Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: implications for neuronal development and trafficking
 Carlos Wilson
Carlos Wilson Christian González-Billault
Christian González-Billault- Department of Biology, Faculty of Sciences, Universidad de Chile, Santiago, Chile
A proper balance between chemical reduction and oxidation (known as redox balance) is essential for normal cellular physiology. Deregulation in the production of oxidative species leads to DNA damage, lipid peroxidation and aberrant post-translational modification of proteins, which in most cases induces injury, cell death and disease. However, physiological concentrations of oxidative species are necessary to support important cell functions, such as chemotaxis, hormone synthesis, immune response, cytoskeletal remodeling, Ca2+ homeostasis and others. Recent evidence suggests that redox balance regulates actin and microtubule dynamics in both physiological and pathological contexts. Microtubules and actin microfilaments contain certain amino acid residues that are susceptible to oxidation, which reduces the ability of microtubules to polymerize and causes severing of actin microfilaments in neuronal and non-neuronal cells. In contrast, inhibited production of reactive oxygen species (ROS; e.g., due to NOXs) leads to aberrant actin polymerization, decreases neurite outgrowth and affects the normal development and polarization of neurons. In this review, we summarize emerging evidence suggesting that both general and specific enzymatic sources of redox species exert diverse effects on cytoskeletal dynamics. Considering the intimate relationship between cytoskeletal dynamics and trafficking, we also discuss the potential effects of redox balance on intracellular transport via regulation of the components of the microtubule and actin cytoskeleton as well as cytoskeleton-associated proteins, which may directly impact localization of proteins and vesicles across the soma, dendrites and axon of neurons.
The Nervous System as a Target for Oxidative Species
The chemical reduction-oxidation (redox) balance commands physiological and pathological responses at different levels ranging from cells to tissues to biological systems. Among organs, the brain is especially vulnerable to oxidation for three main reasons. First, the brain consumes high levels of O2—up to 20% of the amount used by the entire body (Sparaco et al., 2006). Given that the brain represents only 2% of the total body mass, metabolites derived from O2 in the brain are highly concentrated in a restricted space, increasing the risk of oxidation. Second, Fe2+ is abundant in many specific areas of the brain (Gerlach et al., 1994), contributing to non-reversible oxidation. Fe2+, the redox-active form of iron, catalyzes the conversion of hydrogen peroxide (H2O2) into the hydroxyl radical (HO⊕) through the Fenton reaction. The hydroxyl radical represents the most chemically reactive of all reactive oxygen species (ROS; Núñez et al., 2012). Finally, the brain lacks effective mechanisms to remove accumulated pro-oxidative molecules (Halliwell, 1992). Together, these factors necessitate that the brain must utilize effective biochemical measures to counter oxidative stress.
Increased oxidation of molecules in both central and peripheral neurons is often associated with aging, oxidative stress and disease (Andersen, 2004). The term oxidative stress was coined to reflect an imbalance between oxidative and reductive molecules that leads to increased accumulation of pro-oxidant species, with deleterious consequences in most cases (Sies, 1997). This is the case for several neuronal pathologies, including Parkinson’s disease, Alzheimer’s disease, Huntington’s disease and amyotrophic lateral sclerosis (Andersen, 2004). Most sporadic versions of these pathologies are linked to aging and have a documented positive correlation between oxidative stress and development of pathology.
Although oxidation of intracellular components owing to increased oxidative stress is a natural consequence of long-term exposure to pro-oxidant conditions, it is important to remember that physiological synthesis of oxidative species is required for normal cellular function (Rhee, 2006; Dáux and Toledano, 2007). The innate immune response depends on proper synthesis of ROS produced by NADPH oxidases (NOXs). Particularly, phagocytosis, chemotaxis and cellular locomotion of immune cells require basal ROS synthesis (Lambeth, 2004). Other processes requiring physiological concentrations of ROS include thyroid hormone synthesis, Ca2+ homeostasis, ion channel dynamics and cytoskeletal remodeling (Bedard and Krause, 2007; Hidalgo and Nunez, 2007; Espinosa et al., 2009; Sakai et al., 2012; Contreras-Ferrat et al., 2014).
In the central nervous system (CNS), enzymatic production of physiological levels of ROS contributes to synaptic plasticity and memory consolidation (Knapp and Klann, 2002; Massaad and Klann, 2011). Genetic models in which gp91phox and p47phox proteins are inactivated (the catalytic and one of the regulatory subunits of the NOX complex, respectively) exhibit abnormal long-term potentiation responses after electrical stimulation, which is an ex vivo paradigm to evaluate synaptic plasticity in hippocampal neurons (Kishida et al., 2006). gp91phox and p47phox knockout mice also have decreased consolidation of spatial memory, suggesting a neuronal disorder owing to impaired NOX activity and ROS signaling (Kishida et al., 2006). Humans with chronic granulomatous disease (CGD), an inherited syndrome caused by point mutations in the NOXs proteins gp91phox, p47phox, p67phox and p22phox, affecting the immune response to pathogens, develop cognitive impairments and reduced intellectual coefficients compared with healthy control individuals (Pao et al., 2004). CGD develops during childhood and often requires repeated long-term hospitalization throughout life. It has been proposed that the intellectual deficits in CGD children could be linked to irregular school attendance and that this could even be the main cause for their low intellectual coefficients. However, other infectious diseases with similar periods of hospitalization do not result in cognitive deficits, suggesting that CGD patients develop specific neuronal alterations mainly attributable to reduced NOX activity and decreased ROS synthesis (Pao et al., 2004).
Taken together, these observations suggest that basal physiological ROS synthesis is required for normal cellular function, including the regulation of neurotransmission, but that high and unregulated ROS concentrations lead to oxidative stress and disease.
General Overview of Redox Balance
Intracellular Sources of ROS
The main oxidative species derived from O2 are H2O2, HO⊕ and the superoxide anion (O2⊕–) and are collectively called ROS (Bedard and Krause, 2007). Nitrogen (N2), the principal gas in the atmosphere we breathe, also induces intracellular oxidation via the production of physiologically reactive nitrogen species (Weidinger and Kozlov, 2015).
The main sources of intracellular ROS are mitochondria and NOXs. In mitochondria, complexes I and III of the electron transfer chain produce the short-lived O2⊕–, a radical derived from O2 (Murphy, 2009; Bigarella et al., 2014). No intracellular signaling pathway that regulates mitochondrial superoxide synthesis has yet been described, suggesting that mitochondria could be a source of constitutive ROS production. Synthesis and release of ROS from mitochondria depend on the tissue and its intrinsic metabolism. Mitochondrial dysfunction quickly leads to oxidative stress that targets DNA, membrane lipids and proteins, directly affecting cell physiology (Tahara et al., 2009).
NOXs represent the other major cellular source of ROS (Bedard and Krause, 2007). The NOX family includes seven members that catalyze the production of O2⊕– in an NADPH-dependent reaction. The family is composed of five canonical NOXs (NOX1 to NOX5) and two dual oxidases (Duox1 and Duox2; Lambeth et al., 2007). NOXs represent the main enzymatic source of ROS, and several signal transduction pathways are involved in their regulation (Dang et al., 2001; Park et al., 2001; Chen et al., 2003; Hoyal et al., 2003). NOX1, NOX2 and NOX4 are expressed in the CNS (Sorce and Krause, 2009), with NOX2 being the principal enzyme expressed in neurons. NOX2 can produce superoxide by itself but requires interaction with regulatory proteins for stabilization and to increase ROS levels under physiological circumstances. Together with its partners p22phox, p47phox, p67phox and p40phox, NOX2 synthesizes superoxide to meet the physiological requirements of neurons (Bokoch and Diebold, 2002; Glogauer et al., 2003; Nauseef, 2004; Decoursey and Ligeti, 2005).
ROS as Signaling Molecules
Superoxide reactivity is fairly low, mainly owing to its short life-time and restricted diffusion area (Weidinger and Kozlov, 2015). However, superoxide can be converted to H2O2 either spontaneously or enzymatically via superoxide dismutase (Núñez et al., 2012). H2O2, the most stable ROS, is converted to H2O by several antioxidant enzymes, e.g., glutathione peroxidase and catalase, and this is likely the reason why oxidative modifications induced by H2O2 are transient and reversible (Weidinger and Kozlov, 2015). Thus, under normal conditions, the synthesis of superoxide and H2O2 are enzymatically regulated and their levels remain under a physiological threshold. In the presence of Fe2+, however, H2O2 is rapidly converted to HO⊕ through the Fenton reaction (Núñez et al., 2012). Hydroxyl radicals modify molecules in a non-reversible way, leading to permanent modifications of proteins and other targets. To consider ROS as signaling molecules, they should meet certain spatial and regulatory criteria, namely they should be produced locally and their levels regulated by intracellular molecular systems. According to these criteria, superoxide and H2O2, but not HO⊕, are considered signaling molecules (Dáux and Toledano, 2007; Janssen-Heininger et al., 2008; Gerich et al., 2009).
Regulation of Protein Function by Oxidation via Post-Translational Modification
Cysteine thiol groups (SH) of proteins are the main targets for oxidation (Stadtman and Berlett, 1997). In general terms, oxidation of SH groups leads to glutathionylation, nitrosylation and disulfide bond formation. These modifications are enzymatically reversed through the glutaredoxin (Grx), thioredoxin and peroxiredoxin systems, among others (Ghezzi, 2005; Shelton et al., 2005; Janssen-Heininger et al., 2008). Oxidation also leads to sulfenic acid formation and protein carbonylation, two non-reversible modifications that permanently affect protein structure and function (Bigarella et al., 2014).
The functions of several proteins, including cytoskeletal proteins, depend on ROS signaling and oxidation (Sparaco et al., 2006). Several studies have shown that redox balance affects both in vitro and in vivo cytoskeletal dynamics, which directly impacts cell morphology and morphometrics. In neurons, cytoskeletal rearrangement commands cell development, polarization and neurotransmission (Jaworski et al., 2009; Hoogenraad and Akhmanova, 2010; Stiess and Bradke, 2011; Caceres et al., 2012; Gonzalez-Billault et al., 2012). Neurons are highly polarized cells, having a cell body from which emerge several dendrites and an axon to establish functional communication networks with other neurons and glial cells (Caceres et al., 2012). Acquisition of this morphology depends directly on the dynamics of actin microfilaments and microtubules (Neukirchen and Bradke, 2011). Given the influence of the redox state on neuronal function and its potential role in modifying the cytoskeleton, it is interesting to review the contribution of the redox balance to cytoskeletal organization in neurons.
Contribution of Redox Balance to Organization of the Neuronal Cytoskeleton
Redox State of Neuronal Cytoskeleton Proteins
The main components of the neuronal cytoskeleton network, namely actin microfilaments, microtubules and neurofilaments, are susceptible to oxidation (Sparaco et al., 2006). Post-mortem histological studies from non-pathological human samples have revealed a basal pool of glutathionylated proteins in the prefrontal cortex, cerebellum and spinal cord. Cellular analysis of the prefrontal cortex revealed that neurons are more highly glutathionylated than oligodendrocytes and astrocytes. Biochemical analysis revealed that actin, tubulin and neurofilaments are glutathionylated, suggesting that the redox state of neurons and cytoskeletal proteins under basal conditions is slanted toward oxidation (Sparaco et al., 2006). The relevance of cytoskeleton oxidation depends on the spatiotemporal context in which a defined modification occurs as well as the source of the ROS. Whereas physiological ROS production is needed for proper cytoskeleton polymerization, oxidation tends to disrupt polymerization and impair cytoskeletal dynamics under oxidative stress conditions (Munnamalai and Suter, 2009; Hung et al., 2010; Morinaka et al., 2011; Wilson et al., 2015).
Actin Modification and Regulation of F-Actin Dynamics by Oxidative Species
Intracellular ROS production is needed for proper cell migration and chemotaxis, which are actin-dependent processes (Roberts et al., 1999; Ambruso et al., 2000; Kim and Dinauer, 2001). Actin monomers contain 5 Cys and 16 Met residues (Dalle-Donne et al., 2002, 2003). Of these, only Cys 374 is fully exposed to the cytoplasm (Dalle-Donne et al., 2003). By contrast, Met 44, 47 and 355 are prominently exposed to the cytoplasm and Met 176, 190, 227 and 260 are also susceptible to the action of oxidative molecules (Dalle-Donne et al., 2002). In vitro assays suggested that an oxidative environment inhibits actin polymerization (Dalle-Donne et al., 2003). In addition, both Cys and Met residues can be carbonylated after in vitro treatment with hypochlorous acid (HOCl), a common derivative product released by leukocytes during the initial phase of the immune response. This polymerization assay was designed to recapitulate in vitro the exact oxidative environment of the immune cell response; importantly, this environment inhibits actin polymerization (Dalle-Donne et al., 2001). This analysis also revealed that specific glutathionylation at Cys 374 decreases actin polymerization (Dalle-Donne et al., 2003). In neutrophils, glutaredoxin 1 (Grx1), a deglutathionylating enzyme that reduces oxidized Cys residues, is needed to maintain actin dynamics (Sakai et al., 2012). ROS depletion using the NOX inhibitor diphenyleneiodonium or Grx1 overexpression decreases actin glutathionylation, increases the amount of filamentous actin (F-actin) and impairs proper cellular migration. In contrast, loss of Grx1 function via knockdown and knockout strategies increases the amount of glutathionylated actin and decreases the level of F-actin. Sakai et al. (2012) proposed that physiological levels of ROS and redox balance regulate actin dynamics, which are required for chemotaxis and migration of immune cells. On the other hand, physiological ROS levels induce local membrane protrusions in marsupial kidney epithelium Ptk1 cells in a mechanism that involves cofilin, the actin-related protein 2/3 complex (Arp2/3) and the extracellular signal–regulated kinase (ERK), thereby enhancing the retrograde flow of actin at the leading edge of these migrating cells (Taulet et al., 2012). Figure 1 summarizes the effect of the oxidative power on actin polymerization and F-actin dynamics.
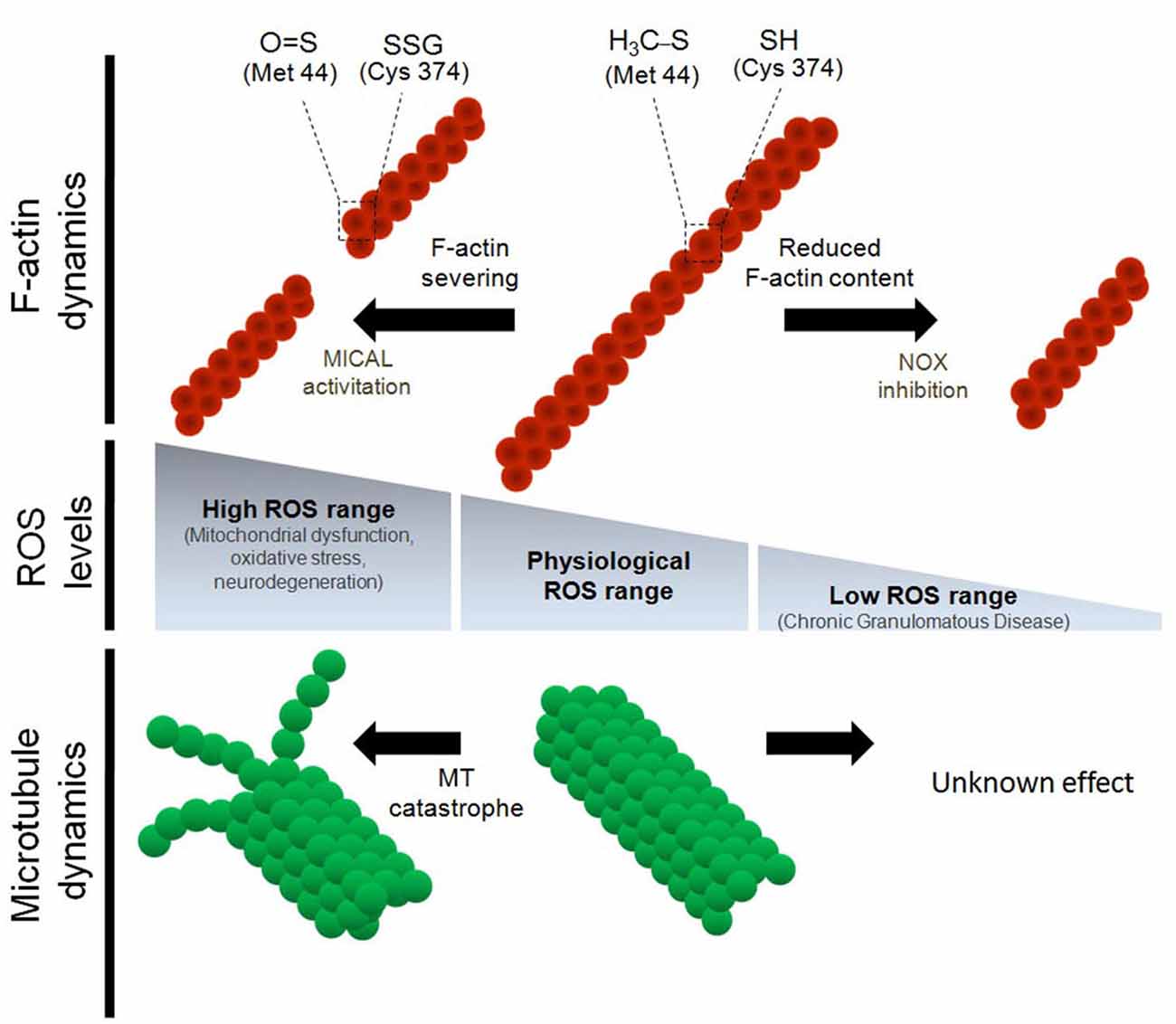
Figure 1. Regulation of the cytoskeleton dynamics by differential reactive oxygen species (ROS) levels. Physiological concentrations of ROS are needed for proper F-actin and microtubule dynamics both in vitro and in vivo. Oxidative stress induces the severing of F-actin and impairs microtubule polymerization. In the particular case of actin monomers, both Cys 374 and Met 42 residues are targets for oxidative molecules during oxidative stress conditions. Oxidation of Cys 374 leads to actin glutathionylation. On the other hand, oxidation of Met 44, which occurs specifically by MICAL1, leads to Met oxidation and severing of the actin filaments (Hung et al., 2011). Despite both α and β tubulin have several Cys residues that are susceptible to oxidation, it has not been described which Cys residues of tubulin monomers are modified by oxidation. In contrast, down-regulation of ROS synthesis, mainly through NOX inhibition, decreases the F-actin content and alters the dynamic properties of the actin cytoskeleton. None of the post-translational modification of actin monomers have been described after NOX inhibition and ROS down-regulation. Finally, the effect of the inhibition of ROS synthesis (below physiological levels) on microtubule dynamics has not been explored.
ROS also target the actin cytoskeleton in neuronal cells. The Semaphorin/Plexin signaling pathway represents a major repulsive cue for axonal guidance (Hung and Terman, 2011). Semaphorin 3A (Sema3A) signaling induces a local increase in H2O2 at the growth cones of dorsal root ganglion neurons by activating MICAL1 and MICAL3 (Morinaka et al., 2011). MICAL1 is a binding partner for the cytosolic domain of Plexin A, a Sema3A receptor (Terman et al., 2002). The principal target of MICAL1 is actin (Hung et al., 2010, 2011; Giridharan and Caplan, 2014). In bristle cells of Drosophila melanogaster, oxidation of actin at Met 44 by MICAL1 severs actin filaments (Hung et al., 2011). MICAL1-dependent actin oxidation is reversed by Selenoprotein R (SelR) activation, restoring F-actin dynamics and polymerization (Hung et al., 2013). After neuronal injury, Sema3A levels rise above normal levels to inhibit the ability of axons to regenerate and regrow. These findings represent a redox mechanism by which Sema3A induces the collapse of axonal growth cones and impedes axon guidance, supporting the hypothesis that redox imbalance—particularly oxidative stress—leads to neuronal damage. Therefore, MICAL1 contributes to axonal pathfinding by regulating ROS signaling at dorsal root ganglion growth cones. In addition to directly regulating the actin redox balance, MICAL1 may modify actin dynamics by promoting interaction between proteins. MICAL1 interacts with Cas and CasL proteins, which may be involved in cross-talk between actin and intermediate filaments (Suzuki et al., 2002). Moreover, MICAL1 negatively regulates the nuclear dbf2-related kinase NDR2 in non-neuronal cells (Zhou et al., 2011). NDR1 and NDR2 contribute to targeting the Par3/Par6/aPKC complex to growing axons, a mechanism that promotes neuronal polarity (Yang et al., 2014). Together, these findings suggest that MICAL1 both directly and indirectly regulates the neuronal actin cytoskeleton, contributing to neuronal function and development under both normal and stress conditions.
In the marine mollusk Aplysia, NOX-derived ROS are required to maintain proper F-actin dynamics in the growth cones of bag cells (Munnamalai and Suter, 2009; Munnamalai et al., 2014). Inhibition of NOX activity using pharmacological inhibitors like apocynin, diphenyleneiodonium and VAS2870 reduce F-actin content in these growth cones and reduce both retrograde actin flow and neurite outgrowth, supporting the idea that actin dynamics and neurite elongation require basal NOX activity (Munnamalai and Suter, 2009). Cultured embryonic hippocampal neurons that express the mutant P156Q p22phox, which down-regulates ROS synthesis by the NOX complex, show a decrease in the number, length and lifetime of filopodia at axonal growth cones (Wilson et al., 2015). Moreover, lamellar actin organization of stage 1 neurons, the initial morphology from which neurons develop, is disrupted after loss of NOX function (Wilson et al., 2015). Together, this evidence supports the hypothesis that local ROS signaling is needed to maintain normal F-actin dynamics in neurons and is consistent with other reports proposing that ROS are needed to support membrane protrusions, lamellar structures and filopodia at the leading edge of migrating cells (Taulet et al., 2012). Interestingly, NOX2 and p40phoxco-distribute with F-actin at the growth cone of neuronal bag cells in Aplysia, suggesting that local ROS production may be involved in neurite outgrowth (Munnamalai et al., 2014). NOXs are expressed in axons and dendrites of embryonic and adult neurons, suggesting that local ROS synthesis that may be involved in filopodial dynamics and neurite growth (Tejada-Simon et al., 2005; Wilson et al., 2015). Figure 2 summarizes the differential effect of ROS on the organization of the growth cones of axons.
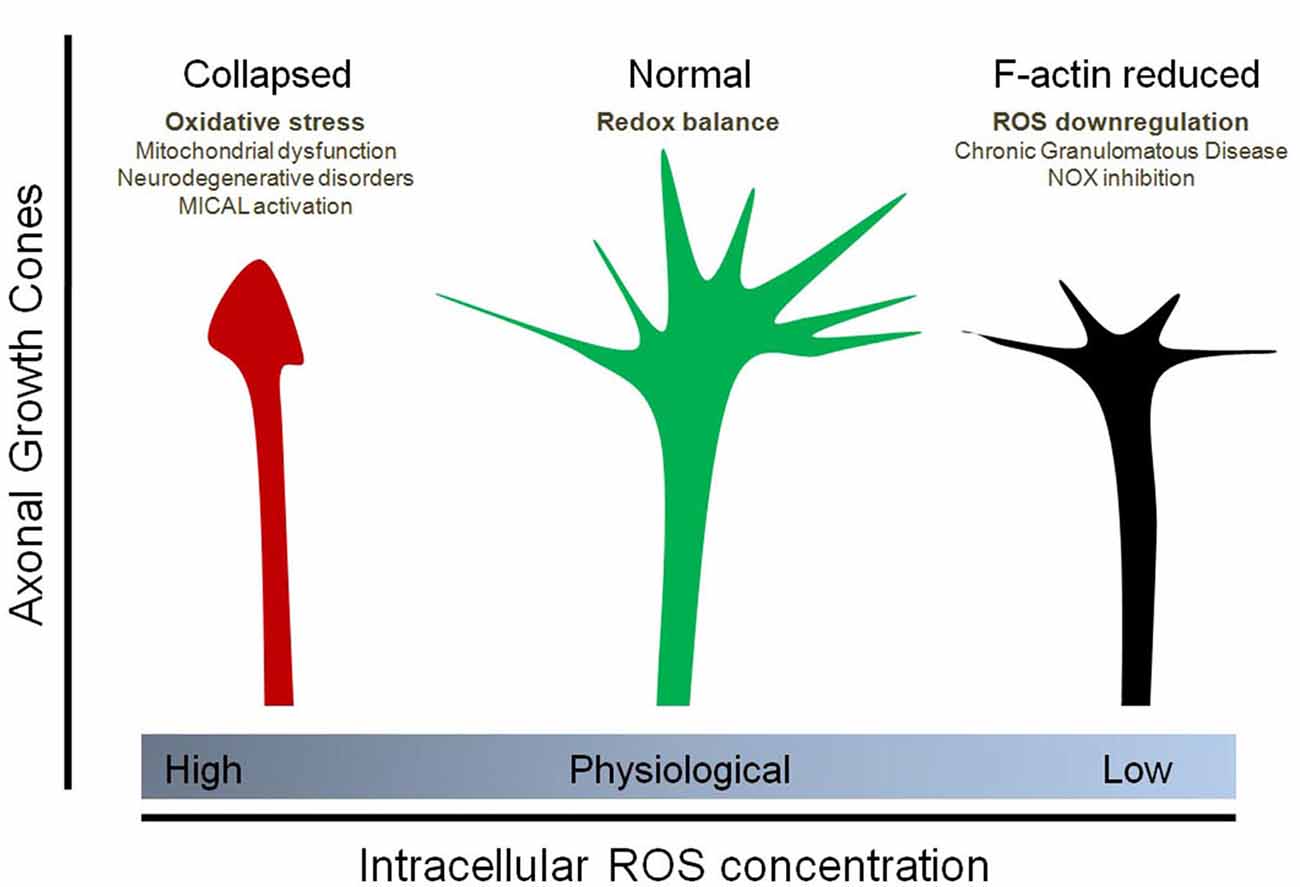
Figure 2. Effect of physiological and non-physiological ROS on axonal growth cone organization. Oxidative stress (due to mitochondrial dysfunction, Sema3A/Plexin signaling and MICAL1 activation, neurodegenerative disorders, among others) induces the collapse of the axonal growth cones of neurons. In contrast, down-regulation of ROS synthesis, particularly due to NOX inhibition as well as Chronic Granulomatous Disease (CGD), decreases the F-actin content at the growth cone and affect filopodial dynamics by decreasing the number and length of filopodia at the axonal growth cone. Physiological concentrations of ROS, principally maintained by NOX enzymes and basal mitochondrial activity, are needed for a proper organization of the growth cones. In this context, the implicances for vesicular trafficking due to altered cytoskeleton dynamics has not been explored.
Tubulin Modification and Regulation of Microtubule Dynamics by Oxidative Species
α-Tubulin and β-tubulin contain 12 and 8 Cys residues, respectively, and each of these residues can be oxidized by endogenous and exogenous oxidizing agents (Luduena and Roach, 1991; Löwe et al., 2001; Landino et al., 2002, 2004a). The functions of these Cys residues are linked to GTP binding, microtubule polymerization and drug response (Mellon and Rebhun, 1976; Luduena et al., 1985; Luduena and Roach, 1991). In in vitro polymerization assays using purified tubulin from adult bovine brain, oxidative species added to the reaction medium dramatically reduced tubulin polymerization. Peroxynitrite (ONOO–), a ROS produced from the reaction between superoxide and nitric oxide (NO⊕), progressively oxidizes the thiol groups of tubulin monomers, thereby decreasing the ability of microtubules to polymerize in vitro (Landino et al., 2007). The same results were obtained with NO⊕ and nitroxyl donors. Moreover, ONOO– promotes disulfide bond formation between α- and β-tubulin (Landino et al., 2004a). In addition, in vitro assays revealed that tubulin is glutathionylated after treatment with ONOO–, and that this modification is reversed by the glutathione/glutathione reductase system, composed of glutathione, glutathione reductase, Grx and NADPH (Landino et al., 2004a). The reversal of tubulin glutathionylation by this system is interesting because intracellular signaling pathways may modulate microtubule polymerization in a reversible manner. Figure 1 summarizes the effect of high oxidative power on microtubule dynamics. However, the inhibition of ROS synthesis below a physiological range has not been explored in terms of tubulin modifications nor microtubule dynamics.
Another layer of regulation is provided by proteins that stabilize or destabilize microtubules. Microtubule-associated protein 2 (MAP2) and tau are MAPS that specifically regulate MT polymerization in dendrites and axon. MAP2 and tau contain one and seven Cys residues, respectively (Lewis et al., 1988). Oxidation of MAP2 and tau Cys residues decreases microtubule polymerization in vitro, suggesting that redox balance regulates tubulin not only through direct interaction but also by regulating their stabilization by MAPs (Landino et al., 2004b). It is plausible that oxidized/reduced MAPs present differential microtubule stabilization. Moreover, binding of MAPs to microtubules may promote differential regulation of molecular motors in axons and dendrites (Dixit et al., 2008), affecting trafficking and cargo destination. Therefore, redox-dependent MAP modifications may be an additional mechanism for regulating cytoskeletal dynamics in neurons. Indeed, increased nitrosylation of MAP1B at Cys 2457 is involved in neurite retraction through a mechanism that couples microtubule stability and dynein function (Stroissnigg et al., 2007; Villarroel-Campos and Gonzalez-Billault, 2014).
Microtubule function depends on its intrinsic polymerization properties (Mitchison and Kirschner, 1984, 1988) as well as the specific tubulin isotype (Kavallaris, 2010) and post-translational modifications (Janke, 2014). Microtubule proteins can be modified by redox state, but understanding the functional consequences of such modifications can be challenging. For example, tubulin modifications induced by ONOO– treatment in vitro can be difficult to interpret because ONOO–is unstable at physiological pH, and thus in vitro microtubule polymerization assays are performed at basic pH (typically pH 10). In addition, tubulin is glutathionylated in both cell-specific and tissue-specific ways (Sparaco et al., 2006, 2009). Prefrontal cortex, cerebellum and spinal cord tissue samples from non-pathological humans exhibit tubulin glutathionylation under basal conditions. In addition, after treatment with oxidized glutathione, neurons are preferentially glutathionylated compared with astrocytes and oligodendrocytes (Sparaco et al., 2006). Thus, it seems that tubulin Cys residues can be modified by redox balance in both in vitro and in vivo contexts.
Neurite outgrowth is affected by tubulin oxidation in cellular models. In the motor neuron-derived neuroblastoma cell line NSC34, oxidation induced by oxidized glutathione promote the formation of retraction bulbs and thin axon-like processes (Carletti et al., 2011). Under these conditions, glutathionylated tubulin levels are increased and interestingly, tyrosination of α-tubulin is simultaneously decreased. These findings suggest that an oxidative cytoplasmic environment induces tubulin glutathionylation, leading to neurite retraction and degeneration. Moreover, in Friedrich’s ataxia, a neuropathological condition characterized by degeneration of spinal cord pathways, immunohistochemical studies in motor neurons revealed co-distribution of tyrosinated tubulin and glutathionylated proteins (Sparaco et al., 2009). In the future, it will be necessary to establish whether there is a causal relationship between tubulin glutathionylation and changes in microtubule dynamics.
Studies in cultured primary neurons exploring phenotypes and mechanisms underlying the regulation of microtubule dynamics by redox state are still preliminary. However, there is some indirect evidence suggesting a putative link between microtubule polymerization and ROS balance. CRMP-2, a molecular regulator of microtubule polymerization, is oxidized at Cys 504, inducing homodimerization. These dimers can form a transient complex with thioredoxin, which creates a docking site for the protein kinase GSK3-β. GSK3-β-dependent CRMP-2 phosphorylation is linked to growth cone collapse in cultured dorsal root ganglion cells, recapitulating some molecular pathways involved in the initial steps of neurodegeneration (Morinaka et al., 2011). Therefore, new research may help establish a direct link between regulation of microtubule dynamics and redox balance in neuronal systems.
Participation of Oxidative Species in the Central Nervous System and Neuronal Development
Genetic models that reduce ROS production in the nervous system, such as gp91phox and p47phox knockout mice, are characterized by a macroscopically normal brain overall, including the cerebral cortex and hippocampus (Kishida et al., 2006). However, they display cognitive impairments as well as impaired synaptic plasticity, a phenomenon that involves, among other molecular events, cytoskeletal remodeling (Kishida et al., 2006). Therefore, a complete understanding of the contribution of ROS to normal nervous system physiology is important. Some insights have emerged in the last few years. For example, ROS levels have been shown to contribute to the commitment of neuronal progenitors to differentiate into mature neurons (Forsberg et al., 2013; Quadrato and Di Giovanni, 2013; Forsberg and Di Giovanni, 2014). Along the same line, loss of NOX function (using either neurons from NOX2 knockout mice or NOX2 knockdown in cultured neurons) has been shown to decrease the proliferation rate and number of neural stem cells in vivo and in vitro, suggesting that physiological ROS levels derived from NOX2 are needed to maintain a basal population of adult hippocampal neuronal progenitors (Dickinson et al., 2011). It was recently observed that inhibition of NOX functions alter normal neuronal polarization and reduces axonal growth (Wilson et al., 2015). Similarly, differentiation of cerebellar granule neurons involves glutathione homeostasis and NOX activity (Olguín-Albuerne and Morán, 2015). Of note, MICAL1 knockout mice exhibit abnormal mossy fiber lamination, aberrant F-actin content and decreased Rab6 trafficking to the growth cones of hippocampal neurons, suggesting a role in the development of mossy fiber axons and of specific sub-areas of the hippocampus and supporting the notion that redox balance is needed for development of brain tissues (Van Battum et al., 2014). Together, these lines of evidence support a new hypothesis in the field of ROS, that the physiological and controlled production of ROS is needed for signaling and development in neurons. In the future, it will be important to address in vivo functions for ROS from various sources and their involvement in neuronal differentiation, migration and axonal guidance.
A major challenge in this field is to understand the specificity that redox imbalance have on cytoskeleton proteins compared to DNA or lipids. This is especially important considering that modifications on these molecules could also affect neuronal functions and morphology. In fact, ROS contributes to the transcription of several genes associated with metabolism, cell cycle and development (Bigarella et al., 2014), supporting the notion that ROS have pleiotropic effects on sub-cellular components. Moreover, there are technical concerns about the quantification of ROS in cells. Both ROS and cytoskeleton proteins are dynamic cellular elements with short life-times. Thus, it is hard to establish a correlation between local synthesis of ROS and the oxidation of actin filaments and microtubules in a living cell. New genetic tools based on fluorescence microscopy have emerged in the last years to measure ROS levels (Mishin et al., 2015). Genetically encoded probes to measure ROS combined with cytoskeleton biosensors will be necessary to define the spatial and temporal association between ROS synthesis and cytoskeleton remodeling.
Emerging Concepts in the Contribution of Redox Balance to Vesicle Trafficking
Intracellular trafficking is highly dependent on actin microfilaments, microtubules and molecular motors such as myosin, kinesin and dynein. In addition, members of the Rab family of small GTPases are essential for targeting components to discrete domains within cells. Several Rab proteins derived from the trans-Golgi network, the early/late endosome and recycling endosomes regulate neurite outgrowth and development (Villarroel-Campos et al., 2014). In preceding sections, we discussed how changes in ROS content may target tubulin and actin dynamics to regulate the tracks for intracellular trafficking. However, the link between redox balance and the vesicle components involved in neuronal trafficking remains poorly understood.
Several studies correlate MICAL activity with trafficking. It has been recovered as an interacting partner for several members of the Rab family in yeast two-hybrid experiments (Fukuda et al., 2008). MICAL1 deletion leads to aberrant destination of the IgCAM cell adhesion molecules to the growth cones of cultured hippocampal neurons in a Rab6- and actin-dependent mechanism, establishing a link between redox state and vesicle trafficking in neurons (Van Battum et al., 2014). Interestingly, MICAL1 interacts with Rab1, which is involved in vesicle trafficking from the endoplasmic reticulum to the Golgi (Fischer et al., 2005). Additionally, MICAL3 interacts with Rab8, which in turns interacts with Rab6 to promote exocytosis of secretory vesicles (Van Battum et al., 2014). Moreover, expression of a mutant isoform of MICAL3 in HeLa cells induces accumulation of vesicles at the cell cortex by inhibiting vesicle docking with the plasma membrane, ultimately decreasing release of vesicle contents (Grigoriev et al., 2011). This suggests a link between Rab-dependent vesicle trafficking and ROS. Zinc deficiency has been shown to decrease tubulin polymerization via oxidation of tubulin thiol groups (Mackenzie et al., 2011). Interestingly, tubulin oxidation also impairs translocation of the transcription factor NFκB to the nucleus, suggesting a link between redox state and microtubule-dependent trafficking in a cellular model (Mackenzie et al., 2011).
In addition to ROS, NO also plays a role in terms of neuronal function and vesicle trafficking. A recent report suggest that NO reduces the expression of the molecular motors KIF5 and KIF21B and it decreases the length of the axons of cultured cortical neurons (Redondo et al., 2015). Authors hypothesize that NO exposure could affect KIF-dependent vesicle trafficking required for normal axonal growth. In fact, axonal retraction and NO release are key issues in some neurodegenerative disorders, like Parkinson’s disease (More et al., 2013; Tripathy et al., 2015). However, authors did not explored vesicle movement after NO exposure and this issue does not allow to conclude a direct effect of NO on axonal trafficking through cytoskeleton regulation. Moreover, G-protein coupled receptors that respond to NO (NO/CG) contributes positively to the physiology of neurons and neurotransmission (Hardingham et al., 2013; Russwurm et al., 2013). In summary, oxidative molecules signaling is an emerging concept in the field of cytoskeleton regulation and further studies will be required to understand the contribution to the vesicle trafficking and its impact on the physiology of neurons.
Concluding Remarks and Future Perspectives
ROS influence many different cellular functions under both physiological and pathological conditions. The targets of ROS include DNA, lipids and proteins. Among these, cytoskeletal proteins can be modified in vitro and in vivo by redox molecules. An imbalance between oxidative and reductive species leads to oxidative stress, which affects the polymerization of both F-actin and microtubules. In contrast, down-regulation of ROS also affects normal cytoskeletal organization, impacting the morphology, development and physiology of cells and neurons. Neuronal development and specification of neuronal compartments depend on redox homeostasis, in a mechanism that involves regulation of the actin cytoskeleton by the NOX complex. Further exploration of the role of redox balance in regulating microtubule dynamics in cellular models is required. Abnormal polymerization of actin microfilaments and microtubules directly affects vesicle trafficking and specific cargo delivery throughout the soma, dendrites and axon. However, the regulation of vesicle trafficking and protein sorting by redox balance represents an unexplored field despite strong evidence in several cellular contexts that cytoskeletal proteins are targets of oxidative species. Moreover, the contribution of redox balance to the interaction between the cytoskeleton and cytoskeleton-associated proteins such as myosins, dyneins and kinesin molecular motors has not been studied, and such analysis may reveal direct effects on vesicle trafficking and cargo destination. New evidence has emerged concerning the dissection of the cellular sources of ROS that can modulate cytoskeletal dynamics. The development of new ratiometric microscopy tools to characterize the spatiotemporal production of ROS may give other important clues about how redox balance controls neuronal physiology.
Funding
This work was funded by CONICYT doctoral fellowship 21120221 to CW and by grants ACT-1114 and Fondecyt 1140325 to CG-B.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ambruso, D. R., Knall, C., Abell, A. N., Panepinto, J., Kurkchubasche, A., Thurman, G., et al. (2000). Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc. Natl. Acad. Sci. U S A 97, 4654–4659. doi: 10.1073/pnas.080074897
Andersen, J. K. (2004). Oxidative stress in neurodegeneration: cause or consequence?. Nat. Med. 10(Supplement), S18–S25. doi: 10.1038/nrn1434
Bedard, K., and Krause, K. H. (2007). The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313. doi: 10.1152/physrev.00044.2005
Bigarella, C. L., Liang, R., and Ghaffari, S. (2014). Stem cells and the impact of ROS signaling. Development 141, 4206–4218. doi: 10.1242/dev.107086
Bokoch, G. M., and Diebold, B. A. (2002). Current molecular models for NADPH oxidase regulation by Rac GTPase. Blood 100, 2692–2696. doi: 10.1182/blood-2002-04-1149
Caceres, A., Ye, B., and Dotti, C. G. (2012). Neuronal polarity: demarcation, growth and commitment. Curr. Opin. Cell Biol. 24, 547–553. doi: 10.1016/j.ceb.2012.05.011
Carletti, B., Passarelli, C., Sparaco, M., Tozzi, G., Pastore, A., Bertini, E., et al. (2011). Effect of protein glutathionylation on neuronal cytoskeleton: a potential link to neurodegeneration. Neuroscience 192, 285–294. doi: 10.1016/j.neuroscience.2011.05.060
Chen, Q., Powell, D. W., Rane, M. J., Singh, S., Butt, W., Klein, J. B., et al. (2003). Akt phosphorylates p47phox and mediates respiratory burst activity in human neutrophils. J. Immunol. 170, 5302–5308. doi: 10.4049/jimmunol.170.10.5302
Contreras-Ferrat, A., Llanos, P., Vasquez, C., Espinosa, A., Osorio-Fuentealba, C., Arias-Calderon, M., et al. (2014). Insulin elicits a ROS-activated and an IP(3)-dependent Ca(2)(+) release, which both impinge on GLUT4 translocation. J. Cell Sci. 127, 1911–1923. doi: 10.1242/jcs.138982
Dalle-Donne, I., Giustarini, D., Rossi, R., Colombo, R., and Milzani, A. (2003). Reversible S-glutathionylation of Cys 374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic. Biol. Med. 34, 23–32. doi: 10.1016/s0891-5849(02)01182-6
Dalle-Donne, I., Rossi, R., Giustarini, D., Gagliano, N., Di Simplicio, P., Colombo, R., et al. (2002). Methionine oxidation as a major cause of the functional impairment of oxidized actin. Free Radic. Biol. Med. 32, 927–937. doi: 10.1016/s0891-5849(02)00799-2
Dalle-Donne, I., Rossi, R., Giustarini, D., Gagliano, N., Lusini, L., Milzani, A., et al. (2001). Actin carbonylation: from a simple marker of protein oxidation to relevant signs of severe functional impairment. Free Radic. Biol. Med. 31, 1075–1083. doi: 10.1016/s0891-5849(01)00690-6
Dang, P. M., Fontayne, A., Hakim, J., El Benna, J., and Perianin, A. (2001). Protein kinase C zeta phosphorylates a subset of selective sites of the NADPH oxidase component p47phox and participates in formyl peptide-mediated neutrophil respiratory burst. J. Immunol. 166, 1206–1213. doi: 10.4049/jimmunol.166.2.1206
Dáux, B., and Toledano, M. B. (2007). ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 8, 813–824. doi: 10.1038/nrm2256
Decoursey, T. E., and Ligeti, E. (2005). Regulation and termination of NADPH oxidase activity. Cell. Mol. Life Sci. 62, 2173–2193. doi: 10.1007/s00018-005-5177-1
Dickinson, B. C., Peltier, J., Stone, D., Schaffer, D. V., and Chang, C. J. (2011). Nox2 redox signaling maintains essential cell populations in the brain. Nat. Chem. Biol. 7, 106–112. doi: 10.1038/nchembio.497
Dixit, R., Ross, J. L., Goldman, Y. E., and Holzbaur, E. L. (2008). Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089. doi: 10.1126/science.1152993
Espinosa, A., Garcia, A., Hartel, S., Hidalgo, C., and Jaimovich, E. (2009). NADPH oxidase and hydrogen peroxide mediate insulin-induced calcium increase in skeletal muscle cells. J. Biol. Chem. 284, 2568–2575. doi: 10.1074/jbc.m804249200
Fischer, J., Weide, T., and Barnekow, A. (2005). The MICAL proteins and rab1: a possible link to the cytoskeleton?. Biochem. Biophys. Res. Commun. 328, 415–423. doi: 10.1016/j.bbrc.2004.12.182
Forsberg, K., and Di Giovanni, S. (2014). Cross talk between cellular redox status, metabolism and p53 in neural stem cell biology. Neuroscientist 20, 326–342. doi: 10.1177/1073858413514634
Forsberg, K., Wuttke, A., Quadrato, G., Chumakov, P. M., Wizenmann, A., and Di Giovanni, S. (2013). The tumor suppressor p53 fine-tunes reactive oxygen species levels and neurogenesis via PI3 kinase signaling. J. Neurosci. 33, 14318–14330. doi: 10.1523/jneurosci.1056-13.2013
Fukuda, M., Kanno, E., Ishibashi, K., and Itoh, T. (2008). Large scale screening for novel rab effectors reveals unexpected broad Rab binding specificity. Mol. Cell. Proteomics 7, 1031–1042. doi: 10.1074/mcp.m700569-mcp200
Gerich, F. J., Funke, F., Hildebrandt, B., Fasshauer, M., and Muller, M. (2009). H(2)O(2)-mediated modulation of cytosolic signaling and organelle function in rat hippocampus. Pflugers Arch. 458, 937–952. doi: 10.1007/s00424-009-0672-0
Gerlach, M., Ben-Shachar, D., Riederer, P., and Youdim, M. B. (1994). Altered brain metabolism of iron as a cause of neurodegenerative diseases? J. Neurochem. 63, 793–807. doi: 10.1046/j.1471-4159.1994.63030793.x
Ghezzi, P. (2005). Regulation of protein function by glutathionylation. Free Radic. Res. 39, 573–580. doi: 10.1080/10715760500072172
Giridharan, S. S., and Caplan, S. (2014). MICAL-family proteins: complex regulators of the actin cytoskeleton. Antioxid. Redox Signal. 20, 2059–2073. doi: 10.1089/ars.2013.5487
Glogauer, M., Marchal, C. C., Zhu, F., Worku, A., Clausen, B. E., Foerster, I., et al. (2003). Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J. Immunol. 170, 5652–5657. doi: 10.4049/jimmunol.170.11.5652
Gonzalez-Billault, C., Munoz-Llancao, P., Henriquez, D. R., Wojnacki, J., Conde, C., and Caceres, A. (2012). The role of small GTPases in neuronal morphogenesis and polarity. Cytoskeleton (Hoboken) 69, 464–485. doi: 10.1002/cm.21034
Grigoriev, I., Yu, K. L., Martinez-Sanchez, E., Serra-Marques, A., Smal, I., Meijering, E., et al. (2011). Rab6, Rab8 and MICAL3 cooperate in controlling docking and fusion of exocytotic carriers. Curr. Biol. 21, 967–974. doi: 10.1016/j.cub.2011.04.030
Halliwell, B. (1992). Reactive oxygen species and the central nervous system. J. Neurochem. 59, 1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x
Hardingham, N., Dachtler, J., and Fox, K. (2013). The role of nitric oxide in pre-synaptic plasticity and homeostasis. Front. Cell. Neurosci. 7:190. doi: 10.3389/fncel.2013.00190
Hidalgo, C., and Nunez, M. T. (2007). Calcium, iron and neuronal function. IUBMB Life 59, 280–285. doi: 10.1080/15216540701222906
Hoogenraad, C. C., and Akhmanova, A. (2010). Dendritic spine plasticity: new regulatory roles of dynamic microtubules. Neuroscientist 16, 650–661. doi: 10.1177/1073858410386357
Hoyal, C. R., Gutierrez, A., Young, B. M., Catz, S. D., Lin, J. H., Tsichlis, P. N., et al. (2003). Modulation of p47PHOX activity by site-specific phosphorylation: Akt-dependent activation of the NADPH oxidase. Proc. Natl. Acad. Sci. U S A 100, 5130–5135. doi: 10.1073/pnas.1031526100
Hung, R. J., Pak, C. W., and Terman, J. R. (2011). Direct redox regulation of F-actin assembly and disassembly by Mical. Science 334, 1710–1713. doi: 10.1126/science.1211956
Hung, R. J., Spaeth, C. S., Yesilyurt, H. G., and Terman, J. R. (2013). SelR reverses Mical-mediated oxidation of actin to regulate F-actin dynamics. Nat. Cell Biol. 15, 1445–1454. doi: 10.1038/ncb2871
Hung, R. J., and Terman, J. R. (2011). Extracellular inhibitors, repellents and semaphorin/plexin/MICAL-mediated actin filament disassembly. Cytoskeleton (Hoboken) 68, 415–433. doi: 10.1002/cm.20527
Hung, R. J., Yazdani, U., Yoon, J., Wu, H., Yang, T., Gupta, N., et al. (2010). Mical links semaphorins to F-actin disassembly. Nature 463, 823–827. doi: 10.1038/nature08724
Janke, C. (2014). The tubulin code: molecular components, readout mechanisms and functions. J. Cell Biol. 206, 461–472. doi: 10.1083/jcb.201406055
Janssen-Heininger, Y. M., Mossman, B. T., Heintz, N. H., Forman, H. J., Kalyanaraman, B., Finkel, T., et al. (2008). Redox-based regulation of signal transduction: principles, pitfalls and promises. Free Radic. Biol. Med. 45, 1–17. doi: 10.1016/j.freeradbiomed.2008.03.011
Jaworski, J., Kapitein, L. C., Gouveia, S. M., Dortland, B. R., Wulf, P. S., Grigoriev, I., et al. (2009). Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron 61, 85–100. doi: 10.3410/f.1145052.605000
Kavallaris, M. (2010). Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 10, 194–204. doi: 10.1038/nrc2830
Kim, C., and Dinauer, M. C. (2001). Rac2 is an essential regulator of neutrophil nicotinamide adenine dinucleotide phosphate oxidase activation in response to specific signaling pathways. J. Immunol. 166, 1223–1232. doi: 10.4049/jimmunol.166.2.1223
Kishida, K. T., Hoeffer, C. A., Hu, D., Pao, M., Holland, S. M., and Klann, E. (2006). Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol. Cell. Biol. 26, 5908–5920. doi: 10.1128/mcb.00269-06
Knapp, L. T., and Klann, E. (2002). Role of reactive oxygen species in hippocampal long-term potentiation: contributory or inhibitory? J. Neurosci. Res. 70, 1–7. doi: 10.1002/jnr.10371
Lambeth, J. D. (2004). NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 4, 181–189. doi: 10.1038/nri1312
Lambeth, J. D., Kawahara, T., and Diebold, B. (2007). Regulation of Nox and Duox enzymatic activity and expression. Free Radic. Biol. Med. 43, 319–331. doi: 10.1016/j.freeradbiomed.2007.03.028
Landino, L. M., Hasan, R., Mcgaw, A., Cooley, S., Smith, A. W., Masselam, K., et al. (2002). Peroxynitrite oxidation of tubulin sulfhydryls inhibits microtubule polymerization. Arch. Biochem. Biophys. 398, 213–220. doi: 10.1006/abbi.2001.2729
Landino, L. M., Koumas, M. T., Mason, C. E., and Alston, J. A. (2007). Modification of tubulin cysteines by nitric oxide and nitroxyl donors alters tubulin polymerization activity. Chem. Res. Toxicol. 20, 1693–1700. doi: 10.1021/tx7001492
Landino, L. M., Moynihan, K. L., Todd, J. V., and Kennett, K. L. (2004a). Modulation of the redox state of tubulin by the glutathione/glutaredoxin reductase system. Biochem. Biophys. Res. Commun. 314, 555–560. doi: 10.1016/j.bbrc.2003.12.126
Landino, L. M., Robinson, S. H., Skreslet, T. E., and Cabral, D. M. (2004b). Redox modulation of tau and microtubule-associated protein-2 by the glutathione/glutaredoxin reductase system. Biochem. Biophys. Res. Commun. 323, 112–117. doi: 10.1016/j.bbrc.2004.08.065
Lewis, S. A., Wang, D. H., and Cowan, N. J. (1988). Microtubule-associated protein MAP2 shares a microtubule binding motif with tau protein. Science 242, 936–939. doi: 10.1126/science.3142041
Löwe, J., Li, H., Downing, K. H., and Nogales, E. (2001). Refined structure of alpha beta-tubulin at 3. 5 A resolution. J. Mol. Biol. 313, 1045–1057. doi: 10.2210/pdb1jff/pdb
Luduena, R. F., and Roach, M. C. (1991). Tubulin sulfhydryl groups as probes and targets for antimitotic and antimicrotubule agents. Pharmacol. Ther. 49, 133–152. doi: 10.1016/0163-7258(91)90027-j
Luduena, R. F., Roach, M. C., Jordan, M. A., and Murphy, D. B. (1985). Different reactivities of brain and erythrocyte tubulins toward a sulfhydryl group-directed reagent that inhibits microtubule assembly. J. Biol. Chem. 260, 1257–1264.
Mackenzie, G. G., Salvador, G. A., Romero, C., Keen, C. L., and Oteiza, P. I. (2011). A deficit in zinc availability can cause alterations in tubulin thiol redox status in cultured neurons and in the developing fetal rat brain. Free Radic. Biol. Med. 51, 480–489. doi: 10.1016/j.freeradbiomed.2011.04.028
Massaad, C. A., and Klann, E. (2011). Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox Signal. 14, 2013–2054. doi: 10.1089/ars.2010.3208
Mellon, M. G., and Rebhun, L. I. (1976). Sulfhydryls and the in vitro polymerization of tubulin. J. Cell Biol. 70, 226–238. doi: 10.1083/jcb.70.1.226
Mishin, A. S., Belousov, V. V., Solntsev, K. M., and Lukyanov, K. A. (2015). Novel uses of fluorescent proteins. Curr. Opin. Chem. Biol. 27, 1–9. doi: 10.1016/j.cbpa.2015.05.002
Mitchison, T., and Kirschner, M. (1984). Dynamic instability of microtubule growth. Nature 312, 237–242. doi: 10.1038/312237a0
Mitchison, T., and Kirschner, M. (1988). Cytoskeletal dynamics and nerve growth. Neuron 1, 761–772. doi: 10.1016/0896-6273(88)90124-9
More, S. V., Kumar, H., Kim, I. S., Song, S. Y., and Choi, D. K. (2013). Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediators Inflamm. 2013:952375. doi: 10.1155/2013/952375
Morinaka, A., Yamada, M., Itofusa, R., Funato, Y., Yoshimura, Y., Nakamura, F., et al. (2011). Thioredoxin mediates oxidation-dependent phosphorylation of CRMP2 and growth cone collapse. Sci. Signal 4:ra26. doi: 10.1126/scisignal.2001127
Munnamalai, V., and Suter, D. M. (2009). Reactive oxygen species regulate F-actin dynamics in neuronal growth cones and neurite outgrowth. J. Neurochem. 108, 644–661. doi: 10.1111/j.1471-4159.2008.05787.x
Munnamalai, V., Weaver, C. J., Weisheit, C. E., Venkatraman, P., Agim, Z. S., Quinn, M. T., et al. (2014). Bidirectional interactions between NOX2-type NADPH oxidase and the F-actin cytoskeleton in neuronal growth cones. J. Neurochem. 130, 526–540. doi: 10.1111/jnc.12734
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/bj20081386
Nauseef, W. M. (2004). Assembly of the phagocyte NADPH oxidase. Histochem. Cell Biol. 122, 277–291. doi: 10.1007/s00418-004-0679-8
Neukirchen, D., and Bradke, F. (2011). Neuronal polarization and the cytoskeleton. Semin. Cell Dev. Biol. 22, 825–833. doi: 10.1016/j.semcdb.2011.08.007
Núñez, M. T., Urrutia, P., Mena, N., Aguirre, P., Tapia, V., and Salazar, J. (2012). Iron toxicity in neurodegeneration. Biometals 25, 761–776. doi: 10.1007/s10534-012-9523-0
Olguín-Albuerne, M., and Morán, J. (2015). ROS produced by NOX2 control in vitro development of cerebellar granule neurons development. ASN Neuro. 7:1759091415578712. doi: 10.1177/1759091415578712
Pao, M., Wiggs, E. A., Anastacio, M. M., Hyun, J., Decarlo, E. S., Miller, J. T., et al. (2004). Cognitive function in patients with chronic granulomatous disease: a preliminary report. Psychosomatics 45, 230–234. doi: 10.1176/appi.psy.45.3.230
Park, H. S., Lee, S. M., Lee, J. H., Kim, Y. S., Bae, Y. S., and Park, J. W. (2001). Phosphorylation of the leucocyte NADPH oxidase subunit p47(phox) by casein kinase 2: conformation-dependent phosphorylation and modulation of oxidase activity. Biochem. J. 358, 783–790. doi: 10.1042/0264-6021:3580783
Quadrato, G., and Di Giovanni, S. (2013). Waking up the sleepers: shared transcriptional pathways in axonal regeneration and neurogenesis. Cell. Mol. Life Sci. 70, 993–1007. doi: 10.1007/s00018-012-1099-x
Redondo, J., Hares, K., Wilkins, A., Scolding, N., and Kemp, K. (2015). Reductions in kinesin expression are associated with nitric oxide-induced axonal damage. J. Neurosci. Res. 93, 882–892. doi: 10.1002/jnr.23556
Rhee, S. G. (2006). Cell signaling. H2O2, a necessary evil for cell signaling. Science 312, 1882–1883. doi: 10.1126/science.1130481
Roberts, A. W., Kim, C., Zhen, L., Lowe, J. B., Kapur, R., Petryniak, B., et al. (1999). Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity 10, 183–196. doi: 10.1016/s1074-7613(00)80019-9
Russwurm, M., Russwurm, C., Koesling, D., and Mergia, E. (2013). NO/cGMP: the past, the present and the future. Methods Mol. Biol. 1020, 1–16. doi: 10.1007/978-1-62703-459-3_1
Sakai, J., Li, J., Subramanian, K. K., Mondal, S., Bajrami, B., Hattori, H., et al. (2012). Reactive oxygen species-induced actin glutathionylation controls actin dynamics in neutrophils. Immunity 37, 1037–1049. doi: 10.1016/j.immuni.2012.08.017
Shelton, M. D., Chock, P. B., and Mieyal, J. J. (2005). Glutaredoxin: role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid. Redox Signal. 7, 348–366. doi: 10.1089/ars.2005.7.348
Sies, H. (1997). Oxidative stress: oxidants and antioxidants. Exp. Physiol. 82, 291–295. doi: 10.1113/expphysiol.1997.sp004024
Sorce, S., and Krause, K. H. (2009). NOX enzymes in the central nervous system: from signaling to disease. Antioxid. Redox Signal. 11, 2481–2504. doi: 10.1089/ars.2009.2578
Sparaco, M., Gaeta, L. M., Santorelli, F. M., Passarelli, C., Tozzi, G., Bertini, E., et al. (2009). Friedreich’s ataxia: oxidative stress and cytoskeletal abnormalities. J. Neurol. Sci. 287, 111–118. doi: 10.1016/j.jns.2009.08.052
Sparaco, M., Gaeta, L. M., Tozzi, G., Bertini, E., Pastore, A., Simonati, A., et al. (2006). Protein glutathionylation in human central nervous system: potential role in redox regulation of neuronal defense against free radicals. J. Neurosci. Res. 83, 256–263. doi: 10.1002/jnr.20729
Stadtman, E. R., and Berlett, B. S. (1997). Reactive oxygen-mediated protein oxidation in aging and disease. Chem. Res. Toxicol. 10, 485–494. doi: 10.1021/tx960133r
Stiess, M., and Bradke, F. (2011). Neuronal polarization: the cytoskeleton leads the way. Dev. Neurobiol. 71, 430–444. doi: 10.1002/dneu.20849
Stroissnigg, H., Trancíková, A., Descovich, L., Fuhrmann, J., Kutschera, W., Kostan, J., et al. (2007). S-nitrosylation of microtubule-associated protein 1B mediates nitric-oxide-induced axon retraction. Nat. Cell Biol. 9, 1035–1045. doi: 10.1038/ncb1625
Suzuki, T., Nakamoto, T., Ogawa, S., Seo, S., Matsumura, T., Tachibana, K., et al. (2002). MICAL, a novel CasL interacting molecule, associates with vimentin. J. Biol. Chem. 277, 14933–14941. doi: 10.1074/jbc.M111842200
Tahara, E. B., Navarete, F. D., and Kowaltowski, A. J. (2009). Tissue-, substrate-and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic. Biol. Med. 46, 1283–1297. doi: 10.1016/j.freeradbiomed.2009.02.008
Taulet, N., Delorme-Walker, V. D., and Dermardirossian, C. (2012). Reactive oxygen species regulate protrusion efficiency by controlling actin dynamics. PLoS One 7:e41342. doi: 10.1371/journal.pone.0041342
Tejada-Simon, M. V., Serrano, F., Villasana, L. E., Kanterewicz, B. I., Wu, G. Y., Quinn, M. T., et al. (2005). Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol. Cell. Neurosci. 29, 97–106. doi: 10.1016/j.mcn.2005.01.007
Terman, J. R., Mao, T., Pasterkamp, R. J., Yu, H. H., and Kolodkin, A. L. (2002). MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell 109, 887–900. doi: 10.1016/s0092-8674(02)00794-8
Tripathy, D., Chakraborty, J., and Mohanakumar, K. P. (2015). Antagonistic pleiotropic effects of nitric oxide in the pathophysiology of Parkinson’s disease. Free Radic. Res. 49, 1129–1139. doi: 10.3109/10715762.2015.1045505
Van Battum, E. Y., Gunput, R. A., Lemstra, S., Groen, E. J., Yu, K. L., Adolfs, Y., et al. (2014). The intracellular redox protein MICAL-1 regulates the development of hippocampal mossy fibre connections. Nat. Commun. 5:4317. doi: 10.1038/ncomms5317
Villarroel-Campos, D., Gastaldi, L., Conde, C., Caceres, A., and Gonzalez-Billault, C. (2014). Rab-mediated trafficking role in neurite formation. J. Neurochem. 129, 240–248. doi: 10.1111/jnc.12676
Villarroel-Campos, D., and Gonzalez-Billault, C. (2014). The MAP1B case: an old MAP that is new again. Dev. Neurobiol. 74, 953–971. doi: 10.1002/dneu.22178
Weidinger, A., and Kozlov, A. V. (2015). Biological activities of reactive oxygen and nitrogen species: oxidative stress versus signal transduction. Biomolecules 5, 472–484. doi: 10.3390/biom5020472
Wilson, C., Núñez, M. T., and González-Billault, C. (2015). Contribution of NADPH-oxidase to the establishment of hippocampal neuronal polarity in culture. J. Cell Sci. 128, 2989-2995. doi: 10.1242/jcs.168567
Yang, R., Kong, E., Jin, J., Hergovich, A., and Püschel, A. W. (2014). Rassf5 and Ndr kinases regulate neuronal polarity through Par3 phosphorylation in a novel pathway. J. Cell Sci. 127, 3463–3476. doi: 10.1242/jcs.146696
Keywords: redox, cytoskeleton, neurons, development, trafficking
Citation: Wilson C and González-Billault C (2015) Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: implications for neuronal development and trafficking. Front. Cell. Neurosci. 9:381. doi: 10.3389/fncel.2015.00381
Received: 26 June 2015; Accepted: 11 September 2015;
Published: 30 September 2015.
Edited by:
Daniel Marcel Suter, Purdue University, USAReviewed by:
Laura Anne Lowery, Boston College, USAMary Halloran, University of Wisconsin, USA
Stephanie L. Gupton, University of North Carolina at Chapel Hill, USA
Copyright © 2015 Wilson and González-Billault. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian González-Billault, Department of Biology, Faculty of Sciences, Universidad de Chile, Las Palmeras 3425, 7800024 Santiago, Chile, chrgonza@uchile.cl