Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases
 Miguel Chin-Chan
Miguel Chin-Chan Juliana Navarro-Yepes
Juliana Navarro-Yepes Betzabet Quintanilla-Vega
Betzabet Quintanilla-Vega- Department of Toxicology, CINVESTAV-IPN, Mexico City, Mexico
Neurodegenerative diseases including Alzheimer (AD) and Parkinson (PD) have attracted attention in last decades due to their high incidence worldwide. The etiology of these diseases is still unclear; however the role of the environment as a putative risk factor has gained importance. More worryingly is the evidence that pre- and post-natal exposures to environmental factors predispose to the onset of neurodegenerative diseases in later life. Neurotoxic metals such as lead, mercury, aluminum, cadmium and arsenic, as well as some pesticides and metal-based nanoparticles have been involved in AD due to their ability to increase beta-amyloid (Aβ) peptide and the phosphorylation of Tau protein (P-Tau), causing senile/amyloid plaques and neurofibrillary tangles (NFTs) characteristic of AD. The exposure to lead, manganese, solvents and some pesticides has been related to hallmarks of PD such as mitochondrial dysfunction, alterations in metal homeostasis and aggregation of proteins such as α-synuclein (α-syn), which is a key constituent of Lewy bodies (LB), a crucial factor in PD pathogenesis. Common mechanisms of environmental pollutants to increase Aβ, P-Tau, α-syn and neuronal death have been reported, including the oxidative stress mainly involved in the increase of Aβ and α-syn, and the reduced activity/protein levels of Aβ degrading enzyme (IDE)s such as neprilysin or insulin IDE. In addition, epigenetic mechanisms by maternal nutrient supplementation and exposure to heavy metals and pesticides have been proposed to lead phenotypic diversity and susceptibility to neurodegenerative diseases. This review discusses data from epidemiological and experimental studies about the role of environmental factors in the development of idiopathic AD and PD, and their mechanisms of action.
Introduction
Life expectancy has increased in last decades and health care improvements have contributed to people living longer. However, this has also contributed to increase the number of people with chronic disabling diseases such as Alzheimer (AD) and Parkinson (PD). Genesis of both neurodegenerative diseases has not been elucidated and several endogenous (genetic) and exogenous (environment) factors contribute to the onset and/or development of these illnesses, which highlights the necessity to expand the research on identifying the environmental risk factors that predispose to the development of these neurodegenerative diseases.
It is known that the etiology of neurodegenerative diseases is multifactorial, and there is evidence that potential external factors including lifestyle and chemical exposures are linked with the risk of the onset of these diseases. Since the vast majority of AD and PD cases are observed in elderly populations, yet the exposure to risk factors occurred years or decades before the diagnosis, the assessment of chronic exposures is difficult to perform in retrospective studies to associate them with the onset/development of the disease. Therefore more research for better definition of exposure, as well as for the identification of early specific biomarkers for the diagnosis of these diseases is needed. Attention is now focused on environmental factors that potentially damage the developing nervous system through epigenetic mechanisms, resulting in neurodegenerative diseases later in life. In this review we briefly examined the evidence of environmental etiologies related to two of the most common neurodegenerative diseases, AD and PD, from epidemiological as well as experimental studies.
Alzheimer’s Disease
Alzheimer’s disease is the major form of dementia in elderly and possibly contributes to 60–70% of cases. It is a progressive, disabling and irreversible disease (Goedert and Spillantini, 2006). There are two recognized forms of AD. The first one is named familial or of early onset (EOAD), which is directly related to specific gene mutations in the amyloid precursor protein (APP) and presenilin (PSEN) 1 and 2 genes, both related to the amyloid-beta (Aβ) peptide synthesis (Piaceri et al., 2013). The EOAD begins at early age, less than 65 years, and only explains 5% of all cases. The second one, the late-onset or sporadic AD (LOAD) is the most common form of AD with 95% of all cases. This form of AD is not caused by punctual mutations, but some genetic risk factors have also been described such as polymorphisms in ApoE (encoding for apolipoprotein E), SORL1 (encoding for neuronal receptor of ApoE), and GSK3 (encoding for glycogen synthase kinase 3 beta) genes. The ApoE gene is the strongest genetic risk factor for LOAD, although it is not sufficient to explain the occurrence of the disease (Godfrey et al., 2003). Therefore, the etiology of LOAD remains unclear, and it is suggested that it has a multifactorial etiology.
Two hypotheses have been most studied for AD development. One is related to the overproduction of the Aβ peptide. According to this, neurofibrillary tangles (NFTs) result from the onset of amyloid deposits as Aβ plaques. While the second hypothesis suggests that the hyperphosphorylation of the Tau protein and its subsequent deposition as NFTs is the ultimate responsible for the disease. The amyloid cascade hypothesis establishes that Aβ aggregation initiates the brain damage leading to memory loss and to AD (Hardy and Higgins, 1992). Aβ is normally produced during aging, mediated by the proteolytic processing of the APP by the amyloidogenic enzymatic pathway. In this pathway, APP is processed by β- and γ-secretase complexes producing Aβ, soluble APPβ (sAPPβ) and the amyloid intracellular domain (AICD). Alternatively, APP can be processed by the non-amyloidogenic pathway leading to the production of AICD, sAPPα but not Aβ (Thinakaran and Koo, 2008). Thus Aβ increased levels in the brain of LOAD patients could be mediated by: (i) an increase in APP expression; (ii) an increase in the amyloidogenic pathway; or (iii) a reduction in the non-amyloidogenic pathway. It is stablished that the increase of a member of the β secretase complex, BACE1 (beta-site APP cleaving enzyme 1) produces high brain Aβ levels (Sun et al., 2012). On the other hand, the reduction on the activity of ADAM10 (a desintegrin and metalloproteinase domain-containing protein 10) could also lead to the overproduction of Aβ (Kojro and Fahrenholz, 2005). Furthermore, some mutations such as those in PSEN 1 or 2, a catalytic member of the γ-secretase complex, can also increase the production of Aβ (Piaceri et al., 2013).
Another mechanism to increase brain Aβ levels is through a reduction in the Aβ degradation. There are proteins collectively known as Aβ-degrading enzyme (IDE)s that have the ability to degrade Aβ, including insulin-like IDE, angiotensin-converting enzyme (ACE), endothelin-converting enzyme (ECE), plasmin, cathepsin B, aminopeptidase A, matrix metalloproteinase (MMP) 2 and 9, neprilysin (NEP, neutral endopeptidase) and others. These enzymes have been suggested as viable therapeutic targets for AD treatment (Nalivaeva et al., 2012). Finally, a reduced brain clearance of Aβ can be another pathway for the brain accumulation of Aβ. Some cholesterol transporters such as the low density lipoprotein receptor-related protein 1 (LRP1) are involved in the Aβ export from the brain to the cerebrospinal fluid (CSF). This receptor links the imbalance of cholesterol homeostasis with AD pathogenesis (Zlokovic et al., 2010).
On the other hand, aggregates of the microtubule (MT)-associated protein Tau observed in cell bodies and apical dendrites as NFTs cause neurofibrillary lesions associated with AD. Tau is a phosphoprotein mainly localized in the axon of neurons for the stabilization of MTs; it contains a high number of serine and threonine residues, and is therefore a substrate of many kinases (Goedert et al., 1988). The abnormal aggregation of Tau into insoluble paired helical filaments (PHFs), which are the major component of NFTs found in cell bodies and apical dendrites of neurons are lesions associated with AD (Friedhoff et al., 2000). Under pathological conditions, Tau is hyperphosphorylated at “pathological” sites leading to MTs depolymerization, axonal transport disruption and aggregation (Götz, 2001). It has been proposed that repeat domains (RD) of the MT-binding domain (MBD) in the C-terminal structure of Tau can rapidly form PHFs compared with the complete protein, suggesting that RDs are indispensable for its aggregation (Wille et al., 1992), and for Tau filament formation (Tokimasa et al., 2005).
There is no cure for AD, and therapeutic treatments are basically to ameliorate the symptoms. Therefore, an early and opportune diagnosis is indispensable to slow the progression of the disease. Currently, the determination of Tau and Aβ levels in blood and CSF are broadly used for the diagnosis of AD, and several medical tools are also used to confirm the diagnosis including the medical history, mental status tests, and evaluations of the brain structure and function with neuroimaging techniques (Lewczuk et al., 2014). However, these biomarkers are not sensitive nor specific for AD. Interestingly, an emerging body of evidence suggests that micro RNAs (miRNAs, small non-coding RNAs involved in the post-transcriptional regulation of gene expression) could be putative biomarkers for detecting neurodegeneration Thus, recent reviews have shown that some miRNAs are differentially associated with AD by modulating the expression of important genes involved in Aβ production (e.g., BACE1) or inflammation (Goodall et al., 2013; Van den Hove et al., 2014).
Aβ Homeostasis as a Target of Environmental Factors
Environmental factors such as diet (fat-rich), heavy metals, biogenic metals and pesticides have been involved in AD development due to their ability to disrupt metabolic pathways involved in the homeostasis of Aβ. In addition, factors such as lifestyle (antioxidants and exercise) can prevent AD development (Figure 1). Many of these environmental factors are oxidative agents acting through different mechanisms as discussed later. The brain is particularly vulnerable to oxidative stress do to its high glucose-based metabolic rate, low levels of antioxidants, high levels of polyunsaturated fatty acids, and high enzymatic activities related to transition metals that catalyze the formation of free radicals (Halliwell et al., 1992). In addition, micromolar concentrations of Aβ induce the formation of H2O2 in culture cells leading to neurotoxicity, and the presence of some antioxidant enzymes prevents the toxicity of the peptide (Butterfield et al., 2001). The mechanism by which Aβ generates free radicals is not known, and other endogenous factors also generate reactive oxygen species (ROS) in AD. For instance, the ion Fe3+, which is at high concentrations in NFTs and Aβ-aggregates, catalyzes the formation of reactive species such as H2O2, as well as advanced glycation end products (AGE) that are related to neurodegeneration (Smith et al., 1997b). On the other hand, activated microglia that surrounds the senile plaques is a source of NO and O2 (Cras et al., 1990), which can react to form the peroxinitrite radical (ONOO-) (Smith et al., 1997a). Likewise, inflammation has gained importance in AD pathogenesis (Tuppo and Arias, 2005). The central nervous system is considered a privileged site with its own immune system and microglia and astrocytes are the principal cells involved in the inflammatory response. It is accepted the microglial chemotaxis of Aβ and the phagocytosis of amyloid fibrils, effects that produce an increase in the secretion of pro-inflammatory cytokines and ROS, which in consequence produces neuronal loss (Rogers and Lue, 2001). In agreement, astrocytes are also recruited into amyloid plaques for Aβ degradation (Wyss-Coray et al., 2003), and it is possible that the activation of microglia and astrocytes is a consequence of Aβ aggregation. The role of inflammatory processes in AD is supported by the use of non-steroidal anti-inflammatory drugs (NSAIDs) to reduce the Aβ levels (Weggen et al., 2001), and the risk of AD (Etminan et al., 2003).
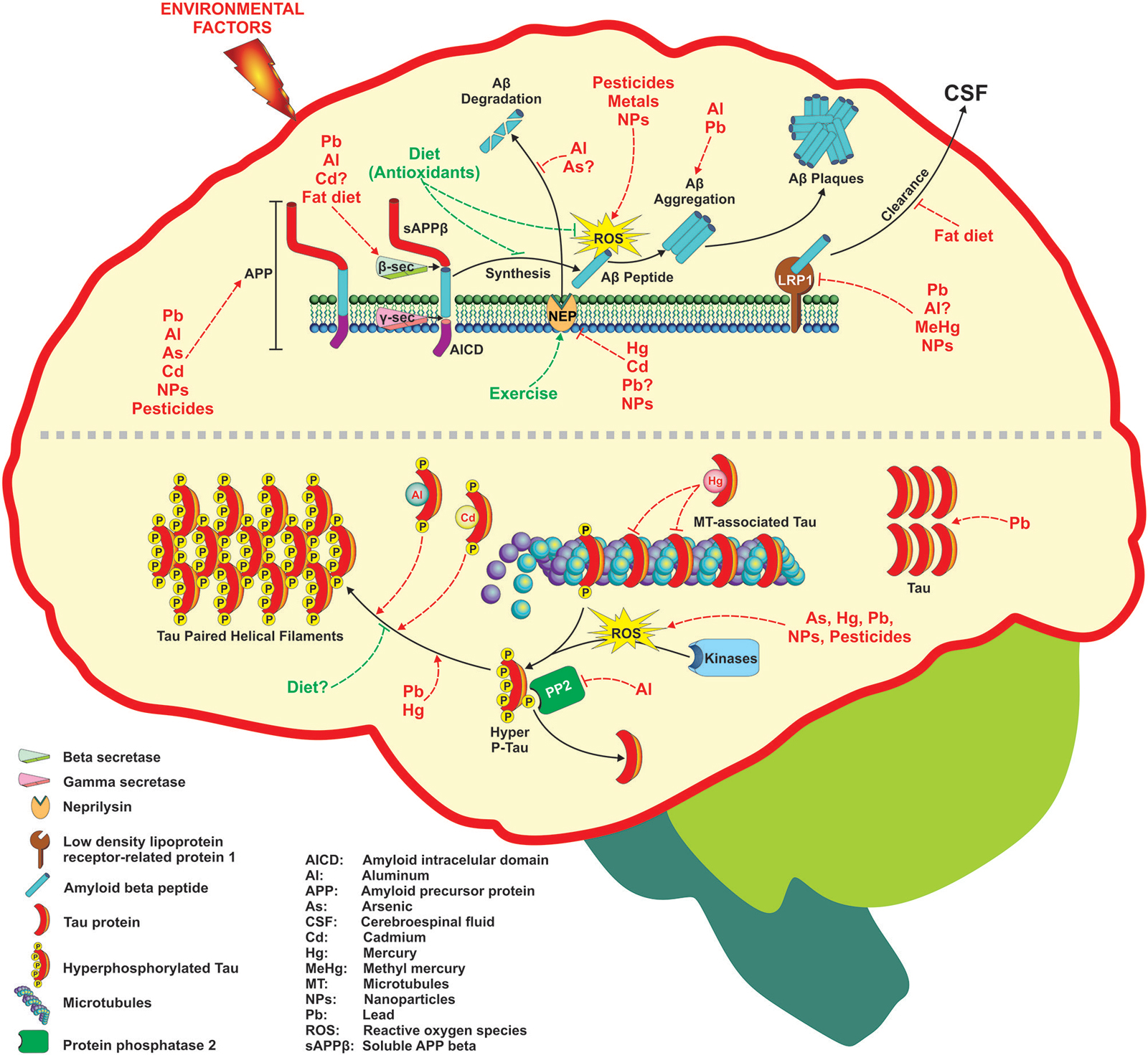
Figure 1. Environmental factors associated with Alzheimer’s disease (AD) development through different mechanisms. Several factors including metals, pesticides, nanoparticles, and diet can affect the two targets of AD such as Aβ generation and Tau phosphorylation. The figure depicts the molecular targets than can be modified at different levels following the amyloid hypothesis that ends in Aβ senile plaques formation (upper part) or the hyperphosphorylation of Tau protein and its subsequent deposition as neurofibrillary tangles (NFTs) (lower part). For more detail see the text.
Metals. Lead (Pb) is a heavy metal well known by its neurological toxic effects, although a direct association with AD development has not been reported. Pb affects cognitive abilities, intelligence, memory, speed processing and motor functions in children (Mason et al., 2014), while studies in elderly are limited. A cohort study reported that bone Pb levels were associated with poor cognitive performance scores in old workers, suggesting that past Pb exposure can contribute to late cognitive deterioration (Dorsey et al., 2006). However, a recent study reported no association between serum Pb levels and AD (Park et al., 2014). Despite the few epidemiological studies relating Pb exposure with AD, the evidence is more solid in experimental studies. The influence of Pb in AD was initially suggested from results in rats early exposed (from postnatal day 1–20) or late exposed (at 18–20 months of age) to Pb (200 ppm, drinking water). Results showed an increase in the APP mRNA expression late in life after the neonatal exposure, but not in rats exposed as adults (Basha et al., 2005). Similarly, a study performed in non-human primates (Macaca fascicularis) exposed to Pb (1.5 mg/Kg/day, from birth to 400 days) showed that monkeys exposed at a young age had an increased number of amyloid plaques late in life (at 23 years old) (Wu et al., 2008). The increased Aβ levels appear to be mediated by an augmented expression of APP and BACE1 (Wu et al., 2008). These effects were also observed in differentiated SH-SY5Y cells incubated with Pb (5–100 µM/48 h) and analyzed 6 days later (Bihaqi and Zawia, 2012). Another study performed in differentiated SH-SY5Y cells showed an increase in Aβ secretion and APP expression, as well as reduced expression and protein levels of NEP (an Aβ IDE), suggesting that Pb can target both the synthesis and degradation of Aβ (Huang et al., 2011). However, in a recent work conducted in our laboratory, Pb did not show changes in NEP expression in differentiated SH-SY5Y cells exposed to 50 µM Pb, but an increase in APP levels (Chin-Chan et al., 2015). Another mechanism by which Pb increases Aβ levels is by reducing the brain Aβ clearance. A recent study showed that acute Pb exposure (27 mg/Kg, i.p.) to APP transgenic mice (V717F) reduced the expression of LRP1, resulting in the accumulation of Aβ in the hippocampus and cortex of treated mice (Gu et al., 2011). Studies from this group support that Pb can disrupt the brain export of Aβ leading to its accumulation and plaques formation (Behl et al., 2009, 2010).
Exposure to Pb during development is a good example of an environmental contaminant as a risk factor to promote neurodegenerative diseases such as AD, supporting the hypothesis that many adult diseases have a fetal origin (FeBAD) (Basha et al., 2005). The group of Zawia has extensively worked on latent responses to prenatal and early postnatal exposures to Pb. Authors exposed male neonatal rats to Pb (200 ppm, drinking water) from postnatal day 1–20, or to aging animals (18–20 months of age), and observed an increase in the APP mRNA expression as well as in the activity of the transcription factor Sp1 (one of the regulators of APP) in the cortex of neonates, and after 20 months of Pb exposure had ceased. They observed a concomitant increase in Aβ levels in old animals exposed to Pb at birth. Interestingly, APP and Aβ protein levels did not respond to Pb exposure at old age (Basha et al., 2005). Similarly, a study conducted in cynomolgus monkeys exposed to Pb (1.5 mg/Kg/day, via infant formula) from birth to 400 days of age, and terminated 23 years later showed increased mRNA levels of APP and Sp1 in the frontal cortex compared with control animals, and high levels of the biomarker of oxidative DNA damage, 8-oxo-dG, suggesting an oxidative mechanism (Wu et al., 2008). Aβ1–42 and Aβ-1–40 levels in aged monkeys were also increased, as well as the intracellular Aβ staining and dense-plaques compared with age-matched controls (Wu et al., 2008). In addition, a study reported that gestational exposure to Pb (0.1, 0.5 and 1%, drinking water) in mice led to increased brain levels of Aβ and worst spatial memory performance, as well as increased levels of pro-inflammatory agents such as interleukin-1 (IL-1) and tumor necrosis factor alpha (TNF-α; Li et al., 2014a).
Mercury (Hg) is a heavy metal with a high potential to cause neurotoxicity. Early studies about Minamata and Iraq disasters led us to understand the neurotoxicity of this metal. It is widely accepted that Hg disrupts the brain development and produces cognitive and motor disabilities (Johansson et al., 2007), and in adults, Hg exposure produces memory loss and cognitive alterations (Wojcik et al., 2006; Chang et al., 2008). An early study suggested a link between Hg exposure and AD. Authors reported increased levels of Hg (in microsomes) and bromide (in the whole brain), and reduced levels of rubidium (in the whole brain, microsomes, and nuclei), selenium (Se; in microsomes) and zinc (Zn; in nuclei) in AD brains compared with controls (Wenstrup et al., 1990). A subsequent work reported more than a 2-fold increase in blood Hg levels in AD patients (n = 33) compared with a control group (n = 65), as well as a positive correlation between blood Hg concentration and CSF levels of Aβ (n = 15, Pearson’s correlation r = 0.7440, p = 0.0015) (Hock et al., 1998). More recently, several metals, including manganese (Mn), nickel, cadmium (Cd), Pb and Hg were determined in plasma and CSF of AD patients (n = 173) and healthy controls (n = 54), however only plasma Mn and Hg concentrations were significantly higher in AD patients (Gerhardsson et al., 2008). On the other hand, ApoE4 is a risk factor for AD, probably because this protein does not have sulfhydryl (SH) groups to scavenge heavy metals like Hg, whereas ApoE2 has four SH groups and then the ability to reduce the metal toxicity in the brain (Mutter et al., 2004). Therefore, ApoEε2 is considered a protective genotype for AD development (Suri et al., 2013). Similarly, Godfrey et al. (2003) observed a shift toward the risky ApoEε4 allele in patients with presumptive Hg-related neuropsychiatric symptoms with an elevated Hg body burden (n = 400; Godfrey et al., 2003).
The ability of Hg to increase Aβ levels has been studied in vitro and in vivo, and the suggested mechanisms include an increased production, a reduced degradation and/or a diminished brain clearance of the peptide. Olivieri et al. (2000) reported an increased secretion of both Aβ-40 and Aβ-42 when neuroblastoma cells were exposed to 50 µg/dL of inorganic Hg concomitant with ROS overproduction (Olivieri et al., 2000), and a study conducted in aggregating brain-cell cultures of fetal rat telencephalon showed that MeHg (non-cytotoxic concentrations/10–50 days) produced increased APP levels accompanied by ROS production and glia activation (Monnet-Tschudi et al., 2006). Rat pheochromocytoma (PC12) cells exposed to both inorganic and organic (MeHg) Hg (10–1000 nM) also showed a dose-dependent overproduction of Aβ-40 probably by an increase in APP levels as well as to a reduction in Aβ degradation by NEP (Song and Choi, 2013). However, an increase in Aβ-42 levels was observed in differentiated SH-SY5Y cells exposed to Hg (10 and 20 µM) but not in the APP expression, and rather a reduced activity of the Aβ-degrading enzyme, NEP was observed (Chin-Chan et al., 2015). Negative effects of Hg on Aβ aggregation have also been published, for example, Atwood et al. (1998) studied the role of the pH and the ability of various metals to aggregate Aβ; Zn, Cu and Fe showed the highest potential on Aβ aggregation, and Hg did not show an important effect (Atwood et al., 1998). Regarding in vivo studies, oral administration of 20–2000 µg/Kg/day/4 weeks of MeHg produced a dose-dependent increase in Aβ-42 in the hippocampus of male rats, but not significant changes in APP or NEP protein levels (Kim et al., 2014). Interestingly, authors observed a reduced brain expression of the LRP1 receptor, which was positively correlated with increased Aβ levels in the hippocampus and reduced levels in the CSF, suggesting a reduced clearance of the pathogenic peptide from the brain (Kim et al., 2014).
Inorganic arsenic (As) is a known neurotoxic metalloid with adverse effects in both the neurodevelopment and cognitive function (Tyler and Allan, 2014), although its effects in elderly have been less studied. There are few papers evaluating the role of As exposure as a risk factor for AD development. A study conducted in rural-dwelling adults and elders in Texas, US (FRONTIER project) reported that long-term exposure to low As levels (3–15 µg/L As in water) correlated (after adjustment by confounders, including the ApoEε4 presence) with a poor score of cognitive abilities and memory, which reflects the earliest manifestations of AD (O’Bryant et al., 2011). Similarly, a positive correlation was observed between serum As levels and the cognitive ability in AD patients from Hong Kong (n = 44, Pearson’s correlation coefficient r = 0.55, p < 0.0001) compared with matched controls (n = 41), lower serum Zn concentrations were observed in AD patients as well (Baum et al., 2010). On the other hand, the study conducted in several European countries showed a higher prevalence of AD and other dementias in those countries with As levels in topsoils about 18 ppm such as Italy, Switzerland, Spain, France, Belgium and Norway, compared with countries with lower As levels (in the range of 9 ppm), including Luxemburg, Denmark, Finland, UK and Nertherlands (Dani, 2010). In experimental studies, the administration of inorganic As (20 mg/L, drinking water during the gestation and early postnatal life) to mice produced a significant loss of spatial memory (Ramos-Chávez et al., 2015).
A plausible mechanism for the cognitive and memory alterations induced by As exposure is by alterating the amyloid pathway. Zarazúa et al. (2011) reported that cholinergic SN56.B5.G4 cells incubated with sodium arsenite or the organic form dimethylarsinic acid (DMA) (5–10 µM/12–24 h) showed an increase in APP and sAPPβ levels, and consequently an increase in Aβ only with DMA. Similar effects were observed in neurons from Tg2576 mice (a murine model that overexpresses a mutant form of APP most used in AD). They suggest that DMA-induced effects may be due by an increased Aβ anabolism (enhanced APP expression), although authors did not discard an alteration in the Aβ degradation pathway (Zarazúa et al., 2011). The mechanism by which As causes Aβ overproduction has not been determined, but As exposure has been associated with brain inflammatory responses and oxidative stress, which is in agreement with the inflammatory and oxidative hypotheses of AD (Gong and O’Bryant, 2010).
Cadmium is another toxic heavy metal associated with neurological alterations including memory loss and mental retardation (Wang and Du, 2013). An early study observed higher plasma levels of various metals including Cd, aluminum (Al), As, and Se in AD patients (n = 24) compared with healthy volunteers (n = 28) (Basun et al., 1991). Also, the liver from autopsied AD patients (n = 17) had significant higher Cd levels compared with age- and sex-matched control subjects (n = 17) (Lui et al., 1990). However, Gerhardsson et al. (2008) did not observe significant differences in Cd concentrations in plasma or CSF in patients with AD (n = 173) compared with healthy control subjects (n = 54) (Gerhardsson et al., 2008). There is evidence linking Cd exposure with Aβ overproduction. Li et al. (2012) observed cognitive alterations accompanied by an increased production of Aβ-42 and increased size and number of senile plaques in the cerebral cortex and hippocampus from APP/PSEN1 transgenic mice treated with Cd (2.5 mg/Kg/4 days, drinking water). These effects were attributed to a reduced expression of ADAM10, sAPPα and NEP proteins, suggesting that the non-amyloidogenic pathway as well as Aβ degradation are target of Cd exposure (Li et al., 2012). Additionally, authors reported an increase in free-Zn levels, suggesting that Cd displaces Zn from its native enzymes, including NEP.
Recently, Ashok et al. (2015) investigated the role of the exposure to individual metals (As, Pb and Cd) and their combination in the AD-amyloid pathway in male rats exposed from gestational day 5 to postnatal day 80 through drinking water. They reported that metals activated the synthesis of Aβ in the frontal cortex and/or hippocampus, mediated by an increase in APP, and in APP-processing enzymes such as beta secretases BACE1 and PSEN at postnatal days 24 (post-weaning) and 90 (adulthood). Pb was the most potent metal to induce Aβ, followed by Cd, and As had the smallest effect, however all did increase the APP production. Interestingly, they demonstrated a synergic effect of metals mainly due to As, the exposure to these three cations produced a dramatic increase in Aβ, PSEN1, BACE1 and APP, suggesting an enhanced amyloidogenic processing (Ashok et al., 2015). Authors also observed (Ashok et al., 2015) increased levels of malondialdehyde (MDA), reduced activity of antioxidant enzymes, and the induction of 1L-1α and IL-1β in the frontal cortex and hippocampus of rats exposed to As + Pb + Cd mixture. Authors suggest that ROS-induced IL-1 overproduction was responsible for the APP expression. This is supported by the fact that the 5’ÚTR region of the mRNA of APP has a responsive element for IL-1 (Rogers et al., 1999; Ashok et al., 2015).
Aluminum is a neurotoxic element involved in the etiology of neurodegenerative disorders such as AD; however, there is no consistent evidence. The incident of Al pollution in Cornwall, UK (1998) gave evidence of Al potential neurotoxicity. Similar brain pathological characteristic found in AD patients were observed in subjects exposed to Al in this region (Exley and Esiri, 2006), as well as alterations in brain functions (Altmann et al., 1999). A recent study conducted in China reported a marginal positive association between Al levels in soil and the mortality caused by AD (Shen et al., 2014); while other studies reported no association. Experimental evidence appears to be more consistent. Chronic oral Al administration in rats (20 g/day in the food/twice weekly from 6 months of age to the end of their lives) increases the Aβ production by raising the levels of APP in hippocampal and cortical tissues (brain regions important for the memory process) (Walton and Wang, 2009). Cultured rat cortical neurons exposed to Al (50 µM/48 days) showed an accumulation of Aβ; furthermore, Al induced conformational changes in Aβ and enhanced its aggregation forming fibrillar deposits on the surface of cultured neurons. The aggregated Aβ was dissolved by the addition of desferroxamine, a chelator of Al (Exley et al., 1993; Kawahara et al., 2001). The administration of Al plus D-galactose (an animal model for AD) produced the impairment of memory and increased the production of Aβ in the cortex and hippocampus. Additionally, an augmented expression of BACE1 and a reduction of NEP were observed in this co-treatment (Luo et al., 2009). Another study showed that Al reduced the Aβ degradation by decreasing the activity of cathepsin B, suggesting the activation of the amyloidogenic pathway and a reduction of the catabolism of Aβ (Sakamoto et al., 2006). In addition, a reduced expression of LRP1 was also observed in these mice administered with Al plus D-galactose, indicating a possible reduction of the clearance of Aβ as well (Luo et al., 2009). Transgenic mice (Tg2576) fed with Al (2 mg/Kg, in the diet/9 months) showed an increased production of Aβ and proteins involved in its anabolism; the accumulation of amyloid plaques were reversed by the treatment with vitamin E, suggesting the contribution of Al-induced oxidative mechanism (Praticò et al., 2002).
As mentioned earlier, miRNA can be biomarkers of early diagnosis of AD, however few studies have reported the involvement of pollutants in the miRNAs homeostasis (Ray et al., 2014). Interestingly, Tg2576 mice exposed to Al (2 mg/Kg/9 months, through the diet) showed an increased expression of miRNAs (e.g., miR146a and miR125b) involved in a pro-inflammatory response similar to that observed in AD brains (Bhattacharjee et al., 2014; Zhao et al., 2014), and treatment of primary human astroglial (HAG) cells with 100 nM of Al + Fe increased the expression of NFκB-induced miR-125b and miR-146a (Pogue et al., 2011); these miRNAs are reported in AD pathology (Lukiw, 2012). Further studies are necessary to look for a possible relation between xenobiotic exposures and the deregulation of miRNA expression involved in neurodegeneration.
Pesticides. The association between chronic pesticide exposure and the prevalence of dementias, including AD has not been as well studied as with other environmental risk factors, and results are often inconsistent. This is mainly because the difficulty on getting adequate data on the levels of exposure of individual pesticides, which is often indirectly evaluated by structure questionnaires (exposure index). Some of the studies with positive associations include the one performed in the agricultural Cache County, Utah, US in about 3000 occupationally pesticides-exposed participants who were followed-up for 3, 7 and 10 years. The hazard ratio for developing AD was slightly higher for organophosphate (OP) pesticides exposure (HR = 1.53, 95% CI, 1.05–2.23) than to organochlorines (OCl) (HR = 1.49, 95% CI, 0.99–2.24), after adjusting for some variables, including ApoE genotype (Hayden et al., 2010). Similarly, a recent case-control study observed a 3.8-fold increase in the OCl metabolite DDE in serum from AD patients (n = 79) compared with control participants (n = 86); in addition, authors reported that the highest tertile of DDE levels was associated with an increased risk for AD development (odds ratio-OR = 4.18, 95% CI, 2.54–5.82), and carriers of an ApoEε4 allele may be more susceptible (Richardson et al., 2014). Baldi et al. (2003) evaluated the association between lifelong cumulative exposure to pesticides and neurodegenerative diseases in a subsample from a cohort of elderly people (aged 65 years or older) (PAQUID study) in southwestern France. Authors analyzed 96 incident cases of AD (71 women and 25 men) in a 5-year follow-up approach, and observed a significant association between AD and occupational exposure to pesticides in men with a relative risk of 2.4 (95% CI, 1.0–5.6) after adjusting by education and smoking. Results were not significant in women (Baldi et al., 2003).
The role of pesticides in alterations observed in cognitive functions and AD has been suggested based on epidemiological studies, but the mechanisms have been poorly explored. In vitro studies performed in differentiated SH-SY5Y cells incubated with OCl pesticides such as DDE and its parent compound DDT (1 µM/48 h) showed increased APP protein levels, although authors did not evaluate Aβ levels (Richardson et al., 2014). A recent study reported that DDT augmented Aβ levels by increasing APP and BACE1 levels in human neuroglioma H4-AβPPswe cells, as well as by reducing the clearance and degradation of the peptide by targeting the Aβ-degrading enzyme, IDE and the ATP-binding cassette transporter A1 (ABC1; Li et al., 2015). Regarding in vivo data, chlorpyrifos (CPF), an OP insecticide associated with cognitive impairment, oxidative stress and neuronal damage caused a significant increase in Aβ levels in the cortex and hippocampus, as well as increased memory loss and reduced motor activity in Tg2576 mice 6 months after an acute subcutaneous administration of 50 mg/Kg of CPF (Salazar et al., 2011). However, Peris-Sampedro et al. (2014) did not find increased Aβ levels neither significant changes in memory acquisition in Tg2576 mice treated with CPF (25 mg/Kg/twice weekly/4 weeks, intragastric) and analyzed 6 months later (Peris-Sampedro et al., 2014). More studies are needed to better understand the mechanisms by which OCl, OP and other insecticides are linked to AD.
Paraquat (PQ), is a common used herbicide that has been suggested to be involved in AD development. A recent study showed that treatment of wild type and APP transgenic (Tg2576) mice with PQ (10 mg/Kg/twice a week/3 weeks) produced a significant increase in Aβ levels in transgenic mice that was associated with mitochondrial oxidative damage in cerebral cortex leading to the impairment of learning and memory. Interestingly, the overexpression of peroxiredoxin 3, a mitochondrial antioxidant defense enzyme produced an improvement in cognitive functions and a significant reduction in Aβ levels in APP transgenic mice exposed to PQ (Chen et al., 2012a), suggesting that pro-oxidant xenobiotics like PQ can contribute to AD.
Nanoparticles. As the synthesis of NPs for different applications, including drug delivery strategies in the treatment of AD is growing, it is necessary to study the potential toxic effects on proteins related to AD development.
There are not epidemiological studies associating the exposure to NPs with AD. However, there is increasing experimental evidence suggesting the potential role of NPs in brain damage. A recent study reported that nasal administration of TiO2-NPs (2.5–10 mg/Kg/90 days) to mice caused neuronal death in the hippocampus, oxidative stress and gliosis, and microarray analysis revealed a decline of genes associated with memory and cognition (Ze et al., 2014). Similarly, rats exposed to CuO-NPs (0.5 mg/Kg/day/14 days, i.p.) showed worst spatial cognition and a reduction in electrophysiological endpoints such as long-term potentiation, which matched with augmented levels of ROS and lipid peroxidation products (MDA and 4-hydroxinonenal-HNE), and reduced levels of antioxidants enzymes (An et al., 2012). Studies of NPs of Al, Cu and Ag administered at different doses and routes in rats and mice showed that they produce brain alterations such as motor, sensory and cognitive deteriorations (Sharma et al., 2009; Sharma and Sharma, 2012). However a recent study did not observe memory loss in adult mice administered with Ag-NPs (10, 25, and 50 mg/Kg/7 days) (Liu et al., 2013). Regarding in vitro studies, the exposure of human (SK-N-SH) and mouse (Neuro-2a) neuroblastoma cells to silica NPs (SiNPs) (10 µg/mL/24 h) raised the intracellular content of Aβ in both cell lines, which was associated with increased APP and reduced NEP protein levels. These effects may be mediated by ROS production, since SiNPs increased the production of intracellular ROS (Yang et al., 2014). Likewise, treatment of Neuro-2a cells to silver NPs (AgNPs, 12.5 µg/mL/24 h) showed the deposition of Aβ plaques and an increased expression of APP, while NEP and LPR1 (or LDLR) expression and protein levels were reduced, suggesting that AgNPs can induce AD by altering the amyloidogenic pathway: Aβ synthesis, degradation or clearance (Huang et al., 2015). Interestingly, authors also reported an increased expression of genes involved in the inflammatory response such as IL-1, C-X-C motif chemokine 13 (CXCL13), macrophage receptor with collagenous structure (MARCO), and glutathione synthetase (GSS) (Huang et al., 2015).
Tau Hyperphosphorylation by Environmental Factors
Several environmental factors have shown to mediate AD development through alterations on Tau phosphorylation and/or aggregation (Figure 1).
Metals. In vivo and in vitro studies have suggested the potential of Hg to induce P-Tau. Fujimura et al. (2009) reported an increased neuronal death and more migrating astrocytes in cerebral cortex of male mice exposed to MeHg (30 ppm, drinking water), as well as increased levels of P-Tau mediated by c-jun N-terminal kinase (JNK; Fujimura et al., 2009). An in vitro study showed that inorganic Hg (50 µg/dL/30 min) increased P-Tau in SH-SY5Y cells by a ROS-dependent mechanism, which was reverted by the co-treatment with the antioxidant melatonin (Olivieri et al., 2000). Another study demonstrated that Hg ions coordinate with Cys291 of the second repeated (R2) of the MT-binding domain of Tau increasing the heparin-induced aggregation, and a conformational change in Tau demonstrated by circular dicroism (CD) analysis (Yang et al., 2010). On the other hand, Cd appears to play a role in Tau hypothesis since it promotes the aggregation of this protein. It was shown that Cd (II) accelerates heparin-induced aggregation of the third repeated (R3) of Tau. The binding of Cd (II) to the dimeric R3 produces changes on its conformation demonstrated by CD (Jiang et al., 2007). Subchronic As administration to rats (NaAsO2 at 3 and 10 mg/Kg/day/4–12 weeks, intragastric) induced P-Tau, suggesting that As-destabilization and disruption of the cytoskeletal framework may lead to axonal degeneration (Vahidnia et al., 2008). Regarding Pb, it was reported that infantile Pb exposure in cynomolgus monkeys elevated mRNA and protein levels of Tau as well as its transcriptional regulators (Sp1 and Sp3) in aged primates (23 years old). An increase in P-Tau phosphorylation and mRNA and protein levels of cyclin dependent kinase 5 (cdk5, a kinase that phosphorylates Tau) were also observed (Bihaqi and Zawia, 2013). Other studies also reported that maternal (Li et al., 2010a) and early postnatal exposures (Liu et al., 2014) to Pb produced significant increased P-Tau levels and cognitive impairment in mice. Finally, chronic Al exposure caused Tau aggregation, and it was suggested that Al is bound to P-Tau in the Al-NFTs lesions (Singer et al., 1997; Shin et al., 2003). Also, a study showed that Al is able to confer resistance to the degradation of PHFs both in vivo and in vitro (Shin et al., 1994), and it can inhibit the activity of the protein phosphatase 2 (PP2), which is involved in P-Tau de-phosphorylation (Yamamoto et al., 1990).
Pesticides. There is some evidence suggesting that pesticide exposure can disrupt Tau function. A recent study showed that the administration of the insecticides deltamethrin (pyethroid) and carbofuran (carbamate) to rats (daily administration by gavage/28 days) produced neuronal death in the cortex and hippocampus and a dysfunction in the spatial memory and learning. These alterations were attributed to a reduced expression of synaptic proteins involved in the memory consolidation. Additionally, P-Tau and activation of p-GSK3β (a major kinase that phosphorylates Tau) were observed (Chen et al., 2012b). Similarly, Wills et al. (2012) showed P-Tau in the striatum, through the activation of p-GSK3β, as well as hyperacetylation of α-tubulin in mice treated with PQ (10 mg/Kg, i.p., twice weekly/6 weeks), suggesting a cytoskeleton remodeling (Wills et al., 2012).
Nanoparticles. The effect of NPs on Tau phosphorylation has not been extensively studied. Silica NPs (siNPs) used in medicine are also able to increase P-Tau at Ser262 and Ser396, two phosphorylation sites characteristic of AD. It was demonstrated that this effect was dependent on the activation of the kinase GSK3β in human SK-N-SH and mouse Neuro-2a cells by a mechanism probably mediated by oxidative stress, since ROS were increased in cells exposed to these NPs (Yang et al., 2014).
Parkinson’s Disease
Parkinson Disease is a chronic and progressive neurological disorder characterized by the selective loss of dopaminergic neurons of the substantia nigra pars compacta (SNpc). The cardinal features of the syndrome are related to motor dysfunction including tremor at rest, rigidity, akinesia (or bradykinesia), and postural instability. The motor symptoms appear when at least 60% of dopaminergic neurons are lost and 80–85% of dopamine content in the striatum is depleted (Jankovic, 2008; Wirdefeldt et al., 2011). Additional to the neuronal loss, the main neuropathological hallmark of PD is the presence of Lewy bodies (LB) in the surviving neurons, which are eosinophilic cytoplasmic inclusions containing aggregates of protein such as α-synuclein (α-syn) (Gibb and Lees, 1988; Spillantini et al., 1998). PD is the second most common neurodegenerative disorder after AD. Due to the lack of specific/differential diagnostic biomarkers, the diagnosis of PD is based on clinical criteria of specific cardinal motor signs of the disease and on the response to levodopa. PD diagnosis is confirmed by the depletion of brain stem pigmented neurons and the presence of LB at necropsy, this is the reason of the misclassification of PD cases (about 10–15%) (Schrag et al., 2002; Jankovic, 2008). There is no cure for PD, and the existing therapies only provide brief relief of motor symptoms through improving the dopamine deficit or by surgical methods. This highlights the need of research on early specific/differential biomarkers to have more accurate diagnosis of neurodegenerative disorders, as well as biomarkers for the identification of populations at risk to implement neuroprotective therapies (Jankovic, 2008).
As in the case of AD, circulating miRNAs are being studied as differential biomarkers for PD. Some reviews have recently addressed this topic, showing the association of specific miRNAs for some genes involved in PD, such as SNCA and LRRK2 (encoding for leucine–rich repeat kinase 2) with PD development (Goodall et al., 2013; Maciotta et al., 2013). Some studies have reported differentially expressed miRNAs in serum of PD patients not observed in control subjects or in other diseases. For example, Vallelunga et al. (2014) reported two differentially expressed miRNAs (miR-30c and miR-148b) in Italian PD patients (n = 25 vs. 25 healthy controls) (Vallelunga et al., 2014), and another study found that serum levels of miR-29c, miR-29a, and miR-19b were down-regulated in PD patients (n = 65 vs. 65 healthy controls) from Barcelona, Spain (Botta-Orfila et al., 2014). Also, a reduced expression of miR-34b and miR-34c in several brain areas including the substantia nigra of PD patients (n = 11 vs. 6 healthy controls) was detected; interestingly the misregulation of miR-34b/c was observed in patients in pre-motor stages of the disease. Additionally, these miRNAs were deregulated in differentiated SH-SY5Y dopaminergic neuronal cells, which was associated with altered mitochondrial function, oxidative stress and ATP depletion, as well as decreased protein levels of DJ1 (a mitochondrial peroxidase) and Parkin (an E3 ubiquitin ligase) that are associated with the familial form of PD (Miñones-Moyano et al., 2011).
Although the research on PD has rapidly advanced, the molecular mechanisms involved are still unclear and its etiology is complex. Several molecular mechanisms of neuronal death in PD pathogenesis have been described including mitochondrial dysfunction, impairment of protein quality pathways, oxidative/nitrative stress, microglia activation and inflammation. These mechanisms converge and are consistent with a major role of oxidative stress in PD, which damage organelles and proteins leading to increased protein aggregates (e.g., α-syn), that in turn overwhelms the degradation systems leading to a self-perpetuating cycle and further oxidative stress (Wirdefeldt et al., 2011; Goldman, 2014). The evidence in postmortem PD brains supports these mechanisms, as well as a decreased in reduced GSH levels, α-syn aggregation, proteasome impairment and autophagy dysfunction (review in Navarro-Yepes et al., 2014).
A fraction of PD occurrence has a clear familial inheritance and it is related to mutations in at least 6 genes that have been associated with PD onset. The identification of genes such as SNCA or PARK1 encoding for α-syn (maybe involved in the regulation of dopamine release and transport), LRRK2 or PARK8 encoding for LRRK2 (or Dardarin), PARK7 encoding for DJ1, PARK6 or PINK1 encoding for PTEN-induced putative kinase 1 (PINK1, a mitochondrial kinase), and PARK2 encoding for Parkin have provided clues about the molecular mechanisms involved in its pathogenesis (Corti et al., 2011; Cookson, 2012). However, 90% of PD cases are sporadic and cannot be attributed only to genetic factors, which suggests that PD have a multifactorial etiology (Goldman, 2014). In addition to the aging, which is the main risk factor for PD (Tanner and Goldman, 1996), epidemiological evidence suggests that the exposure to environmental toxicants, mainly pesticides, metals and solvents could increase the risk of developing PD, and factors such as tobacco consumption can protect against PD development (Figure 2; Hatcher et al., 2008; Gao and Hong, 2011).
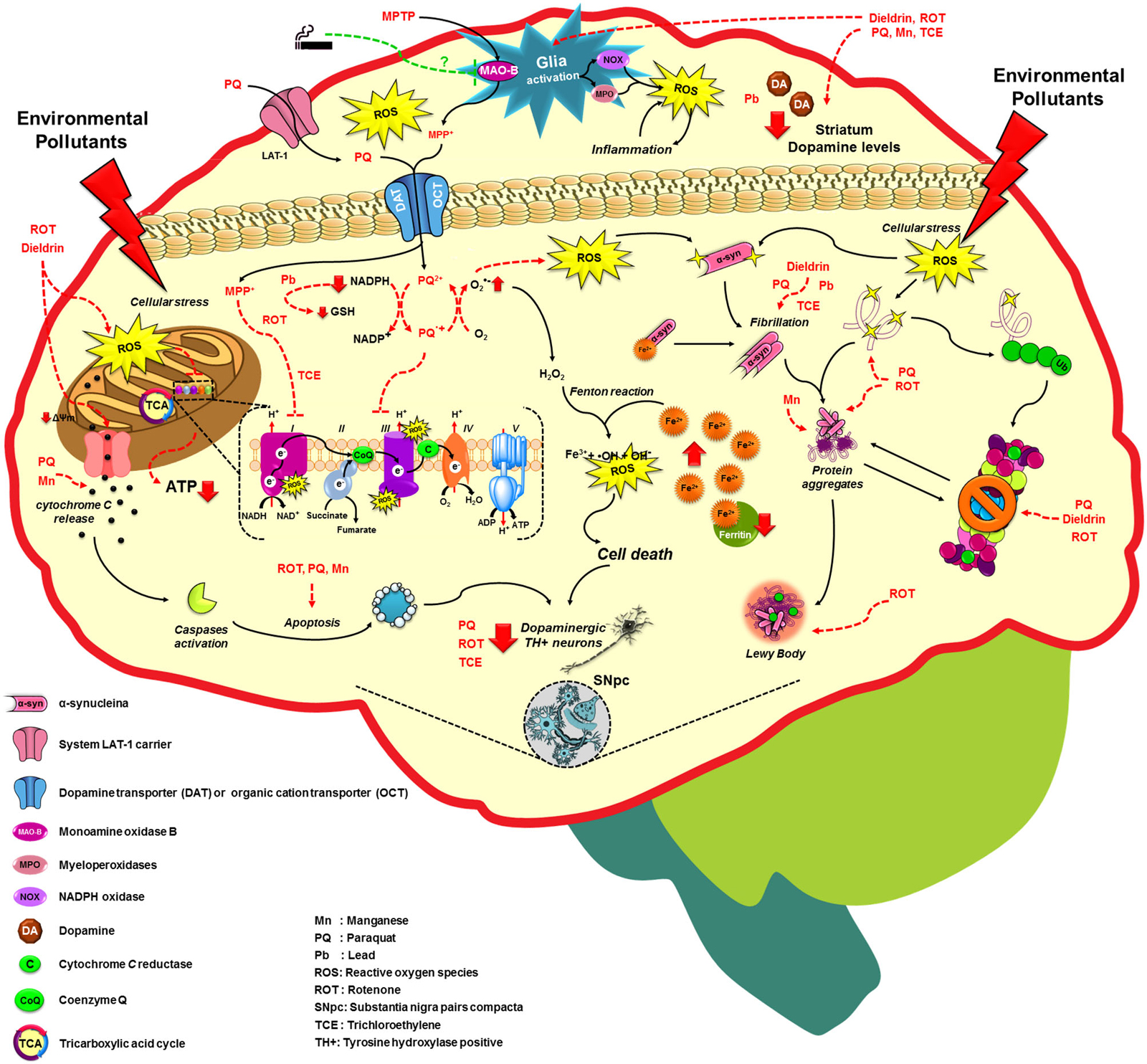
Figure 2. Molecular mechanisms altered by environmental factors related to increased Parkinson’s disease risk. Exposure to environmental toxicants mainly pesticides, metals and solvents may lead to the selective loss of dopaminergic neurons on the substantia nigra pars compacta (SNpc) through the dysregulation/alteration of the molecular mechanisms implicated on PD development such as mitochondrial dysfunction, impairment of protein quality pathways, microglia activation and inflammation, which converge in the production of oxidative stress as the main factor in PD. For more detail see the text.
Metals
It has been proposed that chronic exposure to heavy metals such as iron, Pb and Mn and their combinations can be associated with an increased risk of developing PD, since they accumulate in the substantia nigra and generate oxidative stress. However, epidemiological evidence is controversial (Lai et al., 2002). The epidemiological evidence of Pb association with PD is more consistent because the accumulative lifetime exposure can be estimated through Pb concentration in bone that has a half–life of years to decades. Initially, Kuhn et al. (1998) reported that 7 out of 9 postal workers exposed to lead-sulfate batteries for up to 30 years developed parkinsonian symptoms, suggesting that Pb intoxication may play a role in the occurrence of these symptoms (Kuhn et al., 1998). Coon et al. (2006) evaluated this association in 121 PD patients vs. 414 controls and found that chronic Pb exposure (evaluated by Pb concentrations in tibial and calcaneal bones) increased 2–fold the risk of PD (OR = 2.27, 95% CI, 1.13–4.55) for individuals in the highest quartile of lifetime Pb exposure relative to those in the lowest quartile (Coon et al., 2006). In the same way, it was reported that the cumulative exposure to Pb increases the risk of PD (OR = 3.21, 95% CI, 1.17–8.83) in 330 PD patients (vs. 308 controls) recruited from 4 clinics for movement disorders in Boston, MA area (Weisskopf et al., 2010b), and the exposure to Pb for more than 20 years showed a stronger association with PD risk in a health system population-based case-control study (144 cases vs. 464 controls) from the metropolitan Detroit area (Gorell et al., 2004). At the molecular level, Pb exposure significantly decreases the dopamine release and the dopamine D1 receptor sensitivity post-synaptically in microdialysate samples from rats subchronically exposed to Pb (50 ppm/90 days) (Kala and Jadhav, 1995), and in rats treated with 250 ppm of Pb for 3–6 weeks through drinking water (Tavakoli-Nezhad and Pitts, 2005). Furthermore, it increases the lipid peroxidation and reduces the antioxidant cell capacity (Sandhir et al., 1994), and causes fibrillation and aggregation of α-syn (Yamin et al., 2003), which induces hippocampal injury and decreases the ability of learning and memory in rats exposed to 0–300 ppm of Pb (Yamin et al., 2003; Zhang et al., 2012).
Manganese is an essential element with important physiological functions for cellular homeostasis. The epidemiologic evidence does not provide sufficient support for an association between Mn exposure and PD risk (Wirdefeldt et al., 2011; Mortimer et al., 2012). Only one case–control study (144 cases vs. 464 controls) in a population from the metropolitan Detroit area reported an increase of PD risk when the exposure to Mn was over 20 years (OR = 10.63, 95% CI, 1.07–105.99) (Gorell et al., 2004). However, occupational or environmental exposures to Mn have been associated with a neurological syndrome that include cognitive deficits, neuropsychological abnormalities and Parkinsonism (Guilarte, 2013). Mn was related to PD since 1837, when it was noted that high Mn exposures caused a severe and debilitating disorder known as “manganism” or manganese–induced Parkinsonism, which consists on an extra pyramidal syndrome that resembles the dystonic movements associated with parkinsonian symptoms (Couper, 1837; Jankovic, 2005), but it is clinically distinct from PD since patients do not respond to dopamine replacement therapies (Cook et al., 1974; Huang et al., 1993; Lu et al., 1994). Several cases of Mn–induced Parkinsonism have been reported in individuals whose professions involve prolonged contact with high atmospheric levels of Mn such as welders, miners and smelters (Rodier, 1955; Wang et al., 1989; Lee, 2000). Several investigations have shown that sustained exposure to low-concentrations (below the current US standard of 5.0 mg/m3) is consistent with early manganism, suggesting that Mn is a neurotoxic chemical (Park, 2013). Patients with manganism and primates experimentally intoxicated with Mn consistently show damage to the globus pallidus, which is in contrast with PD where there is a preferential degeneration of dopamine neurons in the SNpc and preservation of the pallidum (Perl and Olanow, 2007). Likewise, it was observed microglia activation in the substantia nigra pars reticulate (SNpr) and SNpc in Cynomolgus macaques exposed to Mn (5–6.7 mg/Kg/week/10 months) (Verina et al., 2011). In vitro, it has been observed that Mn treatment (50–300 µM MnCl2/ 3–48 h) induces cytochrome C release and activation of caspases 9 and 3, as well as protein aggregation in N27 dopaminergic neuronal cells that stably express α-syn (Harischandra et al., 2015).
Iron is an essential element transported into the brain through the transferrin receptor and divalent metal transporter 1 (DMT1; Zheng and Monnot, 2012). It has been evaluated in relation to the risk of PD in few epidemiological studies without convincing evidence (Rybicki et al., 1993; Logroscino et al., 2008; Miyake et al., 2011; Abbott et al., 2012). However, iron and its deregulated homeostasis have been proposed to have a role in the pathogenesis of PD because of its pro-oxidants characteristics that may lead to ROS generation via Fenton and Haber–Weiss reactions (Stohs and Bagchi, 1995; Sian-Hulsmann et al., 2011). The substantia nigra has the highest levels of iron in the human brain, probably due to the presence of neuromelanin in pigmented SNpc dopaminergic neurons that have an impressive capacity of chelating metals, iron in particular; however, this may be a dual-edged sword that may increase their vulnerability since iron may react with ROS produced from dopamine metabolism and promote the further generation of highly toxic radicals (Zecca et al., 2002, 2004). Alterations in iron distribution have been observed in the substantia nigra of PD postmortem brains (Dexter et al., 1987, 1991; Hirsch et al., 1991). On the other hand, it was observed in postmortem samples that although the total iron concentration in the whole substantia nigra was not significantly different between parkinsonian and control samples, there was an increase in the free-iron concentration and a decrease in iron–binding ferritin levels, ferritin sequestrates the excess of iron under physiological conditions (Wypijewska et al., 2010). Likewise, it was reported that free-iron induces fibrillation and aggregation of α-syn in a dose- and time-dependent way in SK-N-SH cells incubated with ferric iron (1–10 mmol/L/24–48 h) (Li et al., 2010b). Mice administered with iron (120 µg/g of carbonyl iron, oral gavage) at a dose equivalent to that found in iron-fortified human infant formula (12 mg/L of iron) from days 10 to 17 post-partum (an equivalent period to the first human year of life) showed a progressive midbrain neurodegeneration and enhanced vulnerability to toxic injury at 12 and 24 months of age (Kaur et al., 2007).
Pesticides
The hypothesis that pesticide exposures may be related to PD development was prompted by the discovery that intravenous injection of 1-methy l-4pheny l-1, 2, 3, 6-tetrahydropyridine (MPTP), a byproduct of the synthesis of heroin, developed a Parkinson syndrome clinically indistinguishable from PD (Langston et al., 1983); subsequent findings showed that MPTP selectively damaged dopaminergic neurons in the substantia nigra (Langston and Ballard, 1983; Langston et al., 1984). Since then, environmental factors with similar toxicological profiles have received attention as potential risk factors for PD.
A meta-analysis conducted in 2000 evaluated the association between pesticide exposures and PD in 19 case–control studies published between 1989 and 1999. Authors showed that most studies found an elevated risk of PD with the exposure to pesticides, the calculated combined OR was 1.94 (95% CI, 1.49–2.53); similar ORs were observed in studies conducted in United States, Asia, Europa and Canada. Additionally, it was observed that the risk of PD increased with longer exposure times, with an OR of 5.81 (95% CI, 1.99–16.97) for ≥10 years of exposure; however, specific types of pesticides were not identified (Priyadarshi et al., 2000). Subsequently, Brown et al. (2006) reviewed 31 case–control studies published until 2003, and found that about half of them reported significant associations between pesticide exposure and PD risk with ORs from 1.6–7. Interestingly, in most studies, authors observed a positive association between the exposure to herbicides and insecticides and PD risk, but not with the exposure to fungicides alone (Brown et al., 2006).
In line with this, a recent review by Freire and Koifman (2012) analyzed the epidemiological evidence published between 2000 and 2011, including ecological, cross–sectional, prospective and case–control studies. They found that 7 out of the 8 prospective (cohort) studies provided evidence of an association between pesticide exposure and PD, reporting risk estimates of 2-fold or higher. Among 23 case–control studies, 13 studies reported a significant increased risk of PD for the professional use of pesticides in comparison with unexposed controls, with ORs ranging from 1.1 to 1.4, which is in agreement with the review of Priyadarshi in the 1990’s (Freire and Koifman, 2012). Furthermore, van der Mark et al. (2012) performed a systematic review and calculated the summary risk ratio (sRR) from 39 case–control studies, 4 cohort studies and 3 cross–sectional studies. When a job–exposure matrix was constructed, a higher sRR (2.5, 95% CI, 1.5–4.1) was observed compared with self–reported exposure evaluation (1.5, 95% CI, 1.3–1.8). This meta–analysis found a positive association between PD and insecticides (sRR = 1.50, 95% CI, 1.07–2.11), and herbicides (sRR = 1.40, 95% C, 1.08–1.81), but not with fungicides (sRR = 0.99, 95% CI, 0.71–1.40) (van der Mark et al., 2012), in agreement with Brown et al. (2006) and Freire and Koifman (2012). Other factors related to pesticide exposure such as well–water consumption, farming, and rural living have been associated with an increased PD risk. The meta–analysis of Priyadarshi et al. (2001) found a combined OR of 1.56 (95% CI, 1.18–2.07) for rural living, 1.42 (95% CI, 1.05–1.91) for farming and 1.26 (95% CI, 0.97–1.64) for well–water consumption. However, whether of these factors are independent risk factors or correlated with pesticide exposure could not be determined (Priyadarshi et al., 2001).
In support to epidemiological evidence, increased levels of some pesticides have been quantified in postmortem brains from PD patients. High concentrations of some OCl pesticides have been observed in PD cases compared with controls, including dieldrin, lindane, and p-p-DDE (Fleming et al., 1994; Corrigan et al., 1998, 2000). In the same way, 2 epidemiologic studies reported a significant association (OR ranging from 1.3 to 1.8) between dieldrin use and PD in farmers participants in the Agricultural Health Study (AHS; Kamel et al., 2007; Tanner et al., 2011). Another nested case–control study within the Finnish Mobile Clinic Health Examination Survey in Finland, with serum samples collected during 1968–1972, observed that increasing serum concentrations of dieldrin were associated with an increased PD risk (OR = 1.95, 95% CI, 1.26–3.02) in 68 cases vs. 183 controls restricted to never smokers, while no other OCl pesticide showed an association (Weisskopf et al., 2010a).
The epidemiologic evidence that dieldrin exposure may be associated with PD is supported by toxicological data at molecular level. Dieldrin may cross the blood–brain barrier and remains in lipid-rich tissues such as the brain (Kanthasamy et al., 2005), and it has been shown that it is selectively toxic to dopaminergic neurons and could induce several of the pathologic mechanisms of PD including the depletion of brain dopamine levels, increased ROS in nigral dopaminergic neurons, inhibition of mitochondrial oxidative phosphorylation that lead to a reduction of cellular ATP production, alteration of the mitochondrial membrane potential and cytochrome C release in animal models such as rats and mice chronically exposed to dieldrin (0.3–3 mg/Kg/day in the diet) (Bergen, 1971; Wagner and Greene, 1978; Purkerson-Parker et al., 2001; Hatcher et al., 2007), and in primary mesencephalic cultures or dopaminergic cell lines (0.01–300 µM) (Sanchez-Ramos et al., 1998; Kitazawa et al., 2001, 2003; Kanthasamy et al., 2005). Aggregation of α–syn, ubiquitin–proteasome impairment function (Uversky et al., 2001; Sun et al., 2005) and microglia activation (Mao and Liu, 2008) have also been observed.
Paraquat is a quaternary nitrogen herbicide used worldwide. Due to its structural similarity to MPP (the active metabolite of MPTP), it was thought to be toxic to dopaminergic neurons and thus might be related to PD. The possible association between PQ and PD received attention from the study of Liou et al. (1997) performed in PD patients (120 patients and 240 controls) in Taiwan, in which the pesticide use was associated with an increased risk of developing PD, being higher for those individuals who reported using PQ (Liou et al., 1997). Likewise, Tanner et al. (2011) reported a significant association between PD and the use of oxidative pesticides, including PQ (OR = 2.5, 95% CI, 1.4–4.7) in professional pesticide applicators (110 cases and 358 controls) (Tanner et al., 2011). Similarly, other epidemiologic studies have associated the exposure to PQ with PD (Hertzman et al., 1990; Ascherio et al., 2006; Kamel et al., 2007; Wang et al., 2011).
Paraquat is taken up into dopaminergic terminals by the dopamine transport and organic cation transporter 3 (Rappold et al., 2011), and causes cellular toxicity by oxidative stress through the cellular redox cycling generating superoxide radical by the oxidation of NADPH, which in turn impairs the restauration of GSH levels and thus the activity of several antioxidant systems (Berry et al., 2010; Franco et al., 2010). It has been observed that repeated administrations of PQ to adult mice and rats (5–10 mg/Kg/ week/at least 3 weeks, i.p.) increase ROS levels in the striatal homogenate, induce a dose-dependent decrease in dopaminergic neurons from the substantia nigra, a decline in striatal dopamine nerve terminal density, and a neurobehavioral syndrome characterized by reduced ambulatory activity (Brooks et al., 1999; McCormack et al., 2002; Kuter et al., 2010). PQ also reproduces other biochemical and neuropathological characteristics of human Parkinsonism such as microglia activation (Wu et al., 2005; Purisai et al., 2007), α-syn up-regulation and fibrillation (Uversky et al., 2001; Manning-Bog et al., 2002), increases lipid peroxidation (increase of 4-hydroxynonenals) (McCormack et al., 2005), alters parkin solubility promoting its intracellular aggregation (Wang et al., 2005), induces a proteasome dysfunction in SH-SY5Y cells (Ding and Keller, 2001; Yang and Tiffany-Castiglioni, 2007), as well as in homogenates from postmortem PD brains (McNaught and Jenner, 2001; McNaught et al., 2002), impairs mitochondrial function at the level of complex III to generate ROS (Castello et al., 2007; Drechsel and Patel, 2009), promotes cytochrome C release (González-Polo et al., 2004; Fei et al., 2008), induces GSH depletion (Schmuck et al., 2002; Kang et al., 2009), and causes cell injury leading to apoptotic cell death (Berry et al., 2010; Franco et al., 2010). PQ has been used as a toxicological model for PD that has permitted getting important information about the mechanisms involved in the neurodegeneration associated with PD (Gao and Hong, 2011).
Rotenone, an OP insecticide has also been associated with an increased risk of PD. Two epidemiological studies found an association between rotenone exposure and PD risk, reporting an increased risk of 10–fold (OR = 10.0, 95% CI, 2.9–34.3) in East Texas farmers (Dhillon et al., 2008), and 2.5-fold (OR = 2.5, 95% CI, 1.3–4.7) in PD cases (n = 110) compared with controls (n = 358) from professional pesticide applicators participants in the AHS (Tanner et al., 2011). Rotenone can freely cross the blood–brain barrier and is a well-established mitochondrial toxin that specifically inhibits the complex I (NADH–dehydrogenase) of the electron transport chain leading to ATP depletion, energy failure and mitochondrial ROS production, which in turn induces cytochrome C release and apoptotic cell death (Clayton et al., 2005; Radad et al., 2006; Sherer et al., 2007). It has been shown that, like MPTP, rotenone treatment in animal models (1.5–3 mg/Kg/day/up to 3 weeks) reproduces features of PD such as bradykinesia, postural instability and/or rigidity, reduces the tyrosine hydroxylase-positive neurons in the substance nigra, induces a loss of striatal dopamine, and the accumulation of α-syn and poly-ubiquitin positive aggregates in remaining dopaminergic neurons (Betarbet et al., 2000; Sherer et al., 2003; Cannon et al., 2009). Likewise, Betarbet et al. (2006) observed that chronic administration of 3.0 mg/Kg/day of rotenone for up to 5 weeks to male rats caused the oxidation of DJ-1, accumulation of α-syn, and proteasomal impairment (Betarbet et al., 2006). These effects were also observed in neuroblastoma SK-N-MC cells treated with rotenone (5 nM/4 weeks), as well as a loss of GSH, oxidative DNA and protein damage and caspase-dependent death (Sherer et al., 2002; Betarbet et al., 2006). Rotenone has also the capacity to activate microglia (Sherer et al., 2003); Gao et al. (2002) demonstrated that the addition of microglia to primary neuron-enriched cultures (neuron/glia cultures) markedly increased the dopaminergic neurodegeneration induced by rotenone (1 nM/8 days), and this neurotoxicity was attenuated by the inhibition of NADPH oxidase or scavenging the superoxide radical that is liberated from the microglia (Gao et al., 2002). Since rotenone recapitulates several mechanisms of PD pathogenesis, this pesticide is currently used as a toxicological model to study the underlying mechanisms on the PD development.
Despite the widespread use of OP insecticides such as malathion, methyl parathion, chlorpyriphos and diazinon, not many studies have evaluated the association between specific OP and PD risk. Dhillon et al. (2008) found a 2–fold increase (OR = 2.0, 95% CI, 1.02–3.8) in the risk of PD in Texan agricultural workers exposed to chlorpyriphos (cases = 100, controls = 84) (Dhillon et al., 2008). An increased risk of PD was also observed in rural residents from California possibly exposed to high levels of chlorpyriphos (OR = 1.87, 95% CI, 1.05–3.31) and diazinon (OR = 1.75, 95% CI, 1.12–2.76) through the consumption of contaminated well–water (Gatto et al., 2009). One study conducted in a population from the Group Health Cooperative (GHC) in Western Washington State occupationally exposed to methyl parathion found a high risk of PD (OR = 8.08, 95% CI, 0.92–70.85), although the association was not statistically significant (Firestone et al., 2005). This is particularly relevant, because parkinsonian effects have been reported in cases of patients intoxicated with OP (Bhatt et al., 1999).
Solvents
Solvents are widespread used due to their commercial applications, including metal degreasing, dry cleaning, and as ingredients of paint thinners and detergents. Some solvents are lipophilic and thus easily absorbed by the central and peripheral nervous system tissues (Lock et al., 2013). There are isolated cases of acute Parkinsonism associated with large solvent exposures such as in workers exposed to n-hexane (Pezzoli et al., 1989), and toluene (Papageorgiou et al., 2009), among others. There is no consistent evidence of the association of solvent exposure and PD (Wirdefeldt et al., 2011). One case-control study based on a questionnaire reported an increased risk of PD by the exposure to organic solvents (OR = 2.78, 95% CI, 1.23–6.26) in 86 PD patients and 86 controls from the Emilia-Romagna region in Italy (Smargiassi et al., 1998). Another case–control study reported an increased risk of PD when the exposure to solvents was above 20 years (OR = 3.59, 95% CI, 1.26–19.26) in 182 cases (vs. 422 controls) identified through death certificates of the Rolls-Royce PLC national pension fund archive from employees of five manufacturing locations in United Kingdom who had any mention of PD (McDonnell et al., 2003).
Trichloroethylene (TCE) is one of the specific solvents that has been investigated in detail (Goldman, 2014). Some clinical case reports have reported the onset of PD in workers exposed to TCE through chronic inhalation and dermal exposure by handling TCE, suggesting a potential link between the exposure to TCE and PD (Kochen et al., 2003; Gash et al., 2008). More recently, an epidemiologic study in 99 twin pairs discordant for PD showed that the exposure to TCE was associated with a 6–fold increased risk of PD (OR = 6.1, 95% CI, 1.2–33) (Goldman, 2014). In animal models, TCE may recapitulate several key pathological features of PD. The systemic exposure of adult rats to TCE (1000 mg/Kg/day/5 days a week/2 and 6 weeks, oral gavage) inhibits mitochondrial complex I enzyme activity, increases oxidative stress markers, activates the microglia, induces nigral α-syn accumulation and a significant loss of dopaminergic neurons on the SNpc in a dose-dependent manner, as well as defects in the rotarod behavior test (Liu et al., 2010). In a similar way, the administration of n-hexane and its metabolite 2, 5-hexanedione (400 mg/Kg/day/5 days a week/6 weeks, i.p.) to mice caused that both chemicals reduced the striatal dopamine concentration by 38 and 33%, respectively, but neuronal cell loss was not confirmed (Pezzoli et al., 1990). On the other hand, there is no evidence that acute or subchronic exposure to toluene promotes the degeneration of the nigrostriatal dopamine system (Lock et al., 2013).
Nanoparticles and PD
Nanoparticles are an important alternative in the development of treatment strategies for neurodegenerative diseases due to their small particle size, large surface and high drug loading efficiency, which allow them to cross the blood-brain barrier and efficiently release specific drugs (Li et al., 2014b; Leyva-Gómez et al., 2015). However, their small size allows them to penetrate the cell and organelles, disrupting their normal function (Buzea et al., 2007).
Although some NPs are being used in therapies for PD, no epidemiological studies are available associating them with PD risk. However, there is evidence suggesting that they could contribute to alter the molecular mechanisms involved in the pathogenesis of PD. Thus, it was reported that intranasal instillation of SiO2-NPs (20 µg/day/1–7 days) to rats resulted in their presence in the striatum, the induction of oxidative damage, an inflammatory response, and depleted dopamine concentration and tyrosine hydroxylase levels, suggesting that these NPs have a negative impact on striatal dopaminergic neurons (Wu et al., 2011). Another report in adult zebrafish exposed to SiO2-NPs (300 and 1000 µg/mL; 15 and 50-nm of size) showed alterations in neurobehavioral parameters (general, cognitive behavior and locomotive activity), with the most significant effects observed with the smallest NPs, similar to those observed in neurodegenerative diseases (Li et al., 2014b). In vitro studies also support the potential contribution of NPs in PD development. The exposure of dopaminergic neurons (PC12 cells) to SiO2-NPs (25–200 µg/mL/24 h) triggered an oxidative stress, disturbed the cell cycle, induced apoptosis, and activated the p53-mediated signaling pathway (Wu et al., 2011); while the exposure of these cells to TiO2-NPs (50, 100 and 200 mg/mL/24 h) induced a dose–dependent increase in the expression and aggregation of α-syn, as well as a reduction of the expressions of Parkin (E3 ligase), and the ubiquitin C-terminal hydrolase (UCH-L1), these events were associated with increased oxidative stress (Wu and Xie, 2014). Also, the exposure PC12 cells to iron oxide (Fe2O3-NPs; 0.15–15 mM) decreased the neurite growth in response to the nerve growth factor (NGF) (Pisanic et al., 2007). Likewise, citrate-capped gold nanoparticles (Au-NPs; 0.3–32 nM, 10–22 nm) produced a dose-dependent aggregation of purified α-syn, being strongest for the smallest NPs (Alvarez et al., 2013). In contrast, the administration of Neurotensin (NTS)-polyplex NPs (8.5 nmol/Kg, i.v), a nanocarrier gene with a potential for nanomedicine-based applications for PD treatment, to BALB/c mice does not produce systemic inflammatory (up to 24 h after treatment) nor hepatic cytotoxicity (at 24 and 96 h after treatment), supporting the safety of these NTS-polyplex NPs as a potential therapeutic approach (Hernandez et al., 2014).
Early Exposure to Environmental Factors and AD or PD Development: Epigenetic Evidence
Epigenetic DNA modifications include DNA methylation, histone post-translational modifications (mainly acetylation) and miRNAs (Holliday, 2006). DNA methylation is one of the most studied epigenetic modifications that influence the gene expression. It involves the addition of methyl groups to cytosine bases located at cytosine–phosphate–guanine (CpG) sites by the action of DNA methyltransferases (DNMTs). Alterations in DNA methylation on the promoter regions of genes regulate the gene expression of important processes such as embryonic development, cellular differentiation and aging (Bird, 2002). Increasing evidence suggests that epigenetic changes in the developing embryo that may play important roles in the susceptibility to diseases in later life (imprinted disease phenotypes) result from maternal exposures to environmental stimuli at critical periods of development. This suggests that a short exposure to chemicals could be memorized through epigenetic mechanisms long after the chemical trigger has gone (Jang and Serra, 2014), and recent studies have suggested that an epigenetic component could be involved in neurodegenerative diseases related to environmental factors (Marques et al., 2011).
The latent brain expression of genes observed in animals developmentally exposed to an environmental contaminant may be mediated through epigenetic pathways that are regulated via the DNA methylation. While the conditions leading to early life hypo- or hyper-methylation of specific genes are not known, both can induce oxidative DNA damage; for instance the hypo-methylation of APP gene increases its expression driving the overproduction of APP and Aβ levels, which in turn facilitate the ROS production damaging the DNA, and producing neuronal loss. While the hyper-methylation affects the gene transcription and DNA repair pathways. Therefore, both changes in DNA methylation can impact gene expression and imprint susceptibility to oxidative DNA damage in the aged brain (Zawia et al., 2009). Thus, it is suggested that Pb interferes with the DNA methylating capacity, thus altering the expression of AD-related genes. The study performed in aged monkeys developmentally exposed to Pb revealed a reduced activity of brain Dnmt, and the exposure of mouse primary cells from the cerebral cortex to Pb (0.1 µM) resulted in a similar effect on Dnmt1 activity a week after 24 h-treatment (Wu et al., 2008). Also, Bihaqi and Zawia (2012) showed a significant latent increase in AD biomarkers an a reduction in the protein and mRNA levels of DNA methylating enzymes Dnmt1 and Dnmt3a, and methyl CpG binding protein 2 (MeCP2) in differentiated SH-SY5Y cells treated with Pb (5–100 µM/48 h) and analyzed 6 days later (Bihaqi and Zawia, 2012). Aberrant CpG methylation in APP, Tau and GSK3β genes was reported in post-mortem brains (Iwata et al., 2014). In addition, it suggested that reduced levels of CpG methylation in the promoter of APP could be mediated by the oxidation of guanine (8-oxdG) (Zawia et al., 2009); this is because the oxidation of guanine in CpG dinucleotides inhibits adjacent cytosine methylation (Weitzman et al., 1994). On the other hand, Cd, another metal involved in AD pathology, reduces the enzymatic activity of Dnmt in rat liver cell cultures (Poirier and Vlasova, 2002), but this effect has not been evaluated in cerebral cells. While a study showed that subchronic As exposure (3 and 36 ppm/from gestation until 4 months of age) altered the methylation of genes involved in neuronal plasticity, including reelin (RELN) and protein phosphatase 1 (PP1), which was associated with memory deficits (Martínez et al., 2011). Regarding other compounds, the perinatal exposure to permethrin (34 mg/Kg/daily, by gavage from postnatal day 6–21) to mice showed altered brain functions including biomarkers of maintenance of dopaminergic neurons, and impairment of spatial memory at 6 months of age (Nasuti et al., 2013).
The relation between epigenetic modifications and PD has been less studied; however, a potential role of DNA methylation in the promoter of α-syn encoding gene (SNCA) in the neuropathogenesis of PD has been suggested, considering that α-syn is a fundamental component of LB, the main hallmark of PD (Lu et al., 2013). A DNA hypomethylation of SNCA was reported in the substantia nigra of sporadic PD patients, suggesting that it might contribute to the dysregulation of SNCA expression in PD (Jowaed et al., 2010; Matsumoto et al., 2010). In addition, increased SNCA mRNA levels were observed in SNpc of PD (Chiba-Falek et al., 2006), and reduced levels of Dnmt1 have been observed in postmortem brains from PD and dementia with LB (DLB) patients, as well as in brains of α-syn transgenic mice; authors suggest that this effect could be a novel mechanism of epigenetic dysregulation in LB-related diseases such as PD (Desplats et al., 2011). Finally, a lesser degree of methyation of the TNFα promoter, a key inflammatory cytokine associated with dopaminergic cell death was observed in the SNpc from PD patients, predisposing to an increase neuronal vulnerability to inflammatory reactions (Mogi et al., 1996; Pieper et al., 2008).
Environmental factors associated with an increased risk of PD such as pesticides can alter the expression of genes by epigenetic mechanisms (Kwok, 2010). It was reported that pre-treatment with 5-aza-2’deoxycytidine (5’-aza-dC, a DNMT inhibitor) exacerbated the dopaminergic neuron damage induced by PQ, MPP+, 6-hydroxydopamine (6-OHDA) and rotenone treatment, and induced oxidative stress, the transcriptional up-regulation of α-syn, and demethylation of the α-syn promoter (Wang et al., 2013). Likewise, the folate deficiency sensitizes mice to MPTP-induced PD-like pathology and motor dysfunction (Duan et al., 2002); it is well known that folate deficiency alters the development of human nervous system (Greenblatt et al., 1994).
On the other hand, it was reported that the exposure to environmental neurotoxicants associated with PD during early life or pregnancy can determine the progressive damage of the substantia nigra years before the onset of clinical parkinsonism, as well as to increase the vulnerability to effects of a second environmental factor (two–hit model) (Logroscino, 2005). A study in C57BL/6 mice daily treated with pQ (0.3 mg/Kg) or maneb (1 mg/Kg) or PQ + maneb from postnatal day 5–19 and then re-exposed as adults to PQ (10 mg/Kg) or maneb (30 mg/Kg) or PQ + maneb (twice a week/3 weeks) showed that dopaminergic cell loss and decreased dopamine levels were amplified by the adult re-challenge to the pesticides, suggesting that the developmental exposure to neurotoxins enhanced the adult susceptibility to a new toxic insult (Thiruchelvam et al., 2002). Similarly, prenatal exposure of pregnant C57BL/6J mice to PQ (0.3 mg/Kg) or maneb (1 mg/Kg) altered the development of the nigrostriatal system and enhanced its vulnerability to neurotoxins later in life, which could contribute with the development of PD during aging (Barlow et al., 2004).
Although there is no direct evidence linking early exposure to environmental pollutants and epigenetic changes with increased susceptibility to LOPD, there is a plausible association based on the following considerations: (1) epigenetic alterations have been observed in PD brains; (2) the exposure to environmental factors is associated with an increased risk of LOPD development and factors such as pesticides and metals can alter mechanisms of epigenetic regulation such as DNA methylation; and (3) early exposure to environmental pollutants might be associated with LOPD later in life. Further studies are needed to confirm this hypothesis in this promising research field to understand the mechanisms underlying the long-term effects of the environment on the PD development.
Concluding Remarks
The emerging association between exposures to several toxic compounds with neurodegenerative diseases is of considerable public health importance, given the increasing dementia prevalence, the negative social and economic consequences of neurodegeneration-related disabilities, and the increasing environmental pollution in some geographic areas worldwide. Some of the epidemiological studies show not consistent results on getting significant estimates of hazard risk for AD or PD, mainly due to some limitations that include the difficulty on accurate diagnosis for AD or PD cases due to the lack of specific biomarkers, the deficiency to accurately assess chronic exposures, and/or the lack of inclusion of important confounding variables such as co-exposure to toxic compounds, genetic variants and lifestyle among others. Nevertheless, epidemiological studies along with experimental data have led to highlight the potential risk to develop these degenerative diseases due to the exposure to environmental pollutants such as metals, NPs and pesticides, among others. Interestingly, these pollutants show similar mechanisms of toxicity, which converge in a generalized mechanism based on the generation of oxidative stress that leads to common hallmarks of both neurodegenerative disorders. For example, the generation of oxidative stress by increasing the production of ROS and/or deregulating the antioxidant enzymes promotes the formation of protein aggregates such as Tau, Aβ or α-syn. This in turn overwhelms the degradation systems, and produces the activation of the glia inducing neuroinflammation, a process that per se increases the generation of further oxidative stress leading to a self-perpetuating cycle, and finally to neuronal loss of specific brain region such as the hippocampus and cerebral cortex in AD and substantia nigra in PD. The oxidative stress induced by these neurotoxicants activates/inhibits signaling pathways leading to augmented/diminished activity of enzymes that promote the accumulation of toxic materials in neural cells such as damaged/aberrant proteins, Aβ in AD or α–syn in PD and oxidative byproducts, or the oxidation of DNA that can alter genetic or epigenetic regulation. Furthermore, the link between early life exposure to environmental factors and the origin of neurodegenerative diseases is getting attention and can help to clarify the role of the environment on the development of these degenerative diseases. On the other hand, the lack of specific/differential biomarkers for AD or PD limits the early diagnosis and then the timely treatment. In this regard, specific circulating miRNAs have been associated with pathological processes such as AD and PD, therefore they are promising non-invasive biomarkers for these neurological diseases. Additionally, the identification of biomarkers to determine the past exposure to environmental pollutants is of vital importance for a better and opportune management of these diseases. Thus, as we have more knowledge of the risk from the exposure to environmental pollutants, more well-designed epidemiological studies (controlling for as many variables as possible and with high sample sizes) are necessary to improve the quality of life of elderly and to prevent the development of neurodegenerative diseases worldwide.
Abbreviations
Aβ, amyloid beta peptide; AICD, amyloid intracellular domain; AD, Alzheimer’s disease; Al, aluminum; AGE, advanced glycation end products; ApoE, apolipoprotein E; APP, amyloid precursor protein; As, arsenic; α-syn, alpha-synuclein; BACE1, beta-site APP cleaving enzyme 1; Cd, cadmium; CSF, cerebrospinal fluid; EOAD, early-onset Alzheimer’ disease; Hg, mercury; HR, hazard ratio; IDE, insulin-like degrading enzyme; LB, Lewy bodies; LOAD, late-onset Alzheimer’ disease; LRP1, low density lipoprotein receptor-related protein 1; Me-Hg, methyl mercury; Mn, manganese; MTs, microtubules; NEP, neprilysin; NFTs, neurofibrillary tangles; NPs, nanoparticles; OP, organophosphates; OCl, organochlorines; PHFs, paired helical filaments; Pb, lead; PD, Parkinson’s disease; PQ, paraquat; PS, presenilin; ROS, reactive oxygen species; RR, relative risk; sAPPβ, soluble APP beta; SNpc, substantia nigra pars compacta; SNpr, substantia nigra pars reticulate; Tau, tau protein; P-Tau, phosphorylated Tau; TCE, trichloroethylene; Zn, zinc.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbott, R. D., Ross, G. W., Tanner, C. M., Andersen, J. K., Masaki, K. H., Rodriguez, B. L., et al. (2012). Late-life hemoglobin and the incidence of Parkinson’s disease. Neurobiol. Aging 33, 914–920. doi: 10.1016/j.neurobiolaging.2010.06.023
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Altmann, P., Cunningham, J., Dhanesha, U., Ballard, M., Thompson, J., and Marsh, F. (1999). Disturbance of cerebral function in people exposed to drinking water contaminated with aluminium sulphate: retrospective study of the Camelford water incident. BMJ 319, 807–811. doi: 10.1136/bmj.319.7213.807
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Alvarez, Y. D., Fauerbach, J. A., Pellegrotti, J. V., Jovin, T. M., Jares-Erijman, E. A., and Stefani, F. D. (2013). Influence of gold nanoparticles on the kinetics of alpha-synuclein aggregation. Nano Lett. 13, 6156–6163. doi: 10.1021/nl403490e
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
An, L., Liu, S., Yang, Z., and Zhang, T. (2012). Cognitive impairment in rats induced by nano-CuO and its possible mechanisms. Toxicol. Lett. 213, 220–227. doi: 10.1016/j.toxlet.2012.07.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ascherio, A., Chen, H., Weisskopf, M. G., O’Reilly, E., McCullough, M. L., Calle, E. E., et al. (2006). Pesticide exposure and risk for Parkinson’s disease. Ann. Neurol. 60, 197–203. doi: 10.1002/ana.20904
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ashok, A., Rai, N. K., Tripathi, S., and Bandyopadhyay, S. (2015). Exposure to As-, Cd- and Pb-mixture induces Aβ, amyloidogenic APP processing and cognitive impairments via oxidative stress-dependent neuroinflammation in young rats. Toxicol. Sci. 143, 64–80. doi: 10.1093/toxsci/kfu208
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Atwood, C. S., Moir, R. D., Huang, X., Scarpa, R. C., Bacarra, N. M., Romano, D. M., et al. (1998). Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis. J. Biol. Chem. 273, 12817–12826. doi: 10.1074/jbc.273.21.12817
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baldi, I., Lebailly, P., Mohammed-Brahim, B., Letenneur, L., Dartigues, J. F., and Brochard, P. (2003). Neurodegenerative diseases and exposure to pesticides in the elderly. Am. J. Epidemiol. 157, 409–414. doi: 10.1093/aje/kwf216
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barlow, B. K., Richfield, E. K., Cory-Slechta, D. A., and Thiruchelvam, M. (2004). A fetal risk factor for Parkinson’s disease. Dev. Neurosci. 26, 11–23. doi: 10.1159/000080707
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Basha, M. R., Wei, W., Bakheet, S. A., Benitez, N., Siddiqi, H. K., Ge, Y. W., et al. (2005). The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J. Neurosci. 25, 823–829. doi: 10.1523/jneurosci.4335-04.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Basun, H., Forssell, L. G., Wetterberg, L., and Winblad, B. (1991). Metals and trace elements in plasma and cerebrospinal fluid in normal aging and Alzheimer’s disease. J. Neural Transm. Park. Dis. Dement. Sect. 3, 231–258.
Baum, L., Chan, I. H., Cheung, S. K., Goggins, W. B., Mok, V., Lam, L., et al. (2010). Serum zinc is decreased in Alzheimer’s disease and serum arsenic correlates positively with cognitive ability. Biometals 23, 173–179. doi: 10.1007/s10534-009-9277-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Behl, M., Zhang, Y., Monnot, A. D., Jiang, W., and Zheng, W. (2009). Increased β-amyloid levels in the choroid plexus following lead exposure and the involvement of low-density lipoprotein receptor protein-1. Toxicol. Appl. Pharmacol. 240, 245–254. doi: 10.1016/j.taap.2009.05.024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Behl, M., Zhang, Y., Shi, Y., Cheng, J., Du, Y., and Zheng, W. (2010). Lead-induced accumulation of beta-amyloid in the choroid plexus: role of low density lipoprotein receptor protein-1 and protein kinase C. Neurotoxicology 31, 524–532. doi: 10.1016/j.neuro.2010.05.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bergen, W. G. (1971). The in vitro effect of dieldrin on respiration of rat liver mitochondria. Proc. Soc. Exp. Biol. Med. 136, 732–735. doi: 10.3181/00379727-136-35352
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Berry, C., La Vecchia, C., and Nicotera, P. (2010). Paraquat and Parkinson’s disease. Cell Death Differ. 17, 1115–1125. doi: 10.1038/cdd.2009.217
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Betarbet, R., Canet-Aviles, R. M., Sherer, T. B., Mastroberardino, P. G., McLendon, C., Kim, J. H., et al. (2006). Intersecting pathways to neurodegeneration in Parkinson’s disease: effects of the pesticide rotenone on DJ-1, alpha-synuclein and the ubiquitin-proteasome system. Neurobiol. Dis. 22, 404–420. doi: 10.1016/j.nbd.2005.12.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Betarbet, R., Sherer, T. B., MacKenzie, G., Garcia-Osuna, M., Panov, A. V., and Greenamyre, J. T. (2000). Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 3, 1301–1306. doi: 10.1038/81834
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bhatt, M. H., Elias, M. A., and Mankodi, A. K. (1999). Acute and reversible parkinsonism due to organophosphate pesticide intoxication: five cases. Neurology 52, 1467–1471. doi: 10.1212/wnl.52.7.1467
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bhattacharjee, S., Zhao, Y., Hill, J. M., Percy, M. E., and Lukiw, W. J. (2014). Aluminum and its potential contribution to Alzheimer’s disease (AD). Front. Aging Neurosci. 6:62. doi: 10.3389/fnagi.2014.00062
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bihaqi, S. W., and Zawia, N. H. (2012). Alzheimer’s disease biomarkers and epigenetic intermediates following exposure to Pb in vitro. Curr. Alzheimer Res. 9, 555–562. doi: 10.2174/156720512800617964
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bihaqi, S. W., and Zawia, N. H. (2013). Enhanced taupathy and AD-like pathology in aged primate brains decades after infantile exposure to lead (Pb). Neurotoxicology 39, 95–101. doi: 10.1016/j.neuro.2013.07.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bird, A. (2002). DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21. doi: 10.1101/gad.947102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Botta-Orfila, T., Morató, X., Compta, Y., Lozano, J. J., Falgàs, N., Valldeoriola, F., et al. (2014). Identification of blood serum micro-RNAs associated with idiopathic and LRRK2 Parkinson’s disease. J. Neurosci. Res. 92, 1071–1077. doi: 10.1002/jnr.23377
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brooks, A. I., Chadwick, C. A., Gelbard, H. A., Cory-Slechta, D. A., and Federoff, H. J. (1999). Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Res. 823, 1–10. doi: 10.1016/s0006-8993(98)01192-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brown, T. P., Rumsby, P. C., Capleton, A. C., Rushton, L., and Levy, L. S. (2006). Pesticides and Parkinson’s disease–is there a link? Environ. Health Perspect. 114, 156–164. doi: 10.1289/ehp.8095
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Butterfield, D. A., Drake, J., Pocernich, C., and Castegna, A. (2001). Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid β-peptide. Trends Mol. Med. 7, 548–554. doi: 10.1016/s1471-4914(01)02173-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buzea, C., Pacheco, I. I., and Robbie, K. (2007). Nanomaterials and nanoparticles: sources and toxicity. Biointerphases 2, MR17–MR71. doi: 10.1116/1.2815690
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cannon, J. R., Tapias, V., Na, H. M., Honick, A. S., Drolet, R. E., and Greenamyre, J. T. (2009). A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 34, 279–290. doi: 10.1016/j.nbd.2009.01.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Castello, P. R., Drechsel, D. A., and Patel, M. (2007). Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain. J. Biol. Chem. 282, 14186–14193. doi: 10.1074/jbc.m700827200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chang, J. W., Pai, M. C., Chen, H. L., Guo, H. R., Su, H. J., and Lee, C. C. (2008). Cognitive function and blood methylmercury in adults living near a deserted chloralkali factory. Environ. Res. 108, 334–339. doi: 10.1016/j.envres.2008.06.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, N. N., Luo, D. J., Yao, X. Q., Yu, C., Wang, Y., Wang, Q., et al. (2012b). Pesticides induce spatial memory deficits with synaptic impairments and an imbalanced tau phosphorylation in rats. J. Alzheimers Dis. 30, 585–594. doi: 10.3233/JAD-2012-111946
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, L., Yoo, S. E., Na, R., Liu, Y., and Ran, Q. (2012a). Cognitive impairment and increased Aβ levels induced by paraquat exposure are attenuated by enhanced removal of mitochondrial H2O2. Neurobiol. Aging 33, 432.e15–432.e26. doi: 10.1016/j.neurobiolaging.2011.01.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chiba-Falek, O., Lopez, G. J., and Nussbaum, R. L. (2006). Levels of alpha-synuclein mRNA in sporadic Parkinson disease patients. Mov. Disord. 21, 1703–1708. doi: 10.1002/mds.21007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chin-Chan, M., Segovia, J., Quintanar, L., Arcos-López, T., Hersh, L., Chow, K., et al. (2015). Mercury reduces the enzymatic activity of neprilysin in differentiated SH-SY5Y cells. Toxicol. Sci. doi: 10.1093/toxsci/kfv037. [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Clayton, R., Clark, J. B., and Sharpe, M. (2005). Cytochrome c release from rat brain mitochondria is proportional to the mitochondrial functional deficit: implications for apoptosis and neurodegenerative disease. J. Neurochem. 92, 840–849. doi: 10.1111/j.1471-4159.2004.02918.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cook, D. G., Fahn, S., and Brait, K. A. (1974). Chronic manganese intoxication. Arch. Neurol. 30, 59–64. doi: 10.1001/archneur.1974.00490310061010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cookson, M. R. (2012). Parkinsonism due to mutations in PINK1, parkin and DJ-1 and oxidative stress and mitochondrial pathways. Cold Spring Harb. Perspect. Med. 2:a009415. doi: 10.1101/cshperspect.a009415
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Coon, S., Stark, A., Peterson, E., Gloi, A., Kortsha, G., Pounds, J., et al. (2006). Whole-body lifetime occupational lead exposure and risk of Parkinson’s disease. Environ. Health Perspect. 114, 1872–1876. doi: 10.1289/ehp.9102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Corrigan, F. M., Murray, L., Wyatt, C. L., and Shore, R. F. (1998). Diorthosubstituted polychlorinated biphenyls in caudate nucleus in Parkinson’s disease. Exp. Neurol. 150, 339–342. doi: 10.1006/exnr.1998.6776
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Corrigan, F. M., Wienburg, C. L., Shore, R. F., Daniel, S. E., and Mann, D. (2000). Organochlorine insecticides in substantia nigra in Parkinson’s disease. J. Toxicol. Environ. Health A 59, 229–234. doi: 10.1080/009841000156907
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Corti, O., Lesage, S., and Brice, A. (2011). What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol. Rev. 91, 1161–1218. doi: 10.1152/physrev.00022.2010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Couper, J. (1837). On the effects of black oxide of manganese when inhaled into the lungs. Br. Ann. Med. Pharmacol. 1, 41–42.
Cras, P., Kawai, M., Siedlak, S., Mulvihill, P., Gambetti, P., Lowery, D., et al. (1990). Neuronal and microglial involvement in beta-amyloid protein deposition in Alzheimer’s disease. Am. J. Pathol. 137, 241–246.
Dani, S. U. (2010). Arsenic for the fool: an exponential connection. Sci. Total Environ. 408, 1842–1846. doi: 10.1016/j.scitotenv.2010.01.027
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Desplats, P., Spencer, B., Coffee, E., Patel, P., Michael, S., Patrick, C., et al. (2011). Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 286, 9031–9037. doi: 10.1074/jbc.C110.212589
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dexter, D. T., Carayon, A., Javoy-Agid, F., Agid, Y., Wells, F. R., Daniel, S. E., et al. (1991). Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 114(Pt. 4), 1953–1975. doi: 10.1093/brain/114.4.1953
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dexter, D. T., Wells, F. R., Agid, F., Agid, Y., Lees, A. J., Jenner, P., et al. (1987). Increased nigral iron content in postmortem parkinsonian brain. Lancet 2, 1219–1220. doi: 10.1016/s0140-6736(87)91361-4
Dhillon, A. S., Tarbutton, G. L., Levin, J. L., Plotkin, G. M., Lowry, L. K., Nalbone, J. T., et al. (2008). Pesticide/environmental exposures and Parkinson’s disease in east texas. J. Agromedicine 13, 37–48. doi: 10.1080/10599240801986215
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ding, Q., and Keller, J. N. (2001). Proteasome inhibition in oxidative stress neurotoxicity: implications for heat shock proteins. J. Neurochem. 77, 1010–1017. doi: 10.1046/j.1471-4159.2001.00302.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dorsey, C. D., Lee, B. K., Bolla, K. I., Weaver, V. M., Lee, S. S., Lee, G. S., et al. (2006). Comparison of patella lead with blood lead and tibia lead and their associations with neurobehavioral test scores. J. Occup. Environ. Med. 48, 489–496. doi: 10.1097/01.jom.0000199678.86629.3b
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Drechsel, D. A., and Patel, M. (2009). Differential contribution of the mitochondrial respiratory chain complexes to reactive oxygen species production by redox cycling agents implicated in parkinsonism. Toxicol. Sci. 112, 427–434. doi: 10.1093/toxsci/kfp223
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duan, W., Ladenheim, B., Cutler, R. G., Kruman, I. I., Cadet, J. L., and Mattson, M. P. (2002). Dietary folate deficiency and elevated homocysteine levels endanger dopaminergic neurons in models of Parkinson’s disease. J. Neurochem. 80, 101–110. doi: 10.1046/j.0022-3042.2001.00676.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Etminan, M., Gill, S., and Samii, A. (2003). Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: systematic review and meta-analysis of observational studies. BMJ 327:128. doi: 10.1136/bmj.327.7407.128
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Exley, C., and Esiri, M. M. (2006). Severe cerebral congophilic angiopathy coincident with increased brain aluminium in a resident of Camelford, Cornwall, UK. J. Neurol. Neurosurg. Psychiatry 77, 877–879. doi: 10.1136/jnnp.2005.086553
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Exley, C., Price, N. C., Kelly, S. M., and Birchall, J. D. (1993). An interaction of beta-amyloid with aluminium in vitro. FEBS Lett. 324, 293–295. doi: 10.1016/0014-5793(93)80137-j
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fei, Q., McCormack, A. L., Di Monte, D. A., and Ethell, D. W. (2008). Paraquat neurotoxicity is mediated by a Bak-dependent mechanism. J. Biol. Chem. 283, 3357–3364. doi: 10.1074/jbc.m708451200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Firestone, J. A., Smith-Weller, T., Franklin, G., Swanson, P., Longstreth, W. T. Jr., and Checkoway, H. (2005). Pesticides and risk of Parkinson disease: a population-based case-control study. Arch. Neurol. 62, 91–95. doi: 10.1001/archneur.62.1.91
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fleming, L., Mann, J. B., Bean, J., Briggle, T., and Sanchez-Ramos, J. R. (1994). Parkinson’s disease and brain levels of organochlorine pesticides. Ann. Neurol. 36, 100–103. doi: 10.1002/ana.410360119
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Franco, R., Li, S., Rodriguez-Rocha, H., Burns, M., and Panayiotidis, M. I. (2010). Molecular mechanisms of pesticide-induced neurotoxicity: relevance to Parkinson’s disease. Chem. Biol. Interact. 188, 289–300. doi: 10.1016/j.cbi.2010.06.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Freire, C., and Koifman, S. (2012). Pesticide exposure and Parkinson’s disease: epidemiological evidence of association. Neurotoxicology 33, 947–971. doi: 10.1016/j.neuro.2012.05.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Friedhoff, P., von Bergen, M., Mandelkow, E. M., and Mandelkow, E. (2000). Structure of tau protein and assembly into paired helical filaments. Biochim. Biophys. Acta 1502, 122–132. doi: 10.1016/S0925-4439(00)00038-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fujimura, M., Usuki, F., Sawada, M., and Takashima, A. (2009). Methylmercury induces neuropathological changes with tau hyperphosphorylation mainly through the activation of the c-jun-N-terminal kinase pathway in the cerebral cortex, but not in the hippocampus of the mouse brain. Neurotoxicology 30, 1000–1007. doi: 10.1016/j.neuro.2009.08.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gao, H. M., and Hong, J. S. (2011). Gene-environment interactions: key to unraveling the mystery of Parkinson’s disease. Prog. Neurobiol. 94, 1–19. doi: 10.1016/j.pneurobio.2011.03.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gao, H. M., Hong, J. S., Zhang, W., and Liu, B. (2002). Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J. Neurosci. 22, 782–790.
Gash, D. M., Rutland, K., Hudson, N. L., Sullivan, P. G., Bing, G., Cass, W. A., et al. (2008). Trichloroethylene: Parkinsonism and complex 1 mitochondrial neurotoxicity. Ann. Neurol. 63, 184–192. doi: 10.1002/ana.21288
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gatto, N. M., Cockburn, M., Bronstein, J., Manthripragada, A. D., and Ritz, B. (2009). Well-water consumption and Parkinson’s disease in rural California. Environ. Health Perspect. 117, 1912–1918. doi: 10.1289/ehp.0900852
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gerhardsson, L., Lundh, T., Minthon, L., and Londos, E. (2008). Metal concentrations in plasma and cerebrospinal fluid in patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 25, 508–515. doi: 10.1159/000129365
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gibb, W. R., and Lees, A. J. (1988). The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 51, 745–752. doi: 10.1136/jnnp.51.6.745
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Godfrey, M. E., Wojcik, D. P., and Krone, C. A. (2003). Apolipoprotein E genotyping as a potential biomarker for mercury neurotoxicity. J. Alzheimers Dis. 5, 189–195.
Goedert, M., and Spillantini, M. G. (2006). A century of Alzheimer’s disease. Science 314, 777–781. doi: 10.1126/science.1132814
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goedert, M., Wischik, C. M., Crowther, R. A., Walker, J. E., and Klug, A. (1988). Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. U S A 85, 4051–4055. doi: 10.1073/pnas.85.11.4051
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goldman, S. M. (2014). Environmental toxins and Parkinson’s disease. Annu. Rev. Pharmacol. Toxicol. 54, 141–164. doi: 10.1146/annurev-pharmtox-011613-135937
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goldman, S. M., Quinlan, P. J., Ross, G. W., Marras, C., Meng, C., Bhudhikanok, G. S., et al. (2012). Solvent exposures and Parkinson disease risk in twins. Ann. Neurol. 71, 776–784. doi: 10.1002/ana.22629
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gong, G., and O’Bryant, S. E. (2010). The arsenic exposure hypothesis for Alzheimer disease. Alzheimer Dis. Assoc. Disord. 24, 311–316. doi: 10.1097/WAD.0b013e3181d71bc7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
González-Polo, R. A., Rodríguez-Martín, A., Morán, J. M., Niso, M., Soler, G., and Fuentes, J. M. (2004). Paraquat-induced apoptotic cell death in cerebellar granule cells. Brain Res. 1011, 170–176. doi: 10.1016/s0006-8993(04)00466-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goodall, E. F., Heath, P. R., Bandmann, O., Kirby, J., and Shaw, P. J. (2013). Neuronal dark matter: the emerging role of microRNAs in neurodegeneration. Front. Cell. Neurosci. 7:178. doi: 10.3389/fncel.2013.00178
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gorell, J. M., Peterson, E. L., Rybicki, B. A., and Johnson, C. C. (2004). Multiple risk factors for Parkinson’s disease. J. Neurol. Sci. 217, 169–174. doi: 10.1016/j.jns.2003.09.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Götz, J. (2001). Tau and transgenic animal models. Brain Res. Brain Res. Rev. 35, 266–286. doi: 10.1016/s0165-0173(01)00055-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Greenblatt, J. M., Huffman, L. C., and Reiss, A. L. (1994). Folic acid in neurodevelopment and child psychiatry. Prog. Neuropsychopharmacol. Biol. Psychiatry 18, 647–660. doi: 10.1016/0278-5846(94)90074-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gu, H., Wei, X., Monnot, A. D., Fontanilla, C. V., Behl, M., Farlow, M. R., et al. (2011). Lead exposure increases levels of beta-amyloid in the brain and CSF and inhibits LRP1 expression in APP transgenic mice. Neurosci. Lett. 490, 16–20. doi: 10.1016/j.neulet.2010.12.017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guilarte, T. R. (2013). Manganese neurotoxicity: new perspectives from behavioral, neuroimaging and neuropathological studies in humans and non-human primates. Front. Aging Neurosci. 5:23. doi: 10.3389/fnagi.2013.00023
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Halliwell, B., Gutteridge, J. M., and Cross, C. E. (1992). Free radicals, antioxidants and human disease: where are we now? J. Lab. Clin. Med. 119, 598–620.
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Harischandra, D. S., Jin, H., Anantharam, V., Kanthasamy, A., and Kanthasamy, A. G. (2015). α-Synuclein protects against manganese neurotoxic insult during the early stages of exposure in a dopaminergic cell model of Parkinson’s disease. Toxicol. Sci. 143, 454–468. doi: 10.1093/toxsci/kfu247
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hatcher, J. M., Pennell, K. D., and Miller, G. W. (2008). Parkinson’s disease and pesticides: a toxicological perspective. Trends Pharmacol. Sci. 29, 322–329. doi: 10.1016/j.tips.2008.03.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hatcher, J. M., Richardson, J. R., Guillot, T. S., McCormack, A. L., Di Monte, D. A., Jones, D. P., et al. (2007). Dieldrin exposure induces oxidative damage in the mouse nigrostriatal dopamine system. Exp. Neurol. 204, 619–630. doi: 10.1016/j.expneurol.2006.12.020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hayden, K. M., Norton, M. C., Darcey, D., Ostbye, T., Zandi, P. P., Breitner, J. C., et al. (2010). Occupational exposure to pesticides increases the risk of incident AD: the Cache County study. Neurology 74, 1524–1530. doi: 10.1212/WNL.0b013e3181dd4423
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hernandez, M. E., Rembao, J. D., Hernandez-Baltazar, D., Castillo-Rodriguez, R. A., Tellez-Lopez, V. M., Flores-Martinez, Y. M., et al. (2014). Safety of the intravenous administration of neurotensin-polyplex nanoparticles in BALB/c mice. Nanomedicine 10, 745–754. doi: 10.1016/j.nano.2013.11.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hertzman, C., Wiens, M., Bowering, D., Snow, B., and Calne, D. (1990). Parkinson’s disease: a case-control study of occupational and environmental risk factors. Am. J. Ind. Med. 17, 349–355. doi: 10.1002/ajim.4700170307
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hirsch, E. C., Brandel, J. P., Galle, P., Javoy-Agid, F., and Agid, Y. (1991). Iron and aluminum increase in the substantia nigra of patients with Parkinson’s disease: an X-ray microanalysis. J. Neurochem. 56, 446–451. doi: 10.1111/j.1471-4159.1991.tb08170.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hock, C., Drasch, G., Golombowski, S., Müller-Spahn, F., Willershausen-Zonnchen, B., Schwarz, P., et al. (1998). Increased blood mercury levels in patients with Alzheimer’s disease. J. Neural Transm. 105, 59–68. doi: 10.1007/s007020050038
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Holliday, R. (2006). Epigenetics: a historical overview. Epigenetics 1, 76–80. doi: 10.4161/epi.1.2.2762
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huang, H., Bihaqi, S. W., Cui, L., and Zawia, N. H. (2011). In vitro Pb exposure disturbs the balance between Aβ production and elimination: the role of AbetaPP and neprilysin. Neurotoxicology 32, 300–306. doi: 10.1016/j.neuro.2011.02.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huang, C. L., Hsiao, I. L., Lin, H. C., Wang, C. F., Huang, Y. J., and Chuang, C. Y. (2015). Silver nanoparticles affect on gene expression of inflammatory and neurodegenerative responses in mouse brain neural cells. Environ. Res. 136, 253–263. doi: 10.1016/j.envres.2014.11.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huang, C. C., Lu, C. S., Chu, N. S., Hochberg, F., Lilienfeld, D., Olanow, W., et al. (1993). Progression after chronic manganese exposure. Neurology 43, 1479–1483. doi: 10.1212/WNL.43.8.1479
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iwata, A., Nagata, K., Hatsuta, H., Takuma, H., Bundo, M., Iwamoto, K., et al. (2014). Altered CpG methylation in sporadic Alzheimer’s disease is associated with APP and MAPT dysregulation. Hum. Mol. Genet. 23, 648–656. doi: 10.1093/hmg/ddt451
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jang, H., and Serra, C. (2014). Nutrition, epigenetics and diseases. Clin. Nutr. Res. 3, 1–8. doi: 10.7762/cnr.2014.3.1.1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jankovic, J. (2005). Searching for a relationship between manganese and welding and Parkinson’s disease. Neurology 64, 2021–2028. doi: 10.1212/01.wnl.0000166916.40902.63
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jankovic, J. (2008). Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376. doi: 10.1136/jnnp.2007.131045
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jiang, L. F., Yao, T. M., Zhu, Z. L., Wang, C., and Ji, L. N. (2007). Impacts of Cd(II) on the conformation and self-aggregation of Alzheimer’s tau fragment corresponding to the third repeat of microtubule-binding domain. Biochim. Biophys. Acta 1774, 1414–1421. doi: 10.1016/j.bbapap.2007.08.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Johansson, C., Castoldi, A. F., Onishchenko, N., Manzo, L., Vahter, M., and Ceccatelli, S. (2007). Neurobehavioural and molecular changes induced by methylmercury exposure during development. Neurotox. Res. 11, 241–260. doi: 10.1007/bf03033570
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jowaed, A., Schmitt, I., Kaut, O., and Wüllner, U. (2010). Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J. Neurosci. 30, 6355–6359. doi: 10.1523/jneurosci.6119-09.2010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kala, S. V., and Jadhav, A. L. (1995). Low level lead exposure decreases in vivo release of dopamine in the rat nucleus accumbens: a microdialysis study. J. Neurochem. 65, 1631–1635. doi: 10.1046/j.1471-4159.1995.65041631.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kamel, F., Tanner, C., Umbach, D., Hoppin, J., Alavanja, M., Blair, A., et al. (2007). Pesticide exposure and self-reported Parkinson’s disease in the agricultural health study. Am. J. Epidemiol. 165, 364–374. doi: 10.1093/aje/kwk024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kang, M. J., Gil, S. J., and Koh, H. C. (2009). Paraquat induces alternation of the dopamine catabolic pathways and glutathione levels in the substantia nigra of mice. Toxicol. Lett. 188, 148–152. doi: 10.1016/j.toxlet.2009.03.026
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kanthasamy, A. G., Kitazawa, M., Kanthasamy, A., and Anantharam, V. (2005). Dieldrin-induced neurotoxicity: relevance to Parkinson’s disease pathogenesis. Neurotoxicology 26, 701–719. doi: 10.1016/j.neuro.2004.07.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kaur, D., Peng, J., Chinta, S. J., Rajagopalan, S., Di Monte, D. A., Cherny, R. A., et al. (2007). Increased murine neonatal iron intake results in Parkinson-like neurodegeneration with age. Neurobiol. Aging 28, 907–913. doi: 10.1016/j.neurobiolaging.2006.04.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kawahara, M., Kato, M., and Kuroda, Y. (2001). Effects of aluminum on the neurotoxicity of primary cultured neurons and on the aggregation of beta-amyloid protein. Brain Res. Bull. 55, 211–217. doi: 10.1016/s0361-9230(01)00475-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, D. K., Park, J. D., and Choi, B. S. (2014). Mercury-induced amyloid-beta (Aβ) accumulation in the brain is mediated by disruption of Aβ transport. J. Toxicol. Sci. 39, 625–635. doi: 10.2131/jts.39.625
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kitazawa, M., Anantharam, V., and Kanthasamy, A. G. (2001). Dieldrin-induced oxidative stress and neurochemical changes contribute to apoptopic cell death in dopaminergic cells. Free Radic. Biol. Med. 31, 1473–1485. doi: 10.1016/s0891-5849(01)00726-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kitazawa, M., Anantharam, V., and Kanthasamy, A. G. (2003). Dieldrin induces apoptosis by promoting caspase-3-dependent proteolytic cleavage of protein kinase Cdelta in dopaminergic cells: relevance to oxidative stress and dopaminergic degeneration. Neuroscience 119, 945–964. doi: 10.1016/s0306-4522(03)00226-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kochen, W., Kohlmüller, D., De Biasi, P., and Ramsay, R. (2003). The endogeneous formation of highly chlorinated tetrahydro-beta-carbolines as a possible causative mechanism in idiopathic Parkinson’s disease. Adv. Exp. Med. Biol. 527, 253–263. doi: 10.1007/978-1-4615-0135-0_29
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kojro, E., and Fahrenholz, F. (2005). The non-amyloidogenic pathway: structure and function of alpha-secretases. Subcell Biochem. 38, 105–127. doi: 10.1007/0-387-23226-5_5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kuhn, W., Winkel, R., Woitalla, D., Meves, S., Przuntek, H., and Müller, T. (1998). High prevalence of parkinsonism after occupational exposure to lead-sulfate batteries. Neurology 50, 1885–1886. doi: 10.1212/wnl.50.6.1885
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kuter, K., Nowak, P., Golembiowska, K., and Ossowska, K. (2010). Increased reactive oxygen species production in the brain after repeated low-dose pesticide paraquat exposure in rats. A comparison with peripheral tissues. Neurochem. Res. 35, 1121–1130. doi: 10.1007/s11064-010-0163-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kwok, J. B. (2010). Role of epigenetics in Alzheimer’s and Parkinson’s disease. Epigenomics 2, 671–682. doi: 10.2217/epi.10.43
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lai, B. C., Marion, S. A., Teschke, K., and Tsui, J. K. (2002). Occupational and environmental risk factors for Parkinson’s disease. Parkinsonism Relat. Disord. 8, 297–309. doi: 10.1016/s1353-8020(01)00054-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Langston, J. W., and Ballard, P. A. Jr. (1983). Parkinson’s disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N. Engl. J. Med. 309, 310. doi: 10.1056/nejm198308043090511
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Langston, J. W., Ballard, P., Tetrud, J. W., and Irwin, I. (1983). Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980. doi: 10.1126/science.6823561
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Langston, J. W., Forno, L. S., Rebert, C. S., and Irwin, I. (1984). Selective nigral toxicity after systemic administration of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyrine (MPTP) in the squirrel monkey. Brain Res. 292, 390–394. doi: 10.1016/0006-8993(84)90777-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, J. W. (2000). Manganese intoxication. Arch. Neurol. 57, 597–599. doi: 10.1001/archneur.57.4.597
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lewczuk, P., Mroczko, B., Fagan, A., and Kornhuber, J. (2014). Biomarkers of Alzheimer’s disease and mild cognitive impairment: a current perspective. Adv. Med. Sci. 60, 76–82. doi: 10.1016/j.advms.2014.11.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Leyva-Gómez, G., Cortés, H., Magaña, J. J., Leyva-García, N., Quintanar-Guerrero, D., and Florán, B. (2015). Nanoparticle technology for treatment of Parkinson’s disease: the role of surface phenomena in reaching the brain. Drug Discov. Today. doi: 10.1016/j.drudis.2015.02.009. [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, W. J., Jiang, H., Song, N., and Xie, J. X. (2010b). Dose- and time-dependent alpha-synuclein aggregation induced by ferric iron in SK-N-SH cells. Neurosci. Bull. 26, 205–210. doi: 10.1007/s12264-010-1117-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, G., Kim, C., Kim, J., Yoon, H., Zhou, H., and Kim, J. (2015). Common pesticide, dichlorodiphenyltrichloroethane (DDT), increases amyloid-beta levels by impairing the function of ABCA1 and IDE: implication for Alzheimer’s disease. J. Alzheimers Dis. doi: 10.3233/JAD-150024. [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, X., Liu, B., Li, X. L., Li, Y. X., Sun, M. Z., Chen, D. Y., et al. (2014b). SiO2 nanoparticles change colour preference and cause Parkinson’s-like behaviour in zebrafish. Sci. Rep. 4:3810. doi: 10.1038/srep03810
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, N., Liu, F., Song, L., Zhang, P., Qiao, M., Zhao, Q., et al. (2014a). The effects of early life Pb exposure on the expression of IL1-beta, TNF-alpha and Abeta in cerebral cortex of mouse pups. J. Trace Elem. Med. Biol. 28, 100–104. doi: 10.1016/j.jtemb.2013.07.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, X., Lv, Y., Yu, S., Zhao, H., and Yao, L. (2012). The effect of cadmium on Abeta levels in APP/PS1 transgenic mice. Exp. Ther. Med. 4, 125–130. doi: 10.3892/etm.2012.562
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, N., Yu, Z. L., Wang, L., Zheng, Y. T., Jia, J. X., Wang, Q., et al. (2010a). Increased tau phosphorylation and beta amyloid in the hipocampus of mouse pups by early life lead exposure. Acta Biol. Hung. 61, 123–134. doi: 10.1556/abiol.61.2010.2.1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liou, H. H., Tsai, M. C., Chen, C. J., Jeng, J. S., Chang, Y. C., Chen, S. Y., et al. (1997). Environmental risk factors and Parkinson’s disease: a case-control study in Taiwan. Neurology 48, 1583–1588. doi: 10.1212/wnl.48.6.1583
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, M., Choi, D. Y., Hunter, R. L., Pandya, J. D., Cass, W. A., Sullivan, P. G., et al. (2010). Trichloroethylene induces dopaminergic neurodegeneration in Fisher 344 rats. J. Neurochem. 112, 773–783. doi: 10.1111/j.1471-4159.2009.06497.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, P., Huang, Z., and Gu, N. (2013). Exposure to silver nanoparticles does not affect cognitive outcome or hippocampal neurogenesis in adult mice. Ecotoxicol. Environ. Saf. 87, 124–130. doi: 10.1016/j.ecoenv.2012.10.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, F., Xue, Z., Li, N., Huang, H., Ying, Y., Li, J., et al. (2014). Effects of lead exposure on the expression of amyloid beta and phosphorylated tau proteins in the C57BL/6 mouse hippocampus at different life stages. J. Trace Elem. Med. Biol. 28, 227–232. doi: 10.1016/j.jtemb.2014.01.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lock, E. A., Zhang, J., and Checkoway, H. (2013). Solvents and Parkinson disease: a systematic review of toxicological and epidemiological evidence. Toxicol. Appl. Pharmacol. 266, 345–355. doi: 10.1016/j.taap.2012.11.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Logroscino, G. (2005). The role of early life environmental risk factors in Parkinson disease: what is the evidence? Environ. Health Perspect. 113, 1234–1238. doi: 10.1289/ehp.7573
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Logroscino, G., Gao, X., Chen, H., Wing, A., and Ascherio, A. (2008). Dietary iron intake and risk of Parkinson’s disease. Am. J. Epidemiol. 168, 1381–1388. doi: 10.1093/aje/kwn273
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lu, C. S., Huang, C. C., Chu, N. S., and Calne, D. B. (1994). Levodopa failure in chronic manganism. Neurology 44, 1600–1602. doi: 10.1212/wnl.44.9.1600
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lu, H., Liu, X., Deng, Y., and Qing, H. (2013). DNA methylation, a hand behind neurodegenerative diseases. Front. Aging Neurosci. 5:85. doi: 10.3389/fnagi.2013.00085
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lui, E., Fisman, M., Wong, C., and Diaz, F. (1990). Metals and the liver in Alzheimer’s disease. An investigation of hepatic zinc, copper, cadmium and metallothionein. J. Am. Geriatr. Soc. 38, 633–639.
Lukiw, W. J. (2012). NF-kappaB-regulated, proinflammatory miRNAs in Alzheimer’s disease. Alzheimers Res. Ther. 4:47. doi: 10.1186/alzrt150
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Luo, Y., Niu, F., Sun, Z., Cao, W., Zhang, X., Guan, D., et al. (2009). Altered expression of Abeta metabolism-associated molecules from D-galactose/AlCl(3) induced mouse brain. Mech. Ageing Dev. 130, 248–252. doi: 10.1016/j.mad.2008.12.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maciotta, S., Meregalli, M., and Torrente, Y. (2013). The involvement of microRNAs in neurodegenerative diseases. Front. Cell. Neurosci. 7:265. doi: 10.3389/fncel.2013.00265
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Manning-Bog, A. B., McCormack, A. L., Li, J., Uversky, V. N., Fink, A. L., and Di Monte, D. A. (2002). The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J. Biol. Chem. 277, 1641–1644. doi: 10.1074/jbc.c100560200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mao, H., and Liu, B. (2008). Synergistic microglial reactive oxygen species generation induced by pesticides lindane and dieldrin. Neuroreport 19, 1317–1320. doi: 10.1097/wnr.0b013e32830b3677
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marques, S. C., Oliveira, C. R., Pereira, C. M., and Outeiro, T. F. (2011). Epigenetics in neurodegeneration: a new layer of complexity. Prog. Neuropsychopharmacol. Biol. Psychiatry 35, 348–355. doi: 10.1016/j.pnpbp.2010.08.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Martínez, L., Jiménez, V., García-Sepúlveda, C., Ceballos, F., Delgado, J. M., Niño-Moreno, P., et al. (2011). Impact of early developmental arsenic exposure on promotor CpG-island methylation of genes involved in neuronal plasticity. Neurochem. Int. 58, 574–581. doi: 10.1016/j.neuint.2011.01.020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mason, L. H., Harp, J. P., and Han, D. Y. (2014). Pb neurotoxicity: neuropsychological effects of lead toxicity. Biomed. Res. Int. 2014:840547. doi: 10.1155/2014/840547
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Matsumoto, L., Takuma, H., Tamaoka, A., Kurisaki, H., Date, H., Tsuji, S., et al. (2010). CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS One 5:e15522. doi: 10.1371/journal.pone.0015522
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McCormack, A. L., Atienza, J. G., Johnston, L. C., Andersen, J. K., Vu, S., and Di Monte, D. A. (2005). Role of oxidative stress in paraquat-induced dopaminergic cell degeneration. J. Neurochem. 93, 1030–1037. doi: 10.1111/j.1471-4159.2005.03088.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McCormack, A. L., Thiruchelvam, M., Manning-Bog, A. B., Thiffault, C., Langston, J. W., Cory-Slechta, D. A., et al. (2002). Environmental risk factors and Parkinson’s disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol. Dis. 10, 119–127. doi: 10.1006/nbdi.2002.0507
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McDonnell, L., Maginnis, C., Lewis, S., Pickering, N., Antoniak, M., Hubbard, R., et al. (2003). Occupational exposure to solvents and metals and Parkinson’s disease. Neurology 61, 716–717. doi: 10.1212/wnl.61.5.716
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McNaught, K. S., Belizaire, R., Jenner, P., Olanow, C. W., and Isacson, O. (2002). Selective loss of 20S proteasome alpha-subunits in the substantia nigra pars compacta in Parkinson’s disease. Neurosci. Lett. 326, 155–158. doi: 10.1016/s0304-3940(02)00296-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McNaught, K. S., and Jenner, P. (2001). Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 297, 191–194. doi: 10.1016/s0304-3940(00)01701-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Miñones-Moyano, E., Porta, S., Escaramis, G., Rabionet, R., Iraola, S., Kagerbauer, B., et al. (2011). MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum. Mol. Genet. 20, 3067–3078. doi: 10.1093/hmg/ddr210
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Miyake, Y., Tanaka, K., Fukushima, W., Sasaki, S., Kiyohara, C., Tsuboi, Y., et al. (2011). Dietary intake of metals and risk of Parkinson’s disease: a case-control study in Japan. J. Neurol. Sci. 306, 98–102. doi: 10.1016/j.jns.2011.03.035
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mogi, M., Harada, M., Narabayashi, H., Inagaki, H., Minami, M., and Nagatsu, T. (1996). Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci. Lett. 211, 13–16. doi: 10.1016/0304-3940(96)12706-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Monnet-Tschudi, F., Zurich, M. G., Boschat, C., Corbaz, A., and Honegger, P. (2006). Involvement of environmental mercury and lead in the etiology of neurodegenerative diseases. Rev. Environ. Health 21, 105–117. doi: 10.1515/reveh.2006.21.2.105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mortimer, J. A., Borenstein, A. R., and Nelson, L. M. (2012). Associations of welding and manganese exposure with Parkinson disease: review and meta-analysis. Neurology 79, 1174–1180. doi: 10.1212/WNL.0b013e3182698ced
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mutter, J., Naumann, J., Sadaghiani, C., Schneider, R., and Walach, H. (2004). Alzheimer disease: mercury as pathogenetic factor and apolipoprotein E as a moderator. Neuro. Endocrinol. Lett. 25, 331–339.
Nalivaeva, N. N., Beckett, C., Belyaev, N. D., and Turner, A. J. (2012). Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer’s disease? J. Neurochem. 120(Suppl. 1), 167–185. doi: 10.1111/j.1471-4159.2011.07510.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nasuti, C., Carloni, M., Fedeli, D., Gabbianelli, R., Di Stefano, A., Serafina, C. L., et al. (2013). Effects of early life permethrin exposure on spatial working memory and on monoamine levels in different brain areas of pre-senescent rats. Toxicology 303, 162–168. doi: 10.1016/j.tox.2012.09.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Navarro-Yepes, J., Zavala-Flores, L., Anandhan, A., Wang, F., Skotak, M., Chandra, N., et al. (2014). Antioxidant gene therapy against neuronal cell death. Pharmacol. Ther. 142, 206–230. doi: 10.1016/j.pharmthera.2013.12.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
O’Bryant, S. E., Edwards, M., Menon, C. V., Gong, G., and Barber, R. (2011). Long-term low-level arsenic exposure is associated with poorer neuropsychological functioning: a Project FRONTIER study. Int. J. Environ. Res. Public Health 8, 861–874. doi: 10.3390/ijerph8030861
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Olivieri, G., Brack, C., Müller-Spahn, F., Stähelin, H. B., Herrmann, M., Renard, P., et al. (2000). Mercury induces cell cytotoxicity and oxidative stress and increases beta-amyloid secretion and tau phosphorylation in SHSY5Y neuroblastoma cells. J. Neurochem. 74, 231–236. doi: 10.1046/j.1471-4159.2000.0740231.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Papageorgiou, S. G., Karantoni, E., Pandis, D., Kouzoupis, A. V., Kalfakis, N., and Limouris, G. S. (2009). Severe dopaminergic pathways damage in a case of chronic toluene abuse. Clin. Neurol. Neurosurg. 111, 864–867. doi: 10.1016/j.clineuro.2009.07.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Park, R. M. (2013). Neurobehavioral deficits and parkinsonism in occupations with manganese exposure: a review of methodological issues in the epidemiological literature. Saf. Health Work 4, 123–135. doi: 10.1016/j.shaw.2013.07.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Park, J. H., Lee, D. W., Park, K. S., and Joung, H. (2014). Serum trace metal levels in Alzheimer’s disease and normal control groups. Am. J. Alzheimers Dis. Other Demen. 29, 76–83. doi: 10.1177/1533317513506778
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Peris-Sampedro, F., Salazar, J. G., Cabré, M., Reverte, I., Domingo, J. L., Sánchez-Santed, F., et al. (2014). Impaired retention in AbetaPP Swedish mice six months after oral exposure to chlorpyrifos. Food Chem. Toxicol. 72, 289–294. doi: 10.1016/j.fct.2014.07.036
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Perl, D. P., and Olanow, C. W. (2007). The neuropathology of manganese-induced Parkinsonism. J. Neuropathol. Exp. Neurol. 66, 675–682. doi: 10.1097/nen.0b013e31812503cf
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pezzoli, G., Barbieri, S., Ferrante, C., Zecchinelli, A., and Foà, V. (1989). Parkinsonism due to n-hexane exposure. Lancet 2:874. doi: 10.1016/s0140-6736(89)93050-x
Pezzoli, G., Ricciardi, S., Masotto, C., Mariani, C. B., and Carenzi, A. (1990). n-hexane induces parkinsonism in rodents. Brain Res. 531, 355–357. doi: 10.1016/0006-8993(90)90801-h
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Piaceri, I., Nacmias, B., and Sorbi, S. (2013). Genetics of familial and sporadic Alzheimer’s disease. Front. Biosci. (Elite Ed.) 5, 167–177. doi: 10.2741/e605
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pieper, H. C., Evert, B. O., Kaut, O., Riederer, P. F., Waha, A., and Wullner, U. (2008). Different methylation of the TNF-alpha promoter in cortex and substantia nigra: implications for selective neuronal vulnerability. Neurobiol. Dis. 32, 521–527. doi: 10.1016/j.nbd.2008.09.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pisanic, T. R. 2nd, Blackwell, J. D., Shubayev, V. I., Fiñones, R. R., and Jin, S. (2007). Nanotoxicity of iron oxide nanoparticle internalization in growing neurons. Biomaterials 28, 2572–2581. doi: 10.1016/j.biomaterials.2007.01.043
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pogue, A. I., Percy, M. E., Cui, J. G., Li, Y. Y., Bhattacharjee, S., Hill, J. M., et al. (2011). Up-regulation of NF-kB-sensitive miRNA-125b and miRNA-146a in metal sulfate-stressed human astroglial (HAG) primary cell cultures. J. Inorg. Biochem. 105, 1434–1437. doi: 10.1016/j.jinorgbio.2011.05.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Poirier, L. A., and Vlasova, T. I. (2002). The prospective role of abnormal methyl metabolism in cadmium toxicity. Environ. Health Perspect. 110(Suppl. 5), 793–795. doi: 10.1289/ehp.02110s5793
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Praticò, D., Uryu, K., Sung, S., Tang, S., Trojanowski, J. Q., and Lee, V. M. (2002). Aluminum modulates brain amyloidosis through oxidative stress in APP transgenic mice. FASEB J. 16, 1138–1140. doi: 10.1096/fj.02-0012fje
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Priyadarshi, A., Khuder, S. A., Schaub, E. A., and Priyadarshi, S. S. (2001). Environmental risk factors and Parkinson’s disease: a metaanalysis. Environ. Res. 86, 122–127. doi: 10.1006/enrs.2001.4264
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Priyadarshi, A., Khuder, S. A., Schaub, E. A., and Shrivastava, S. (2000). A meta-analysis of Parkinson’s disease and exposure to pesticides. Neurotoxicology 21, 435–440.
Purisai, M. G., McCormack, A. L., Cumine, S., Li, J., Isla, M. Z., and Di Monte, D. A. (2007). Microglial activation as a priming event leading to paraquat-induced dopaminergic cell degeneration. Neurobiol. Dis. 25, 392–400. doi: 10.1016/j.nbd.2006.10.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Purkerson-Parker, S., McDaniel, K. L., and Moser, V. C. (2001). Dopamine transporter binding in the rat striatum is increased by gestational, perinatal and adolescent exposure to heptachlor. Toxicol. Sci. 64, 216–223. doi: 10.1093/toxsci/64.2.216
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Radad, K., Rausch, W. D., and Gille, G. (2006). Rotenone induces cell death in primary dopaminergic culture by increasing ROS production and inhibiting mitochondrial respiration. Neurochem. Int. 49, 379–386. doi: 10.1016/j.neuint.2006.02.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ramos-Chávez, L. A., Rendón-López, C. R., Zepeda, A., Silva-Adaya, D., Del Razo, L. M., and Gonsebatt, M. E. (2015). Neurological effects of inorganic arsenic exposure: altered cysteine/glutamate transport, NMDA expression and spatial memory impairment. Front. Cell. Neurosci. 9:21. doi: 10.3389/fncel.2015.00021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rappold, P. M., Cui, M., Chesser, A. S., Tibbett, J., Grima, J. C., Duan, L., et al. (2011). Paraquat neurotoxicity is mediated by the dopamine transporter and organic cation transporter-3. Proc. Natl. Acad. Sci. U S A 108, 20766–20771. doi: 10.1073/pnas.1115141108
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ray, P. D., Yosim, A., and Fry, R. C. (2014). Incorporating epigenetic data into the risk assessment process for the toxic metals arsenic, cadmium, chromium, lead and mercury: strategies and challenges. Front. Genet. 5:201. doi: 10.3389/fgene.2014.00201
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Richardson, J. R., Roy, A., Shalat, S. L., von Stein, R. T., Hossain, M. M., Duan, B., et al. (2014). Elevated serum pesticide levels and risk for Alzheimer disease. JAMA Neurol. 71, 284–290. doi: 10.1001/jamaneurol.2013.6030
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rodier, J. (1955). Manganese poisoning in Moroccan miners. Br. J. Ind. Med. 12, 21–35. doi: 10.1136/oem.12.1.21
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rogers, J. T., Leiter, L. M., McPhee, J., Cahill, C. M., Zhan, S. S., Potter, H., et al. (1999). Translation of the alzheimer amyloid precursor protein mRNA is up-regulated by interleukin-1 through 5′-untranslated region sequences. J. Biol. Chem. 274, 6421–6431. doi: 10.1074/jbc.274.10.6421
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rogers, J., and Lue, L. F. (2001). Microglial chemotaxis, activation and phagocytosis of amyloid beta-peptide as linked phenomena in Alzheimer’s disease. Neurochem. Int. 39, 333–340. doi: 10.1016/s0197-0186(01)00040-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rybicki, B. A., Johnson, C. C., Uman, J., and Gorell, J. M. (1993). Parkinson’s disease mortality and the industrial use of heavy metals in Michigan. Mov. Disord. 8, 87–92. doi: 10.1002/mds.870080116
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sakamoto, T., Saito, H., Ishii, K., Takahashi, H., Tanabe, S., and Ogasawara, Y. (2006). Aluminum inhibits proteolytic degradation of amyloid beta peptide by cathepsin D: a potential link between aluminum accumulation and neuritic plaque deposition. FEBS Lett. 580, 6543–6549. doi: 10.1016/j.febslet.2006.10.075
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Salazar, J. G., Ribes, D., Cabré, M., Domingo, J. L., Sanchez-Santed, F., and Colomina, M. T. (2011). Amyloid beta peptide levels increase in brain of AbetaPP Swedish mice after exposure to chlorpyrifos. Curr. Alzheimer Res. 8, 732–740. doi: 10.2174/156720511797633197
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sanchez-Ramos, J., Facca, A., Basit, A., and Song, S. (1998). Toxicity of dieldrin for dopaminergic neurons in mesencephalic cultures. Exp. Neurol. 150, 263–271. doi: 10.1006/exnr.1997.6770
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sandhir, R., Julka, D., and Gill, K. D. (1994). Lipoperoxidative damage on lead exposure in rat brain and its implications on membrane bound enzymes. Pharmacol. Toxicol. 74, 66–71. doi: 10.1111/j.1600-0773.1994.tb01077.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schmuck, G., Röhrdanz, E., Tran-Thi, Q. H., Kahl, R., and Schlüter, G. (2002). Oxidative stress in rat cortical neurons and astrocytes induced by paraquat in vitro. Neurotox. Res. 4, 1–13. doi: 10.1080/10298420290007574
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schrag, A., Ben-Shlomo, Y., and Quinn, N. (2002). How valid is the clinical diagnosis of Parkinson’s disease in the community? J. Neurol. Neurosurg. Psychiatry 73, 529–534. doi: 10.1136/jnnp.73.5.529
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sharma, H. S., Ali, S. F., Hussain, S. M., Schlager, J. J., and Sharma, A. (2009). Influence of engineered nanoparticles from metals on the blood-brain barrier permeability, cerebral blood flow, brain edema and neurotoxicity. An experimental study in the rat and mice using biochemical and morphological approaches. J. Nanosci. Nanotechnol. 9, 5055–5072. doi: 10.1166/jnn.2009.gr09
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sharma, H. S., and Sharma, A. (2012). Neurotoxicity of engineered nanoparticles from metals. CNS Neurol. Disord. Drug Targets 11, 65–80. doi: 10.2174/187152712799960817
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shen, X. L., Yu, J. H., Zhang, D. F., Xie, J. X., and Jiang, H. (2014). Positive relationship between mortality from Alzheimer’s disease and soil metal concentration in mainland China. J. Alzheimers Dis. 42, 893–900. doi: 10.3233/JAD-140153
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sherer, T. B., Betarbet, R., Stout, A. K., Lund, S., Baptista, M., Panov, A. V., et al. (2002). An in vitro model of Parkinson’s disease: linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J. Neurosci. 22, 7006–7015.
Sherer, T. B., Kim, J. H., Betarbet, R., and Greenamyre, J. T. (2003). Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp. Neurol. 179, 9–16. doi: 10.1006/exnr.2002.8072
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sherer, T. B., Richardson, J. R., Testa, C. M., Seo, B. B., Panov, A. V., Yagi, T., et al. (2007). Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson’s disease. J. Neurochem. 100, 1469–1479. doi: 10.1111/j.1471-4159.2006.04333.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shin, R. W., Kruck, T. P., Murayama, H., and Kitamoto, T. (2003). A novel trivalent cation chelator Feralex dissociates binding of aluminum and iron associated with hyperphosphorylated tau of Alzheimer’s disease. Brain Res. 961, 139–146. doi: 10.1016/s0006-8993(02)03893-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shin, R. W., Lee, V. M., and Trojanowski, J. Q. (1994). Aluminum modifies the properties of Alzheimer’s disease PHF tau proteins in vivo and in vitro. J. Neurosci. 14, 7221–7233.
Sian-Hulsmann, J., Mandel, S., Youdim, M. B., and Riederer, P. (2011). The relevance of iron in the pathogenesis of Parkinson’s disease. J. Neurochem. 118, 939–957. doi: 10.1111/j.1471-4159.2010.07132.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Singer, S. M., Chambers, C. B., Newfry, G. A., Norlund, M. A., and Muma, N. A. (1997). Tau in aluminum-induced neurofibrillary tangles. Neurotoxicology 18, 63–76.
Smargiassi, A., Mutti, A., De Rosa, A., De Palma, G., Negrotti, A., and Calzetti, S. (1998). A case-control study of occupational and environmental risk factors for Parkinson’s disease in the Emilia-Romagna region of Italy. Neurotoxicology 19, 709–712.
Smith, M. A., Harris, P. L., Sayre, L. M., and Perry, G. (1997a). Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. U S A 94, 9866–9868. doi: 10.1073/pnas.94.18.9866
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Smith, M. A., Richey Harris, P. L., Sayre, L. M., Beckman, J. S., and Perry, G. (1997b). Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J. Neurosci. 17, 2653–2657.
Song, J. W., and Choi, B. S. (2013). Mercury induced the accumulation of amyloid beta (Abeta) in PC12 cells: the role of production and degradation of abeta. Toxicol. Res. 29, 235–240. doi: 10.5487/tr.2013.29.4.235
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998). alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. U S A 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stohs, S. J., and Bagchi, D. (1995). Oxidative mechanisms in the toxicity of metal ions. Free Radic. Biol. Med. 18, 321–336. doi: 10.1016/0891-5849(94)00159-h
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sun, F., Anantharam, V., Latchoumycandane, C., Kanthasamy, A., and Kanthasamy, A. G. (2005). Dieldrin induces ubiquitin-proteasome dysfunction in alpha-synuclein overexpressing dopaminergic neuronal cells and enhances susceptibility to apoptotic cell death. J. Pharmacol. Exp. Ther. 315, 69–79. doi: 10.1124/jpet.105.084632
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sun, X., Bromley-Brits, K., and Song, W. (2012). Regulation of beta-site APP-cleaving enzyme 1 gene expression and its role in Alzheimer’s disease. J. Neurochem. 120(Suppl. 1), 62–70. doi: 10.1111/j.1471-4159.2011.07515.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Suri, S., Heise, V., Trachtenberg, A. J., and Mackay, C. E. (2013). The forgotten APOE allele: a review of the evidence and suggested mechanisms for the protective effect of APOE varepsilon2. Neurosci. Biobehav. Rev. 37, 2878–2886. doi: 10.1016/j.neubiorev.2013.10.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tanner, C. M., and Goldman, S. M. (1996). Epidemiology of Parkinson’s disease. Neurol. Clin. 14, 317–335. doi: 10.1016/S0733-8619(05)70259-0
Tanner, C. M., Kamel, F., Ross, G. W., Hoppin, J. A., Goldman, S. M., Korell, M., et al. (2011). Rotenone, paraquat and Parkinson’s disease. Environ. Health Perspect. 119, 866–872. doi: 10.1289/ehp.1002839
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tavakoli-Nezhad, M., and Pitts, D. K. (2005). Postnatal inorganic lead exposure reduces midbrain dopaminergic impulse flow and decreases dopamine D1 receptor sensitivity in nucleus accumbens neurons. J. Pharmacol. Exp. Ther. 312, 1280–1288. doi: 10.1124/jpet.104.076166
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thinakaran, G., and Koo, E. H. (2008). Amyloid precursor protein trafficking, processing and function. J. Biol. Chem. 283, 29615–29619. doi: 10.1074/jbc.r800019200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thiruchelvam, M., Richfield, E. K., Goodman, B. M., Baggs, R. B., and Cory-Slechta, D. A. (2002). Developmental exposure to the pesticides paraquat and maneb and the Parkinson’s disease phenotype. Neurotoxicology 23, 621–633. doi: 10.1016/s0161-813x(02)00092-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tokimasa, M., Minoura, K., Hiraoka, S., Tomoo, K., Sumida, M., Taniguchi, T., et al. (2005). Importance of local structures of second and third repeat fragments of microtubule-binding domain for tau filament formation. FEBS Lett. 579, 3481–3486. doi: 10.1016/j.febslet.2005.05.020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tuppo, E. E., and Arias, H. R. (2005). The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 37, 289–305. doi: 10.1016/j.biocel.2004.07.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tyler, C. R., and Allan, A. M. (2014). The effects of arsenic exposure on neurological and cognitive dysfunction in human and rodent studies: a review. Curr. Environ. Health Rep. 1, 132–147. doi: 10.1007/s40572-014-0012-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Uversky, V. N., Li, J., and Fink, A. L. (2001). Pesticides directly accelerate the rate of alpha-synuclein fibril formation: a possible factor in Parkinson’s disease. FEBS Lett. 500, 105–108. doi: 10.1016/s0014-5793(01)02597-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vahidnia, A., Romijn, F., Van der Voet, G. B., and de Wolff, F. A. (2008). Arsenic-induced neurotoxicity in relation to toxicokinetics: effects on sciatic nerve proteins. Chem. Biol. Interact. 176, 188–195. doi: 10.1016/j.cbi.2008.07.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vallelunga, A., Ragusa, M., Di Mauro, S., Iannitti, T., Pilleri, M., Biundo, R., et al. (2014). Identification of circulating microRNAs for the differential diagnosis of Parkinson’s disease and multiple system atrophy. Front. Cell. Neurosci. 8:156. doi: 10.3389/fncel.2014.00156
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van den Hove, D. L., Kompotis, K., Lardenoije, R., Kenis, G., Mill, J., Steinbusch, H. W., et al. (2014). Epigenetically regulated microRNAs in Alzheimer’s disease. Neurobiol. Aging 35, 731–745. doi: 10.1016/j.neurobiolaging.2013.10.082
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
van der Mark, M., Brouwer, M., Kromhout, H., Nijssen, P., Huss, A., and Vermeulen, R. (2012). Is pesticide use related to Parkinson disease? Some clues to heterogeneity in study results. Environ. Health Perspect. 120, 340–347. doi: 10.1289/ehp.1103881
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Verina, T., Kiihl, S. F., Schneider, J. S., and Guilarte, T. R. (2011). Manganese exposure induces microglia activation and dystrophy in the substantia nigra of non-human primates. Neurotoxicology 32, 215–226. doi: 10.1016/j.neuro.2010.11.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wagner, S. R., and Greene, F. E. (1978). Dieldrin-induced alterations in biogenic amine content of rat brain. Toxicol. Appl. Pharmacol. 43, 45–55. doi: 10.1016/s0041-008x(78)80031-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Walton, J. R., and Wang, M. X. (2009). APP expression, distribution and accumulation are altered by aluminum in a rodent model for Alzheimer’s disease. J. Inorg. Biochem. 103, 1548–1554. doi: 10.1016/j.jinorgbio.2009.07.027
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, A., Costello, S., Cockburn, M., Zhang, X., Bronstein, J., and Ritz, B. (2011). Parkinson’s disease risk from ambient exposure to pesticides. Eur. J. Epidemiol. 26, 547–555. doi: 10.1007/s10654-011-9574-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, B., and Du, Y. (2013). Cadmium and its neurotoxic effects. Oxid. Med. Cell. Longev. 2013:898034. doi: 10.1155/2013/898034
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, J. D., Huang, C. C., Hwang, Y. H., Chiang, J. R., Lin, J. M., and Chen, J. S. (1989). Manganese induced parkinsonism: an outbreak due to an unrepaired ventilation control system in a ferromanganese smelter. Br. J. Ind. Med. 46, 856–859. doi: 10.1136/oem.46.12.856
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, C., Ko, H. S., Thomas, B., Tsang, F., Chew, K. C., Tay, S. P., et al. (2005). Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin’s protective function. Hum. Mol. Genet. 14, 3885–3897. doi: 10.1093/hmg/ddi413
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Y., Wang, X., Li, R., Yang, Z. F., Wang, Y. Z., Gong, X. L., et al. (2013). A DNA methyltransferase inhibitor, 5-aza-2′-deoxycytidine, exacerbates neurotoxicity and upregulates Parkinson’s disease-related genes in dopaminergic neurons. CNS Neurosci. Ther. 19, 183–190. doi: 10.1111/cns.12059
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weggen, S., Eriksen, J. L., Das, P., Sagi, S. A., Wang, R., Pietrzik, C. U., et al. (2001). A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414, 212–216. doi: 10.1038/35102591
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weisskopf, M. G., Knekt, P., O’reilly, E. J., Lyytinen, J., Reunanen, A., Laden, F., et al. (2010a). Persistent organochlorine pesticides in serum and risk of Parkinson disease. Neurology 74, 1055–1061. doi: 10.1212/WNL.0b013e3181d76a93
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weisskopf, M. G., Weuve, J., Nie, H., Saint-Hilaire, M. H., Sudarsky, L., Simon, D. K., et al. (2010b). Association of cumulative lead exposure with Parkinson’s disease. Environ. Health Perspect. 118, 1609–1613. doi: 10.1289/ehp.1002339
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weitzman, S. A., Turk, P. W., Milkowski, D. H., and Kozlowski, K. (1994). Free radical adducts induce alterations in DNA cytosine methylation. Proc. Natl. Acad. Sci. U S A 91, 1261–1264. doi: 10.1073/pnas.91.4.1261
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wenstrup, D., Ehmann, W. D., and Markesbery, W. R. (1990). Trace element imbalances in isolated subcellular fractions of Alzheimer’s disease brains. Brain Res. 533, 125–131. doi: 10.1016/0006-8993(90)91804-p
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wille, H., Drewes, G., Biernat, J., Mandelkow, E. M., and Mandelkow, E. (1992). Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J. Cell Biol. 118, 573–584. doi: 10.1083/jcb.118.3.573
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wills, J., Credle, J., Oaks, A. W., Duka, V., Lee, J. H., Jones, J., et al. (2012). Paraquat, but not maneb, induces synucleinopathy and tauopathy in striata of mice through inhibition of proteasomal and autophagic pathways. PLoS One 7:e30745. doi: 10.1371/journal.pone.0030745
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wirdefeldt, K., Adami, H. O., Cole, P., Trichopoulos, D., and Mandel, J. (2011). Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur. J. Epidemiol. 26(Suppl. 1), S1–S58. doi: 10.1007/s10654-011-9581-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wojcik, D. P., Godfrey, M. E., Christie, D., and Haley, B. E. (2006). Mercury toxicity presenting as chronic fatigue, memory impairment and depression: diagnosis, treatment, susceptibility and outcomes in a New Zealand general practice setting(1994–2006). Neuro. Endocrinol. Lett. 27, 415–423.
Wu, J., Basha, M. R., Brock, B., Cox, D. P., Cardozo-Pelaez, F., Mcpherson, C. A., et al. (2008). Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. J. Neurosci. 28, 3–9. doi: 10.1523/jneurosci.4405-07.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, X. F., Block, M. L., Zhang, W., Qin, L., Wilson, B., Zhang, W. Q., et al. (2005). The role of microglia in paraquat-induced dopaminergic neurotoxicity. Antioxid. Redox Signal. 7, 654–661. doi: 10.1089/ars.2005.7.654
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, J., Wang, C., Sun, J., and Xue, Y. (2011). Neurotoxicity of silica nanoparticles: brain localization and dopaminergic neurons damage pathways. ACS Nano 5, 4476–4489. doi: 10.1021/nn103530b
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, J., and Xie, H. (2014). Effects of titanium dioxide nanoparticles on alpha-synuclein aggregation and the ubiquitin-proteasome system in dopaminergic neurons. Artif. Cells Nanomed. Biotechnol. 1–5. doi: 10.3109/21691401.2014.980507. [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wypijewska, A., Galazka-Friedman, J., Bauminger, E. R., Wszolek, Z. K., Schweitzer, K. J., Dickson, D. W., et al. (2010). Iron and reactive oxygen species activity in parkinsonian substantia nigra. Parkinsonism Relat. Disord. 16, 329–333. doi: 10.1016/j.parkreldis.2010.02.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wyss-Coray, T., Loike, J. D., Brionne, T. C., Lu, E., Anankov, R., Yan, F., et al. (2003). Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 9, 453–457. doi: 10.1038/nm838
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yamamoto, H., Saitoh, Y., Yasugawa, S., and Miyamoto, E. (1990). Dephosphorylation of tau factor by protein phosphatase 2A in synaptosomal cytosol fractions and inhibition by aluminum. J. Neurochem. 55, 683–690. doi: 10.1111/j.1471-4159.1990.tb04187.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yamin, G., Glaser, C. B., Uversky, V. N., and Fink, A. L. (2003). Certain metals trigger fibrillation of methionine-oxidized alpha-synuclein. J. Biol. Chem. 278, 27630–27635. doi: 10.1074/jbc.m303302200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, X., He, C., Li, J., Chen, H., Ma, Q., Sui, X., et al. (2014). Uptake of silica nanoparticles: neurotoxicity and Alzheimer-like pathology in human SK-N-SH and mouse neuro2a neuroblastoma cells. Toxicol. Lett. 229, 240–249. doi: 10.1016/j.toxlet.2014.05.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, D. J., Shi, S., Zheng, L. F., Yao, T. M., and Ji, L. N. (2010). Mercury(II) promotes the in vitro aggregation of tau fragment corresponding to the second repeat of microtubule-binding domain: coordination and conformational transition. Biopolymers 93, 1100–1107. doi: 10.1002/bip.21527
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, W., and Tiffany-Castiglioni, E. (2007). The bipyridyl herbicide paraquat induces proteasome dysfunction in human neuroblastoma SH-SY5Y cells. J. Toxicol. Environ. Health A 70, 1849–1857. doi: 10.1080/15287390701459262
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zarazúa, S., Bürger, S., Delgado, J. M., Jiménez-Capdeville, M. E., and Schliebs, R. (2011). Arsenic affects expression and processing of amyloid precursor protein (APP) in primary neuronal cells overexpressing the Swedish mutation of human APP. Int. J. Dev. Neurosci. 29, 389–396. doi: 10.1016/j.ijdevneu.2011.03.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zawia, N. H., Lahiri, D. K., and Cardozo-Pelaez, F. (2009). Epigenetics, oxidative stress and Alzheimer disease. Free Radic. Biol. Med. 46, 1241–1249. doi: 10.1016/j.freeradbiomed.2009.02.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ze, Y., Hu, R., Wang, X., Sang, X., Ze, X., Li, B., et al. (2014). Neurotoxicity and gene-expressed profile in brain-injured mice caused by exposure to titanium dioxide nanoparticles. J. Biomed. Mater. Res. A 102, 470–478. doi: 10.1002/jbm.a.34705
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zecca, L., Tampellini, D., Gatti, A., Crippa, R., Eisner, M., Sulzer, D., et al. (2002). The neuromelanin of human substantia nigra and its interaction with metals. J. Neural Transm. 109, 663–672. doi: 10.1007/s007020200055
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zecca, L., Youdim, M. B., Riederer, P., Connor, J. R., and Crichton, R. R. (2004). Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 5, 863–873. doi: 10.1038/nrn1537
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, J., Cai, T., Zhao, F., Yao, T., Chen, Y., Liu, X., et al. (2012). The role of alpha-synuclein and tau hyperphosphorylation-mediated autophagy and apoptosis in lead-induced learning and memory injury. Int. J. Biol. Sci. 8, 935–944. doi: 10.7150/ijbs.4499
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhao, Y., Bhattacharjee, S., Jones, B. M., Hill, J., Dua, P., and Lukiw, W. J. (2014). Regulation of neurotropic signaling by the inducible, NF-kB-sensitive miRNA-125b in Alzheimer’s disease (AD) and in primary human neuronal-glial (HNG) cells. Mol. Neurobiol. 50, 97–106. doi: 10.1007/s12035-013-8595-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zheng, W., and Monnot, A. D. (2012). Regulation of brain iron and copper homeostasis by brain barrier systems: implication in neurodegenerative diseases. Pharmacol. Ther. 133, 177–188. doi: 10.1016/j.pharmthera.2011.10.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zlokovic, B. V., Deane, R., Sagare, A. P., Bell, R. D., and Winkler, E. A. (2010). Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling Alzheimer’s amyloid beta-peptide elimination from the brain. J. Neurochem. 115, 1077–1089. doi: 10.1111/j.1471-4159.2010.07002.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: Alzheimer’s diseases, Parkinson’s disease, neurodegenerative disorders, beta-amyloid, tau protein, alpha-synuclein, metals, pesticides
Citation: Chin-Chan M, Navarro-Yepes J and Quintanilla-Vega B (2015) Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 9:124. doi: 10.3389/fncel.2015.00124
Received: 10 January 2015; Accepted: 17 March 2015;
Published online: 10 April 2015.
Edited by:
Victoria Campos-Peña, Instituto Nacional De Neurologia Y Neurocirugia, MexicoCopyright © 2015 Chin-Chan, Navarro-Yepes and Quintanilla-Vega. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Betzabet Quintanilla-Vega, Department of Toxicology, CINVESTAV-IPN, Ave. IPN 2508, Mexico City, 07360 Mexico Tel: 52+55-57473310, Fax: 52+55-57473395 mquintan@cinvestav.mx
† These authors have contributed equally to this work.