Neuroprotective effect of bexarotene in the SOD1G93A mouse model of amyotrophic lateral sclerosis
 Javier Riancho
Javier Riancho María Ruiz-Soto
María Ruiz-Soto María T. Berciano
María T. Berciano José Berciano
José Berciano Miguel Lafarga
Miguel Lafarga- 1Neurology Service, Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED), Instituto de Investigación Marqués de Valdecilla (IDIVAL), University Hospital Marqués de Valdecilla, University of Cantabria, Santander, Spain
- 2Department of Anatomy and Cell Biology, Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED), Instituto de Investigación Marqués de Valdecilla (IDIVAL), University of Cantabria, Santander, Spain
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by progressive weakness and muscle atrophy related to the loss of upper and lower motor neurons (MNs) without a curative treatment. There is experimental evidence suggesting that retinoids may be involved in ALS pathogenesis. Bexarotene (Bxt) is a retinoid-X receptor agonist used in the treatment of cutaneous lymphoma with a favorable safety profile whose effects have been recently investigated in other neurodegenerative diseases. In this study, we analyze the potential therapeutic effect of Bxt in the SOD1G93A mouse model of ALS. Mice were treated with Bxt or vehicle five times per week from day 60 onward. Survival, weight, and neuromuscular function studies together with histological and biochemical analyses were performed. Bxt significantly delayed motor function deterioration, ameliorated the loss of body weight, and extended mice survival up to 30% of the symptomatic period. Histological analyses of the lumbosacral spinal cord revealed that Bxt markedly delayed the early motor-neuron degeneration occurring at presymptomatic stages in ALS-transgenic mice. Bxt treatment contributed to preserve the MN homeostasis in the SOD1G93A mice. Particularly, it reduced the neuronal loss and the chromatolytic response, induced nucleolar hypertrophy, decreased the formation of ubiquitylated inclusions, and modulated the lysosomal response. As an agonist of the retinoic-X receptor (RXR) pathway, Bxt notably increased the nuclear expression of the RXRα throughout transcriptionally active euchromatin domains. Bxt also contributed to protect the MN environment by reducing reactive astrogliosis and preserving perisomatic synapsis. Overall, these neuroprotective effects suggest that treatment with Bxt could be useful in ALS, particularly in those cases related to SOD1 mutations.
Introduction
Amyotrophic lateral sclerosis (ALS) is the most common disease affecting the motor neurons (MNs) with an annual incidence that ranges from 2 to 4 cases per 100,000 people. It is characterized by progressive loss of upper and lower MNs, accompanied by neuromuscular junction denervation. Death occurs within 2–5 years after diagnosis, usually due to respiratory failure (Kunst, 2004). Although several therapies have been tried, riluzole (a glutamate antagonist) is the only drug currently approved by the U.S. Food and Drug Administration (FDA) for ALS treatment. However, it has shown a very poor effect in patients’ survival (Cheah et al., 2010; Miller et al., 2012).
Approximately 90% of ALS cases are sporadic (sALS), whereas the remaining 10% have a hereditary origin and tend to cluster into families (fALS). Up to 20% of fALS cases are due to mutations of the gene encoding superoxide dismutase 1 (SOD1) (Pasinelli and Brown, 2006; Rothstein, 2009; Ferraiuolo et al., 2011). This is also the basis of a commonly used transgenic mouse model expressing the human SOD1 gene with the G93A mutation (Gurney et al., 1994). High-copy SOD1G93A transgenic mice recapitulate much of the pathophysiology of human ALS, including progressive MN degeneration, functional impairment, and reduced lifespan (Gurney et al., 1994). Furthermore, a relationship between SOD1 mutations and sALS has recently been reported supporting the translational value of studies using transgenic SOD1 mice (Bosco et al., 2010).
The mechanisms involved in ALS pathogenesis remain unclear and are thought to be multiple (Robberecht and Philips, 2013). Disturbances of RNA processing, oxidative and endoplasmic reticulum (ER) stress, impaired protein degradation with protein aggregation, axonal transport defects, and glial cell dysfunction represent some of the mechanisms that have been related to ALS (Corona et al., 2007; Schmidt et al., 2009; Lemmens et al., 2010; Andersen and Al Chalabi, 2011; Philips and Robberecht, 2011; Bendotti et al., 2012; Majounie et al., 2012; Hetz and Mollereau, 2014).
Biochemical analyses have also identified some pathways potentially involved in ALS pathogenesis, including retinoid pathways. Thus, it has been reported that wild type mice fed with a retinoid-free diet develop a clinical phenotype resembling ALS with loss of MNs in the anterior horn and forelimb paralysis (Corcoran et al., 2002). Additionally, Kolarcik and Bowser (2012) have suggested that retinoid signaling is altered in ALS. These authors showed that the pre-treatment of primary MN-enriched cultures with adapalene, a retinoic acid receptor beta (RARβ) agonist, protects MNs from cell death induced by oxidative stress. Furthermore, some reports propose that retinoic acid (RA) could modulate both the ubiquitin proteasome system (UPS) and the autophagy-lysosome pathways helping to maintain intracellular proteostasis (Rajawat et al., 2010; Anguiano et al., 2013; Cheng et al., 2013). Overall, these studies suggest a potential beneficial effect of retinoid pathway activation on neuron survival in MN-related diseases.
Bexarotene (Bxt) is a highly selective retinoid-X receptor (RXR) agonist with a favorable safety profile. As occurs with most of retinoids, hypothirodism, liver toxicity, and cutaneous disorders are the most common adverse reactions observed with its use (Duvic et al., 2001a,b). Bxt has been approved by the FDA for the treatment of cutaneous T-cell lymphoma, and is currently used as a long-term therapy in the clinical practice requiring periodic blood test monitoring (Farol and Hymes, 2004). This drug has already been tested in some mice models of neurodegenerative diseases. Thus, it has been shown that Bxt reduces the amyloid load in an Alzheimer disease mouse model and consequently induced a marked amelioration in the cognitive, social, and olfactory deficits (Cramer et al., 2012). However, other investigators have only partially reproduced these results (Fitz et al., 2013; Price et al., 2013; Veeraraghavalu et al., 2013). On the other hand, Bxt has also been tested in a mouse model of Parkinson’s disease exhibiting beneficial effects (McFarland et al., 2013). Therefore, based on the potential neuroprotective effects of retinoid pathway activation (Mey and Rombach, 1999; Schrage et al., 2006; Kolarcik and Bowser, 2012), we hypothesized that Bxt could be useful in ALS. We planned this study to investigate its effects on both motor function and MN degeneration and survival in the transgenic SOD1G93A mouse model of ALS. Furthermore, we analyzed some potential neuroprotective mechanisms of Bxt including (i) cellular structures involved in proteostasis, such as protein synthesis machinery, nucleolus, and lysosomes, (ii) nuclear expression of the transcription factor RXRα, and (iii) reactive astroglial MN environment.
Materials and Methods
Animals and Drug
Transgenic mice (B6SJLTg [SOD1-G93A] 1Gur/J) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and maintained at the Animal Service of the University of Cantabria. The colony was maintained by mating heterozygous transgenic males with B6SJLF1/J hybrid females. Real time quantitative PCR (rtqPCR) of DNA obtained from tail tissue was used for genotyping, with specific primers detecting human SOD1 and the housekeeping mouse gene ApoB. Primer sequences were SOD1:GGG AAG CTG TTG TCC CAA G and CAA GGG GAG GTA AAA GAG AGC; ApoB: TCA CCA GTC ATT TCT GCC TTT G and GGG AAG CTG TTG TCC CAA G. Transgenic mice and control littermates were housed under controlled temperature and humidity, with a 12-h-light/12-h-dark cycle and free access to water and food. From day 105 of life, both transgenic and control mice were fed with Nutragel® (Bio-serv Frenchtown, NJ, USA). The experimental protocol was approved by the Ethics Committee of the University of Cantabria following the Spanish legislation. Animals were equally distributed into the experimental groups taking into account littermate origin, sex, weight, neuromuscular score baseline values, and transgenic load.
Micronized Bxt (Targretin®, Eisai, Japan) was water dissolved (30 mg/ml) and administered to transgenic SOD1G93A mice 5 days/week at a dose of 100 mg/kg/day by gastric gavage. Another group of mice were treated with vehicle (water) using the same protocol that Bxt-treated mice. The experimental groups included in the study are summarized in Table 1.
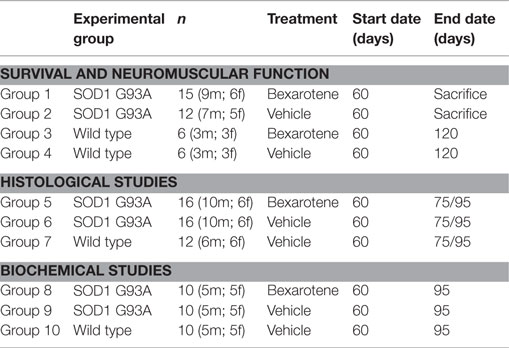
Table 1. Experimental design.
Survival, Weight, and Neuromuscular Tests
To investigate long-term effects of Bxt administration, 27 transgenic mice were divided into two groups. Fifteen (9 males, 6 females) and 12 (7 males, 5 females) mice were treated with Bxt and vehicle, respectively. Additionally, 12 wild type mice were also divided into two groups and treated with Bxt or vehicle under the regimen previously described. Treatment begun on day 60 of life and continued until sacrifice. Animals were weighted three times per week beginning at day 60. After a 2-week training period, neuromuscular tests (Rotarod and hanging test) were recorded twice a week beginning at 9 weeks of age until the end of the experiment. A cylinder spinning at a constant speed of 12 rpm was used for the Rotarod test (Mouse Rota-Rod 47600, Ugo Basile, Argentina). On the other hand, a semi-spheric grid of 20 cm in diameter was used for the hanging test. The best of two attempts, which were separated by 10 min rest, was recorded. Following current recommendations (Vargas et al., 2013), animals were euthanized by the time they developed severe paralysis, when they were not able to straighten up after turning them on their back in the following 15 s.
Histological Analysis
To study histological changes, 2 groups of 16 transgenic mice each were treated with Bxt or vehicle from day 60 until day 95. Eight mice of each group were euthanized on days 75 and 95, respectively, and their spinal cords were processed for light or electron microscopy. The results were compared with those obtained in age-matched wild type mice.
Light and Confocal Microscopy
After deep anesthesia with pentobarbital (50 mg/kg) mice were perfused with 3.7% paraformaldehyde in PBS (pH 7.4) for 15 min. The lumbosacral enlargement of the spinal cord (L2-S1) was dissected and post-fixed for 2 h. Spinal cord fragments were either dehydrated and embedded in diethylene glycol or processed for mechanical dissociation of MNs as previously reported (Pena et al., 2001).
For neuronal dissociation, small tissue fragments from the anterior horn were transferred to a drop of PBS on a siliconized slide. Then, a coverslip was applied on top of the slide and the tissue was squashed by percussion with a histologic needle in order to dissociate neuronal cell bodies. The preparation was then frozen in dry ice, and the coverslip removed using a razor blade. Using this procedure, most MNs remained adhered to the slide. Cell samples were processed in 96% ethanol at 4°C for 10 min, which increases the adhesion of cells to the slide, and rehydrated progressively in 70% ethanol and PBS. These squash preparations were used for propidium iodide (PI), a fluorescent staining of nucleic acids, and immunofluorescence.
Anterior horn cross sections, 2 μm thick, were processed for conventional hematoxylin–eosin staining, PI cytochemical staining, and immunofluorescence. The early loss of MN was evaluated at day 75 and 95 of life. The number of MNs was counted in four sections of each animal at segment L4 using hematoxylin–eosin staining. Only polygonal-shaped neurons present in ventral horns, larger than 20 μm and with prominent nucleoli, were used for the quantitative analysis as previously reported (Mancuso et al., 2014).
For immunofluorescence, tissue sections and squash preparations were sequentially treated with 0.5% Triton X-100 in PBS, 0.1M glycine in PBS containing 1% bovine serum albumin and incubated with the primary antibody overnight at 4°C. Then, the sections were incubated with the specific secondary antibody conjugated with FITC or Cy3 and mounted with Vectashield (Vector, USA). Confocal images were obtained with a LSM510 (Zeiss, Germany) laser scanning microscope using 40× oil or 63× oil (1.4 NA) objectives. In order to avoid overlapping signals, images were obtained by sequential excitation at 488 and 543 nm, to detect FITC and Cy3, respectively. Images were processed using Photoshop software.
To quantify astrogliosis, confocal images of the ventral horn’s gray matter immunostained for glial fibrillary acidic protein (GFAP) were recorded by using a 40× oil objective and the same confocal settings. Images were background corrected by reference regions outside the tissue and fluorescence intensities were estimated by using the ImageJ software (NIH, Bethesda, MD, USA; http://rsb.info.nih.gov/ij/).
The relative nuclear levels of RXRα in MNs of wild type, Bxt-treated and vehicle-treated transgenic SOD1G93A mice were determined on 5 μm thick cryostat sections immunostained with the anti-RXRα antibody. Only anterior horn neurons larger than 20 μm and with prominent nucleoli were sampled. We used three animals per experimental group and at least 35 neurons per animal were sampled. Confocal images were recorded by using a 63× oil objective (NA 1.4) and the same confocal settings at a resolution of 1024 × 1024 pixels. The same immunostaining and image processing procedures were carried out for all the experimental groups. Images were background corrected by reference regions outside the tissue and fluorescence intensities of nuclear RXRα signal, excluding the nucleolus, were estimated by using the ImageJ software (NIH, Bethesda, MD, USA; http://rsb.info.nih.gov/ij/).
The following primary antibodies were used: goat polyclonal antibodies anti-choline acetyltransferase (ChAT) (Millipore, Billerica, MA, USA) and anti-cathepsin D (Santa Cruz Biotechnology, USA), and rabbit polyclonal antibodies anti-GFAP (Dako, Glostrup, Denmark), anti-SOD1 (Enzo Life Sciences, Switzerland), anti-ubiquitin-protein conjugates (Santa Cruz Biotechnology, USA), and anti-RXRα (D-20, Santa Cruz Biotechnology, USA).
Electron Microscopy
For electron microscopy, transgenic mice were perfused with 1% glutaraldehyde and 1% paraformaldehyde in 0.12M phosphate buffer. Tissue samples of the anterior horn were dehydrated and embedded in Araldite. Ultrathin sections were examined with a Jeol 201 electron microscope.
To determine the ultrastructural localization of cathepsin D in lysosomes, we performed immunogold electron microscopy. Tissue samples of the anterior horn fixed with 3.7% paraformaldehyde were dehydrated in methanol and embedded in Lowicryl K4M at −20°C. Ultrathin sections were sequentially incubated with 0.1M glycine in PBS (15 min), 5% BSA in PBS (30 min), and the anti-cathepsin D antibody diluted in PBS containing 0.1M glycine and 1% BSA (1 h at room temperature). After washing, the sections were incubated with an anti-goat secondary antibody conjugated to 10 nm gold-particles (BioCell, UK) diluted 1:25 in 1% BSA in PBS (45 min at room temperature). After washing, the sections were stained with uranyl acetate and lead citrate. Control sections were treated as described but omitting the primary antibody.
Western Blotting and Real Time Quantitative PCR
Ten transgenic mice, treated with Bxt or vehicle from day 60 until day 95 of life, were used in these studies. For each biochemical assay (rtqPCR or WB) at least three mice of each animal group were used. Age-matched wild type mice receiving vehicle were used for comparison. After being anesthetized, they were decapitated and the lumbar spinal cord quickly removed and frozen in liquid nitrogen.
Gene expression was assessed by rtqPCR. RNA from spinal cord samples was isolated with Trizol following the manufacturer’s instructions (Invitrogen, Carlsbad) and purified with the RNeasy kit (Qiagen, Hilden, Germany) as previously reported (Delgado-Calle et al., 2011). Aliquots of RNA were reverse-transcribed with the PrimeScrip RT kit (Takara) using random hexamers as primers. Then, the expression of cathepsin D was determined rtqPCR using gene-specific primers and TaqMan probes (Life Technologies, Foster City, CA, USA). The threshold cycle (Ct) for each well was determined. The results were normalized to hypoxanthine phosphoribosyltransferase 1 (HPRT1). Relative gene expression was calculated as 2−ΛCT, where ΛCT is the difference between the gene of interest threshold cycle and the HPRT1 threshold cycle.
For WB analysis, the spinal cord samples (the ventral region of lumbosacral enlargement) were homogenized in a lysis buffer (100 mM NaCl, 10 mM Tris-HCl, pH 7.5, 1 mM EDTA, and 1 μg/ml aprotinin) and the homogenates were centrifuged. The protein in the supernatants were subjected to SDS-PAGE electrophoresis and afterwards transferred to polyvinylidene difluoride membranes, stained with antibodies, and visualized with the Odyssey system (LI-COR Biotechnology). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and tubulin were used as protein loading controls. The antibodies used were a goat polyclonal anti-cathepsin D (Santa Cruz Biotechnology, Europe) and mouse monoclonal anti-GAPDH (Abcam, Cambridge, MA, USA). ImageJ software (U. S. National Institutes of Health, Bethesda, MD, USA) was used to quantify the density and size of the blots.
Statistical Analysis
The significance of the differences in weight was tested by unpaired Student’s t-test, while non-parametric Mann–Whitney U test was used to compare motor test scores of the Bxt- and vehicle-treated groups. The overall survival and time-course of motor deterioration plots were obtained with the Kaplan–Meier procedure, followed by between-group curve comparisons with the Gehan–Breslow–Wilcoxon test.
The number of MNs in the anterior horn was tested with ANOVA test. Intracellular ubiquitin aggregates, the relative nuclear levels of RXRα, and both the integrated GFAP and RXRα signal assessing astrogliosis were also tested with ANOVA test. The significance of differences of WB and rtqPCR was analyzed with non-parametric Kruskal–Wallis test. p-Values <0.05 were considered as significant. All analyses were two-tailed.
Results
Bxt Treatment Delays Weight Loss and Extends Survival in SOD1G93A Mice
Bxt was well tolerated with no obvious signs of toxicity or drug-related deaths; in fact, the general condition was better preserved in the transgenic mice treated with Bxt than in vehicle-treated transgenic mice. Animals that received vehicle started to lose weight at day 80, while those receiving Bxt continued gaining weight until day 110. Bxt-treated mice weighed up to 20% more than controls. Differences in weight were significant from day 90 onward (Figure 1A). In comparison with wild type mice, the weight of transgenic mice was 75 and 89% in the vehicle-treated and Bxt-treated groups, respectively. Bxt-treated transgenic animals remained more physically active than controls receiving vehicle. However, Bxt administration to wild type mice induced no significant changes in either body weight or neuromuscular tests.
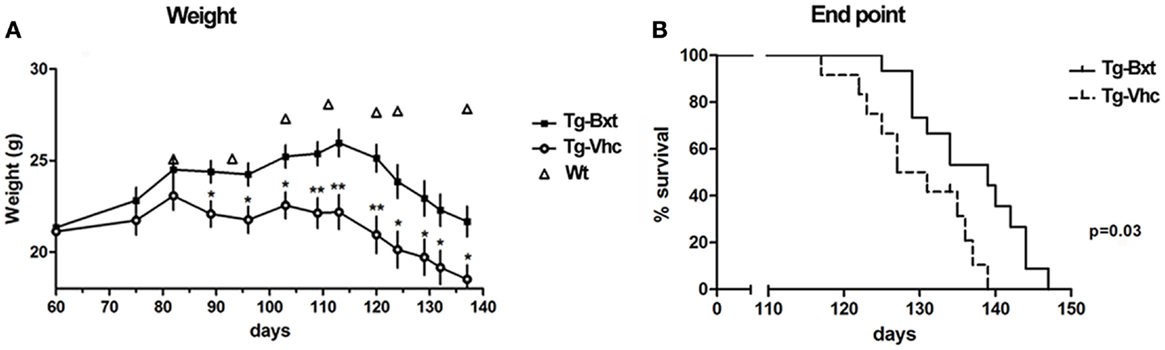
Figure 1. Effects of bexarotene on weight and survival. (A) Bexarotene treatment (Bxt) delays the start-up of weight loss as compared with the vehicle-treated (Vhc) transgenic SOD1G93A mice. (B) Survival analysis showing increase of lifespan in the Bxt group as compared with the Vhc group. WT, wild type mice.
Bxt treatment also prolonged the survival of SOD1G93A transgenic mice. The first death occurred on day 117 in the control group and on day 125 in the Bxt group. The median survival was 10 days longer in the group of animals treated with Bxt [139 vs. 129 days; Gehan–Breslow–Wilcoxon test, p = 0.03; hazard ratio = 0.29, 95% confidence intervals (CI): 0.11–0.80] (Figure 1B).
Bxt Preserves the Neuromuscular Function Delaying the Disease Onset in SOD1G93A Mice
Disease onset assessed by neuromuscular tests was also significantly delayed in Bxt-treated mice. Wild type mice and transgenic mice prior to disease onset were consistently able to stand for more than 1500 s on the Rotarod spinning wheels. The Rotarod median performance in the vehicle-treated transgenic mice started to decrease on day 96, while in the Bxt-treated animals did not change till day 113 (Figure 2A). The median time to reach a threshold of marked deterioration (established at 150 s in the present study) was 110 days in the vehicle-treated group and 119 days in the Bxt-treated group (Gehan–Breslow–Wilcoxon test, p = 0.033; hazard ratio = 0.42; 95% CI: 0.16–1.07) (Figure 2B). Similarly, wild type mice and presymptomatic transgenic mice were consistently able to hang for more than 600 s. Deterioration of the four-limb hanging test scores appeared to begin in both transgenic mice groups approximately on day 100. However, vehicle-treated mice showed more rapid deterioration than Bxt-treated animals. From day 110 to day 124, Bxt-treated animals were able to cling for twice as much time in comparison to those receiving vehicle (p-values 0.03–0.05). Significant between-group differences also existed regarding the time to reach stages of moderate or advanced deterioration. Thus, the median age to hang <50 s was 112 days in vehicle-treated mice and 120 days in the Bxt-treated group (Gehan–Breslow–Wilcoxon test, p = 0.01; hazard ratio = 0.36; 95% CI: 0.13–0.96) (Figure 2C). Similarly, significant between-group differences existed when a 10-s score was considered (p = 0.006).
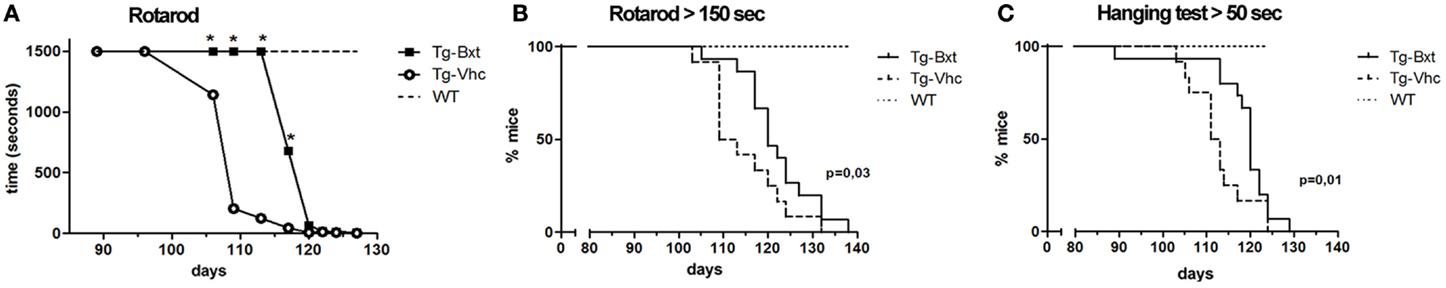
Figure 2. Bexarotene treatment enhances motor performance in SOD1G93A mice. Bexarotene treatment (Bxt) delays neuromuscular function deterioration as assessed by Rotarod (A,B) and hanging (C) tests.
Bxt Partially Preserves MN Cytology and Ameliorates the Spinal MN Loss in SOD1G93A Mice
As expected, the ongoing expression of the mutant transgene induced the development of neuromuscular changes in both groups of transgenic mice. However, our results showed that chronic administration of Bxt delayed the appearance of clinical manifestations of the disease. Therefore, we decided to explore whether the drug also delay histological alterations at presymptomatic stages. At day 75, marked abnormalities were observed in transgenic mice receiving vehicle, by contrast no gross abnormalities were observed in their behavior or motor performance. By conventional hematoxylin–eosin staining, the anterior horn of control wild type mice showed the typical organization of MNs with prominent nucleoli and Nissl bodies in addition to a compact neuropile (Figures 3A,D). Conversely, degenerative signs were observed in the anterior horn of vehicle-treated SOD1G93A mice. They consisted of cytoplasmic vacuolization of some MNs and spongiform changes of the neuropile (Figures 3B,E). These alterations were much less pronounced in transgenic mice treated with Bxt, where both MNs and neuropile generally appeared well preserved (Figures 3C,F).
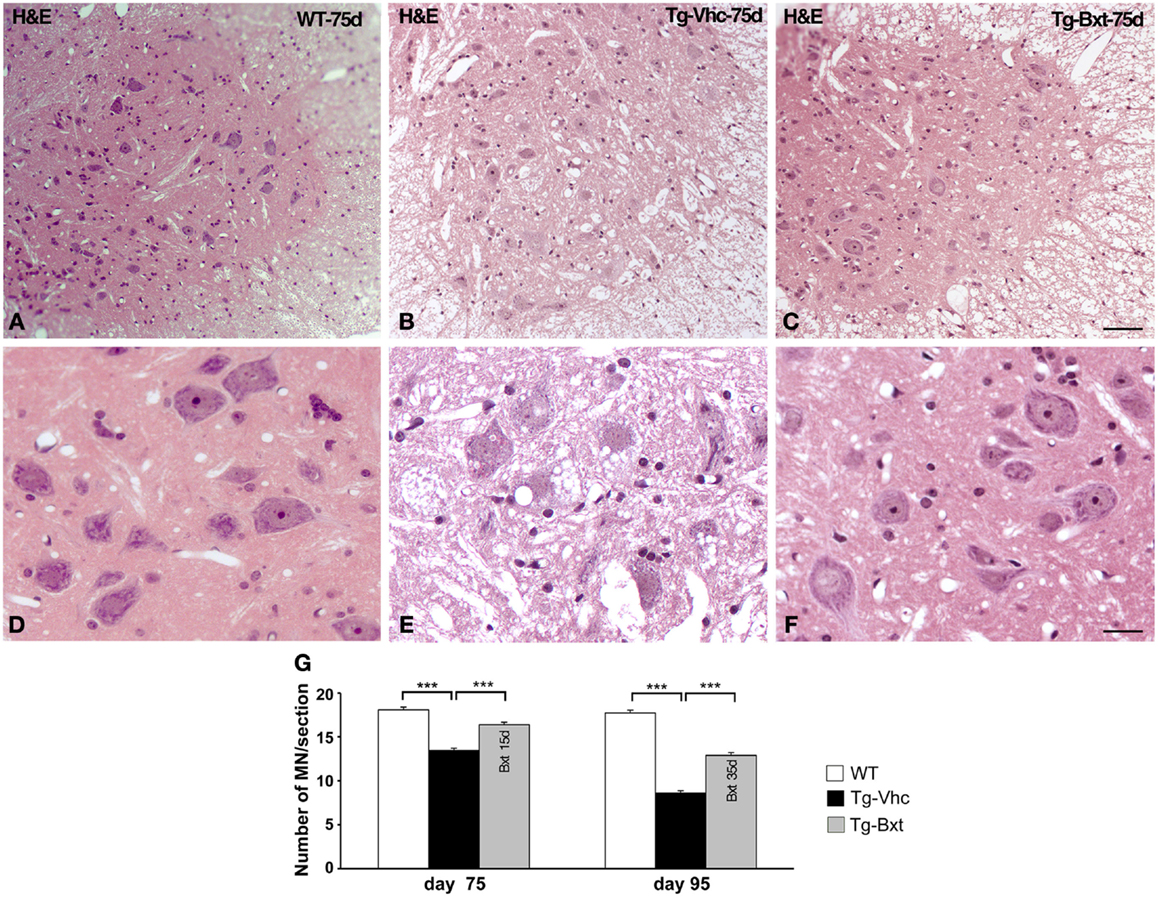
Figure 3. Light microscopy findings in the anterior horn. (A–C) Representative examples of anterior horn cross sections stained with hematoxylin–eosin from 75 day-old wild type [WT (A)] mice and vehicle-treated [Tg-Vhc (B)] and Bxt-treated [Tg-Bxt (C)] transgenic SOD1 G93A mice. The gray matter in the WT mouse shows the typical organization of MNs and a compact neuropile. Note that while MNs appear well preserved in the Tg-Bxt image, the Tg-Vhc image exhibits a reduced number of MNs and a spongiform alteration of the neuropile. Scale bar: 50 μm. (D–F) Higher magnification images illustrate the prominent vacuolar degeneration of MNs in the Tg-Vhc transgenic mice. Scale bar: 25 μm. (F) Quantitative analyses of spinal cord MNs at 75 and 95 days of age (G) (***p < 0.001).
We next analyzed the MN loss at both presymptomatic and early-symptomatic stages. This was assessed by evaluating the number of MN cell bodies in cryosections of the anterior horn (segments L4 and L5) stained with PI that fulfilled the previously referred criteria (Mancuso et al., 2014). On day 75, vehicle-treated SOD1G93A mice had 13.5 ± 0.23 MNs per section, while Bxt-treated SOD1 mice had 16.4 ± 0.25 MNs per section (74 and 90%, respectively, of MNs in wild type mice) (Figure 3G). The differences were more marked, at day 95 of life, when in the vehicle-treated group there were 8.6 ± 0.3 MNs per section, whereas 12.9 ± 0.30 MNs were present in Bxt-treated mice (48 and 72%, respectively, of MNs in wild type mice). Differences were significant at both stages (p < 0.001) (Figure 3G).
The morphological changes observed by conventional hematoxylin–eosin staining were further characterized by PI staining and ultrastructural analysis. On day 75, fluorescent staining of nucleic acid with PI revealed prominent nucleoli and Nissl bodies in healthy wild type MNs (Figures 4A,D) and different levels of chromatolysis in MNs of the vehicle-treated transgenic SOD1G93A mice (Figures 4B,E,F). Large cytoplasmic vacuoles unstained with PI were commonly observed in advanced stages of MN degeneration (Figure 4F). In contrast, most MNs in Bxt-treated SOD1G93A mice exhibited well-preserved Nissl bodies and prominent nucleoli (Figures 4C,G). Electron microscopy analysis on day 75 confirmed that the preservation of the basic cellular organization in MNs of Bxt-treated SOD1G93A mice. This included the normal structure of the nucleolus and chromatin as well as the cytoplasmic domains enriched in rough endoplasmic reticulum (RER) and free polyribosomes (Figure 4H). In contrast, a severe disruption of RER (chromatolysis), associated with the presence of abnormal and swelling mitochondria and different degrees of vacuolar degeneration, was frequently observed in MNs of the vehicle-treated SOD1G93A mice (Figures 4I–J).
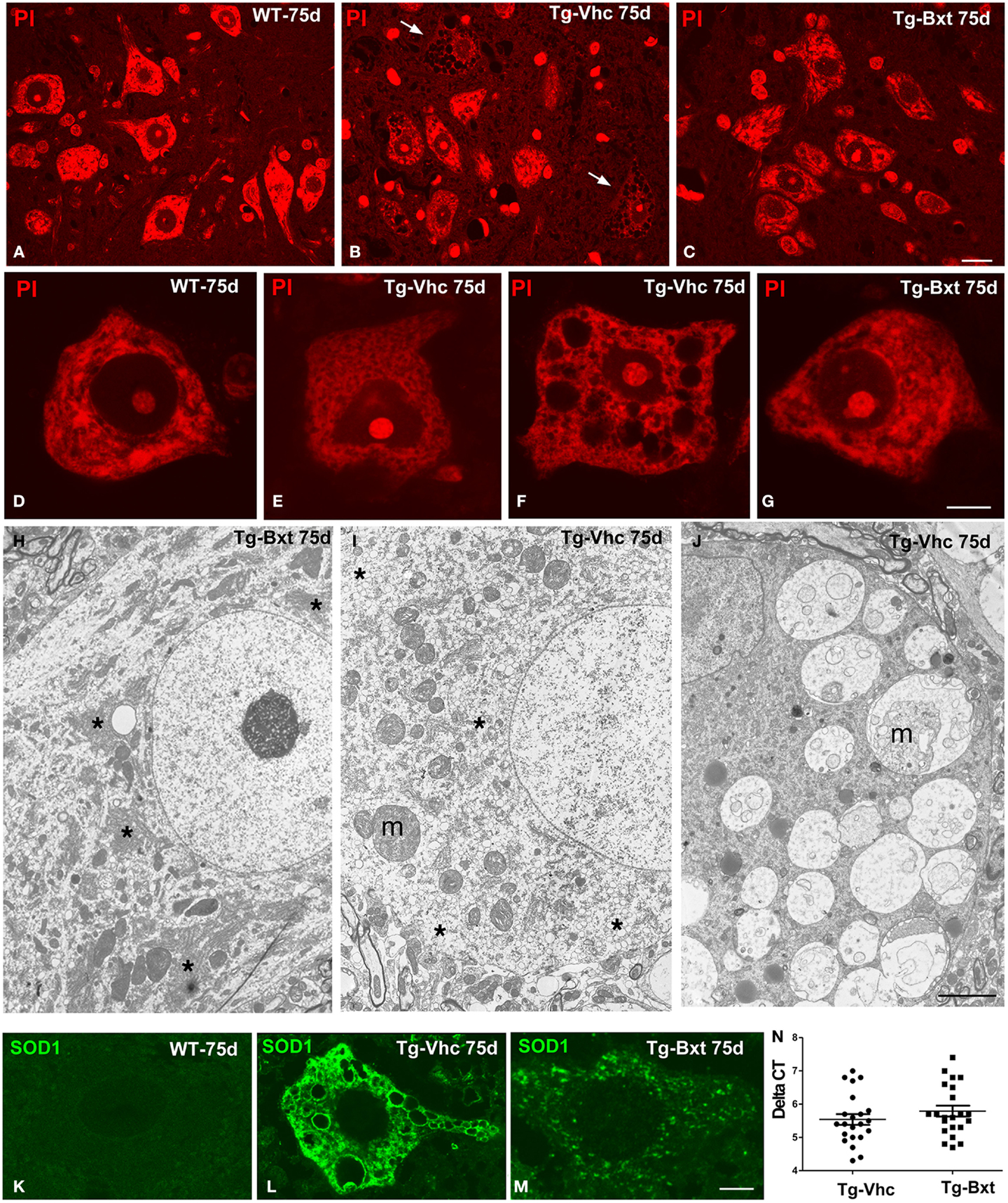
Figure 4. Morphological alterations of MNs. Representative examples of anterior horn cross cryosections stained with PI from 75 days old wild type [WT (A)] mice and vehicle-treated [Tg-Vhc (B)] and Bxt-treated [Tg-Bxt (C)] transgenic SOD1G93A mice. Note that the prominent nucleoli in all MNs and the well-preserved Nissl bodies in both WT and Tg-Bxt animals. Several MNs in the anterior horn of the Tg-Vhc mouse exhibit a severe cytoplasmic disruption with vacuolar degeneration and chromatolysis (white arrows). Scale bar: 20 μm. (D–G) Higher magnification images of dissociated MNs stained with PI from WT (D), Tg-Vhc (E–F), and Tg-Bxt mice (G). Note that the typical distribution of Nissl bodies in the WT MN (D) and different stages of chromatolysis and vacuolar degeneration in MNs from the Tg-Vhc mouse (E,F). Scale bar: 7.5 μm. (H–J) Electron micrographs of MNs from Tg-Bxt (H) and Tg-Vhc transgenic SOD1G93A mice (I,J). The MN from the Tg-Bxt mouse exhibits a well preserved nuclear and cytoplasmic structure, including typical arrays of RER cisterns (asterisks). MNs from the Tg-Vhc mice show severe chromatolysis (asterisks), extensive cytoplasmic vesiculation, mitochondrial abnormalities (m), and accumulation of large vacuoles derived from altered mitochondria (m). Scale bars: (H), 4 μm; (I,J), 3 μm. (K–M) Representative confocal microscopy images of MNs immunolabeled for SOD1 from the WT (K) and Tg-Vhc (L) and Tg-Bxt (M) SOD1G93A mice. Strong immunostaining signal of SOD1 appears throughout the cytoplasm, excluding the lumen of large vacuoles, in the MN of the Tg-Vhc mouse. Scale bar: 5 μm. (N) Transgene expression in Bxt-treated and Vhc-treated transgenic mice.
The pathogenic basis of the transgenic SOD1G93A mouse model lies on the over-expression of the toxic SOD1G93A protein (Jaarsma et al., 2000; Sau et al., 2007; Brotherton et al., 2012). To confirm the validity of the experimental model, we decided to investigate if chronic administration of Bxt modulated the expression of the transgene. Interestingly, rtqPCR analysis revealed no significant differences in human SOD1 mRNA levels between Bxt-treated and vehicle-treated transgenic mice (Figure 4N). Additionally, we also assessed the mutant SOD1 protein distribution by immunofluorescence. While MNs from wild type mice showed a weak diffuse staining of the endogenous SOD1 (Figure 4K), a strong fluorescent signal corresponding to the mutant SOD1 was clearly detected in the cytoplasm of MNs from SOD1G93A mice. Moreover, SOD1 immunoreactivity was absent from the nucleus (Figures 4L,M). At day 75, accumulations of mutant SOD1 were commonly observed throughout the cytoplasm, excluding vacuoles, in degenerating MNs from vehicle-treated transgenic mice (Figure 4L). However, a lower cytoplasmic concentration of this mutant protein, visible as a diffuse staining and small aggregates, was found in MNs of Bxt-treated mice (Figure 4M). These findings suggest that Bxt partially prevents mutant protein aggregation and preserves neuronal proteostasis until more advanced stages.
The histological analysis of the anterior horn at symptomatic stages of disease (95 days old) shows morphological alterations in both groups of transgenic SOD1G93A mice. Thus, light microscopy analysis of toluidine blue stained semithin sections clearly demonstrated the presence of degenerating MNs (Figures 5A,B). However, whereas most MNs exhibited severe vacuolar degeneration in vehicle-treated mice (Figure 5A), some well-preserved MNs coexisted with others that displayed different levels of vacuolar degeneration in Bxt-treated mice (Figure 5B). In both transgenic groups of animals, the ultrastructural analysis of MNs in advanced stages of neurodegeneration revealed (i) mitochondrial alterations, including dilation of cristae, presence of dense granules in the matrix, and global swelling resulting in the formation of large vacuoles, (ii) disruption of the RER, (iii) presence of lipid droplets, (iv) proliferation of neurofilaments, and (v) frequently nuclear eccentricity (Figures 5C–E). Moreover, axonal degeneration of myelinated fibers was a common finding in the neuropile at symptomatic stages (Figure 5D, inset). As expected, histological analysis of animals euthanized at the full paralysis stage showed similar abnormalities in both groups.
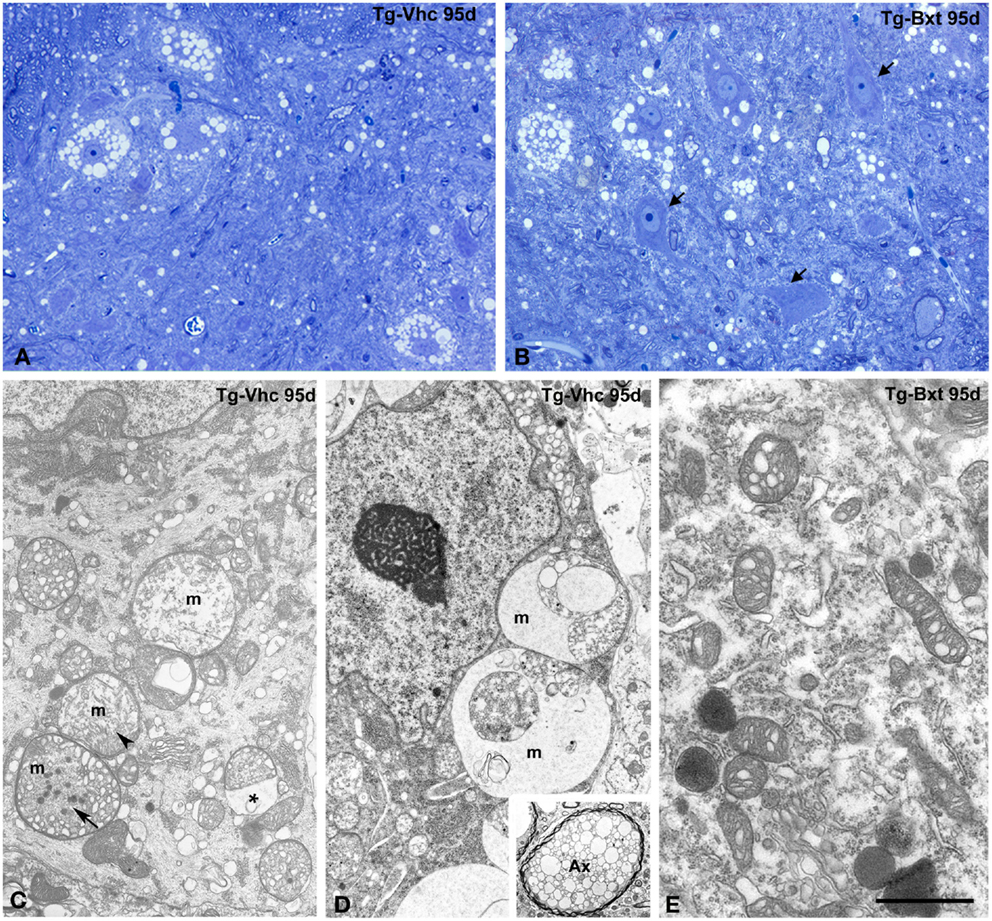
Figure 5. Morphological alterations of MNs at symptomatic stages of neurodegeneration. (A,B) Toluidine blue staining of gray matter sections from vehicle-treated [Tg-Vhc (A)] and Bxt-treated [Tg-Bxt (B)] transgenic SOD1G93A mice at day 95. The (A) shows MN loss and prominent vacuolar degeneration of remnant cell bodies. The (B) illustrates well-preserved neuronal bodies (arrows) coexisting with other ones exhibiting different levels of vacuolar degeneration. Scale bar: 20 μm. (C–E) Representative electron micrographs of MNs from Tg-Vhc (C,D) and Tg-Bxt (E) mice at advanced stages of neurodegeneration. (C,D) The cell bodies show large vacuoles containing remnants of degenerated mitochondria (m), with dilatation of the intermembranous space (asterisk) and presence of dense granules (arrows), in addition to cytoplasmic accumulation of small vesicles and nuclear eccentricity. The inset illustrates a typical axonal vacuolar degeneration of a myelinated fiber (Ax). (E) Detail of cytoplasm from a Bxt-treated MN illustrating the initial dilation of mitochondrial cristae. Scale bars: (C–E) 3 μm, inset 2.6 μm.
Bxt Induces Nucleolar Hypertrophy in Spinal Cord MNs of SOD1G93A Mice
It is well established that the nucleolus plays an essential role in the synthesis of rRNAs and molecular assembly of pre-ribosomal particles (Raska et al., 2006; Boisvert et al., 2007). Indeed, the nucleolar configuration and size correlated with the transcriptional activity of the ribosomal genes in mammalian neurons (Kinderman and Jones, 1993; Berciano et al., 2007; Hetman and Pietrzak, 2012; Palanca et al., 2014). To determine the possible contribution of the nucleolus to the neuroprotective effects of Bxt, we have analyzed the structural organization and size of MN nucleoli. Squash preparations of MNs stained with PI revealed the euchromatic nuclear configuration and the presence of prominent nucleoli with a high intensity of rRNA signal in wild type mice as well as in both vehicle-treated and Bxt-treated transgenic mice (Figures 6A–C). Morphometric analysis of nucleolar diameter on presymptomatic day 60 (before starting Bxt treatment) did not reveal significant differences between wild type and transgenic SOD1G93A mice. However, at an early-symptomatic stage (95 days old), the nucleolar diameter of MNs was significantly higher in MNs of SOD1G93A mice treated with Bxt for 30 days than those in MNs of both wild type and vehicle-treated SOD1G93A mice (Figure 6F). Electron microscopy analysis confirmed this nucleolar hypertrophy, and also revealed the reticulated nucleolar configuration, with numerous fibrillar centers, in most of MNs of both vehicle-treated and Bxt-treated SOD1G93A mice (Figures 6D,E). The Bxt-induced increase in nucleolar size is consistent with a neuroprotectetive response of the nucleolus for enhancing ribosome biogenesis.
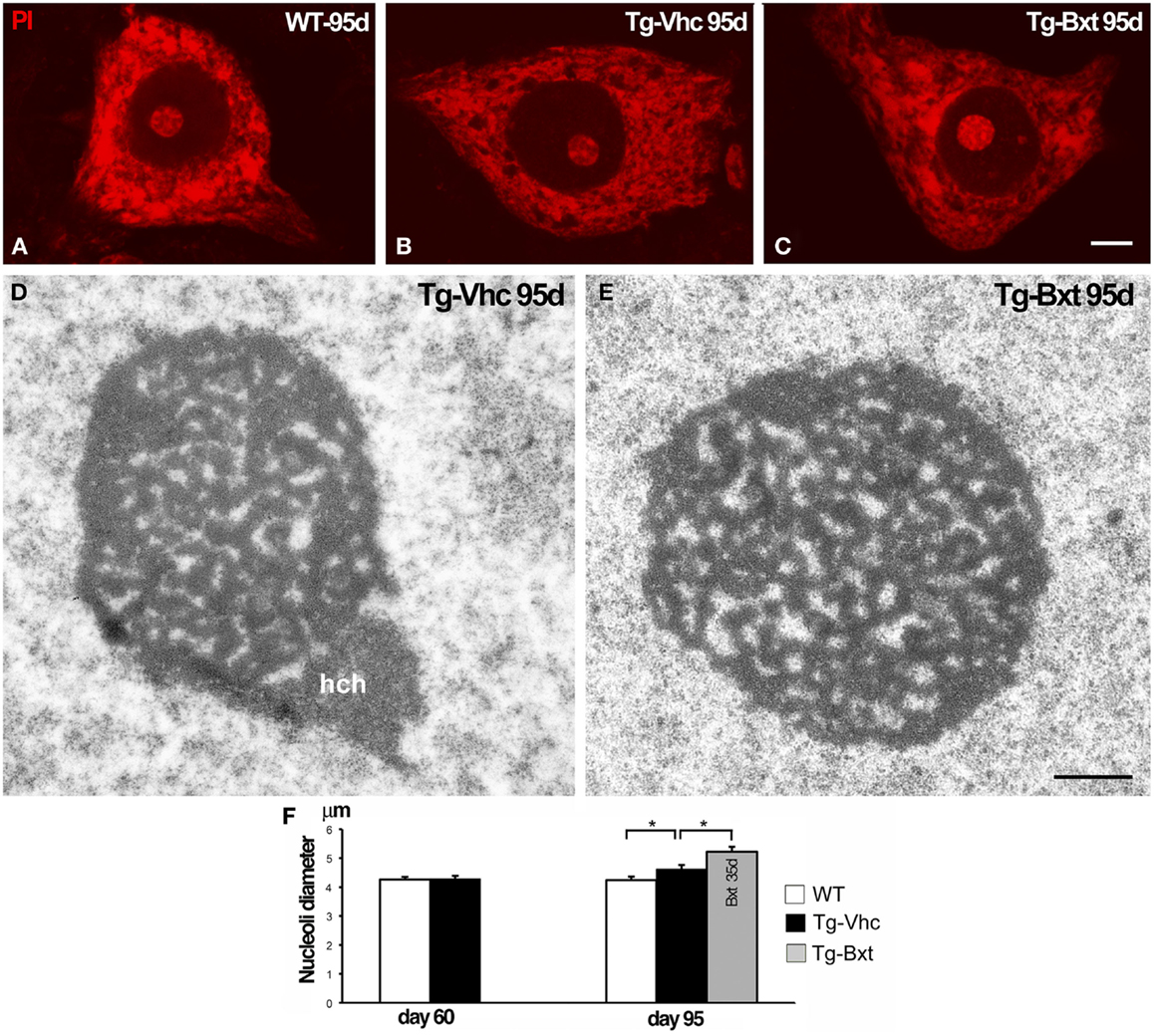
Figure 6. Nucleolar response of MNs in transgenic SOD1G93A mice. Higher magnification images of dissociated MNs stained with PI from wild type [WT (A)], vehicle-treated [Tg-Vhc (B)], and Bxt-treated [Tg-Bxt (C)] mice at day 95. MNs from Bxt-treated mice have a larger nucleolus. Scale bar: 5 μm. (D,E) Representative electron micrographs of MN nucleoli from [Tg-Vhc (D)] and [Tg-Bxt (E)] illustrating their reticulated configuration (hch: heterochromatin mass). Scale bar (D,E): 1 μm. (F) Morphometric analysis of nucleolar diameters of MNs at 60 and 95 days old (*p < 0.05).
Bxt Contributes to Modulate Cellular Proteostasis in the Spinal Cord MNs of SOD1G93A Mice
Protein aggregation is a common feature of many neurodegenerative diseases (Ross and Poirier, 2004). The presence of ubiquitylated protein aggregates has been widely described in ALS mice, consistent with a dysfunction of cellular proteostasis (Robberecht and Philips, 2013). This prompted us to investigate whether Bxt reduces protein aggregation into ubiquitylated cytoplasmic inclusions. Immunostaining of semithin sections of the anterior horn with an antibody that recognizes ubiquitin-protein conjugates revealed a high-nuclear concentration of ubiquitylated proteins, excluding the nucleolus, and a diffuse cytoplasmic signal in MNs from both wild type and transgenic SOD1G93A mice (Figures 7A–C). Ubiquitin-positive cytoplasmic inclusions were commonly found in MNs exhibiting different levels of vacuolar degeneration and chromatolysis in vehicle-treated SOD1G93A mice (Figures 7B,D,E). The quantitative analysis of the proportion of MNs containing ubiquitin-positive cytoplasmic inclusions (Figure 7F) showed a significant reduction in Bxt-treated SOD1G93A mice as compared with vehicle-treated mice (14% vs. 43%, p < 0.001). These findings strongly support a Bxt-induced enhancement of proteostasis resulting in lower levels of protein aggregation.
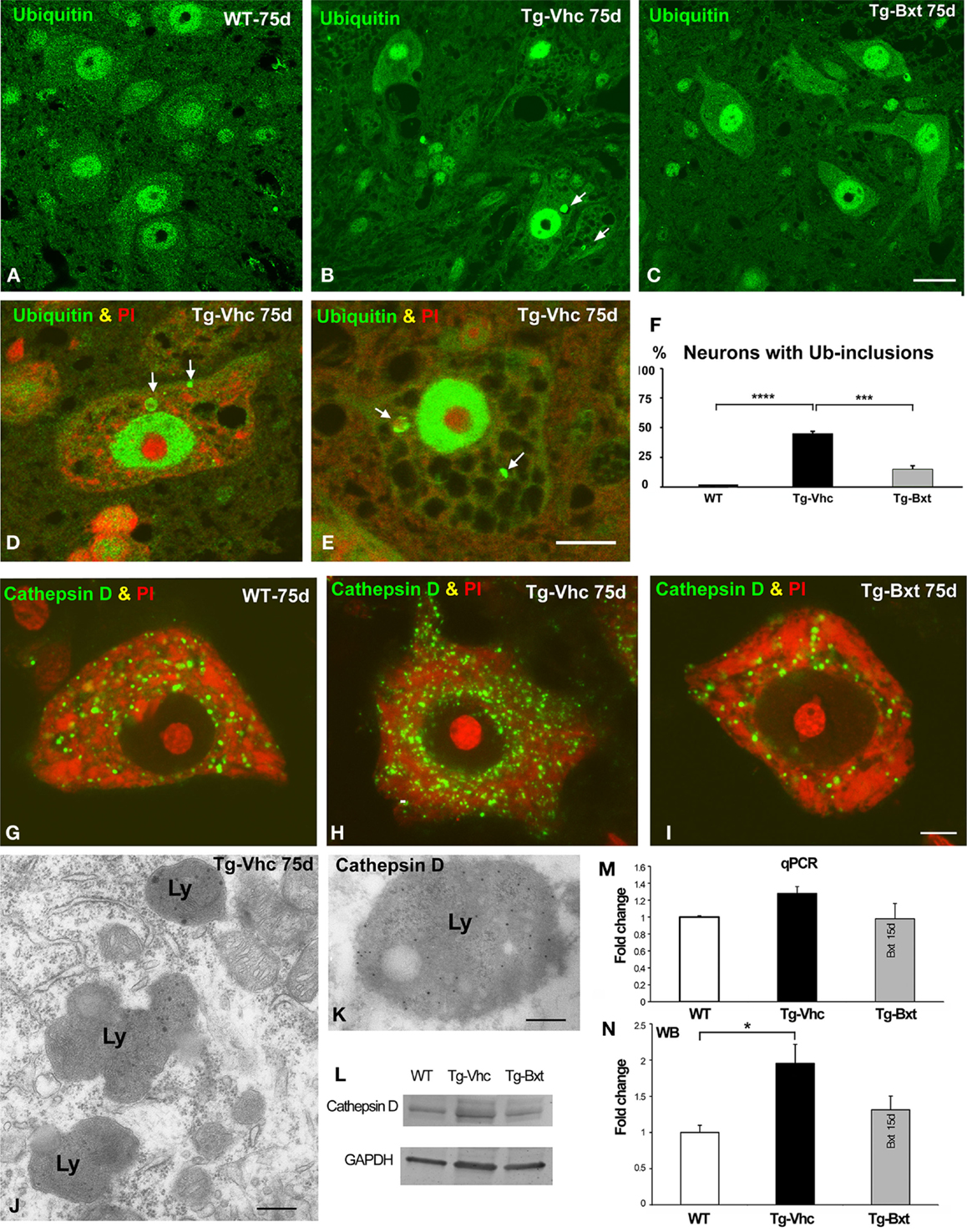
Figure 7. Alterations of proteostasis in MNs: the UPS and lysosome pathway. (A–C) Representative confocal images of anterior horn semithin sections immunostained for ubiquitin-protein conjugate from wild type mice [WT (A)], vehicle-treated [Tg-Vhc (B)], and Bxt-treated [Tg-Bxt (C)] transgenic SOD1G93A mice. Note that the presence of ubiquitin-positive cytoplasmic inclusions (white arrows) in MNs from the Tg-Vhc mouse (B). Scale bar: 25 μm. (D,E) Detail of MNs from Tg-Vhc mice immunostained for ubiquitin and counterstained with PI. The MN in (D) shows the presence of ubiquitin-positive cytoplasmic inclusion (arrows) and small Nissl bodies. A prominent vacuolar degeneration and ubiquitin-positive inclusions (arrows) are observed in the MN of (E). Scale bar (D,E): 10 μm. (F) Quantitative analyses of the proportion of MNs containing ubiquitylated inclusions (****p < 0.0001; ***p < 0.001). (G–I) Representative examples of dissociated MNs immunostained for cathepsin D and counterstained with PI from WT (G), Tg-Vhc (H), and Tg-Bxt SOD1G93A mice (I). The MN of the Tg-Vhc mouse shows greater accumulation of lysosomes (H) than those observed in both WT and Tg-Bxt (G,I). Scale bar (G–I): 5 μm. (J) Electron micrograph of the cytoplasm of a MN from Tg-Vhc mouse illustrating the accumulation of lysosomes. Scale bar: 750 nm. (K) Immunogold electron microscopy image illustrating the localization of the cathepsin D in the electron-dense lysosomal matrix. Scale bar: 500 nm. (L–N) Western blot and real time rtqPCR analysis of cathepsin D (*p < 0.05).
Together with the UPS, the autophagy-lysosome pathway is the other major proteolytic system (Korac et al., 2013). To evaluate the lysosomal response in transgenic SOD1G93A mice, we investigated the expression pattern of the protease cathepsin D, as a molecular marker of lysosomes. Immunostaining for cathepsin D in squash preparations counterstained with PI revealed the abundance of cathepsin D-positive lysosomes interspersed between Nissl bodies in wild type MNs (Figure 7G). However, a notable greater accumulation of lysosomes, particularly at the perinuclear cytoplasm, was clearly visible in vehicle-treated transgenic mice (Figure 7H). Interestingly, most MNs in Bxt-treated transgenic mice displayed a cathepsin D pattern similar to that observed in wild type mice (Figure 7I). Conventional electron microscopy analysis showed the accumulation of lysosomes in MNs of vehicle-treated transgenic mice (Figure 7J). Their lysosomal nature was confirmed with immunogold electron microscopy for cathepsin D. Gold particles decorated the cathepsin D-rich electron-dense matrix while the lipid-rich electron-lucent areas were free of immunolabeling (Figure 7K). Biochemical analyses with rtqPCR and WB demonstrated an increase in cathepsin D mRNA and protein levels in the anterior horn of vehicle-treated transgenic SOD1G93A mice as compared with both Bxt-treated SOD1G93A and wild type mice (Figures 7L–N).
Bxt Treatment Increases Nuclear Expression Levels of RXRα in MNs
On the basis that Bxt is an agonist of nuclear RXR receptors, which are ligand-dependent transcription factors, we investigated whether the neuroprotective effect of Bxt treatment could be partially mediated by an increase in the nuclear expression of RXRα in MNs. At day 75, confocal microscopy images of immunolabeled MNs revealed a weak diffuse nuclear pattern of RXRα expression, excluding the negative nucleolus, in wild type mice (Figure 8A). A similar pattern but with the presence of some microfoci of low RXRα immunoreactivity was found in MNs of transgenic SOD1G93A mice (Figure 8B). Interestingly, Bxt-treatment induced a remarkable increase in nuclear RXRα immunolabeling with a non-homogeneous distribution. Thus, RXRα appeared concentrated in numerous microfoci with higher fluorescence intensities than those observed in vehicle-treated MNs. These bright microfoci stood out over the diffuse nuclear staining (Figure 8C). In addition to the negative nucleolus, several irregular areas with very low or no RXRα signal were observed throughout de nucleus (Figure 8C). The measurement of nuclear (excluding nucleolus) fluorescent intensity of RXRα on confocal images, using the ImageJ software, showed a significant increase of this nuclear receptor signal in Bxt-treated MNs as compared with both wild type and vehicle-treated transgenic SOD1G93A mice (Figure 8D).
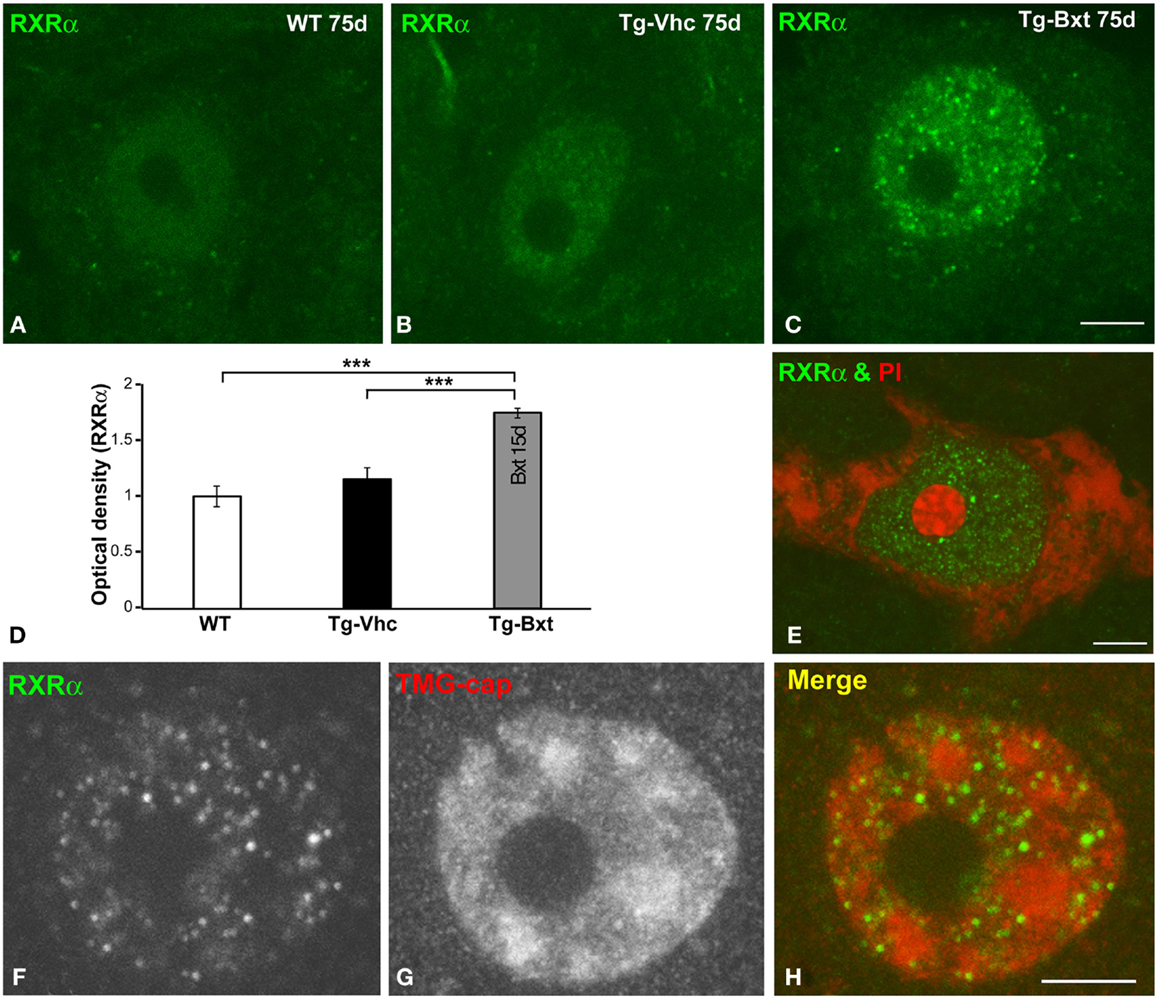
Figure 8. Nuclear expression of the RXRα in MNs. (A–C) Immunofluorescence for RXRα in MN nuclei from wild type [WT (A)], vehicle-treated [Tg-Vhc (B)], and Bxt-treated [Tg-Bxt (C)] transgenic SOD1G93A mice. (D) Densitometric analysis of nuclear fluorescence intensity of RXRα signal in MNs (p < 0.001). (E) Double labeling for RXRα and PI in a MN from Bxt-treated SOD1G93A mouse revealed the absence of RXRα signal in the nucleolus. (F–H) Co-immunostaining for RXRα and TMG-cap in a MN nucleus from Bxt-treated SOD1G93A mouse. Note that the distribution of RXRα microfoci in euchromatin domains and their absence within the nuclear speckles. Scale bars: 4 μm.
To further characterize the nuclear compartmentalization of RXRα in Bxt-treated neurons, we performed double labeling experiments combining RXRα detection with PI, as a marker of the neuronal nucleolus (Palanca et al., 2014), demonstrating the absence of RXRα in this nuclear compartment (Figure 8E). Moreover, we performed co-immunostaining for RXRα and the TMG-cap of spliceosomal small nuclear ribonucleoproteins (snRNPs), as a marker of nuclear speckles of splicing factors. These nuclear structures are DNA free nuclear domains involved in the storage and processing of pre-mRNA splicing factors (Lamond and Spector, 2003). Interestingly, nuclear microfoci of RXRα appeared distributed in euchromatin regions and were conspicuously absent from the DNA free nuclear speckles immunolabeled with the anti-TMG-cap antibody (Figures 8F–H). However, RXRα microfoci were frequently observed at the periphery of nuclear speckles (Figure 8H).
Bxt Contributes to Preserve the MN Environment in Spinal Cord of SOD1G93A Mice
It is well accepted that astrocytes are involved in ALS pathogenesis in both human patients (Haidet-Phillips et al., 2011) and rodent models (Yamanaka et al., 2008). To determine the potential neuroprotective effects of Bxt treatment on the reactive astroglial response to MN neurodegeneration, we performed GFAP immunostaining on semithin (2 μm thick) sections of the anterior horn. As illustrated in Figures 9A–E, reactive astrogliosis with hypertrophic astrocytes, which is a common feature of SOD1G93A mice (Hall et al., 1998), was less pronounced in the anterior horn of Bxt-treated mice. Thus, at day 75, the densitometric analysis of GFAP fluorescence intensity measured in the gray matter of vehicle-treated and Bxt-treated SOD1G93A mice increased by 3.5 and 2.3 times, respectively, respect to wild type mice (p < 0.001, wt vs. both Tg-Vhc and Tg-Bxt; p < 0.001, Tg-Vhc vs. Tg-Bxt) (Figure 9F). These findings reflect a Bxt-induced down-regulation of the reactive astroglial response.
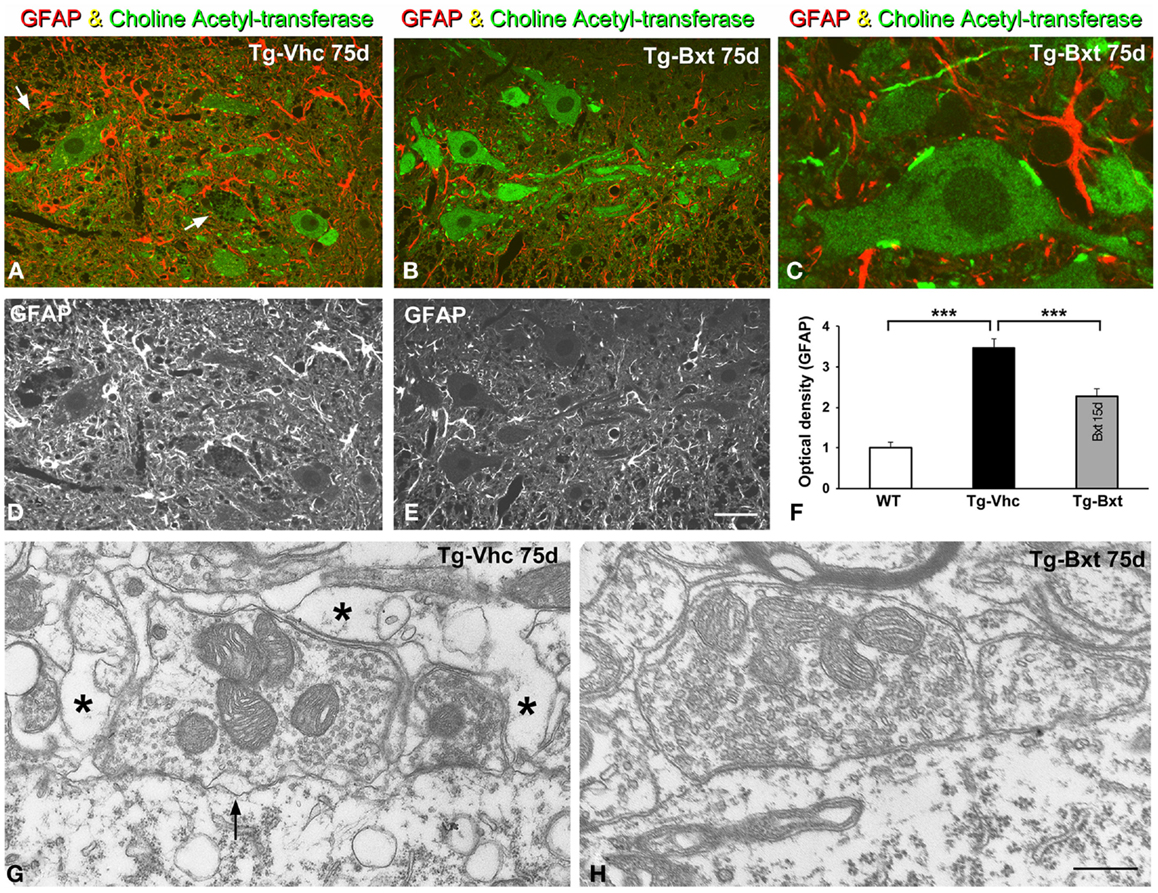
Figure 9. Reactive astrogliosis and synaptic alterations. (A–E) Representative confocal images of anterior horn sections coimmunolabeled for ChAT and GFAP from Vhc-treated [Tg-Vhc (A,D)] and Bxt-treated [Tg-Bxt (B,C,E)] transgenic SOD1G93A mice. Note that the prominent reactive astrogliosis and vacuolar degeneration of MNs (white arrows) in the Tg-Vhc mouse (A,D). Scale bar: (A,B,D,E), 30 μm; (C), 10 μm. (F) Densitometric analysis of fluorescence intensity of GFAP (***p < 0.001). (G,H) Electron micrographs of perisomatic synapses with their associated perisynaptic glia from Tg-Vhc (G) and Tg-Bxt (H) MNs. Note that the well preserved synaptic structure in the MN from the Tg-Bxt mouse (H) and the disruption of the synaptic cleft (arrow) and perisynaptic glia (asterisks) in the MN from the Tg-Vhc mouse (G). Scale bar: 1 μm.
Another important point of the astroglial response is the reorganization of the perisynaptic glia that enwraps synapses, excluding the synaptic cleft, and modulates synaptic transmission (Reichenbach et al., 2010). Cholinergic synaptic boutons on MN cell bodies and dendrites were immunolabeled with the anti-ChAT antibody. Double immunostaining for GFAP and ChAT revealed the well-preserved morphology of the ChAT-positive MNs and their synaptic boutons in Bxt-treated SOD1G93A mice (Figures 9B,C). In contrast, the vacuolar degeneration and the loss of ChAT-positive synapses were prominent features in MNs from vehicle-treated SOD1G93A mice (Figure 9A). By day 75, electron microscopy analysis of perisomatic synapses confirmed the well preserved structure of both synaptic boutons and perisynaptic glia in Bxt-treated mice (Figure 9H). Conversely, severe structural alterations, namely dilatation of the synaptic cleft, detachment of synapses, and severe disruption of the perisynaptic glia, were found in the majority of MNs of mice treated with vehicle (Figure 9G).
Discussion
Amyotrophic lateral sclerosis is one of the most disabling and ominous neurodegenerative diseases, being Riluzole, with a modest effect on survival, the only drug that can be currently offered to patients. Therefore, there is a pressing need to find new therapies to cure or at least ameliorate the severe course of the disease. Our study was undertaken to contribute to the search of new drugs for ALS patients.
Our results indicate that Bxt has a beneficial effect in transgenic SOD1G93A mice. In particular, treatment with this retinoid agonist significantly delayed the disease onset and extended lifespan. As expected, Bxt did not prevent the development of the disease, since it did not block the expression of the mutant transgene. This fact increases the external viability of our findings. Overall, our results suggest that Bxt exerts a neuroprotective effect by interacting with multiple cellular pathways. The main target of Bxt, the RXR, appeared to be overexpressed in MN nuclei of mice treated with the drug, confirming its target enhancement in the spinal cord after oral administration.
Some of early cellular alterations reported here and in previous studies (Chiu et al., 1995; Bendotti et al., 2001; Pasinelli et al., 2004; Zang et al., 2005; Martin et al., 2007; Koval et al., 2013; Vinsant et al., 2013a,b) in MNs of the SOD1G93A mouse include vacuolar degeneration of cell bodies and neuronal processes, mitochondrial alterations, presence of intracellular protein aggregates, synapse disruption, progressive loss of MNs, and astrogliosis in the anterior horn. All these changes reveal a severe neuronal dysfunction preceding clinical manifestations. Our histological results are consistent with the notion that Bxt, although unable to block the main pathophysiological mechanism, since it did not modify the mutant transgene expression, delayed disease onset by preserving MN homeostasis until more advanced stages.
A main target of Bxt, the RXRα, appeared overexpressed in MN nuclei of mice treated with the drug, suggesting that the neuroprotective effect of Bxt was partially mediated by the activation of the RA signaling pathway. A similar neuronal nuclear localization of RXRα has been previously reported after spinal cord contusion injury, suggesting that the participation of the RA signaling in the physiological response to CNS injury (Schrage et al., 2006). When activated, RXR are known to promote transcription of numerous target genes (Evans and Mangelsdorf, 2014). Our results show a significant increase in the nuclear expression of RXRα in MNs of Bxt-treated SOD1G93A mice in comparison with MNs of vehicle-treated SOD1G93A mice, suggesting a Bxt-induced up-regulation of protein-coding target genes. In this vein, RXRα appears distributed throughout the extensive nuclear domains of euchromatin (active chromatin), which is a chromatin configuration characteristic of MNs (Riancho et al., 2014), but it is excluded from the nucleolus and DNA free nuclear speckles of splicing factors (Lamond and Spector, 2003). Moreover, the numerous nuclear microfoci of RXRα induced by Bxt treatment have an organization and spatial distribution similar to those of transcription factories identified in neurons using the in situ transcription assay with 5′-fluorouridine incorporation into nascent RNA (Casafont et al., 2006). Interestingly, some of these RXRα microfoci appear in the periphery of nuclear speckles, where specific highly active genes have been shown to localize (Xing et al., 1993; Moen et al., 2004). Taken together, the extensive nuclear localization of RXRα in MNs of Bxt-treated SOD1G93A mice supports that this nuclear receptor is actively engaged in transcriptional activation of target genes and presumably in epigenetic changes via chromatin remodeling (Evans and Mangelsdorf, 2014). This interpretation is also consistent with the recent RXR cistrome data indicating the existence of many thousands of genomic binding sites for a single nuclear receptor affecting elaborate networks of genes (Tang et al., 2011). In fact, several reports have suggested that retinoids play a protective role in degenerative and acquired neurological diseases, such as Alzheimer or stroke (Sato et al., 2008; Crockett et al., 2011; Obulesu et al., 2011; Shimada et al., 2013).
Retinoids modulate the expression of hundreds of genes, including a large number of neuronal genes (Lane and Bailey, 2005; Evans and Mangelsdorf, 2014). In the case of ALS, retinoid-regulated neuronal genes might impact on important cellular processes, such as the antioxidant response (SOD1), neuroinflammation and immune modulation (VEGF, IL2, p65 subunit of NFkβ), cytoskeletal organization (neurofilament L, M, H proteins), ion transport (K+ channel Kir2.1, L-type and N-type Ca++ channels), intracelular signaling (phospholipase A2, CREB, PKA), and synaptic homeostasis (ChAT, vesicular ACh and GABA transporters, NMDA receptor NR1 and kainate receptor GluR6) (Lane and Bailey, 2005; Kolarcik and Bowser, 2012). Overall, these effects could be partially responsible of the neuroprotective effect of Bxt in the transgenic SOD1 mouse model of ALS. However, the precise mechanism of action of retinoids in ALS remains to be elucidated. Although in a preliminary study Crochemore et al. (2009) reported that chronic administration of all-trans RA had a negative effect in transgenic ALS mice by reducing lifespan, our results are consistent with several studies supporting the neuroprotective role of retinoids. First, the administration of a retinoid-free diet in rats induced a phenotype similar to ALS, indicating that a defect in retinoid signaling could promote MN disease (Corcoran et al., 2002). Second, gene expression profiling in both ALS animal models and sALS patients have demonstrated altered transcription of genes related to retinoid pathways (Malaspina et al., 2001; Jokic et al., 2007; Malaspina and Turkheimer, 2007). Third, it has also been reported that RA signaling facilitates axon outgrowth and nerve regeneration, whereas its disruption leads to MN degeneration (Maden, 2007). Moreover, Kolarcik and Bowser (2012) have recently analyzed the expression of genes of the retinoid pathway in the spinal cord of ALS patients and found that different isoforms of RAR correlated with the survival of MNs. In line with this, the authors demonstrated that pre-treatment of primary MNs-enriched cultures with a pan-RAR or RARβ-specific agonist decreased MN cell death associated with oxidative stress, while a RARβ-specific antagonist enhanced cell death. On the basis that RXR can also act as heterodimers with peroxisome proliferator-activated receptor (PPAR) and liver X receptor (LXR), future experimental studies combining treatments with PPAR/LXR agonists and Bxt could be considered. This combined therapy might potentially enhance the neuroprotective effect of Bxt on MN survival.
As in other neurodegenerative diseases, oxidative and ER stresses with disturbed proteostasis are considered relevant features in ALS (Rothstein, 2009; Parakh et al., 2013; An et al., 2014). Consistent with a proteostasis dysfunction, we have observed lysosomal proliferation, with up-regulation of cathepsin D expression and increased cytoplasmic aggregation of aberrant proteins into ubiquitylated inclusions in MNs of the vehicle-treated SOD1G93A mice. Several studies correlate the disturbance of neuronal proteostasis with an impairment in the UPS and secondary dysfunction of autophagy-lysosome pathway (Martinez-Vicente and Cuervo, 2007). Although this pathway constitutes a physiological mechanism to degrade proteins that “escape” from UPS and is beneficial for cell welfare (Tan et al., 2014), dysfunction of lysosomal proteolysis can be extremely harmful for neuronal function and survival (Ghavami et al., 2014). Indeed, up-regulation of the neuronal lysosomal system with increased levels of capthesin D is a prominent finding in Alzheimer’s disease (Cataldo et al., 1995; Adamec et al., 2000). Similarly, altered mRNA and protein levels of cathepsin D have been correlated with more severe MN degeneration in a transgenic mouse model of ALS (Wootz et al., 2006). The up-regulation of the lysosomal system reported here, in the SOD1G93A mouse, may simply reflect a compensatory response of MNs to the accumulation of aberrant proteins. However, several studies support a potential direct role of cathepsin D as mediator of neurotoxicity and cell death through abnormal permeabilization of lysosome and enzyme leakage into cytoplasm (Adamec et al., 2000; Boya and Kroemer, 2008).
As expected, when compared with wild type mice, our transgenic SOD1G93A mice showed protein aggregates, identified as ubiquitylated inclusions, which are a pathological hallmark of ALS (Ferraiuolo et al., 2011; Bendotti et al., 2012). There is previous evidence that over-expression of mutant SOD1 protein tends to collapse the UPS by inducing ER stress, resulting in secondary accumulation of misfolded proteins and a severe disturbance of neuronal proteostasis (Saxena et al., 2009). The higher cytoplasmic accumulation of ubiquitylated inclusions and lysosomes in MNs from the vehicle-treated SOD1G93A than in MNs from Bxt-treated SOD1G93A mice suggests that Bxt treatment reduces proteostasis dysfunction. Consistent with this potential neuroprotective effect of Bxt on neuronal proteostasis, Rajawat et al. (2011) have recently reported that RA promotes the autophagy-lysosome pathway by inducing the acidification and maturation of the autophagosome through a mechanism independent of the classic retinoid nuclear receptors. Moreover, a recent study indicates that the administration of RA has a neuroprotective effect under conditions of UPS inhibition (Cheng et al., 2013). In line with this concept, our results suggest that MNs from Bxt-treated SOD1G93A mice could have a greater ability to control proteostasis, and particularly to clear aberrant proteins, than MNs from vehicle-treated SOD1G93A mice. Upon detection of a pathobiological signal, neurons commonly activate a neuroprotective compensatory response that, if detrimental conditions persist, precedes the activation of the neurodegeneration and cell death. In this context, our results support that Bxt treatment delays the intricate progression of neuroprotection to neurodegeneration in ALS spinal cord MNs.
Spinal cord MNs from both wild type and transgenic SOD1G93A mice had prominent nucleoli with a reticular configuration (Riancho et al., 2014; present results). The nucleolar hypertrophy detected in MNs of Bxt-treated transgenic SOD1G93A mice seems to reflect a reactive nucleolar response to enhance nucleolar transcription and ribosome biogenesis. It is well established that nucleolar size positively correlates with both cell size and the transcription rate of rDNA genes (Raska et al., 2006; Berciano et al., 2007; Palanca et al., 2014). Enlargement of nucleolar size with up-regulation of ribosomal (rRNA) gene expression has previously been reported in experimental models of neuronal injury, such as axotomy and treatment with proteasome inhibitors (Kinderman and Jones, 1993; Palanca et al., 2014). Nucleolar enlargement also occurs in MNs of ALS patients with very rapid clinical course (Wakayama, 1992). The higher nucleolar size of the MNs of Bxt-treated mice, as compared with those of vehicle-treated mice, suggests an enhanced Bxt-induced compensatory response of the nucleolus to the ER stress caused by the mutant SOD1 protein (Kikuchi et al., 2006; Saxena et al., 2009). This neuroprotective response could sustain ribosome biogenesis and translational activity in order to delay the disruption of the protein synthesis machinery (chromatolysis) in transgenic SOD1G93A mice.
A remarkable finding here is the Bxt-induced reduction of astrogliosis combined with better preservation of the perisynaptic glia in transgenic SOD1G93A mice. Both cellular events also support a neuroprotective response. Reactive astrogliosis with hypertrophy and hyperplasia of astrocytes is a well-known response to degeneration and neuron loss. In the case of ALS, astrogliosis has been reported in both patients and murine models of the disease (Hall et al., 1998; Fischer et al., 2004). The slowdown of the astrogliosis response in Bxt-treated mice is consistent with the minor neurodegenerative signs observed in MNs. The involvement of astrogliosis in ALS pathogenesis is also supported by a recent study showing that spinal cord astrocytes expressing ALS-linked mutant SOD1 release cytotoxic factors to MNs (Nagai et al., 2007). Regarding the possible cellular effects of Bxt on astrocyte behavior, a previous study in an experimental model of astrocyte–neuron co-culture demonstrated that treatment with all-trans RA induced the differentiation and attenuated the astrocyte proliferation (Wohl and Weiss, 1998). This finding suggests that retinoids can contribute to down-regulate the proliferation (hyperplasia) of astrocytes in the transgenic SOD1G93Amice.
Synaptic alterations and their associated excitotoxicity in MNs have been widely studied in ALS as pathogenic mechanisms (Van Den et al., 2006; Sunico et al., 2011; Casas et al., 2013). In addition to the ultrastructural alterations in synaptic boutons previously reported in the SOD1G93A mice (Sunico et al., 2011), our results show a severe disruption of the perisynaptic glia that surround perisomatic MN synapses. Perisynaptic glia plays an important role in maintaining synaptic homeostasis through different functions, such as neurotransmitter traffic regulation, energy recycle, and ion homeostasis (Belanger and Magistretti, 2009; Reichenbach et al., 2010). The current findings support that these regulatory mechanisms may be overwhelmed in the SOD1G93A mice, contributing to disease progression. In this vein, preservation or delayed disruption of perisynaptic glia in Bxt-treated SOD1G93A mice could also represent an untapped potential mechanism of neuroprotection.
Several authors have reported a variety of treatments in the SOD1G93A mouse model of ALS. In view of our results, Bxt seems to be slightly better than riluzole and similar to other promising drugs recently tested (Gurney et al., 1998; Mancuso et al., 2014). In the future, mixed approaches combining drugs with synergic therapeutic effects will be worthwhile. In our view, one advantage of Bxt is that is a drug currently used in the clinical practice, a fact that would definitely facilitate future therapeutic trials.
In conclusion, the results of the present study demonstrate that chronic treatment with Bxt delayed disease onset, preserved motor function and extended survival in transgenic SOD1G93A mice. Multiple mechanisms may be involved in this neuroprotective effect, including the activation of the RXR target genes. Bxt ameliorated the MN loss in the ALS mice by preserving the organization of the cellular structures directly involved in proteostasis, such as the protein synthesis machinery and the lysosomes. The Bxt-induced amelioration of proteostasis disturbance was also supported by the decrease of ubiquitylated cytoplasmic inclusions and by the neuroprotective nucleolar response. Additionally, Bxt contributed to protect MN environment by reducing reactive astrogliosis and synaptic alterations. Although further studies are needed to fully elucidate the molecular mechanisms involved in neuroprotective effects of Bxt, this drug appears to be a promising neuroprotective therapy for ALS, especially for those cases related to SOD1 mutations.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Mrs Raquel Ceballos and Jana Arozamena for technical assistance. This work was supported by the following grants: “Dirección General de Investigación” of Spain (BFU2011-23983) and “Centro de Investigación en Red sobre Enfermedades Neurodegenerativas (CIBERNED; CB06/05/0037)” from Spain.
References
Adamec, E., Mohan, P. S., Cataldo, A. M., Vonsattel, J. P., and Nixon, R. A. (2000). Up-regulation of the lysosomal system in experimental models of neuronal injury: implications for Alzheimer’s disease. Neuroscience 100, 663–675. doi: 10.1016/S0306-4522(00)00281-5
An, T., Shi, P., Duan, W., Zhang, S., Yuan, P., Li, Z., et al. (2014). Oxidative stress and autophagic alteration in brainstem of SOD1-G93A mouse model of ALS. Mol. Neurobiol. 49(3):1435–48 doi:10.1007/s12035-013-8623-3
Andersen, P. M., and Al Chalabi, A. (2011). Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol. 7, 603–615. doi:10.1038/nrneurol.2011.150
Anguiano, J., Garner, T. P., Mahalingam, M., Das, B. C., Gavathiotis, E., and Cuervo, A. M. (2013). Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat. Chem. Biol. 9, 374–382. doi:10.1038/nchembio.1230
Belanger, M., and Magistretti, P. J. (2009). The role of astroglia in neuroprotection. Dialogues. Clin. Neurosci. 11, 281–295.
Bendotti, C., Calvaresi, N., Chiveri, L., Prelle, A., Moggio, M., Braga, M., et al. (2001). Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J. Neurol. Sci. 191, 25–33. doi:10.1016/S0022-510X(01)00627-X
Bendotti, C., Marino, M., Cheroni, C., Fontana, E., Crippa, V., Poletti, A., et al. (2012). Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: implication for protein aggregation and immune response. Prog. Neurobiol. 97, 101–126. doi:10.1016/j.pneurobio.2011.10.001
Berciano, M. T., Novell, M., Villagra, N. T., Casafont, I., Bengoechea, R., Val-Bernal, J. F., et al. (2007). Cajal body number and nucleolar size correlate with the cell body mass in human sensory ganglia neurons. J. Struct. Biol. 158, 410–420. doi:10.1016/j.jsb.2006.12.008
Boisvert, F. M., van Koningsbruggen, S., Navascues, J., and Lamond, A. I. (2007). The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 8, 574–585. doi:10.1038/nrm2184
Bosco, D. A., Morfini, G., Karabacak, N. M., Song, Y., Gros-Louis, F., Pasinelli, P., et al. (2010). Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403. doi:10.1038/nn.2660
Boya, P., and Kroemer, G. (2008). Lysosomal membrane permeabilization in cell death. Oncogene 27, 6434–6451. doi:10.1038/onc.2008.310
Brotherton, T. E., Li, Y., Cooper, D., Gearing, M., Julien, J. P., Rothstein, J. D., et al. (2012). Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. Proc. Natl. Acad. Sci. U.S.A. 109, 5505–5510. doi:10.1073/pnas.1115009109
Casafont, I., Navascues, J., Pena, E., Lafarga, M., and Berciano, M. T. (2006). Nuclear organization and dynamics of transcription sites in rat sensory ganglia neurons detected by incorporation of 5’-fluorouridine into nascent RNA. Neuroscience 140, 453–462. doi:10.1016/j.neuroscience.2006.02.030
Casas, C., Herrando-Grabulosa, M., Manzano, R., Mancuso, R., Osta, R., and Navarro, X. (2013). Early presymptomatic cholinergic dysfunction in a murine model of amyotrophic lateral sclerosis. Brain Behav. 3, 145–158. doi:10.1002/brb3.104
Cataldo, A. M., Barnett, J. L., Berman, S. A., Li, J., Quarless, S., Bursztajn, S., et al. (1995). Gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron 14, 671–680. doi:10.1016/0896-6273(95)90324-0
Cheah, B. C., Vucic, S., Krishnan, A. V., and Kiernan, M. C. (2010). Riluzole, neuroprotection and amyotrophic lateral sclerosis. Curr. Med. Chem. 17, 1942–1199. doi:10.2174/092986710791163939
Cheng, B., Martinez, A. A., Morado, J., Scofield, V., Roberts, J. L., and Maffi, S. K. (2013). Retinoic acid protects against proteasome inhibition associated cell death in SH-SY5Y cells via the AKT pathway. Neurochem. Int. 62, 31–42. doi:10.1016/j.neuint.2012.10.014
Chiu, A. Y., Zhai, P., Dal Canto, M. C., Peters, T. M., Kwon, Y. W., Prattis, S. M., et al. (1995). Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol. Cell Neurosci. 6, 349–362. doi:10.1006/mcne.1995.1027
Corcoran, J., So, P. L., and Maden, M. (2002). Absence of retinoids can induce motoneuron disease in the adult rat and a retinoid defect is present in motoneuron disease patients. J. Cell Sci. 115, 4735–4741. doi:10.1242/jcs.00169
Corona, J. C., Romo, L. B., and Tapia, R. (2007). Glutamate excitotoxicity and therapeutic targets for amyotrophic lateral sclerosis. Expert. Opin. Ther. Targets. 11, 1415–1428. doi:10.1517/14728222.11.11.1415)
Cramer, P. E., Cirrito, J. R., Wesson, D. W., Lee, C. Y., Karlo, J. C., Zinn, A. E., et al. (2012). ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506. doi:10.1126/science.1217697
Crochemore, C., Virgili, M., Bonamassa, B., Canistro, D., Pena-Altamira, E., Paolini, M., et al. (2009). Long-term dietary administration of valproic acid does not affect, while retinoic acid decreases, the lifespan of G93A mice, a model for amyotrophic lateral sclerosis. Muscle Nerve 39, 548–552. doi:10.1002/mus.21260
Crockett, S., Clarke, M., Reeves, S., and Sims, B. (2011). Cystine glutamate exchanger upregulation by retinoic acid induces neuroprotection in neural stem cells. Neuroreport 22, 598–602. doi:10.1097/WNR.0b013e3283494359
Delgado-Calle, J., Arozamena, J., Garcia-Renedo, R., Garcia-Ibarbia, C., Pascual-Carra, M. A., Gonzalez-Macias, J., et al. (2011). Osteocyte deficiency in hip fractures. Calcif. Tissue Int. 89, 327–334. doi:10.1007/s00223-011-9522-0
Duvic, M., Hymes, K., Heald, P., Breneman, D., Martin, A. G., Myskowski, P., et al. (2001a). Bexarotene is effective and safe for treatment of refractory advanced-stage cutaneous T-cell lymphoma: multinational phase II-III trial results. J. Clin. Oncol. 19, 2456–2471.
Duvic, M., Martin, A. G., Kim, Y., Olsen, E., Wood, G. S., Crowley, C. A., et al. (2001b). Phase 2 and 3 clinical trial of oral bexarotene (Targretin capsules) for the treatment of refractory or persistent early-stage cutaneous T-cell lymphoma. Arch. Dermatol. 137, 581–593. doi:10-1001/pubs.Arch Dermatol.-ISSN-0003-987x-137-5-dst0039
Evans, R. M., and Mangelsdorf, D. J. (2014). Nuclear receptors, RXR, and the big bang. Cell 157, 255–266. doi:10.1016/j.cell.2014.03.012
Farol, L. T., and Hymes, K. B. (2004). Bexarotene: a clinical review. Expert. Rev. Anticancer Ther. 4, 180–188. doi:10.1586/14737140.4.2.180
Ferraiuolo, L., Kirby, J., Grierson, A. J., Sendtner, M., and Shaw, P. J. (2011). Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 616–630. doi:10.1038/nrneurol.2011.152
Fischer, L. R., Culver, D. G., Tennant, P., Davis, A. A., Wang, M., Castellano-Sanchez, A., et al. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240. doi:10.1016/j.expneurol.2003.10.004
Fitz, N. F., Cronican, A. A., Lefterov, I., and Koldamova, R. (2013). Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science 340, 924c–992c. doi:10.1126/science.1235809
Ghavami, S., Shojaei, S., Yeganeh, B., Ande, S. R., Jangamreddy, J. R., Mehrpour, M., et al. (2014). Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 112, 24–49. doi:10.1016/j.pneurobio.2013.10.004
Gurney, M. E., Fleck, T. J., Himes, C. S., and Hall, E. D. (1998). Riluzole preserves motor function in a transgenic model of familial amyotrophic lateral sclerosis. Neurology 50, 62–66. doi:10.1212/WNL.50.1.62
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775. doi:10.1126/science.8209258
Haidet-Phillips, A. M., Hester, M. E., Miranda, C. J., Meyer, K., Braun, L., Frakes, A., et al. (2011). Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 29, 824–828. doi:10.1038/nbt.1957
Hall, E. D., Oostveen, J. A., and Gurney, M. E. (1998). Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia 23, 249–256. doi:10.1002/(SICI)1098-1136(199807)23:3<249::AID-GLIA7>3.0.CO;2-#
Hetman, M., and Pietrzak, M. (2012). Emerging roles of the neuronal nucleolus. Trends Neurosci. 35, 305–314. doi:10.1016/j.tins.2012.01.002
Hetz, C., and Mollereau, B. (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249. doi:10.1038/nrn3689
Jaarsma, D., Haasdijk, E. D., Grashorn, J. A., Hawkins, R., van Duijn, W., Verspaget, H. W., et al. (2000). Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol. Dis. 7, 623–643. doi:10.1006/nbdi.2000.0299
Jokic, N., Ling, Y. Y., Ward, R. E., Michael-Titus, A. T., Priestley, J. V., and Malaspina, A. (2007). Retinoid receptors in chronic degeneration of the spinal cord: observations in a rat model of amyotrophic lateral sclerosis. J. Neurochem. 103, 1821–1833. doi:10.1111/j.1471-4159.2007.04893.x
Kikuchi, H., Almer, G., Yamashita, S., Guegan, C., Nagai, M., Xu, Z., et al. (2006). Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc. Natl. Acad. Sci. U.S.A. 103, 6025–6030. doi:10.1073/pnas.0509227103
Kinderman, N. B., and Jones, K. J. (1993). Testosterone enhancement of the nerve cell body response to injury: evidence using in situ hybridization and ribosomal DNA probes. J. Neurosci. 13, 1523–1532.
Kolarcik, C. L., and Bowser, R. (2012). Retinoid signaling alterations in amyotrophic lateral sclerosis. Am. J. Neurodegener. Dis. 1, 130–145.
Korac, J., Schaeffer, V., Kovacevic, I., Clement, A. M., Jungblut, B., Behl, C., et al. (2013). Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J. Cell Sci. 126, 580–592. doi:10.1242/jcs.114926
Koval, E. D., Shaner, C., Zhang, P., du Maine, X., Fischer, K., Tay, J., et al. (2013). Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum. Mol. Genet. 22, 4127–4135. doi:10.1093/hmg/ddt261
Kunst, C. B. (2004). Complex genetics of amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75, 933–947. doi:10.1086/426001
Lamond, A. I., and Spector, D. L. (2003). Nuclear speckles: a model for nuclear organelles. Nat. Rev. Mol. Cell Biol. 4, 605–612. doi:10.1038/nrm1172
Lane, M. A., and Bailey, S. J. (2005). Role of retinoid signalling in the adult brain. Prog. Neurobiol. 75, 275–293. doi:10.1016/j.pneurobio.2005.03.002
Lemmens, R., Moore, M. J., Al Chalabi, A., Brown, R. H. Jr., and Robberecht, W. (2010). RNA metabolism and the pathogenesis of motor neuron diseases. Trends Neurosci. 33, 249–258. doi:10.1016/j.tins.2010.02.003
Maden, M. (2007). Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci. 8, 755–765. doi:10.1038/nrn2212
Majounie, E., Renton, A. E., Mok, K., Dopper, E. G., Waite, A., Rollinson, S., et al. (2012). Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330. doi:10.1016/S1474-4422(12)70043-1
Malaspina, A., Kaushik, N., and de Belleroche, J. (2001). Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. J. Neurochem. 77, 132–145. doi:10.1046/j.1471-4159.2001.00231.x
Malaspina, A., and Turkheimer, F. (2007). A review of the functional role and of the expression profile of retinoid signaling and of nuclear receptors in human spinal cord. Brain Res. Bull. 71, 437–446. doi:10.1016/j.brainresbull.2006.10.032
Mancuso, R., Del Valle, J., Modol, L., Martinez, A., Granado-Serrano, A. B., Ramirez-Nunez, O., et al. (2014). Resveratrol improves motoneuron function and extends survival in SOD1 ALS mice. Neurotherapeutics 11, 419–432. doi:10.1007/s13311-013-0253-y
Martin, L. J., Liu, Z., Chen, K., Price, A. C., Pan, Y., Swaby, J. A., et al. (2007). Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: mechanisms of mitochondriopathy and cell death. J. Comp. Neurol. 500, 20–46. doi:10.1002/cne.21160
Martinez-Vicente, M., and Cuervo, A. M. (2007). Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 6, 352–361. doi:10.1016/S1474-4422(07)70076-5
McFarland, K., Spalding, T. A., Hubbard, D., Ma, J. N., Olsson, R., and Burstein, E. S. (2013). Low dose bexarotene treatment rescues dopamine neurons and restores behavioral functions in models of Parkinson’s disease. ACS Chem. Neurosci. 4, 1430–1438. doi:10.1021/cn400100f1
Mey, J., and Rombach, N. (1999). Retinoic acid increases BDNF-dependent regeneration of chick retinal ganglion cells in vitro. Neuroreport 10, 3573–3577. doi:10.1097/00001756-199911260-00020
Miller, R. G., Mitchell, J. D., and Moore, D. H. (2012). Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database. Syst. Rev. 3, CD001447. doi:10.1002/14651858.CD001447.pub3
Moen, P. T. Jr., Johnson, C. V., Byron, M., Shopland, L. S., de la Serna, I. L., Imbalzano, A. N., et al. (2004). Repositioning of muscle-specific genes relative to the periphery of SC-35 domains during skeletal myogenesis. Mol. Biol. Cell 15, 197–206. doi:10.1091/mbc.E03-06-0388
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi:10.1038/nn1876
Obulesu, M., Dowlathabad, M. R., and Bramhachari, P. V. (2011). Carotenoids and Alzheimer’s disease: an insight into therapeutic role of retinoids in animal models. Neurochem. Int. 59, 535–541. doi:10.1016/j.neuint.2011.04.004
Palanca, A., Casafont, I., Berciano, M. T., and Lafarga, M. (2014). Reactive nucleolar and Cajal body responses to proteasome inhibition in sensory ganglion neurons. Biochim. Biophys. Acta 1842, 848–859. doi:10.1016/j.bbadis.2013.11.016
Parakh, S., Spencer, D. M., Halloran, M. A., Soo, K. Y., and Atkin, J. D. (2013). Redox regulation in amyotrophic lateral sclerosis. Oxid. Med. Cell Longev. 2013, 408681. doi:10.1155/2013/408681
Pasinelli, P., Belford, M. E., Lennon, N., Bacskai, B. J., Hyman, B. T., Trotti, D., et al. (2004). Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 43, 19–30. doi:10.1016/j.neuron.2004.06.021
Pasinelli, P., and Brown, R. H. (2006). Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 7, 710–723. doi:10.1038/nrn1971
Pena, E., Berciano, M. T., Fernandez, R., Ojeda, J. L., and Lafarga, M. (2001). Neuronal body size correlates with the number of nucleoli and Cajal bodies, and with the organization of the splicing machinery in rat trigeminal ganglion neurons. J. Comp Neurol. 430, 250–263. doi:10.1002/1096-9861(20010205)430:2<250::AID-CNE1029>3.0.CO;2-L
Philips, T., and Robberecht, W. (2011). Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 10, 253–263. doi:10.1016/S1474-4422(11)70015-1
Price, A. R., Xu, G., Siemienski, Z. B., Smithson, L. A., Borchelt, D. R., Golde, T. E., et al. (2013). Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science 340, 924d–992d. doi:10.1126/science.1234089
Rajawat, Y., Hilioti, Z., and Bossis, I. (2010). Autophagy: a target for retinoic acids. Autophagy 6, 1224–1226. doi:10.4161/auto.6.8.13793
Rajawat, Y., Hilioti, Z., and Bossis, I. (2011). Retinoic acid induces autophagosome maturation through redistribution of the cation-independent mannose-6-phosphate receptor. Antioxid. Redox. Signal. 14, 2165–2177. doi:10.1089/ars.2010.3491
Raska, I., Shaw, P. J., and Cmarko, D. (2006). New insights into nucleolar architecture and activity. Int. Rev. Cytol. 255, 177–235. doi:10.1016/S0074-7696(06)55004-1
Reichenbach, A., Derouiche, A., and Kirchhoff, F. (2010). Morphology and dynamics of perisynaptic glia. Brain Res. Rev. 63, 11–25. doi:10.1016/j.brainresrev.2010.02.003
Riancho, J., Ruiz-Soto, M., Villagrá, N. T., Berciano, J., Berciano, M. T., and Lafarga, M. (2014). Compensatory motor neuron response to chromatolysis in the murine hSOD1 G93A model of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 22:346. doi:10.3389/fncel.2014.00346
Robberecht, W., and Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264. doi:10.1038/nrn3430
Ross, C. A., and Poirier, M. A. (2004). Protein aggregation and neurodegenerative disease. Nat. Med. 10(Suppl.), S10–S17. doi:10.1038/nm1066
Rothstein, J. D. (2009). Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol. 65(Suppl. 1), S3–S9. doi:10.1002/ana.21543
Sato, Y., Meller, R., Yang, T., Taki, W., and Simon, R. P. (2008). Stereo-selective neuroprotection against stroke with vitamin A derivatives. Brain Res. 1241, 188–192. doi:10.1016/j.brainres.2008.09.020
Sau, D., De Biasi, S., Vitellaro-Zuccarello, L., Riso, P., Guarnieri, S., Porrini, M., et al. (2007). Mutation of SOD1 in ALS: a gain of a loss of function. Hum. Mol. Genet. 16, 1604–1618. doi:10.1093/hmg/ddm110
Saxena, S., Cabuy, E., and Caroni, P. (2009). A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636. doi:10.1038/nn.2297
Schmidt, E. R., Pasterkamp, R. J., and van den Berg, L. H. (2009). Axon guidance proteins: novel therapeutic targets for ALS? Prog. Neurobiol. 88, 286–301. doi:10.1016/j.pneurobio.2009.05.004
Schrage, K., Koopmans, G., Joosten, E. A., and Mey, J. (2006). Macrophages and neurons are targets of retinoic acid signaling after spinal cord contusion injury. Eur. J. Neurosci. 23, 285–295. doi:10.1111/j.1460-9568.2005.04534.x
Shimada, J., Taniguchi, J., Mori, M., Sato, Y., Takuwa, H., Ito, H., et al. (2013). Retinol palmitate prevents ischemia-induced cell changes in hippocampal neurons through the Notch1 signaling pathway in mice. Exp. Neurol. 247, 182–187. doi:10.1016/j.expneurol.2013.04.014
Sunico, C. R., Dominguez, G., Garcia-Verdugo, J. M., Osta, R., Montero, F., and Moreno-Lopez, B. (2011). Reduction in the motoneuron inhibitory/excitatory synaptic ratio in an early-symptomatic mouse model of amyotrophic lateral sclerosis. Brain Pathol. 21, 1–15. doi:10.1111/j.1750-3639.2010.00417.x
Tan, C. C., Yu, J. T., Tan, M. S., Jiang, T., Zhu, X. C., and Tan, L. (2014). Autophagy in aging and neurodegenerative diseases: implications for pathogenesis and therapy. Neurobiol. Aging 35, 941–957. doi:10.1016/j.neurobiolaging.2013.11.019
Tang, Q., Chen, Y., Meyer, C., Geistlinger, T., Lupien, M., Wang, Q., et al. (2011). A comprehensive view of nuclear receptor cancer cistromes. Cancer Res. 71, 6940–6947. doi:10.1158/0008-5472.CAN-11-2091
Van Den, B. L., Van Damme, P., Bogaert, E., and Robberecht, W. (2006). The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 1068–1082. doi:10.1016/j.bbadis.2006.05.002
Vargas, M. R., Burton, N. C., Gan, L., Johnson, D. A., Schafer, M., Werner, S., et al. (2013). Absence of Nrf2 or its selective overexpression in neurons and muscle does not affect survival in ALS-linked mutant hSOD1 mouse models. PLoS One 8:e56625. doi:10.1371/journal.pone.0056625
Veeraraghavalu, K., Zhang, C., Miller, S., Hefendehl, J. K., Rajapaksha, T. W., Ulrich, J., et al. (2013). Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science 340, 924f–992f. doi:10.1126/science.1235505
Vinsant, S., Mansfield, C., Jimenez-Moreno, R., Del, G. M., Yoshikawa, V. M., Hampton, T. G., et al. (2013a). Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part I, background and methods. Brain Behav. 3, 335–350. doi:10.1002/brb3.143
Vinsant, S., Mansfield, C., Jimenez-Moreno, R., Del, G. M., Yoshikawa, V. M., Hampton, T. G., et al. (2013b). Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part II, results and discussion. Brain Behav. 3, 431–457. doi:10.1002/brb3.142
Wakayama, I. (1992). Morphometry of spinal motor neurons in amyotrophic lateral sclerosis with special reference to chromatolysis and intracytoplasmic inclusion bodies. Brain Res. 586, 12–18. doi:10.1016/0006-8993(92)91365-L
Wohl, C. A., and Weiss, S. (1998). Retinoic acid enhances neuronal proliferation and astroglial differentiation in cultures of CNS stem cell-derived precursors. J. Neurobiol. 37, 281–290. doi:10.1002/(SICI)1097-4695(19981105)37:2<281::AID-NEU7>3.0.CO;2-J
Wootz, H., Weber, E., Korhonen, L., and Lindholm, D. (2006). Altered distribution and levels of cathepsinD and cystatins in amyotrophic lateral sclerosis transgenic mice: possible roles in motor neuron survival. Neuroscience 143, 419–430. doi:10.1016/j.neuroscience.2006.07.048
Xing, Y., Johnson, C. V., Dobner, P. R., and Lawrence, J. B. (1993). Higher level organization of individual gene transcription and RNA splicing. Science 259, 1326–1330. doi:10.1126/science.8446901
Yamanaka, K., Boillee, S., Roberts, E. A., Garcia, M. L., McAlonis-Downes, M., Mikse, O. R., et al. (2008). Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl. Acad. Sci. U.S.A. 105, 7594–7599. doi:10.1073/pnas.0802556105
Keywords: amyotrophic lateral sclerosis, bexarotene, G93ASOD1, mouse, proteostasis, retinoid-X receptor
Citation: Riancho J, Ruiz-Soto M, Berciano MT, Berciano J and Lafarga M (2015) Neuroprotective effect of bexarotene in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 9:250. doi: 10.3389/fncel.2015.00250
Received: 21 April 2015; Accepted: 18 June 2015;
Published: 01 July 2015
Edited by:
Pier Giorgio Mastroberardino, Erasmus MC University Medical Center Rotterdam, NetherlandsReviewed by:
Gunnar P. H. Dietz, Schwabe Pharma Deutschland, GermanyKhursheed Wani, University of Michigan, USA
Copyright: © 2015 Riancho, Ruiz-Soto, Berciano, Berciano and Lafarga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel Lafarga, Department of Anatomy and Cell Biology, University of Cantabria, Av Cardenal Herrera Oria s/n 39008, Santander 39011, Spain, lafargam@unican.es