Reelin and Neuropsychiatric Disorders
 Kazuhiro Ishii
Kazuhiro Ishii Ken-ichiro Kubo
Ken-ichiro Kubo Kazunori Nakajima
Kazunori Nakajima- Department of Anatomy, Keio University School of Medicine, Tokyo, Japan
Proper neuronal migration and laminar formation during corticogenesis is essential for normal brain function. Disruption of these developmental processes is thought to be involved in the pathogenesis of some neuropsychiatric conditions. Especially, Reelin, a glycoprotein mainly secreted by the Cajal-Retzius cells and a subpopulation of GABAergic interneurons, has been shown to play a critical role, both during embryonic and postnatal periods. Indeed, animal studies have clearly revealed that Reelin is an essential molecule for proper migration of cortical neurons and finally regulates the cell positioning in the cortex during embryonic and early postnatal stages; by contrast, Reelin signaling is closely involved in synaptic function in adulthood. In humans, genetic studies have shown that the reelin gene (RELN) is associated with a number of psychiatric diseases, including Schizophrenia (SZ), bipolar disorder (BP) and autistic spectrum disorder. Indeed, Reln haploinsufficiency has been shown to cause cognitive impairment in rodents, suggesting the expression level of the Reelin protein is closely related to the higher brain functions. However, the molecular abnormalities in the Reelin pathway involved in the pathogenesis of psychiatric disorders are not yet fully understood. In this article, we review the current progress in the understanding of the Reelin functions that could be related to the pathogenesis of psychiatric disorders. Furthermore, we discuss the basis for selecting Reelin and molecules in its downstream signaling pathway as potential therapeutic targets for psychiatric illnesses.
Introduction
Falconer (1951) first described the mutant reeler mouse, which is characterized by reeling gait caused by dysregulation of motor coordination and ataxia. More than four decades later, the gene responsible for the reeler phenotype was identified and the protein encoded by the gene was named Reelin (Bar et al., 1995; D’Arcangelo et al., 1995; Hirotsune et al., 1995; Ogawa et al., 1995). Until date, much work has been carried out towards understanding the functions of Reelin during cortical development, because the Reelin-deficient mutant mouse, reeler, shows largely inverted cortical layers (Caviness and Sidman, 1973; Tissir and Goffinet, 2003; Honda et al., 2011; Sekine et al., 2014).
Reelin is a glycoprotein secreted mainly from the Cajal-Retzius cells in the developing cerebral cortex and hippocampus, and acts as a key regulator of various aspects of laminar formation, including neuronal migration, cell aggregation and dendrite formation (Nakajima et al., 1997; Kubo et al., 2010; Franco et al., 2011; Jossin and Cooper, 2011; Sekine et al., 2014; Kohno et al., 2015). Many downstream molecules and several pathways involved in Reelin signaling during development have been elucidated from animal studies (Howell et al., 1997; Hiesberger et al., 1999; Ballif et al., 2004; Honda et al., 2011; Jossin and Cooper, 2011; Sekine et al., 2012; Figure 1).
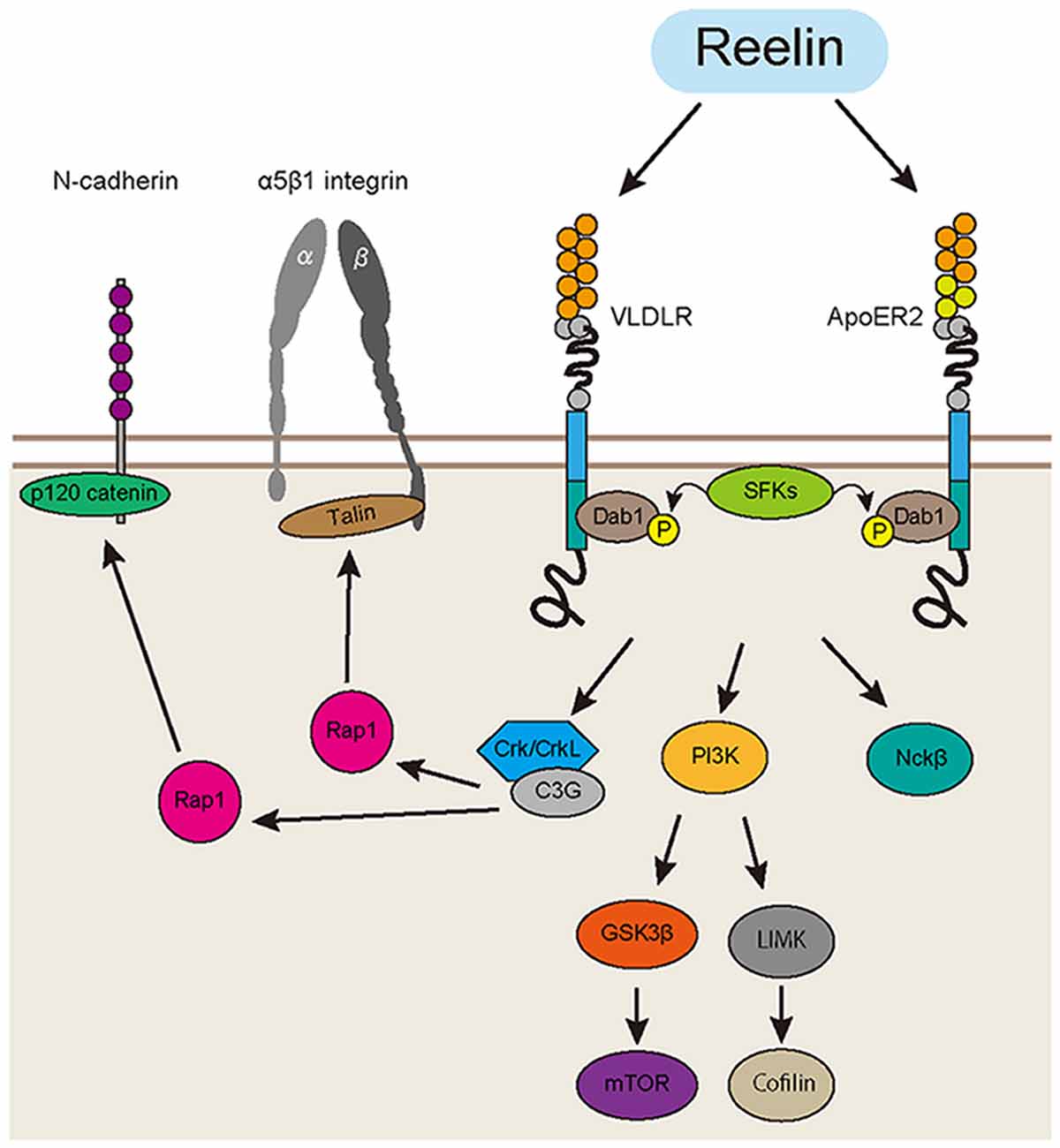
Figure 1. Reelin signaling pathway in the developing cortex. The Reelin protein binds to very-low-density lipoprotein receptor (VLDLR) and apolipoprotein E receptor 2 (ApoER2). Then, Dab1 is phosphorylated by the Src-family tyrosine kinases (SFKs). Phosphorylated Dab1 recruits downstream molecules such as Crk/CrkL, phosphatidylinositol 3-kinase (PI3K) and Nckβ. CrkL binds to C3G, an effector protein. C3G promotes the formation of Rap1-GTP, which activates cell adhesion molecules, including α5β1 integrin and N-cadherin. There are also Dab1-PI3K-mTOR, Dab1-PI3K-cofilin and Dab1-Nckβ pathways, which regulate the actin cytoskeleton relevant to dendrite formation and neuronal migration in the developing cortex.
In the postnatal period, the distribution and expression patterns of Reelin are dramatically changed as compared to those during the embryonic period (Alcántara et al., 1998). This suggests that the roles of Reelin in the postnatal brain might also be changed. Intriguingly, a number of lines of evidence indicate that Reelin signaling modulates synaptic function in the adult brain (Herz and Chen, 2006; Figure 2).
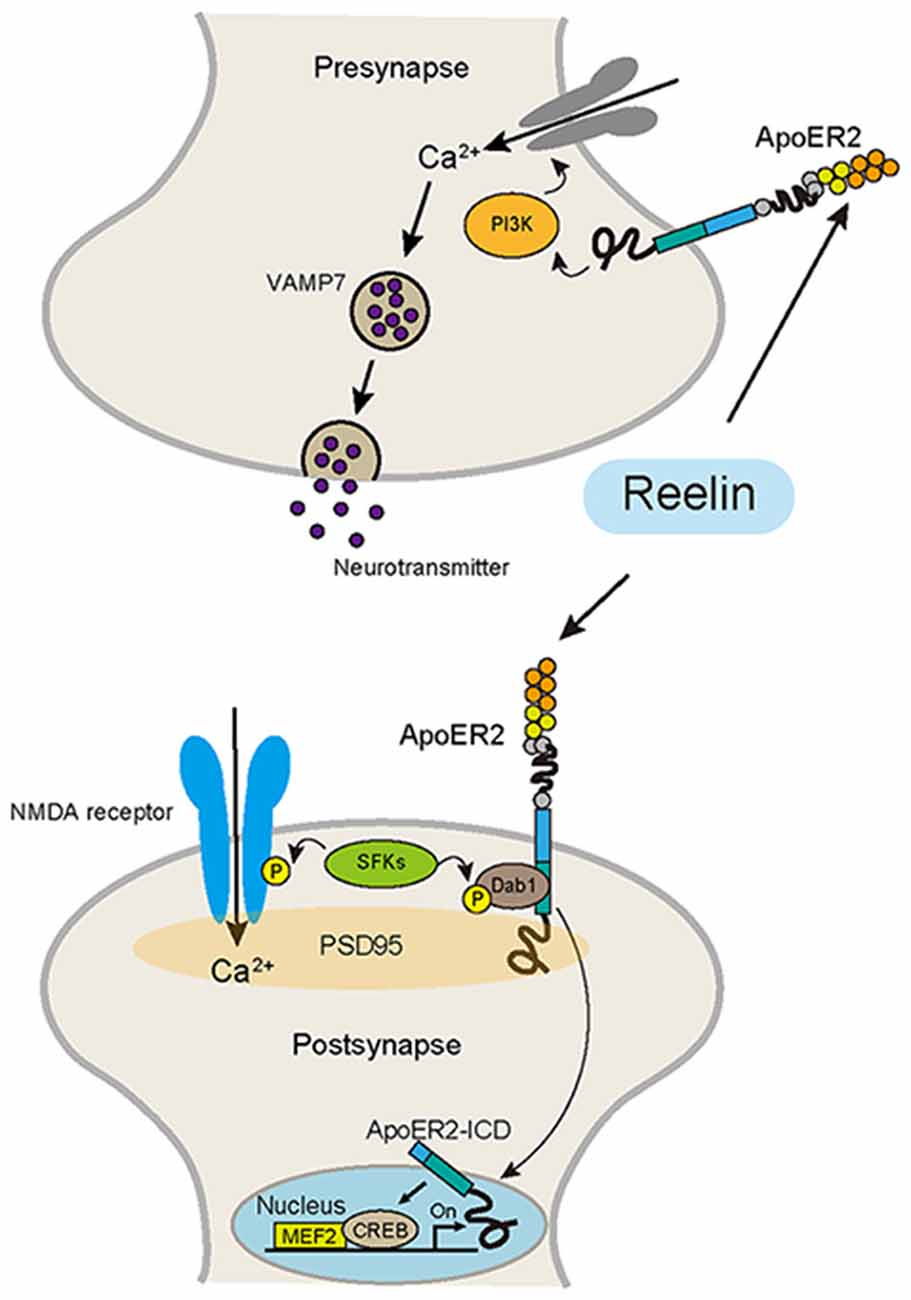
Figure 2. Reelin signaling pathway in the adult brain, which regulates synaptic function. Reelin binds to ApoER2 and activates SFKs, which phosphorylate a tyrosine of the NR2 subunit of the N-methyl-D-aspartate (NMDA) receptor, resulting in the potentiation of NMDA receptor-mediated Ca2+ influx. ApoER2 binds to PSD-95, a scaffolding protein in the post synapse. ApoER2 is proteolytically cleaved by γ-secretase activity, which is activated by NMDA receptor signaling. Then, the intracellular domain of ApoER2 (ApoER2-ICD) is released from the cell membrane and translocates into the nucleus and activates transcriptional factors, including MEF2 and CREB. In the presynapse, the Reelin-ApoER2-PI3K pathway promotes Ca2+ influx, which leads to the selective mobilization of VAMP7-enriched synaptic vesicles, promoting neurotransmitter release.
In humans, genetic studies have reported that the RELN locus is associated with neuropsychiatric disorders, such as Schizophrenia (SZ), bipolar disorder (BP), autism spectrum disorder (ASD) and Alzheimer’s disease (AD; Ovadia and Shifman, 2011; Wang et al., 2014; Bufill et al., 2015; Li et al., 2015). Indeed, homozygous (rl/rl) and heterozygous reeler (rl/-) mice haploinsufficient for Reelin show cognitive and behavioral abnormalities (Tueting et al., 1999; Qiu et al., 2006a), supporting the notion that RELN haploinsufficiency may lead to higher brain dysfunctions relevant to neuropsychiatric disorders in humans. A number of studies have revealed that Reelin also plays a pivotal role in regulating synaptic functions, including N-methyl-d-aspartate (NMDA) receptor signaling (Beffert et al., 2005; Qiu et al., 2006b), and these findings may give a clue to uncover the impairments in the Reelin signaling pathway underlying the development of psychiatric disorders.
In this article, we first provide an overview of the current progress in Reelin research at the molecular, cellular and tissue levels. Second, we review human genetic studies of neuropsychiatric disorders associated with mutations in the RELN gene locus. Then, we focus on how Reelin dysfunction leads to the behavioral abnormalities relevant to neuropsychiatric diseases in mouse models. Finally, we discuss the basis for selecting Reelin and molecules in its downstream signaling pathway as potential therapeutic targets for neuropsychiatric disorders.
Reelin has an Essential Role in Cortical Development
In mammals, cortical expansion is thought to contribute to the acquisition of higher brain functions during the course of evolution (Molnar et al., 2006). The neocortex is composed of a well-organized six-layer structure, including excitatory and inhibitory neurons (Rakic, 2009). The excitatory neurons, born directly or indirectly from the radial glia (Tabata et al., 2009; Sekine et al., 2013), which are neural progenitor cells located in the ventricular zone, migrate radially towards the brain surface, and eventually reach their final destinations (Rakic, 1972). Since the late-born neurons migrate past their predecessors, the earlier-born neurons are placed at a deeper position and the later-born neurons at a more superficial position in the cortical plate (CP; Caviness, 1982; Takahashi et al., 1999). This pattern of cell alignment is called a birth-date-dependent “inside-out” pattern. By contrast, inhibitory interneurons are born in the ganglionic eminences (GEs) in the ventral telencephalon and preoptic area in the rostral diencephalon that are located far from their final destination, and migrate tangentially for a long distance towards the dorsal pallium (Anderson et al., 1997; Tamamaki et al., 1997; Yozu et al., 2005; Kanatani et al., 2008, 2015; Gelman et al., 2009, 2011; Marin et al., 2010). Correct neuronal migration and laminar formation are essential for the establishment of proper brain functions. Indeed, disruption of the cortical architecture has been observed in various neuropsychiatric disorders (Arnold, 2000; Wegiel et al., 2010).
Reelin is a glycoprotein that is mainly secreted from the Cajal-Retzius cells located in the marginal zone (MZ), and a subpopulation of GABAergic interneurons (Rice and Curran, 2001). Reelin-deficient mice, reeler, show largely inverted cortical layers, which strongly supports the notion that Reelin is a key regulator of cortical development. A recent study showed that cortical laminar formation in reeler brains exhibit a more complex pattern than previously thought. Boyle et al. (2011) reported that the layer formation is severely disorganized in the reeler cortex, with layer II/III neurons located in the middle of the cortex and cells from other layers (layer IV, V and VI) split between the deep and superficial layers in a mirror image fashion. The secreted Reelin protein binds to its receptors, apolipoprotein E receptor 2 (ApoER2) and very-low-density lipoprotein receptor (VLDLR), which are mainly expressed on the cell membrane of cortical neurons (Trommsdorff et al., 1999; Hirota et al., 2015). Then, disabled homolog 1 (Dab1), an intracellular adaptor protein, is phosphorylated via Fyn and Src, the Src-family tyrosine kinases (SFKs; Hiesberger et al., 1999; Howell et al., 1999). Phosphorylated Dab1 recruits and binds with several downstream molecules, including Crk, phosphatidylinositol 3-kinase (PI3K), and Nckβ (Beffert et al., 2002; Pramatarova et al., 2003; Park and Curran, 2008). The Crk family proteins (Crk and CrkL) are adaptor proteins that bind to phosphorylated Dab1 and recruit their effector proteins (Chen et al., 2004). Park and Curran (2008) reported that conditional double-knockout mice of Crk and CrkL showed the major anatomic features of reeler, including disruption of layer formation in the cerebral cortex and hippocampus, absence of preplate splitting, impaired dendrite formation, and cerebellar hypofoliation. The brains of mice deficient in C3G, an effector protein of CrkL, also show impaired preplate splitting (Voss et al., 2008). These findings indicate the Reelin-Dab1-Crk/CrkL pathway plays a critical role in controlling layer formation in the cortex, including preplate splitting. Sekine et al. (2012) recently reported that Reelin activates integrin α5β1 through an intracellular Dab1-Crk/CrkL-C3G-Rap1 pathway. Furthermore, they showed that activated integrin α5β1 controls the terminal translocation, a final mode of neuronal movement beneath the MZ (Nadarajah et al., 2001; Sekine et al., 2011, 2012). Other groups reported that Reelin signaling regulates the function of another cell adhesion molecule, N-cadherin. Franco et al. (2011) reported that Dab1 acts on the migratory neurons to stabilize their leading processes in a Rap1-dependent manner. They showed that Rap1 regulates the function of N-cadherin and finally controls the somal translocation (Franco et al., 2011). The same group also reported that N-cadherin activated through the Reelin-Rap1 pathway enhances heterotypic cell-cell contact between the Cajar-Retzius cells and migratory neurons (Gil-Sanz et al., 2013). Jossin and Cooper (2011) demonstrated that the Rap1-N-cadherin pathway also regulates the transition of migratory neurons from multipolar cells to bipolar cells beneath the CP; this mode change is crucial for the migratory neurons to enter the CP (Figure 1).
Reelin signaling plays a role in the processes of dendrite development (Olson et al., 2006; Jossin and Goffinet, 2007). Olson et al. (2006) found that Dab1-suppressed migrating neurons showed simplified leading processes that were less likely to attach to the MZ and exhibited abnormal cell positioning. Jossin and Goffinet (2007) reported that Reelin promoted dendritogenesis through activation of mammalian target of rapamycin (mTOR) and S6 kinase 1 (S6K1) in a Dab1-, PI3K- and Akt-dependent manner. In regard to axon formation, by ablation of Cajal-Retzius cells in a slice and analysis of reeler mice, Del Rio et al. (1997) demonstrated abnormalities in the development of axon fibers from the entorhinal cortex to the hippocampus. However, using reeler cortical explants, Jossin and Goffinet (2001) found that Reelin itself did not exhibit any significant attraction or repulsion to cortical axons.
In the adult brain, the main source of Reelin is no longer the Cajal-Retzius cells, but a subpopulation of GABAergic interneurons, a change that is also associated with a change in the distribution pattern of Reelin (Alcántara et al., 1998; Pesold et al., 1998). These findings suggest that the roles of Reelin in the adult brain may also be different from those in the developing brain. We shall review and discuss the functions of Reelin in the adult brain in “Reelin Regulation of Brain Function and Behavior” Section.
Genetic Study of RELN in Neuropsychiatric Disorders
In humans, the mutations of the RELN have been shown to be associated with autosomal recessive lissencephaly with cerebellar hypoplasia (Hong et al., 2000). The brain phenotypes in these patients are similar to those found in the reeler mice, including abnormal laminar formation and cerebellar hypoplasia. In addition to this severe brain malformation, mutations in the RELN locus are also associated with neuropsychiatric disorders, with no apparent abnormalities in the brain structure. In this section, we provide an overview of the recent genetic studies carried out to examine the association of RELN with neuropsychiatric disorders.
Schizophrenia
SZ is a devastating psychiatric disease that affects approximately 1% of the population and is characterized by hallucinations, delusions and cognitive disturbances. The first clinical features of SZ typically emerge between early childhood and adolescence, with many patients experiencing chronic SZ symptoms (Sawa and Snyder, 2002). The clinical symptoms are based on brain dysfunctions attributed to genetic and environmental factors (Insel, 2010). As SZ shows high heritability, the risk genes with the greatest impact on the predisposition to SZ have been pursued by many researchers (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). As for the RELN gene, Li et al. (2013) conducted a case-control association study and identified six single-nucleotide polymorphisms (SNPs; rs2237628, rs362626, rs362814, rs362813, rs362731, rs362726) which are located in the intron 29 of the RELN gene that were significantly associated with the risk of SZ in the Chinese population. Several groups have reported that the existence of a gender-specific (women) association between SNPs in RELN and SZ (Shifman et al., 2008; Kuang et al., 2011; Li et al., 2011). Li et al. (2011) also reported the association of another SNP (rs12705169) in the RELN locus with SZ. Although rs12705169 is located in the intron of RELN and does not lead to any change of the protein structure or function, it may be involved in alternative RNA splicing and miRNA generation. When they divided the subjects by gender, they found that this SNP was positively associated with SZ only in women. Similarly, Kuang et al. (2011) demonstrated an association of rs362719 of RELN, which is located in the exon 42 and contributes to the EGF-like domain of the fifth Reelin repeat, with susceptibility to SZ in their female, but not male, participants. Shifman et al. (2008) performed a genome-wide association study (GWAS) for SZ in Ashkenazi Jews and showed that rs7341475, a SNP in the intron four of the RELN, was associated with SZ only in women. However, the association of this SNP (rs7341475) with SZ was not replicated in any other study in the Chinese population (Liu et al., 2011). Subsequently, Ben-David and Shifman (2010) performed a meta-analysis of four GWAS studies to assess the association of rs7341475 with SZ, and suggested this locus might have a small effect on the SZ risk. Therefore, the association of rs7341475 with SZ may remain inconclusive. Wedenoja et al. (2008) replicated the previous linkage of SZ on 7q22 in an independent subject. They also studied four candidate genes, including RELN. Although the selected candidate genes showed no association to the clinical diagnosis of SZ, the quantifiable trait component analysis revealed that allelic variants of RELN contributed to the endophenotypes of SZ, including working memory and executive function (Wedenoja et al., 2008).
Other Neuropsychiatric Disorders and Neurological Diseases
ASD is characterized by impairments of social interaction and communication, as well as repetitive behaviors and restricted interests (Maenner et al., 2014). Twin and family studies have revealed the important role of genetic factors in ASD, with a heritability value of as high as 90% (Freitag et al., 2007). Although many studies have been conducted to explore the genetic association between RELN and ASD, the results have been inconclusive. Persico et al. (2001) was the first to report that polymorphic GGC repeats located in the 5′ untranslated region (5′ UTR) of the RELN were associated with autistic disorder, finding that was subsequently replicated in three studies (Zhang et al., 2002; Skaar et al., 2005; Dutta et al., 2007). However, other groups have failed to show an association between the triplet repeats in the 5′ UTR of the RELN and autism (Krebs et al., 2002; Bonora et al., 2003; Devlin et al., 2004; Li et al., 2004). In the family-based association analyses carried out by Serajee et al. (2006), the most significant results were the apparent association of autism, in a broad diagnosis of the disease, with rs736707 in intron 59 and rs362691 in exon 22 of the RELN. Li et al. (2008) also showed a significant genetic association between rs736707 in intron 59 and ASD in a Han Chinese population. On the other hand, other groups have failed to detect these associations (Dutta et al., 2008; He et al., 2011). Dutta et al. (2008) carried out case-control and family-based association studies for six SNPs (rs727531, rs2072403, rs2072402, rs362691, rs362719, rs736707), and found that these SNPs of RELN were unlikely to be associated with ASD. Recently, a Chinese group conducted a meta-analysis for case-control and transmission disequilibrium test (TDT) studies published from 2001 to 2013 and concluded that rs362691 might contribute significantly to the risk of ASD (Wang et al., 2014).
There are only a few reports that suggested the existence of an association between the RELN and BP (Goes et al., 2010; Ovadia and Shifman, 2011). Goes et al. (2010) found that the rs362719 of RELN was associated with susceptibility to BP, particularly in females.
Several groups have also reported an association between AD and the RELN gene (Seripa et al., 2008; Antoniades et al., 2011; Kramer et al., 2011; Bufill et al., 2015; Fehér et al., 2015). AD is a neurodegenerative disease characterized by the formation of neurofibrillary tangles and beta-amyloid plaques in the brain, and patients clinically manifest progressive impairments of memory and cognition (Scheltens et al., 2016). Several SNPs in the RELN gene have been reported to be associated with the risk of AD. Seripa et al. (2008) showed an association between a triplet tandem repeat in the 5′UTR and rs607755 with AD, particularly in females. Another group also showed significant association between rs607755 and the risk of AD (Fehér et al., 2015). However, their results were inconsistent with the conclusion by Seripa et al. (2008) that rs607755 was significantly associated with AD only in males (Fehér et al., 2015). Antoniades et al. (2011) reported that an SNP in exon 22 of RELN (rs362691) was significantly associated with the risk of AD in a Greek population. Bufill et al. (2015) reported that SNPs in the RELN gene (rs528528 and rs2299356) and two genes (PLK2 and CAMK2A) related to the Reelin signaling pathway were associated with AD and mild cognitive impairment (MCI).
Reelin Regulation of Brain Function and Behavior
Expression Level of Reelin and Vulnerability to Neuropsychiatric Disorders
Many studies have provided evidence for altered Reelin expression in rodents with cognitive dysfunction, which may relate to neuropsychiatric diseases. Heterozygous reeler mice (HRM), in which the amount of Reelin protein is approximately 50% as compared to that in the wild type mice, exhibit behavioral abnormalities (Costa et al., 2002). Thus, HRM has been of interest as an animal model of psychiatric diseases. In spite of the initial inconsistent findings on the occurrence of behavioral deficits in HRM (Salinger et al., 2003; Podhorna and Didriksen, 2004), many groups have reported behavioral traits associated with Reln haploinsufficiency in HRM (Tueting et al., 1999; Qiu et al., 2006a; Barr et al., 2007, 2008; Teixeira et al., 2011; Kutiyanawalla et al., 2012; Iafrati et al., 2014). Several groups have also reported defect in the prepulse inhibition (PPI), an impairment of sensory motor gating that is associated with SZ, in HRM (Tueting et al., 1999; Barr et al., 2008; Teixeira et al., 2011; Kutiyanawalla et al., 2012). Iafrati et al. (2014) showed that Reelin deficiency in HRM caused defects in the juvenile morphofunctional properties of excitatory synapses in the prefrontal cortex (PFC) and behavioral dysfunction in prefrontal circuits. Furthermore, Barr et al. (2007) showed that the Reelin receptors VLDLR and ApoER2 regulated sensory motor gating. They demonstrated that acoustic PPI was intact in both Vldlr- and Apoer2-mutant mice. However, Vldlr-homozygous knockouts mice exhibited deficits in crossmodal PPI, while Apoer2-heterozygous and homozygous knockouts mice exhibited increased crossmodal PPI (Barr et al., 2007). Qiu et al. (2006a) demonstrated that HRM exhibited hippocampus-dependent learning deficit underlying the impairment of hippocampal plasticity. There are several interesting studies to show that mouse behavioral alterations are manifested as a consequence of gene-environment interaction, similar to many psychiatric disorders in humans (Laviola et al., 2009; Romano et al., 2014; Howell and Pillai, 2016). There have also been a number of reports indicating that in humans, the amount of Reelin protein in the brain, plasma and cerebrospinal fluid (CSF) are associated with neuropsychiatric conditions. Almost all studies have shown that the reduced amount of Reelin in the brain and blood are associated with a high risk of development of neuropsychiatric disorders (Guidotti et al., 2000; Fatemi et al., 2005; Eastwood and Harrison, 2006). Interestingly, however, increased expression levels of Reelin in the CSF (Sáez-Valero et al., 2003) and frontal cortex (Botella-López et al., 2006) were found in case of AD. Accumulation of Reelin has also been reported in amyloid-like plaques, with a decline in Reelin-positive neurons, which might represent a risk factor for AD (Knuesel et al., 2009). Reelin protein itself is thought to be protective against AD-like neuropathology, since the reduced Reelin expression in a transgenic AD background markedly elevated amyloid-β plaque formation (Kocherhans et al., 2010).
The Expression Level of Reelin is Regulated by Multiple Mechanisms
In addition to genetic mutations/polymorphisms, various mechanisms may lead to aberrant expression of Reelin. In the postnatal brain, the main source of Reelin is shifted from Cajal-Retzius cells to a subpopulation of GABAergic interneurons (Alcántara et al., 1998; Pesold et al., 1998). Using in situ hybridization analysis combined with immunohistochemistry, Pesold et al. (1998) showed that Reelin is preferentially expressed in GABAergic interneurons in the adult rat cortex and hippocampus. Furthermore, they found Reelin immunoreactivity not only in neurons, but also in the extracellular space, suggesting that the GABAergic interneurons secrete Reelin into the extracellular matrix (Pesold et al., 1998). However, how Reelin is secreted by these specialized interneurons in the postnatal brain remains an unresolved question, although a constitutive mechanism dependent on a specific sequence of positively charged amino acids in the Reelin carboxy terminus domain has been reported (Rodriguez et al., 2000). Thus, in mature brains, the amount of secreted Reelin protein may depend on the number and distribution of Reelin-positive interneurons.
Several studies have shown the involvement of epigenetic mechanisms in the transcriptional regulation of RELN (Abdolmaleky et al., 2005; Grayson et al., 2005). Grayson et al. (2005) analyzed the pattern of methylation within the CpG island of the RELN promoter in human SZ brains obtained from a brain bank. They showed hypermethylation of the promoter region of RELN in the SZ brains (Grayson et al., 2005). Abdolmaleky et al. (2005) also demonstrated hypermethylation of a CpG island containing CRE and an SP1-binding site in the promoter region of RELN in post-mortem examination of the brains of SZ patients. However, Tochigi et al. (2008) reanalyzed this RELN promoter region by using the same brain samples as in the previous study, and showed that the extent of methylation in this promoter region did not differ significantly between the SZ patients and controls. Furthermore, Mill et al. (2008) prepared a microarray-based comprehensive epigenomic scan and found epigenetic changes in the loci associated with glutamatergic and GABAergic neurotransmitter pathways. However, they did not detect any association between hypermethylation in the promoter region of RELN and major psychosis. Recently, a systematic review of DNA methylation in SZ and BP has been reported (Teroganova et al., 2016). Although a number of differentially methylated genes were detected, including RELN, diverse methodologies used across studies hampered the reliability of the meta-analysis (Teroganova et al., 2016). Thus, in regard to the epigenetic regulation of RELN expression, further investigation is needed to confirm the association of hypermethylation in the RELN promoter region with the predisposition to major mental illnesses.
Perturbation of transcriptional regulation could affect the Reelin protein distribution as well as expression levels. Baek et al. (2015) reported that RELN transcription was derepressed in ectopic cortical regions, mediated by activation of the transcription factor FOXG1 in human focal cortical malformations. They found that misexpression of Reelin in the progenitor cells caused non-cell autonomous neuronal migration defects, resulting in focal cortical malformations. These results may, at least to some extent, resemble the effects of ectopic Reelin overexpression in the mouse brain (Kubo et al., 2010).
Reelin Signaling is Involved in Synaptic Functions and Behavior
During cortical development, the main source of Reelin is the Cajal-Retzius cells located in the MZ. Subsequently, these cells gradually disappeared, and the main source of Reelin finally shifts to a subtype of GABAergic interneurons (Miyoshi et al., 2010). Thus, during this period, the expression pattern of Reelin changes dynamically. This suggests that the role of Reelin in the postnatal brain also changes dramatically. Indeed, in adulthood, there is much evidence to indicate that Reelin modulates NMDA receptor-mediated synaptic functions (Beffert et al., 2005; Iafrati et al., 2014; Figure 2). Dysfunction of NMDA receptor signaling plays a key role in the pathogenesis of major neuropsychiatric disorders, including SZ and AD (Zhou and Sheng, 2013). Recently, it has been reported that hundreds of genetic loci were associated with SZ in a large-scale GWAS (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). In this analysis, the glutamatergic neurotransmission-related genes including GRIN2A, a NMDA receptor subunit, were highly associated with SZ. NMDA dysfunction has also been reported to be related to cognitive impairment in AD. In addition to the widely used drug, cholinesterase inhibitors, for AD, a new drug that modulates glutamate signaling has also become available recently for AD (Scarpini et al., 2003). Also, it has been shown that Reelin acts on NMDA receptor signaling via ApoER2, a Reelin receptor (Beffert et al., 2005; Chen et al., 2005; Herz and Chen, 2006). Beffert et al. (2005) demonstrated that an alternative splicing variant of ApoER2 modulated the NMDA receptor activity through Reelin signaling. They found that alternatively spliced exon 19, which encodes amino acids in the intracellular domain of ApoER2 (ApoER2-ICD), is necessary for the stimulation of tyrosine phosphorylation of the NMDA receptor subunit NR2 in hippocampal slices in response to Reelin. Furthermore, they confirmed that the mice expressing a mutant ApoER2 lacking exon 19 showed severe cognitive disturbances related to the NMDA receptor function (Beffert et al., 2005). Interestingly, birds do not have exon 19 (Brandes et al., 1997), suggesting that adding this new exon may partly contribute to the acquisition of higher brain functions in mammals. Chen et al. (2010) further showed that the Reelin-ApoER2-NMDA receptor pathway is involved in the pathophysiology of AD. They demonstrated that ApoE4, which is associated with an earlier average age at onset of dementia, depletes ApoER2 from the neuronal surface after ligand-induced endocytosis by Reelin, which impairs synaptic plasticity and NMDA phosphorylation induced by Reelin (Chen et al., 2010). Moreover, Lane-Donovan et al. (2015) demonstrated that Reelin signaling protects against the Aβ toxicity that induces synaptic dysfunction in the early stages of aging. Using Reelin conditional knockout mice, they found that the loss of Reelin rendered excitatory synapses susceptible to functional suppression by Aβ, which resulted in impaired learning and memory. Iafrati et al. (2014) demonstrated that in vivo injection with ketamine, an NMDA receptor antagonist, or Ro25-6981, an inhibitor of GluN2B-NMDA receptors, in the juvenile period rescued the behavioral abnormalities, reduced the dendritic spine density, and anomalous long-term potentiation (LTP) in the PFC of HRM. Their results suggest Reelin is essential for proper functional and behavioral development of juvenile prefrontal circuits through modulating the NMDAR-mediated signaling pathway.
Reelin signaling is also involved in the presynaptic functions (Figure 2). Reelin acts presynaptically in mature neurons to rapidly enhance neurotransmitter release. This role of Reelin depends on the function of VAMP7, a vesicular SNARE protein (Bal et al., 2013). Telese et al. (2015) reported interesting findings; they reported that the Reelin pathway controls learning and memory through activation of the transcriptional factors. Proteolytic cleavage of ApoER2 is a crucial component of the synapse-to-nuclear signaling triggered by Reelin. When Reelin binds to its receptor ApoER2, nuclear translocation of the ApoER2-ICD is triggered. Then, ApoER2-ICD binds to the transcriptional factors MEF2 and CREB, and as a result, expressions of some genes involved in synaptic plasticity are activated (Telese et al., 2015).
Observations in animal studies using reeler and Reelin receptor mutant mice support the notion that Reelin-synapse dysfunction leads to cognitive dysfunction and neuropsychiatric symptoms (Weeber et al., 2002; Qiu et al., 2006a,b; Pujadas et al., 2010; Trotter et al., 2013). Several groups have reported that Reelin plays an important role in synaptic plasticity, mainly dependent on the NMDA receptor dysfunction in the hippocampal region (Weeber et al., 2002; Qiu et al., 2006b; Pujadas et al., 2010). Weeber et al. (2002) reported that Reelin signaling is crucial for memory formation and synaptic plasticity in the hippocampal CA1 region. They demonstrated using hippocampal slices, that mice with KO of each of the Reelin receptors, VLDLR and ApoER2, exhibited defects in LTP. They further showed that treatment of the hippocampal slices obtained from wild-type mice with Reelin significantly enhanced the LTP in the CA1 region, which was abolished in mice deficient in either of the receptors. Qiu et al. (2006b) replicated Weeber’s findings by using HRM. They also observed defect of LTP in the CA1 region by electrophysiological analysis. In addition, according to a report by Pujadas et al. (2010), overexpression of Reelin enhanced the synaptic function in the hippocampus. They generated transgenic mice that overexpressed Reelin under the control of the CaMKIIα promoter. The mice showing Reelin overexpression exhibited increased spine hypertrophy and LTP in the hippocampus (Pujadas et al., 2010). Trotter et al. (2013) generated Dab1-conditional knockout mice and examined the spine morphology and synaptic functions. They conditionally deleted Dab1 protein from the excitatory neurons of the adult forebrain using Dab1flox/flox; CaMKII-Cre mice. This Cre driver line exhibits Cre expression in the forebrain, starting at approximately P19. They showed that loss of Dab1 led to a reduction of the spine size, suppression of Akt and ERK signaling, loss of hippocampal LTP, and deficits in hippocampus-dependent learning and memory. These results obtained using transgenic mice are consistent with the previous results obtained using reeler mice (Pujadas et al., 2010; Trotter et al., 2013). Furthermore, recently, Imai et al. (2016) generated dorsal forebrain-specific Dab1 conditional knockout mice (Dab1flox/flox; Emx1-Cre mice), in which Cre is expressed specifically in the dorsal forebrain from the beginning of corticogenesis, and performed behavioral tests. These Dab1-conditional knockout mice showed normal motor functions, but exhibited hyperactivity, decreased anxiety-like behavior, and deficits in spatial reference and working memory (Imai et al., 2016). In these mice, in addition to the deficient Reelin-Dab1 signaling, the disorganized laminar structure in the cerebral cortex might also have contributed to the observed behavioral abnormalities. The best way to understand Reelin function in adulthood is to analyze conditional Reelin gene (Reln) knockout mice. Lane-Donovan et al. (2015) generated inducible conditional Reln knockout mice and analyzed them. After Reelin inactivation at 2 months of age, they found that while the mice had no apparent abnormalities of the cortical structure, they did exhibit mild behavioral changes and electrophysiological phenotype. Importantly, the behavioral phenotypes in the conditional Reln knockout mice may be somewhat different from those observed in Reln-deficienct (homozygous and heterozygous reeler) mice, which may be based on the degree of structural abnormalities. In the mature brain, the structure would not be seriously affected by Reelin deficiency. By contrast, Reelin deficiency during the developmental and early postnatal stages would severely affect the brain structure, which may be related to the development of some types of young-onset mental disorders. Further investigations in conditional Reln knockout mice are needed to clarify this issue.
In humans, dysfunction of the PFC, which plays crucial roles in cognitive functions, has been implicated in many psychiatric disorders (Weinberger et al., 1988). Analogous to the human PFC, the medial PFC (mPFC) is involved in various cognitive functions and social behaviors in rodents (Ishii et al., 2015a). Brosda et al. (2011) demonstrated that Reelin knockdown in the mPFC resulted in behavioral alterations in young adult rats, including disruption of sensorimotor gaiting and deficits in spatial working memory as well as object recognition. Sui et al. (2012) also reported that the expression level of Reelin was related to the regulation of LTP in the rat mPFC. They found that epigenetic regulation of Reln was involved in the induction of LTP in the mPFC, which resulted in behavioral alterations. Iafrati et al. (2014) also reported that HRM showed reduced dendritic spine density and abnormal LTP in the PFC. Interestingly, Ishii et al. (2015b) demonstrated dominant expression of VLDLR in both excitatory pyramidal neurons and GABAergic inhibitory interneurons, except for the migrating interneurons in the rostral migratory stream in the postnatal mPFC. As compared to the findings in the hippocampus, there is not much evidence to support the importance of Reelin signaling in the synaptic functions in the mPFC. Further investigations are needed to elucidate the role of Reelin in the functions of the mPFC relevant to mental illnesses.
Reelin and its Downstream Signaling Molecules as a Target of Therapeutic Intervention
The findings described in the above sections suggest that Reelin pathways could be potentially useful as targets of therapeutic interventions for neuropsychiatric disorders. Indeed, several groups have directly assessed this possibility by examining the effects of administration of Reelin protein into the mouse brain (Rogers et al., 2011, 2013; Hethorn et al., 2015; Ishii et al., 2015b). Ishii et al. (2015b) demonstrated that Reelin exerted a preventive effect on phencyclidine (PCP)-induced behavioral deficits. They injected a conditioned medium containing Reelin protein into the mouse cerebral ventricles before administering PCP, and assessed the behavior of the animals. The group of mice that had received prior administration of Reelin showed normal cognitive and sensory-motor gating, indicating that the PCP-induced brain dysfunction was prevented by the Reelin injection. This study is based on their previous findings that prior transplantation of GABAergic neuronal progenitors into the mPFC of mice prevented the behavioral deficits induced by PCP and that the transplanted progenitors preferentially differentiated into Reelin/somatostatin-double positive GABAergic neurons specifically in the mPFC (Tanaka et al., 2011). Another group demonstrated that in vivo injection of Reelin into the mouse cerebral ventricle affected the synaptic functions and cognitive functions in wild-type mice and HRM (Rogers et al., 2011, 2013). The same group further showed that Reelin administration ameliorated both the synaptic plasticity and the cognitive behavioral deficits in a mouse model of Angelman syndrome, which is characterized by mental retardation, absence of speech, seizures and motor dysfunction (Hethorn et al., 2015).
Although we cannot directly apply this strategy of directly injecting Reelin into the human brain, modulating the activities of the signaling molecules downstream of Reelin could be a potential therapeutic approach. Reelin has been shown to activate PI3K and Akt (protein kinase B), to inhibit glycogen synthase kinase 3β (GSK3 β; Beffert et al., 2002) and activate the mTOR-S6K1 pathway (Jossin and Goffinet, 2007). Among them, GSK3 is already known as a target for mood stabilizers (De Sarno et al., 2002) and antipsychotics (Emamian et al., 2004; Li et al., 2007). In addition, animal models with disrupted mTOR signaling exhibit cognitive and behavioral deficits mimicking neuropsychiatric symptoms (Gururajan and van den Buuse, 2014). On the other hand, rapamycin, a well-established mTORC1 inhibitor, has been demonstrated to rescue cognitive impairments in several neurodevelopmental models of neuropsychiatric disorders, such as those of serotonin receptor (5-HT6) activation, neonatal PCP treatment, and post-weaning social isolation (Meffre et al., 2012), indicating overstimulation of mTOR signaling in these models. Surprisingly, rapamycin treatment also rescues lamination defects in tuberous sclerosis complex (TSC) 2-deficient mice, in which loss of TSC2 leads to activation of mTOR signaling and aberrant regulation of Reelin-Dab1 signaling (Moon et al., 2015). This inconsistency in the mTOR activities between different models might be explained by the varied activities of mTOR signaling as well as Reelin signaling among different brain regions and/or neuronal populations. Since such variation of Reelin (and its downstream) signaling activities could underlie the complex behavioral phenotypes of human neuropsychiatric disorders, further studies are required to distinguish clinical phenotypes and/or subpopulations that reflect disrupted Reelin signaling.
Conclusion
In the developing cortex, Reelin is a key regulator of neuronal migration and laminar formation, which are essential for achieving higher brain functions in mammals. Intriguingly, however, the expression pattern of Reelin and its functions are dramatically different in the postnatal brain as compared to the embryonic brain. Indeed, Reelin modulates synaptic functions, which may also be based on the structural changes of the dendrites and spines in the postnatal period, which are closely related to cognitive behaviors and predisposition to neuropsychiatric symptoms. Although genetic studies have lent support to the notion of an association between the RELN gene and neuropsychiatric disorders, recent large-scale genome-wide analyses have revealed that the contribution of RELN mutations/polymorphisms to the development of neuropsychiatric disorders is not much higher than previously thought. However, many studies have indicated multiple mechanisms underlying the regulation of Reelin production, e.g., epigenetic regulation, cleavage events, and subtype specification of GABAergic interneurons. Perturbation of these regulatory mechanisms can also lead to brain dysfunction. The findings of animal experiments suggest that Reelin and its downstream signaling are closely related to the synaptic functions that underlie the mouse behaviors relevant to neuropsychiatric disorders. Therefore, the Reelin pathway is a potential therapeutic intervention target for neuropsychiatric disorders. Further investigations are needed to clarify how Reelin signaling regulates the higher brain functions and how it is involved in the development of neuropsychiatric disorders.
Author Contributions
All authors contributed equally to the ideas and editing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Nakajima Laboratory for valuable discussions. This work supported by grants form the MEXT/JSPS KAKENHI (JP 16H06482, JP 15H01586, JP 15H02355, JP 15H01293, JP 26430075, JP 15K09723, JP 16K09997), Keio Gijuku Academic Development Funds, Keio Gijuku Fukuzawa Memorial Fund for the Advancement of Education and Research, Program for the Advancement of Keio Next Generation Research Projects, Takeda Science Foundation, and The Naito Foundation.
References
Abdolmaleky, H. M., Cheng, K. H., Russo, A., Smith, C. L., Faraone, S. V., Wilcox, M., et al. (2005). Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am. J. Med. Genet. B Neuropsychiatr. Genet. 134B, 60–66. doi: 10.1002/ajmg.b.30140
Alcántara, S., Ruiz, M., D’Arcangelo, G., Ezan, F., de Lecea, L., Curran, T., et al. (1998). Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J. Neurosci. 18, 7779–7799.
Anderson, S. A., Eisenstat, D. D., Shi, L., and Rubenstein, J. L. (1997). Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science 278, 474–476. doi: 10.1126/science.278.5337.474
Antoniades, D., Katopodi, T., Pappa, S., Lampropoulos, A., Konsta, V., Frydas, E., et al. (2011). The role of reelin gene polymorphisms in the pathogenesis of Alzheimer’s disease in a Greek population. J. Biol. Regul. Homeost. Agents 25, 351–358.
Arnold, S. E. (2000). Cellular and molecular neuropathology of the parahippocampal region in schizophrenia. Ann. N Y Acad. Sci. 911, 275–292. doi: 10.1111/j.1749-6632.2000.tb06732.x
Baek, S. T., Copeland, B., Yun, E. J., Kwon, S. K., Guemez-Gamboa, A., Schaffer, A. E., et al. (2015). An AKT3-FOXG1-reelin network underlies defective migration in human focal malformations of cortical development. Nat. Med. 21, 1445–1454. doi: 10.1038/nm.3982
Bal, M., Leitz, J., Reese, A. L., Ramirez, D. M., Durakoglugil, M., Herz, J., et al. (2013). Reelin mobilizes a VAMP7-dependent synaptic vesicle pool and selectively augments spontaneous neurotransmission. Neuron 80, 934–946. doi: 10.1016/j.neuron.2013.08.024
Ballif, B. A., Arnaud, L., Arthur, W. T., Guris, D., Imamoto, A., and Cooper, J. A. (2004). Activation of a Dab1/CrkL/C3G/Rap1 pathway in Reelin-stimulated neurons. Curr. Biol. 14, 606–610. doi: 10.1016/j.cub.2004.03.038
Bar, I., Lambert De Rouvroit, C., Royaux, I., Krizman, D. B., Dernoncourt, C., Ruelle, D., et al. (1995). A YAC contig containing the reeler locus with preliminary characterization of candidate gene fragments. Genomics 26, 543–549. doi: 10.1016/0888-7543(95)80173-j
Barr, A. M., Fish, K. N., and Markou, A. (2007). The reelin receptors VLDLR and ApoER2 regulate sensorimotor gating in mice. Neuropharmacology 52, 1114–1123. doi: 10.1016/j.neuropharm.2006.11.011
Barr, A. M., Fish, K. N., Markou, A., and Honer, W. G. (2008). Heterozygous reeler mice exhibit alterations in sensorimotor gating but not presynaptic proteins. Eur. J. Neurosci. 27, 2568–2574. doi: 10.1111/j.1460-9568.2008.06233.x
Beffert, U., Morfini, G., Bock, H. H., Reyna, H., Brady, S. T., and Herz, J. (2002). Reelin-mediated signaling locally regulates protein kinase B/Akt and glycogen synthase kinase 3β. J. Biol. Chem. 277, 49958–49964. doi: 10.1074/jbc.m209205200
Beffert, U., Weeber, E. J., Durudas, A., Qiu, S., Masiulis, I., Sweatt, J. D., et al. (2005). Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron 47, 567–579. doi: 10.1016/j.neuron.2005.07.007
Ben-David, E., Shifman, S., and International Schizophrenia Consortium. (2010). Further investigation of the association between rs7341475 and rs17746501 and schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B, 1244–1247. doi: 10.1002/ajmg.b.31093
Bonora, E., Beyer, K. S., Lamb, J. A., Parr, J. R., Klauck, S. M., Benner, A., et al. (2003). Analysis of reelin as a candidate gene for autism. Mol. Psychiatry 8, 885–892. doi: 10.1038/sj.mp.4001310
Botella-López, A., Burgaya, F., Gavín, R., Garcia-Ayllón, M. S., Gómez-Tortosa, E., Peña-Casanova, J., et al. (2006). Reelin expression and glycosylation patterns are altered in Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 103, 5573–5578. doi: 10.1073/pnas.0601279103
Boyle, M. P., Bernard, A., Thompson, C. L., Ng, L., Boe, A., Mortrud, M., et al. (2011). Cell-type-specific consequences of Reelin deficiency in the mouse neocortex, hippocampus and amygdala. J. Comp. Neurol. 519, 2061–2089. doi: 10.1002/cne.22655
Brandes, C., Novak, S., Stockinger, W., Herz, J., Schneider, W. J., and Nimpf, J. (1997). Avian and murine LR8B and human apolipoprotein E receptor 2: differentially spliced products from corresponding genes. Genomics 42, 185–191. doi: 10.1006/geno.1997.4702
Brosda, J., Dietz, F., and Koch, M. (2011). Impairment of cognitive performance after reelin knockdown in the medial prefrontal cortex of pubertal or adult rats. Neurobiol. Dis. 44, 239–247. doi: 10.1016/j.nbd.2011.07.008
Bufill, E., Roura-Poch, P., Sala-Matavera, I., Antón, S., Lleó, A., Sánchez-Saudinós, B., et al. (2015). Reelin signaling pathway genotypes and Alzheimer disease in a Spanish population. Alzheimer Dis. Assoc. Disord. 29, 169–172. doi: 10.1097/WAD.0000000000000002
Caviness, V. S. Jr. (1982). Neocortical histogenesis in normal and reeler mice: a developmental study based upon [3H]thymidine autoradiography. Brain Res. 256, 293–302. doi: 10.1016/0165-3806(82)90141-9
Caviness, V. S. Jr., and Sidman, R. L. (1973). Time of origin or corresponding cell classes in the cerebral cortex of normal and reeler mutant mice: an autoradiographic analysis. J. Comp. Neurol. 148, 141–151. doi: 10.1002/cne.901480202
Chen, Y., Beffert, U., Ertunc, M., Tang, T. S., Kavalali, E. T., Bezprozvanny, I., et al. (2005). Reelin modulates NMDA receptor activity in cortical neurons. J. Neurosci. 25, 8209–8216. doi: 10.1523/jneurosci.1951-05.2005
Chen, Y., Durakoglugil, M. S., Xian, X., and Herz, J. (2010). ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc. Natl. Acad. Sci. U S A 107, 12011–12016. doi: 10.1073/pnas.0914984107
Chen, K., Ochalski, P. G., Tran, T. S., Sahir, N., Schubert, M., Pramatarova, A., et al. (2004). Interaction between Dab1 and CrkII is promoted by Reelin signaling. J. Cell Sci. 117, 4527–4536. doi: 10.1242/jcs.01320
Costa, E., Davis, J., Pesold, C., Tueting, P., and Guidotti, A. (2002). The heterozygote reeler mouse as a model for the development of a new generation of antipsychotics. Curr. Opin. Pharmacol. 2, 56–62. doi: 10.1016/s1471-4892(01)00121-7
D’Arcangelo, G., Miao, G. G., Chen, S. C., Soares, H. D., Morgan, J. I., and Curran, T. (1995). A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374, 719–723. doi: 10.1038/374719a0
Del Rio, J. A., Heimrich, B., Borrell, V., Förster, E., Drakew, A., Alcántara, S., et al. (1997). A role for Cajal-Retzius cells and reelin in the development of hippocampal connections. Nature 385, 70–74. doi: 10.1038/385070a0
De Sarno, P., Li, X., and Jope, R. S. (2002). Regulation of Akt and glycogen synthase kinase-3β phosphorylation by sodium valproate and lithium. Neuropharmacology 43, 1158–1164. doi: 10.1016/s0028-3908(02)00215-0
Devlin, B., Bennett, P., Dawson, G., Figlewicz, D. A., Grigorenko, E. L., McMahon, W., et al. (2004). Alleles of a reelin CGG repeat do not convey liability to autism in a sample from the CPEA network. Am. J. Med. Genet. B Neuropsychiatr. Genet. 126B, 46–50. doi: 10.1002/ajmg.b.20125
Dutta, S., Guhathakurta, S., Sinha, S., Chatterjee, A., Ahmed, S., Ghosh, S., et al. (2007). Reelin gene polymorphisms in the Indian population: a possible paternal 5′UTR-CGG-repeat-allele effect on autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 106–112. doi: 10.1002/ajmg.b.30419
Dutta, S., Sinha, S., Ghosh, S., Chatterjee, A., Ahmed, S., and Usha, R. (2008). Genetic analysis of reelin gene (RELN) SNPs: no association with autism spectrum disorder in the Indian population. Neurosci. Lett. 441, 56–60. doi: 10.1016/j.neulet.2008.06.022
Eastwood, S. L., and Harrison, P. J. (2006). Cellular basis of reduced cortical reelin expression in schizophrenia. Am. J. Psychiatry 163, 540–542. doi: 10.1176/appi.ajp.163.3.540
Emamian, E. S., Hall, D., Birnbaum, M. J., Karayiorgou, M., and Gogos, J. A. (2004). Convergent evidence for impaired AKT1-GSK3β signaling in schizophrenia. Nat. Genet. 36, 131–137. doi: 10.1038/ng1296
Falconer, D. S. (1951). Two new mutants, ‘trembler’ and ‘reeler’, with neurological actions in the house mouse (Mus musculus L.). J. Genet. 50, 192–205. doi: 10.1007/bf02996215
Fatemi, S. H., Snow, A. V., Stary, J. M., Araghi-Niknam, M., Reutiman, T. J., Lee, S., et al. (2005). Reelin signaling is impaired in autism. Biol. Psychiatry 57, 777–787. doi: 10.1016/j.biopsych.2004.12.018
Fehér, A., Juhász, A., Pákáski, M., Kálmán, J., and Janka, Z. (2015). Genetic analysis of the RELN gene: gender specific association with Alzheimer’s disease. Psychiatry Res. 230, 716–718. doi: 10.1016/j.psychres.2015.09.021
Franco, S. J., Martinez-Garay, I., Gil-Sanz, C., Harkins-Perry, S. R., and Müller, U. (2011). Reelin regulates cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron 69, 482–497. doi: 10.1016/j.neuron.2011.01.003
Freitag, C. M., Kleser, C., Schneider, M., and von Gontard, A. (2007). Quantitative assessment of neuromotor function in adolescents with high functioning autism and Asperger Syndrome. J. Autism Dev. Disord. 37, 948–959. doi: 10.1007/s10803-006-0235-6
Gelman, D., Griveau, A., Dehorter, N., Teissier, A., Varela, C., Pla, R., et al. (2011). A wide diversity of cortical GABAergic interneurons derives from the embryonic preoptic area. J. Neurosci. 31, 16570–16580. doi: 10.1523/jneurosci.4068-11.2011
Gelman, D. M., Martini, F. J., Nóbrega-Pereira, S., Pierani, A., Kessaris, N., and Marín, O. (2009). The embryonic preoptic area is a novel source of cortical GABAergic interneurons. J. Neurosci. 29, 9380–9389. doi: 10.1523/jneurosci.0604-09.2009
Gil-Sanz, C., Franco, S. J., Martinez-Garay, I., Espinosa, A., Harkins-Perry, S., and Müller, U. (2013). Cajal-Retzius cells instruct neuronal migration by coincidence signaling between secreted and contact-dependent guidance cues. Neuron 79, 461–477. doi: 10.1016/j.neuron.2013.06.040
Goes, F. S., Willour, V. L., Zandi, P. P., Belmonte, P. L., MacKinnon, D. F., Mondimore, F. M., et al. (2010). Sex-specific association of the Reelin gene with bipolar disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B, 549–553. doi: 10.1002/ajmg.b.31018
Grayson, D. R., Jia, X., Chen, Y., Sharma, R. P., Mitchell, C. P., Guidotti, A., et al. (2005). Reelin promoter hypermethylation in schizophrenia. Proc. Natl. Acad. Sci. U S A 102, 9341–9346. doi: 10.1073/pnas.0503736102
Guidotti, A., Auta, J., Davis, J. M., Di-Giorgi-Gerevini, V., Dwivedi, Y., Grayson, D. R., et al. (2000). Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch. Gen. Psychiatry 57, 1061–1069. doi: 10.1001/archpsyc.57.11.1061
Gururajan, A., and van den Buuse, M. (2014). Is the mTOR-signalling cascade disrupted in Schizophrenia? J. Neurochem. 129, 377–387. doi: 10.1111/jnc.12622
He, Y., Xun, G., Xia, K., Hu, Z., Lv, L., Deng, Z., et al. (2011). No significant association between RELN polymorphism and autism in case-control and family-based association study in Chinese Han population. Psychiatry Res. 187, 462–464. doi: 10.1016/j.psychres.2010.04.051
Herz, J., and Chen, Y. (2006). Reelin, lipoprotein receptors and synaptic plasticity. Nat. Rev. Neurosci. 7, 850–859. doi: 10.1038/nrn2009
Hethorn, W. R., Ciarlone, S. L., Filonova, I., Rogers, J. T., Aguirre, D., Ramirez, R. A., et al. (2015). Reelin supplementation recovers synaptic plasticity and cognitive deficits in a mouse model for Angelman syndrome. Eur. J. Neurosci. 41, 1372–1380. doi: 10.1111/ejn.12893
Hiesberger, T., Trommsdorff, M., Howell, B. W., Goffinet, A., Mumby, M. C., Cooper, J. A., et al. (1999). Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 24, 481–489. doi: 10.1016/s0896-6273(00)80861-2
Hirota, Y., Kubo, K., Katayama, K., Honda, T., Fujino, T., Yamamoto, T. T., et al. (2015). Reelin receptors ApoER2 and VLDLR are expressed in distinct spatiotemporal patterns in developing mouse cerebral cortex. J. Comp. Neurol. 523, 463–478. doi: 10.1002/cne.23691
Hirotsune, S., Takahara, T., Sasaki, N., Hirose, K., Yoshiki, A., Ohashi, T., et al. (1995). The reeler gene encodes a protein with an EGF-like motif expressed by pioneer neurons. Nat. Genet. 10, 77–83. doi: 10.1038/ng0595-77
Honda, T., Kobayashi, K., Mikoshiba, K., and Nakajima, K. (2011). Regulation of cortical neuron migration by the Reelin signaling pathway. Neurochem. Res. 36, 1270–1279. doi: 10.1007/s11064-011-0407-4
Hong, S. E., Shugart, Y. Y., Huang, D. T., Shahwan, S. A., Grant, P. E., Hourihane, J. O., et al. (2000). Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet. 26, 93–96. doi: 10.1038/79246
Howell, B. W., Hawkes, R., Soriano, P., and Cooper, J. A. (1997). Neuronal position in the developing brain is regulated by mouse disabled-1. Nature 389, 733–737. doi: 10.1038/39607
Howell, B. W., Herrick, T. M., and Cooper, J. A. (1999). Reelin-induced tyrosine [corrected] phosphorylation of disabled 1 during neuronal positioning. Genes Dev. 13, 643–648. doi: 10.1101/gad.13.6.643
Howell, K. R., and Pillai, A. (2016). Long-term effects of prenatal hypoxia on schizophrenia-like phenotype in heterozygous reeler mice. Mol. Neurobiol. 53, 3267–3276. doi: 10.1007/s12035-015-9265-4
Iafrati, J., Orejarena, M. J., Lassalle, O., Bouamrane, L., Gonzalez-Campo, C., and Chavis, P. (2014). Reelin, an extracellular matrix protein linked to early onset psychiatric diseases, drives postnatal development of the prefrontal cortex via GluN2B-NMDARs and the mTOR pathway. Mol. Psychiatry 19, 417–426. doi: 10.1038/mp.2013.66
Imai, H., Shoji, H., Ogata, M., Kagawa, Y., Owada, Y., Miyakawa, T., et al. (2016). Dorsal forebrain-specific deficiency of reelin-Dab1 signal causes behavioral abnormalities related to psychiatric disorders. Cereb. Cortex doi: 10.1093/cercor/bhv334 [Epub ahead of print].
Ishii, K., Kubo, K., Endo, T., Yoshida, K., Benner, S., Ito, Y., et al. (2015a). Neuronal heterotopias affect the activities of distant brain areas and lead to behavioral deficits. J. Neurosci. 35, 12432–12445. doi: 10.1523/jneurosci.3648-14.2015
Ishii, K., Nagai, T., Hirota, Y., Noda, M., Nabeshima, T., Yamada, K., et al. (2015b). Reelin has a preventive effect on phencyclidine-induced cognitive and sensory-motor gating deficits. Neurosci. Res. 96, 30–36. doi: 10.1016/j.neures.2014.12.013
Jossin, Y., and Cooper, J. A. (2011). Reelin, Rap1 and N-cadherin orient the migration of multipolar neurons in the developing neocortex. Nat. Neurosci. 14, 697–703. doi: 10.1038/nn.2816
Jossin, Y., and Goffinet, A. M. (2001). Reelin does not directly influence axonal growth. J. Neurosci. 21:RC183.
Jossin, Y., and Goffinet, A. M. (2007). Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Mol. Cell. Biol. 27, 7113–7124. doi: 10.1128/mcb.00928-07
Kanatani, S., Honda, T., Aramaki, M., Hayashi, K., Kubo, K., Ishida, M., et al. (2015). The COUP-TFII/Neuropilin-2 is a molecular switch steering diencephalon-derived GABAergic neurons in the developing mouse brain. Proc. Natl. Acad. Sci. U S A 112, E4985–E4994. doi: 10.1073/pnas.1420701112
Kanatani, S., Yozu, M., Tabata, H., and Nakajima, K. (2008). COUP-TFII is preferentially expressed in the caudal ganglionic eminence and is involved in the caudal migratory stream. J. Neurosci. 28, 13582–13591. doi: 10.1523/jneurosci.2132-08.2008
Knuesel, I., Nyffeler, M., Mormède, C., Muhia, M., Meyer, U., Pietropaolo, S., et al. (2009). Age-related accumulation of Reelin in amyloid-like deposits. Neurobiol. Aging 30, 697–716. doi: 10.1016/j.neurobiolaging.2007.08.011
Kocherhans, S., Madhusudan, A., Doehner, J., Breu, K. S., Nitsch, R. M., Fritschy, J. M., et al. (2010). Reduced Reelin expression accelerates amyloid-β plaque formation and tau pathology in transgenic Alzheimer’s disease mice. J. Neurosci. 30, 9228–9240. doi: 10.1523/JNEUROSCI.0418-10.2010
Kohno, T., Honda, T., Kubo, K., Nakano, Y., Tsuchiya, A., Murakami, T., et al. (2015). Importance of Reelin C-terminal region in the development and maintenance of the postnatal cerebral cortex and its regulation by specific proteolysis. J. Neurosci. 35, 4776–4787. doi: 10.1523/JNEUROSCI.4119-14.2015
Kramer, P. L., Xu, H., Woltjer, R. L., Westaway, S. K., Clark, D., Erten-Lyons, D., et al. (2011). Alzheimer disease pathology in cognitively healthy elderly: a genome-wide study. Neurobiol. Aging 32, 2113–2122. doi: 10.1016/j.neurobiolaging.2010.01.010
Krebs, M. O., Betancur, C., Leroy, S., Bourdel, M. C., Gillberg, C., Leboyer, M., et al. (2002). Absence of association between a polymorphic GGC repeat in the 5′ untranslated region of the reelin gene and autism. Mol. Psychiatry 7, 801–804. doi: 10.1038/sj.mp.4001071
Kuang, W. J., Sun, R. F., Zhu, Y. S., and Li, S. B. (2011). A new single-nucleotide mutation (rs362719) of the reelin (RELN) gene associated with schizophrenia in female Chinese Han. Genet. Mol. Res. 10, 1650–1658. doi: 10.4238/vol10-3gmr1343
Kubo, K., Honda, T., Tomita, K., Sekine, K., Ishii, K., Uto, A., et al. (2010). Ectopic Reelin induces neuronal aggregation with a normal birthdate-dependent “inside-out” alignment in the developing neocortex. J. Neurosci. 30, 10953–10966. doi: 10.1523/JNEUROSCI.0486-10.2010
Kutiyanawalla, A., Promsote, W., Terry, A., and Pillai, A. (2012). Cysteamine treatment ameliorates alterations in GAD67 expression and spatial memory in heterozygous reeler mice. Int. J. Neuropsychopharmacol. 15, 1073–1086. doi: 10.1017/s1461145711001180
Lane-Donovan, C., Philips, G. T., Wasser, C. R., Durakoglugil, M. S., Masiulis, I., Upadhaya, A., et al. (2015). Reelin protects against amyloid β toxicity in vivo. Sci. Signal. 8:ra67. doi: 10.1126/scisignal.aaa6674
Laviola, G., Ognibene, E., Romano, E., Adriani, W., and Keller, F. (2009). Gene-environment interaction during early development in the heterozygous reeler mouse: clues for modelling of major neurobehavioral syndromes. Neurosci. Biobehav. Rev. 33, 560–572. doi: 10.1016/j.neubiorev.2008.09.006
Li, W., Guo, X., and Xiao, S. (2015). Evaluating the relationship between reelin gene variants (rs7341475 and rs262355) and schizophrenia: a meta-analysis. Neurosci. Lett. 609, 42–47. doi: 10.1016/j.neulet.2015.10.014
Li, H., Li, Y., Shao, J., Li, R., Qin, Y., Xie, C., et al. (2008). The association analysis of RELN and GRM8 genes with autistic spectrum disorder in Chinese Han population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 194–200. doi: 10.1002/ajmg.b.30584
Li, M., Luo, X. J., Xiao, X., Shi, L., Liu, X. Y., Yin, L. D., et al. (2013). Analysis of common genetic variants identifies RELN as a risk gene for schizophrenia in Chinese population. World J. Biol. Psychiatry 14, 91–99. doi: 10.3109/15622975.2011.587891
Li, J., Nguyen, L., Gleason, C., Lotspeich, L., Spiker, D., Risch, N., et al. (2004). Lack of evidence for an association between WNT2 and RELN polymorphisms and autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 126B, 51–57. doi: 10.1002/ajmg.b.20122
Li, X., Rosborough, K. M., Friedman, A. B., Zhu, W., and Roth, K. A. (2007). Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int. J. Neuropsychopharmacol. 10, 7–19. doi: 10.1017/s1461145706006547
Li, W., Song, X., Zhang, H., Yang, Y., Jiang, C., Xiao, B., et al. (2011). Association study of RELN polymorphisms with schizophrenia in Han Chinese population. Prog. Neuropsychopharmacol. Biol. Psychiatry 35, 1505–1511. doi: 10.1016/j.pnpbp.2011.04.007
Liu, X. Y., Li, M., Yang, S. Y., Su, B., and Yin, L. D. (2011). Association of RELN SNP rs7341475 with schizophrenia in the Chinese population. Dongwuxue Yanjiu 32, 499–503. doi: 10.3724/SP.J.1141.2011.05499
Maenner, M. J., Rice, C. E., Arneson, C. L., Cunniff, C., Schieve, L. A., Carpenter, L. A., et al. (2014). Potential impact of DSM-5 criteria on autism spectrum disorder prevalence estimates. JAMA Psychiatry 71, 292–300. doi: 10.1001/jamapsychiatry.2013.3893
Marin, O., Valiente, M., Ge, X., and Tsai, L. H. (2010). Guiding neuronal cell migrations. Cold Spring Harb. Perspect. Biol. 2:a001834. doi: 10.1101/cshperspect.a001834
Meffre, J., Chaumont-Dubel, S., Mannoury la Cour, C., Loiseau, F., Watson, D. J., Dekeyne, A., et al. (2012). 5-HT(6) receptor recruitment of mTOR as a mechanism for perturbed cognition in schizophrenia. EMBO Mol. Med. 4, 1043–1056. doi: 10.1002/emmm.201201410
Mill, J., Tang, T., Kaminsky, Z., Khare, T., Yazdanpanah, S., Bouchard, L., et al. (2008). Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am. J. Hum. Genet. 82, 696–711. doi: 10.1016/j.ajhg.2008.01.008
Miyoshi, G., Hjerling-Leffler, J., Karayannis, T., Sousa, V. H., Butt, S. J., Battiste, J., et al. (2010). Genetic fate mapping reveals that the caudal ganglionic eminence produces a large and diverse population of superficial cortical interneurons. J. Neurosci. 30, 1582–1594. doi: 10.1523/JNEUROSCI.4515-09.2010
Molnar, Z., Metin, C., Stoykova, A., Tarabykin, V., Price, D. J., Francis, F., et al. (2006). Comparative aspects of cerebral cortical development. Eur. J. Neurosci. 23, 921–934. doi: 10.1111/j.1460-9568.2006.04611.x
Moon, U. Y., Park, J. Y., Park, R., Cho, J. Y., Hughes, L. J., McKenna, J., et al. (2015). Impaired reelin-Dab1 signaling contributes to neuronal migration deficits of tuberous sclerosis complex. Cell Rep. 12, 965–978. doi: 10.1016/j.celrep.2015.07.013
Nadarajah, B., Brunstrom, J. E., Grutzendler, J., Wong, R. O., and Pearlman, A. L. (2001). Two modes of radial migration in early development of the cerebral cortex. Nat. Neurosci. 4, 143–150. doi: 10.1038/83967
Nakajima, K., Mikoshiba, K., Miyata, T., Kudo, C., and Ogawa, M. (1997). Disruption of hippocampal development in vivo by CR-50 mAb against reelin. Proc. Natl. Acad. Sci. U S A 94, 8196–8201. doi: 10.1073/pnas.94.15.8196
Ogawa, M., Miyata, T., Nakajima, K., Yagyu, K., Seike, M., Ikenaka, K., et al. (1995). The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron 14, 899–912. doi: 10.1016/0896-6273(95)90329-1
Olson, E. C., Kim, S., and Walsh, C. A. (2006). Impaired neuronal positioning and dendritogenesis in the neocortex after cell-autonomous Dab1 suppression. J. Neurosci. 26, 1767–1775. doi: 10.1523/JNEUROSCI.3000-05.2006
Ovadia, G., and Shifman, S. (2011). The genetic variation of RELN expression in schizophrenia and bipolar disorder. PLoS One 6:e19955. doi: 10.1371/journal.pone.0019955
Park, T. J., and Curran, T. (2008). Crk and Crk-like play essential overlapping roles downstream of disabled-1 in the Reelin pathway. J. Neurosci. 28, 13551–13562. doi: 10.1523/JNEUROSCI.4323-08.2008
Persico, A. M., D’Agruma, L., Maiorano, N., Totaro, A., Militerni, R., Bravaccio, C., et al. (2001). Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol. Psychiatry 6, 150–159. doi: 10.1038/sj.mp.4000850
Pesold, C., Impagnatiello, F., Pisu, M. G., Uzunov, D. P., Costa, E., Guidotti, A., et al. (1998). Reelin is preferentially expressed in neurons synthesizing γ-aminobutyric acid in cortex and hippocampus of adult rats. Proc. Natl. Acad. Sci. U S A 95, 3221–3226. doi: 10.1073/pnas.95.6.3221
Podhorna, J., and Didriksen, M. (2004). The heterozygous reeler mouse: behavioural phenotype. Behav. Brain Res. 153, 43–54. doi: 10.1016/j.bbr.2003.10.033
Pramatarova, A., Ochalski, P. G., Chen, K., Gropman, A., Myers, S., Min, K. T., et al. (2003). Nck β interacts with tyrosine-phosphorylated disabled 1 and redistributes in Reelin-stimulated neurons. Mol. Cell. Biol. 23, 7210–7221. doi: 10.1128/mcb.23.20.7210-7221.2003
Pujadas, L., Gruart, A., Bosch, C., Delgado, L., Teixeira, C. M., Rossi, D., et al. (2010). Reelin regulates postnatal neurogenesis and enhances spine hypertrophy and long-term potentiation. J. Neurosci. 30, 4636–4649. doi: 10.1523/JNEUROSCI.5284-09.2010
Qiu, S., Korwek, K. M., Pratt-Davis, A. R., Peters, M., Bergman, M. Y., and Weeber, E. J. (2006a). Cognitive disruption and altered hippocampus synaptic function in Reelin haploinsufficient mice. Neurobiol. Learn. Mem. 85, 228–242. doi: 10.1016/j.nlm.2005.11.001
Qiu, S., Zhao, L. F., Korwek, K. M., and Weeber, E. J. (2006b). Differential reelin-induced enhancement of NMDA and AMPA receptor activity in the adult hippocampus. J. Neurosci. 26, 12943–12955. doi: 10.1523/JNEUROSCI.2561-06.2006
Rakic, P. (1972). Mode of cell migration to the superficial layers of fetal monkey neocortex. J. Comp. Neurol. 145, 61–83. doi: 10.1002/cne.901450105
Rakic, P. (2009). Evolution of the neocortex: a perspective from developmental biology. Nat. Rev. Neurosci. 10, 724–735. doi: 10.1038/nrn2719
Rice, D. S., and Curran, T. (2001). Role of the reelin signaling pathway in central nervous system development. Annu. Rev. Neurosci. 24, 1005–1039. doi: 10.1146/annurev.neuro.24.1.1005
Rodriguez, M. A., Pesold, C., Liu, W. S., Kriho, V., Guidotti, A., Pappas, G. D., et al. (2000). Colocalization of integrin receptors and reelin in dendritic spine postsynaptic densities of adult nonhuman primate cortex. Proc. Natl. Acad. Sci. U S A 97, 3550–3555. doi: 10.1073/pnas.050589797
Rogers, J. T., Rusiana, I., Trotter, J., Zhao, L., Donaldson, E., Pak, D. T., et al. (2011). Reelin supplementation enhances cognitive ability, synaptic plasticity and dendritic spine density. Learn. Mem. 18, 558–564. doi: 10.1101/lm.2153511
Rogers, J. T., Zhao, L., Trotter, J. H., Rusiana, I., Peters, M. M., Li, Q., et al. (2013). Reelin supplementation recovers sensorimotor gating, synaptic plasticity and associative learning deficits in the heterozygous reeler mouse. J. Psychopharmacol. 27, 386–395. doi: 10.1177/0269881112463468
Romano, E., De Angelis, F., Ulbrich, L., De Jaco, A., Fuso, A., and Laviola, G. (2014). Nicotine exposure during adolescence: cognitive performance and brain gene expression in adult heterozygous reeler mice. Psychopharmacology (Berl) 231, 1775–1787. doi: 10.1007/s00213-013-3388-y
Sáez-Valero, J., Costell, M., Sjögren, M., Andreasen, N., Blennow, K., and Luque, J. M. (2003). Altered levels of cerebrospinal fluid reelin in frontotemporal dementia and Alzheimer’s disease. J. Neurosci. Res. 72, 132–136. doi: 10.1002/jnr.10554
Salinger, W. L., Ladrow, P., and Wheeler, C. (2003). Behavioral phenotype of the reeler mutant mouse: effects of RELN gene dosage and social isolation. Behav. Neurosci. 117, 1257–1275. doi: 10.1037/0735-7044.117.6.1257
Sawa, A., and Snyder, S. H. (2002). Schizophrenia: diverse approaches to a complex disease. Science 296, 692–695. doi: 10.1126/science.1070532
Scarpini, E., Scheltens, P., and Feldman, H. (2003). Treatment of Alzheimer’s disease: current status and new perspectives. Lancet Neurol. 2, 539–547. doi: 10.1016/s1474-4422(03)00502-7
Scheltens, P., Blennow, K., Breteler, M. M., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s disease. Lancet 388, 505–517. doi: 10.1016/s0140-6736(15)01124-1
Schizophrenia Working Group of the Psychiatric Genomics Consortium. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Sekine, K., Honda, T., Kawauchi, T., Kubo, K., and Nakajima, K. (2011). The outermost region of the developing cortical plate is crucial for both the switch of the radial migration mode and the Dab1-dependent “inside-out” lamination in the neocortex. J. Neurosci. 31, 9426–9439. doi: 10.1523/JNEUROSCI.0650-11.2011
Sekine, K., Kawauchi, T., Kubo, K., Honda, T., Herz, J., Hattori, M., et al. (2012). Reelin controls neuronal positioning by promoting cell-matrix adhesion via inside-out activation of integrin α5β1. Neuron 76, 353–369. doi: 10.1016/j.neuron.2012.07.020
Sekine, K., Kubo, K., and Nakajima, K. (2014). How does Reelin control neuronal migration and layer formation in the developing mammalian neocortex? Neurosci. Res. 86, 50–58. doi: 10.1016/j.neures.2014.06.004
Sekine, K., Tabata, H., and Nakajima, K. (2013). “Cell polarity and initiation of migration (Chapter 12),” in Comprehensive Developmental Neuroscience: Cellular Migration and Formation of Neuronal Connections, eds J. L. R. Rubenstein and P. Rakic (Amsterdam: Academic Press), 231–244.
Serajee, F. J., Zhong, H., and Mahbubul Huq, A. H. (2006). Association of Reelin gene polymorphisms with autism. Genomics 87, 75–83. doi: 10.1016/j.ygeno.2005.09.008
Seripa, D., Matera, M. G., Franceschi, M., Daniele, A., Bizzarro, A., Rinaldi, M., et al. (2008). The RELN locus in Alzheimer’s disease. J. Alzheimers Dis. 14, 335–344.
Shifman, S., Johannesson, M., Bronstein, M., Chen, S. X., Collier, D. A., Craddock, N. J., et al. (2008). Genome-wide association identifies a common variant in the reelin gene that increases the risk of schizophrenia only in women. PLoS Genet. 4:e28. doi: 10.1371/journal.pgen.0040028
Skaar, D. A., Shao, Y., Haines, J. L., Stenger, J. E., Jaworski, J., Martin, E. R., et al. (2005). Analysis of the RELN gene as a genetic risk factor for autism. Mol. Psychiatry 10, 563–571. doi: 10.1038/sj.mp.4001614
Sui, L., Wang, Y., Ju, L. H., and Chen, M. (2012). Epigenetic regulation of reelin and brain-derived neurotrophic factor genes in long-term potentiation in rat medial prefrontal cortex. Neurobiol. Learn. Mem. 97, 425–440. doi: 10.1016/j.nlm.2012.03.007
Tabata, H., Kanatani, S., and Nakajima, K. (2009). Differences of migratory behavior between direct progeny of apical progenitors and basal progenitors in the developing cerebral cortex. Cereb. Cortex 19, 2092–2105. doi: 10.1093/cercor/bhn227
Takahashi, T., Goto, T., Miyama, S., Nowakowski, R. S., and Caviness, V. S. Jr. (1999). Sequence of neuron origin and neocortical laminar fate: relation to cell cycle of origin in the developing murine cerebral wall. J. Neurosci. 19, 10357–10371.
Tamamaki, N., Fujimori, K. E., and Takauji, R. (1997). Origin and route of tangentially migrating neurons in the developing neocortical intermediate zone. J. Neurosci. 17, 8313–8323.
Tanaka, D. H., Toriumi, K., Kubo, K., Nabeshima, T., and Nakajima, K. (2011). GABAergic precursor transplantation into the prefrontal cortex prevents phencyclidine-induced cognitive deficits. J. Neurosci. 31, 14116–14125. doi: 10.1523/JNEUROSCI.2786-11.2011
Teixeira, C. M., Martin, E. D., Sahun, I., Masachs, N., Pujadas, L., Corvelo, A., et al. (2011). Overexpression of Reelin prevents the manifestation of behavioral phenotypes related to schizophrenia and bipolar disorder. Neuropsychopharmacology 36, 2395–2405. doi: 10.1038/npp.2011.153
Telese, F., Ma, Q., Perez, P. M., Notani, D., Oh, S., Li, W., et al. (2015). LRP8-reelin-regulated neuronal enhancer signature underlying learning and memory formation. Neuron 86, 696–710. doi: 10.1016/j.neuron.2015.03.033
Teroganova, N., Girshkin, L., Suter, C. M., and Green, M. J. (2016). DNA methylation in peripheral tissue of schizophrenia and bipolar disorder: a systematic review. BMC Genet. 17:27. doi: 10.1186/s12863-016-0332-2
Tissir, F., and Goffinet, A. M. (2003). Reelin and brain development. Nat. Rev. Neurosci. 4, 496–505. doi: 10.1038/nrn1113
Tochigi, M., Iwamoto, K., Bundo, M., Komori, A., Sasaki, T., Kato, N., et al. (2008). Methylation status of the reelin promoter region in the brain of schizophrenic patients. Biol. Psychiatry 63, 530–533. doi: 10.1016/j.biopsych.2007.07.003
Trommsdorff, M., Gotthardt, M., Hiesberger, T., Shelton, J., Stockinger, W., Nimpf, J., et al. (1999). Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97, 689–701. doi: 10.1016/s0092-8674(00)80782-5
Trotter, J., Lee, G. H., Kazdoba, T. M., Crowell, B., Domogauer, J., Mahoney, H. M., et al. (2013). Dab1 is required for synaptic plasticity and associative learning. J. Neurosci. 33, 15652–15668. doi: 10.1523/JNEUROSCI.2010-13.2013
Tueting, P., Costa, E., Dwivedi, Y., Guidotti, A., Impagnatiello, F., Manev, R., et al. (1999). The phenotypic characteristics of heterozygous reeler mouse. Neuroreport 10, 1329–1334. doi: 10.1097/00001756-199904260-00032
Voss, A. K., Britto, J. M., Dixon, M. P., Sheikh, B. N., Collin, C., Tan, S. S., et al. (2008). C3G regulates cortical neuron migration, preplate splitting and radial glial cell attachment. Development 135, 2139–2149. doi: 10.1242/dev.016725
Wang, Z., Hong, Y., Zou, L., Zhong, R., Zhu, B., Shen, N., et al. (2014). Reelin gene variants and risk of autism spectrum disorders: an integrated meta-analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 165B, 192–200. doi: 10.1002/ajmg.b.32222
Wedenoja, J., Loukola, A., Tuulio-Henriksson, A., Paunio, T., Ekelund, J., Silander, K., et al. (2008). Replication of linkage on chromosome 7q22 and association of the regional Reelin gene with working memory in schizophrenia families. Mol. Psychiatry 13, 673–684. doi: 10.1038/sj.mp.4002047
Weeber, E. J., Beffert, U., Jones, C., Christian, J. M., Forster, E., Sweatt, J. D., et al. (2002). Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J. Biol. Chem. 277, 39944–39952. doi: 10.1074/jbc.M205147200
Wegiel, J., Kuchna, I., Nowicki, K., Imaki, H., Wegiel, J., Marchi, E., et al. (2010). The neuropathology of autism: defects of neurogenesis and neuronal migration and dysplastic changes. Acta. Neuropathol. 119, 755–770. doi: 10.1007/s00401-010-0655-4
Weinberger, D. R., Berman, K. F., and Illowsky, B. P. (1988). Physiological dysfunction of dorsolateral prefrontal cortex in schizophrenia. III. A new cohort and evidence for a monoaminergic mechanism. Arch. Gen. Psychiatry 45, 609–615. doi: 10.1001/archpsyc.1988.01800310013001
Yozu, M., Tabata, H., and Nakajima, K. (2005). The caudal migratory stream: a novel migratory stream of interneurons derived from the caudal ganglionic eminence in the developing mouse forebrain. J. Neurosci. 25, 7268–7277. doi: 10.1523/JNEUROSCI.2072-05.2005
Zhang, H., Liu, X., Zhang, C., Mundo, E., Macciardi, F., Grayson, D. R., et al. (2002). Reelin gene alleles and susceptibility to autism spectrum disorders. Mol. Psychiatry 7, 1012–1017. doi: 10.1038/sj.mp.4001124
Keywords: reelin, psychiatric disorder, schizophrenia, animal model, reeler
Citation: Ishii K, Kubo K-i and Nakajima K (2016) Reelin and Neuropsychiatric Disorders. Front. Cell. Neurosci. 10:229. doi: 10.3389/fncel.2016.00229
Received: 11 April 2016; Accepted: 22 September 2016;
Published: 18 October 2016.
Edited by:
Gabriella D’Arcangelo, Rutgers University, USAReviewed by:
Reno Cervo Reyes, University of California, San Francisco, USATheresa Pohlkamp, UT Southwestern Medical Center, USA
Copyright © 2016 Ishii, Kubo and Nakajima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazunori Nakajima, kazunori@keio.jp
†Present address: Kazuhiro Ishii, Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore, MD, USA