22585
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bas, E., Martinez-Soriano, F., Láinez, J. M., and Marco, J. (2009). An experimental comparative study of dexamethasone, melatonin and tacrolimus in noise-induced hearing loss. Acta. Otolaryngol. 129, 385–389. doi: 10.1080/00016480802566279
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bas, E., Van De Water, T. R., Gupta, C., Dinh, J., Vu, L., Martínez-Soriano, F., et al. (2012b). Efficacy of three drugs for protecting against gentamicin-induced hair cell and hearing losses. Br. J. Pharmacol. 166, 1888–1904. doi: 10.1111/j.1476-5381.2012.01890.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Beckman, K. B., and Ames, B. N. (1997). Oxidative decay of DNA. J. Biol. Chem. 272, 19633–19636. doi: 10.1074/jbc.272.32.19633
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Beckman, J. S., and Koppenol, W. H. (1996). Nitric oxide, superoxide and peroxynitrite: the good, the bad and ugly. Am. J. Physiol. 271, C1424–C1437.
Bodmer, J. L., Burns, K., Schneider, P., Hofmann, K., Steiner, V., Thome, M., et al. (1997). TRAMP, a novel apoptosis-mediating receptor with sequence homology to tumor necrosis factor receptor 1 and Fas (Apo-1/CD95). Immunity 6, 79–88. doi: 10.1016/s1074-7613(00)80244-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bratton, S. B., Walker, G., Srinivasula, S. M., Sun, X. M., Butterworth, M., Alnemri, E. S., et al. (2001). Recruitment, activation and retention of caspases-9 and -3 by Apaf-1 apoptosome and associated XIAP complexes. EMBO J. 20, 998–1009. doi: 10.1093/emboj/20.5.998
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brenner, C., and Grimm, S. (2006). The permeability transition pore complex in cancer cell death. Oncogene 25, 4744–4756. doi: 10.1038/sj.onc.1209609
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brough, D., and Rothwell, N. J. (2007). Caspase-1-dependent processing of pro-interleukin-1beta is cytosolic and precedes cell death. J. Cell Sci. 120, 772–781. doi: 10.1242/jcs.03377
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Budhram-Mahadeo, V., Morris, P. J., Smith, M. D., Midgley, C. A., Boxer, L. M., and Latchman, D. S. (1999). P53 suppresses the activation of the Bcl-2 promoter by the Brn-3a POU family transcription factor. J. Biol. Chem. 274, 15237–15244. doi: 10.1074/jbc.274.21.15237
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cai, B., Chang, S. H., Becker, E. B., Bonni, A., and Xia, Z. (2006). p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65. J. Biol. Chem. 281, 25215–25222. doi: 10.1074/jbc.m512627200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cain, K., Bratton, S. B., Langlais, C., Walker, G., Brown, D. G., Sun, X. M., et al. (2000). Apaf-1 oligomerizes into biologically active approximately 700-kDa and inactive approximately 1.4-MDa apoptosome complexes. J. Biol. Chem. 275, 6067–6070. doi: 10.1074/jbc.275.9.6067
Capano, M., and Crompton, M. (2006). Bax translocates to mitochondria of heart cells during simulated ischaemia: involvement of AMP-activated and p38 mitogen-activated protein kinases. Biochem. J. 395, 57–64. doi: 10.1042/bj20051654
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chai, J., Du, C., Wu, J. W., Kyin, S., Wang, X., and Shi, Y. (2000). Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 406, 855–862. doi: 10.1038/35022514
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chai, J., Shiozaki, E., Srinivasula, S. M., Wu, Q., Datta, P., Alnemri, E. S., et al. (2001). Structural basis of caspase-7 inhibition by XIAP. Cell 104, 769–780. doi: 10.1016/s0092-8674(02)02034-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chandler, J. M., Cohen, G. M., and MacFarlane, M. (1998). Different subcellular distribution of caspase-3 and caspase-7 following Fas-induced apoptosis in mouse liver. J. Biol. Chem. 273, 10815–10818. doi: 10.1074/jbc.273.18.10815
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, Z., Gibson, T. B., Robinson, F., Silvestro, L., Pearson, G., Xu, B., et al. (2001). MAP kinases. Chem. Rev. 101, 2449–2476. doi: 10.1021/cr000241p
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chinnaiyan, A. M., O’Rourke, K., Tewari, M., and Dixit, V. M. (1995). FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 81, 505–512. doi: 10.1016/0092-8674(95)90071-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Choung, Y. H., Taura, A., Pak, K., Choi, S. J., Masuda, M., and Ryan, A. F. (2009). Generation of highly-reactive oxygen species is closely related to hair cell damage in rat organ of Corti treated with gentamicin. Neuroscience 161, 214–226. doi: 10.1016/j.neuroscience.2009.02.085
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Coffin, A. B., Rubel, E. W., and Raible, D. W. (2013). Bax, Bcl2 and p53 differentially regulate neomycin- and gentamicin-induced hair cell death in the zebrafish lateral line. J. Assoc. Res. Otolaryngol. 14, 645–659. doi: 10.1007/s10162-013-0404-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Coleman, J. N. K., Littlesunday, C., Jackson, R., and Meyer, T. (2007). AM-111 protects against permanent hearing loss from impulse noise trauma. Hear. Res. 226, 70–78. doi: 10.1016/j.heares.2006.05.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cotanche, D. A., Hennig, A. K., Riedl, A. E., and Messana, E. P. (1997). “Hair cell regeneration in the chick cochlea – were we stand after 10 years of work,” in Psychophysical and Physiological Advances in Hearing: Proceedings of the 11th International Symposium on Hearing, eds A. R. Palmer, A. Rees, A. Q. Summerfield and A. Meddis (London: Whurr), 109–115.
Daugas, E., Susin, S. A., Zamzami, N., Ferri, K. F., Irinopoulou, T., Larochette, N., et al. (2000). Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 14, 729–739.
Deng, Y., Ren, X., Yang, L., Lin, Y., and Wu, X. (2003). A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell 115, 61–70. doi: 10.1016/s0092-8674(03)00757-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Deshmane, S. L., Kremlev, S., Amini, S., and Sawaya, B. E. (2009). Monocyte chemoattractant protein-1 (MCP-1): an overview. J. Interferon Cytokine Res. 29, 313–326. doi: 10.1089/jir.2008.0027
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Deveraux, Q. L., Leo, E., Stennicke, H. R., Welsh, K., Salvesen, G. S., and Reed, J. C. (1999). Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 18, 5242–5251. doi: 10.1093/emboj/18.19.5242
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dhanasekaran, D. N., and Reddy, E. P. (2008). JNK signaling in apoptosis. Oncogene 27, 6245–6251. doi: 10.1038/onc.2008.301
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ding, D., Jiang, H., and Salvi, R. J. (2010). Mechanisms of rapid sensory hair-cell death following co-administration of gentamicin and ethacrynic acid. Hear. Res. 259, 16–23. doi: 10.1016/j.heares.2009.08.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dinh, C. T., Bas, E., Chan, S. S., Dinh, J. N., Vu, L., and Van De Water, T. R. (2011). Dexamethasone treatment of tumor necrosis factor-alpha challenged organ of Corti explants activates nuclear factor kappa B signaling that induces changes in gene expression that favor hair cell survival. Neuroscience 188, 157–167. doi: 10.1016/j.neuroscience.2011.04.061
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dinh, C., Bas, E., Dinh, J., Vu, L., Gupta, C., and Van De Water, T. R. (2013). Short interfering RNA against Bax attenuates TNFα-induced ototoxicity in rat organ of Corti explants. Otolaryngol. Head Neck Surg. 148, 834–840. doi: 10.1177/0194599813477631
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dinh, C. T., Haake, S., Chen, S., Hoang, K., Nong, E., Eshraghi, A. A., et al. (2008b). Dexamethasone protects organ of corti explants against tumor necrosis factor-alpha-induced loss of auditory hair cells and alters the expression levels of apoptosis-related genes. Neuroscience 157, 405–413. doi: 10.1016/j.neuroscience.2008.09.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dinh, C., Hoang, K., Haake, S., Chen, S., Angeli, S., Nong, E., et al. (2008a). Biopolymer-released dexamethasone prevents tumor necrosis factor alpha-induced loss of auditory hair cells in vitro: implications toward the development of a drug-eluting cochlear implant electrode array. Otol. Neurotol. 29, 1012–1019. doi: 10.1097/mao.0b013e3181859a1f
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nanopoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Donovan, N., Becker, E. B., Konishi, Y., and Bonni, A. (2002). JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J. Biol. Chem. 277, 40944–40949. doi: 10.1074/jbc.m206113200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dworakowski, R., Anilkumar, N., Zhang, M., and Shah, A. M. (2006). Redox signaling involving NADPH oxidase-derived reactive oxygen species. Biochem. Soc. Trans. 34, 960–964. doi: 10.1042/bst0340960
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eichhorst, S. T., Müller, M., Li-Weber, M., Schulze-Bergkamen, H., Angel, P., and Krammer, P. H. (2000). A novel AP-1 element in the CD95 ligand promoter is required for induction of apoptosis in hepatocellular carcinoma cells upon treatment with anticancer drugs. Mol. Cell. Biol. 20, 7826–7837. doi: 10.1128/mcb.20.20.7826-7837.2000
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Enari, M., Sakahira, H., Yokoyama, H., Okawa, K., Iwamatsu, A., and Nagata, S. (1998). A caspase-activated DNase that degrades DNA during apoptosis and its inhibitos ICAD. Nature 391, 43–50. doi: 10.1038/34112
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eshraghi, A. A., He, J., Mou, C. H., Polak, M., Zine, A., Bonny, C., et al. (2006). D-JNKI-1 treatment prevents the progression of hearing loss in a model of cochlear implantation trauma. Otol. Neurotol. 27, 504–511. doi: 10.1097/00129492-200606000-00012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eshraghi, A. A., Hoosien, G., Ramsay, S., Dinh, C. T., Bas, E., Balkany, T. J., et al. (2010). Inhibition of the JNK signal cascade conserves hearing against electrode insertion trauma-induced loss. Cochlear Implants Int. 11, 104–109. doi: 10.1179/146701010x12671177544104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Evans, P., and Halliwell, B. (1999). Free radicals and hearing. Cause, consequence and criteria. Ann. N Y Acad. Sci. 884, 19–40. doi: 10.1111/j.1749-6632.1999.tb08633.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fan, M., Goodwin, M. E., Birrer, M. J., and Chambers, T. C. (2001). The c-Jun NH(2)-terminal protein kinase/AP-1 pathway is required for efficient apoptosis induced by vinblastine. Cancer Res. 61, 4450–4458.
Faris, M., Kokot, N., Latinis, K., Kasibhatla, S., Green, D. R., Koretzky, G. A., et al. (1998). The c-Jun N-terminal kinase cascade plays a role in stress-induced apoptosis in Jurkat cells by up-regulating Fas ligand expression. J. Immunol. 160, 134–144.
Fernandes-Alnemri, T., Wu, J., Yu, J. W., Datta, P., Miller, B., Jankowski, W., et al. (2007). The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14, 1590–1604. doi: 10.1038/sj.cdd.4402194
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fetoni, A. R., Piacentini, R., Fiorita, A., Paludetti, G., and Troiani, D. (2009). Water-solube Coenzyme Q10 formulation (Q-ter) promotes outer hair cell survival in a guinea pig model of noise-induced hearing loss (NIHL). Brain Res. 1257, 108–116. doi: 10.1016/j.brainres.2008.12.027
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Figari, I. S., Mori, N. A., and Palladino, M. A. Jr. (1987). Regulation of neutrophil migration and superoxide production by recombinant tumor necrosis factors-alpha and -beta: comparison to recombinant interferon-gamma and interleukin-1 alpha. Blood 70, 979–984.
Fink, S. L., and Cookson, B. T. (2006). Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 8, 1812–1825. doi: 10.1111/j.1462-5822.2006.00751.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fischer, U., Stroh, C., and Schulze-Osthoff, K. (2006). Unique and overlapping substrate specificities of caspase-8 and caspase-10. Oncogene 25, 152–159. doi: 10.1038/sj.onc.1209015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fujioka, M., Kanzaki, S., Okano, H. J., Masuda, M., Ogawa, K., and Okano, H. (2006). Proinflammatory cytokines expression in noise-induced damaged cochlea. J. Neurosci. Res. 83, 575–583. doi: 10.1002/jnr.20764
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Galluzzi, L., Vitale, I., Abrams, J. M., Alnemri, E. S., Baehreck, E. H., Blagosklonny, M. V., et al. (2012). Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 19, 107–120. doi: 10.1038/cdd.2011.96
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guicciardi, M. E., and Gores, G. J. (2009). Life and death by death receptors. FASEB J. 23, 1625–1637. doi: 10.1096/fj.08-111005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Haake, S. M., Dinh, C. T., Chen, S., Eshraghi, A. A., and Van De Water, T. R. (2009). Dexamethasone protects auditory hair cells against TNFalpha-initiated apoptosis via activation of PI3K/Akt and NFkappaB signaling. Hear. Res. 255, 22–32. doi: 10.1016/j.heares.2009.05.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hagenbuchner, J., Kuznetsov, A., Hermann, M., Hausott, B., Obexer, P., and Ausserlechner, M. J. (2012). FOXO3-induced reactive oxygen species are regulated by BCL2L11 (Bim) and SESN3. J. Cell Sci. 125, 1191–1203. doi: 10.1242/jcs.092098
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Han, W., Shi, X., and Nuttall, A. L. (2006). AIF and endoG translocation in noise exposure induced hair cell death. Hear. Res. 211, 85–95. doi: 10.1016/j.heares.2005.10.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
He, S., Wang, L., Miao, L., Wang, T., Du, F., Zhao, L., et al. (2009). Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111. doi: 10.1016/j.cell.2009.05.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hegde, R., Srinivasula, S. M., Zhang, Z., Wassell, R., Mukattash, R., Cilenti, L., et al. (2002). Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. 277, 432–438. doi: 10.1074/jbc.M109721200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Henderson, D., Bielefeld, E. C., Harris, K. C., and Hu, B. H. (2006). The role of oxidative stress in noise-induced hearing loss. Ear Hear. 27, 1–19. doi: 10.1097/01.aud.0000191942.36672.f3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hirose, K., and Liberman, M. C. (2003). Lateral wall histophatology and endoncochlear potential in the noise-damage mouse cochlea. J. Assoc. Res. Otolaryngol. 4, 339–352. doi: 10.1007/s10162-002-3036-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hirose, K., Westrum, L. E., Cunningham, D. E., and Rubel, E. W. (2004). Electron microscopy of degenerative changes in the chick basilar papilla after gentamicin exposure. J. Comp. Neurol. 470, 164–180. doi: 10.1002/cne.11046
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hsu, S. C., Gavrilin, M. A., Tsai, M. H., Han, J., and Lai, M. Z. (1999). p38 mitogen-activated protein kinase is involved in Fas ligand expression. J. Biol. Chem. 274, 25769–25776. doi: 10.1074/jbc.274.36.25769
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hsu, H., Xiong, J., and Goeddel, D. V. (1995). The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 81, 495–504. doi: 10.1016/0092-8674(95)90070-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hu, B. H., Henderson, D., and Nicotera, T. M. (2006). Extremely rapid induction of outer hair cell apoptosis in the chinchilla cochlea following exposure to impulse noise. Hear. Res. 211, 16–25. doi: 10.1016/j.heares.2005.08.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huang, T., Cheng, A. G., Stupak, H., Liu, W., Kim, A., Staecker, H., et al. (2000). Oxidative stress-induced apoptosis of cochlear sensory cells: otoprotective strategies. Int. J. Dev. Neurosci. 18, 259–270. doi: 10.1016/s0736-5748(99)00094-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ichimiya, I., Yoshida, K., Hirano, T., Suzuki, M., and Mogi, G. (2000). Significance of spiral ligament fibrocytes with cochlear inflammation. Int. J. Pediatr. Otorhinolaryngol. 56, 45–51. doi: 10.1016/s0165-5876(00)00408-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ichimiya, I., Yoshida, K., Suzuki, M., and Mogi, G. (2003). Expression of adhesion molecules by cultured spiral ligament fibrocytes stimulated with proinflammatory cytokines. Ann. Otol. Rhinol. Laryngol. 112, 722–728. doi: 10.1177/000348940311200813
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Infante, E. B., Channer, G. A., Telischi, F. F., Gupta, C., Dinh, J. T., Vu, L., et al. (2012). Mannitol protects hair cells against tumor necrosis factor α-induced loss. Otol. Neurotol. 33, 1656–1663. doi: 10.1097/mao.0b013e31826bedd9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Itoh, N., and Nagata, S. (1993). A novel protein domain required for apoptosis mutational analysis of human Fas antigen. J. Biol. Chem. 268, 10932–10937.
Jamesdaniel, S., Hu, B., Kermany, M. H., Jiang, H., Ding, D., Coling, D., et al. (2011). Noise induced changes in the expression of p38/MAPK signaling proteins in the sensory epithelium of the inner ear. J. Proteomics 75, 410–424. doi: 10.1016/j.jprot.2011.08.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jiang, H., Sha, S. H., Forge, A., and Schacht, J. (2006). Caspase-independent pathways of hair cell death induced by kanamycin in vivo. Cell Death Differ. 13, 20–30. doi: 10.1038/sj.cdd.4401706
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kagan, V. E., Tyurin, V. A., Jiang, J., Tyurina, Y. Y., Ritov, V. B., Amoscato, A. A., et al. (2005). Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 1, 223–232. doi: 10.1038/nchembio727
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kamata, H., Honda, S., Maeda, S., Chang, L., Hirata, H., and Karin, M. (2005). Reactive oxygen species promotes TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120, 649–661. doi: 10.1016/j.cell.2004.12.041
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kandasamy, K., Srinivasula, S. M., Alnemri, E. S., Thompson, C. B., Korsmeyer, S. J., Bryant, J. L., et al. (2003). Involvement of proapoptotic molecules Bax and Bak in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced mitochondrial disruption and apoptosis: differential regulation of cytochrome c and Smac/DIABLo release. Cancer Res. 63, 1712–1721.
Keithley, E. M., Wang, X., and Barkdull, G. C. (2008). Tumor necrosis factor alpha can induce recruitment of inflammatory cells to the cochlea. Otol. Neurotol. 29, 854–859. doi: 10.1097/mao.0b013e31818256a9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kepp, O., Galluzzi, L., Zitvogel, L., and Kroemer, G. (2010). Pyroptosis: a cell death modality of its kind. Eur. J. Immunol. 40, 627–630. doi: 10.1002/eji.200940160
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keshet, Y., and Seger, R. (2010). The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 661, 3–38. doi: 10.1007/978-1-60761-795-2_1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Khawaja, N. R., Carré, M., Kovacic, H., Estève, M. A., and Braguer, D. (2008). Patupilone-induced apoptosis is mediated by mitochondrial reactive oxygen species through Bim relocalization to mitochondria. Mol. Pharmacol. 74, 1072–1083. doi: 10.1124/mol.108.048405
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, J., Cho, H. J., Sagong, B., Kim, S. J., Lee, J. T., So, H. S., et al. (2014). Alpha-lipoic acid protects against cisplatin-induced ototoxicity via the regulat of MAPKs and proinflammatory cytokines. Biochem. Biophys. Res. Commun. 449, 183–189. doi: 10.1016/j.bbrc.2014.04.118
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kohen, R., and Nyska, A. (2002). Oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions and methods for their quantification. Toxicol. Pathol. 30, 620–650. doi: 10.1080/01926230290166724
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Korsmeyer, S. J., Wei, M. C., Saito, M., Weiler, S., Oh, K. J., and Schlesinger, P. H. (2000). Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 7, 1166–1173. doi: 10.1038/sj.cdd.4400783
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kovacsovics, M., Martinon, F., Micheau, O., Bodmer, J. L., Hofmann, K., and Tschopp, J. (2002). Overexpression of Helicard, a CARD-containing helicase cleaved during apoptosis, accelerates DNA degradation. Curr. Biol. 12, 838–843. doi: 10.1016/s0960-9822(02)00842-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kroemer, G., Galluzzi, L., and Brenner, C. (2007). Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99–163. doi: 10.1152/physrev.00013.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kuhn, M., Heman-Ackah, S. E., Shaikh, J. A., and Roehm, P. C. (2011). Sudden sensorineural hearing loss: a review of diagnosis, treatment and prognosis. Trends Amplif. 15, 91–105. doi: 10.1177/1084713811408349
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kujawa, S. G., and Liberman, M. C. (2009). Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J. Neurosci. 29, 14077–14085. doi: 10.1523/JNEUROSCI.2845-09.2009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kurioka, T., Matsunobu, T., Niwa, K., Tamura, A., Satoh, Y., and Shiotani, A. (2014). Activated protein C rescues the cochlea from noise-induced hearing loss. Brain Res. 1583, 201–210. doi: 10.1016/j.brainres.2014.07.052
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kuwana, T., Mackey, M. R., Perkins, G., Ellisman, M. H., Latterich, M., Schneiter, R., et al. (2002). Bid, Bax and lipids cooperat to form supramolecula ropenings in the outer mitochondrial membrane. Cell 111, 331–342. doi: 10.1016/s0092-8674(02)01036-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kyriakis, J. M., and Avruch, J. (2001). Mammalian mitogen-activated protein kinase signal transcution pathways activated by stress and inflammation. Physiol. Rev. 81, 807–869.
Lamkanfi, M., Kanneganti, T. D., Van Damme, P., Vanden Berghe, T., Vanoverberghe, I., Vandekerckhove, J., et al. (2008). Targeted peptidecentic proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol. Cell. Proteomics 7, 2350–2363. doi: 10.1074/mcp.m800132-mcp200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lei, K., and Davis, R. J. (2003). JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. U S A 100, 2432–2437. doi: 10.1073/pnas.0438011100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lei, K., Nimnual, A., Zong, W. X., Kennedy, N. J., Flavell, R. A., Thompson, C. B., et al. (2002). The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction of c-Jun NH(2)-terminal kinase. Mol. Cell. Biol. 22, 4929–4942. doi: 10.1128/mcb.22.13.4929-4942.2002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Le Prell, C. G., Yamashita, D., Minami, S. B., Yamasoba, T., and Miller, J. M. (2007). Mechanisms of noise-induced hearing loss indicated multiple methods of prevention. Hear. Res. 226, 22–43. doi: 10.1016/j.heares.2006.10.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, Y., Johnson, N., Capano, M., Edwards, M., and Crompton, M. (2004). Cyclophilin-D promotes the mitochondrial permeability transition but has opposite effects on apoptosis and necrosis. Biochem. J. 383, 101–109. doi: 10.1042/bj20040669
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, L. Y., Luo, X., and Wang, X. (2001). Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412, 95–99. doi: 10.1038/35083620
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, W., Srinivasula, S. M., Chai, J., Li, P., Wu, J. W., Zhang, Z., et al. (2002). Structural insights into the pro-apoptotic function of mitochondrial serine protease HtrA2/Omi. Nat. Struct. Biol. 9, 436–441. doi: 10.1038/nsb795
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, H., Zhu, H., Xu, C. J., and Yuan, J. (1998). Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94, 491–501. doi: 10.1016/s0092-8674(00)81590-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lin, F. R., Niparko, J. K., and Ferrucci, L. (2011). Hearing loss prevalence in the United States. Arch. Intern. Med. 171, 1851–1852. doi: 10.1001/archinternmed.2011.506
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, J., and Lin, A. (2005). Role of JNK activation in apoptosis: a double-edged sword. Cell Res. 15, 36–42. doi: 10.1038/sj.cr.7290262
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, X., Zou, H., Slaughter, C., and Wang, X. (1997). DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 89, 175–184. doi: 10.1016/s0092-8674(00)80197-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lorenzo, H. K., Susin, S. A., Penninger, J., and Kroemer, G. (1999). Apoptosis inducing factor (AIF): a phylogenetically old caspase-independent effector of cell death. Cell Death Differ. 6, 516–524. doi: 10.1038/sj.cdd.4400527
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Low, W. K., Tan, M. G., Chua, A. W., Sun, L., and Wang, D. Y. (2009). 12th Yahya Cohen memorial lecture: the cellular and molecular basis of radiation-induced sensorineural hearing loss. Ann. Acad. Med. Singapore 38, 91–94.
Luo, X., Budihardjo, I., Zou, H., Slaughter, C., and Wang, X. (1998). Bid, a Bcl-2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94, 481–490. doi: 10.1016/s0092-8674(00)81589-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Madesh, M., Antonsson, B., Srinivasula, S. M., Alnemri, E. S., and Hajnoczky, G. (2002). Rapid kinetics of tBid-induced cytochrome c and Smac/DIABLO release and mitochondrial depolarization. J. Biol. Chem. 277, 5651–5659. doi: 10.1074/jbc.m108171200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maeda, Y., Fukushima, K., Omichi, R., Kariya, S., and Nishizaki, K. (2013). Time courses of changes in phospho- and total-MAP kinases in the cochlea after intense noise exposure. PLoS One 8:e58775. doi: 10.1371/journal.pone.0058775
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maeda, K., Yoshida, K., Ichimiya, I., and Suzuki, M. (2005). Dexamethasone inhibits tumor necrosis factor-alpha-induced cytokine secretion from spiral ligament fibrocytes. Hear. Res. 202, 154–160. doi: 10.1016/j.heares.2004.08.022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mahdavian Delavary, B., van der Veer, W. M., van Egmond, M., Niessen, F. B., and Beelen, R. H. (2011). Macrophages in skin injury and repair. Immunobiology 216, 753–762. doi: 10.1016/j.imbio.2011.01.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mangiardi, D. A., McLaughlin-Williamson, K., May, K. E., Messana, E. P., Mountain, D. C., and Cotanche, D. A. (2004). Progression of hair cell ejection and molecular markers of apoptosis in the avian cochlea following gentamicin treatment. J. Comp. Neurol. 475, 1–8. doi: 10.1002/cne.20129
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marchetti, P., Castedo, M., Susin, S. A., Zamzami, N., Hirsch, T., Macho, A., et al. (1996). Mitochondrial permeability transition is a central coordinating event in apoptosis. J. Exp. Med. 184, 1155–1160. doi: 10.1084/jem.184.3.1155
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Markou, T., Dowling, A. A., Kelly, T., and Lazou, A. (2009). Regulation of Bcl-2 phosphorylation in response to oxidative stress in cardiac myocytes. Free Radic. Res. 42, 809–816. doi: 10.1080/10715760903071649
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marshall, K. D., and Baines, C. P. (2014). Necroptosis: is there a role for mitochondria? Front. Physiol. 5:323. doi: 10.3389/fphys.2014.00323
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marzo, I., Brenner, C., and Kroemer, G. (1998). The central role of the mitochondrial megachannel in apoptosis: evidence obtained with intact cells, isolated mitochondria and purified protein complexes. Biomed. Pharmacother. 52, 248–251. doi: 10.1016/s0753-3322(98)80009-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Matsui, J. I., Gale, J. E., and Warchol, M. E. (2004). Critical signaling events during the aminoglycoside-induced death of sensory hair cells in vitro. J. Neurobiol. 61, 250–266. doi: 10.1002/neu.20054
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McComb, S., Cheung, H. H., Korneluk, R. G., Wang, S., Krishnan, L., and Sad, S. (2012). cIAP1 and cIAP2 limit macrophage necroptosis by inhibiting Rip1 and Rip3 activation. Cell Death Differ. 19, 1791–1801. doi: 10.1038/cdd.2012.59
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Milhas, D., Cuvillier, O., Therville, N., Clavé, P., Thomsen, M., Levade, T., et al. (2005). Caspase-10 triggers Bid cleavage and caspase cascade activation in FasL-induced apoptosis. J. Biol. Chem. 280, 19836–19842. doi: 10.1074/jbc.m414358200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Murai, N., Kirkegaard, M., Järlebark, L., Risling, M., Suneson, A., and Ulfendahl, M. (2008). Activation of JNK in the inner ear following impulse noise exposure. J. Neurotrauma 25, 72–77. doi: 10.1089/neu.2007.0346
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mutsaers, S. E., Bishop, J. E., McGrouther, G., and Laurent, G. J. (1997). Mechanisms of tissue repair: from wound healing to fibrosis. Int. J. Biochem. Cell Biol. 29, 5–17. doi: 10.1016/s1357-2725(96)00115-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nagai, H., Noguchi, T., Takeda, K., and Ichijo, H. (2007). Pathophysiological roles of ASK1-MAP kinase signaling pathways. J. Biochem. Mol. Biol. 40, 1–6. doi: 10.5483/bmbrep.2007.40.1.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nagata, S. (1999). Fas ligand-induced apoptosis. Annu. Rev. Genet. 33, 29–55. doi: 10.1146/annurev.genet.33.1.29
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakagawa, T., Shimizu, S., Watanabe, T., Yamaguchi, O., Otsu, K., Yamagata, H., et al. (2005). Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434, 652–658. doi: 10.1038/nature03317
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakamagoe, M., Tabuchi, K., Uemaetomari, I., Nishimura, B., and Hara, A. (2010). Estradiol protects the cochlea against gentamicin ototoxicity through inhibition of the JNK pathway. Hear. Res. 261, 67–74. doi: 10.1016/j.heares.2010.01.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ng, P. W., Porter, A. G., and Jänicke, R. U. (1999). Molecular cloning and characterization of two novel pro-apoptotic isoforms of caspase-10. J. Biol. Chem. 274, 10301–10308. doi: 10.1074/jbc.274.15.10301
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nicotera, T. M., Ding, D., McFadden, S. L., Salvemini, D., and Salvi, R. (2004). Paraquat-induced hair cell damage and protection with the superoxide dismutase mimetic m40403. Audiol. Neurootol. 9, 353–362. doi: 10.1159/000081284
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nicotera, T. M., Hu, B. H., and Henderson, D. (2003). The caspase pathway in noise-induced apoptosis of the chinchilla cochlea. J. Assoc. Res. Otolaryngol. 4, 466–477. doi: 10.1007/s10162-002-3038-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nordang, L., and Anniko, M. (2005). Nitro-L-arginine methyl ester: a potential protector against gentamicin ototoxicity. Acta Otolaryngol. 125, 1033–1038. doi: 10.1080/00016480510038022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ohlemiller, K. K., McFadden, S. L., Ding, D. L., Lear, P. M., and Ho, Y. S. (2000). Targeted mutation of the gene for cellular glutathione peroxidase (Gpx1) increases noise-induced hearing loss in mice. J. Assoc. Res. Otolaryngol. 1, 243–254. doi: 10.1007/s101620010043
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ohlemiller, K. K., Wright, J. S., and Dugan, L. L. (1999). Early elevation of cochlear reactive oxygen species following noise exposure. Audiol. Neurootol. 4, 229–236. doi: 10.1159/000013846
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Oleinik, N. V., Krupenko, N. I., and Krupenko, S. A. (2007). Cooperation between JNK1 and JNK2 in activation of p53 apoptotic pathway. Oncogene 26, 7222–7230. doi: 10.1038/sj.onc.1210526
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Op de Beeck, K., Schacht, J., and Van Camp, G. (2011). Apoptosis in acquired and genetic hearing impairment: the programmed death of the hair cell. Hear. Res. 281, 18–27. doi: 10.1016/j.heares.2011.07.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ottilie, S., Diaz, J. L., Horne, W., Chang, J., Wang, Y., Wilson, G., et al. (1997). Dimerization properties of human BAD. Identification of a BH-3 domain and analysis of its binding to mutant BCL-2 and BCL-XL proteins. J. Biol. Chem. 272, 30866–30872. doi: 10.1074/jbc.272.49.30866
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Owens, T. W., Valentijn, A. J., Upton, J. P., Keeble, J., Zhang, L., Lindsay, J., et al. (2009). Apoptosis commitment and activation of mitochondrial Bax during anoikis is regulated by p38MAPK. Cell Death Differ. 16, 1551–1562. doi: 10.1038/cdd.2009.102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pahl, H. L. (1999). Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18, 6853–6866. doi: 10.1038/sj.onc.1203239
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Park, H. Y., Lee, M. H., Kang, S. U., Hwang, H. S., Park, K., Choung, Y. H., et al. (2012). Nitric oxide mediates TNF-α-induced apoptosis in the auditory cell line. Laryngoscope 122, 2256–2264. doi: 10.1002/lary.23444
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pathak, S., Goldofsky, E., Vivas, E. X., Bonagura, V. R., and Vambutas, A. (2011). IL-1β is overexpressed and aberrantly regulated in corticosteroid nonresponders with autoimmune inner ear disease. J. Immunol. 186, 1870–1879. doi: 10.4049/jimmunol.1002275
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pirvola, U., XingQun, L., Virkkala, J., Saarma, M., Murakata, C., Camoratto, A. M., et al. (2000). Rescue of hearing, auditory hair cells and neurons by CEP1347/KT7515, an inhibitor of cJun Nterminal kinase activation. J. Neurosci. 20, 43–50.
Porras, A., Zuluaga, S., Black, E., Valladares, A., Alvarez, A. M., Ambrosino, C., et al. (2004). P38 alpha mitogen-activated protein kinase sensitizes cells to apoptosis induced by different stimuli. Mol. Biol. Cell. 15, 922–933. doi: 10.1091/mbc.E03-08-0592
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Proost, P., Wuyts, A., and van Damme, J. (1996). The role of chemokines in inflammation. Int. J. Clin. Lab. Res. 26, 211–223. doi: 10.1007/BF02602952
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pujol, R., Rebillard, G., Puel, J. L., Lenoir, M., Eybalin, M., and Recasens, M. (1990). Glutamate neurotoxicity in the cochlea: a possible consequence of ischaemic or anoxic conditions accurring in ageing. Acta Otolaryngol. Suppl. 476, 32–36. doi: 10.3109/00016489109127253
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Puthalakath, H., Huang, D. C., O’Reilly, L. A., King, S. M., and Strasser, A. (1999). The proapoptotic activity of the Bcl-2 family member Bim is regulated by interation with the dynein motor complex. Mol. Cell 3, 287–296. doi: 10.1016/s1097-2765(00)80456-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pyun, J. H., Kang, S. U., Hwang, H. S., Oh, Y. T., Kang, S. H., Lim, Y. A., et al. (2011). Epicatechin inhibits radiation-induced auditory cell death by suppression of reactive oxygen species generation. Neuroscience 199, 410–420. doi: 10.1016/j.neuroscience.2011.09.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rewerska, A., Pawelczyk, M., Rajkowska, E., Politanski, P., and Sliwinska-Kowalska, M. (2013). Evaluating D-methionine dose to attenuate oxidative stress-mediated hearing loss following overexposure to noise. Eur. Arch. Otorhinolaryngol. 270, 1513–1520. doi: 10.1007/s00405-012-2265-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roux, P. P., and Blenis, J. (2004). ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 68, 320–344. doi: 10.1128/mmbr.68.2.320-344.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roy, S., and Nicholson, D. W. (2000). Cross-talk in cell death signaling. J. Exp. Med. 192, f21–f26. doi: 10.1084/jem.192.8.f21
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rybak, L. P., Whitworth, C. A., Mukherjea, D., and Ramkumar, V. (2007). Mechanisms of cisplatin-induced ototoxicity and prevention. Hear. Res. 226, 157–167. doi: 10.1016/j.heares.2006.09.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sahara, A., Aoto, M., Eguchi, Y., Imamoto, N., Yoneda, Y., and Tsujimoto, Y. (1999). Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation. Nature 401, 168–173. doi: 10.1038/43678
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sakon, S., Xue, X., Takekawa, M., Sasazuki, T., Okazaki, T., Kojima, Y., et al. (2003). NFkappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J. 22, 3898–3909. doi: 10.1093/emboj/cdg379
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Samson, J., Wiktorek-Smagur, A., Politanski, P., Rajkowska, E., Pawlaczyk-Luszczynska, M., Dudarewicz, A., et al. (2008). Noise-induced time-dependent changes in oxidative stress in the mouse cochlea and attenuation by D-methionine. Neuroscience 152, 146–150. doi: 10.1016/j.neuroscience.2007.11.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Satoh, H., Firestein, G. S., Billings, P. B., Harris, J. P., and Keithley, E. M. (2002). Tumor necrosis factor-alpha, an initiator and etanercept and inhibitor of cochlear inflammation. Laryngoscope 112, 1627–1634. doi: 10.1097/00005537-200209000-00019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Satoh, H., Firestein, G. S., Billings, P. B., Harris, J. P., and Keithley, E. M. (2003). Proinflammatory cytokine expression in the endolymphatic sac during inner ear inflammation. J. Assoc. Res. Otolaryngol. 4, 139–147. doi: 10.1007/s10162-002-3025-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Saunders, J. C., Dear, S. P., and Schneider, M. E. (1985). The anatomical consequences of acoustic injury: a review and tutorial. J. Acoust. Soc. Am. 78, 833–860. doi: 10.1121/1.392915
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Scarlett, J. L., and Murphy, M. P. (1997). Release of apoptogenic proteins form the mitochondrial intermembrane space during the mitochondrial permeability transition. FEBS Lett. 418, 282–286. doi: 10.1016/s0014-5793(97)01391-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sedlak, T. W., Oltvai, Z. N., Yang, E., Wang, K., Boise, L. H., Thompson, C. B., et al. (1995). Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proc. Natl. Acad. Sci. U S A 92, 7834–7838. doi: 10.1073/pnas.92.17.7834
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Seidman, M., Babu, S., Tang, W., Naem, E., and Quirk, W. S. (2003). Effects of resveratrol on acoustic trauma. Otolaryngol. Head Neck Surg. 129, 463–470. doi: 10.1016/s0194-5998(03)01586-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shen, H. M., and Pervaiz, S. (2006). TNF receptor superfamily-induced cell death: redox-dependent execution. FASEB J. 20, 1589–1598. doi: 10.1096/fj.05-5603rev
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shin, Y. S., Hwang, H. S., Kang, S. U., Chang, J. W., Oh, Y. T., and Kim, C. H. (2014). Inhibition of p38 mitogen-activated protein kinase ameliorates radiation-induced ototoxicity in zebrafish and cochlea-derived cell lines. Neurotoxicology 40, 111–122. doi: 10.1016/j.neuro.2013.12.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Silke, J., and Strasser, A. (2013). The FLIP side of life. Sci. Signal. 6:pe2. doi: 10.1126/scisignal.2003845
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sinha, K., Das, J., Pal, P. B., and Sil, P. C. (2013). Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 87, 1157–1180. doi: 10.1007/s00204-013-1034-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Smith, D. I., Lawrence, M., and Hawkins, J. E. Jr. (1985). Effects of noise and quinine on the vessels of the stria vascularis: an image analysis study. Am. J. Otolaryngol. 6, 280–289. doi: 10.1016/s0196-0709(85)80056-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
So, H., Kim, H., Lee, J. H., Park, C., Kim, Y., Kim, E., et al. (2007). Cisplatin cytotoxicity of auditory cells requires secretions of proinflammatory cytokines via activation of ERK and NF-kappaB. J. Assoc. Res. Otolaryngol. 8, 338–355. doi: 10.1007/s10162-007-0084-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Spoendlin, H. (1971). Primary structural changes in the organ of Corti after acoustic overstimulation. Acta Otolaryngol. 71, 166–176. doi: 10.3109/00016487109125346
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stephanou, A., Scarabelli, T. M., Brar, B. K., Nakanishi, Y., Matsumura, M., Knight, R. A., et al. (2001). Induction of apoptosis and Fas receptor/Fas ligand expression by ischemi/reperfusion in cardiac myocytes requires serine 727 of the STAT-1 trancription factor but not tyrosine 701. J. Biol. Chem. 276, 28340–28347. doi: 10.1074/jbc.m101177200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Suckfuell, M., Canis, M., Strieth, S., Scherer, H., and Haisch, A. (2007). Intratympanic treatment of acute acoustic trauma with a cell-permeable JNK ligand: a prospective randomized phase I/II study. Acta Otolaryngol. 127, 938–942. doi: 10.1080/00016480601110212
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Suzuki, Y., Imai, Y., Nakayama, H., Takahashi, K., Takio, K., and Takahashi, R. (2001). A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol. Cell 8, 613–621. doi: 10.1016/s1097-2765(01)00341-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tabuchi, K., Oikawa, K., Hoshino, T., Nishimura, B., Hayashi, K., Yanagawa, T., et al. (2010). Cochlear protection from acoustic injury by inhibitors of p38 mitogen-activated protein kinase and sequestosome 1 stress protein. Neuroscience 166, 665–670. doi: 10.1016/j.neuroscience.2009.12.038
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tabuchi, K., Pak, K., Chavez, E., and Ryan, A. F. (2007). Role of inhibitor of apoptosis protein in gentamicin-induced cohlear hair cell damage. Neuroscience 149, 213–222. doi: 10.1016/j.neuroscience.2007.06.061
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tahera, Y., Meltser, I., Johansson, P., Bian, Z., Stierna, P., Hansson, A. C., et al. (2006). NF-kappaB mediated glucocorticoid response in the inner ear after acoustic trauma. J. Neurosci. Res. 83, 1066–1076. doi: 10.1002/jnr.20795
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tartaglia, L. A., Ayres, T. M., Wong, G. H., and Goeddel, D. V. (1993). A novel domain within the 55 kd TNF receptor signals cell death. Cell 74, 845–853. doi: 10.1016/0092-8674(93)90464-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Taub, D. D., Proost, P., Murphy, W. J., Anver, M., Longo, D. L., van Damme, J., et al. (1995). Monocyte chemotactic protein-1 (MCP-1), -2 and -3 are chemotactic for human T lymphocytes. J. Clin. Invest. 95, 1370–1376. doi: 10.1172/jci117788
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Taylor, C. T. (2008). Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem. J. 409, 19–26. doi: 10.1042/bj20071249
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Teranishi, M., Nakashima, T., and Wakabayashi, T. (2001). Effects of alpha-tocopherol on cisplatin-induced ototoxicity in guinea pigs. Hear. Res. 151, 61–70. doi: 10.1016/s0300-2977(00)00080-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tourian, L. Jr., Zhao, H., and Srikant, C. B. (2004). P38alpha, but not p38beta, inhibits the phosphorylation and presence of c-FLIPS in DISC to potentiate Fas-mediated caspase-8 activation and type I apoptotic signaling. J. Cell Sci. 117, 6459–6471. doi: 10.1242/jcs.01573
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tournier, C., Hess, P., Yang, D. D., Xu, J., Turner, T. K., Nimnual, A., et al. (2000). Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288, 870–874. doi: 10.1126/science.288.5467.870
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tsujimoto, Y., and Shimizu, S. (2007). Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 12, 835–840. doi: 10.1007/s10495-006-0525-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tsujimoto, M., Yokota, S., Vilcek, J., and Weissmann, G. (1986). Tumor necrosis factor provokes superoxide anion generation from neutrophils. Biochem. Biophys. Res. Commun. 137, 1094–1100. doi: 10.1016/0006-291x(86)90337-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Turrens, J. F. (2003). Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335–344. doi: 10.1111/j.1469-7793.2003.00335.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vandenabeele, P., Galluzzi, L., Vanden Berghe, T., and Kroemer, G. (2010). Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714. doi: 10.1038/nrm2970
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vanden Berghe, T., Linkermann, A., Jouan-Lanhouet, S., Walczak, H., and Vandenabeele, P. (2014). Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev. Mol. Cell Biol. 15, 135–147. doi: 10.1038/nrm3737
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van Wijk, F., Staecker, H., Keithley, E., and Lefebvre, P. P. (2006). Local perfusion of the tumor necrosis factor alpha blocker infliximab to the inner ear improves autoimmune neurosensory hearing loss. Audiol. Neurootol. 11, 357–365. doi: 10.1159/000095897
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ventura, J. J., Cogswell, P., Flavell, R. A., Baldwin, A. S. Jr., and Davis, R. J. (2004). JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. 18, 2905–2915. doi: 10.1101/gad.1223004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wajant, H., Pfizenmaier, K., and Scheurich, P. (2003). Tumor necrosis factor signaling. Cell Death Differ. 10, 45–65. doi: 10.1038/sj.cdd.4401189
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, J., Chun, H. J., Wong, W., Spencer, D. M., and Lenardo, M. J. (2001). Caspase-10 is an initiator caspase in death receptor signaling. Proc. Natl. Acad. Sci. U S A 98, 13884–13888. doi: 10.1073/pnas.241358198
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, J., Ding, D., Shulman, A., Stracher, A., and Salvi, R. J. (1999). Leupeptin protects sensory hair cells from acoustic trauma. Neuroreport 10, 811–816. doi: 10.1097/00001756-199903170-00027
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, A., Hou, N., Bao, D., Liu, S., and Xu, T. (2012a). Mechanism of alpha-lipoic acid in attenuating kanamycin-induced ototoxiciy. Neural Regen. Res. 7, 2793–2800. doi: 10.3969/j.issn.1673-5374.2012.35.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Z., Jiang, H., Chen, S., Du, F., and Wang, X. (2012b). The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243. doi: 10.1016/j.cell.2011.11.030
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Y., Kim, N. S., Haince, J. F., Kang, H. C., David, K. K., Andrabi, S. A., et al. (2011). Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1 dependent cell death (parthanatos). Sci. Signal. 4:ra20. doi: 10.1126/scisignal.2000902
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, X. T., Pei, D. S., Xu, J., Guan, Q. H., Sun, Y. F., Liu, X. M., et al. (2007a). Opposing effects of Bad phosphorylation at two distinct sites by Akt1 and JNK1/2 on ischemic brain injury. Cell. Signal. 19, 1844–1856. doi: 10.1016/j.cellsig.2007.04.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, J., Ruel, J., Ladrech, S., Bonny, C., Van de Water, T. R., and Puel, J. L. (2007b). Inhibition of the c-Jun N-terminal kinase-mediated mitochondrial cell death pathway restores auditory function in sound-exposed animals. Mol. Pharmacol. 71, 654–666. doi: 10.1124/mol.106.028936
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, J., Van De Water, T. R., Bonny, C., de Ribaupierre, F., Puel, J. L., and Zine, A. (2003). A peptide inhibitor of c-Jun N-terminal kinase protects against both aminoglycoside and acoustic trauma-induced auditory hair cell death and hearing loss. J. Neurosci. 23, 8596–8607.
Wei, X., Zhao, L., Liu, J., Dodel, R. C., Farlow, M. R., and Du, Y. (2005). Minocycline prevents gentamicin-induced ototoxicity by inhibiting p38 MAP kinase phosphorylation and caspase 3 activation. Neuroscience 131, 513–521. doi: 10.1016/j.neuroscience.2004.11.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Whitfield, J., Neame, S. J., Paquet, L., Bernard, O., and Ham, J. (2001). Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 29, 629–643. doi: 10.1016/s0896-6273(01)00239-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Widlak, P., Li, L. Y., Wang, X., and Garrard, W. T. (2001). Action of recombinant human apoptotic endonuclease G on naked DNA and chromatin substrates: cooperation with exonuclease and DNase I. J. Biol. Chem. 276, 48404–48409.
Yamane, H., Nakai, Y., Takayama, M., Konishi, K., Iguchi, H., Nakagawa, T., et al. (1995). The emergence of free radicals after acoustic trauma and strial blood flow. Acta Otolaryngol. Suppl. 519, 87–92. doi: 10.3109/00016489509121877
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yamashita, D., Jiang, H. Y., Schacht, J., and Miller, J. M. (2004a). Delayed production of free radicals following noise exposure. Brain Res. 1019, 201–209. doi: 10.1016/j.brainres.2004.05.104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yamashita, D., Miller, J. M., Jiang, H. Y., Minami, S. B., and Schacht, J. (2004b). AIF and EndoG in noise-induced hearing loss. Neuroreport 15, 2719–2722.
Yang, W. P., Henderson, D., Hu, B. H., and Nicotera, T. M. (2004). Quantitative analysis of apoptotic and necrotic outer hair cells after exposure to different levels of continuous noise. Hear. Res. 196, 69–76. doi: 10.1016/j.heares.2004.04.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ylikoski, J., Xing-Qun, L., Virkkala, J., and Pirvola, U. (2002). Blockade of c-Jun N-terminal kinase pathway attenuates gentamicin-induced cochlear and vestibular hair cell death. Hear. Res. 163, 71–81. doi: 10.1016/s0378-5955(01)00380-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yoshida, K., Ichimiya, I., Suzuki, M., and Mogi, G. (1999). Effect of proinflammatory cytokines on cultured spiral ligament fibrocytes. Hear. Res. 137, 155–159. doi: 10.1016/s0378-5955(99)00134-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yu, S. W., Andrabi, S. A., Wang, H., Kim, N. S., Poirier, G. G., Dawson, T. M., et al. (2006). Apoptosis-inducing factor mediates poly(ADP-ribose)(PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. U S A 103, 18314–18319. doi: 10.1073/pnas.0606528103
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, D. W., Shao, J., Lin, J., Zhang, N., Lu, B. J., Lin, S. C., et al. (2009). RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336. doi: 10.1126/science.1172308
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zheng, H. W., Chen, J., and Sha, S. H. (2014). Receptor-interacting protein kinases modulate noise induced sensory hair cell death. Cell Death Dis. 5:e1262. doi: 10.1038/cddis.2014.177
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zou, J., Pyykkö, I., Sutinen, P., and Toppila, E. (2005). Vibration induced hearing loss in guinea pig cochlea: expression of TNF-alpha and VEGF. Hear. Res. 202, 13–20. doi: 10.1016/j.heares.2004.10.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: trauma, cochlea, auditory hair cells, apoptosis, inflammation, necrosis, otoprotection, repair
Citation: Dinh CT, Goncalves S, Bas E, Van De Water TR and Zine A (2015) Molecular regulation of auditory hair cell death and approaches to protect sensory receptor cells and/or stimulate repair following acoustic trauma. Front. Cell. Neurosci. 9:96. doi: 10.3389/fncel.2015.00096
Received: 22 November 2014; Accepted: 03 March 2015;
Published online: 31 March 2015.
Edited by:
Allison B. Coffin, Washington State University, USAReviewed by:
Richardson N. Leão, Brain Institute, BrazilDale Warren Hailey, University or Washington, USA
Copyright © 2015 Dinh, Goncalves, Bas, Van De Water and Zine. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Azel Zine, Integrative and Adaptive Neurosciences, Aix-Marseille Université, CNRS, UMR 7260, 3 Place Victor Hugo, 13331 Marseille Cedex 03, France azel.zine@univ-amu.fr