Riluzole But Not Melatonin Ameliorates Acute Motor Neuron Degeneration and Moderately Inhibits SOD1-Mediated Excitotoxicity Induced Disrupted Mitochondrial Ca2+ Signaling in Amyotrophic Lateral Sclerosis
 Manoj Kumar Jaiswal
Manoj Kumar Jaiswal- Center of Physiology, University of Göttingen, Göttingen, Germany
Selective motoneurons (MNs) degeneration in the brain stem, hypoglossal motoneurons (HMNs), and the spinal cord resulting in patients paralysis and eventual death are prominent features of amyotrophic lateral sclerosis (ALS). Previous studies have suggested that mitochondrial respiratory impairment, low Ca2+ buffering and homeostasis and excitotoxicity are the pathological phenotypes found in mice, and cell culture models of familial ALS (fALS) linked with Cu/Zn-superoxide dismutase 1 (SOD1) mutation. In our study, we aimed to understand the impact of riluzole and melatonin on excitotoxicity, neuronal protection and Ca2+ signaling in individual HMNs ex vivo in symptomatic adult ALS mouse brain stem slice preparations and in WT and SOD1-G93A transfected SH-SY5Y neuroblastoma cell line using fluorescence microscopy, calcium imaging with high speed charged coupled device camera, together with immunohistochemistry, cell survival assay and histology. In our experiments, riluzole but not melatonin ameliorates MNs degeneration and moderately inhibit excitotoxicity and cell death in SH-SY5YWT or SH-SY5YG93A cell lines induced by complex IV blocker sodium azide. In brain stem slice preparations, riluzole significantly inhibit HMNs cell death induced by inhibiting the mitochondrial electron transport chain by Na-azide. In the HMNs of brainstem slice prepared from adult (14–15 weeks) WT, and corresponding symptomatic SOD1G93A mice, we measured the effect of riluzole and melatonin on [Ca2+]i using fura-2 AM ratiometric calcium imaging in individual MNs. Riluzole caused a significant decrease in [Ca2+]i transients and reversibly inhibited [Ca2+]i transients in Fura-2 AM loaded HMNs exposed to Na-azide in adult symptomatic SOD1G93A mice. On the contrary, melatonin failed to show similar effects in the HMNs of WT and SOD1G93A mice. Intrinsic nicotinamide adenine dinucleotide (NADH) fluorescence, an indicator of mitochondrial metabolism and health in MNs, showed enhanced intrinsic NADH fluorescence in HMNs in presence of riluzole when respiratory chain activity was inhibited by Na-azide. Riluzole’s inhibition of excitability and Ca2+ signaling may be due to its multiple effects on cellular function of mitochondria. Therefore formulating a drug therapy to stabilize mitochondria-related signaling pathways using riluzole might be a valuable approach for cell death protection in ALS. Taken together, the pharmacological profiles of the riluzole and melatonin strengthen the case that riluzole indeed can be used as a therapeutic agent in ALS whereas claims of the efficacy of melatonin alone need further investigation as it fail to show significant neuroprotection efficacy.
Introduction
One of the hallmarks of ALS is extensive loss of MNs in the brain stem, hypoglossal nucleus, facial nucleus, and the spinal cord (Spalloni et al., 2004; Von Lewinski and Keller, 2005a,b; Spiller et al., 2016). Evidence is increasing that Ca2+ dysregulation and mitochondrial dysfunction is involved in the SOD1 G93A (SOD1G93A) mouse model (Kong and Xu, 1998; Jaiswal and Keller, 2009; Jaiswal, 2013, 2014; Chang and Martin, 2016; Paine and Jaiswal, 2016). The precise mechanisms leading to the selective loss of MNs in ALS patients as well as a possible determinant of selective MNs death in transgenic (Tg) mouse models of this disorder remain elusive. Many findings indicated two important characteristic features of this disorder: (1) the presence of low level of calcium binding proteins in combination with high buffering capacity in affected MNs (Alexianu et al., 1994; Kaal et al., 1998; Laslo et al., 2000; Jaiswal et al., 2009; Jaiswal, 2014) and (2) involvement of AMPA receptors (AMPAR; Rothstein, 1995; Van Den Bosch et al., 2000; Udagawa et al., 2015; Chang and Martin, 2016).
Riluzole is the only FDA-approved neuroprotective compound used in clinics for the treatment of ALS/MN disease (Bensimon et al., 1994; Jehle et al., 2000; Obinu et al., 2002; Gutierrez et al., 2016). Riluzole’s neuroprotective properties appear to arise from both presynaptic and postsynaptic mechanisms for attenuating glutamatergic neurotransmission, allowing riluzole to act as an antiglutamate drug (Bryson et al., 1996; Doble, 1996; Wokke, 1996; Geevasinga et al., 2016) and it block depolarization-evoked Ca2+ transients in MNs (Hubert et al., 1998). Moreover, many groups reported that riluzole works by inhibiting Na+ channels (Stefani et al., 1997; Yokoo et al., 1998; Zona et al., 1998; Geevasinga et al., 2016), inhibits Cl- channels (Bausch and Roy, 1996), increases glutamate uptake and has an impact on glutamate receptors (Azbill et al., 2000) and inhibits GABA reuptake (Kretschmer et al., 1998). Additionally, riluzole was shown to attenuate neurotoxicity in tissue preparations and an animal model as well (Lang-Lazdunski et al., 2000; Mu et al., 2000). Earlier it was shown that an increase in [Ca2+]i is a critical messenger for diverse pathophysiological events or in case of ALS and other neurodegenerative disorder and generally abnormal [Ca2+]i increases are cytotoxic (Bootman et al., 1993; Berridge, 1997; Budd, 1998). Riluzole is known to block persistent sodium currents and earlier it was shown that riluzole block the Trpm4 (pore-forming subunits of Sur1-regulated NCCa-ATP channels), a key mechanism for the beneficial effect of riluzole in spinal cord injury (Simard et al., 2012). Moreover, Trpm4 is a Ca2+-activated cation channel that were found to expressed at higher levels in Th2 cells and inhibition of Trpm4 expression increased Ca2+ influx (Weber et al., 2010) via Ca2+ release-activated Ca2+ (CRAC) channel and Ca2+-activated K+ current (Kca) channels (Dolmetsch and Lewis, 1994). More recently it was discovered that Ca2+ influx and oscillations are also regulated by Trpm4 in Jurkat T cells (Launay et al., 2004). Work from all these previous studies indicated three compelling reasons for the beneficial effects of riluzole (i) block of persistent Na+ currents (molecular mechanism not known), (ii) inhibition of glutamatergic signaling pathways and (iii) an indirect neurotrophic effect. In spite of all these discoveries, it is not precisely established yet the precise mechanism through which riluzole acts on ion channels either directly or through second messenger signaling cascades, which attenuate ion influx-induced neuronal death.
Another effective cellular antioxidant known to be efficiently scavenging toxic free radicals, ROS and associated reactants is Melatonin (Jou et al., 2004; Reiter et al., 2016). Recent findings in which azides/CN-induced cell death via blocking complex IV of the mitochondrial ETC is reversed by melatonin and prevention of mitochondrial damage induced by ruthenium red (inhibits the mitochondrial Ca2+ uniporter for Ca2+ uptake), suggest that melatonin may act at the mitochondrial subcellular level and reduce ROS induced damage (Yamamoto and Tang, 1996; Martin et al., 2000). Furthermore, administrations of melatonin repair mitochondrial functioning in aging mice and shown to be neuroprotective in in vitro models of Alzheimer’s disease (Pappolla et al., 1997; Okatani et al., 2003).
Collectively, these results demonstrate the protective effects of riluzole and melatonin in neurodegenerative diseases involving mitochondrial dysfunction. However, we do not know if the therapeutic effects of these drugs in various pre-clinical mitochondria related neurodegenerative diseases also effective in ALS. Moreover, since mitochondrial ROS-induced cell death and excitotoxicity play a prominent role in ALS disease, specific neuroprotection mechanisms of riluzole and melatonin at the mitochondrial sub cellular level involving excitotxicity and Ca2+ signaling need to be further investigated. In this context, we determine whether treatment with riluzole or melatonin could attenuate sodium-azide (Na-azide) induced ROS and eventually cell death. Additionally, we investigated the impact of riluzole and melatonin on vulnerable HMNs of adult symptomatic SOD1G93A mice and corresponding wild type (WT) littermates. Imaging experiments were performed on adult brain stem slices where mitochondrial function of HMNs was disturbed by a bath application of 3 mM Na-azide, which inhibits complex IV and thereby disturbs mitochondrial metabolism. Riluzole and melatonin protection studies were similarly designed where application of sodium azide was performed because of, (1) its quick and reversible action, and (2) In both sALS and fALS a decrease in mitochondrial complex IV observed (Nowicky and Duchen, 1998; Menzies et al., 2002). The cell death, cell survival and Ca2+ signals were characterized and evaluated in the presence or absence of drugs.
We found that riluzole moderately ameliorates MN degeneration, inhibits excitotoxicity and mildly and reversibly inhibits Ca2+ signaling. However, melatonin fails to show significant MN protection and significant impact on Ca2+ signaling both in adult WT and corresponding SOD1G93A mice as well as in cell culture model of ALS. We anticipate that this experimental system can be used to screen the drugs targets either alone or in combination of drug cocktails that can be used for large-scale compound screening.
Materials and Methods
SH-SY5YWT and SH-SY5YG93A Neuroblastoma Cell Lines
SH-SY5Y human neuroblastoma cell line transfected with either WT or G93A mutant linked with fALS were routinely cultured in growth medium and were used as a valid and robust in vitro system to investigate the cellular excitotoxicity and mitochondria mediated alterations associated with SOD1-G93A mutations (Carri et al., 1997; Goos et al., 2007; Jaiswal et al., 2009). Detail descriptions about the transfection strategy, cell culture maintenance, cell culture growth medium and procedures are described earlier (Jaiswal et al., 2009).
Measurement of Cell Viability Using Mitochondrial Toxin Sodium Azide and Neuroprotection by Riluzole and Melatonin
Toxicity of Na-azide induced mitochondrial inhibition and thereby cell death assay in SH-SY5YWT and SH-SY5YG93A was done using bright field microscopy, hematoxylin and eosin (H&E) histochemistry and WST-1 assay. Neuroprotection study was done using similar method. SH-SY5YWT and SH-SY5YG93A transfected cells on glass coverslips were treated for 3 min (acute), 30 min (moderate) and 60 min (chronic) exposure with 3 mM Na-azide (60 min data not shown), cells were fixed with 4% paraformaldehyde (PFA), dehydrated through water/ethanol and histolene and then stained with Meyer’s hemalum reagent (1:1 in H2O). While choosing the Na-azide concentrations we followed the previous literature and accordingly we selected three different Na-azide concentrations, 1, 3, and 10 mM for cytotoxicity induced cell death assays and neuroprotection study (Hunter et al., 1959; Harvey et al., 1999). Concentration of 1 mM Na-azide not always leads to swelling of mitochondria for acute (3 min) exposure. Also shown by other groups at 0.1 mM or less than 2 mM sodium cyanide (NaCN) was not always adequate to prevent completely spontaneous swelling or swelling induced by low concentrations of phosphate (Hunter et al., 1959; Harvey et al., 1999). Higher concentration, e.g., 10 mM Na-azide might react with components other than cytochrome oxidase, therefore based on our pilot toxicity experiments and previous literature, we have chosen 3 mM Na-azide concentration suitable for our acute toxicity (3 min exposure) and neuroprotection assays. To study the differences in impact of Na-azide–induced cell death and neuroprotection mediated by riluzole and melatonin, SY5YWT and SH-SY5YG93A cells at a density of 105 cells/cm2 were seeded into 24-well plates. SH-SY5YWT and SH-SY5YG93A cells were exposed to culture medium with 3 mM Na-azide for 3 min (acute), 30 and 60 min (chronic). After 3, 30, and 60 min (60 min data not shown) of exposure, cell photomicrograph was acquired using brightfield microscope. Furthermore, for neuroprotection assay by riluzole and melatonin, both cell lines were exposed to DMEM growth medium with 3 mM Na-azide+100 μM riluzole or 3 mM Na-azide+100 μM melatonin for 3 min (acute). After rinsing the cover slips twice with water, cover slips were dehydrated and Vectashield Medium (Vector Laboratories, Burlingame, CA, USA) was used for mounting the cover slips.
To further investigate the impact of Na-azide exposure on cell viability and neuroprotection by riluzole and melatonin, cells at a density of 105 cells/cm2 in 24-well plates were treated with culture medium or medium containing 3 mM Na-azide final concentration. After 3, 30, and 60 min (60 min data not shown) of incubation with 3 mM Na-azide, WST-1 cell proliferation reagent (Roche Applied Science, Mannheim, Germany) was used to determine cell viability. For neuroprotection, similar WST-1 assay were done using 3 mM Na-azide+100 μM riluzole or 3 mM Na-azide+100 μM melatonin. The WST-1 assay is comprises a cleavage of the tetrazolium salt by healthy mitochondria to soluble orange formazan, by the enzyme succinate dehydrogenase present in the ETS of healthy mitochondria. The formazan dye formed after incubation of cells with WST-1 for 2 h was quantified by measuring the absorbance at 490 nm using the Genios multiplate reader (Tecan, Crailsheim, Germany) and correlates with the total number of metabolically viable cells.
Animals and Genotyping
Superoxide dismutase 1G93A Tg 1Gur (Fast Line) mice strain is considered a well-established animal model for human ALS. This mice strain acquired from the Jackson Laboratory (Bar Harbor, ME, USA) and in-house breeding was carried out in our animal facility. The SOD1G93A Tg mouse develop paralysis in the limb and eventually dies at 4–5 months of age due to the loss of MNs in the brain stem, HMNs, FMNs, and the spinal cord whereas WT littermates were unaffected. The Tg mouse carries a variation of the human mutant G93A-SOD1 gene in which at position 93 glycine is substituted by alanine (Gurney et al., 1994). Breeding and genotyping procedures were adapted from Hoyaux et al. (2000; for further details also see Jaiswal and Keller, 2009). Animal breeding and experimental procedures were approved and carried out in accordance with the guidelines of the ethics committee of the Medical Faculty of Georg-August University, Göttingen, Germany.
Preparation of Acute Brain Stem Slices from Adult SOD1G93A/WT Mice Littermates
Mice were anesthetized until the paw withdrawal reflex disappeared in a chamber containing an ether vapor-enriched atmosphere and immediately sacrificed. Mice brains were quickly removed and then put in 4°C ice-cold aCSF. Transverse 250 μm thick acute brain stem slices were prepared from the 14–15 weeks old WT and symptomatic SOD1G93A mice in late stage of motor dysfunction according to the procedures described earlier (Ladewig and Keller, 2000; Jaiswal and Keller, 2009) using a vibroslicer (Leika VT 1000S, Göttingen, Germany). Selectively vulnerable brain stem HMNs (Nuc. hypoglossus; XII) region were visually identified by their location nearby to the IV ventricle during the brain stem slice preparations transversely (Figure 3). To achieve maximum oxygen supply to slices, aCSF (in mM: NaCl 118, KCl 3, NaH2PO4 1, CaCl2 1.5, MgCl2 1, NaHCO3 25, glucose 20; pH 7.4; 320 mOsm) was constantly simmered with a mixture of 95% O2 and 5% CO2 (carbogen). An essential requirement for the microfluorometric measurements was the preservation of MNs in adult slices near to the slice surface in a physiologically viable and intact condition. This challenging requirement for Ca2+ imaging in MNs was accomplished by reducing mechanical tissue disturbances during the thick adult brain slice preparations, slice cutting at 4°C temperature and regular maximum oxygen delivery to keep metabolic conditions optimum for cells viability (For details, see Jaiswal and Keller, 2009). Slices were kept at ∼27°C in continuous carbogen-bubbled aCSF solution at pH 7⋅4 for 30 min and then let it cool down to room temperature (RT, ∼20–21°C) prior to dye loading. Unless indicated otherwise, all experiments were performed at RT.
Charge Coupled Device (CCD) Fast Optical Imaging
Intracellular calcium [Ca2+]i fluorescence measurement were achieved using an optical recording system including an upright Axioscope microscope (Zeiss, Göttingen, Germany) equipped with a computer-operated monochromator (TILL Photonics, Martinsried, Germany) built on a galvanometric scanner (Polychrome II, TILL Photonics, Martinsried, Germany). Briefly, a modified version of the CCD camera system (TILL Photonics, Planegg, Germany) and Achroplan W 63×, 0.9W objective were used to collect fluorescent signal changes in defined “regions of interest” (ROIs) in cell soma. Fluorescence signals changes were digitized using a 12-bit CCD camera (PCO, Kelheim, Germany), binning of which was put to 4 × 4, with a sampling rate between 3 and 13 Hz using TILL Vision Software (TILL Vision Software V3.3, TILL Photonics, Germany). Dichroic mirror with mid reflection at 425 nm was used for Fura-2 AM calcium dye (Invitrogen, Carlsbad, CA, USA) to direct the emitted light. To achieve the ratiometric calcium imaging, swapping between wavelengths (360 and 390 nm) was achieved in ∼3 ms, thereby allowing fast ratiometric Ca2+ measurements.
Microfluorometric Ca2+ Measurements in SH-SY5Y Transfected Cells and HMNs from WT and SOD1G93A Brain Stem Slice Preparations
WT (SH-SY5YWT) and SOD1 (SH-SY5YG93A) transfected cell layers on cover slips were stained with membrane-permeable Fura-2 AM (Kd ∼ 0.2 μM) dye by incubating with RPMI-1640 medium having 10% FCS (Invitrogen, Carlsbad, CA, USA) and 0.846 mM Ca2+ (5 μM Fura-2 AM dissolved in DMSO containing 10% pluronic acid for 30 min at 37°C and continuous bubbled with 95% O2 and 5% CO2). After the dye incubation cells were washed with RPMI and incubated in culture medium for ∼20 min at 37°C to permit de-esterification of dye. Baseline [Ca2+]i were measured in transfected cells 2–5 days in culture. Chemical induction of Ca2+ measurements were done by incubating the cells for 3 min with 3 mM Na-azide and effects of 100 μM riluzole and 100 μM melatonin were measured by inhibition of Ca2+ with similar measurements (data not shown).
In HMNS of brain stem slice, calcium signals were measured by defining appropriate ROIs of MN somas as previously described (Lips and Keller, 1998). [Ca2+]i signals were monitored by bath-loading of the brain stem slices with 5 μM final dye concentration of Fura-2 AM for 40 min at 27°C with continuous carbogen bubble (5% CO2 and 95% O2). After the dye incubation, prior to onset of imaging slices were washed with aCSF and incubated another 30 min in aCSF for de-esterification of Fura-2 AM. Excitation of Fura-2 was done alternately at 360 and 390 nm by UV light and emitted light was directed to a dichroic mirror having mid-reflection at 425 nm and filtered by a band pass filter of 505–530 nm (Zeiss, Goettingen, Germany) using a computer-controlled monochromator. Changes in [Ca2+]i are denoted as variations in Fura-2 ratio (F/F0) where F = fluorescence at different time points of experiment and F0 = baseline fluorescence value before drug/chemical applications (for details see Jaiswal and Keller, 2009). Further calculations and analysis of [Ca2+]i signals were performed offline using IGOR Pro (Wavemetrics, Lake Oswego, OR, USA) and OriginPro 6.0 (OriginLab Corporation, Northampton, MA, USA) Software.
Intrinsic nicotinamide adenine dinucleotide (NADH) Fluorescence
The metabolic state of HMNs from SOD1G93A mice in the presence and absence of 100 μm riluzole was monitored by measuring and monitoring intrinsic nicotinamide adenine dinucleotide (NADH) fluorescence of HMNs in the slice preparations using a high-speed CCD camera. Briefly, NAD(P)H excitation at 360 nm was carried via a computer-controlled monochromator (TILL Photonics, Martinsried, Germany) reflected onto the surface of the slice using dichroic mirror (400 nm, Zeiss, Goettingen, Germany). The hypoglossal nucleus was easily recognized by a high level of intrinsic fluorescence. Fluorescence emission was collected by using a CCD camera (TILL Photonics, Planegg, Germany). All experiments used a 410 nm LP filter between the dichroic mirror and camera to maximize light capture. Imaging was performed after focusing onto the surface of slices in hypoglossal area using 63× (0.9 NA) Achroplan water objective (Zeiss, Göttingen, Germany) and collected by a 12-bit CCD camera (PCO, Kelheim, Germany) described earlier. Calculations and analysis of NAD(P)H signals were performed offline after the experiment using IGOR Pro (Wavemetrics, Lake Oswego, OR, USA) and OriginPro 6.0 (OriginLab Corporation, Northampton, MA, USA) Software.
Chemical Reagents
Fura-2 AM dye (50mg/vial) was purchased from Invitrogen (Carlsbad, CA, USA) and dissolved in 50 μL DMSO containing 20% Pluronic F-127 to a concentration of 1 mM. Na-azide, riluzole, melatonin and all other chemicals were purchased from Sigma–Aldrich (St. Louis, MO, USA). To prepare the stock solutions, riluzole and melatonin were dissolved in 100% ethanol to make 10 mM stock solutions and all the time kept at 4°C to avoid ethanol evaporation. Na-azide was dissolved in distilled water. To keep cells viable drug solutions when mixed in the perfusate always simmered with carbogen (95% O2, 5% CO2) during the experiments.
Statistical Analysis
SH-SY5YWT and SH-SY5YG93A transfected cells attached with cover slip was used for single experiments and replicate in five separate experiments (five field of view) for each condition in cell viability assay (cell survival/death) and seven separate experiments (seven field of view for each cover slides for SH-SY5YWT and SH-SY5YG93A) for neuroprotection assay. All slices were used for a sole experiment and 5–6 HMNs taken from each slice in Fura-2 calcium imaging data experiments. 2–3 slices were used per mouse and 5–6 HMNs selected from each slice for bar graph presentation. In slice experiments, bar diagram represents 12 imaging experiments recorded from 12 separate slices obtained from 5 WT mice and 10 imaging experiments recorded from 10 slices obtained from 5 SOD1G93A mice in acute Na-azide experiments. In case of riluzole experiments, bar diagram represent 14 imaging experiments recorded from 14 separate slices obtained from 6 WT mice and 13 imaging experiments recorded from 13 slices obtained from 6 SOD1G93A mice. In case of NADH fluorescence imaging, 5–6 cells from slice for five separate experiments for each condition from five different mice of same genotypes was used for bar diagram representation. Values in the text and error bars in experimental figures are represented as mean ± SD. Significance for all the in vitro assay, slice imaging and pharmacological intervention was calculated using the two-tailed unpaired student t-test and a p-value < 0.05 was considered statistically significant. One-way analysis of variance (ANOVA) with Bonferroni’s method was used as the post hoc test used to recognize statistically significant Na-azide effects, and neuroprotection effects of riluzole and melatonin exposed cells as compared with controls with in groups.
Results
Cell Death Assessment by Inhibition of Mitochondria and Neuroprotective Actions of Riluzole and Melatonin against Sodium Azide-Induced Cell Death in In vitro SH-SY5Y Cell Culture Model of ALS
We utilized differentiated SH-SY5YWT and SH-SY5YG93A cells used previously to generate WT and SOD1G93A cell culture model of ALS (Jaiswal et al., 2009). Cell death, cell viability and anti-aggregate formation were assessed using bright field microscopy, immunocytochemistry and WST-1 cell proliferation assay (Figures 1 and 2). SH-SY5YG93A cells showed slightly higher levels of cell death compared to SH-SY5YWT after 3 and 30 min (Figures 1A–Da–c and 2A) exposure to mitochondrial toxin Na-azide (Figures 1A–Da–c and 2A). A similar pattern was observed while investigating the apoptosis and aggregation assessed by bright field microscopy demonstrating typical apoptotic morphology, e.g., condensation/fragmentation of nuclear material and elongation of cell size and cell membranes. Cells displaying fragmentation of nuclear materials (pyknosis)/cell membrane elongation were classified as apoptotic and showed aggregates formation with both cell lines after incubation for 30 and 60 min (Figures 1Aa–c,Ca–c; data not shown for 60 min exposure) but not with acute 3 min (acute) induction (Figures 1Aa–c,Ca–c). We have not quantified the cell numbers after 30 and 60 min exposure to Na-azide due to lump formation after 30 min exposure and inability to separate each cells with in aggregates and almost complete cell death 60 min after the inhibition of mitochondria by Na-azide (Figures 1Ac,Cc). Cell survival/death was assessed by H&E staining and WST-1 test. Cell count for H&E staining for both SH-SY5YWT and SH-SY5YG93A cells treated with Na-azide for 3 min (77.0 ± 4.7 for WT and 71.5 ± 6.1 for SOD1G93A) and 30 min (59.2 ± 5.8 for WT and 54.0 ± 5.5 for SOD1G93A) were significantly lower compared to untreated cells with Na-azide (114.5 ± 2.8 for WT and 107.0 ± 5.1 for SOD1G93A; Figures 1A–Da–c and 2A; N = 5; ∗∗∗P < 0.001, ∗∗P < 0.005, Students two sample t-test, Table 1).
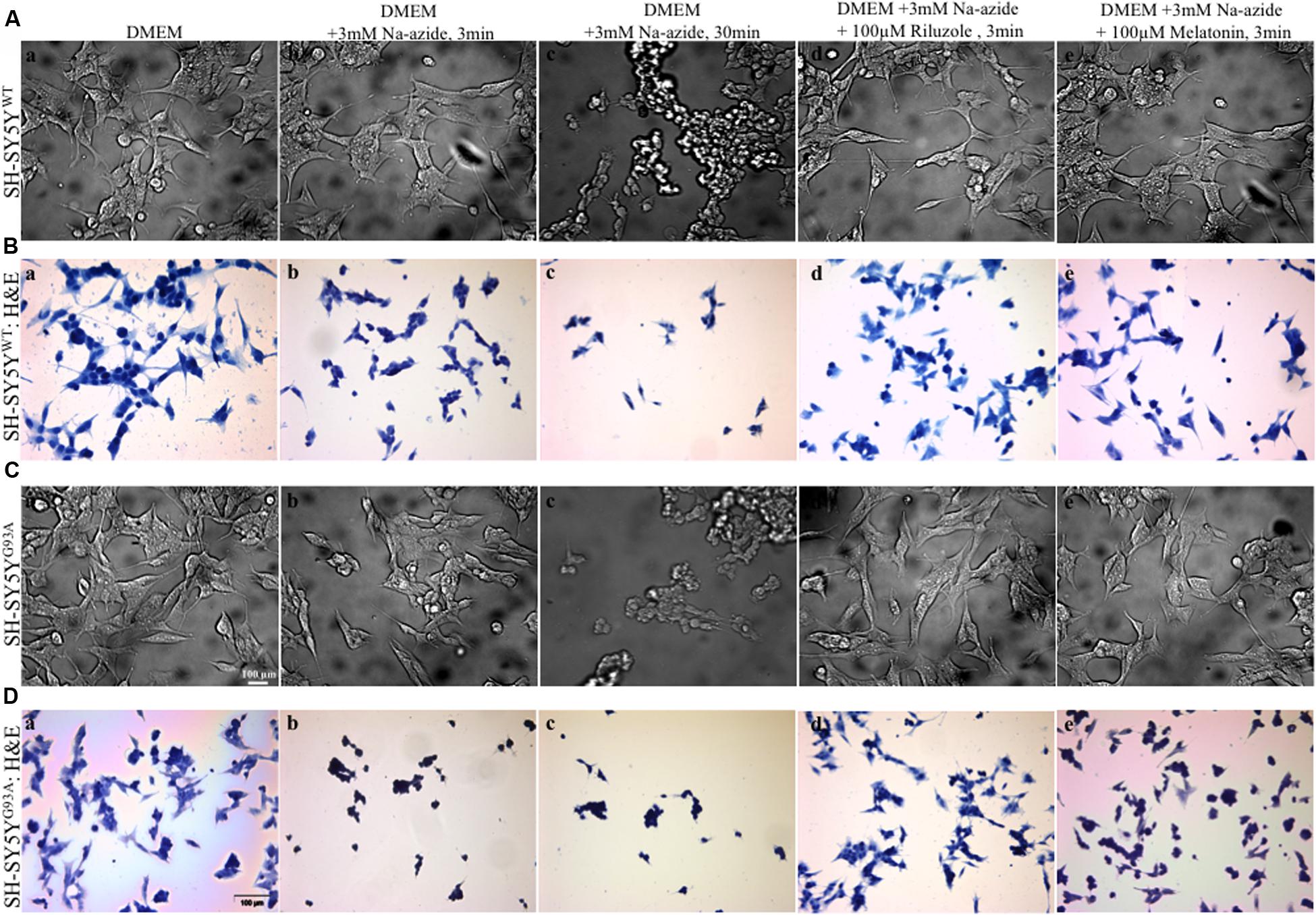
FIGURE 1. Motor neurons cell death assessment using mitochondrial toxin Sodium azide and neuroprotection assay to access efficacy of riluzole and melatonin. (A) Photomicrograph of bright field images of differentiated SH-SY5YWT cells exposed to growth medium DMEM (a), DMEM+3 mM Na-azide for 3 min (b), DMEM+3 mM Na-azide for 30 min (c), DMEM+3 mM Na-azide+3 mM riluzole for 3 min (d), and DMEM+3 mM Na-azide+3 mM melatonin for 3 min (e). Note the decrease in cell density after Na-azide exposure, which is ameliorated in presence of riluzole, but not melatonin. (B) H&E staining showed a substantially higher density of living SH-SY5YWT cells exposed to DMEM (a) compared to SH-SY5YWT cells exposed to DMEM+3 mM Na-azide for 3 min (acute, b) and 30 min (chronic, c). Note the shrinkage, clustering, and reductions in cell number of severely damaged/dead SH-SY5YWT neuroblastoma cells in b and c. H&E staining showed 3 min acute exposure of 3 mM Na-azide induced cell death is partially ameliorated in the presence of 3 mM riluzole (d), but not in the presence of 3 mM melatonin (e). (C) Bright field images of differentiated SH-SY5YG93A cells exposed to DMEM (a), DMEM+3 mM Na-azide for 3 min (b), DMEM+3 mM Na-azide for 30 min (c), DMEM+3 mM Na-azide+3 mM riluzole for 3 min (d), and DMEM+3 mM Na-azide+3 mM melatonin for 3 min (e). Note the decrease in cell density in presence of 3 mM (acute) and 30 min (chronic) Na-azide exposure. Note the increase in cell density of SH-SY5YWT cells compared to SH-SY5YG93A cells in presence of 3 mM riluzole suggesting greater susceptibility of SH-SY5YG93A to mitochondrial-induced damage. (D) H&E staining showed a similar substantially higher density of living SH-SY5YG93A cells like SH-SY5YWT cells exposed to DMEM (a) compared to cells exposed to 3 min (acute, b) and 30 min (chronic, c) exposure with 3 mM Na-azide. Cell death of H&E stained SH-SY5YG93A cells in presence of 3 mM Na-azide exposure is partially ameliorated in presence of 3 mM riluzole (d), but not in presence of 3 mM melatonin exposure (e). Scale bar for (A) and (C) = 20 μm and (B) and (D) = 100 μm.
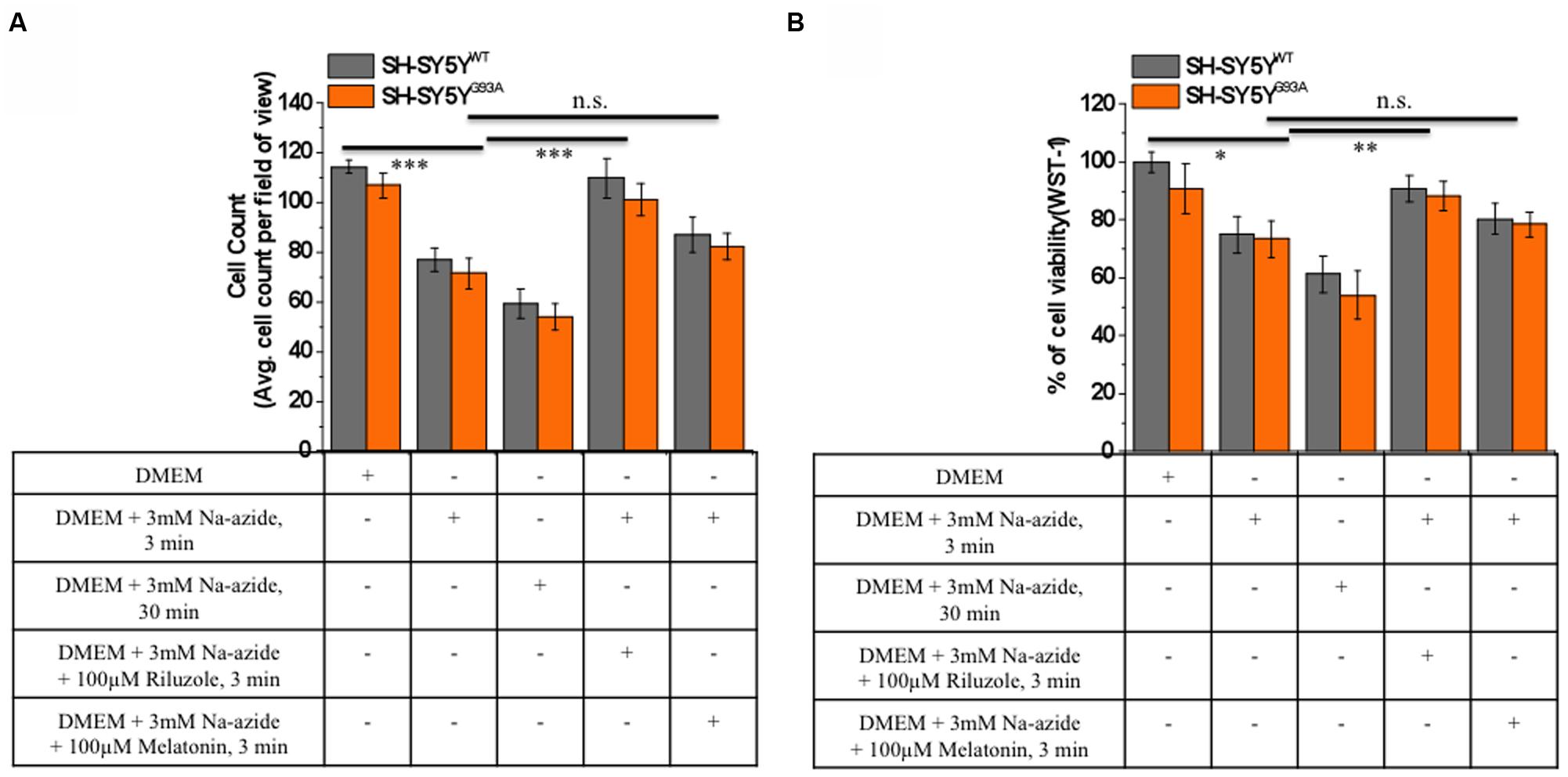
FIGURE 2. Riluzole but not melatonin stimulation increased cells survival and neuroprotective against sodium azide induced mitochondrial ETC blockade both in SH-SY5YWT and SH-SY5YG93A cells. (A) Live cell count of SH-SY5YWT (gray bar) and SH-SY5YG93A (red bar) neuroblastoma cells after incubation with DMEM, DMEM+3 mM Na-azide for 3 min, DMEM+3 mM Na-azide for 30 min, DMEM+3 mM Na-azide+3 mM riluzole for 3 min and DMEM+3 mM Na-azide+3 mM melatonin for 3 min (average of cell counts for seven field of view in each conditions). Please note the significant difference between riluzole-treated and non-treated SH-SY5YWT and SH-SY5YG93A cells exposed to Na-azide (p < 0.005). (B) Toxicity of Na-azide for SH-SY5YWT and SH-SY5YG93A cells was measured using the WST-1 cell viability test in presence of DMEM, DMEM+3 mM Na-azide for 3 min, DMEM+3 mM Na-azide for 30 min, DMEM+3 mM Na-azide+3 mM riluzole for 3 min and DMEM+3 mM Na-azide+3 mM melatonin for 3 min. SH-SY5YWT and SH-SY5YG93A cells were more vulnerable to the action of Na-azide in absence of riluzole compared to 3 mM riluzole presence in DMEM growth medium (p < 0.01). Data are expressed as the mean ± SEM. ∗∗∗P < 0.005, ∗∗P < 0.01, ∗P < 0.05 two-tailed unpaired student t-test.
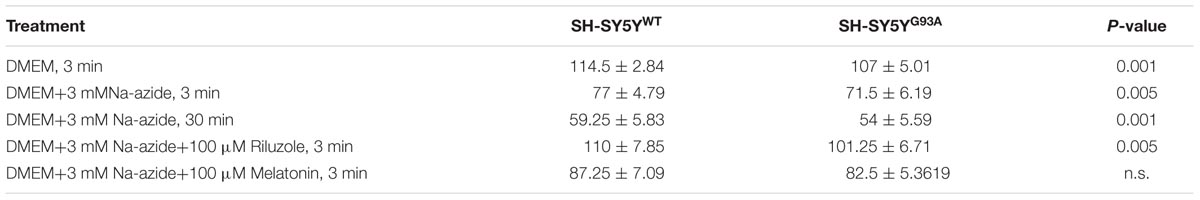
TABLE 1. Cell count of H&E staining for SH-SY5YWT and SH-SY5YG93A cells treated with sodium azide in presence and absence of riluzole and melatonin.
It was previously reported by several groups that riluzole and melatonin ameliorates ALS progression in SOD1G93A mouse model and extend morbidity of hALS patients. We next attempted to determine whether riluzole and melatonin confers neuroprotection and ameliorate cell death in ALS cell culture model and if yes at what extent. Previously it was shown that riluzole up to 10 μM have no effect on cell survival in DA neurons and SH-SY5Y cells. However, concentrations greater than 50 μM have a remarkable effect on cell survival (Storch et al., 2000); therefore, here we used 100 μM riluzole and melatonin exposure with superfusate and evaluate the protective effects of riluzole on cell survival in SH-SY5YWT and SH-SY5YG93A cells in vitro (Figures 1A–Dd,e and 2A). Our findings show that the acute inhibition of mitochondrial metabolism and function and thereby cell death by Na-azide is ameliorated by riluzole but there is no significant effects of melatonin in both SH-SY5YWT and SH-SY5YG93A cells (Figures 1A–Dd,e and 2A). The average number of H&E positive cells in a given field of view (seven field of view) after the treatment of 100 μM riluzole to SH-SY5YWT and SH-SY5YG93A cells is significantly higher (Figure 2A; 110.0 ± 7.8 for WT and 101.2 ± 6.7 for SOD1G93A) compared to non-treated cells stimulated with 3 mM Na-azide (Figure 2A; 77.0 ± 4.7 for WT and 71.5 ± 6.1 for SOD1G93A; for riluzole treated and non-treated cells; ∗∗P < 0.005, Students two sample t-test; Table 1). In addition to decreasing the cell death, riluzole had significant protective effects on the neuritis, fine cell processes and cell membrane integrity being preserve from mitochondrial inhibition and the effect was most pronounced in SH-SY5YWT compared to SH-SY5YG93A cells (Figures 1Ad–Dd). However, impact of 100 μM melatonin treatment on Na-azide induced inhibition of mitochondria is not significantly different then non-treated cells in both SH-SY5YWT and SH-SY5YG93A cells though there are minor increase in cell count of WT and SOD1G93A cells (Figures 1A–De and 2A; 87.2 ± 7.0 for WT and 82.5 ± 5.3 for SOD1G93A, Table 1).
Cell viability assessed by WST-1 assay showed significant dose dependent ROS-induced mitochondrial toxicity and cell death. Cell viability of SH-SY5YWT and SH-SY5YG93A cells in presence of Na-azide for 3 and 30 min leads to a decrease in cell survival (75.0 ± 3.4 and 61.2 ± 6.1% for 3 and 30 min incubation, respectively, for WT; Figure 2B) and were significantly not different with SOD1G93A cells (73.5 ± 6.2 and 54.0 ± 8.4% for 3 and 30 min incubation, respectively; Figure 2B; Table 2). However, cell viability for both WT and SOD1G93A treated cells were much lower and significantly different compared to untreated cells without Na-azide toxicity (100 ± 3.4% for WT and 91 ± 8.6% for SOD1G93A; Figures 1A–Da–c and 2A; N = 5; ∗∗∗P < 0.001, Students two sample t-test; Table 2). Similarly % of SH-SY5YWT and SH-SY5YG93A viable cells after the treatment of 100 μM riluzole is significantly higher (Figure 2B; 91.0 ± 4.5 for WT and 88.2 ± 5.2 for SOD1G93A; Table 2) compared to non-treated cells stimulated with 3mM Na-azide for 3 min (Figure 2A; 75 ± 6.3% for WT and 73.5 ± 6.3% for SOD1G93A; ∗∗P < 0.01, Students two sample t-test; Table 2). There is no significant increase in cell survival after melatonin treatment (Figure 2B; 80.2 ± 5.3 for WT and 78.5 ± 4.2 for SOD1G93A; Table 2).
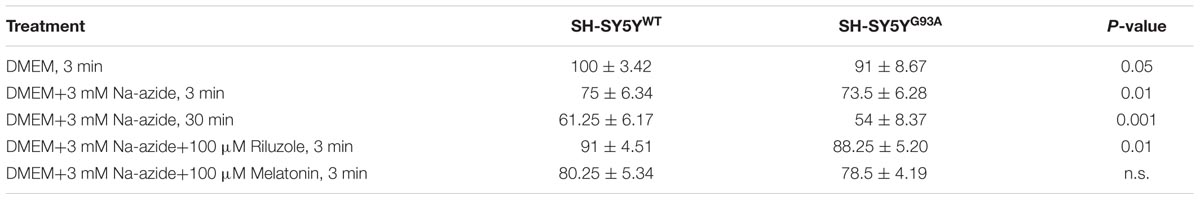
TABLE 2. WST-1 cell viability assay in SH-SY5YWT and SH-SY5YG93A cells treated with sodium azide in presence and absence of riluzole and melatonin.
Impact of Riluzole on Sodium Azide-Induced Mitochondrial [Ca2+]i Release and Excitotoxicity
To test further the efficacy of riluzole on Na+-azide induced mitochondrial Ca2+ signaling, Fura-2 AM stained HMNs of brain stem slices were exposed to Na-azide in presence and absence of 100 μM riluzole. Sketch diagram of hypoglossal MNs in brainstem slice preparations (Figure 3A) and representative photomicrograph of fura-2 AM stained HMNs in WT and (left) and symptomatic SOD1G93A mice (right) shown in Figure 3B. We evaluated the protective impact of riluzole on calcium signaling of HMNs of adult WT (Figures 3C,E) and symptomatic SOD1G93A mice brain stem slices in vitro (Figures 3D,F). We found that riluzole has a moderate effect on inhibition of Na-azide (3 mM) induced [Ca2+]i signaling of MN’s mediated by Na+-azide induced excitotoxicity. At higher concentration (>50 μM) riluzole moderately inhibited [Ca2+]i fluorescence in both WT and selectively impaired HMNs of symptomatic SOD1G93A mice whereas at lower concentrations (<50 μM) there is no significant effect (data not shown). Sodium azide-induced mitochondrial [Ca2+]i release response is significantly different between WT and symptomatic SOD1G93A mice. The impact of riluzole inhibition is not specific to only SOD1G93A mice and it also moderately inhibits [Ca2+]i signaling in WT mice as well (Figure 3; Table 3).
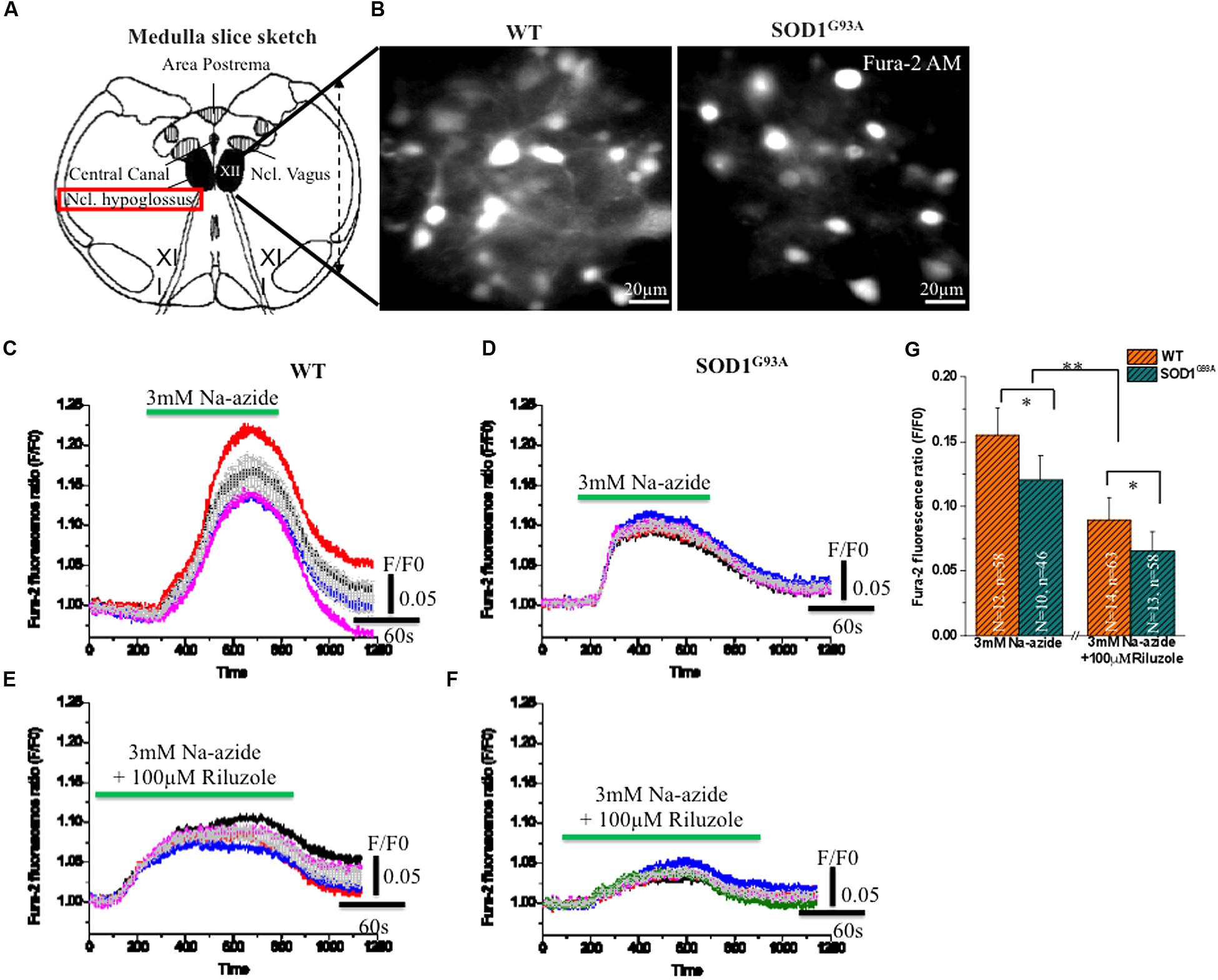
FIGURE 3. Riluzole induce inhibition of [Ca2+]i in fura-2 AM loaded HMNs exposed to 2 mM Na-azide in adult WT and symptomatic SOD1G93A mice. (A) A graphical illustration of the adult mouse brainstem slice containing hypoglossal motoneurons (HMNs) marked with black colors [Modified from Jaiswal, 2009. Optical analysis of [Ca2+]i and mitochondrial signaling pathways: implications for the selective vulnerability of motoneurons in amyotrophic lateral sclerosis (ALS). Goöttingen, Univ., Diss., 2008. Copyright© 2009 Jaiswal, M. K. ediss.uni-goettingen.de. Used with permission]. (B) A CCD camera images (4 × 4 binning) showing 15 weeks adult WT (left) and symptomatic SOD1 (right) mice HMNs stained with ratiometric calcium dye fura-2 AM (excitation at 360 nm). (C) 3 mM Na-azide was added in aCSF superfusate and the calcium fluorescence signal was recorded in 14–15 weeks old WT mice HMNs. (D) 3 mM Na-azide was added in aCSF superfusate and the calcium fluorescence signal was recorded in 14–15 weeks old symptomatic SOD1G93A mice HMNs. (E) 3 mM Na-azide was added in aCSF superfusate together with 100 μM riluzole and the calcium fluorescence signal was recorded in 14–15 weeks old WT mice HMNs. (F) 3 mM Na-azide was added in aCSF superfusate together with 100 μM riluzole and the calcium fluorescence signal was recorded in 14–15 weeks old symptomatic SOD1G93A mice HMNs. Average peak amplitude (gray color) shown as means from 5 to 6 cells in an imaging field (C–F). (G) Bar diagram of effect of 100 μM riluzole on Na-azide induced blockade of [Ca2+]i measured in adult WT (Light gray) and symptomatic SOD1G93A (Gray sparse) mice HMNs. Data are expressed as means 12 imaging experiments from five WT mice and corresponding 10 imaging experiments from five symptomatic SOD1G93A mice in acute 3 mM Na-azide experiments. In case of riluzole experiments, bar diagram represent 14 imaging experiments obtained from six WT mice and corresponding 13 imaging experiments obtained from six symptomatic SOD1G93A mice. The changes in peak amplitude of [Ca2+]I were calculated as F/F0 (360/390). ∗P < 0.05, ∗∗P < 0.01 compared with adult WT and symptomatic SOD1G93A mice after riluzole application. Data are expressed as the mean ± SD.

TABLE 3. Sodium azide-induced mitochondrial [Ca2+]i release and excitotoxicity in absence and presence of riluzole.
As illustrated in Figures 3C,D,G; 3 mM Na-azide caused a rise in the [Ca2+]i transient by mitochondrial complex IV inhibition, with average peak amplitudes (F/F0) reaching 0.15 ± 0.02 and 0.11 ± 0.01 (Figures 3C,D,G; Table 3) respectively, in HMNs of adult WT (5 mice/N = 12 slice; n = 58 HMNs) and symptomatic SOD1G93A mice (5 mice/N = 10 slice; n = 46 HMNs) and subsequently comes to baseline over 2–3 min wash out with aCSF. In the presence of 100 μM riluzole, application of 3 mM Na-azide caused a rise in the [Ca2+]i transient, with average of the peak amplitudes (F/F0) reaching 0.089 ± 0.01 and 0.065 ± 0.01 (Figures 3E–G; Table 3) respectively, in HMNs of adult WT (6 mice/N = 14 slice; n = 63 HMNs) and symptomatic SOD1G93A mice (6 mice/N = 13 slice; n = 58 HMNs). The inhibition of the peak amplitude of [Ca2+]i in SOD1G93A mice (F/F0 = 0.065 ± 0.01) is prominent compared to WT mice (F/F0 = 0.089 ± 0.01; P < 0.05, Students two sample t-test; quantitative fluorescence value compared in Table 3). The inhibition of [Ca2+]i peak amplitude in SOD1G93A mice by riluzole might be attributed to a decrease in the entry of Ca2+ through a voltage dependent calcium channel (VDCC) and need further investigations. We conclude that riluzole inhibits Na-azide-induced mitochondrial Ca2+ signaling and it modestly ameliorate MN degeneration by inhibition of excitotoxicity trigger by [Ca2+]i efflux.
Impact of Riluzole on Metabolic State of HMNs in Presence of Sodium Azide-Induced Mitochondrial Respiratory Chain Inhibition and Excitotoxicity
Mitochondrial ETC associated proton efflux causes accumulation of negative charges in the matrix and electrochemical gradient across the mitochondrial membrane. Intrinsic NADH fluorescence in MNs has been shown to serve as a valuable tool to characterize the metabolic signature of intrinsic energy profiles. Accordingly, evaluation of intrinsic NADH fluorescence after inhibition of mitochondrial complex IV with Na-azide indicated that the mitochondrial respiratory chain was significantly disturbed in SOD1 G93A animals which is rescues in acute condition by riluzole by yet unknown mechanism (Figure 4). To confirm that impact of riluzole on mitochondrial NADH fluorescence, experiments were performed, in which respiratory chain activity of HMNs was inhibited by 3 mM Na-azide in absence (aCSF; Figure 4A) and presence of riluzole (100 μM riluzole; Figure 4B). Kinetic profile of HMNs (n = 5) NADH fluorescence in single slice in aCSF (black) and in presence of riluzole (red) shown in Figure 4C for three consecutive application.
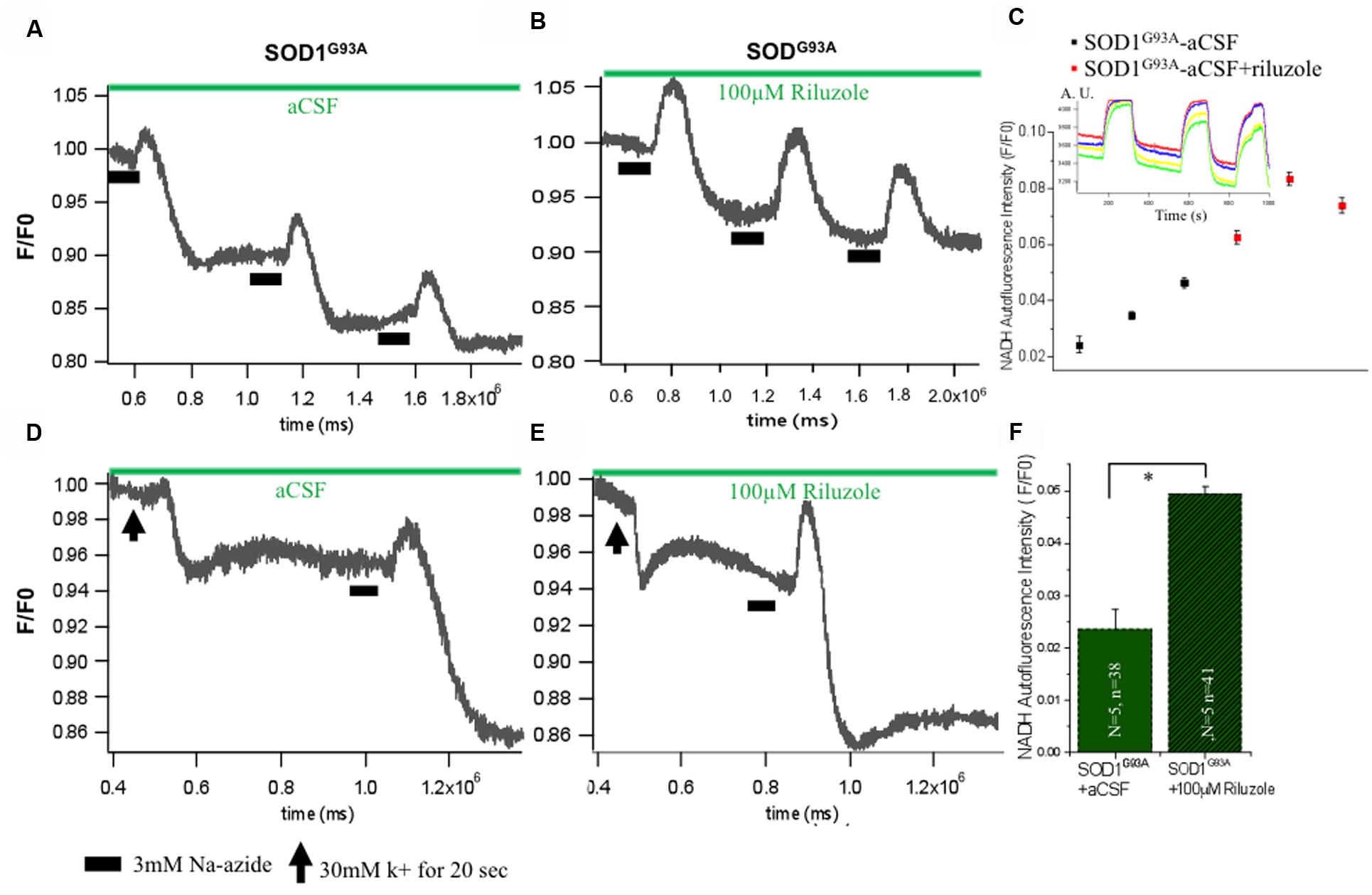
FIGURE 4. Impact of riluzole on metabolic state of HMNs measured by NADH fluorescence. (A) Mitochondrial activity of SOD1G93A mice HMNs was inhibited by 3 mM Na-azide in presence of aCSF. (B) Mitochondrial activity of SOD1G93A mice HMNs was inhibited by 3 mM Na-azide in presence of 100 μM riluzole. (C) NADH fluorescence intensity profile of HMNs (n = 5) measured in a single slice in aCSF (black) and in presence of riluzole (red) after three consecutive application of 3 mM Na-azide. Time series experiment for single measurement shown in inset. (D) NADH fluorescence after K+ induced plasma membrane depolarization and Na-azide-evoked NADH response in HMNs of SOD1G93A mice in presence of aCSF. (E) NADH fluorescence after K+ induced plasma membrane depolarization and Na-azide-evoked NADH response in HMNs of SOD1G93A mice in presence of 100 μM riluzole. Horizontal black bars indicate the duration of application of 3 mM Na-azide. The black arrow indicates the 30 mM K+ induced plasma membrane for 30 s. (F) Graphical representation of the effect of 3 mM Na-azide in presence of aCSF (N = 5 slice; n = 38 HMNs) and 100 μM riluzole (N = 5 slice; n = 41) SOD1G93A mice. The data are expressed as mean ± SD; N, number of slices/experiments, n, number of cells. ∗p < 0.05.
To analyze the comparative efficiency of mitochondria of SOD1G93A mice, HMNs were evoked by a 30 mM K+ depolarizing stimulus. When SOD1G93A mice HMNs were exposed to 30 mM K+ for 20 s, there is immediate and slow plateau shape decrease in the NADH fluorescence and subsequently returned to the basal level. To determine whether the mitochondrial depolarization by 30 mM K+ has any apparent influence on the lifetime of the NADH fluorescence and riluzole still rescues HMNs metabolism, 100 μM riluzole was added to the aCSF superfusate after K+-induced depolarization (Figures 4D,E). Application of 3 mM Na-azide after 20 s depolarization induced by K+ resulted in an increase of NADH fluorescence of 0.024 ± 0.003 and 0.049 ± 0.001 in the presence of aCSF (N = 5 slice; n = 38 HMNs) and aCSF+100 μM riluzole (N = 5 slice; n = 41 HMNs), respectively, in SOD1G93A mice (Figure 4F). Following previous trends there was a slight increase in the NADH fluorescence in presence of 100 μM riluzole but there was no significant difference in NADH fluorescence mean value after 3 mM Na-azide-evoked responses (normalized, F/F0) post K+-induced depolarization with or without 100 μM riluzole.
Discussion
We have shown earlier that [Ca2+]i in HMNs increases in response to stimulation with mitochondrial electron transport complex IV inhibitors Na-azide/cyanide and differentially regulated in WT and SOD1G93A mice and cell culture model of SOD1 (Bergmann and Keller, 2004; Jaiswal and Keller, 2009; Jaiswal et al., 2009). Given that riluzole and melatonin has been shown to avert excitotoxicity induced cell death by various mechanism, including oxidative stress and calcium signaling (Wang, 2009; Mazzone and Nistri, 2011; Jaiswal, 2012; Geevasinga et al., 2016; Gutierrez et al., 2016; Reiter et al., 2016), inhibition of persistent sodium currents and Trpm4 (Simard et al., 2012), Trpm4 mediated increase of Ca2+ influx (Weber et al., 2010) via Ca2+ CRAC and Kca channels (Dolmetsch and Lewis, 1994), we examined whether riluzole and melatonin affects mtSOD1-mediated cell death and excitotoxicity and improve cell survival. Furthermore, since Na+-azide blocks the mitochondrial ETC in HMNs of ALS, we further analyzed whether riluzole and melatonin protect the HMNs from Na-azide induced MN death induced by excitotoxicity and mediated by inhibition of [Ca2+]mito signaling cascades. The effect of drug riluzole and melatonin on the excitotoxicity-induced cell death and [Ca2+]i concentration induced by mitochondrial blocker was measured, and the impact of riluzole and melatonin-modulated Ca2+ influx and Ca2+ release were evaluated. We found that riluzole but not melatonin, moderately inhibits excitotoxicity-induced [Ca2+]mito signaling and thereby modestly protects the cell death in WT and SOD1G93A cell culture model of ALS and SOD1G93A mice model (Figures 1–3). Previously, it was shown that melatonin enhance mitochondrial ETC I and IV and thereby improving mitochondrial respiration, ATP synthesis and energy metabolism under stress circumstances (Leon et al., 2005). However, in our experiments melatonin fail to show neuroprotective action on either cell death (Figures 1 and 2) or Na+-azide induced [Ca2+]i signaling (data not shown) in adult WT and corresponding symptomatic SOD1G93A mice. Our results indicate that the melatonin mitochondrial neuroprotective action might be governed by respiratory complex I inhibition or by yet unknown unidentified mechanism(s). We therefore believe that the riluzole-targeted inhibition of mitochondrial signaling could be beneficial in ALS. In this paper, we provide evidence using three independent methods about inhibition of depolarization-evoked calcium transients and excitotoxcity in healthy and diseased MNs and cell death/survival assay in cell culture model of MNs by neuropharmacological substrates riluzole to protects cells against excitotoxic damage and hence its therapeutic effects in ALS. These observations may be relevant to understand the vulnerability of MNs to the excitotoxic insult that may contribute to the aetiopathology of ALS.
We found a substantial increase in cell viability and inhibition of excitotoxicity induced cell death in MNs by riluzole but not by melatonin (Figures 1 and 2), thus providing evidence for the efficacy of riluzole in in vitro transfected cell culture model in the treatment of ALS. Riluzole has been shown to alleviate neurological symptoms in the SOD1G93A Tg mouse model of ALS by suppressing oxidative stress (Seo et al., 2015). Indeed, we observed that riluzole reduced Na-azide induced and SOD1G93A-mediated cell death (Figure 1) and increase cell viability (Figure 2) in SH-SY5YWT and SH-SY5YG93A cells, suggesting that mitochondrial excitotoxicity and ROS is associated with the mechanism of SOD1G93A-induced cytotoxicity. However, compared to riluzole, melatonin fail to prevent Na-azide induced and SOD1G93A-mediated cell death (Figure 1) and there is no significant increase in cell viability (Figure 2) in SH-SY5YWT and SH-SY5YG93A cells, suggesting that mechanism of cell death protection by melatonin might adopt different cellular mechanism as it did not affect cell viability induced by Na-azide induced ROS generation through mitochondrial inhibition.
Furthermore, we demonstrated that the inhibition of peak amplitudes of [Ca2+]i in symptomatic SOD1G93A and adult WT mice is significant when measured with application of Na+-azide together with riluzole compared to of Na+-azide alone (Figure 3, p < 0.05). The overall inhibition of the rise in peak [Ca2+]i transients in the presence of riluzole after a Na+-azide application in adult WT and symptomatic SOD1G93A might be due to the potential inhibition of complex IV and decrease in the entry of Ca2+ through VDCC. Additionally, in symptomatic SOD1G93A mice, the presence of riluzole slightly diminished the Na+-azide induced [Ca2+]mito increase and delayed the baseline recovery by up to 30–60 s. Our results therefore indicate that riluzole’s moderate inhibition of [Ca2+]mito signaling may be because of a partial blockade of intracellular Ca2+ release from the mitochondria. Our finding further supports the assumption that the riluzole ameliorates excitability and Ca2+ signaling and this pathway may have role in its manifold effects on mitochondria mediated cellular metabolism in ALS.
NADH fluorescence is a good experimental measure to monitor the metabolic profile of the MNs. In the intact HMNs besetting the mitochondria by Na-azide adversely affects the production of NADH in the mitochondrial metabolism. Our data shows slight increase in the NADH fluorescence in presence of 100 μM riluzole compared to aCSF but there was no significant difference in NADH fluorescence mean value (Figure 4). This implies that there is a significant mitochondrial metabolism dysfunction and mitochondria in SOD1G93A mice might not able to sequester enough calcium after K+-induced depolarization (dysfunctional Ca2+ accumulation activity) or Na-azide-evoked release response is hampered in HMNs of SOD1G93A mice.
Overall, our results indicate that the inhibition and stabilization of Ca2+ transient by riluzole under physiological situation, e.g., diffusion-restricted and in homeostatically controlled mitochondrial domains might indeed have numerous functional advantages (Figure 5). Accordingly, drugs targeting protection of mitochondrial function and homeostasis could be useful in various forms of ALS by utilizing multidrug therapy where combined pharmacological intervention target different domains of mitochondria and Mito-cycle which ameliorates excitotoxicity both at the MNs and at the neighboring cells and most likely prolong survival of ALS patients. Our experiments promise a limited but effective and methodological way to test the multiple drugs and their targets most likely tested with different drug cocktails to check the efficacy of multi-drug therapy. However, more detailed study allowing identification of mechanism and molecular targets would be necessary.
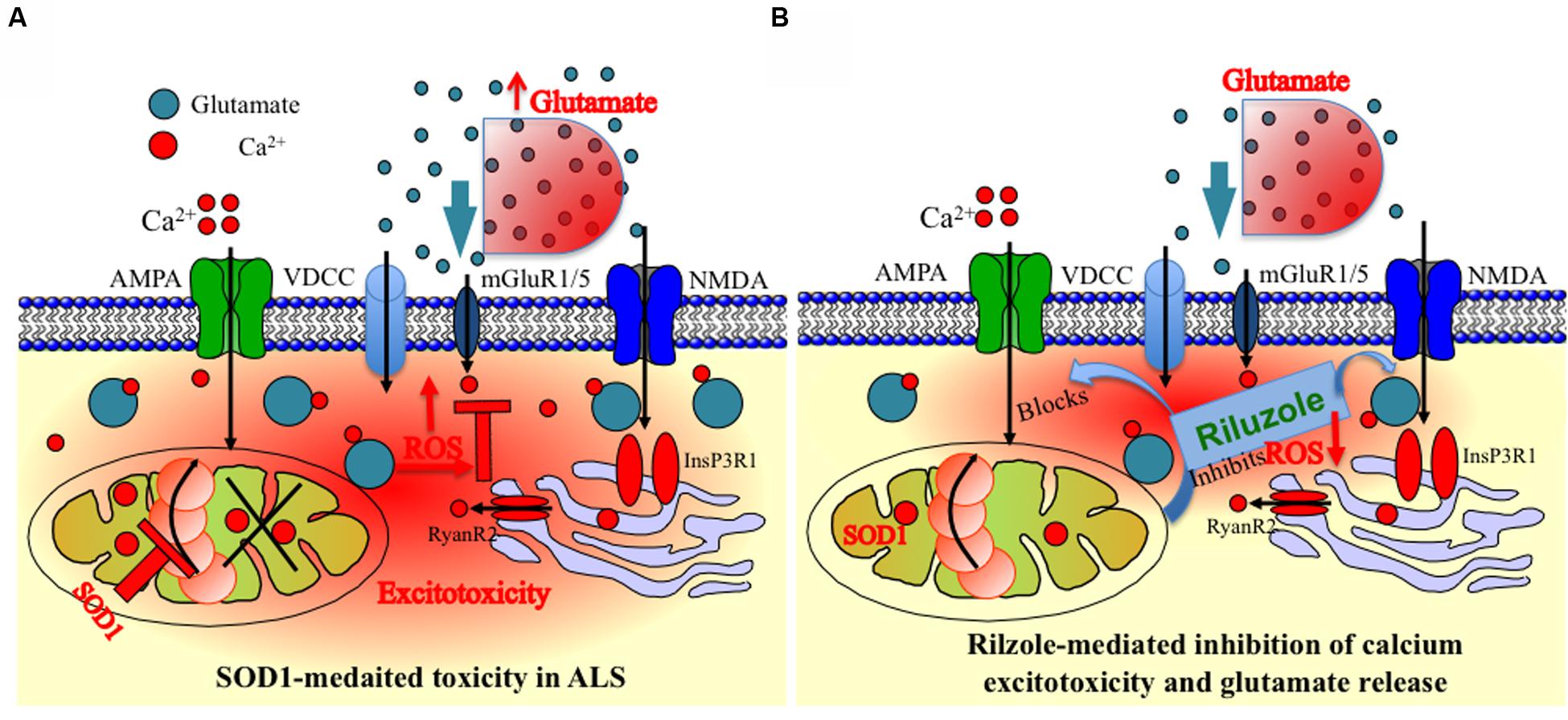
FIGURE 5. Schematic presentation of the model of mitochondria-mediated sodium-azide induced excitotoxicity inhibition by Riluzole in ALS. (A) Mitochondrial disturbance and calcium excitotoxicity associated with SOD1 mediated toxicity are predicted to be the key trigger to ALS etiology. The production of ROS by disturbed mitochondrial metabolism can be readily cytotoxic as ROS can destroy the membrane integrity by averting the specificity of membrane channels or triggering the opening of particular leaky channels as well as by destroying the lipid components of the membrane. Mitochondrial inhibition additionally decreases cellular ATP levels, and this further enhances accumulation of intracellular Ca2+. In addition, inhibition of the respiratory chain furthermore decreases the ΔΨm leading to reduced Ca2+ uptake into the mitochondrial matrix and potential release of Ca2+ into the cytoplasm leading to Ca2+ excitotoxicity. Furthermore, it is also hypothesized and estimated that ROS produced in MNs can escape into the extracellular environment and can damage the glutamate transporters on astrocytes. (B) An event following a mitochondrial malfunction in MNs inhibits complex IV of the ETC which leads to ROS generation seen in SOD1-mediated toxicity can be chemically trigger by Na-azide. This phenomenon can be partially reversed by blocking ROS production by riluzole in MNs or by inhibition of Ca2+ efflux at synapse sites. In addition, ROS generated via mitochondria are mostly neutralized by the inhibition of glutamate release trigger by riluzole. The impact of ROS for the cell becomes more severe when mitochondria are placed cardinally to buffer the calcium and to control the subsequent metabolic pathways, since an uncontrolled elevation in the cytosolic Ca2+ can lead to immediate cell death. The observed schematic diagram provide a potential mechanism of how mitochondrial inhibition can lead to selective MN degeneration and reverse by riluzole.
Author Contributions
MKJ established the experimental protocol, designed the experiments, carried out all of the experiments, analyzed the data, and wrote the manuscript. Bernhard U. Keller provides laboratory support and financial support for research work.
Funding
This study was partially supported by the Bundesministerium für Bildung und Forschung (BioRegioN/ERANET) [Grant 0313610A] and the Göttingen Center for Molecular Physiology of the Brain [Grant 1356834].
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to thank Prof. Dr. Bernhard U. Keller for comments on the manuscript, valuable discussions and mentorship and financial help. I would like to thank Dr. Diane M. Jaworski from University of Vermont and Drs. Namboodri and Arun from USUHS/Walter reed army institute for SH-SY5Y cell line, Cornelia Hühne for excellent technical assistance and Dr. Ananta Paine for comments on the manuscript.
Abbreviations
aCFS, artificial cerebrospinal fluid; ALS, amyotrophic lateral sclerosis; AM, acetoxy methyl ester; CCD, charge coupled device; CRAC, Ca2+ release-activated Ca2+; DMSO, dimethyl sulfoxide; ER, endoplasmic reticulum; ETC, electron transport chain; F, fluorescence; fALS, familial amyotrophic lateral sclerosis; HMNs, hypoglossal motoneurons; MNs, motor neurons; NADH, nicotinamide adenine dinucleotide; ROI, regions of interest; ROS, reactive oxygen species; SD, standard deviation; SEM, standard error of mean; SOD1, superoxide dismutase 1; WT, wild-type; [Ca2+]i, cytosolic calcium; [Ca2+]mito, mitochondrial calcium; ΔΨm, mitochondrial membrane potential.
References
Alexianu, M. E., Ho, B. K., Mohamed, A. H., La Bella, V., Smith, R. G., and Appel, S. H. (1994). The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann. Neurol. 36, 846–858. doi: 10.1002/ana.410360608
Azbill, R. D., Mu, X., and Springer, J. E. (2000). Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res. 871, 175–180. doi: 10.1016/S0006-8993(00)02430-6
Bausch, A. R., and Roy, G. (1996). Volume-sensitive chloride channels blocked by neuroprotective drugs in human glial cells (U-138MG). Glia 18, 73–77. doi: 10.1002/(SICI)1098-1136(199609)18:1<73::AID-GLIA8>3.0.CO;2-4
Bensimon, G., Lacomblez, L., and Meininger, V. (1994). A controlled trial of riluzole in amyotrophic lateral sclerosis ALS/Riluzole Study Group. N. Engl. J. Med. 330, 585–591. doi: 10.1056/NEJM199403033300901
Bergmann, F., and Keller, B. U. (2004). Impact of mitochondrial inhibition on excitability and cytosolic Ca2+ levels in brainstem motoneurons from mouse. J. Physiol. 555, 45–59. doi: 10.1113/jphysiol.2003.053900
Berridge, M. J. (1997). Elementary and global aspects of calcium signaling. J. Physiol. 499, 291–306. doi: 10.1113/jphysiol.1997.sp021927
Bootman, M. D., Berridge, M. J., and Lipp, P. (1993). Cooking with calcium: The recipes for composing global signals from elementary events. Cell 91, 367–373. doi: 10.1016/S0092-8674(00)80420-1
Bryson, H., Fulton, B., and Benfield, P. (1996). Riluzole: A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in amyotrophic lateral sclerosis. Drugs 52, 549–563. doi: 10.2165/00003495-199652040-00010
Budd, S. L. (1998). Mechanisms of neuronal damage in brain hypoxia/ischemia: Focus on the role of mitochondrial calcium accumulation. Pharmacol. Ther. 80, 203–229. doi: 10.1016/S0163-7258(98)00029-1
Carri, M. T., Ferri, A., Battistoni, A., Famhy, L., Gabbianelli, R., Poccia, F., et al. (1997). Expression of a Cu, Zn superoxide dismutase typical of familial amyotrophic lateral sclerosis induces mitochondrial alteration and increase of cytosolic Ca2+ concentration in transfected neuroblastoma SH-SY5Y cells. FEBS Lett. 414, 365–368.
Chang, Q., and Martin, L. J. (2016). Voltage-gated calcium channels are abnormal in cultured spinal motoneurons in the G93A-SOD1 transgenic mouse model of ALS. Neurobiol. Dis. 93, 78–95. doi: 10.1016/j.nbd.2016.04.009
Doble, A. (1996). The pharmacology and mechanism of action of riluzole. Neurology (suppl. 4) 47, S233–S241. doi: 10.1212/WNL.47.6_Suppl_4.233S
Dolmetsch, R. E., and Lewis, R. S. (1994). Signaling between intracellular Ca2+ stores and depletion-activated Ca2+ channels generates [Ca2+]i oscillations in T lymphocytes. J. Gen. Physiol. 103, 365–388. doi: 10.1085/jgp.103.3.365
Geevasinga, N., Menon, P., Ng, K., Van Den Bos, M., Byth, K., Kiernan, M. C., et al. (2016). Riluzole exerts transient modulating effects on cortical and axonal hyperexcitability in ALS. Amyotroph. Lateral Scler. Frontotemporal Degener. 17, 580–588.
Goos, M., Zech, W. D., Jaiswal, M. K., Balakrishnan, S., Ebert, S., Mitchell, T., et al. (2007). Expression of a Cu, Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of neuroblastoma cells to infectious injury. BMC Infect. Dis. 12:131. doi: 10.1186/1471-2334-7-131
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase. Science 264, 1772–1775.
Gutierrez, J., Federici, T., Peterson, B., Bartus, R., Betourne, A., Boulis, N. M., et al. (2016). Development of intrathecal Riluzole: a new route of administration for the treatment of amyotrophic lateral sclerosis patients. Neurosurgery 63 (Suppl. 1), 193. doi: 10.1227/01.neu.0000489810.52605.80
Harvey, J., Hardy, H. C., and Ashford, M. L. J. (1999). Dual actions of the metabolic inhibitor, sodium azide on KATP channel currents in the rat CRI-G1 insulinoma cell line. Br. J. Pharmacol. 126, 51–60. doi: 10.1038/sj.bjp.0702267
Hoyaux, D., Alao, J., Fuchs, J., Kiss, R., Keller, B., Heizmann, C. W., et al. (2000). S100A6, a calcium- and zinc-binding protein, is overexpressed in SOD1 mutant mice, a model for amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1498, 264–272. doi: 10.1016/S0167-4889(00)00101-4
Hubert, J. P., Burgevin, M. C., Terro, F., Hugon, J., and Doble, A. (1998). Effects of depolarizing stimuli on calcium homeostasis in cultured rat motoneurons. Br. J. Pharmacol. 125, 1421–1428. doi: 10.1038/sj.bjp.0702207
Hunter, F. E. Jr., Levy, J. F., Fink, J., Schutz, B., Guerra, F., and Hurwitz, F. (1959). Studies on the mechanism by which anaerobiosis prevents swelling of mitochondria in vitro: effect of electron transport chain inhibitors. J. Biol. Chem. 234, 2176–2186.
Jaiswal, M. K. (2009). Optical Analysis of [Ca2+]i and Mitochondrial Signaling Pathways: Implications for the Selective Vulnerability of Motoneurons in Amyotrophic Lateral Sclerosis (ALS). Available at: https://ediss.uni-goettingen.de/handle/11858/00-1735-0000-0006-B65A-C
Jaiswal, M. K. (2012). “Motoneuron specific calcium dysregulation and perturbed cellular calcium homestasis in amyotrophic lateral sclerosis: recent advances gained from genetically modified animals and cell culture models,” in Motor Neuron Diseases: Causes, Classification and Treatments, 1st Edn, eds B. J. Turner and J. B. Atkin (New York, NY: Nova Publishers), 87–114.
Jaiswal, M. K. (2013). Calcium, mitochondria and the pathogenesis of als: the good, the bad and the ugly. Front. Cell. Neurosci. 7:199. doi: 10.3389/fncel.2013.00199
Jaiswal, M. K. (2014). Selective vulnerability of motoneuron and perturbed mitochondrial calcium homeostasis in amyotrophic lateral sclerosis: implications for motoneurons specific calcium dysregulation. Mol. Cel. Ther 2, 26. doi: 10.1186/2052-8426-2-26
Jaiswal, M. K., and Keller, B. U. (2009). Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol. Pharmacol. 75, 478–489. doi: 10.1124/mol.108.050831
Jaiswal, M. K., Zech, W. D., Goos, M., Leutbecher, C., Ferri, A., Zippelius, A., et al. (2009). Impairment of mitochondrial calcium handling in a mtSOD1 cell culture model of motoneuron disease. BMC Neurosci. 10:64. doi: 10.1186/1471-2202-10-64
Jehle, T., Bauer, J., Blauth, E., Hummet, A., Darstein, M., Freiman, T. M., et al. (2000). Effects of riluzole on electrically evoked neurotransmitter release. Br. J. Pharmacol. 130, 1227–1234. doi: 10.1038/sj.bjp.0703424
Jou, M. J., Peng, T. I., Reiter, R. J., Jou, S. B., Wu, H. Y., and Wen, S. T. (2004). Visualization of the antioxidative effects of melatonin at the mitochondrial level during oxidative stress-induced apoptosis of rat brain astrocytes. J. Pineal Res. 37, 55–70. doi: 10.1111/j.1600-079X.2004.00140.x
Kaal, E. C. A., Veldman, H., Sodaar, P., Joosten, E. A. J., and Bär, P. R. (1998). Oxidant treatment causes a dose-dependent phenotype of apoptosis in cultered motoneurons. J. Neurosci. Res. 54, 778–786. doi: 10.1002/(SICI)1097-4547(19981215)54:6<778::AID-JNR5>3.0.CO;2-0
Kong, J., and Xu, Z. (1998). Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 18, 3241–3250.
Kretschmer, B. D., Kratzer, U., and Schmidt, W. J. (1998). Riluzole, a glutamate release inhibitor and motor behavior. Naunyn Schmiedebergs Arch. Pharmacol. 358, 181–190. doi: 10.1007/PL00005241
Ladewig, T., and Keller, B. U. (2000). Simultaneous patch-clamp recording and calcium imaging in a rhythmically active neuronal network in the brainstem slice preparation from mouse. Pflugers. Arch. 440, 322–332. doi: 10.1007/s004240051055
Lang-Lazdunski, L., Heurteaux, C., Dupont, H., Widmann, C., and Lazdunski, M. (2000). Prevention of ischemic spinal cord injury: Comparative effects of magnesium sulfate and riluzole. J. Vasc. Surg. 32, 179–189. doi: 10.1067/mva.2000.105960
Laslo, P., Lipski, J., Nicholson, L. F., Miles, G. B., and Funk, G. D. (2000). Calcium binding proteins in motoneurons at low and high risk for degeneration in ALS. Neuroreport 20, 3305–3308.
Launay, P., Cheng, H., Srivatsan, S., Penner, R., Fleig, A., and Kinet, J. P. (2004). TRPM4 regulates calcium oscillations after T cell activation. Science 306, 1374–1377. doi: 10.1126/science.1098845
Leon, J., Acuna-Castroviejo, D., Escames, G., Tan, D. X., and Reiter, R. J. (2005). Melatonin mitigates mitochondrial malfunction. J. Pineal Res. 38, 1–9. doi: 10.1111/j.1600-079X.2004.00181.x
Lips, M. B., and Keller, B. U. (1998). Endogenous calcium buffering in motoneurones of the nucleus hypoglossus from mouse. J. Physiol. 511, 105–117. doi: 10.1111/j.1469-7793.1998.105bi.x
Martin, M., Macias, M., Escames, G., Reiter, R. J., Agapito, M. T., Ortiz, G. G., et al. (2000). Melatonin-induced increased activity of the respiratory chain complexes I and IV can prevent mitochondrial damage induced by ruthenium red in vivo. J. Pineal Res. 28, 242–248. doi: 10.1034/j.1600-079X.2000.280407.x
Mazzone, G. L., and Nistri, A. (2011). Delayed neuroprotection by riluzole against excitotoxic damage evoked by kainate on rat organotypic spinal cord cultures. Neuroscience 190, 318–327. doi: 10.1016/j.neuroscience.2011.06.013
Menzies, F. M., Cookson, M. R., Taylor, R. W., Turnbull, D. M., Chrzanowska-Lightowlers, Z. M., Dong, L., et al. (2002). Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain 125, 1522–1533. doi: 10.1093/brain/awf167
Mu, X., Azbill, R. D., and Springer, J. E. (2000). Riluzole improves measures of oxidative stress following traumatic spinal cord injury. Brain Res. 870, 66–72. doi: 10.1016/S0006-8993(00)02402-1
Nowicky, A. V., and Duchen, M. R. (1998). Changes in [Ca2+]i and membrane currents during impaired mitochondrial metabolism in dissociated rat hippocampal neurons. J. Physiol. 507, 131–145. doi: 10.1111/j.1469-7793.1998.131bu.x
Obinu, M. C., Reibaud, M., Blanchard, V., Moussaoui, S., and Mperato, A. (2002). Neuroprotective effect of riluzole in a primate model of Parkinson’s disease: behavioral and histological evidence. Mov. Disord. 17, 13–19. doi: 10.1002/mds.1272
Okatani, Y., Wakatsuki, A., Reiter, R. J., Enzan, H., and Miyahara, Y. (2003). Protective effect of melatonin against mitochondrial injury induced by ischemia and reperfusion of rat liver. Eur. J. Pharmacol. 469, 145–152. doi: 10.1016/S0014-2999(03)01643-1
Paine, A., and Jaiswal, M. K. (2016). Promise and pitfalls of mitochondrial replacement for prevention and cure of heritable neurodegenerative diseases caused by deleterious mutations in mitochondrial DNA. Front. Cell. Neurosci. 10:219. doi: 10.3389/fncel.2016.00219
Pappolla, M. A., Sos, M., Omar, R. A., Bick, R. J., Hickson-Bick, D. L. M., Reiter, R. J., et al. (1997). Melatonin prevents death of neuroblastoma cells exposed to the Alzheimer amyloid peptide. J. Neurosci. 17, 1683–1690.
Reiter, R. J., Mayo, J. C., Tan, D. X., Sainz, R. M., Alatorre-Jimenez, M., and Qin, L. (2016). Melatonin as an antioxidant: under promises but over delivers. J. Pineal Res. 61, 253–278. doi: 10.1111/jpi.12360
Rothstein, J. D. (1995). Excitotoxicity and neurodegeneration in amyotrophic lateral sclerosis. Clin. Neurosci. 3, 348–359.
Seo, J. S., Choi, J., Leem, Y. H., and Han, P. L. (2015). Rosmarinic acid alleviates neurological symptoms in the g93a-sod1 transgenic mouse model of amyotrophic lateral sclerosis. Exp. Neurobiol. 24, 341–350. doi: 10.5607/en.2015.24.4.341
Simard, J. M., Tsymbalyuk, O., Keledjian, K., Ivanov, A., Ivanova, S., and Gerzanich, V. (2012). Comparative effects of glibenclamide and riluzole in a rat model of severe cervical spinal cord injury. Exp. Neurol. 233, 566–574. doi: 10.1016/j.expneurol.2011.11.044
Spalloni, A., Albo, F., Ferrari, F., Mercuri, N., Bernardi, G., Zona, C., et al. (2004). Cu/Zn-superoxide dismutase (GLY93?ALA) mutation alters AMPA receptor subunit expression and function and potentiates kainate-mediated toxicity in motor neurons in culture. Neurobiol. Dis. 15, 340–350.
Spiller, K. J., Cheung, C. J., Restrepo, C. R., Kwong, L. K., Stieber, A. M., Trojanowski, J. Q., et al. (2016). Selective motor neuron resistance and recovery in an jnducible mouse model of TDP-43 proteinopathy. J. Neurosci. 36, 7707–7717. doi: 10.1523/JNEUROSCI.1457-16.2016
Stefani, A., Spadoni, F., and Bernardi, G. (1997). Differential inhibition by riluzole, lamotrigine, and phenytoin of sodium and calcium currents in cortical neurons: implications for neuroprotective strategies. Exp. Neurol. 147, 115–122. doi: 10.1006/exnr.1997.6554
Storch, A., Burkhardt, K., Ludolph, A. C., and Schwarz, J. (2000). Protective effects of riluzole on dopamine neurons: involvement of oxidative stress and cellular energy metabolism. J. Neurochem. 75, 2259–2269. doi: 10.1046/j.1471-4159.2000.0752259.x
Udagawa, T., Fujioka, Y., Tanaka, M., Honda, D., Yokoi, S., Riku, Y., et al. (2015). FUS regulates AMPA receptor function and FTLD/ALS-associated behaviour via GluA1 mRNA stabilization. Nat. Commun. 6, 7098. doi: 10.1038/ncomms8098
Van Den Bosch, L., Vandenberghe, W., Klaassen, H., Van Houtte, E., and Robberecht, W. (2000). Ca (2+)-permeable AMPA receptors and selective vulnerability of motor neurons. J. Neurol. Sci. 180, 29–34. doi: 10.1016/S0022-510X(00)00414-7
Von Lewinski, F., and Keller, B. U. (2005a). Ca2+, mitochondria and selective motoneuron vulnerability: implications for ALS. Trends Neurosci. 28, 494–500. doi: 10.1016/j.tins.2005.07.001
Von Lewinski, F., and Keller, B. U. (2005b). Mitochondrial Ca2+ buffering in hypoglossal motoneurons from mouse. Neurosci. Lett. 380, 203–208. doi: 10.1016/j.neulet.2005.01.010
Wang, X. (2009). The anti-apoptotic activity of melatonin in neurodegenerative diseases. CNS Neurosci. Ther. 15, 345–357. doi: 10.1111/j.1755-5949.2009.00105.x
Weber, K. S., Hildner, K., Murphy, K. M., and Allen, P. M. (2010). Trpm4 differentially regulates Th1 and Th2 function by altering calcium signaling and NFAT localization. J. Immunol. 185, 2836–2846. doi: 10.4049/jimmunol.1000880
Yamamoto, H. A., and Tang, H. W. (1996). Preventive effect of melatonin against cyanide-induced seizures and lipid peroxidation in mice. Neurosci. Lett. 207, 89–92. doi: 10.1016/0304-3940(96)12493-9
Yokoo, H., Shiraishi, S., Kobayashi, H., Yanagita, T., Yamamoto, R., and Wada, A. (1998). Selective inhibition by riluzole of voltage-dependent sodium channels and catecholamine secretion in adrenal chromaffin cells. Naunyn-Schmiedebergs Arch. Pharmacol. 357, 526–531. doi: 10.1007/PL00005203
Keywords: ALS, SOD1G93A, riluzole, melatonin, mitochondria, excitotoxicity, cell death, Ca2+ signaling
Citation: Jaiswal MK (2017) Riluzole But Not Melatonin Ameliorates Acute Motor Neuron Degeneration and Moderately Inhibits SOD1-Mediated Excitotoxicity Induced Disrupted Mitochondrial Ca2+ Signaling in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 10:295. doi: 10.3389/fncel.2016.00295
Received: 11 July 2016; Accepted: 12 December 2016;
Published: 06 January 2017.
Edited by:
Chao Deng, University of Wollongong, AustraliaReviewed by:
Raman Chandrasekar, Kansas State University, USAGundars Goldsteins, University of Eastern Finland, Finland
Copyright © 2017 Jaiswal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manoj Kumar Jaiswal, mj2750@cumc.columbia.edu
†Present address: Manoj Kumar Jaiswal, Divisions of Molecular Imaging and Neuropathology, New York State Psychiatry Institute, New York, NY, USA; Department of Psychiatry, Columbia University, New York, NY, USA