Mitochondria, Bioenergetics and Excitotoxicity: New Therapeutic Targets in Perinatal Brain Injury
 Bryan Leaw
Bryan Leaw Syam Nair
Syam Nair Rebecca Lim
Rebecca Lim Claire Thornton
Claire Thornton Carina Mallard
Carina Mallard Henrik Hagberg4,5
Henrik Hagberg4,5- 1The Ritchie Centre, Hudson Institute of Medical Research, Clayton, VIC, Australia
- 2Perinatal Center, Institute of Physiology and Neuroscience, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden
- 3Department of Obstetrics and Gynaecology, Monash University Clayton, Clayton, VIC, Australia
- 4Centre for the Developing Brain, Division of Imaging Sciences and Biomedical Engineering, King’s College London, King’s Health Partners, St. Thomas’ Hospital, London, United Kingdom
- 5Perinatal Center, Department of Clinical Sciences, Sahlgrenska Academy, Gothenburg University, Gothenburg, Sweden
Injury to the fragile immature brain is implicated in the manifestation of long-term neurological disorders, including childhood disability such as cerebral palsy, learning disability and behavioral disorders. Advancements in perinatal practice and improved care mean the majority of infants suffering from perinatal brain injury will survive, with many subtle clinical symptoms going undiagnosed until later in life. Hypoxic-ischemia is the dominant cause of perinatal brain injury, and constitutes a significant socioeconomic burden to both developed and developing countries. Therapeutic hypothermia is the sole validated clinical intervention to perinatal asphyxia; however it is not always neuroprotective and its utility is limited to developed countries. There is an urgent need to better understand the molecular pathways underlying hypoxic-ischemic injury to identify new therapeutic targets in such a small but critical therapeutic window. Mitochondria are highly implicated following ischemic injury due to their roles as the powerhouse and main energy generators of the cell, as well as cell death processes. While the link between impaired mitochondrial bioenergetics and secondary energy failure following loss of high-energy phosphates is well established after hypoxia-ischemia (HI), there is emerging evidence that the roles of mitochondria in disease extend far beyond this. Indeed, mitochondrial turnover, including processes such as mitochondrial biogenesis, fusion, fission and mitophagy, affect recovery of neurons after injury and mitochondria are involved in the regulation of the innate immune response to inflammation. This review article will explore these mitochondrial pathways, and finally will summarize past and current efforts in targeting these pathways after hypoxic-ischemic injury, as a means of identifying new avenues for clinical intervention.
Introduction
Perinatal brain injury remains a significant cause of long-term neurological and physical disability, and a significant socioeconomic burden in both developed and developing countries (Kruse et al., 2009; Lawn et al., 2011). While preterm birth carries a significantly increased risk of brain injury, babies born term are also at risk of exposure to insults including hypoxic-ischemeic encephalopathy (HIE), metabolic disease, or neonatal stroke (Hagberg et al., 2015). Moderate hypothermia is a validated and increasingly used intervention in infants that develop brain injury, including from hypoxia-ischemia (HI; Hutchison et al., 2008; Azzopardi et al., 2013). However, its availability is typically restricted to developed countries due to the cost of equipment needed as well as the need for a stable supply of electricity to maintain a therapeutic level of cooling (Kumar et al., 2009). While there is recognition that cooling may still be effective in low resource settings that do not have tertiary intensive care, there remains the ethical dilemma of performing clinical trials on neonatal encephalopathy with the addition of normothermia groups (Montaldo et al., 2015; Tagin et al., 2015). Furthermore, hypothermia does not always confer neuroprotection (Azzopardi et al., 2009). There is a need to better understand the underlying pathogenesis of perinatal brain injury to be able to identify new therapeutic targets, and thus reduce the prevalence of neurological impairment and associated lifelong physical disability such as cerebral palsy.
Mitochondria are the powerhouses of the cell, primarily responsible for the production of adenosine triphosphate (ATP) as well as playing regulatory roles in cell death, including autophagy and apoptosis (Nunnari and Suomalainen, 2012). Advances in genomic and proteomic sequencing have provided irrefutable evidence that these vestiges of bacterial ancestry also perform more diverse roles, particularly in disease (Hagberg et al., 2014). These range from early work showing that mutations in mitochondrial DNA (mtDNA) are implicated in diseases such as Parkinson’s disease (Swerdlow et al., 1996), to research on the epigenetic modulation of mtDNA, including methylation by DNA methyltransferases, which add yet another layer in which mitochondria can influence and contribute to disease (van der Wijst and Rots, 2015). There is thus a growing appreciation that identifying and targeting mitochondrial pathways thought to be responsible for the manifestation of initial injury post asphyxia as well as long-term neurodevelopment holds great promise in the field of perinatal medicine. In addition, recent work suggests that mitochondria, and reactive oxygen species (ROS) derived from mitochondria, have important roles in the regulation of inflammation both in response to sterile and microbial insults (Suliman and Piantadosi, 2016). This review article seeks to summarize past therapeutic interventions, as well as current efforts to target these mitochondrial pathways during the fragile early period of development, and to identify new avenues for therapeutic intervention. First, however it is important to understand the different phases of hypoxic-ischemic injury to identify targets for intervention.
The Phases of Hypoxic-Ischemic Injury
The understanding of the pathogenesis of perinatal brain injury has evolved rapidly in recent years. Initial thoughts centered on a cascade of energy and nutrient deficiency, inflammation, oxidative stress and subsequent cell death and neuronal loss (Volpe, 2001; Hagberg et al., 2015). Currently it is well accepted that HI triggers an acute series of events, as well as a tertiary phase of injury response, which extends days to months after the initial insult (Fleiss and Gressens, 2012). Briefly, the events preceding these later outcomes can be divided into three distinct periods—the primary, secondary and tertiary phases, each identified by different molecular processes as well as the involvement of different cellular subtypes (Fleiss and Gressens, 2012). During the primary phase in the minutes-to-hours following asphyxia, neural energy failure occurs through a rapid decrease of high energy phosphates (phosphocreatine (pCr), ATP). This is followed by a transient recovery to baseline levels during reperfusion generating a “latent” phase therapeutic window. A secondary “delayed” phase subsequently occurs from hours to days post injury characterized by an apoptotic-necroptotic cell death phenotype (Northington et al., 2011). The tertiary phase is thought to be responsible for persisting effects years after injury, including immune memory (Hoeijmakers et al., 2016) and proper development of white matter tracts in adulthood (Favrais et al., 2011). This suggests that inflammation during the early perinatal phase can modify later risk to developing neurological and psychiatric disorders (Hagberg et al., 2012). Mitochondria play essential roles in both proper neurodevelopment as well as in response to HI injury pathogenesis (Hagberg et al., 2014), so effective targeting of mitochondria derangement might have therapeutic merit, especially during this open therapeutic window.
Physiological Roles of Mitochondria, Structure, Trafficking
Mitochondria are small membrane-enclosed organelles, remarkably mobile and plastic, associated with ATP generation, calcium regulation and the biosynthesis of amino acids, lipids and nucleotides (Green et al., 2011).
Mitochondria constantly change their shape and undergo fusion, fission, migration biogenesis and degradation (mitophagy); matching the cellular needs (Archer, 2013; Figure 1). In order to understand variations in mitochondria function and consequent selective vulnerability to injury, the organelle must be placed within the context of its cellular, functional, developmental and neuroanatomical environment (Dubinsky, 2009; Rintoul and Reynolds, 2010). The location of mitochondria in the cell varies between cell types, but they are most often localized near sites of high ATP utilization as their major role is to produce and supply energy, ATP, to the cells through the enzyme complexes forming the respiratory chain. Electron flow through the electron transport chain (ETC) generates a proton gradient across the inner mitochondrial membrane, which drives the production of ATP by ATP synthase. Under normal conditions, this machinery provides almost all (>90%) of the ATP in the brain (Hagberg et al., 2014). However, a small proportion of electrons escape the ETC complexes I, II and III under normal conditions to react with oxygen to form superoxide (; Grivennikova et al., 2010). Mitochondrial function is critically important during brain development and throughout life in metabolic tasks and for the regulation of cell death.
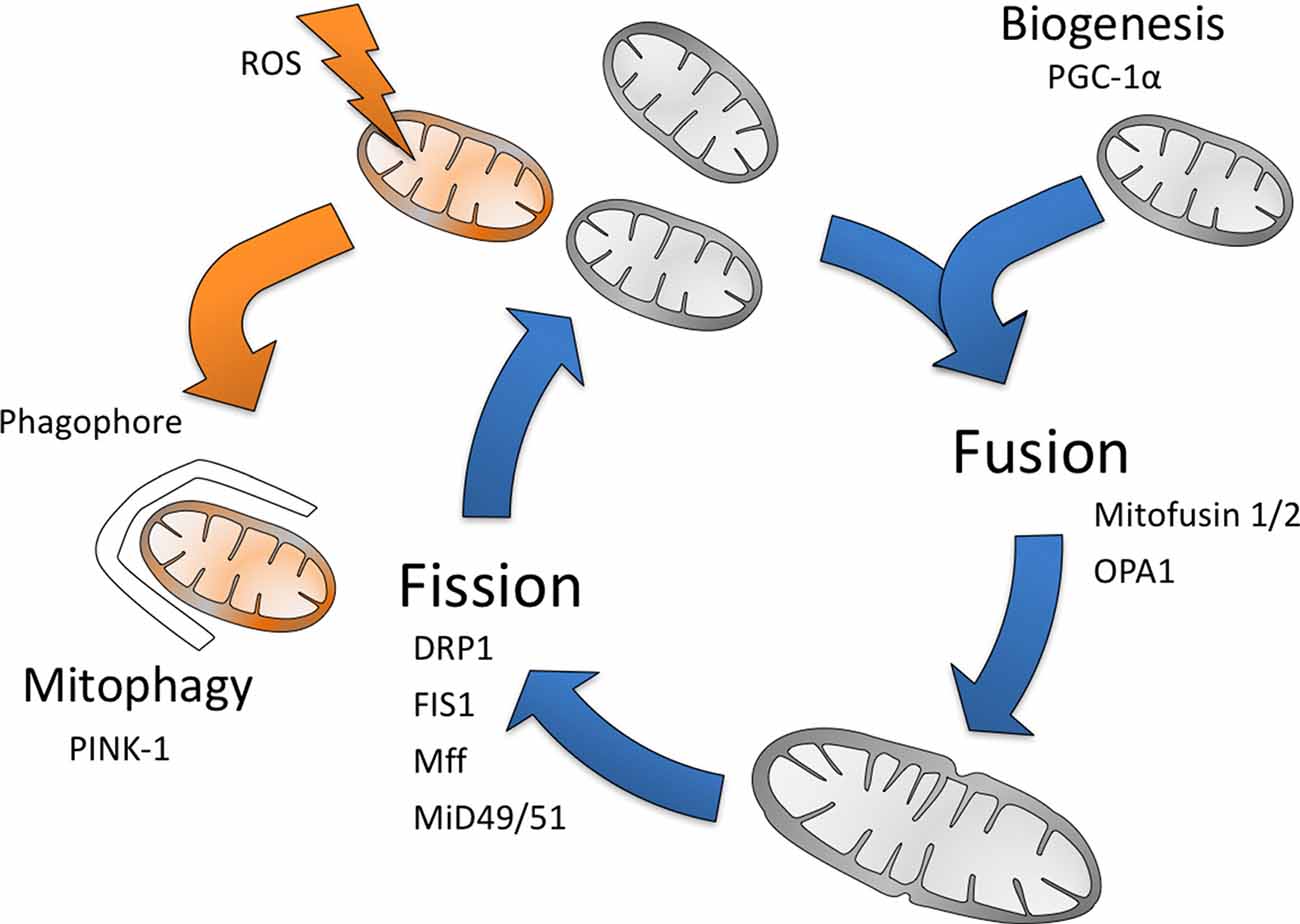
Figure 1. The mitochondrial life cycle. Mitochondria turnover occurs continuously throughout life, with mitochondria constantly being degraded (mitophagy) and replaced (biogenesis). Mitochondria are also able to produce reactive oxygen species (ROS), which causes mitochondrial membrane destabilization and damage to their proteins and DNA, resulting in their degradation. Mitochondria also undergo fragmentation (fission) and fusion, which are vital to many cellular processes and in particular allow adaptation to cellular needs (Gottlieb and Carreira, 2010).
Mitochondrial Morphobiogenesis: Physiological Mechanisms and Brain Development
Mitophagy
The mitochondrial genome is unprotected and mtDNA repair is inefficient compared with that in the nucleus (Hiona et al., 2010). Damaged mitochondria can generate large amounts of ROS which is not only toxic to mtDNA but can promote lipid peroxidation, mitochondrial dysfunction, impaired cellular function and induce apoptosis (Murphy, 2009; Grivennikova et al., 2010). Therefore, damaged and dysfunctional mitochondria are constantly being degraded (mitophagy) to reduce the oxidative load and are replaced by new mitochondria through mitochondrial biogenesis (Gottlieb and Carreira, 2010). This process is particularly important in long-lived non-proliferative cells such as neurons. Dissipation of mitochondrial membrane potential results in accumulation of the kinase PINK-1 at the outer mitochondrial membrane which phosphorylates ubiquitin and parkin resulting in binding of mitophagy receptors (e.g., optineurin, NDP52) to the mitochondrial surface. The latter will recruit the autophagy machinery proteins (e.g., ULK1, DFCP1 and WIPI1) and the mitochondria will be wrapped into the autophagosome and degraded by fusion with lysosomes (Lazarou et al., 2015; Pickrell and Youle, 2015). The importance of mitophagy for brain development remains unknown. However, deletion of the autophagy (Atg5 or Atg7) genes results in neurodegeneration during early life in mice (Hara et al., 2006; Komatsu et al., 2006) and suppression of genes involved in lysosomal function causes severe central nervous system (CNS) abnormalities (Levine and Kroemer, 2008). More specifically to mitophagy, mutations in the human genes Parkin and PINK-1 result in a juvenile form of neurodegeneration (Levine and Kroemer, 2008).
Biogenesis
Mitochondrial biogenesis usually occurs starting from already existing mitochondria and needs proteins encoded by both nuclear and mtDNA (Scarpulla, 2011). Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) is the key regulator in this process, forming complexes with DNA and supporting the function of transcription factors such as nuclear respiratory factors (Wu et al., 1999; Huss et al., 2002). These factors regulate transcription of nuclear genes encoding mitochondrial proteins. Additionally, the mitochondrial transcription factor A (TFAM) is expressed from nuclear DNA, translated in the cytosol, and transported into the mitochondria (Attardi and Schatz, 1988). TFAM drives the transcription of 13 additional key enzymes needed for assembly of the ETC, encoded within the mitochondrial genome, together with the RNA needed for their translation (Larsson et al., 1998). During neuronal differentiation, the number of mitochondria per cell increases, and inhibition of mitochondrial biogenesis impairs neuronal differentiation (Vayssière et al., 1992; Mattson et al., 2008). PGC-1α is expressed at high concentrations in neurons due to their high energy demands (Andersson and Scarpulla, 2001).
Overexpression of PGC-1α in cultured neurons increases the number of dendritic spines and promotes synaptic differentiation, whereas deletion of the gene for PGC-1α, has the opposite effect (Cheng et al., 2012). In addition, mice with knockdown of the PGC-1α gene exhibit progressive neuropathology and abnormal behavior (Lin et al., 2004; Cheng et al., 2012). These data strongly suggest that mitochondrial biogenesis has an important regulatory role in synaptic and brain development.
Fusion/Fission
Mitochondria constantly fuse and divide which appears to be crucial for a number of functions such as the maintenance of organelle function, mediating DNA or protein quality control and repair of injured mitochondria (Tanaka and Youle, 2008). The process is driven by dynamin-related protein 1 (DRP1) recruited to mitochondria by fission proteins (e.g., FIS1, Mff, MiD49/51; Sheridan and Martin, 2010; van der Bliek et al., 2013). Mitofusins 1 and 2 mediate fusion of the outer mitochondrial membrane, whereas Optic atrophy 1 (OPA1) fuses the inner membrane (Benard and Karbowski, 2009). The fusion–fission cycle is critical for embryonic and brain development (Benard and Karbowski, 2009). Mitochondrial fission appears essential for dendritic development. Disruption of DRP1-dependent fission leads to very elongated mitochondria in Purkinje cells resulting in abnormal spines, dendrites and synapses, and ultimately in ataxic behavior (Liu and Shio, 2008). Overexpression of dominant-negative alleles of the gene that encodes DRP1, leads to fewer spines and synapses, reversed by DRP1 which increases the density of dendritic spines (Li et al., 2004). Cultured neurons deficient in DRP1 have decreased numbers of neurites and defective synapses (Ishihara et al., 2009); a dominant-negative mutation of the DRP1 gene was found in a newborn girl with microcephaly and abnormal brain development (Waterham et al., 2007). These results support that fission is an important means to increase the number of mitochondria to meet energy demands during neuronal plasticity. Inability to meet these demands has severe effects on CNS development. Mitochondrial fusion is also critical for brain development. Mitofusin 1 and mitofusin 2 are both essential for embryonic CNS development in mice and for cerebellar development. A mutation in mitofusin 2 causes the neurodegenerative disorder Charcot-Marie-Tooth neuropathy type 2A (Chen et al., 2003; Zuchner et al., 2004). Additionally, overexpression of OPA1 leads to a decrease in dendritic spines, and OPA1 gene mutation causes autosomal-dominant optic atrophy type 1 (Delettre et al., 2000; Li et al., 2004). Mitochondrial fusion proteins may also attenuate apoptosis by inhibiting the release of proapoptotic agents like cytochrome c (cyt c), while mitochondrial fission protein DRP1 promotes apoptosis through Bax, leading to mitochondrial outer membrane permeabilization and cell death (Cassidy-Stone et al., 2008).
Mitochondrial Role in Apoptosis and Secondary Brain Injury
Apoptosis (programmed cell death) is essential for the normal development of tissues and is especially key in neuronal development (Raff et al., 1993). The balance between cell survival and cell death is therefore required to be highly regulated; as such it is unsurprising that aberrant activation of apoptotic pathways occurs in several neurological conditions including stroke and a variety of neurodegenerative diseases (Vila and Przedborski, 2003). Cellular apoptosis can be achieved through two routes, the extrinsic pathway activated in response to extracellular signals such as the cell death receptor Fas and tumor necrosis factor alpha (TNF-α) and mediated by death receptors (Green, 2000) and the intrinsic pathway activated in response to DNA damage or cellular stress. Although each pathway has unique members, both mechanisms may converge downstream at the level of the mitochondrion, where if the insult is severe enough, there is catastrophic permeabilization from which the cell cannot recover. It is important to note that mechanisms of mitochondrial permeabilization are age-dependant and while Cyclophilin D is critical in the adult brain, Bax-dependent mechanisms dominate in the immature brain (Wang et al., 2009).
Mitochondrial permeabilization results in the release of mitochondrial apoptogenic factors into the cytosol including apoptosis-inducing factor (AIF), endonuclease g (Endo G), cyt c and Smac/Diablo. These proteins have a number of pro-apoptotic functions; cyt c interacts with Apaf-1 to form an active apoptosome, providing a platform for procaspase-9 cleavage; Smac/Diablo interacts with inhibitors of apoptosis (IAP) reducing their negative influence on the activity of caspases (Vila and Przedborski, 2003). In contrast with cyt c and Smac/Diablo, AIF and Endo G operate through a caspase-independent pathway. Both are translocated to the nucleus from the mitochondria in response to death—inducing stimuli where they induce fragmentation of nuclear DNA (Susin et al., 1999; Li et al., 2001).
Some plasma membrane receptors contain the so-called death domain in their intracellular domain (e.g., TNF-R1, DR4, DR5, Fas) and are able to trigger apoptosis when activated from the binding of the corresponding ligand (e.g., TNF-α, TRAIL, FasL). This extrinsic pathway of apoptosis continues with the activation of a death-inducing signaling complex (DISC) adjacent to the death domain of the receptor. Activated DISC catalyzes the proteolytic cleavage and transactivation of procaspase-8 (Love, 2003). Activated caspase-8 either directly activates caspase-3 or mediates cleavage of Bcl-2 interacting domain (BID) to truncated Bid (tBid), which integrates different death pathways at the mitochondria (Sugawara et al., 2004). tBid translocates to the mitochondria where it interacts with other proapoptotic proteins and triggers the release of apoptogenic factors like cyt c and AIF from the mitochondria. Apoptosis then proceeds in the same way as for the intrinsic pathway with caspase-dependent and caspase-independent cell death regulated by mitochondria. There is ample evidence to suggest that both the intrinsic (Zhu et al., 2000, 2005) and to some extent the extrinsic pathway (Graham et al., 2004; Kichev et al., 2014) are critical contributors to immature brain injury (see this issue). In addition, AIF is released from mitochondria after neonatal HI (Zhu et al., 2003) and binds to cyclophilin A in the cytosol and the complex translocates to the nucleus and induces DNA damage (Zhu et al., 2007a) and contributes to brain injury (Zhu et al., 2007b). Notably, the caspase-dependent route appears to be more important in females while the AIF pathway is more predominant in males (Zhu et al., 2006; Johnston and Hagberg, 2007). The knowledge about mitochondrial physiology and its role in cell death has expanded dramatically over the past decade. Nonetheless, more detailed information regarding the role of mitochondria in perinatal injury and brain development is urgently needed in order to develop more effective mitoprotective therapies.
Contribution of Mitochondria to Inflammation
Inflammation is an important risk factor for injury in the developing brain (Strunk et al., 2014; Hagberg et al., 2015; Lai et al., 2017). Mitochondria regulate innate immune responses and play a direct role in the assembly of innate sensing machineries that trigger the host immune response. This is achieved mainly through transcriptional regulation of inflammatory cytokines/chemokines and their maturation by inflammasomes (Monlun et al., 2017). Mitochondria converge on signaling pathways involved in inflammation through: (a) mitochondrial ROS (mtROS) production; (b) mtDNA release; and (c) mitochondrial antiviral signaling protein (MAVS). These act as key triggers in the activation of innate immune response following a variety of stress signals that include infection, tissue damage and metabolic dysregulation (Sandhir et al., 2017). ROS produced by mitochondria, are a major host defense mechanism since they act as a crucial signaling molecule and as mediators of inflammation.
The movement of across mitochondrial membranes is highly limited because of its negative charge. However, the presence of transmembrane proteins, such as voltage-dependent anion channels (VDAC) found in mitochondria, allow trans-membrane passage of produced in ETC (Han et al., 2003). Thereby allowing access to cytosolic targets leading to multiple functional outcomes such as activation of redox-sensitive transcription factors like hypoxia inducible factor 1 alpha (HIF-1α) and NF-κB, causing activation of pro-inflammatory cytokines and inflammasomes (Chandel et al., 2000; Wang et al., 2010).
Pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) bind to specific receptors including RIG-I-like receptors (RLRs), NOD-like receptors (NLRs) and Toll-like receptors (TLR), to generate cytokines that are essential for eliminating pathogens or repairing tissue damage (Mogensen, 2009). mtDNA is a rich source of DAMPs which activate several innate immune pathways involving toll-like receptor 9 (TLR9), NLRP3 and STING signaling thereby resulting in effector responses (Weinberg et al., 2015). Systemic activation of toll-like receptor 2 (TLR2) induces brain inflammation and increases the vulnerability to HI, possibly through suppression of mitochondrial respiration (Mottahedin et al., 2017a,b).
During viral infection, the pattern recognition receptors RIG-I and MDA5 attach to viral RNA interacting the mitochondrial polypeptide adaptor, MAVS, which drives the production of type I interferon (Saitoh and Akira, 2010). Studies have demonstrated that viral-mediated disruption of mtDNA homeostasis serves as a cell-intrinsic indicator of infection that works in parallel with virus sensing mechanisms to engage antiviral innate immunity (West et al., 2015). Interestingly, recent studies have demonstrated that mtDNA induces lung and liver inflammation (Zhang et al., 2010). It is also proposed that mitochondrial DAMPs drive hyperactivation of innate immunity, which might underlie systemic inflammatory response syndrome (Tait and Green, 2012).
Effect of Inflammation on Mitochondrial Metabolism
As the site of cellular respiration and energy production, mitochondrial metabolism is one of the central processes affected by inflammation. Acutely triggered immune responses, as well as chronic inflammation, are characterized by significant changes in mitochondrial metabolism. This can result in shifts in energy supply/demand causing metabolic acidosis and hypoxia, thereby triggering phenotypic shifts in immune cells like microglia. Thus, strategies directed at controlling excessive inflammation mediated by mitochondria through metabolic control may represent novel preventive and therapeutic interventions.
Mitochondria can exert immune regulation at different levels by manipulation of metabolic pathways, thereby allowing an appropriate cytokine response to each situation, which is crucial for the correct establishment of immune responses (Monlun et al., 2017; Tur et al., 2017). In macrophages, activation via the lipopolysaccharide (LPS)/Toll-like receptor 4 (TLR4) pathway cause accumulation of Krebs cycle intermediates such as succinate in the mitochondria, resulting in the stabilization of HIF-1α and promoting inflammatory gene expression, such as induction of interleukin-1 beta (IL-1β; Tannahill et al., 2013; Mills et al., 2016).
The switch from oxidative phosphorylation (OXPHOS) to glycolysis, a phenomenon similar to the Warburg effect, is an important concept for understanding metabolic changes occurring in immune cells upon activation (Kelly and O’Neill, 2015). The preferential use of glycolysis over OXPHOS, although inefficient in terms of total energy production, allows immune cells to churn-out ATP and intermediates for cytokine production at a faster rate (Marelli-Berg et al., 2012; Chang et al., 2013). In anti-microbial defense mechanisms, such as neutrophil extracellular traps (NETs), neutrophils derive energy from glycolysis as they contain very few mitochondria. NET formation is inhibited by the glycolysis inhibitor, 2-deoxy-glucose, and to a lesser extent by the ATP synthase inhibitor oligomycin (Rodríguez-Espinosa et al., 2015). These examples provide evidence of cross talk between metabolism and immune response and thus the possibility of metabolism-based immune regulation.
Mitochondrial metabolism is necessary for T cell activation and pharmacologic inhibition of OXPHOS and glycolysis in vitro ablate T cell proliferation (Chang et al., 2013). Activated T cells isolated from a mouse model of systemic lupus erythematosus (SLE) are dependent on mitochondrial metabolism (Wahl et al., 2010) and furthermore, peripheral blood lymphocytes from patients with SLE have increased mitochondrial metabolism and ROS production (Gergely et al., 2002).
Release of mtDNA can cause activation of inflammasomes resulting in caspase-1-dependent secretion of the inflammatory cytokines IL-1β and IL-18, and an inflammatory form of cell death referred to as pyroptosis (Yu et al., 2014). Pyroptosis has been defined as a type of programmed cell death triggered by pathological stimuli and is important in controlling microbial infections (Bergsbaken et al., 2009). NLRP3-mediated inflammatory signaling, IL-1β production and pyroptosis in macrophages causes a disruption of glycolytic flux, an important signal for host-cell response to the intracellular pathogen, which disrupt metabolism by uptake of host-cell glucose (Sanman et al., 2016).
Recent data indicate that TFAM when released from necrotic cells could act as a specific DAMP causing a pro-inflammatory and cytotoxic responses (Little et al., 2014). TFAM exacerbates the inflammatory response in T cells through lysosomal dysfunction (Baixauli et al., 2015). Following necrosis, TFAM acts as a danger signal enhancing the plasmacytoid dendritic cell (pDC) response by binding to the receptor for advanced glycation end products (RAGE) and TLR9 (Cherry et al., 2016).
It is clear from above that there is a link between mitochondrial function, inflammation and energy metabolism in immune cells, however, such effects have thus far remained poorly characterized in the brain. In microglia cells, a short exposure to a low dose of LPS causes a transient increase in OXPHOS (Figure 2). This increase in OXPHOS is however suppressed in response to a prolonged exposure to high dose of LPS, forcing a shift in metabolism towards glycolysis to match the metabolic demand. This LPS-induced metabolic reprogramming is directly related to mitochondrial dynamics as preventing excessive mitochondrial fission in microglia by a DRP-1 inhibitor reverses the LPS induced metabolic switch and reduces the pro-inflammatory cytokine output in microglia (Nair et al., 2016). Thus, in addition to their well-appreciated roles in cellular metabolism and programmed cell death, mitochondria appear to have a cardinal role in regulating the immune system. In summary, patients with inflammatory diseases may benefit from detailed evaluation of mitochondrial function and suitable metabolic support to improve immune dysfunction. Altering mitochondrial dynamics may be a therapeutic modality for preventing neuroinflammation-induced microglia over-activation and may prevent neuronal cell death associated with various neurodegenerative processes. This is of particular importance given the lack of treatment options for perinatal brain injury despite many showing promise at the pre-clinical phases.
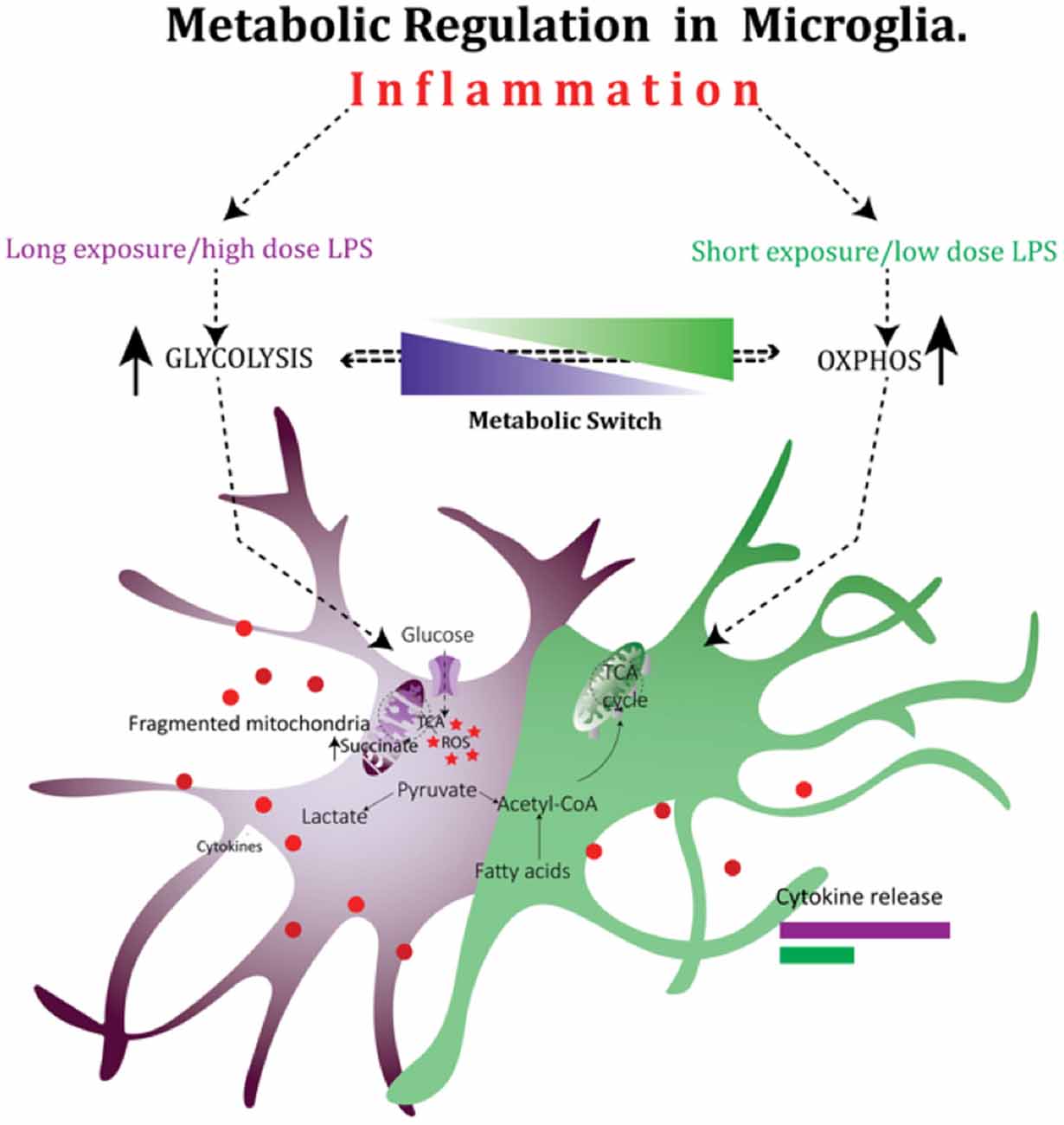
Figure 2. Mitochondria play a critical role in the regulation of cellular metabolism in microglia after exposure to lipopolysaccharide (LPS). Metabolic reprogramming in microglia cells during inflammation: in microglia cells, a short exposure to a low dose of LPS causes a transient increase in oxidative phosphorylation (OXPHOS). This increase in OXPHOS is however suppressed in response to a prolonged exposure to high dose of LPS, forcing a shift in metabolism towards glycolysis to match the metabolic demand.
Therapies for Perinatal Brain Injury
Despite improvements in the standard of care, the only “treatment” available for perinatal brain injury is preventative in nature—clinical hypothermia. Hypothermia, or cooling of the infants’ body temperature to 33–34°C within several hours of an hypoxic-ischemic event, was first shown in a pilot study to reduce both mortality and long term motor outcomes (Eicher et al., 2005), and is now widely established in neonatal care (Shankaran et al., 2005; Azzopardi et al., 2009; Edwards et al., 2010). Despite this, hypothermia is not always effective, e.g., with head cooling studies showing no effect in infants with severe brain injury after HIE (Gluckman et al., 2005). A factor contributing to the variance in therapeutic efficacy is the requirement of maintaining core body temperature within a small and narrow window, which is particularly challenging in low resource settings (Kumar et al., 2009). To overcome these caveats, hypothermia has been used in combination with other experimental therapies that have shown promise in clinical trials.
Combination Treatments Approved for Clinical Trials
Erythropoietin (EPO)
Erythropoietin (EPO) is a naturally produced angiogenic hormone having neuroprotective, neurogenic (Wang et al., 2004) as well as anti-inflammatory (Sun Y. et al., 2005; Rees et al., 2010) and anti-apoptotic (Kellert et al., 2007) properties. Recombinant human EPO (rhEPO) is safe for administration and is currently used to treat patients with low circulating levels as a result of chronic kidney disease or chemotherapy. Importantly, EPO is an activator of mitochondrial biogenesis (Carraway et al., 2010), and within the CNS appears to work by increasing the expression of the mitochondrial regulator PGC-1α in astrocytes and neurons (Horng et al., 2015). In a rodent model of adult stroke, rhEPO also increased vascular endothelial growth factor (VEGF) and brain-derived neurotrophic factor (BDNF), stimulating recovery as well as reducing stroke infarct size and improving functional outcomes (Wang et al., 2004). rhEPO also ameliorated hyperoxia-induced apoptosis and inflammation in neonatal mice (Kaindl et al., 2008; Sifringer et al., 2010), possibly via modulation of key autophagic proteins (Bendix et al., 2012).
In humans, an early clinical trial showed infants with HIE treated with a combination of rhEPO and hypothermia, had improved neurological outcomes, as well as fewer white matter tract abnormalities (Elmahdy et al., 2010). Even though EPO is one of the most promising neuroprotective interventions currently undergoing clinical testing, a recent 2-year follow-up study in very preterm infants treated with EPO (NCT00413946) showed no demonstrable differences in neurodevelopmental outcomes (Natalucci et al., 2016). However, in a Chinese study preterm infants were randomly assigned to receive rhEPO (500 IU/kg; n = 366) or placebo (n = 377) intravenously within 72 h after birth and then once every other day for 2 weeks (Song et al., 2016). The rate of moderate/severe neurological disability in the rhEPO group (7.1%) was significantly lower compared to the placebo group (18.8%; p < 0.001; Song et al., 2016). Furthermore, the considerable variation in between timing and dosage even in pre-clinical studies (see commentary by Juul, 2013) can mean the difference between improved neurological outcomes (Fan et al., 2013) and no therapeutic merit (Fang et al., 2013).
Melatonin
Melatonin, more commonly known as the sleep hormone, has been widely used as an anti-oxidative therapy in experimental models for a number of years (Balduini et al., 2012; Manchester et al., 2015). Melatonin works primarily via scavenging free radicals and stimulating the innate anti-oxidant system (Barlow-Walden et al., 1995). There is evidence that melatonin pre-treatment is neuroprotective in a rat model of middle cerebral artery occlusion (MCAO; Pei and Cheung, 2004), and furthermore, this protection was observed when melatonin was administered to the fetus directly (Welin et al., 2007) or via the maternal circulation (Miller et al., 2005). After umbilical cord occlusion, melatonin significantly reduced DNA and RNA fragmentation, apoptosis, as well as inflammation and astroglial activation (Miller et al., 2005). Crucially, melatonin can also promote white matter maturation, one of the key deficits in diseases of childhood disability such as cerebral palsy (Olivier et al., 2009). Recently mitochondria have been postulated as a major synthesis site for melatonin (He et al., 2016), suggesting that mitochondria may play a critical role in its anti-oxidative and therapeutic properties. Indeed, melatonin administration has been shown to be mitoprotective via acting as a mitochondrial antioxidant (Martin et al., 2000) and can also directly inhibit the opening of the mitochondrial permeability transition pore (mtPTP), responsible for release of calcium and cyt c from the mitochondria (Andrabi et al., 2004). Melatonin also modulates mitochondrial bioenergetics, decreasing mitochondrial fission and increasing mitochondrial fusion after oxidative stress (Parameyong et al., 2015; Chuang et al., 2016), and regulating autophagy and mitophagy (see review by Coto-Montes et al., 2012). In clinical trials, melatonin combined with clinical hypothermia improved white matter tract development and reduced apoptosis in a piglet model of HIE (Robertson et al., 2013) and a phase I clinical trial is currently underway (NCT02621944).
Stem Cell Therapies
There has been a focus on cell therapies in the field of regenerative medicine in recent years (Baraniak and McDevitt, 2010). Cell therapies have been heralded as the next pillar of modern medicine due to their reported multiple modes of action, touted as an advantage over traditional pharmacological agents that typically only target a single pathway in the pathophysiology of perinatal brain injury. Umbilical cord blood cells (UCBs) are one of the most well-studied and characterized cells in perinatal brain injury to date. UCBs consist of a combination of cell types including hematopoietic stem cells, endothelial progenitors, lymphocytes, monocytes and mesenchymal stem cells (MSCs; Pimentel-Coelho et al., 2012). UCBs have anti-inflammatory and anti-apoptotic properties in animal models of HIE, with their primary mode of action being the modulation of immune cells after injury (Schwarting et al., 2008), protecting from hypoxia-induced apoptosis (Hall et al., 2009) and release of trophic factors that promote neural recovery and repair post-ischemia such as BDNF and neurotrophins (Fan et al., 2005; Newman et al., 2006). During injury these UCBs, including MSCs, hone towards sites of injury, their migratory ability powered via interactions with chemokine and cytokine receptors including the fractalkine receptor CX3CR1 (Ji et al., 2004). Once there, MSCs drive neuronal differentiation after injury, promote cellular regeneration (Busch et al., 2011) and confer anti-inflammatory properties which reduce immune cell proliferation and activation (Li et al., 2005), and improve long-term motor outcomes (Van Velthoven et al., 2010). Recent evidence however suggests that MSCs can also participate in transfer of mitochondria via structures termed tunneling nanotubes (TNTs; Hsu et al., 2016). TNT formation is a result of F-actin polymerization, and is driven by activation of the pro-apoptotic p53 signaling within the stressed cell and downstream activation of the Akt/PI3K/mTor pathways (Wang et al., 2011). Co-culture of MSCs and human umbilical vein endothelial cells (HUVECs) exposed to glucose-oxygen deprivation resulted in the formation of TNTs and transfer of mitochondria from MSCs to HUVECs, restoring functional aerobic respiration and reducing apoptosis (Liu et al., 2014). This transfer has also been observed in corneal epithelium (Jiang et al., 2016) or pulmonary epithelium providing protection (Wecht and Rojas, 2016).
Tissue banking following birth is a growing market, and while UCB administration has been proven safe (Sun W. et al., 2005), there is some concern that few patients ever require therapy, with some approximating that 1 in 3000 children will require transfusion (Nietfeld et al., 2008). Together UCBs have been utilized in 30 clinical trials worldwide as an intervention for cerebral palsy (Clinicaltrials.gov, search query “umbilical cord cells” AND “cerebral palsy”), with one study completed (NCT01193660, Clinicaltrials.gov). The study showed that a combination therapy of UCBs and EPO resulted in significant improvements in motor and cognition, with associated improvements in structural and metabolic changes in the brain (Min et al., 2013).
Mitochondrial-Targeted Therapies
Whilst it is clear that broadly targeting excitotoxicity has some merit, it is clear from the mixed success of ion channel blockers that more targeted therapies are required in perinatal brain injury. Mitochondria play a pivotal role in bioenergetics, cell-cycle regulation and the oxidative stress response, and thus represent an emerging target for neuroprotective therapies.
Protecting from Mitochondrial Permeabilization
Mitochondrial Calcium Uniporter (Mcu)
Mitochondrial calcium uniporter (Mcu) is the pore-forming complex located on the mitochondrial inner membrane, and is responsible for fine-tuning the mitochondrial membrane potential (Oxenoid et al., 2016). Mcu plays a crucial role in mediating NMDA-receptor-induced excitotoxic death (Stout et al., 1998), as it allows Ca2+ influx into the mitochondria, perpetuating the downward apoptotic spiral. Crucially, knockdown of Mcu stabilizes the mitochondrial membrane and confers resistance to excitotoxicity, as well as neuroprotection as modulated by synaptic firing by adjacent neurons (Qiu et al., 2013; Utkina-Sosunova et al., 2013). This opens up the possibility that targeting of Mcu by pharmacological agents might have therapeutic merit in clinical conditions (Camara et al., 2010, 2011) where excitotoxicity-induced cell death is a major pathological feature.
Sigma-1 Receptors
Other ways of preventing mitochondrial destabilization are via modulators such as the sigma-1 receptor, located on the endoplasmic reticulum. The administration of 4-Phenyl-1-(4-phenylbutyl)piperidine (PPBP), a sigma-1 agonist, reduced neuronal death in vitro and in an in vivo model of excitotoxic developmental brain injury, with a concomitant decrease in microglial activation and loss of mitochondrial membrane potential (Wegleiter et al., 2014). Other sigma-1 agonists such as dehydroepiandrosterone (DHEA; Hashimoto et al., 2006) and allopregnanolone (Shirayama et al., 2011) have also been shown to improve cognitive deficits and infer anti-depressant effects in models of neuropsychiatric disorders, likely via interactions with NMDA receptors.
Mitochondrial ATP-Sensitive K+ (mitoKATP) Channel Openers
The mitochondrial ATP-sensitive K+ (mitoKATP) channel is essential for the tightly regulated leak of K+ ions across the mitochondrial membrane, allowing precise control over its membrane potential (Facundo et al., 2006). Interestingly, opening mitoKATP channels by compounds such as diazoxide result in protection from ischemic damage as well as reducing mtROS release (Facundo et al., 2007) and cell death (Fornazari et al., 2008). In a piglet model of HIE, diazoxide protects the integrity of the mitochondrial membrane and accumulating Ca2+ levels in CA1 pyramidal neurons (Domoki et al., 2004), and in a mouse model of MCAO confers neuroprotection to hypoxic neurons via modulation of pro- and anti-apoptotic proteins along the Bax-Bcl2 pathways (Liu et al., 2002). Furthermore, opening of the mitoKATP channel seems to be critical for induction of tolerance in the brain, a phenomenon whereby a sub-threshold insult of e.g., hypoxia, ischemia or a drug renders the CNS resistant to a second severe insult (Sanders et al., 2010; Hagberg et al., 2014). However, there is some contention as to whether diazoxide works entirely via mitoKATP channels, as the use of diazoxide in submitochondrial particles from pig heart did not affect both mitochondrial membrane potential and nicotinamide adenine dinucleotide (reduced form) (NADH) oxidation (Hanley et al., 2002). Further research have since corroborated these findings (Anastacio et al., 2013; Coetzee, 2013) and identified numerous other pathways by which diazoxide exerts its physiological effects, including activating cardiovascular and endothelial KATP channels (Coetzee, 2013) and regulating the release of neurotransmitters such as norepinephrine (Mohan and Paterson, 2000) and acetylcholine (Kilbinger et al., 2002).
Directly Targeting Mitochondrial Downstream Apoptotic Pathways
Caspase Inhibitors
Caspases such as caspase-3 and -8 play a pivotal role in the early stages of apoptosis after HI injury. This is particularly true for perinatal brain injury (Hu et al., 2000), as altered caspase activity during this crucial period interrupts the programmed cell death critical for the proper functional development of the CNS (Raff et al., 1993). Early efforts using specific and pan-caspase inhibitors had mixed success, with some studies showing reduced neuronal apoptosis (Cheng et al., 1998) and necrosis (Han et al., 2002) after HIE; however another study reported no benefit of pre-treatment with a pan-caspase inhibitor (Joly et al., 2004). The lack of protection may be due to differences in the bioavailability of the pharmacological compounds, or more likely reflects the importance of caspase-independent death pathways in perinatal brain injury. More recent efforts have turned to the generation of safer, more specific and pharmacokinetically enhanced inhibitors. Both quinoline-Val-Asp(Ome)-CH2-O-phenoxy (Q-VD-OPh) and the methyloxyphenylketone (mOPh) derivative, TRP601, were effective in neonatal models of ischemic injury, reducing infarct size, improving neurological function (Renolleau et al., 2007) and attenuating glial activation and inflammation (Chauvier et al., 2011). TRP601 preferentially inhibits caspase-2, an initiator caspase positioned upstream of mitochondrial permeabilization. TRP601 targets caspase-2 activation preventing truncation of Bid, and translocation of Bax to the mitochondrial membrane for mitochondrial permeabilization and release of pro-apoptotic proteins such as cyt C (Carlsson et al., 2011; Chauvier et al., 2011).
Phosphatase and Tensin Homolog Deleted on Chromosome 10 (PTEN) Modulators
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a negative regulator of members of the PI3K/Akt signaling pathway, responsible for regulating cell growth and survival. PTEN is also thought to couple PI3K/Akt signaling to the pro-death c-Jun N-terminal kinase (JNK) pathway. As PTEN has phosphatase activity, the usage of specific tyrosine phosphatase inhibitors such as bpv(pic) have been used to rescue neuronal cell death after ischemic injury via blocking PTEN-mediated downregulation of the PI3K pathway and decoupling from JNK1/2 signaling (Zhang et al., 2007). In a mouse model of kainate-induced excitotoxicity, bpv(pic) also displayed anti-inflammatory properties, reducing reactive astrogliosis and mitochondria apoptosis (Grande et al., 2014). PTEN inhibition has also been shown to rescue cortical neurons after HI in the immature brain (Zhao et al., 2013) and neuroprotective trophic factors such as IGF-1, growth hormone and hexarelin (Gustafson et al., 1999; Brywe et al., 2005a,b) all seem to act by enhancing PI3K/Akt signaling preventing BAX dependent mitochondrial permeabilization.
JNK Inhibitors
Activation of the JNK pathways leads to further downstream pro-apoptotic pathways including BCL-2 (Jin et al., 2006) and MAPK-activating death domain-containing protein (MADD; Centeno et al., 2007). Interestingly, Jnk3−/− mice are resistant to ischemic (Kuan et al., 2003), hypoxic-ischemic (Pirianov et al., 2007) and excitotoxic brain injury (Brecht et al., 2005), thus raising the possibility of targeting JNK activity as a therapy for perinatal brain injury. The JNK inhibitor SP600125 reduced infarct size and reduced apoptosis primarily via blockade of mitochondrial translocation of pro-apoptotic proteins Bax and Bim, and thus release of cyt c (Gao et al., 2005). Interestingly from a therapeutic standpoint, blockade of JNK activity was neuroprotective when administered either before or after ischemia (Guan et al., 2006) and via either intracerebral or intravenous injection (Guan et al., 2006). JNK inhibitors also have long-lasting activity, improving white matter development as well as cognitive and motor function in rats with HIE up to 14 weeks after injury (Nijboer et al., 2010, 2013).
p53 Inhibitors
p53, along with Ca2+ leak and ROS generation, are the major causes of mitochondrial permeabilization via interactions with the pro-apoptotic BCL-2 family (Galluzzi et al., 2009). Reduction of available p53 also reduces binding to DNA sites responsible for cell death (Leker et al., 2004) as well as reduced expression of BAX and caspase activity (Culmsee et al., 2001). Blocking p53 association with the mitochondrial membrane with pifithrin-μ reduced cerebral damage, ROS production and improved sensorimotor function after 6–10 weeks post-HI (Nijboer et al., 2011). There is some debate however whether the primary mode of action of pifithrin-μ is via p53 blockade as its use accompanied with partial or complete neuronal p53 deletion confer no or limited neuroprotection (Baburamani et al., 2017).
It is evident that directly targeting mitochondrial pathways in pre-clinical studies have shown a plethora of therapeutic merit. However studies have also reported that indirect protection of mitochondria via maintaining the homeostasis of the CNS microenvironment, or blocking subsequent energy failure, could also have merit.
Indirect Protection of Mitochondrial Function
Creatine
Creatine is a vital component in both aerobic respiration and ATP recycling in all tissues of the body. Creatine is used to enhance athletic performance by a biphasic “loading” period involving ingestion of 20 g/day for the first week and then a “maintenance” phase of 2 g/day (Hultman et al., 1996), resulting in sustained improvements in high-intensity exercise performance and lean body mass (Buford et al., 2007). In traumatic brain injury, creatine improved mitochondrial bioenergetics and reduced ROS production (Sullivan et al., 2000), and reduced the severity of cerebral infarcts in a model of unilateral carotid artery ligation (Berger et al., 2004). In perinatal brain injury, creatine supplementation reduces ATP depletion after ischemic injury (Brewer and Wallimann, 2000) thereby protecting neurons from oxygen-glucose deprivation-mediated apoptosis and necrosis (Balestrino et al., 2002). In models of ischemia, creatine supplementation has been shown to reduce severity of infarcts and improving neurological function (Lensman et al., 2006) likely via improving cerebral blood flow (Prass et al., 2007). There is also limited evidence that creatine has anti-oxidant properties, scavenging hydroxyl free radicals and affording cytoprotection (Sestili et al., 2011). From a clinical standpoint, while creatine weakly crosses the blood-brain barrier (BBB), creatine can also be supplemented maternally as creatine can cross the placental barrier, and supplementation via this route is also neuroprotective in a spiny mouse model of birth asphyxia (Ireland et al., 2011). Efforts to create more lipophilic Cr-derived compounds resulted in the generation of PCr-Mg-complex acetate (PCr-Mg-CPLX), which readily crossed the BBB (Lunardi et al., 2006) and when administered in a model of MCAO improved stroke and behavioral outcomes (Perasso et al., 2009).
Dichloroacetate (DCA)
Dichloroacetate (DCA) is a small molecule inhibitor of pyruvate dehydrogenase (PDH) kinase (PDHK), which regulates the activity of PDH. PDH is responsible for the conversion of pyruvate to acetyl-CoA, a critical component of the Krebs cycle. DCA is used in clinic to treat congenital lactic acidosis (Berendzen et al., 2006), of which 90% of reported cases are a result of a genetic defect in the α subunit of PDH (Lissens et al., 2000). Importantly, long-term administration of DCA is well-tolerated in children (Stacpoole et al., 2008) and is able to cross the BBB (Williams et al., 2001). DCA was initially used in models of ischemic-reperfusion injury as a means of reducing secondary energy failure, with administration of DCA increasing levels of ATP and PCr 4 h after ischemic injury (Katayama and Welsh, 1989) and also decreasing levels of lactate within the brain (Kaplan et al., 1987). Recently, intraperitoneal administration of DCA in a model of unilateral HI reduced infarct size and apoptosis via increased mitochondrial biogenesis as a result of increased availability of mitochondrial acetyl-CoA (Sun et al., 2016). More studies are required to assess DCA as a potential therapy after HI injury.
Protecting Astrocytes
Astrocytes are the most abundant cell-type in the brain, crucial for maintenance of optimal bioenergetics in the brain as well as providing trophic support and neurotransmitter release. High densities of mitochondria have been localized in the fine astrocytic processes responsible for regulating cerebrovascular flow (Zonta et al., 2003), and therefore neurovascular coupling (Otsu et al., 2015). However, these processes are damaged after ischemia (Ito et al., 2009), leading to neuronal dysfunction and death (Bambrick et al., 2004). Protecting astrocytes could have therapeutic merit, given their ability to re-uptake glutamate, delaying or preventing excitotoxic injury. Furthermore, astrocytic mitochondrial function is restored between 30 min and 1 h after ischemic injury (Morken et al., 2014), substantially quicker than neuronal mitochondrial recovery, and as such are ideally placed to reduce the impact in the secondary phase of hypoxia-induced injury (Berger et al., 2016). In vivo, astrocytes participate in mitochondrial transfer towards injured neurons, improving cell survival and neurological outcomes, a mechanism which is governed by CD38 signaling (Hayakawa et al., 2016). Modulation of astrocytic signaling, such as inhibition of the SNAP (Soluble NSF Attachment Protein) REceptor (SNARE) pathway also influences NMDA receptor expression and therefore sensitivity towards excitotoxic injury (Hines and Haydon, 2013). Administration of anti-oxidants such as resveratrol, melatonin or nicotinamide adenine dinucleotide phosphate (reduced form) (NADPH) inhibitors are protective towards astrocytic function after ischemia (Fernández-Gajardo et al., 2014), via inhibition of apoptotic pathways and stabilization of mitochondrial function (Lin et al., 2014).
Conclusion
The only clinically approved therapy for a hypoxic-ischemic event, whole-body hypothermia, has been shown to be effective at neuroprotection and reducing mortality rates. However, no therapies exist that target mitochondria and the stages of perinatal brain injury once they have manifested, critically the latent phases are responsible for later neurological impairment and disabilities such as cerebral palsy.
Evidence in recent years has identified mitochondrial dysfunction and downstream activation of pro-apoptotic pathways as a major target for future therapies. In pre-clinical models, these therapies have shown promise at not just reducing inflammation but also improving neurological outcomes weeks after the onset of ischemic injury. Critically, therapies targeting multiple pathogenic pathways, including cell therapies, are currently in late phase clinical trials.
Advances in technology may soon allow researchers to overcome the major problem of CNS therapies, namely the ability to cross the BBB. Nanoparticle-based drug delivery systems including synthetic polyamidoamine dendrimers have been used to great effect in an animal model of cerebral palsy, showing localization to glial cells and suppressing neuroinflammation (Kannan et al., 2012). Compounds such as creatine can also be infused in large amounts into albumin-coated gold nanospheres, providing a quicker and more effective administration route to cross the BBB (López-Viota et al., 2009). However, more research into the exact roles that mitochondria play in disease and subsequent therapy is needed, with a particular emphasis on more accurately mapping the chronological events downstream of perinatal brain injury and thus identifying therapeutic windows for novel intervention.
Author Contributions
All authors contributed to the writing of the manuscript as well as critical revision. All authors read and approved the final manuscript.
Funding
BL is supported by a Cerebral Palsy Alliance Project Grant (PG4615). HH and CT are supported by the Wellcome Trust, WT094823MA Swedish Medical Research Council (VR2012-3500), ALF-LUA (ALFGBG426401), ERANET (MICRO-MET; EU and research councils in Europe), the Leducq Foundation (DSRR_P34404), the Swedish Brain Foundation (FO2013-095), the Byggmästare Olle Engkvist Foundation, the Wilhelm and Martina Lundgren Foundation, the Frimurarna Barnhusdirektionen Foundation, the Åhlen foundation. CM: the Swedish Medical Council (VR 2012-2992); ALF-LUA (ALFGBG-432291); Torsten Söderberg Foundation (M98/15); the Swedish Brain Foundation (FO2015-0190).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AIF, apoptosis-inducing factor; ATP, adenosine triphosphate; BBB, blood-brain barrier; BDNF, brain-derived neurotrophic factor; BID, Bcl-2 interacting domain; CNS, central nervous system; Cyt c, cytochrome c; DAMPs, damage-associated molecular patterns; DCA, dichloroacetate; DHEA, dehydroepiandrosterone; DISC, death-inducing signaling complex; DRP1, dynamin-related protein 1; Endo G, endonuclease G; EPO, erythropoietin; ETC, electron transport chain; HI, hypoxia-ischemeia; HIE, hypoxia-ischemeic encephalopathy; HIF-1α, hypoxia inducible factor 1 alpha; HUVECs, human umbilical vein endothelial cells; IAP, inhibitors of apoptosis; IL-, interleukin; JNK, c-Jun N-terminal kinase; MADD, MAPK-activating death domain-containing protein; MAVS, mitochondrial antiviral signaling protein; MCAO, middle cerebral artery occlusion; Mcu, mitochondrial calcium uniporter; mitoKATP, mitochondrial ATP-sensitive K+; mOPH, methyloxyphenylketone; MSCs, mesenchymal stem cells; mtDNA, mitochondrial DNA; mtPTP, mitochondrial permeability transition pore; mtROS, mitochondrial ROS; NAD, nicotinamide adenine dinucleotide; NADH, nicotinamide adenine dinucleotide (reduced form); NADPH, nicotinamide adenine dinucleotide phosphate (reduced form); NETs, neutrophil extracellular traps; NLRs, NOD-like receptors; , superoxide; OPA1, optic atrophy 1; OXPHOS, oxidative phosphorylation; pCR, phosphocreatine; pDC, plasmacytoid dendritic cell; PDH, pyruvate dehydrogenase; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PPBP, 4-Phenyl-1-(4-phenylbutyl)piperidine; PTEN, phosphatase and tensin homolog deleted on chromosome 10; Q-VD-OPh, quinoline-Val-Asp(Ome)-CH2-O-phenoxy; RAGE, receptor for advanced glycation end products; rhEPO, recombinant human EPO; RLRs, RIG-I-like receptors; ROS, reactive oxygen species; SLE, systemic lupus erythematosus; tBid, truncated Bid; TFAM, mitochondrial transcription factor A; TLR-, Toll-like receptor; TNF-α, tumor necrosis factor alpha; TNTs, tunneling nanotubes; UCBs, umbilical cord blood cells; VDAC, voltage-dependent anion channels; VEGF, vascular endothelial growth factor.
References
Anastacio, M. M., Kanter, E. M., Keith, A. D., Schuessler, R. B., Nichols, C. G., and Lawton, J. S. (2013). Inhibition of succinate dehydrogenase by diazoxide is independent of the ATP-sensitive potassium channel subunit sulfonylurea type 1 receptor. J. Am. Coll. Surg. 216, 1144–1149. doi: 10.1016/j.jamcollsurg.2013.01.048
Andersson, U., and Scarpulla, R. C. (2001). Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol. Cell. Biol. 21, 3738–3749. doi: 10.1128/mcb.21.11.3738-3749.2001
Andrabi, S. A., Sayeed, I., Siemen, D., Wolf, G., and Horn, T. F. (2004). Direct inhibition of the mitochondrial permeability transition pore: a possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J. 18, 869–871. doi: 10.1096/fj.03-1031fje
Archer, S. L. (2013). Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 369, 2236–2251. doi: 10.1056/NEJMra1215233
Attardi, G., and Schatz, G. (1988). Biogenesis of mitochondria. Annu. Rev. Cell Biol. 4, 289–333. doi: 10.1146/annurev.cb.04.110188.001445
Azzopardi, D., Robertson, N. J., Kapetanakis, A., Griffiths, J., Rennie, J. M., Mathieson, S. R., et al. (2013). Anticonvulsant effect of xenon on neonatal asphyxial seizures. Arch. Dis. Child. Fetal Neonatal. Ed. 98, F437–F439. doi: 10.1136/archdischild-2013-303786
Azzopardi, D. V., Strohm, B., Edwards, A. D., Dyet, L., Halliday, H. L., Juszczak, E., et al. (2009). Moderate hypothermia to treat perinatal asphyxial encephalopathy. N. Engl. J. Med. 361, 1349–1358. doi: 10.1056/NEJMoa0900854
Baburamani, A. A., Sobotka, K. S., Vontell, R., Mallard, C., Supramaniam, V. G., Thornton, C., et al. (2017). Effect of Trp53 gene deficiency on brain injury after neonatal hypoxia-ischemia. Oncotarget 8, 12081–12092. doi: 10.18632/oncotarget.14518
Baixauli, F., Acín-Pérez, R., Villarroya-Beltrí, C., Mazzeo, C., Nuñez-Andrade, N., Gabandé-Rodriguez, E., et al. (2015). Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 22, 485–498. doi: 10.1016/j.cmet.2015.07.020
Balduini, W., Carloni, S., Perrone, S., Bertrando, S., Tataranno, M. L., Negro, S., et al. (2012). The use of melatonin in hypoxic-ischemic brain damage: an experimental study. J. Matern. Fetal Neonatal Med. 25, 119–124. doi: 10.3109/14767058.2012.663232
Balestrino, M., Lensman, M., Parodi, M., Perasso, L., Rebaudo, R., Melani, R., et al. (2002). Role of creatine and phosphocreatine in neuronal protection from anoxic and ischemic damage. Amino Acids 23, 221–229. doi: 10.1007/s00726-001-0133-3
Bambrick, L., Kristian, T., and Fiskum, G. (2004). Astrocyte mitochondrial mechanisms of ischemic brain injury and neuroprotection. Neurochem. Res. 29, 601–608. doi: 10.1023/b:nere.0000014830.06376.e6
Baraniak, P. R., and McDevitt, T. C. (2010). Stem cell paracrine actions and tissue regeneration. Regen. Med. 5, 121–143. doi: 10.2217/rme.09.74
Barlow-Walden, L. R., Reiter, R. J., Abe, M., Pablos, M., Menendez-Pelaez, A., Chen, L. D., et al. (1995). Melatonin stimulates brain glutathione peroxidase activity. Neurochem. Int. 26, 497–502. doi: 10.1016/0197-0186(94)00154-m
Benard, G., and Karbowski, M. (2009). Mitochondrial fusion and division: regulation and role in cell viability. Semin. Cell Dev. Biol. 20, 365–374. doi: 10.1016/j.semcdb.2008.12.012
Bendix, I., Schulze, C., Haefen, C. V., Gellhaus, A., Endesfelder, S., Heumann, R., et al. (2012). Erythropoietin modulates autophagy signaling in the developing rat brain in an in vivo model of oxygen-toxicity. Int. J. Mol. Sci. 13, 12939–12951. doi: 10.3390/ijms131012939
Berendzen, K., Theriaque, D. W., Shuster, J., and Stacpoole, P. W. (2006). Therapeutic potential of dichloroacetate for pyruvate dehydrogenase complex deficiency. Mitochondrion 6, 126–135. doi: 10.1016/j.mito.2006.08.033
Berger, R., Middelanis, J., Vaihinger, H. M., Mies, G., Wilken, B., and Jensen, A. (2004). Creatine protects the immature brain from hypoxic-ischemic injury. J. Soc. Gynecol. Investig. 11, 9–15. doi: 10.1016/j.jsgi.2003.07.002
Berger, H. R., Morken, T. S., Vettukattil, R., Brubakk, A.-M., Sonnewald, U., and Widerøe, M. (2016). No improvement of neuronal metabolism in the reperfusion phase with melatonin treatment after hypoxic-ischemic brain injury in the neonatal rat. J. Neurochem. 136, 339–350. doi: 10.1111/jnc.13420
Bergsbaken, T., Fink, S. L., and Cookson, B. T. (2009). Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109. doi: 10.1038/nrmicro2070
Brecht, S., Kirchhof, R., Chromik, A., Willesen, M., Nicolaus, T., Raivich, G., et al. (2005). Specific pathophysiological functions of JNK isoforms in the brain. Eur. J. Neurosci. 21, 363–377. doi: 10.1111/j.1460-9568.2005.03857.x
Brewer, G. J., and Wallimann, T. W. (2000). Protective effect of the energy precursor creatine against toxicity of glutamate and β-amyloid in rat hippocampal neurons. J. Neurochem. 74, 1968–1978. doi: 10.1046/j.1471-4159.2000.0741968.x
Brywe, K. G., Leverin, A. L., Gustavsson, M., Mallard, C., Granata, R., Destefanis, S., et al. (2005a). Growth hormone-releasing peptide hexarelin reduces neonatal brain injury and alters Akt/glycogen synthase kinase-3β phosphorylation. Endocrinology 146, 4665–4672. doi: 10.1210/en.2005-0389
Brywe, K. G., Mallard, C., Gustavsson, M., Hedtjärn, M., Leverin, A. L., Wang, X., et al. (2005b). IGF-I neuroprotection in the immature brain after hypoxia-ischemia, involvement of Akt and GSK3β? Eur. J. Neurosci. 21, 1489–1502. doi: 10.1111/j.1460-9568.2005.03982.x
Buford, T. W., Kreider, R. B., Stout, J. R., Greenwood, M., Campbell, B., Spano, M., et al. (2007). International society of sports nutrition position stand: creatine supplementation and exercise. J. Int. Soc. Sports Nutr. 4:6. doi: 10.1186/1550-2783-4-6
Busch, S. A., Hamilton, J. A., Horn, K. P., Cuascut, F. X., Cutrone, R., Lehman, N., et al. (2011). Multipotent adult progenitor cells prevent macrophage-mediated axonal dieback and promote regrowth after spinal cord injury. J. Neurosci. 31, 944–953. doi: 10.1523/JNEUROSCI.3566-10.2011
Camara, A. K., Bienengraeber, M., and Stowe, D. F. (2011). Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front. Physiol. 2:13. doi: 10.3389/fphys.2011.00013
Camara, A. K., Lesnefsky, E. J., and Stowe, D. F. (2010). Potential therapeutic benefits of strategies directed to mitochondria. Antioxid. Redox Signal. 13, 279–347. doi: 10.1089/ars.2009.2788
Carlsson, Y., Schwendimann, L., Vontell, R., Rousset, C. I., Wang, X., Lebon, S., et al. (2011). Genetic inhibition of caspase-2 reduces hypoxic-ischemic and excitotoxic neonatal brain injury. Ann. Neurol. 70, 781–789. doi: 10.1002/ana.22431
Carraway, M. S., Suliman, H. B., Jones, W. S., Chen, C.-W., Babiker, A., and Piantadosi, C. A. (2010). Erythropoietin activates mitochondrial biogenesis and couples red cell mass to mitochondrial mass in the heart. Circ. Res. 106, 1722–1730. doi: 10.1161/CIRCRESAHA.109.214353
Cassidy-Stone, A., Chipuk, J. E., Ingerman, E., Song, C., Yoo, C., Kuwana, T., et al. (2008). Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 14, 193–204. doi: 10.1016/j.devcel.2007.11.019
Centeno, C., Repici, M., Chatton, J. Y., Riederer, B. M., Bonny, C., Nicod, P., et al. (2007). Role of the JNK pathway in NMDA-mediated excitotoxicity of cortical neurons. Cell Death Differ. 14, 240–253. doi: 10.1038/sj.cdd.4401988
Chandel, N. S., Trzyna, W. C., McClintock, D. S., and Schumacker, P. T. (2000). Role of oxidants in NF-κB activation and TNF-α gene transcription induced by hypoxia and endotoxin. J. Immunol. 165, 1013–1021. doi: 10.4049/jimmunol.165.2.1013
Chang, C. H., Curtis, J. D., Maggi, L. B. Jr., Faubert, B., Villarino, A. V., O’Sullivan, D., et al. (2013). Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. doi: 10.1016/j.cell.2013.05.016
Chauvier, D., Renolleau, S., Holifanjaniaina, S., Ankri, S., Bezault, M., Schwendimann, L., et al. (2011). Targeting neonatal ischemic brain injury with a pentapeptide-based irreversible caspase inhibitor. Cell Death Dis. 2:e203. doi: 10.1038/cddis.2011.87
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200. doi: 10.1083/jcb.200211046
Cheng, Y., Deshmukh, M., D’Costa, A., Demaro, J. A., Gidday, J. M., Shah, A., et al. (1998). Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J. Clin. Invest. 101, 1992–1999. doi: 10.1172/jci2169
Cheng, A., Wan, R., Yang, J. L., Kamimura, N., Son, T. G., Ouyang, X., et al. (2012). Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 3:1250. doi: 10.1038/ncomms2238
Cherry, C., Thompson, B., Saptarshi, N., Wu, J., and Hoh, J. (2016). 2016: a ‘mitochondria’ odyssey. Trends Mol. Med. 22, 391–403. doi: 10.1016/j.molmed.2016.03.009
Chuang, J. I., Pan, I. L., Hsieh, C. Y., Huang, C. Y., Chen, P. C., and Shin, J. W. (2016). Melatonin prevents the dynamin-related protein 1-dependent mitochondrial fission and oxidative insult in the cortical neurons after 1-methyl-4-phenylpyridinium treatment. J. Pineal Res. 61, 230–240. doi: 10.1111/jpi.12343
Coetzee, W. A. (2013). Multiplicity of effectors of the cardioprotective agent, diazoxide. Pharmacol. Ther. 140, 167–175. doi: 10.1016/j.pharmthera.2013.06.007
Coto-Montes, A., Boga, J. A., Rosales-Corral, S., Fuentes-Broto, L., Tan, D. X., and Reiter, R. J. (2012). Role of melatonin in the regulation of autophagy and mitophagy: a review. Mol. Cell. Endocrinol. 361, 12–23. doi: 10.1016/j.mce.2012.04.009
Culmsee, C., Zhu, X., Yu, Q. S., Chan, S. L., Camandola, S., Guo, Z., et al. (2001). A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults and amyloid α-peptide. J. Neurochem. 77, 220–228. doi: 10.1046/j.1471-4159.2001.00220.x
Delettre, C., Lenaers, G., Griffoin, J. M., Gigarel, N., Lorenzo, C., Belenguer, P., et al. (2000). Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 26, 207–210. doi: 10.1038/79936
Domoki, F., Bari, F., Nagy, K., Busija, D. W., and Siklós, L. (2004). Diazoxide prevents mitochondrial swelling and Ca2+ accumulation in CA1 pyramidal cells after cerebral ischemia in newborn pigs. Brain Res. 1019, 97–104. doi: 10.1016/j.brainres.2004.05.088
Dubinsky, J. M. (2009). Heterogeneity of nervous system mitochondria: location, location, location! Exp. Neurol. 218, 293–307. doi: 10.1016/j.expneurol.2009.05.020
Edwards, A. D., Brocklehurst, P., Gunn, A. J., Halliday, H., Juszczak, E., Levene, M., et al. (2010). Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: synthesis and meta-analysis of trial data. BMJ 340:c363. doi: 10.1136/bmj.c363
Eicher, D. J., Wagner, C. L., Katikaneni, L. P., Hulsey, T. C., Bass, W. T., Kaufman, D. A., et al. (2005). Moderate hypothermia in neonatal encephalopathy: efficacy outcomes. Pediatr. Neurol. 32, 11–17. doi: 10.1016/j.pediatrneurol.2004.06.014
Elmahdy, H., El-Mashad, A. R., El-Bahrawy, H., El-Gohary, T., El-Barbary, A., and Aly, H. (2010). Human recombinant erythropoietin in asphyxia neonatorum: pilot trial. Pediatrics 125, e1135–e1142. doi: 10.1542/peds.2009-2268
Facundo, H. T. F., de Paula, J. G., and Kowaltowski, A. J. (2007). Mitochondrial ATP-sensitive K+ channels are redox-sensitive pathways that control reactive oxygen species production. Free Radic. Biol. Med. 42, 1039–1048. doi: 10.1016/j.freeradbiomed.2007.01.001
Facundo, H. T. F., Fornazari, M., and Kowaltowski, A. J. (2006). Tissue protection mediated by mitochondrial K+ channels. Biochim. Biophys. Acta 1762, 202–212. doi: 10.1016/j.bbadis.2005.06.003
Fan, X., van Bel, F., van der Kooij, M. A., Heijnen, C. J., and Groenendaal, F. (2013). Hypothermia and erythropoietin for neuroprotection after neonatal brain damage. Pediatr. Res. 73, 18–23. doi: 10.1038/pr.2012.139
Fan, C. G., Zhang, Q. J., Tang, F. W., Han, Z. B., Wang, G. S., and Han, Z. C. (2005). Human umbilical cord blood cells express neurotrophic factors. Neurosci. Lett. 380, 322–325. doi: 10.1016/j.neulet.2005.01.070
Fang, A. Y., Gonzalez, F. F., Sheldon, R. A., and Ferriero, D. M. (2013). Effects of combination therapy using hypothermia and erythropoietin in a rat model of neonatal hypoxia-ischemia. Pediatr. Res. 73, 12–17. doi: 10.1038/pr.2012.138
Favrais, G., van de Looij, Y., Fleiss, B., Ramanantsoa, N., Bonnin, P., Stoltenburg-Didinger, G., et al. (2011). Systemic inflammation disrupts the developmental program of white matter. Ann. Neurol. 70, 550–565. doi: 10.1002/ana.22489
Fernández-Gajardo, R., Matamala, J. M., Carrasco, R., Gutiérrez, R., Melo, R., and Rodrigo, R. (2014). Novel therapeutic strategies for traumatic brain injury: acute antioxidant reinforcement. CNS Drugs 28, 229–248. doi: 10.1007/s40263-013-0138-y
Fleiss, B., and Gressens, P. (2012). Tertiary mechanisms of brain damage: a new hope for treatment of cerebral palsy? Lancet Neurol. 11, 556–566. doi: 10.1016/s1474-4422(12)70058-3
Fornazari, M., de Paula, J. G., Castilho, R. F., and Kowaltowski, A. J. (2008). Redox properties of the adenoside triphosphate-sensitive K+ channel in brain mitochondria. J. Neurosci. Res. 86, 1548–1556. doi: 10.1002/jnr.21614
Galluzzi, L., Blomgren, K., and Kroemer, G. (2009). Mitochondrial membrane permeabilization in neuronal injury. Nat. Rev. Neurosci. 10, 481–494. doi: 10.1038/nrn2665
Gao, Y., Signore, A. P., Yin, W., Cao, G., Yin, X. M., Sun, F., et al. (2005). Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J. Cereb. Blood Flow Metab. 25, 694–712. doi: 10.1038/sj.jcbfm.9600062
Gergely, P. Jr., Niland, B., Gonchoroff, N., Pullmann, R. Jr., Phillips, P. E., and Perl, A. (2002). Persistent mitochondrial hyperpolarization, increased reactive oxygen intermediate production and cytoplasmic alkalinization characterize altered IL-10 signaling in patients with systemic lupus erythematosus. J. Immunol. 169, 1092–1101. doi: 10.4049/jimmunol.169.2.1092
Gluckman, P. D., Wyatt, J. S., Azzopardi, D., Ballard, R., Edwards, A. D., Ferriero, D. M., et al. (2005). Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet 365, 663–670. doi: 10.1016/s0140-6736(05)17946-x
Gottlieb, R. A., and Carreira, R. S. (2010). Autophagy in health and disease. 5. Mitophagy as a way of life. Am. J. Physiol. Cell Physiol. 299, C203–C210. doi: 10.1152/ajpcell.00097.2010
Graham, E. M., Sheldon, R. A., Flock, D. L., Ferriero, D. M., Martin, L. J., O’Riordan, D. P., et al. (2004). Neonatal mice lacking functional Fas death receptors are resistant to hypoxic-ischemic brain injury. Neurobiol. Dis. 17, 89–98. doi: 10.1016/j.nbd.2004.05.007
Grande, V., Manassero, G., and Vercelli, A. (2014). Neuroprotective and anti-inflammatory roles of the phosphatase and tensin homolog deleted on chromosome ten (PTEN) inhibition in a mouse model of temporal lobe epilepsy. PLoS One 9:e114554. doi: 10.1371/journal.pone.0114554
Green, D. R. (2000). Apoptotic pathways: paper wraps stone blunts scissors. Cell 102, 1–4. doi: 10.1016/S0092-8674(00)00003-9
Green, D. R., Galluzzi, L., and Kroemer, G. (2011). Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333, 1109–1112. doi: 10.1126/science.1201940
Grivennikova, V. G., Kareyeva, A. V., and Vinogradov, A. D. (2010). What are the sources of hydrogen peroxide production by heart mitochondria? Biochim. Biophys. Acta 1797, 939–944. doi: 10.1016/j.bbabio.2010.02.013
Guan, Q. H., Pei, D. S., Zong, Y. Y., Xu, T. L., and Zhang, G. Y. (2006). Neuroprotection against ischemic brain injury by a small peptide inhibitor of c-Jun N-terminal kinase (JNK) via nuclear and non-nuclear pathways. Neuroscience 139, 609–627. doi: 10.1016/j.neuroscience.2005.11.067
Gustafson, K., Hagberg, H., Bengtsson, B.-A., Brantsing, C., and Isgaard, J. (1999). Possible protective role of growth hormone in hypoxia-ischemia in neonatal rats. Pediatr. Res. 45, 318–323. doi: 10.1203/00006450-199903000-00005
Hagberg, H., Gressens, P., and Mallard, C. (2012). Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann. Neurol. 71, 444–457. doi: 10.1002/ana.22620
Hagberg, H., Mallard, C., Ferriero, D. M., Vannucci, S. J., Levison, S. W., Vexler, Z. S., et al. (2015). The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 11, 192–208. doi: 10.1038/nrneurol.2015.13
Hagberg, H., Mallard, C., Rousset, C. I., and Thornton, C. (2014). Mitochondria: hub of injury responses in the developing brain. Lancet Neurol. 13, 217–232. doi: 10.1016/S1474-4422(13)70261-8
Hall, A. A., Leonardo, C. C., Collier, L. A., Rowe, D. D., Willing, A. E., and Pennypacker, K. R. (2009). Delayed treatments for stroke influence neuronal death in rat organotypic slice cultures subjected to oxygen glucose deprivation. Neuroscience 164, 470–477. doi: 10.1016/j.neuroscience.2009.08.051
Han, D., Antunes, F., Canali, R., Rettori, D., and Cadenas, E. (2003). Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 278, 5557–5563. doi: 10.1074/jbc.M210269200
Han, B. H., Xu, D., Choi, J., Han, Y., Xanthoudakis, S., Roy, S., et al. (2002). Selective, reversible caspase-3 inhibitor is neuroprotective and reveals distinct pathways of cell death after neonatal hypoxic-ischemic brain injury. J. Biol. Chem. 277, 30128–30136. doi: 10.1074/jbc.M202931200
Hanley, P. J., Mickel, M., Löffler, M., Brandt, U., and Daut, J. (2002). K(ATP) channel-independent targets of diazoxide and 5-hydroxydecanoate in the heart. J. Physiol. 542, 735–741. doi: 10.1113/jphysiol.2002.023960
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. doi: 10.1038/nature04724
Hashimoto, K., Fujita, Y., and Iyo, M. (2006). Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of fluvoxamine: role of sigma-1 receptors. Neuropsychopharmacology 32, 514–521. doi: 10.1038/sj.npp.1301047
Hayakawa, K., Esposito, E., Wang, X., Terasaki, Y., Liu, Y., Xing, C., et al. (2016). Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555. doi: 10.1038/nature18928
He, C., Wang, J., Zhang, Z., Yang, M., Li, Y., Tian, X., et al. (2016). Mitochondria synthesize melatonin to ameliorate its function and improve mice oocyte’s quality under in vitro conditions. Int. J. Mol. Sci. 17:939. doi: 10.3390/ijms17060939
Hines, D. J., and Haydon, P. G. (2013). Inhibition of a SNARE-sensitive pathway in astrocytes attenuates damage following stroke. J. Neurosci. 33, 4234–4240. doi: 10.1523/JNEUROSCI.5495-12.2013
Hiona, A., Sanz, A., Kujoth, G. C., Pamplona, R., Seo, A. Y., Hofer, T., et al. (2010). Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS One 5:e11468. doi: 10.1371/journal.pone.0011468
Hoeijmakers, L., Heinen, Y., van Dam, A.-M., Lucassen, P. J., and Korosi, A. (2016). Microglial priming and Alzheimer’s disease: a possible role for (Early) immune challenges and epigenetics? Front. Hum. Neurosci. 10:398. doi: 10.3389/fnhum.2016.00398
Horng, L.-Y., Hsu, P.-L., Chen, L.-W., Tseng, W.-Z., Hsu, K.-T., Wu, C.-L., et al. (2015). Activating mitochondrial function and haemoglobin expression with EH-201, an inducer of erythropoietin in neuronal cells, reverses memory impairment. Br. J. Pharmacol. 172, 4741–4756. doi: 10.1111/bph.13248
Hsu, Y.-C., Wu, Y.-T., Yu, T.-H., and Wei, Y.-H. (2016). Mitochondria in mesenchymal stem cell biology and cell therapy: from cellular differentiation to mitochondrial transfer. Semin. Cell Dev. Biol. 52, 119–131. doi: 10.1016/j.semcdb.2016.02.011
Hu, B. R., Liu, C. L., Ouyang, Y., Blomgren, K., and Siesjö, B. K. (2000). Involvement of caspase-3 in cell death after hypoxia-ischemia declines during brain maturation. J. Cereb. Blood Flow Metab. 20, 1294–1300. doi: 10.1097/00004647-200009000-00003
Hultman, E., Söderlund, K., Timmons, J. A., Cederblad, G., and Greenhaff, P. L. (1996). Muscle creatine loading in men. J. Appl. Physiol. 81, 232–237.
Huss, J. M., Kopp, R. P., and Kelly, D. P. (2002). Peroxisome proliferator-activated receptor coactivator-1α (PGC-1β) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-α and -γ. Identification of novel leucine-rich interaction motif within PGC-1α. J. Biol. Chem. 277, 40265–40274. doi: 10.1074/jbc.M206324200
Hutchison, J. S., Ward, R. E., Lacroix, J., Hébert, P. C., Barnes, M. A., Bohn, D. J., et al. (2008). Hypothermia therapy after traumatic brain injury in children. N. Engl. J. Med. 358, 2447–2456. doi: 10.1056/NEJMoa0706930
Ireland, Z., Castillo-Melendez, M., Dickinson, H., Snow, R., and Walker, D. W. (2011). A maternal diet supplemented with creatine from mid-pregnancy protects the newborn spiny mouse brain from birth hypoxia. Neuroscience 194, 372–379. doi: 10.1016/j.neuroscience.2011.05.012
Ishihara, N., Nomura, M., Jofuku, A., Kato, H., Suzuki, S. O., Masuda, K., et al. (2009). Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 11, 958–966. doi: 10.1038/ncb1907
Ito, U., Hakamata, Y., Kawakami, E., and Oyanagi, K. (2009). Degeneration of astrocytic processes and their mitochondria in cerebral cortical regions peripheral to the cortical infarction: heterogeneity of their disintegration is closely associated with disseminated selective neuronal necrosis and maturation of injury. Stroke 40, 2173–2181. doi: 10.1161/STROKEAHA.108.534990
Ji, J. F., He, B. P., Dheen, S. T., and Tay, S. S. W. (2004). Interactions of chemokines and chemokine receptors mediate the migration of mesenchymal stem cells to the impaired site in the brain after hypoglossal nerve injury. Stem Cells 22, 415–427. doi: 10.1634/stemcells.22-3-415
Jiang, D., Gao, F., Zhang, Y., Wong, D. S., Li, Q., Tse, H. F., et al. (2016). Mitochondrial transfer of mesenchymal stem cells effectively protects corneal epithelial cells from mitochondrial damage. Cell Death Dis. 7:e2467. doi: 10.1038/cddis.2016.358
Jin, H. O., Park, I. C., An, S., Lee, H. C., Woo, S. H., Hong, Y. J., et al. (2006). Up-regulation of Bak and Bim via JNK downstream pathway in the response to nitric oxide in human glioblastoma cells. J. Cell. Physiol. 206, 477–486. doi: 10.1002/jcp.20488
Johnston, M. V., and Hagberg, H. (2007). Sex and the pathogenesis of cerebral palsy. Dev. Med. Child Neurol. 49, 74–78. doi: 10.1017/s0012162207000199.x
Joly, L. M., Mucignat, V., Mariani, J., Plotkine, M., and Charriaut-Marlangue, C. (2004). Caspase inhibition after neonatal ischemia in the rat brain. J. Cereb. Blood Flow Metab. 24, 124–131. doi: 10.1097/01.wcb.0000100061.36077.5f
Juul, S. E. (2013). Hypothermia plus erythropoietin for neonatal neuroprotection? Commentary on Fan et al. and Fang et al. Pediatr Res 73, 10–11. doi: 10.1038/pr.2012.148
Kaindl, A. M., Sifringer, M., Koppelstaetter, A., Genz, K., Loeber, R., Boerner, C., et al. (2008). Erythropoietin protects the developing brain from hyperoxia-induced cell death and proteome changes. Ann. Neurol. 64, 523–534. doi: 10.1002/ana.21471
Kannan, S., Dai, H., Navath, R. S., Balakrishnan, B., Jyoti, A., Janisse, J., et al. (2012). Dendrimer-based postnatal therapy for neuroinflammation and cerebral palsy in a rabbit model. Sci. Transl. Med. 4:130ra146. doi: 10.1126/scitranslmed.3003162
Kaplan, J., Dimlich, R. V., and Biros, M. H. (1987). Dichloroacetate treatment of ischemic cerebral lactic acidosis in the fed rat. Ann. Emerg. Med. 16, 298–304. doi: 10.1016/s0196-0644(87)80175-0
Katayama, Y., and Welsh, F. A. (1989). Effect of dichloroacetate on regional energy metabolites and pyruvate dehydrogenase activity during ischemia and reperfusion in gerbil brain. J. Neurochem. 52, 1817–1822. doi: 10.1111/j.1471-4159.1989.tb07262.x
Kellert, B. A., McPherson, R. J., and Juul, S. E. (2007). A comparison of high-dose recombinant erythropoietin treatment regimens in brain-injured neonatal rats. Pediatr. Res. 61, 451–455. doi: 10.1203/pdr.0b013e3180332cec
Kelly, B., and O’Neill, L. A. J. (2015). Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 25, 771–784. doi: 10.1038/cr.2015.68
Kichev, A., Rousset, C. I., Baburamani, A. A., Levison, S. W., Wood, T. L., Gressens, P., et al. (2014). Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) signaling and cell death in the immature central nervous system after hypoxia-ischemia and inflammation. J. Biol. Chem. 289, 9430–9439. doi: 10.1074/jbc.m113.512350
Kilbinger, H., Krause, A., Mang, C. F., Englert, H., and Wirth, K. (2002). Effects of K(ATP) channel modulators on acetylcholine release from guinea-pig isolated atria and small intestine. Naunyn Schmiedebergs Arch. Pharmacol. 365, 371–377. doi: 10.1007/s00210-002-0539-9
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi: 10.1038/nature04723
Kruse, M., Michelsen, S. I., Flachs, E. M., Brønnum-Hansen, H., Madsen, M., and Uldall, P. (2009). Lifetime costs of cerebral palsy. Dev. Med. Child Neurol. 51, 622–628. doi: 10.1111/j.1469-8749.2008.03190.x
Kuan, C. Y., Whitmarsh, A. J., Yang, D. D., Liao, G., Schloemer, A. J., Dong, C., et al. (2003). A critical role of neural-specific JNK3 for ischemic apoptosis. Proc. Natl. Acad. Sci. U S A 100, 15184–15189. doi: 10.1073/pnas.2336254100
Kumar, V., Shearer, J. C., Kumar, A., and Darmstadt, G. L. (2009). Neonatal hypothermia in low resource settings: a review. J. Perinatol. 29, 401–412. doi: 10.1038/jp.2008.233
Lai, J. C. Y., Rocha-Ferreira, E., Ek, C. J., Wang, X., Hagberg, H., and Mallard, C. (2017). Immune responses in perinatal brain injury. Brain Behav. Immun. 63, 210–223. doi: 10.1016/j.bbi.2016.10.022
Larsson, N. G., Wang, J., Wilhelmsson, H., Oldfors, A., Rustin, P., Lewandoski, M., et al. (1998). Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 18, 231–236. doi: 10.1038/ng0398-231
Lawn, J. E., Bahl, R., Bergstrom, S., Bhutta, Z. A., Darmstadt, G. L., Ellis, M., et al. (2011). Setting research priorities to reduce almost one million deaths from birth asphyxia by 2015. PLoS Med. 8:e1000389. doi: 10.1371/journal.pmed.1000389
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Leker, R. R., Aharonowiz, M., Greig, N. H., and Ovadia, H. (2004). The role of p53-induced apoptosis in cerebral ischemia: effects of the p53 inhibitor pifithrin α. Exp. Neurol. 187, 478–486. doi: 10.1016/j.expneurol.2004.01.030
Lensman, M., Korzhevskii, D. E., Mourovets, V. O., Kostkin, V. B., Izvarina, N., Perasso, L., et al. (2006). Intracerebroventricular administration of creatine protects against damage by global cerebral ischemia in rat. Brain Res. 1114, 187–194. doi: 10.1016/j.brainres.2006.06.103
Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27–42. doi: 10.1016/j.cell.2007.12.018
Li, Y., Chen, J., Zhang, C. L., Wang, L., Lu, D., Katakowski, M., et al. (2005). Gliosis and brain remodeling after treatment of stroke in rats with marrow stromal cells. Glia 49, 407–417. doi: 10.1002/glia.20126
Li, L. Y., Luo, X., and Wang, X. (2001). Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412, 95–99. doi: 10.1038/35083620
Li, Z., Okamoto, K., Hayashi, Y., and Sheng, M. (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. doi: 10.1016/j.cell.2004.11.003
Lin, C.-J., Chen, T.-H., Yang, L.-Y., and Shih, C.-M. (2014). Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Dis. 5:e1147. doi: 10.1038/cddis.2014.123
Lin, J., Wu, P. H., Tarr, P. T., Lindenberg, K. S., St-Pierre, J., Zhang, C. Y., et al. (2004). Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α null mice. Cell 119, 121–135. doi: 10.1016/j.cell.2004.09.013
Lissens, W., De Meirleir, L., Seneca, S., Liebaers, I., Brown, G. K., Brown, R. M., et al. (2000). Mutations in the X-linked pyruvate dehydrogenase (E1) α subunit gene (PDHA1) in patients with a pyruvate dehydrogenase complex deficiency. Hum. Mutat. 15, 209–219. doi: 10.1002/(SICI)1098-1004(200003)15:3<209::AID-HUMU1>3.0.CO;2-K
Little, J. P., Simtchouk, S., Schindler, S. M., Villanueva, E. B., Gill, N. E., Walker, D. G., et al. (2014). Mitochondrial transcription factor A (Tfam) is a pro-inflammatory extracellular signaling molecule recognized by brain microglia. Mol. Cell. Neurosci. 60, 88–96. doi: 10.1016/j.mcn.2014.04.003
Liu, K., Ji, K., Guo, L., Wu, W., Lu, H., Shan, P., et al. (2014). Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc. Res. 92, 10–18. doi: 10.1016/j.mvr.2014.01.008
Liu, D., Lu, C., Wan, R., Auyeung, W. W., and Mattson, M. P. (2002). Activation of mitochondrial ATP-dependent potassium channels protects neurons against ischemia-induced death by a mechanism involving suppression of Bax translocation and cytochrome c release. J. Cereb. Blood Flow Metab. 22, 431–443. doi: 10.1097/00004647-200204000-00007
Liu, Q. A., and Shio, H. (2008). Mitochondrial morphogenesis, dendrite development and synapse formation in cerebellum require both Bcl-w and the glutamate receptor δ2. PLoS Genet. 4:e1000097. doi: 10.1371/journal.pgen.1000097
López-Viota, J., Mandal, S., Delgado, A. V., Toca-Herrera, J. L., Moller, M., Zanuttin, F., et al. (2009). Electrophoretic characterization of gold nanoparticles functionalized with human serum albumin (HSA) and creatine. J. Colloid Interface Sci. 332, 215–223. doi: 10.1016/j.jcis.2008.11.077
Love, S. (2003). Apoptosis and brain ischaemia. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 267–282. doi: 10.1016/S0278-5846(03)00022-8
Lunardi, G., Parodi, A., Perasso, L., Pohvozcheva, A. V., Scarrone, S., Adriano, E., et al. (2006). The creatine transporter mediates the uptake of creatine by brain tissue, but not the uptake of two creatine-derived compounds. Neuroscience 142, 991–997. doi: 10.1016/j.neuroscience.2006.06.058
Manchester, L. C., Coto-Montes, A., Boga, J. A., Andersen, L. P. H., Zhou, Z., Galano, A., et al. (2015). Melatonin: an ancient molecule that makes oxygen metabolically tolerable. J. Pineal Res. 59, 403–419. doi: 10.1111/jpi.12267
Marelli-Berg, F. M., Fu, H., and Mauro, C. (2012). Molecular mechanisms of metabolic reprogramming in proliferating cells: implications for T-cell-mediated immunity. Immunology 136, 363–369. doi: 10.1111/j.1365-2567.2012.03583.x
Martin, M., Macias, M., Escames, G., Reiter, R. J., Agapito, M. T., Ortiz, G. G., et al. (2000). Melatonin-induced increased activity of the respiratory chain complexes I and IV can prevent mitochondrial damage induced by ruthenium red in vivo. J. Pineal Res. 28, 242–248. doi: 10.1034/j.1600-079x.2000.280407.x
Mattson, M. P., Gleichmann, M., and Cheng, A. (2008). Mitochondria in neuroplasticity and neurological disorders. Neuron 60, 748–766. doi: 10.1016/j.neuron.2008.10.010
Miller, S. L., Yan, E. B., Castillo-Meléndez, M., Jenkin, G., and Walker, D. W. (2005). Melatonin provides neuroprotection in the late-gestation fetal sheep brain in response to umbilical cord occlusion. Dev. Neurosci. 27, 200–210. doi: 10.1159/000085993
Mills, E. L., Kelly, B., Logan, A., Costa, A. S., Varma, M., Bryant, C. E., et al. (2016). Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167, 457.e413–470.e413. doi: 10.1016/j.cell.2016.08.064
Min, K., Song, J., Kang, J. Y., Ko, J., Ryu, J. S., Kang, M. S., et al. (2013). Umbilical cord blood therapy potentiated with erythropoietin for children with cerebral palsy: a double-blind, randomized, placebo-controlled trial. Stem Cells 31, 581–591. doi: 10.1002/stem.1304
Mogensen, T. H. (2009). Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 22, 240–273. doi: 10.1128/cmr.00046-08
Mohan, R. M., and Paterson, D. J. (2000). Activation of sulphonylurea-sensitive channels and the NO-cGMP pathway decreases the heart rate response to sympathetic nerve stimulation. Cardiovasc. Res. 47, 81–89. doi: 10.1016/s0008-6363(00)00057-2
Monlun, M., Hyernard, C., Blanco, P., Lartigue, L., and Faustin, B. (2017). Mitochondria as molecular platforms integrating multiple innate immune signalings. J. Mol. Biol. 429, 1–13. doi: 10.1016/j.jmb.2016.10.028
Montaldo, P., Pauliah, S. S., Lally, P. J., Olson, L., and Thayyil, S. (2015). Cooling in a low-resource environment: lost in translation. Semin. Fetal Neonatal Med. 20, 72–79. doi: 10.1016/j.siny.2014.10.004
Morken, T. S., Brekke, E., Håberg, A., Widerøe, M., Brubakk, A. M., and Sonnewald, U. (2014). Altered astrocyte-neuronal interactions after hypoxia-ischemia in the neonatal brain in female and male rats. Stroke 45, 2777–2785. doi: 10.1161/STROKEAHA.114.005341
Mottahedin, A., Smith, P. L., Hagberg, H., Ek, C. J., and Mallard, C. (2017a). TLR2-mediated leukocyte trafficking to the developing brain. J. Leukoc. Biol. 101, 297–305. doi: 10.1189/jlb.3A1215-568R
Mottahedin, A., Svedin, P., Nair, S., Mohn, C. J., Wang, X., Hagberg, H., et al. (2017b). Systemic activation of Toll-like receptor 2 suppresses mitochondrial respiration and exacerbates hypoxic-ischemic injury in the developing brain. J. Cereb. Blood Flow Metab. 37, 1192–1198. doi: 10.1177/0271678x17691292
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Nair, S., Mallard, C., Thronton, C., Sobotka, K., and Hagberg, H. (2016). Lipopolysaccharide Induces Mitochondrial Fission and A Metabolic Shift in Microglia. Program No. 230.05/W5 Neuroscience Meeting Planner. San Diego, CA: Society for Neuroscience.
Natalucci, G., Latal, B., Koller, B., Ruegger, C., Sick, B., Held, L., et al. (2016). Effect of early prophylactic high-dose recombinant human erythropoietin in very preterm infants on neurodevelopmental outcome at 2 years: a randomized clinical trial. Jama 315, 2079–2085. doi: 10.1001/jama.2016.5504
Newman, M. B., Willing, A. E., Manresa, J. J., Sanberg, C. D., and Sanberg, P. R. (2006). Cytokines produced by cultured human umbilical cord blood (HUCB) cells: implications for brain repair. Exp. Neurol. 199, 201–208. doi: 10.1016/j.expneurol.2006.04.001
Nietfeld, J. J., Pasquini, M. C., Logan, B. R., Verter, F., and Horowitz, M. M. (2008). Lifetime probabilities of hematopoietic stem cell transplantation in the U.S. Biol. Blood Marrow Transplant. 14, 316–322. doi: 10.1016/j.bbmt.2007.12.493
Nijboer, C. H., Bonestroo, H. J. C., Zijlstra, J., Kavelaars, A., and Heijnen, C. J. (2013). Mitochondrial JNK phosphorylation as a novel therapeutic target to inhibit neuroinflammation and apoptosis after neonatal ischemic brain damage. Neurobiol. Dis. 54, 432–444. doi: 10.1016/j.nbd.2013.01.017
Nijboer, C. H., Heijnen, C. J., van der Kooij, M. A., Zijlstra, J., van Velthoven, C. T., Culmsee, C., et al. (2011). Targeting the p53 pathway to protect the neonatal ischemic brain. Ann. Neurol. 70, 255–264. doi: 10.1002/ana.22413
Nijboer, C. H., van der Kooij, M. A., van Bel, F., Ohl, F., Heijnen, C. J., and Kavelaars, A. (2010). Inhibition of the JNK/AP-1 pathway reduces neuronal death and improves behavioral outcome after neonatal hypoxic-ischemic brain injury. Brain Behav. Immun. 24, 812–821. doi: 10.1016/j.bbi.2009.09.008
Northington, F. J., Chavez-Valdez, R., and Martin, L. J. (2011). Neuronal cell death in neonatal hypoxia-ischemia. Ann. Neurol. 69, 743–758. doi: 10.1002/ana.22419
Nunnari, J., and Suomalainen, A. (2012). Mitochondria: in sickness and in health. Cell 148, 1145–1159. doi: 10.1016/j.cell.2012.02.035
Olivier, P., Fontaine, R. H., Loron, G., Van Steenwinckel, J., Biran, V., Massonneau, V., et al. (2009). Melatonin promotes oligodendroglial maturation of injured white matter in neonatal rats. PLoS One 4:e7128. doi: 10.1371/journal.pone.0007128
Otsu, Y., Couchman, K., Lyons, D. G., Collot, M., Agarwal, A., Mallet, J.-M., et al. (2015). Calcium dynamics in astrocyte processes during neurovascular coupling. Nat. Neurosci. 18, 210–218. doi: 10.1038/nn.3906
Oxenoid, K., Dong, Y., Cao, C., Cui, T., Sancak, Y., Markhard, A. L., et al. (2016). Architecture of the mitochondrial calcium uniporter. Nature 533, 269–273. doi: 10.1038/nature17656
Parameyong, A., Govitrapong, P., and Chetsawang, B. (2015). Melatonin attenuates the mitochondrial translocation of mitochondrial fission proteins and Bax, cytosolic calcium overload and cell death in methamphetamine-induced toxicity in neuroblastoma SH-SY5Y cells. Mitochondrion 24, 1–8. doi: 10.1016/j.mito.2015.07.004
Pei, Z., and Cheung, R. T. F. (2004). Pretreatment with melatonin exerts anti-inflammatory effects against ischemia/reperfusion injury in a rat middle cerebral artery occlusion stroke model. J. Pineal Res. 37, 85–91. doi: 10.1111/j.1600-079x.2004.00138.x
Perasso, L., Adriano, E., Ruggeri, P., Burov, S. V., Gandolfo, C., and Balestrino, M. (2009). In vivo neuroprotection by a creatine-derived compound: phosphocreatine-Mg-complex acetate. Brain Res. 1285, 158–163. doi: 10.1016/j.brainres.2009.06.009
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. doi: 10.1016/j.neuron.2014.12.007
Pimentel-Coelho, P. M., Rosado-de-Castro, P. H., da Fonseca, L. M., and Mendez-Otero, R. (2012). Umbilical cord blood mononuclear cell transplantation for neonatal hypoxic-ischemic encephalopathy. Pediatr. Res. 71, 464–473. doi: 10.1038/pr.2011.59
Pirianov, G., Brywe, K. G., Mallard, C., Edwards, A. D., Flavell, R. A., Hagberg, H., et al. (2007). Deletion of the c-Jun N-terminal kinase 3 gene protects neonatal mice against cerebral hypoxic-ischaemic injury. J. Cereb. Blood Flow Metab. 27, 1022–1032. doi: 10.1038/sj.jcbfm.9600413
Prass, K., Royl, G., Lindauer, U., Freyer, D., Megow, D., Dirnagl, U., et al. (2007). Improved reperfusion and neuroprotection by creatine in a mouse model of stroke. J. Cereb. Blood Flow Metab. 27, 452–459. doi: 10.1038/sj.jcbfm.9600351
Qiu, J., Tan, Y.-W., Hagenston, A. M., Martel, M.-A., Kneisel, N., Skehel, P. A., et al. (2013). Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 4:2034. doi: 10.1038/ncomms3034
Raff, M. C., Barres, B. A., Burne, J. F., Coles, H. S., Ishizaki, Y., and Jacobson, M. D. (1993). Programmed cell death and the control of cell survival: lessons from the nervous system. Science 262, 695–700. doi: 10.1126/science.8235590
Rees, S., Hale, N., De Matteo, R., Cardamone, L., Tolcos, M., Loeliger, M., et al. (2010). Erythropoietin is neuroprotective in a preterm ovine model of endotoxin-induced brain injury. J. Neuropathol. Exp. Neurol. 69, 306–319. doi: 10.1097/nen.0b013e3181d27138
Renolleau, S., Fau, S., Goyenvalle, C., Joly, L. M., Chauvier, D., Jacotot, E., et al. (2007). Specific caspase inhibitor Q-VD-OPh prevents neonatal stroke in P7 rat: a role for gender. J. Neurochem. 100, 1062–1071. doi: 10.1111/j.1471-4159.2006.04269.x
Rintoul, G. L., and Reynolds, I. J. (2010). Mitochondrial trafficking and morphology in neuronal injury. Biochim. Biophys. Acta 1802, 143–150. doi: 10.1016/j.bbadis.2009.09.005
Robertson, N. J., Faulkner, S., Fleiss, B., Bainbridge, A., Andorka, C., Price, D., et al. (2013). Melatonin augments hypothermic neuroprotection in a perinatal asphyxia model. Brain 136, 90–105. doi: 10.1093/brain/aws285
Rodríguez-Espinosa, O., Rojas-Espinosa, O., Moreno-Altamirano, M. M. B., López-Villegas, E. O., and Sánchez-García, F. J. (2015). Metabolic requirements for neutrophil extracellular traps formation. Immunology 145, 213–224. doi: 10.1111/imm.12437
Saitoh, T., and Akira, S. (2010). Regulation of innate immune responses by autophagy-related proteins. J. Cell Biol. 189, 925–935. doi: 10.1083/jcb.201002021
Sanders, R. D., Manning, H. J., Robertson, N. J., Ma, D., Edwards, A. D., Hagberg, H., et al. (2010). Preconditioning and postinsult therapies for perinatal hypoxic-ischemic injury at term. Anesthesiology 113, 233–249. doi: 10.1097/ALN.0b013e3181dc1b84
Sandhir, R., Halder, A., and Sunkaria, A. (2017). Mitochondria as a centrally positioned hub in the innate immune response. Biochim. Biophys. Acta 1863, 1090–1097. doi: 10.1016/j.bbadis.2016.10.020
Sanman, L. E., Qian, Y., Eisele, N. A., Ng, T. M., Van Der Linden, W. A., Monack, D. M., et al. (2016). Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. ELife 5:e13663. doi: 10.7554/eLife.13663
Scarpulla, R. C. (2011). Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 1813, 1269–1278. doi: 10.1016/j.bbamcr.2010.09.019
Schwarting, S., Litwak, S., Hao, W., Bahr, M., Weise, J., and Neumann, H. (2008). Hematopoietic stem cells reduce postischemic inflammation and ameliorate ischemic brain injury. Stroke 39, 2867–2875. doi: 10.1161/strokeaha.108.513978
Sestili, P., Martinelli, C., Colombo, E., Barbieri, E., Potenza, L., Sartini, S., et al. (2011). Creatine as an antioxidant. Amino Acids 40, 1385–1396. doi: 10.1007/s00726-011-0875-5
Shankaran, S., Laptook, A. R., Ehrenkranz, R. A., Tyson, J. E., Mcdonald, S. A., Donovan, E. F., et al. (2005). Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N. Engl. J. Med. 353, 1574–1584. doi: 10.1056/NEJMcps050929
Sheridan, C., and Martin, S. J. (2010). Mitochondrial fission/fusion dynamics and apoptosis. Mitochondrion 10, 640–648. doi: 10.1016/j.mito.2010.08.005
Shirayama, Y., Muneoka, K., Fukumoto, M., Tadokoro, S., Fukami, G., Hashimoto, K., et al. (2011). Infusions of allopregnanolone into the hippocampus and amygdala, but not into the nucleus accumbens and medial prefrontal cortex, produce antidepressant effects on the learned helplessness rats. Hippocampus 21, 1105–1113. doi: 10.1002/hipo.20824
Sifringer, M., Brait, D., Weichelt, U., Zimmerman, G., Endesfelder, S., Brehmer, F., et al. (2010). Erythropoietin attenuates hyperoxia-induced oxidative stress in the developing rat brain. Brain Behav. Immun. 24, 792–799. doi: 10.1016/j.bbi.2009.08.010
Song, J., Sun, H., Xu, F., Kang, W., Gao, L., Guo, J., et al. (2016). Recombinant human erythropoietin improves neurological outcomes in very preterm infants. Ann. Neurol. 80, 24–34. doi: 10.1002/ana.24677
Stacpoole, P. W., Gilbert, L. R., Neiberger, R. E., Carney, P. R., Valenstein, E., Theriaque, D. W., et al. (2008). Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics 121, e1223–e1228. doi: 10.1542/peds.2007-2062
Stout, A. K., Raphael, H. M., Kanterewicz, B. I., Klann, E., and Reynolds, I. J. (1998). Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat. Neurosci. 1, 366–373. doi: 10.1038/1577
Strunk, T., Inder, T., Wang, X., Burgner, D., Mallard, C., and Levy, O. (2014). Infection-induced inflammation and cerebral injury in preterm infants. Lancet Infect. Dis. 14, 751–762. doi: 10.1016/S1473-3099(14)70710-8
Sugawara, T., Fujimura, M., Noshita, N., Kim, G. W., Saito, A., Hayashi, T., et al. (2004). Neuronal death/survival signaling pathways in cerebral ischemia. NeuroRx 1, 17–25. doi: 10.1602/neurorx.1.1.17
Suliman, H. B., and Piantadosi, C. A. (2016). Mitochondrial quality control as a therapeutic target. Pharmacol. Rev. 68, 20–48. doi: 10.1124/pr.115.011502
Sullivan, P. G., Geiger, J. D., Mattson, M. P., and Scheff, S. W. (2000). Dietary supplement creatine protects against traumatic brain injury. Ann. Neurol. 48, 723–729. doi: 10.1002/1531-8249(200011)48:5<723::aid-ana5>3.3.co;2-n
Sun, W., Buzanska, L., Domanska-Janik, K., Salvi, R. J., and Stachowiak, M. K. (2005). Voltage-sensitive and ligand-gated channels in differentiating neural stem-like cells derived from the nonhematopoietic fraction of human umbilical cord blood. Stem Cells 23, 931–945. doi: 10.1634/stemcells.2004-0316
Sun, Y., Calvert, J. W., and Zhang, J. H. (2005). Neonatal hypoxia/ischemia is associated with decreased inflammatory mediators after erythropoietin administration. Stroke 36, 1672–1678. doi: 10.1161/01.str.0000173406.04891.8c
Sun, Y., Li, T., Xie, C., Zhang, Y., Zhou, K., Wang, X., et al. (2016). Dichloroacetate treatment improves mitochondrial metabolism and reduces brain injury in neonatal mice. Oncotarget 7, 31708–31722. doi: 10.18632/oncotarget.9150
Susin, S. A., Lorenzo, H. K., Zamzami, N., Marzo, I., Snow, B. E., Brothers, G. M., et al. (1999). Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397, 441–446. doi: 10.1038/17135
Swerdlow, R. H., Parks, J. K., Miller, S. W., Davis, R. E., Tuttle, J. B., Trimmer, P. A., et al. (1996). Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann. Neurol. 40, 663–671. doi: 10.1002/ana.410400417
Tagin, M., Abdel-Hady, H., Rahman, S. U., Azzopardi, D. V., and Gunn, A. J. (2015). Neuroprotection for perinatal hypoxic ischemic encephalopathy in low- and middle-income countries. J. Pediatr. 167, 25–28. doi: 10.1016/j.jpeds.2015.02.056
Tait, S. W. G., and Green, D. R. (2012). Mitochondria and cell signalling. J. Cell Sci. 125, 807–815. doi: 10.1242/jcs.099234
Tanaka, A., and Youle, R. J. (2008). A chemical inhibitor of DRP1 uncouples mitochondrial fission and apoptosis. Mol. Cell 29, 409–410. doi: 10.1016/j.molcel.2008.02.005
Tannahill, G. M., Curtis, A. M., Adamik, J., Palsson-Mcdermott, E. M., Mcgettrick, A. F., Goel, G., et al. (2013). Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242. doi: 10.1038/nature11986
Tur, J., Vico, T., Lloberas, J., Zorzano, A., and Celada, A. (2017). Macrophages and mitochondria: a critical interplay between metabolism, signaling, and the functional activity. Adv. Immunol. 133, 1–36. doi: 10.1016/bs.ai.2016.12.001
Utkina-Sosunova, I. V., Niatsetskaya, Z. V., Sosunov, S. A., Ratner, V. I., Matsiukevich, D., and Ten, V. S. (2013). Nelfinavir inhibits intra-mitochondrial calcium influx and protects brain against hypoxic-ischemic injury in neonatal mice. PLoS One 8:e62448. doi: 10.1371/journal.pone.0062448
van der Bliek, A. M., Shen, Q., and Kawajiri, S. (2013). Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 5:a011072. doi: 10.1101/cshperspect.a011072
van der Wijst, M. G. P., and Rots, M. G. (2015). Mitochondrial epigenetics: an overlooked layer of regulation? Trends Genet. 31, 353–356. doi: 10.1016/j.tig.2015.03.009
Van Velthoven, C. T. J., Kavelaars, A., Van Bel, F., and Heijnen, C. J. (2010). Mesenchymal stem cell treatment after neonatal hypoxic-ischemic brain injury improves behavioral outcome and induces neuronal and oligodendrocyte regeneration. Brain Behav. Immun. 24, 387–393. doi: 10.1016/j.bbi.2009.10.017
Vayssière, J. L., Cordeau-Lossouarn, L., Larcher, J. C., Basseville, M., Gros, F., and Croizat, B. (1992). Participation of the mitochondrial genome in the differentiation of neuroblastoma cells. In Vitro Cell. Dev. Biol. 28A, 763–772. doi: 10.1007/bf02631065
Vila, M., and Przedborski, S. (2003). Targeting programmed cell death in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 365–375. doi: 10.1038/nrn1100
Volpe, J. J. (2001). Perinatal brain injury: from pathogenesis to neuroprotection. Ment. Retard. Dev. Disabil. Res. Rev. 7, 56–64. doi: 0.1002/1098-2779(200102)7:1<56::AID-MRDD1008>3.0.CO;2-A
Wahl, D. R., Petersen, B., Warner, R., Richardson, B. C., Glick, G. D., and Opipari, A. W. (2010). Characterization of the metabolic phenotype of chronically activated lymphocytes. Lupus 19, 1492–1501. doi: 10.1177/0961203310373109
Wang, X., Carlsson, Y., Basso, E., Zhu, C., Rousset, C. I., Rasola, A., et al. (2009). Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury. J. Neurosci. 29, 2588–2596. doi: 10.1523/jneurosci.5832-08.2009
Wang, Y., Cui, J., Sun, X., and Zhang, Y. (2011). Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 18, 732–742. doi: 10.1038/cdd.2010.147
Wang, D., Malo, D., and Hekimi, S. (2010). Elevated mitochondrial reactive oxygen species generation affects the immune response via hypoxia-inducible factor-1α in long-lived Mclk1+/– mouse mutants. J. Immunol. 184, 582–590. doi: 10.4049/jimmunol.0902352
Wang, L., Zhang, Z., Wang, Y., Zhang, R., and Chopp, M. (2004). Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke 35, 1732–1737. doi: 10.1161/01.str.0000132196.49028.a4
Waterham, H. R., Koster, J., Van Roermund, C. W., Mooyer, P. A., Wanders, R. J., and Leonard, J. V. (2007). A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 356, 1736–1741. doi: 10.1056/nejmoa064436
Wecht, S., and Rojas, M. (2016). Mesenchymal stem cells in the treatment of chronic lung disease. Respirology 21, 1366–1375. doi: 10.1111/resp.12911
Wegleiter, K., Hermann, M., Posod, A., Wechselberger, K., Stanika, R. I., Obermair, G. J., et al. (2014). The sigma-1 receptor agonist 4-phenyl-1–(4-phenylbutyl) piperidine (PPBP) protects against newborn excitotoxic brain injury by stabilizing the mitochondrial membrane potential in vitro and inhibiting microglial activation in vivo. Exp. Neurol. 261, 501–509. doi: 10.1016/j.expneurol.2014.07.022
Weinberg, S. E., Sena, L. A., and Chandel, N. S. (2015). Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417. doi: 10.1016/j.immuni.2015.02.002
Welin, A. K., Svedin, P., Lapatto, R., Sultan, B., Hagberg, H., Gressens, P., et al. (2007). Melatonin reduces inflammation and cell death in white matter in the mid-gestation fetal sheep following umbilical cord occlusion. Pediatr. Res. 61, 153–158. doi: 10.1203/01.pdr.0000252546.20451.1a
West, A. P., Khoury-Hanold, W., Staron, M., Tal, M. C., Pineda, C. M., Lang, S. M., et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. doi: 10.1038/nature14156
Williams, P. J., Lane, J. R., Turkel, C. C., Capparelli, E. V., Dziewanowska, Z., and Fox, A. W. (2001). Dichloroacetate: population pharmacokinetics with a pharmacodynamic sequential link model. J. Clin. Pharmacol. 41, 259–267. doi: 10.1177/00912700122010078
Wu, Z., Puigserver, P., Andersson, U., Zhang, C., Adelmant, G., Mootha, V., et al. (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124. doi: 10.1016/s0092-8674(00)80611-x
Yu, J., Nagasu, H., Murakami, T., Hoang, H., Broderick, L., Hoffman, H. M., et al. (2014). Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. U S A 111, 15514–15519. doi: 10.1073/pnas.1414859111
Zhang, Q., Raoof, M., Chen, Y., Sumi, Y., Sursal, T., Junger, W., et al. (2010). Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107. doi: 10.1038/nature08780
Zhang, Q.-G., Wu, D.-N., Han, D., and Zhang, G.-Y. (2007). Critical role of PTEN in the coupling between PI3K/Akt and JNK1/2 signaling in ischemic brain injury. FEBS Lett. 581, 495–505. doi: 10.1016/j.febslet.2006.12.055
Zhao, J., Qu, Y., Wu, J., Cao, M., Ferriero, D. M., Zhang, L., et al. (2013). PTEN inhibition prevents rat cortical neuron injury after hypoxia-ischemia. Neuroscience 238, 242–251. doi: 10.1016/j.neuroscience.2013.02.046
Zhu, C., Qiu, L., Wang, X., Hallin, U., Candé, C., Kroemer, G., et al. (2003). Involvement of apoptosis-inducing factor in neuronal death after hypoxia-ischemia in the neonatal rat brain. J. Neurochem. 86, 306–317. doi: 10.1046/j.1471-4159.2003.01832.x
Zhu, C., Wang, X., Deinum, J., Huang, Z., Gao, J., Modjtahedi, N., et al. (2007a). Cyclophilin A participates in the nuclear translocation of apoptosis-inducing factor in neurons after cerebral hypoxia-ischemia. J. Exp. Med. 204, 1741–1748. doi: 10.1084/jem.20070193
Zhu, C., Wang, X., Huang, Z., Qiu, L., Xu, F., Vahsen, N., et al. (2007b). Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ. 14, 775–784. doi: 10.1038/sj.cdd.4402053
Zhu, C., Wang, X., Hagberg, H., and Blomgren, K. (2000). Correlation between caspase-3 activation and three different markers of DNA damage in neonatal cerebral hypoxia-ischemia. J. Neurochem. 75, 819–829. doi: 10.1046/j.1471-4159.2000.0750819.x
Zhu, C., Wang, X., Xu, F., Bahr, B. A., Shibata, M., Uchiyama, Y., et al. (2005). The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ. 12, 162–176. doi: 10.1038/sj.cdd.4401545
Zhu, C., Xu, F., Wang, X., Shibata, M., Uchiyama, Y., Blomgren, K., et al. (2006). Different apoptotic mechanisms are activated in male and female brains after neonatal hypoxia-ischaemia. J. Neurochem. 96, 1016–1027. doi: 10.1111/j.1471-4159.2005.03639.x
Zonta, M., Angulo, M. C., Gobbo, S., Rosengarten, B., Hossmann, K.-A., Pozzan, T., et al. (2003). Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci. 6, 43–50. doi: 10.1038/nn980
Keywords: perinatal brain injury, hypoxia-ischemia, mitochondria, neuroprotection
Citation: Leaw B, Nair S, Lim R, Thornton C, Mallard C and Hagberg H (2017) Mitochondria, Bioenergetics and Excitotoxicity: New Therapeutic Targets in Perinatal Brain Injury. Front. Cell. Neurosci. 11:199. doi: 10.3389/fncel.2017.00199
Received: 01 May 2017; Accepted: 26 June 2017;
Published: 12 July 2017.
Edited by:
Pier Giorgio Mastroberardino, Erasmus University Rotterdam, NetherlandsReviewed by:
Amadou K. S. Camara, Medical College of Wisconsin, United StatesAlessia Buso, University of Udine, Italy
Copyright © 2017 Leaw, Nair, Lim, Thornton, Mallard and Hagberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bryan Leaw, bryan.leaw@hudson.org.au