Group II Metabotropic Glutamate Receptors Mediate Presynaptic Inhibition of Excitatory Transmission in Pyramidal Neurons of the Human Cerebral Cortex
 Marco Bocchio1†
Marco Bocchio1†  Istvan P. Lukacs1
Istvan P. Lukacs1  Richard Stacey2 Puneet Plaha2 Vasileios Apostolopoulos2
Richard Stacey2 Puneet Plaha2 Vasileios Apostolopoulos2  Laurent Livermore2
Laurent Livermore2  Arjune Sen3,4 Olaf Ansorge4
Arjune Sen3,4 Olaf Ansorge4  Martin J. Gillies4
Martin J. Gillies4  Peter Somogyi1*
Peter Somogyi1*  Marco Capogna5,6*
Marco Capogna5,6*- 1Department of Pharmacology, University of Oxford, Oxford, United Kingdom
- 2Department of Neurosurgery, John Radcliffe Hospital, Oxford University Hospitals NHS Foundation Trust, Oxford, United Kingdom
- 3Oxford Epilepsy Research Group, NIHR Biomedical Research Centre, Oxford, United Kingdom
- 4Nuffield Department of Clinical Neurosciences, Medical Sciences Division, University of Oxford, Oxford, United Kingdom
- 5Department of Biomedicine, Aarhus University, Aarhus, Denmark
- 6The Danish Research Institute of Translational Neuroscience, Nordic EMBL Partnership for Molecular Medicine – Department of Biomedicine, Aarhus University, Aarhus, Denmark
Group II metabotropic glutamate receptor (mGluR) ligands are potential novel drugs for neurological and psychiatric disorders, but little is known about the effects of these compounds at synapses of the human cerebral cortex. Investigating the effects of neuropsychiatric drugs in human brain tissue with preserved synaptic circuits might accelerate the development of more potent and selective pharmacological treatments. We have studied the effects of group II mGluR activation on excitatory synaptic transmission recorded from pyramidal neurons of cortical layers 2–3 in acute slices derived from surgically removed cortical tissue of people with epilepsy or tumors. The application of a selective group II mGluR agonist, LY354740 (0.1–1 μM) inhibited the amplitude and frequency of action potential-dependent spontaneous excitatory postsynaptic currents (sEPSCs). This effect was prevented by the application of a group II/III mGluR antagonist, CPPG (0.1 mM). Furthermore, LY354740 inhibited the frequency, but not the amplitude, of action potential-independent miniature EPSCs (mEPSCs) recorded in pyramidal neurons. Finally, LY354740 did slightly reduce cells’ input resistance without altering the holding current of the neurons recorded in voltage clamp at -90 mV. Our results suggest that group II mGluRs are mainly auto-receptors that inhibit the release of glutamate onto pyramidal neurons in layers 2–3 in the human cerebral cortex, thereby regulating network excitability. We have demonstrated the effect of a group II mGluR ligand at human cortical synapses, revealing mechanisms by which these drugs could exert pro-cognitive effects and treat human neuropsychiatric disorders.
Introduction
Pre- and postsynaptic metabotropic glutamate receptors (mGluRs) are activated by L-glutamate, the major excitatory transmitter in the mammalian central nervous system (CNS). These receptors regulate neuronal excitability and synaptic plasticity and mediate the actions of neuroactive drugs (Anwyl, 1999; Cartmell and Schoepp, 2002; Niswender and Conn, 2010). Eight subtypes of mGluRs have been identified and these are classified into three groups (mGluRs I, II, and III) according to their amino acid sequence similarities, agonist selectivity, and interactions with transduction mechanisms (Conn and Pin, 1997). Group II receptors (mGlu2 and mGlu3) are intriguing due to their peri- and extra-synaptic location outside the synaptic junction (Shigemoto et al., 1997; Corti et al., 2002), predicting receptor activation in an activity-dependent manner when glutamate spills over from the release site (Scanziani et al., 1997). This process may be of interest not only under physiological but also under pathological conditions (Molinari et al., 2012). Group II mGluRs are often coupled to the cyclic AMP cascade (Conn and Pin, 1997) and are potently activated by a series of compounds developed by several pharmaceutical companies and particularly by Eli Lilly and Company, for example (1)-2-aminobicyclo[3.1.0] hexane-2,6-dicarboxylic acid (LY354740) (Schoepp et al., 1999).
Work performed in rodents shows that the activation of group II mGluRs have pre- and post-synaptic effects, consistent with receptor locations (Anwyl, 1999; Cartmell and Schoepp, 2002; Niswender and Conn, 2010). For example, presynaptic group II mGluRs, expressed by the perforant pathway from the entorhinal cortex depress excitatory synaptic responses recorded from hippocampal CA1 pyramidal cells or interneurons of stratum lacunosum moleculare (Kew et al., 2001; Capogna, 2004; Price et al., 2005). Conversely, activation of postsynaptic mGlu3 expressed by hippocampal CA3 pyramidal neurons enhances excitability by inhibiting a K+ conductance and by activating a calcium-sensitive cationic conductance (Ster et al., 2011). Moreover, mGlu3R activation enhances mGlu5R-mediated somatic Ca2+ mobilization in pyramidal cells of the mouse prefrontal cortex (Di Menna et al., 2018).
Furthermore, pharmacological or genetic manipulation of mGlu2 gate synaptic plasticity, for instance at hippocampal mossy fiber to CA3 pyramidal cell synapses (Yokoi et al., 1996). Thus, the cellular mechanisms by which group II mGluRs regulate network function are heterogeneous. Additional complexity is given by the fact that group II mGluRs are expressed on axons that target specific cell types but not others (Kintscher et al., 2012).
Group II mGluRs can be down-regulated during development (Doherty et al., 2004), indicating that these receptors could be critical for network maturation as well as for neurodevelopmental disorders. Conversely, group II mGluRs are up-regulated following epileptic seizures (Doherty and Dingledine, 2001). These observations suggest that these receptors could play a role in various brain disorders. Indeed, group II mGluRs are currently investigated as potential drug targets for treating neurological and psychiatric disorders. Several agonists have been developed with the aim of improving cognitive dysfunction in schizophrenia, Alzheimer’s disease and anxiety disorders (Caraci et al., 2018; Ferraguti, 2018; Stansley and Conn, 2018). For example, agonists of group II mGluRs show anxiolytic activity in a wide range of animal models of anxiety disorder (Swanson et al., 2005) and improve cognition (Stansley and Conn, 2018). Experimentally, LY354740 rescues deficits in stereotypy, locomotion, spatial working memory and cortical glutamate efflux induced by pharmacological blockade of NMDA receptors in rodents (Moghaddam and Adams, 1998). Furthermore, several mGlu2 positive allosteric modulators (PAMs) have shown efficacy in preclinical animal models of schizophrenia (Spooren et al., 2000; Galici, 2005) (see review: Maksymetz et al., 2017).
Behavioral effects of mGluR activation in humans provide translational validity of the results obtained in animal models. For example, group II mGluR agonists attenuate NMDA receptor activation-induced deficits in working memory in human subjects (Krystal et al., 2005). However, clinical trials using group II mGluR agonists or PAMs in psychiatric patients have not led to the introduction of novel clinical treatments (see reviews: Maksymetz et al., 2017; Ferraguti, 2018). Reasons for translational failure are complex, including species differences, lack of appropriate animal models, insufficient dosing and limited bioavailability of the drugs tested (Jucker, 2010). As for many other receptor systems, there is a gap between the vast amounts of information available on the actions of group II mGluRs in rodent versus human CNS. The aim of the present study is to help closing this gap and to elucidate the action of the potent and selective group II mGluR agonist LY354740 on pyramidal neurons recorded from acute slices derived from surgically removed human cortical tissue.
Materials and Methods
Ethics Statement and Patients
Surgical specimens were obtained from the temporal neocortex of drug resistant temporal lobe epilepsy (TLE) (patients A–E, Table 1A, five females) and from the temporal and frontal neocortex of low grade glioma oncological patients with brain tumors (patients F–H, Table 1B, one female, two males) operated at the John Radcliffe Hospital, Oxford, United Kingdom. All studied tissues were necessarily removed during the surgical procedure and were surplus to diagnostic requirements. Ethics approval was sought and obtained from Research Ethics Committees, National Health Service, Health Research Authority, United Kingdom (NRES Committee South Central – Oxford C: reference 15/SC/0639; NRES Committee East of England – Cambridgeshire and Hertfordshire: reference 14/EE/1098). Fully informed written consent was obtained from each patient who participated.

Table 1(A). Clinical data of temporal lobe epilepsy cases.

Table 1(B). Clinical data of glioma cases.
Slice Preparation
A small piece of cortical tissue (size < 1 cm3) including all layers and some white matter was isolated using a scalpel, removed and immediately immersed in cutting artificial cerebrospinal fluid (ACSF) saturated with carbogen (95% O2/5% CO2), at ∼4°C. The solution containing the block of tissue was continuously bubbled with carbogen. Transportation from the operating theater to the laboratory lasted 15–60 min. The block of tissue was placed in a petri dish containing ice-cold cutting ACSF bubbled with carbogen. In some cases, the block of tissue was divided into smaller pieces before transportation. Next, the block of tissue was glued on a platform of a vibratome (Microm HM 650 V, Thermo Fisher Scientific) and cut into slices at a setting of 325 μm thickness in cutting ACSF at 4°C. The block of tissue was oriented in a way to slice perpendicular to the pia and parallel to the apical dendrites of the pyramidal cells. The cutting ACSF contained the following compounds (in mM): 65 sucrose, 85 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 7 MgCl2, 10 glucose (pH 7.3, ∼300 mOsm/L). Slices were incubated in cutting ACSF at 36°C. After ∼10 min, the cutting ACSF was replaced with recording ACSF at 36°C using a peristaltic pump (Gilson) operated at ∼5 mL/min. The recording ACSF contained the following compounds (in mM): 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 26 NaHCO3, 2 CaCl2, 2 MgCl2, 10 glucose, (pH 7.3, ∼300 mOsm/L). Slices were heated at 36°C for 30 min, after which they were stored at room temperature and continuously bubbled with carbogen.
Electrophysiological Recordings
Slices were submerged in a recording chamber and stabilized with a plastic string harp. The chamber was continuously perfused with oxygenated recording ACSF at a rate of 10 mL/min by a peristaltic pump (Gilson) at a temperature of 33 ± 1°C. Neurons were visualized using a differential interference contrast (DIC) microscope (Olympus BX51WI) using a LUMPlanFL 60× water objective (Olympus) and equipped with a camera (Zyla, ANDOR) connected to a desktop computer. Glass electrodes (4–6 MΩ) were prepared from borosilicate glass capillaries (1.2 mm; GC120F, Harvard Apparatus) using a DMZ Universal puller (Zeitz-Instrument). Electrodes were filled with an intracellular solution composed of the following (in mM): 126 K-gluconate, 4 KCl, 4 ATP-Mg, 0.3 GTP-Na2, 10 Na2-phosphocreatine, 10 HEPES, and 0.05% biocytin, with osmolarity of 270–280 mOsmol/L without biocytin, at pH 7.3 adjusted with KOH. Somatic whole-cell patch-clamp recordings were performed from visualized neurons in cortical layers 2–3. Electrophysiological signals were amplified using an EPC10 triple patch clamp amplifier (HEKA Electronik), digitized at 20 kHz for recordings in voltage-clamp mode and at 5 kHz for recordings in current-clamp mode, and acquired using Patchmaster software (HEKA Electronik). All reported voltage values were compensated for a calculated 16 mV liquid junction potential between the ACSF and the recording pipette. Spontaneous or miniature excitatory postsynaptic currents (sEPSCs or mEPSCs) were recorded in continuous voltage clamp with a holding potential of –90 mV (corresponding to the calculated Cl- reversal potential). The mEPSCs were recorded after the addition of 1 μM tetrodotoxin (TTX) to the recording ACSF. Uncompensated series resistance was monitored using a 20 ms-long -10 mV voltage step applied at the beginning of every sweep (i.e., every minute). The sEPSCs or mEPSCs were recorded continuously for up to ∼30 min. After 3–4 min of baseline recording 0.1–1 μM LY354740 was perfused into the recording chamber for 5 min. The drug was subsequently washed out for 10–20 min.
Signal Analysis and Inclusion Criteria
Analysis of synaptic currents and intrinsic membrane responses was performed using Igor Pro (WaveMetrics). PatchMaster files were loaded into Igor Pro with Patcher’s Power Tools (Max-Planck-Institute, Department of Membrane Biophysics). The input resistance (Rin) was calculated from the slope of steady-state voltage responses to a series of 8–10 subthreshold current injections lasting 400 ms. Spike threshold, half-width and fast after-hyperpolarization (AHPfast, in mV) were determined from the first spike in response to a juxtathreshold positive current injection. The spike half-width was defined as the duration at half-amplitude measured between the threshold potential and the peak of the action potential. The membrane time constant τ was estimated from the monoexponential curve fitting of voltage responses to a -30 pA hyperpolarizing pulse. The membrane capacitance was calculated as the ratio between membrane τ and Rin. The rheobase (in pA) was determined as a 50 ms current injection, able to generate a spike in 50% of the cases in 10 trials. The instantaneous firing rate (in Hz) was defined as the number of action potentials evoked during a 1 s-long depolarizing current pulse of twice the amplitude of the rheobase current. The adaptation index (range, 0–1) was defined as the ratio between the first and last inter-spike intervals (ISIs; in ms) elicited by the same current pulse used to measure the instantaneous firing rate. The resting membrane potential was estimated by averaging a 20 s current-clamp trace recorded at a 0 pA holding current. Changes in Rin evoked by LY354740 application were assessed in voltage clamp mode by applying regular hyperpolarizing voltage steps (-10 mV). For these experiments, Rin was calculated as follows:
where Rtot is the total resistance and Rs is the series resistance. Rs was calculated as follows:
where V is the amplitude of the voltage step and Ip is the amplitude of the peak of the capacitive current transient. Rtot was calculated as follows:
where Is is the amplitude of the steady state current.
The following criteria were determined in order to accept or reject event files: (1) recorded neurons were able to generate at least one spike upon current injection in current clamp mode at the beginning of the recording; (2) the holding current to clamp the cell at -90 mV was <-100 pA; (3) the series resistance was <30 MΩ and did not change more than 20% during the recording. Spontaneous synaptic events were detected using TaroTools toolbox for Igor Pro1. The threshold for event detection was set between -5 and -7 pA depending on the signal-to-noise ratio of the recording. Events with 20–80% rise-time longer than 3 ms and half-width shorter than 1 ms were removed. Subsequently, all events were visually inspected for the entire recording period in order to confirm the reliability of the automatic detection. Events were rejected if they did not display typical fast EPSC kinetic (i.e., ratio between decay time and 20–80% rise-time <3). Statistical analysis was performed using Prism software (GraphPad, San Diego, CA, United States), and the tests used are specified throughout the results. Unless indicated otherwise, values presented in the text and in the figures represent the median and the interquartile range (IQR). All data sets were tested for statistically significant outliers using the Rout test (Q = 1%).
Visualization of Recorded Cells
After recording, slices were immersed in a fixative containing 4% paraformaldehyde, 15% v/v picric acid dissolved in 0.1 M phosphate buffer (PB) pH 7.2–7.4, overnight at 4°C. Slices were then thoroughly washed in 0.1M PB until all fixative was removed from the tissue and were either embedded in 20% gelatin and re-sectioned into 60 μm thickness on a vibratome (VT 1000S, Leica) or were processed further without re-sectioning. After permeabilization with Tris Buffered Saline (TBS), containing 0.3% w/v Triton (Tx) (Sigma), the recorded cells were visualized with overnight incubation in Alexa-488-conjugated streptavidin (Invitrogen, Thermo Fisher Scientific), diluted 1:1000 in the same buffer. Sections were then washed with TBS-Tx three times for 10 min and were mounted in Vectashield (Vector Laboratories) on glass slides for microscopic examination. Visualized neurons were examined using an epifluorescent microscope (Leitz DMRB, Leica) equipped with a camera (ORCA-ER, Hamamatsu) and connected to a desktop computer, using a 480/40 excitation and a 527/30 emission filter, corresponding to the fluorophore used. All visualized cells were confirmed to be pyramidal neurons based on the distribution of dendrites and axon and the presence of dendritic spines. Images of Z-stacks of some of the labeled neurons were made using a laser-scanning microscope (LSM 710, Zeiss) with a Plan-Apochromat 20x/0.8 objective (Zeiss).
Chemicals and Drugs
Salts used in the preparation of the internal recording solution and ACSF were obtained from either BDH or Sigma-Aldrich. LY354740 (1)-2-aminobicyclo[3.1.0] hexane-2,6-dicarboxylic acid, CPPG (RS)-α-cyclopropyl-4-phosphonophenylglycine and tetrodotoxin (TTX) were purchased from Tocris Bioscience, LY354740 was stored as frozen aliquots of 10 mM in DMSO. CPPG was stored as frozen aliquots of 100 mM in 1 M NaOH. TTX was stored as frozen aliquots of 1 μM in 10 mM sodium citrate buffer, pH 4.8.
Results
Identity and Spiking Patterns of the Recorded Neurons
Neurons were recorded under visual control (n = 30; n = 8 patients: five TLE, three tumor) and I/V protocols were performed in whole cell current clamp mode to assess the spiking patterns of the recorded cells in response to depolarizing rectangular current pulses. All neurons included in this study displayed membrane responses, action potential kinetics and discharge patterns consistent with features commonly observed in human cortical pyramidal neurons in layers 2–3 (Molnár et al., 2008) (Table 2 and Figure 1). A subset of slices (16 out 30 from six patients, patient IDs: A–D and G, H) were histologically processed to visualize the biocytin-filled neurons. In 15/16 processed slices, the recorded neuron could be identified as pyramidal cells on the basis of spiny dendrites and axonal distribution, consistent with the electrophysiological properties (Figure 1). In one slice the recorded neuron was not recovered.
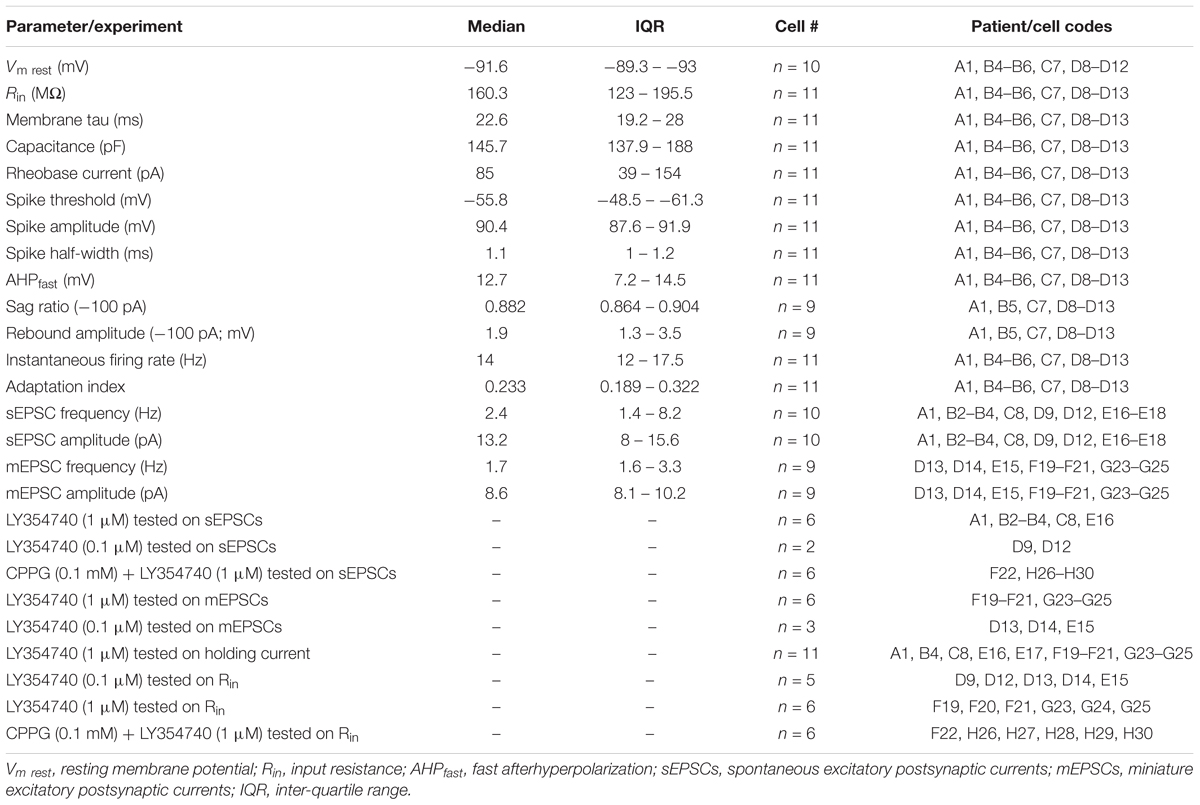
Table 2. Electrophysiological parameters of human cortical pyramidal cells and experimental protocols related to patient and cell codes.
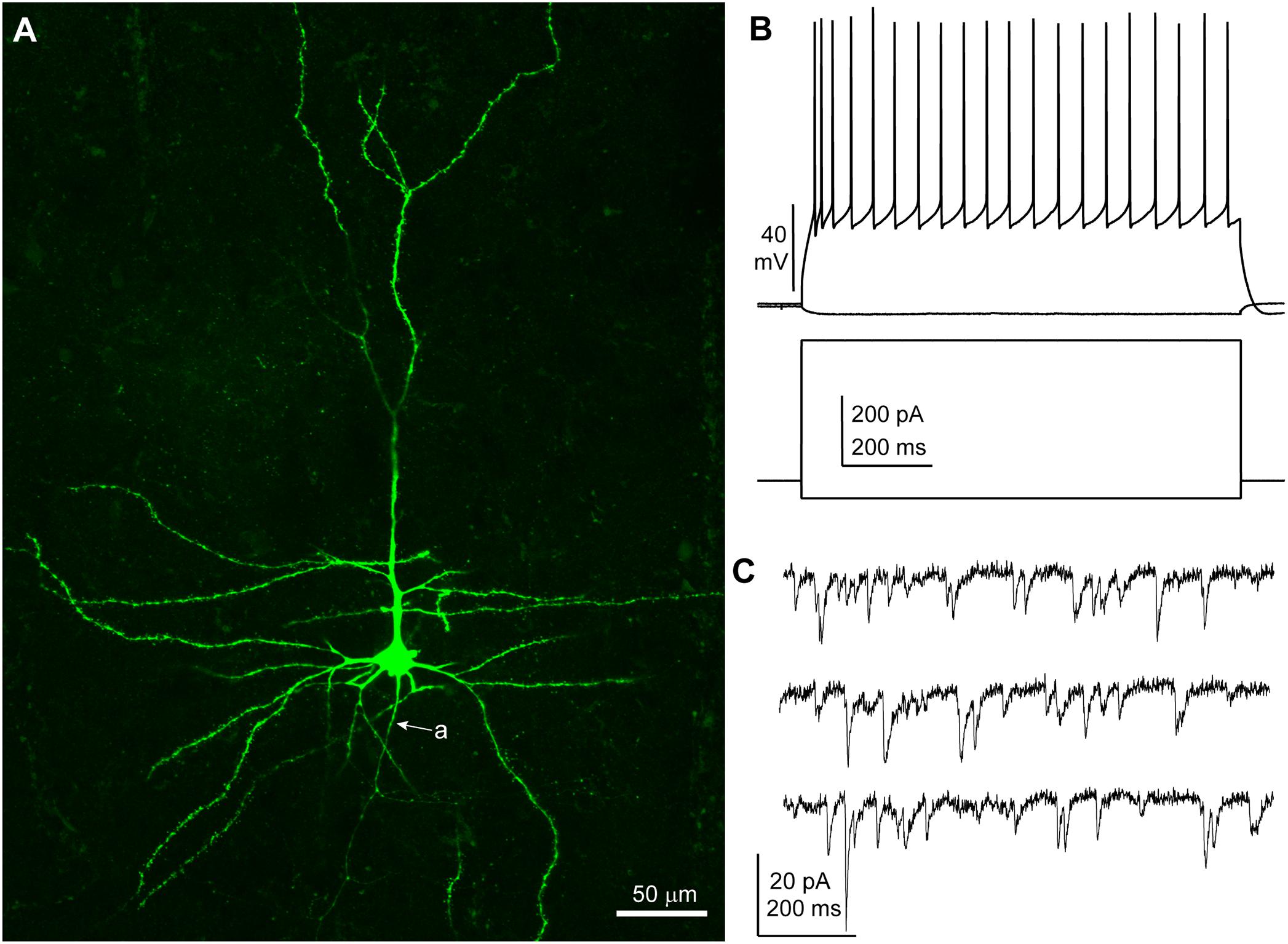
Figure 1. Features of a human cortical pyramidal cell. (A) Confocal microscopic image of a biocytin-filled human cortical pyramidal cell in layer 3 (patient/cell code: H26) showing spiny dendrites, a prominent apical dendrite and axon (a) descending toward the white matter; maximum intensity projection of a z-stack of ∼24 μm thickness; 1.8 μm optical slice thickness; interval 0.9 μm; 25 slices). (B) Voltage responses of the cell shown in (A) recorded in current clamp mode to hyperpolarizing (–50 pA) and depolarizing (+400 pA) current steps (holding potential: –80 mV). (C) Representative traces of spontaneous EPSCs recorded in voltage clamp mode at –90 mV; traces are low-pass filtered at 1 kHz and notch filtered at 50 Hz (width: 0.05 Hz). (A–C) Same cell.
Pharmacological Activation of Group II mGluRs Depresses Excitatory Synaptic Transmission in Human Cortical Pyramidal Cells
Based on the evidence of group II mGluR modulation of glutamatergic transmission and pyramidal cell excitability in the rodent brain (Anwyl, 1999), we tested the effect of these receptors in human cortical pyramidal cells. We recorded sEPSCs from pyramidal cells in voltage clamp mode at -90 mV. The median frequency of sEPSCs was 2.4 Hz (IQR: 1.4–8.2 Hz), whereas the median amplitude was 13.2 (IQR: 8–15.6 pA; n = 10 from five patients).
Application of the selective group II mGluR agonist LY354740 (0.1 μM, n = 2; or 1 μM, n = 6) significantly depressed the frequency and the amplitude of sEPSCs (p = 0.003, Kruskal–Wallis test, pooled n = 8, Figure 2). Specifically, LY354740 reduced sEPSC frequency by 35% (median; IQR: 32–61%; n = 8) from baseline (p < 0.01) and sEPSC amplitude by 12% (median; IQR: 7–21%; n = 8) from baseline (p < 0.01, Dunn’s post hoc test). The depression of sEPSCs partially persisted after 10–20 min post-LY354740 recovery period and could not be fully washed out (baseline vs. washout p < 0.05; LY354740 vs. washout p > 0.05, LY354740 n = 8, washout n = 6, Dunn’s post hoc test).
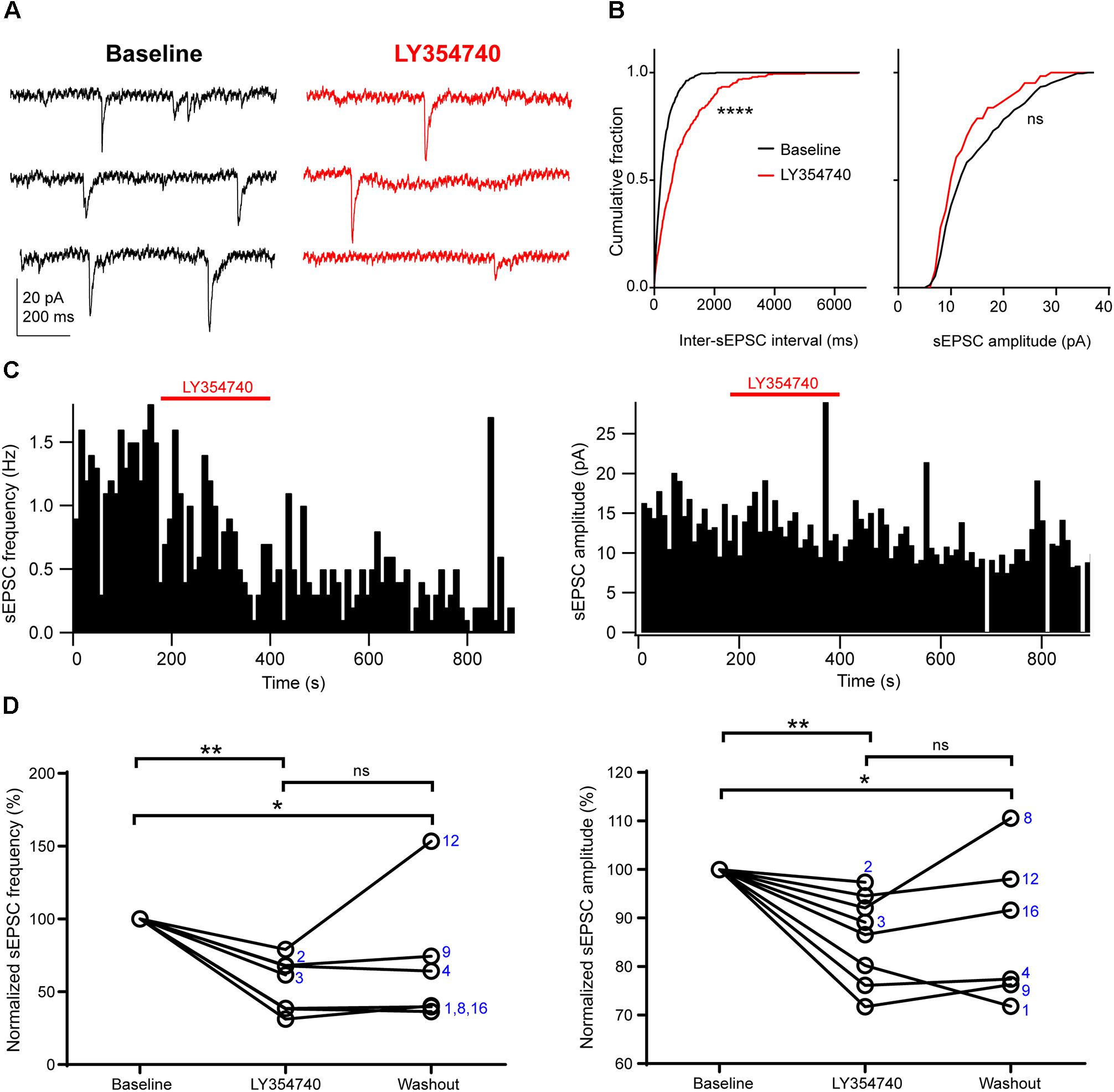
Figure 2. Activation of group II mGluRs depresses excitatory synaptic transmission in human cortical pyramidal cells. (A) Representative traces in voltage clamp mode (–90 mV) during baseline and application of the group II mGluR agonist LY354740 (0.1 μM) to a pyramidal cell. (B) Cumulative probability distributions of the inter-sEPSC intervals and sEPSC amplitudes for the cell shown in (A). LY354740 significantly reduces sEPSC frequency (left, p = 0.0001, Kolmogorov–Smirnov test), and non-significantly decreases sEPSC amplitude (right, p = 0.056, Kolmogorov–Smirnov test). (C) Event time histograms (bin size: 10 ms, cell in A,B) showing the effect of LY354740 application on sEPSC frequency (left) and amplitude (right). (D) Baseline normalized effects of LY354740 (0.1 or 1 μM) on individual cells (see Table 2) show significant reduction of sEPSC frequency (left, p = 0.003 Kruskal–Wallis test; baseline vs. LY354740 p < 0.01, baseline vs. washout p < 0.05, LY354740 vs. washout p > 0.05, Dunn’s post hoc test, n = 8) and sEPSC amplitude (right, p = 0.003 Kruskal–Wallis test; baseline vs. LY354740 p < 0.01, baseline vs. washout p < 0.05, LY354740 vs. washout p > 0.05, Dunn’s post hoc test, n = 8). In some cells, washout could not be analyzed due to changes in series resistance (>20% baseline). Numbers denote codes of individual cells (see Table 2). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
We sought to verify that the inhibition of sEPSCs observed upon application of LY354740 was indeed due to group II mGluRs, and not due to other factors, for example spontaneous sEPSC rundown in the slice. To this end, we pre-incubated slices for ∼10 min with the group II/III mGluR antagonist, CPPG (0.1 mM) prior to the additional application of LY354740 (1 μM). Under these conditions, LY354740 did not trigger significant depression of the frequency (change from baseline: median +9%, IQR: -1 to +22%, p = 0.312, Wilcoxon test, n = 6) or amplitude (change from baseline: median +3%, IQR: -10 to +12%, p > 0.999, Wilcoxon test, n = 6) of sEPSCs (Figure 3). The frequency and amplitude of sEPSCs in control and in the presence of CPPG were not significantly different (frequency in control: median 2.4 Hz, IQR: 1.4–8.2 Hz, n = 10; frequency with CPPG: median 4 Hz, IQR: 1.7–6.5 Hz, n = 6, p = 0.865, Mann–Whitney test; amplitude in control: median 13.2 pA, IQR: 8–15.6 pA, n = 10; amplitude with CPPG: median 10.5, IQR: 9.1–11.7 pA, n = 6, p = 0.534, Mann–Whitney test), suggesting poor or no endogenous activation of group II mGluRs by glutamate under our experimental conditions.
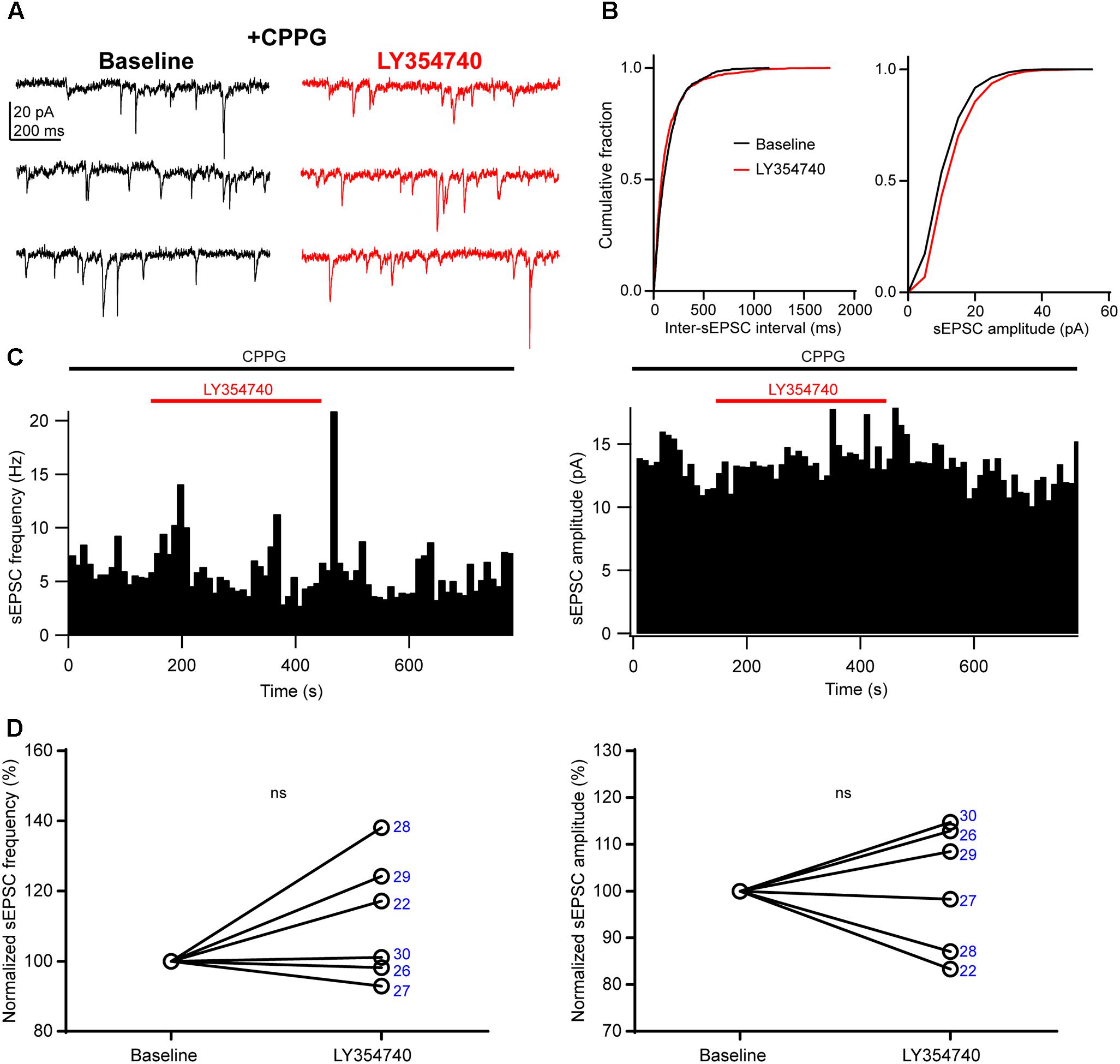
Figure 3. An antagonist of group II/III mGluRs prevents depression of excitatory synaptic transmission by LY354740. (A) Representative traces in voltage clamp mode (–90 mV) during baseline and application of LY354740 (1 μM) in the presence of the group II/III mGluR antagonist CPPG (0.1 mM) in a neuron. (B) Cumulative probability distributions of the inter-sEPSC intervals and sEPSC amplitudes for the cell shown in (A). LY354740 does not reduce sEPSC frequency (p = 0.967, Kolmogorov–Smirnov test) or amplitude (p = 0.999, Kolmogorov–Smirnov test). (C) Event time histograms (bin size: 10 ms, cell in A,B) showing the effect of LY354740 application on sEPSC frequency (left) and amplitude (right) in presence of CPPG. (D), on average, LY354740 (1 μM) does not significantly change sEPSC frequency (left, p = 0.312 Wilcoxon test, n = 6) or sEPSC amplitude (p > 0.999 Wilcoxon test, n = 6). Numbers denote codes of individual cells (see Table 2).
Group II mGluRs Depress Excitatory Synaptic Transmission Mainly via a Presynaptic Effect
In pyramidal cells of the rodent hippocampus (Ster et al., 2011) and primate prefrontal cortex (Jin et al., 2017), the activation of group II mGluRs change postsynaptic conductances. The observed depression of sEPSCs in our experiments may be due to: (1) presynaptic inhibition of glutamate release; (2) a hyperpolarization of other pyramidal cells innervating the recorded pyramidal cell; (3) a purely postsynaptic effects on the recorded cells (e.g., changes of AMPA receptor conductance), or a combination of the above. To discriminate between these scenarios, we tested in voltage clamp mode whether LY354740 application altered the holding current and the Rin of the recorded pyramidal cells (Figure 4). We did not observe a change in the holding current upon the application of LY354740 (1 μM) in neurons at -90 mV (change from baseline: median 2.6 pA, IQR -0.5 to 10.4 pA, p = 0.175, Wilcoxon test, n = 11, Figures 4A,B), suggesting that, at least at this membrane potential, pyramidal cells are not hyperpolarized by this drug. Nonetheless, LY354740 application (0.1 μM, n = 5; or 1 μM, n = 6) triggered a small but significant reduction of Rin (median change -5%, IQR: 3–10%, p = 0.04, Kruskal–Wallis test, pooled n = 11; baseline vs. LY354740 p < 0.05, LY354740 vs. washout p > 0.05, baseline vs. washout p > 0.05, Dunn’s post hoc test). This effect was absent when CPPG (0.1 mM) was applied prior to the additional application of LY354740 (1 μM; p = 0.7, Wilcoxon test, n = 6). The reduction of Rin from baseline triggered by LY354740 was significantly smaller in the presence of CPPG (p = 0.03, Mann–Whitney test), suggesting that this effect was mediated by activation of group II mGluRs. Thus, group II mGluRs appear to trigger small changes in membrane conductance in pyramidal cells.
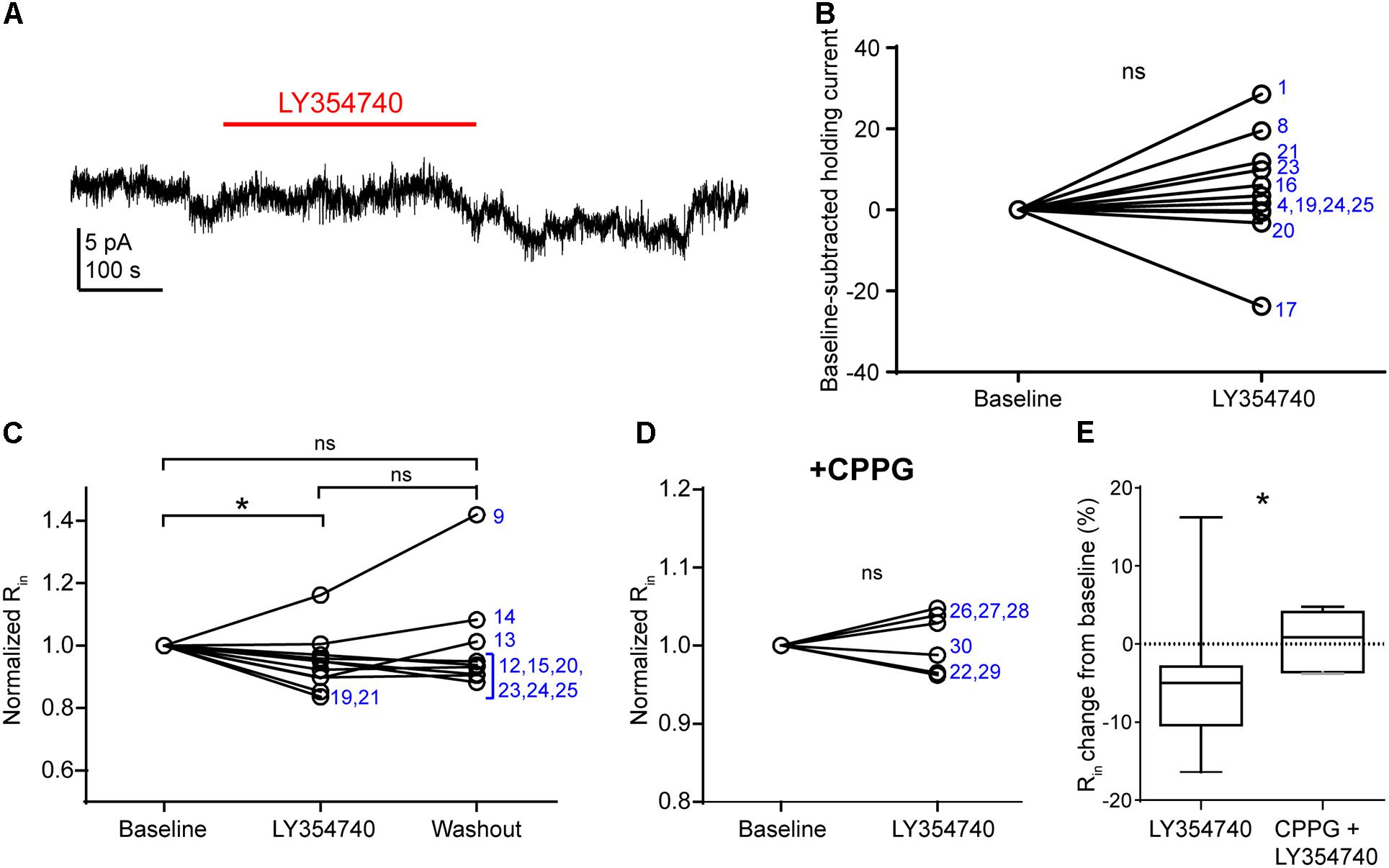
Figure 4. Activation of group II mGluRs does not lead to detectable inward currents in human cortical pyramidal cells. (A) Representative trace in voltage clamp mode (–90 mV) during baseline and application of LY354740 (1 μM). The trace was processed with a low pass filter (5 Hz stop band), a notch filter (50 Hz) and boxcar averaging (window of 50 data points; original sampling rate: 20 kHz) to remove synaptic events and isolate the holding current. (B) LY354740 (1 μM) does not significantly affect the holding current (p = 0.175, Wilcoxon test, n = 11). Numbers denote codes of individual cells (see Table 2). (C) Baseline normalized effects of LY354740 (0.1 or 1 μM) on individual cells (see Table 2) show significant reduction of Rin (median change –5%, IQR: 3–10%, p = 0.04 Kruskal–Wallis test; baseline vs. LY354740 p < 0.05, baseline vs. washout p > 0.05, LY354740 vs. washout p > 0.05, Dunn’s post hoc test, n = 11). (D) On average, LY354740 (1 μM) does not significantly impact pyramidal cells’ Rin when CPPG (0.1 mM) is pre-applied (p = 0.7, Wilcoxon test, n = 6). (E) Boxplot showing significantly bigger reduction of Rin between application of LY354740 only and LY354740 with CPPG (p = 0.03, Mann–Whitney test). In some cells, washout could not be analyzed due to changes in series resistance (>20% baseline). Numbers denote codes of individual cells (see Table 2). ∗p < 0.05.
Next, in order to further discriminate between presynaptic and postsynaptic effects, we tested the drug on spontaneous mEPSCs recorded from pyramidal cells. In the presence of 1 μM TTX to block action potentials, mEPSC frequency had a median value of 1.7 Hz (IQR: 1.6–3.3 Hz) and mEPSC amplitude had a median value of 8.6 pA (IQR: 8.1–10.2 pA, n = 9). Application of 0.1 μM (n = 3 cells) or 1 μM LY354740 (n = 6) caused a significant reduction in the frequency of mEPSCs (change from baseline: median -50%, IQR: -29 to -67%, p = 0.019 Kruskal–Wallis test; baseline vs. LY354740 p < 0.05, Dunn’s post hoc test, n = 9 cells pooled, Figure 5). In contrast, mEPSC amplitude was not significantly altered by LY354740 (change from baseline: median -2%, IQR -4 to +2%, p = 0.239 Kruskal–Wallis test; n = 9, Figure 5). Taken together, these results suggest that activation of group II mGluRs leads to presynaptic and postsynaptic effects in pyramidal cells. However, the lack of changes in holding current (at least at -90 mV) and the modest changes in Rin suggest that the depression of excitatory transmission is predominantly caused by inhibition of glutamate release from glutamatergic terminals innervating pyramidal cells.
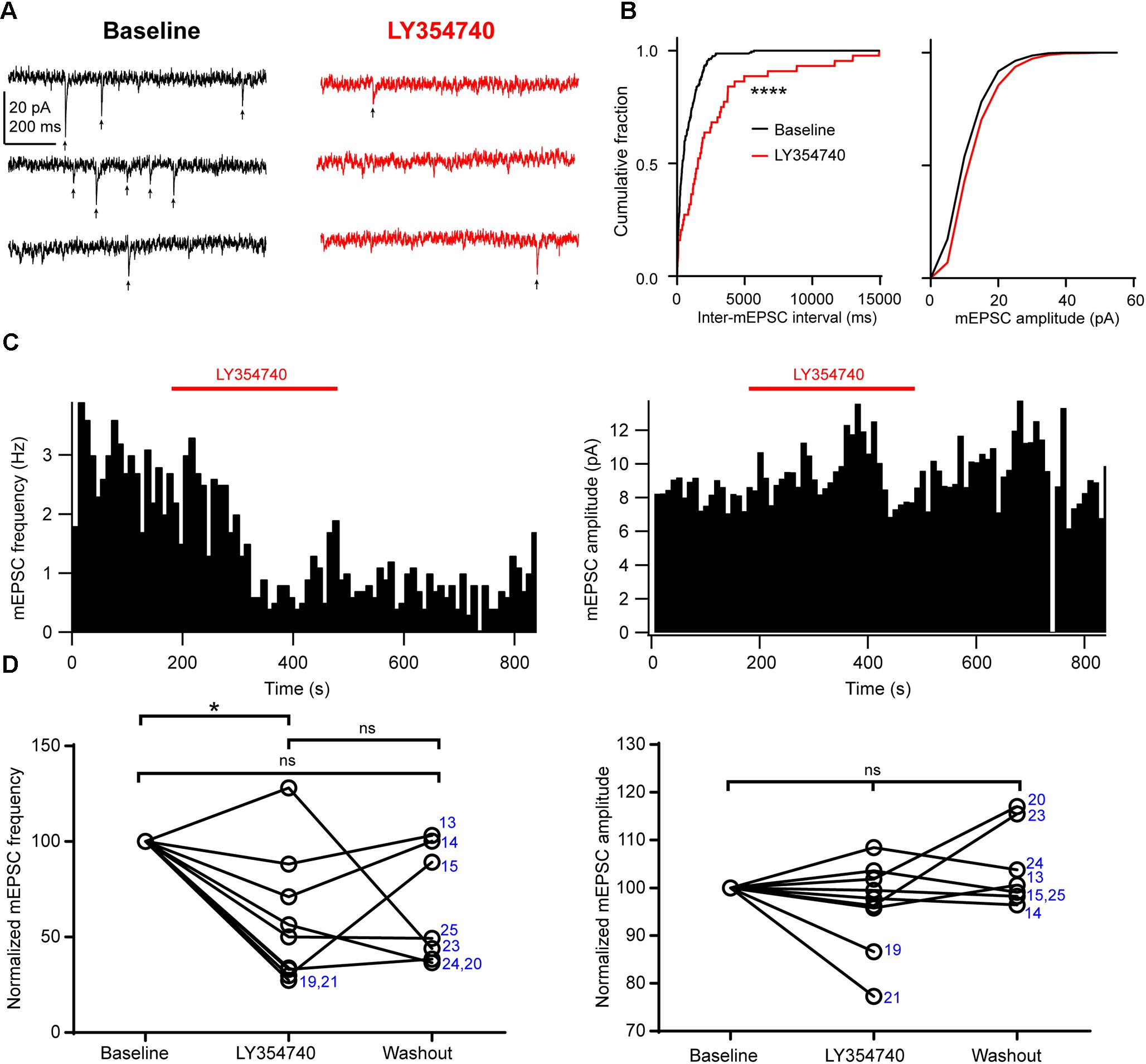
Figure 5. Activation of group II mGluRs depresses excitatory synaptic transmission via a presynaptic effect. (A) Representative traces in voltage clamp mode (–90 mV) from a neuron during baseline and application of the group II mGluR agonist LY354740 (1 μM) in the presence of 1 μM tetrodotoxin; mEPSCs are marked with arrows. (B) Cumulative probability distributions of the inter-mEPSC intervals and mEPSC amplitudes for the cell shown in (A). LY354740 significantly reduces mEPSC frequency (left, p < 0.0001, Kolmogorov–Smirnov test) but not mEPSC amplitude (right, p = 0.999, Kolmogorov–Smirnov test). (C) Event time histograms (bin size: 10 ms, cell in A,B) showing the effect of LY354740 application on mEPSC frequency (left) and amplitude (right). (D) Baseline normalized effect of (0.1 μM, n = 3 cells, or 1 μM, n = 6 cells) on individual cells. LY354740 significantly reduces mEPSC frequency (left, p = 0.019 Kruskal–Wallis test; baseline vs. LY354740 p < 0.05, baseline vs. washout p > 0.05, LY354740 vs. washout p > 0.05, Dunn’s post hoc test, n = 9) but not mEPSC amplitude (right, p = 0.239 Kruskal–Wallis test; n = 9). In some cells, washout could not be analyzed due to change in series resistance (>20% baseline). Numbers denote codes of individual cells (see Table 2). ∗p < 0.05, ∗∗∗∗p < 0.0001.
Discussion
We have demonstrated that activation of group II mGluRs inhibits spontaneous excitatory transmission impinging onto pyramidal neurons in layers 2–3 of the human cerebral cortex. The group II mGluR agonist LY354740 depressed the frequency and amplitude of action potential-dependent sEPSCs but did not alter the holding current of pyramidal cell, although it slightly reduced the cells’ input resistance. It also reduced the frequency but not the amplitude of action potential-independent mEPSCs. Overall, these findings suggest that group II mGluRs act predominantly via a presynaptic effect on glutamate release, rather than via hyperpolarization of layers 2–3 pyramidal cells or postsynaptic effects on ionotropic glutamate receptor conductance. However, we cannot exclude that activation of postsynaptic group II mGluRs in pyramidal cells can lead to network effects. Increases in membrane conductance could lead to membrane hyperpolarization when the cell’s membrane potential is more depolarized and/or to a reduction of the cell’s excitability. If large numbers of pyramidal neurons express group II mGluRs in the human cortex, even a small hyperpolarization or reduction in excitability could alter network dynamics. Our results confirm the negative ‘autoreceptor’ role of this receptor observed in rodent neocortex (e.g., Libri et al., 1997) and hippocampus (e.g., Capogna, 2004). Although group II mGluRs on glutamatergic terminals are activated by glutamate, the source of this transmitter may be either the same terminal that is being suppressed or nearby terminals. In fact, the receptors are widely distributed along the axons and the non-junctional bouton membrane, and glutamate release sites are densely distributed in the neuropil. Therefore, the term ‘autoreceptor’ describes the chemical nature of the terminal and the receptor, and not necessarily a terminal-autonomous regulatory mechanism.
Preclinical studies have demonstrated that group II mGluR agonists exhibit antipsychotic-like properties in animal models of schizophrenia (Stansley and Conn, 2018). However, when these compounds were tested in clinical trials on schizophrenic patients, results were not encouraging (Muguruza et al., 2016). This may be due to patient selection or previous exposure to atypical antipsychotics (Muguruza et al., 2016; Maksymetz et al., 2017). Nonetheless, it is important to acknowledge that rodent animal models do not fully capture the complexities of psychiatric disorders and often show poor predictive power for drug efficacy (Nestler and Hyman, 2010).
Unraveling the cellular effects of group II mGluRs in human cortex could facilitate the identification of causes underlying the poor efficacy of current drugs and help design new, more effective pharmacological treatments. We have tested the effect of a ligand of broad interest for drug development, an agonist for type II mGluRs, on synaptic events recorded from human cortical neurons. Our results demonstrate that this ligand, tested previously in rodents, is effective in inhibiting EPSCs recorded in human pyramidal cells. Future studies using human cortical slices may also help to design novel antipsychotic drugs by shedding light on the physiological effects of PAMs of group II mGluRs (e.g., LY487379 or AZD8529), designed to improve cognition deficits with fewer side effects than more traditional drugs (Singewald et al., 2015).
Our data show that LY354740 inhibited the frequency and amplitude of action potential-dependent sEPSCs as well as the frequency, but not amplitude, of action potential-independent mEPSCs. The latter effect by a drug is usually interpreted as inhibition of spontaneous vesicle fusion and transmitter release (Scanziani et al., 1992). However, LY354740 also appears to trigger postsynaptic effects, as indicated by the small but significant effect of LY354740 on cells’ input resistance. Future studies could confirm the effect of group II mGluRs on neurotransmitter release by examining calcium-dependent release evoked by electrical stimulation of a set of presynaptic fibers. Calcium-dependent and independent neurotransmitter release have been often assumed to share similar mechanisms (Scanziani et al., 1992), although more recent data suggest that the pool of vesicles underlying spontaneous transmitter release can be different from that involved in evoked release (Sara et al., 2005).
In the present study, we could test only a small number of neurons due to limited availability of human cortical tissue. Therefore, we have not explored the subcellular or molecular mechanisms leading to presynaptic depression of glutamatergic transmission. In rodent hippocampus, these receptors are found at pre-terminal axons and on the boutons at some distance from release sites (Shigemoto et al., 1997; Corti et al., 2002), but whether similar localization also occurs in the human cerebral cortex is not known. Based on rodent data, group II mGluR activation could act downstream on N- and P/Q type Ca2+ channels, presynaptic K+ channels, intrinsic release machinery proteins or could be activated by retrograde release of endogenous transmitters from the postsynaptic cells (Niswender and Conn, 2010). Furthermore, our data cannot rule out a contribution of group II mGluRs expressed by pyramidal cells’ dendritic membrane, perhaps at some distance from the soma. Finally, mGlu3 is expressed in human astrocytes where it enhances the uptake of glutamate from the synapse by increasing the expression of glial glutamate transporters (Aronica et al., 2003), and this mechanism could contribute to the inhibition of glutamatergic synaptic events observed in our experiments.
We have only investigated the activation of group II mGluRs by an exogenous ligand. However, it is unclear whether endogenous ligands (likely glutamate) could also modulate neurotransmission. The frequency and amplitude of sEPSCs detected in the presence of the mGluR II/III antagonist CPPG were similar to control conditions, suggesting undetectable levels of group II mGluR activation by endogenous glutamate in acute slices. Future studies should investigate activity-dependent activation of group II mGluRs, as demonstrated in rodents hippocampus (Kew et al., 2001, 2002; Capogna, 2004), which may also be relevant to mechanisms of synaptic plasticity (Tzounopoulos et al., 1998). In the human cortex, group I mGluRs trigger long-term depression of excitatory transmission impinging on fast-spiking GABAergic interneurons (Szegedi et al., 2016), but it is not yet known whether group II mGluRs can mediate analogous effects.
It is yet to be determined whether group II mGluRs depress transmission at all glutamatergic synapses on pyramidal cells or at specific pathways. Spontaneous synaptic events recorded from pyramidal neurons could be mainly due to glutamate released either from thalamus and/or from cortical inputs (Pasquale and Sherman, 2012). Future experiments using selective stimulation of anatomically identified fibers could determine what input(s) physiologically activate group II mGluRs in the human cerebral cortex.
Whether group II mGluR activation leads to depression of most glutamatergic synapses or to suppression of specific pathways, a marked reduction of excitatory transmission in layers 2–3 pyramidal cells is likely to trigger dramatic network effects. Intriguingly, altered activity of cortical neuronal ensembles has been reported in two mouse models of schizophrenia (Hamm et al., 2017), a finding that suggests that group II mGluRs – despite unsuccessful clinical trials to date – could still represent a promising target for this disorder.
It is likely that activation of group II mGluRs causes changes in neurotransmission that are not restricted to glutamatergic synapses onto pyramidal cells. First, these receptors might be located on glutamatergic axons innervating at least some GABAergic interneurons, similar to the modulation of excitability of fast-spiking GABAergic neurons of human cortex by group I mGluRs (Szegedi et al., 2017). Group II mGluRs might be expressed in a cell-type dependent manner, comparable to the expression of mGluR7 (Shigemoto et al., 1996), or other presynaptic metabotropic receptors, e.g., the cannabinoid 1 receptor (Ludanyi et al., 2008). Second, group II mGluRs might also modulate GABAergic transmission (Hayashi et al., 1993; Ohishi et al., 1994). It will be interesting to investigate whether similar mechanisms occur in the human cortex.
Which group II mGluRs were activated in the present experiments? LY354740 is a potent and selective agonist (up to 1 μM) at mGlu2 and mGlu3 receptors with an EC50 of about 10–50 nM in the rat cortex, hippocampus and striatum, and 10 or 30 nM in cells expressing recombinant mGlu2 or mGlu3, respectively (Schoepp et al., 1999). The concentrations used in this study, therefore, were several fold higher than the EC50, in order to ensure activation of mGluRs throughout the entire depth of the slice. Although the concentrations used in this study still predict selectivity over groups I and III mGluRs (EC50: 300 μM and 100 μM, respectively), we did not discriminate between mGlu2 and mGlu3 activation (e.g., Johnson et al., 2013). Thus, future investigations could attempt to discriminate between mGlu2 and mGlu3, also because understanding the different roles of these two receptors could help to design more selective drugs.
The action of group II mGluRs is regulated by development, network events and epileptic-like events in rodents (Doherty et al., 2004). Future studies using tissue closer to pathological focus could test whether pathological processes can cause functional upregulation of receptors.
It is important to acknowledge that the use of tissue from human cerebral cortex of patients subjected to neurosurgery has some methodological limitations. One of these is that the tissue may have some pathological features that remain undetected. We have used cortical tissue from people with epilepsy refractory to medications or from low grade glioma tumor patients (except one patient that was grade III). We have performed the experiments only on cortical tissue that was located outside the focal epileptic region. Accordingly, we have not observed any epileptic-like signal in the human cortical slices used for our study, such as rhythmic spike bursts in current clamp or rhythmic sEPSC bursts in voltage clamp. In the samples obtained from the periphery of diffuse gliomas as assessed by magnetic resonance imaging, variability may be caused by the degree of glial infiltration. Another possible limitation is the variability due to heterogeneity of cortical areas of provenance, different age and sex of the patients, their individual clinical and pharmacological history. Despite this variability, we observed basal functional parameters that were rather homogeneous across samples and patients and consistency in the effects mediated by group II mGluRs. In addition, cortical tissue removed from remote brain tumor sites has been used as control, non-epileptic tissue in a study investigating cellular activities in human epileptic tissue (Jiang et al., 2012).
Conclusion
In conclusion, the present study suggests that the activation of group II mGluRs mainly leads to inhibition of glutamate release at synapses on layers 2–3 pyramidal neurons of human cerebral cortex via presynaptic ‘autoreceptors.’ We have established an experimental framework to test the neurophysiological effects of ligands that are relevant to neuropsychiatric conditions in acute slices of human neocortex. Clarifying the mechanisms of action by these ligands has the potential to shed light on their actions in the human brain and bolster the design of more potent and selective drugs.
Author Contributions
RS, PP, and VA performed neurosurgery. LL, AS, OA, and MHG helped with human tissue collection and regulated procedures. MB, IL, PS, and MC performed the experiments and analyses, and wrote the manuscript with comments from all the authors.
Funding
This work was supported by an ERC grant (ERC-2015-AdG 694988), the Oxford NIHR Biomedical Research Centre, and by the Medical Research Council (MC_UU_12024/4 and MR/R011567/1 to PS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the Oxford Brain Bank, supported by the Medical Research Council (MRC), Brains for Dementia Research (BDR) (Alzheimer Society and Alzheimer Research, United Kingdom), Autistica, United Kingdom, and the NIHR Oxford Biomedical Research Centre. MHG is a Clinical Lecturer funded by the NIHR Academy (National Institute for Health Research Trainee Coordinating Centre) ref CL-2013-13-001. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. We are grateful to Dr. Karri Lamsa for pilot experiments and stimulating discussions that led to this project, Ms. Kristina Wagner, Mr. Michael Howarth, and Ms. Kathryn Newton for excellent technical assistance and to Dr. Jozsef Somogyi for help with microscopy.
Footnotes
References
Anwyl, R. (1999). Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res. Rev. 29, 83–120. doi: 10.1016/S0165-0173(98)00050-2
Aronica, E., Gorter, J. A., Ijlst-Keizers, H., Rozemuller, A. J., Yankaya, B., Leenstra, S., et al. (2003). Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. Eur. J. Neurosci. 17, 2106–2118. doi: 10.1046/j.1460-9568.2003.02657.x
Capogna, M. (2004). Distinct properties of presynaptic group II and III metabotropic glutamate receptor-mediated inhibition of perforant pathway CA1 EPSCs. Eur. J. Neurosci. 19, 2847–2858. doi: 10.1111/j.1460-9568.2004.03378.x
Caraci, F., Nicoletti, F., and Copani, A. (2018). Metabotropic glutamate receptors: the potential for therapeutic applications in Alzheimer ’ s disease. Curr. Opin. Pharmacol. 38, 1–7. doi: 10.1016/j.coph.2017.12.001
Cartmell, J., and Schoepp, D. D. (2002). Regulation of neurotransmitter release bymetabotropic glutamate receptors. J. Neurochem. 75, 889–907. doi: 10.1046/j.1471-4159.2000.0750889.x
Conn, P. J., and Pin, J. (1997). Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 37, 205–237. doi: 10.1146/annurev.pharmtox.37.1.205
Corti, C., Aldegheri, L., Somogyi, P., and Ferraguti, F. (2002). Distribution and synaptic localisation of the metabotropic glutamate receptor 4 (mGluR4) in the rodent CNS. Neuroscience 110, 403–420. doi: 10.1016/S0306-4522(01)00591-7
Di Menna, L., Joffe, M. E., Iacovelli, L., Orlando, R., Lindsley, C. W., Mairesse, J., et al. (2018). Functional partnership between mGlu3 and mGlu5 metabotropic glutamate receptors in the central nervous system. Neuropharmacology 128, 301–313. doi: 10.1016/j.neuropharm.2017.10.026
Doherty, J., and Dingledine, R. (2001). Reduced excitatory drive onto interneurons in the dentate gyrus after status epilepticus. J. Neurosci. 21, 2048–2057. doi: 10.1523/JNEUROSCI.21-06-02048.2001
Doherty, J. J., Alagarsamy, S., Bough, K. J., Conn, P. J., Dingledine, R., and Mott, D. D. (2004). Metabotropic glutamate receptors modulate feedback inhibition in a developmentally regulated manner in rat dentate gyrus. J. Physiol. 2, 395–401. doi: 10.1113/jphysiol.2004.074930
Ferraguti, F. (2018). Metabotropic glutamate receptors as targets for novel anxiolytics. Curr. Opin. Pharmacol. 38, 37–42. doi: 10.1016/j.coph.2018.02.004
Galici, R. (2005). A selective allosteric potentiator of metabotropic glutamate (mGlu) 2 receptors has effects similar to an orthosteric mGlu2/3 receptor agonist in mouse models predictive of antipsychotic activity. J. Pharmacol. Exp. Ther. 315, 1181–1187. doi: 10.1124/jpet.105.091074
Hamm, J. P., Peterka, D. S., Gogos, J. A., and Yuste, R. (2017). Altered cortical ensembles in mouse models of schizophrenia. Neuron 94, 153–167. doi: 10.1016/j.neuron.2017.03.019
Hayashi, Y., Momiyama, A., Takahashi, T., Ohishi, H., Ogawa-Meguro, R., Shigemoto, R., et al. (1993). Role of a metabotropic glutamate receptor in synaptic modulation in the accessory olfactory bulb. Nature 366, 687–690. doi: 10.1038/366687a0
Jiang, M., Zhu, J., Liu, Y., Yang, M., Tian, C., Jiang, S., et al. (2012). Enhancement of asynchronous release from fast-spiking interneuron in human and rat epileptic neocortex. PLoS Biol. 10:e1001324. doi: 10.1371/journal.pbio.1001324
Jin, L. E., Wang, M., Yang, S. T., Yang, Y., Galvin, V. C., Lightbourne, T. C., et al. (2017). MGluR2/3 mechanisms in primate dorsolateral prefrontal cortex: evidence for both presynaptic and postsynaptic actions. Mol. Psychiatry 22, 1615–1625. doi: 10.1038/mp.2016.129
Johnson, P. L., Fitz, S. D., Engleman, E. A., Svensson, K. A., Schkeryantz, J. M., and Shekhar, A. (2013). Group II metabotropic glutamate receptor type 2 allosteric potentiators prevent sodium lactate-induced panic-like response in panic-vulnerable rats. J. Psychopharmacol. 27, 152–161. doi: 10.1177/0269881112454230
Jucker, M. (2010). The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat. Med. 16, 1210–1214. doi: 10.1038/nm.2224
Kew, J. N. C., Ducarre, J., Pflimlin, M., Mutel, V., and Kemp, J. A. (2001). Activity-dependent presynaptic autoinhibition by group II metabotropic glutamate receptors at the perforant path inputs to the dentate gyrus and CA1. Neuropharmacology 40, 20–27. doi: 10.1016/S0028-3908(00)00118-0
Kew, J. N. C., Pflimlin, M. C., Kemp, J. A., and Mutel, V. (2002). Differential regulation of synaptic transmission by mGlu2 and mGlu3 at the perforant path inputs to the dentate gyrus and CA1 revealed in mGlu2 -/- mice. Neuropharmacology 43, 215–221. doi: 10.1016/S0028-3908(02)00084-9
Kintscher, M., Breustedt, J., Miceli, S., Schmitz, D., and Wozny, C. (2012). Group II metabotropic glutamate receptors depress synaptic transmission onto subicular burst firing neurons. PLoS One 7:e45039. doi: 10.1371/journal.pone.0045039
Krystal, J. H., Abi-Saab, W., Perry, E., D’Souza, D. C., Liu, N., Gueorguieva, R., et al. (2005). Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology 179, 303–309. doi: 10.1007/s00213-004-1982-8
Libri, V., Constanti, A., Zibetti, M., and Postlethwaite, M. (1997). Metabotropic glutamate receptor subtypes mediating slow inward tail current (I(ADP)) induction and inhibition of synaptic transmission in olfactory cortical neurones. Br. J. Pharmacol. 120, 1083–1095. doi: 10.1038/sj.bjp.0701021
Ludanyi, A., Eross, L., Czirjak, S., Vajda, J., Halasz, P., Watanabe, M., et al. (2008). Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. J. Neurosci. 28, 2976–2990. doi: 10.1523/JNEUROSCI.4465-07.2008
Maksymetz, J., Moran, S. P., and Conn, P. J. (2017). Targeting metabotropic glutamate receptors for novel treatments of schizophrenia. Mol. Brain 10, 1–19. doi: 10.1186/s13041-017-0293-z
Moghaddam, B., and Adams, B. W. (1998). Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science 281, 1349–1352. doi: 10.1126/science.281.5381.1349
Molinari, F., Cattani, A. A., Mdzomba, J. B., and Aniksztejn, L. (2012). Glutamate transporters control metabotropic glutamate receptors activation to prevent the genesis of paroxysmal burst in the developing hippocampus. Neuroscience 207, 25–36. doi: 10.1016/j.neuroscience.2012.01.036
Molnár, G., Oláh, S., Komlósi, G., Füle, M., Szabadics, J., Varga, C., et al. (2008). Complex events initiated by individual spikes in the human cerebral cortex. PLoS Biol. 6:e222. doi: 10.1371/journal.pbio.0060222
Muguruza, C., Meana, J. J., and Callado, L. F. (2016). Group II Metabotropic glutamate receptors as targets for novel antipsychotic drugs. Front. Pharmacol. 7:130. doi: 10.3389/fphar.2016.00130
Nestler, E. J., and Hyman, S. E. (2010). Animal models of neuropsychiatric disorders. Nat. Neurosci. 13, 1161–1169. doi: 10.1038/nn.2647
Niswender, C. M., and Conn, P. J. (2010). Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322. doi: 10.1146/annurev.pharmtox.011008.145533
Ohishi, H., Ogawa-Meguro, R., Shigemoto, R., Kaneko, T., Nakanishi, S., and Mizuno, N. (1994). Immunohistochemical localization of metabotropic glutamate receptors, mGluR2 and mGluR3, in rat cerebellar cortex. Neuron 13, 55–66. doi: 10.1016/0896-6273(94)90459-6
Pasquale, R. D., and Sherman, S. M. (2012). Modulatory effects of metabotropic glutamate receptors on local cortical circuits. J. Neurosci. 32, 7364–7372. doi: 10.1523/JNEUROSCI.0090-12.2012
Price, C. J., Karayannis, T., and Capogna, M. (2005). Group II and III mGluRs-mediated presynaptic inhibition of EPSCs recorded from hippocampal interneurons of CA1 stratum lacunosum moleculare. Neuropharmacology 49, 45–56. doi: 10.1016/j.neuropharm.2005.05.009
Sara, Y., Virmani, T., Deák, F., Liu, X., and Kavalali, E. T. (2005). An isolated pool of vesicles recycles at rest and drives spontaneous neurotransmission. Neuron 45, 563–573. doi: 10.1016/j.neuron.2004.12.056
Scanziani, M., Capogna, M., Gähwiler, B. H., and Thompson, S. M. (1992). Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron 9, 919–927. doi: 10.1016/0896-6273(92)90244-8
Scanziani, M., Salin, P. A., Vogt, K. E., Malenka, R. C., and Nicoll, R. A. (1997). Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature 385, 630–634. doi: 10.1038/385630a0
Schoepp, D. D., Jane, D. E., and Monn, J. A. (1999). Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology 38, 1431–1476. doi: 10.1016/S0028-3908(99)00092-1
Shigemoto, R., Kinoshita, A., Wada, E., Nomura, S., Ohishi, H., Takada, M., et al. (1997). Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat. Hippocampus 17, 7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997
Shigemoto, R., Kulik, A., Roberts, J. D. B., Ohishi, H., Nusser, Z., Kaneko, T., et al. (1996). Target-cell-specific concentration of a metabotropic glutamate receptor in the presynaptic active zone. Nature 381, 523–525. doi: 10.1038/381523a0
Singewald, N., Schmuckermair, C., Whittle, N., Holmes, A., and Ressler, K. J. (2015). Pharmacology of cognitive enhancers for exposure-based therapy of fear, anxiety and trauma-related disorders. Pharmacol. Ther. 149, 150–190. doi: 10.1016/j.pharmthera.2014.12.004
Spooren, W. P., Gasparini, F., van, der Putten H, Koller, M., Nakanishi, S., and Kuhn, R. (2000). Lack of effect of LY314582 (a group 2 metabotropic glutamate receptor agonist) on phencyclidine-induced locomotor activity in metabotropic glutamate receptor 2 knockout mice. Eur. J. Pharmacol. 397, R1–R2. doi: 10.1016/S0014-2999(00)00269-7
Stansley, B. J., and Conn, P. J. (2018). The therapeutic potential of metabotropic glutamate receptor modulation for schizophrenia. Curr. Opin. Pharmacol. 38, 31–36. doi: 10.1016/j.coph.2018.02.003
Ster, J., Mateos, J. M., Grewe, B. F., Coiret, G., Corti, C., Corsi, M., et al. (2011). Enhancement of CA3 hippocampal network activity by activation of group II metabotropic glutamate receptors. Proc. Natl. Acad. Sci. 1, 8–12. doi: 10.1073/pnas.1100548108
Swanson, C. J., Bures, M., Johnson, M. P., Linden, A. M., Monn, J. A., and Schoepp, D. D. (2005). Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nat. Rev. Drug Discov. 4, 131–144. doi: 10.1038/nrd1630
Szegedi, V., Molnár, G., Paizs, M., Csakvari, E., Barzó, P., Tamás, G., et al. (2017). High-precision fast-spiking basket cell discharges during complex events in the human neocortex. eNeuro 4:ENEURO.0260-17.2017. doi: 10.1523/ENEURO.0260-17.2017
Szegedi, V., Paizs, M., Csakvari, E., Molnar, G., Barzo, P., Tamas, G., et al. (2016). Plasticity in single axon glutamatergic connection to GABAergic interneurons regulates complex events in the human neocortex. PLoS Biol. 14:e2000237. doi: 10.1371/journal.pbio.2000237
Tzounopoulos, T., Janz, R., Su, T. C., Nicoll, R. A., and Malenka, R. C. (1998). A Role for cAMP in long-term depression at hippocampal mossy fiber synapses. Neuron 21, 837–845. doi: 10.1016/S0896-6273(00)80599-1
Keywords: presynaptic receptor, glutamatergic, EPSC, cognitive enhancer, transmitter release, epilepsy, mGluR, human cortex
Citation: Bocchio M, Lukacs IP, Stacey R, Plaha P, Apostolopoulos V, Livermore L, Sen A, Ansorge O, Gillies MJ, Somogyi P and Capogna M (2019) Group II Metabotropic Glutamate Receptors Mediate Presynaptic Inhibition of Excitatory Transmission in Pyramidal Neurons of the Human Cerebral Cortex. Front. Cell. Neurosci. 12:508. doi: 10.3389/fncel.2018.00508
Received: 29 October 2018; Accepted: 07 December 2018;
Published: 08 January 2019.
Edited by:
Enrico Cherubini, Scuola Internazionale Superiore di Studi Avanzati (SISSA), ItalyReviewed by:
Annalisa Scimemi, University at Albany, United StatesHuib Mansvelder, VU University Amsterdam, Netherlands
Copyright © 2019 Bocchio, Lukacs, Stacey, Plaha, Apostolopoulos, Livermore, Sen, Ansorge, Gillies, Somogyi and Capogna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Somogyi, peter.somogyi@pharm.ox.ac.uk Marco Capogna, marco.capogna@biomed.au.dk
†Present address: Marco Bocchio, INMED, INSERM U1249, Marseille, France