Outward currents contributing to inspiratory burst termination in preBötzinger Complex neurons of neonatal mice studied in vitro
- Department of Applied Science, The College of William and Mary, Williamsburg, VA, USA
We studied preBötzinger Complex (preBötC) inspiratory interneurons to determine the cellular mechanisms that influence burst termination in a mammalian central pattern generator. Neonatal mouse slice preparations that retain preBötC neurons generate respiratory motor rhythms in vitro. Inspiratory-related bursts rely on inward currents that flux Na+, thus outward currents coupled to Na+ accumulation are logical candidates for assisting in, or causing, burst termination. We examined Na+/K+ ATPase electrogenic pump current (Ipump), Na+-dependent K+ current (IK–Na), and ATP-dependent K+ current (IK–ATP). The pharmacological blockade of Ipump, IK–Na, or IK–ATP caused pathological depolarization akin to a burst that cannot terminate, which impeded respiratory rhythm generation and reversibly stopped motor output. By simulating inspiratory bursts with current-step commands in synaptically isolated preBötC neurons, we determined that each current generates approximately 3–8 mV of transient post-burst hyperpolarization that decays in 50–1600 ms. Ipump, IK–Na, and – to a lesser extent – IK–ATP contribute to terminating inspiratory bursts in the context of respiratory rhythm generation by responding to activity dependent cues such as Na+ accumulation.
Introduction
Breathing in mammals depends on inspiratory rhythms that originate in the preBötzinger Complex (preBötC) of the ventral medulla (Tan et al., 2008; Bouvier et al., 2010; Gray et al., 2010). The preBötC is unique from an experimental perspective because the cellular mechanisms of rhythm generation can be investigated in slices that retain inspiratory motor function in vitro.
The inward currents that underlie inspiratory bursts include the Ca2+-activated non-specific cation current (ICAN) and the persistent Na+ current (INaP; Feldman and Del Negro, 2006; Pace et al., 2007; Koizumi and Smith, 2008). INaP and ICAN flux Na+ during inspiratory bursts, so burst termination may depend on activity-dependent outward currents linked to Na+ accumulation, as well as the related depletion of ATP due to Na+ pumping. Na/K ATPase electrogenic pump current (Ipump) has been linked to burst termination in spinal locomotor and oral-motor central pattern generator (CPG) networks (Ballerini et al., 1997; Del Negro et al., 1999; Darbon et al., 2003) and midbrain dopamine neurons (Johnson et al., 1992). In addition to generating net outward current, Na/K ATPase pumps consume ATP, which evokes ATP-sensitive K+ current (IK–ATP) in respiratory neurons (Pierrefiche et al., 1996; Mironov et al., 1998; Haller et al., 2001), and thus could influence burst termination. Na+-dependent K+ current (IK–Na) could also be involved because Slack-like IK–Na responds directly to cytosolic Na+ in the lamprey CPG (Yuan et al., 2003; Wallen et al., 2007), and Slick-like IK–Na activates in response to Na+ and is inhibited by ATP (Bhattacharjee et al., 2003). Therefore, depending on subunit composition, IK–Na could act in conjunction with both Ipump and IK–ATP to assist in burst termination.
Burst termination may involve pre- and post-synaptic factors (Rubin et al., 2009), but this study focuses exclusively on post-synaptic mechanisms. Moreover, we concentrate on factors coupled to Na+ because Ca2+-dependent K+ currents (IK–Ca), which often contribute to burst termination in vertebrate CPGs (Grillner, 2003, 2006), are not essential for respiratory rhythmogenesis under standard conditions in vitro (Onimaru et al., 2003; Crowder et al., 2007). Here we show that blocking Ipump, IK–Na, and – to a lesser extent – IK–ATP each causes depolarization and steady-state attenuation of inspiratory bursts, which impedes respiratory rhythmogenesis. In synaptically isolated preBötC neurons, we simulated bursts in current clamp and measured 3–8 mV of transient hyperpolarization attributable to Ipump, IK–Na, and IK–ATP, which contribute to active phase termination during cycles of respiratory network activity in vitro. The data suggest that Na+ and ATP-dependent outward currents contribute to inspiratory burst termination and support rhythmic function of the preBötC.
Materials and Methods
The Institutional Animal Care and Use Committee at The College of William and Mary approved all protocols. Neonatal C57BL/6 mice of post-natal ages 0–4 days were dissected in standard artificial cerebrospinal fluid (ACSF) containing (in mM): 124 NaCl, 3 KCl, 1.5 CaCl2, 1 MgSO4, 25 NaHCO3, 0.5 NaH2PO4, and 30 dextrose, equilibrated with 95% O2–5% CO2 (pH 7.3). Transverse slices (550-μm thick) that expose the preBötC on the rostral surface (Ruangkittisakul et al., 2010) were perfused at 4 ml/min with 27°C ACSF and visualized with Koehler illumination and videomicroscopy. ACSF K+ concentration was raised to 9 mM and respiratory motor output was recorded from the XII nerve roots with suction electrodes.
Patch pipettes (3–4 MΩ) were filled with solution containing (in mM): 140 K-Gluconate, 5 NaCl, 1 EGTA, 10 Hepes, 2 Mg-ATP, and 0.3 Na-GTP (pH 7.3, KOH). Whole-cell current-clamp recordings were performed with a Dagan IX2-700 amplifier (Minneapolis, MN, USA). Data were acquired digitally at 4 kHz after 1 kHz filtering.
We bath-applied strophanthidin (10 μM, in ethanol, 0.01% final concentration in ACSF) to block Ipump, quinidine (100 μM, in DMSO, 0.01% final concentration in ACSF) to block IK–Na, and glibenclamide (500 μM, in DMSO, 0.05% final concentration in ACSF) to block IK–ATP (Sigma-Aldrich, St. Louis, MO, USA). Dose–response curves for quinidine (1, 10, 100, 500, and 1000 μM) and glibenclamide (100, 500, 1000, and 2000 μM) were measured in three or more slices at each dose. The final concentration of ethanol and DMSO never exceeded 0.1% in the ACSF. To assess respiratory network function in dose–response experiments, we measured XII burst amplitude and area, which were normalized with respect to control. We also measured XII burst half width and respiratory frequency, which are reported in millisecond and hertz.
The amplitude, area, and half width of inspiratory bursts were measured during whole-cell recordings in the context of respiratory rhythmic activity. We normalized the burst measurements in steady-state antagonist conditions with respect to control to compute the relative attenuation of the inspiratory burst. The normalization procedure enabled us to study the effects of antagonists across multiple experiments.
We applied 500-ms current pulses to simulate inspiratory-like bursts in synaptically isolated neurons. A cocktail of ionotropic synaptic receptor antagonists was used to stop network activity including: 50 μM DL-2-amino-5-phosphonovaleric acid (APV), 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), 5 μM picrotoxin, and 5 μM strychnine. The amplitude of the current injection was adjusted to evoke at least nine spikes per pulse (∼20 Hz). A transient phase of hyperpolarization was always recorded at the offset of the current pulse, which we used as a model of burst termination in preBötC neurons during endogenous respiratory rhythm generation. The current pulse was then repeated in the presence of strophanthidin, quinidine, or glibenclamide. In most cases, the transient phase of hyperpolarization was reduced, suggesting the removal of an activity-dependent outward current. The antagonist-sensitive response was obtained by digital subtraction, to measure the contribution of Ipump, IK–Na, or IK–ATP to the transient post-pulse hyperpolarization evoked by the original 500-ms current pulse.
Two-tailed t-tests were used to assess changes in XII output (amplitude, area, half width, and frequency), inspiratory burst magnitude (amplitude, area, and half width), as well as the amplitude of transient hyperpolarization responses evoked in synaptically isolated neurons.
Results
Contribution of Ipump
We exposed rhythmically active slice preparations to strophanthidin (10 μM) while recording XII output and inspiratory preBötC neurons (Figure 1). Bias current was adjusted to establish a −65 mV baseline in control, and was not adjusted after strophanthidin application. Within 6 min the XII nerve discharged tonically (n = 11 of 12 slices tested), preBötC neurons depolarized 8.8 ± 1.5 mV (n = 7), and respiratory frequency sped up significantly from 0.23 ± 0.03 to 0.41 ± 0.07 Hz (p < 0.05, n = 7). In addition to increasing frequency, blocking Ipump decreased inspiratory burst amplitude by 52 ± 12% (p < 0.005), decreased area by 68 ± 7% (p < 0.05), and decreased burst half width by 50%, from 245 ± 46 to 122 ± 29 ms (p < 0.05, all n = 7).
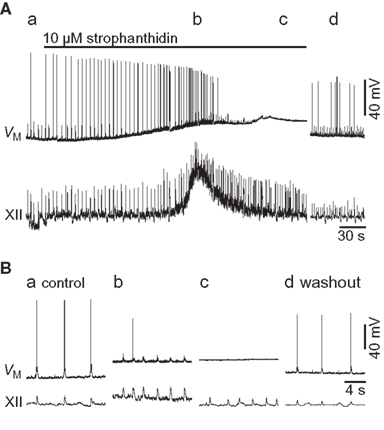
Figure 1. Effects of 10-μM strophanthidin on a preBötC inspiratory neuron (VM) and XII motor output. (A) An overview of the entire experiment showing control (a, 0 min), early strophanthidin (b, 3 min), late strophanthidin (c, 7 min), and washout (d, 35 min). The solid bar shows strophanthidin application. (B) Individual traces (15 s) of the sections shows in (A).
Strophanthidin-induced tonic XII discharge generally lasted 30 s. After a total exposure of 7.7 ± 4.8 min, more than half of the preBötC neurons tested ceased firing and remained quiescent at a membrane potential above −55 mV (n = 4 of 7 tested). In one case the XII output maintained rhythmic function even after the recorded cell fell quiescent (e.g., Figure 1). XII output stopped in four preparations after 8.3 ± 6.0 min of continuous exposure, at which time the recorded preBötC neuron was also quiescent at a depolarized state, without rhythmic drive. Three slices maintained rhythmogenesis throughout the experiment. These effects reversed after 20–40 min of washout.
The experiments shown in Figure 1 provide limited insights into the burst-terminating role of the Na/K ATPase pump because all neurons experienced a profound shift in baseline membrane potential, and inspiratory-related synaptic drive potentials were modified in terms of frequency and magnitude. Thus we sought to study Ipump without the confounding network effects that sped up the rhythm yet diminished synaptic drive. We synaptically isolated preBötC neurons using a cocktail of ionotropic receptor antagonists (Materials and Methods), and simulated burst-like responses with current pulses. Baseline membrane potential was biased to −70 mV, which is very close to EK in 9 mM K+ ACSF (i.e., −71 mV) and thus minimizes K+ currents. We evoked burst-like spiking in control and in the presence of 10-μM strophanthidin. There was a transient phase of hyperpolarization at burst offset in control, which was diminished in the presence of strophanthidin. We computed the Ipump contribution to this post-burst hyperpolarization by digital subtraction of the traces (Figure 2A). The inset in Figure 2A shows the digital subtraction of the raw traces, which represents the membrane trajectory attributable to Ipump for this representative cell. The magnitude of the Ipump mediated post-burst hyperpolarization varied from −14 to −1 mV, Figure 2A (inset) shows a single sweep measuring −3.75 mV, which decayed back to baseline in 1.8 s. In general, Ipump was responsible for −6.4 ± 1.7 mV of post-burst hyperpolarization that decayed back to baseline in 1.6 ± 0.6 s (n = 32 subtractions in nine neurons).
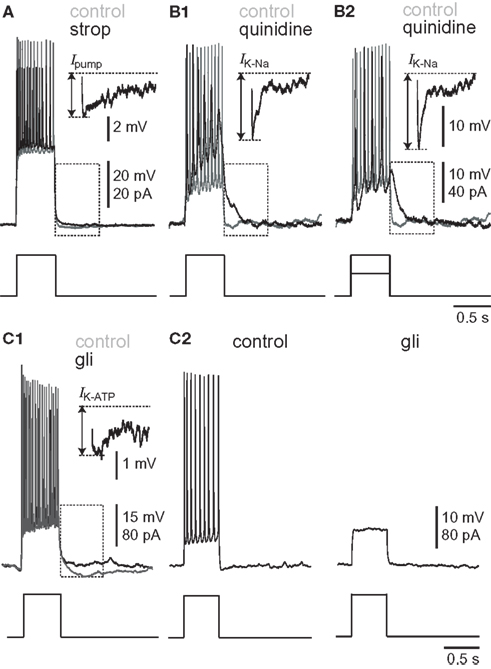
Figure 2. Burst-like responses to current-pulse injections in synaptically isolated preBötC neurons. Broken-line boxes highlight the subtractions that are displayed as insets in each condition. Separate voltage scale bars are given for the inset and (upper) and main voltage trace (lower). Each sweep lasts 3 s; each inset lasts 0.5 s. The broken lines show baseline membrane potential (top) and the nadir of the post-burst hyperpolarization (bottom). The arrow indicates the amplitude of the post-burst hyperpolarization attributable to the pharmacological removal of each outward current. (A) Effects of 10-μM strophanthidin. Baseline membrane potential was adjusted to −70 mV in control (gray) and strophanthidin (black). (B) Effects of 100-μM quinidine. Baseline membrane potential was adjusted to −50 mV in control (gray) and quinidine (black). (B1) Shows the same current magnitude in control and strophanthidin. (B2) shows the response to a diminished current magnitude in quinidine conditions, which was adjusted to evoke approximately the same magnitude of depolarization and spiking as in control. (C) Effects of 500-μM glibenclamide. Baseline membrane potential was adjusted to −50 mV in control (gray) and glibenclamide (black) for (C1), separate black traces for (C2).
Contribution of Ik–Na
We studied the pharmacology of IK–Na by measuring quinidine dose–response curves for respiratory XII output in vitro (Figure 3, Table 1, n ≥ 3 separate slices tested at each concentration). At 1 and 10 μM we observed no significant effects. One hundred micromolar quinidine, and all higher concentrations, significantly attenuated every measure of XII output: amplitude, area, half width, and frequency, before stopping rhythmogenesis. We chose 100 μM for subsequent whole-cell recording experiments because 100 μM exceeds the half-maximal inhibitory concentration (IC50) of 20 μM and its effects on XII output were reversible.
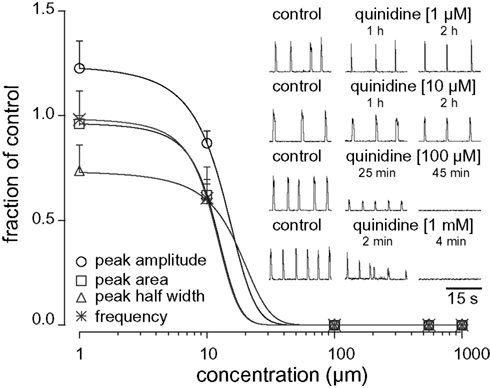
Figure 3. Dose–response curves for quinidine plot XII peak amplitude (circle), area (square), half width (triangle), and respiratory frequency in vitro (asterisk) versus concentration. Inset traces (30 s) show XII activity at the time points and drug conditions as shown.
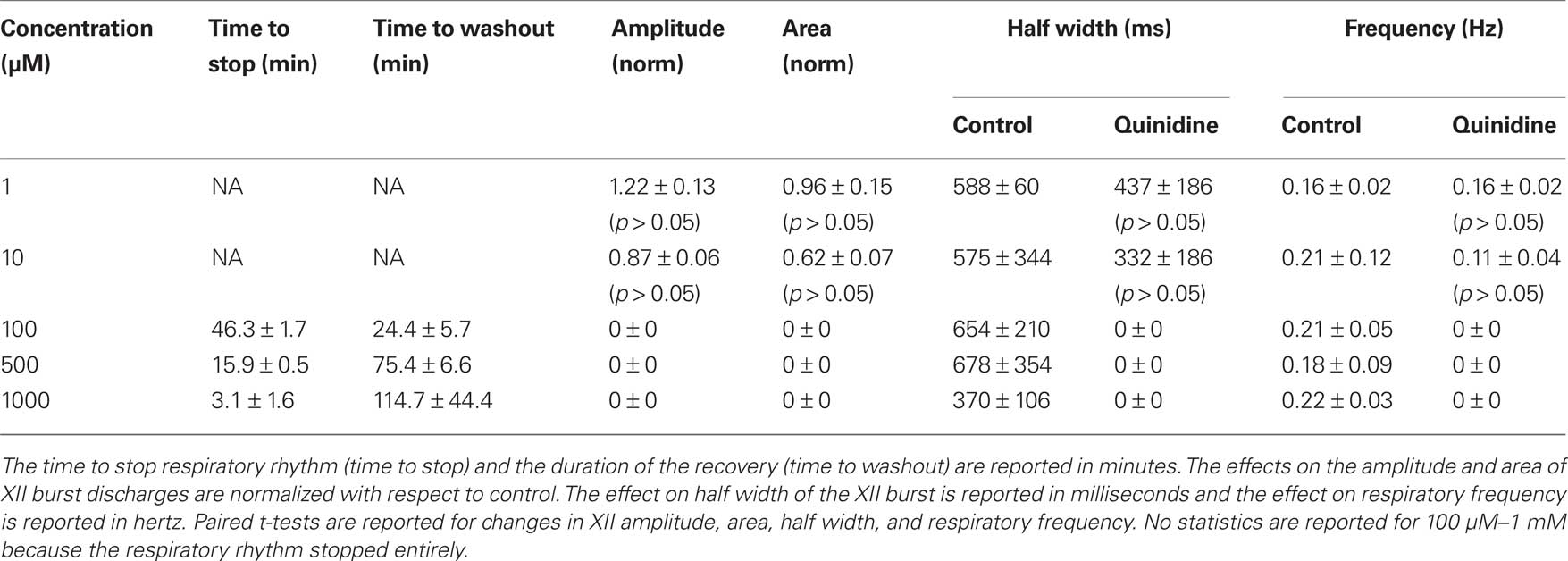
Table 1. Effects of quinidine on respiratory activity in vitro. Slices with respiratory rhythmic motor output were exposed to quinidine at concentrations shown below.
We exposed rhythmic slices to 100 μM quinidine while recording inspiratory preBötC neurons and monitoring XII output (Figure 4). Membrane potential was biased to −65 mV initially and not adjusted further during quinidine application and washout. Ultimately, quinidine depolarized preBötC neurons by 14.1 ± 5.4 mV (n = 4), but had a biphasic effect on inspiratory bursts prior to steady state. First, the decremental burst pattern changed to an incrementing burst pattern with greater amplitude and area (Figures 4 and 5). Baseline membrane potential hyperpolarized −5 to −10 mV during this phase, likely due to accumulation of other activity-dependent outward currents responding to enhanced Na+ influx during the quinidine-enhanced inspiratory bursts. This transient phase lasted less than 2 min so we could not compute burst statistics. After an average of 14.8 ± 4.0 min, the inspiratory burst amplitude decreased by 65 ± 18% and burst area decreased by 74 ± 8% (both p < 0.001, n = 4). Half width increased significantly in two cases from 75.1 ± 13.4 to 95.6 ± 13.4 ms (p < 0.005), decreased significantly in one case from 391 to 99 ms (p < 0.05), and did not change significantly in a fourth case. All inspiratory burst activity at the cellular and network (XII) level stopped after 36.0 ± 5.4 min of quinidine exposure. In a 2 min window preceding rhythmic cessation, respiratory frequency significantly slowed down to 0.07 ± 0.02 Hz, compared to the original control frequency of 0.20 ± 0.03 Hz (p < 0.024, n = 4).
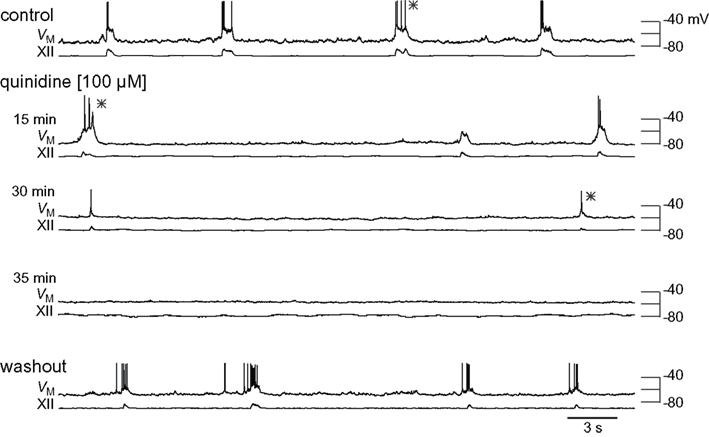
Figure 4. Effects of quinidine on inspiratory bursts and XII motor output. The intracellular and XII responses are shown in control, quinidine (15, 30, and 35 min), and washout. All sweeps (30 s) are scaled to the absolute voltage calibration shown at right. Asterisks (*) denote inspiratory bursts shown at higher resolution in Figure 5.
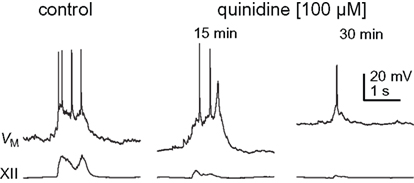
Figure 5. Inspiratory bursts in control and quinidine (15 and 30 min) conditions. Traces are taken from Figure 4, as denoted by asterisks.
We studied IK–Na in synaptically isolated preBötC neurons to circumvent the network-level effects of quinidine, which change inspiratory burst dynamics. Again, we applied current steps to simulate bursts. Since IK–Na is a K+ current we adjusted bias to establish a −50 mV baseline, which exceeds EK by approximately 20 mV. The contribution of IK–Na was obtained by subtraction (control – 100 μM quinidine, Figure 2B). Applying the same magnitude of step current in the presence of quinidine caused depolarization block of spiking because quinidine increases input impedance (Figure 2B1). Therefore, we repeated the experiment but lowered the magnitude of the pulse to evoke burst-like repetitive spiking similar to control (Figure 2B2). In response to the lower magnitude step command, IK–Na caused a −7.7 ± 1.1 mV hyperpolarization lasting 0.54 ± 0.14 s (11 subtractions in three neurons). In response to the larger amplitude step (equivalent to the pulse magnitude in control) IK–Na caused a −7.6 ± 1.2 mV hyperpolarization lasting 0.74 ± 0.13 s (53 subtractions in five neurons). Despite the disparity in the magnitudes of the injected currents and the amount of depolarization they evoked, the amplitude and duration of the IK–Na-mediated hyperpolarization responses were statistically indistinguishable (p > 0.05).
Contribution of Ik–Atp
We studied the pharmacology of IK–ATP by measuring glibenclamide dose–response curves for respiratory XII output in vitro (Figure 6, Table 2, n ≥ 3 separate slices tested at each concentration). One hundred micromolar glibenclamide significantly slowed respiratory frequency from 0.29 ± 0.09 to 0.26 ± 0.05 (p < 0.05) but had no other significant effect on XII output. Five hundred micromolar significantly decreased XII burst amplitude by 54 ± 23% and attenuated burst area by 47 ± 23% (both p < 0.001). XII half width decreased from 305 ± 59 to 163 ± 44 ms, but this was not statistically significant. Respiratory frequency slowed significantly from 0.26 ± 0.03 to 0.06 ± 0.03 Hz (p < 0.01). One to two millimolar significantly perturbed all measures of XII output before ultimately stopping respiratory rhythmogenesis.
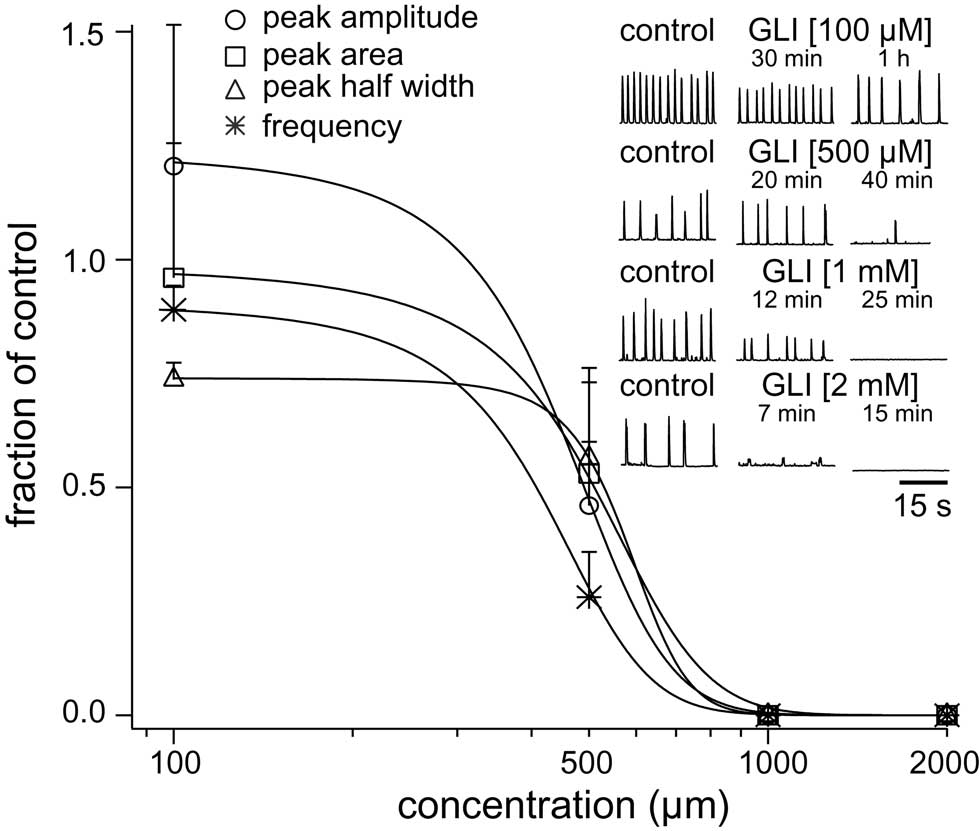
Figure 6. Dose–response curves for glibenclamide (GLI) plot XII peak amplitude (circle), area (square), half width (triangle), and respiratory frequency in vitro (asterisk) versus concentration. Inset traces (30 s) show XII activity at the time points and drug conditions as shown.
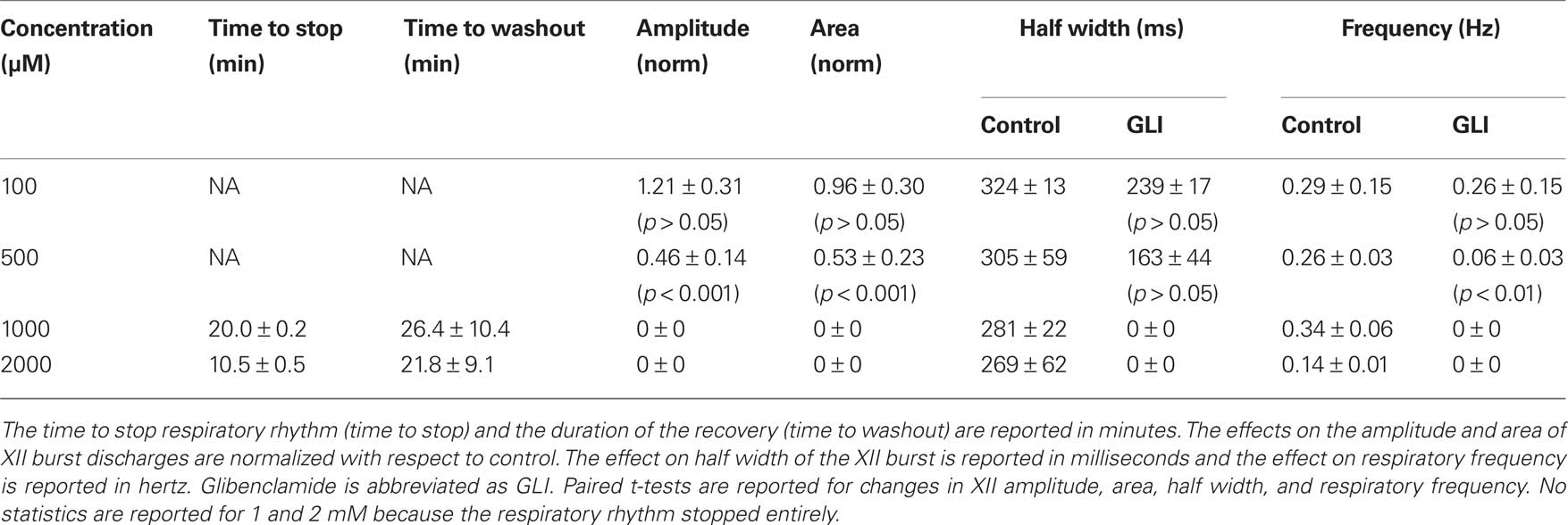
Table 2. Effects of glibenclamide on respiratory activity in vitro. Slices with respiratory rhythmic motor output were exposed to glibenclamide at concentrations shown below.
We performed whole-cell recordings in the context of network activity while applying the IC50, 500-μM. Glibenclamide precipitated unless stirred constantly, which often caused transient blockage of the ACSF perfusion lines and perturbed laminar solution flow through the recording chamber. This limitation does not affect XII recordings but often dislodges whole-cell recording pipettes and precludes long-lasting recordings that include control, glibenclamide, and washout conditions. Nevertheless, Figure 7 shows a complete experiment in which the neuron depolarized nearly 30 mV, inspiratory burst amplitude decreased by 49%, burst area decreased by 55%, and half width decreased from 185 to 85 ms. Respiratory frequency slowed from 0.21 to 0.09 Hz. Inspiratory bursts transiently increased (Figure 7 row 2), but subsequently decreased (rows 3 and 4). Ultimately the neuron locked into a non-terminating plateau-like state akin to burst that cannot self-terminate. This is shown in row 4 of Figure 7, where membrane potential is ∼0 mV prior to the start of washout (<44 min). In washout conditions we terminated the plateau with −20 pA of current bias, and inspiratory bursts recovered substantially. XII output returned to control.
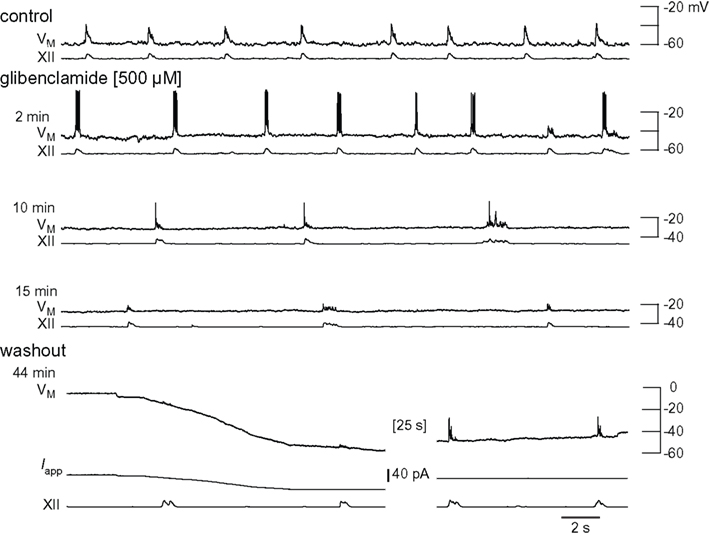
Figure 7. Effects of glibenclamide on inspiratory bursts and XII motor output. Intracellular and XII responses are shown in control and glibenclamide (2, 10, and 15 min). All sweeps (30 s) are scaled to an absolute voltage calibration shown at right. Applied current (Iapp) is shown for washout conditions because it was used to bias the membrane potential transiently.
We also measured IK–ATP using step currents. Solubility problems prematurely terminated 12 out of 15 experiments, but we obtained reliable measurements in control, glibenclamide, and washout conditions in three cases. Current pulses were applied from a baseline membrane potential of −50 mV to measure IK–ATP, to maximize the driving force for K+ current. The IK–ATP contribution was obtained by subtraction (500 μM glibenclamide – control, Figure 2C). Two neurons showed a glibenclamide-sensitive post-burst hyperpolarization (Figure 2C1), which measured −3.2 ± 0.6 mV and decayed in 41.7 ± 7.5 ms (57 subtractions in three cells). One neuron tested showed no effect. After prolonged exposure, glibenclamide abolished repetitive spiking in two of three neurons tested (Figure 2C2).
Discussion
Post-synaptic mechanisms that contribute to inspiratory burst termination include Ipump, IK–Na, and IK–ATP, outward currents evoked by the accumulation of Na+ or the depletion of ATP due to Na+ pumping. Since inspiratory bursts employ ICAN and INaP as inward charge carriers (Del Negro et al., 2005; Pace et al., 2007; Koizumi and Smith, 2008), both of which flux Na+, outward currents that are directly or indirectly sensitive to Na+ are well suited for a role in terminating the active phase of the respiratory cycle.
The pharmacological blockade of Ipump, IK–Na, and IK–ATP in vitro reversibly depolarized the baseline membrane potential, which led to depolarization block of spiking, and then ultimately the attenuation of inspiratory bursts. Respiratory frequency either decreased (quinidine and glibenclamide) or increased (strophanthidin) before stopping altogether. These data suggest direct effects on the rhythmogenic mechanism or effects on sources of excitatory drive to the rhythm generator, e.g., tonic serotonergic drive to the preBötC from the midline raphé (Ptak et al., 2009), rather than effects on premotor or motor neurons (Koizumi et al., 2008). These data further suggest that the activity dependent outward currents Ipump and IK–Na, as well as IK–ATP to a lesser extent, play a significant role in maintaining proper inspiratory burst dynamics, and that these dynamics are important to maintain rhythmogenic function in preBötC.
Caveats and Limitations of Pharmacology
Bath-applied drugs affect the entire respiratory network, so it is hard to separate their effects on cellular burst-terminating mechanisms from network-wide effects on membrane excitability and synaptic drive. To minimize this inherent ambiguity, we performed pharmacological experiments in rhythmically active slices (Figures 1, 4, and 7) and in synaptically isolated neurons (Figure 2). We infer the roles of Ipump, IK–Na, and IK–ATP from both experiments.
We selected antagonist concentration(s) to block Ipump, IK–Na, and IK–ATP while minimizing undesired effects on membrane or network properties.
Strophanthidin blocks Ipump at low micromolar concentrations in spinal locomotor interneurons (10 μM, Ballerini et al., 1997; Darbon et al., 2003), brain stem masticatory motoneurons (4 μM, Del Negro et al., 1999), and midbrain dopamine neurons (2 μM, Johnson et al., 1992). We previously used 10 μM to block Ipump in a study of respiratory frequency regulation (Del Negro et al., 2009).
One millimolar quinidine blocks IK–Na in lamprey CPG interneurons (Wallen et al., 2007) and cloned human IK–Na (Bhattacharjee et al., 2003). We applied 100 μM because this concentration exceeded the IC50 (20 μM, Figure 3), was reversible, and higher doses increased input impedance.
Five hundred micromolar is the IC50 for glibenclamide effects on respiratory motor output (Figure 6). A prior report shows that 50 μM glibenclamide enhances inspiratory bursts, but does not change respiratory rate (Haller et al., 2001). Therefore, the extent of the IK–ATP blockade at 50 μM is unclear. Intracellular application of 100 μM glibenclamide fully occluded K–ATP channel agonist effects in expiratory neurons (Pierrefiche et al., 1996). However, intracellular application does not allow one to assess the cellular and network effects of the drug simultaneously, which was necessary for the present study (e.g., Figures 1, 4, and 7). Bath-applied 500-μM glibenclamide affected respiratory rhythm in vitro, but also abolished repetitive spiking capabilities in two out of three neurons tested (Figure 2C2). Therefore, we cannot exclude the possibility that glibenclamide impedes rhythmogenesis, at least in part, because it impairs the ability to spike repetitively.
Inspiratory Burst Termination: The Role of Calcium-Dependent Potassium Currents?
Burst termination in vertebrate CPGs often involves IK–Ca (Grillner, 2003, 2006). Large- (BK) and small-conductance (SK) forms of IK–Ca influence respiratory regulation and gasping in vivo and in vitro (Büsselberg et al., 2003; Zavala-Tecuapetla et al., 2008), but IK–Ca is not essential for rhythmogenesis in vitro, and removing IK–Ca from the preBötC causes no consistent change in inspiratory burst characteristics (Onimaru et al., 2003; Crowder et al., 2007).
Inspiratory Burst Termination: Activity Dependent Outward Currents
A model of rhythmogenesis in the preBötC posits that synaptic depression and activity dependent outward currents terminate inspiratory bursts (Feldman and Del Negro, 2006; Rubin et al., 2009). Eliminating or attenuating outward currents results in depolarization block and bursts that cannot self-terminate. This experimental study focuses on “activity dependent” outward currents that respond to Na+ accumulation or ATP depletion due to Na+ pumping, which occurs during the inspiratory burst. The role(s) of synaptic depression, voltage-dependent outward currents, as well as the deactivation or inactivation of inward currents (ICAN and INaP) are beyond the scope of this work but by no means ruled out as contributors to burst termination in the preBötC.
In response to blocking Ipump, IK–Na, and IK–ATP, the net decrease in burst magnitude and duration was the opposite outcome predicted by the model. The straightforward model prediction does not take into account the reaction of the network to pharmacological blockade of outward currents. In the real system, depolarization of membrane potentials, combined with depolarization block of spiking, exert persistent and pathological effects on network function. Individual preBötC neurons become impaired at quiescent depolarized states and thus cannot sustain phasic inspiratory burst activity. Initially, blocking Ipump and IK–ATP increased respiratory frequency (Figures 1 and 7). This can be attributed to constituent rhythm-generating neurons depolarizing closer to burst threshold and firing at a faster rate, which may speed the process of recurrent excitation in the rhythm-generating network. Recurrent excitation influences the interburst interval and respiratory frequency (Rekling et al., 1996; Rekling and Feldman, 1998; Mironov, 2008, 2009). However, as preBötC neurons continually depolarize (which was common in steady state, see Figures 1, 4, and 7), then fewer and fewer neurons would be able to participate in rhythmogenesis. The net synaptic drive would decrease, resulting in smaller bursts. This general attrition from the network may ultimately abolish the ability to generate rhythmic activity and XII motor output.
Role of Ipump
Ipump has a tonic component that is constitutively active to maintain ion gradients, as well as a transient component that is activity dependent. Transient Ipump has been measured in motoneurons (Del Negro et al., 1999), dopamine neurons (Johnson et al., 1992), and preBötC neurons (Del Negro et al., 2009), all of which show rhythmic bursting activity in networks. The transient component of Ipump can reach 600 pA and last 50–150 ms (Del Negro et al., 2009). Here the transient component of Ipump caused a −6 mV after-hyperpolarization lasting 0.1–1.6 s (Figure 2A). These results suggest that the transient Ipump exerts a profound but short-lasting effect that may help stop the inspiratory burst and then rapidly deactivate. In the context of ongoing respiratory rhythm, blocking Ipump causes, on average, 8.8 mV of depolarization and depolarization block of spiking. This steady depolarization of the baseline membrane potential we largely attribute to loss of the tonic component of Ipump, which can be as high as 200 pA during the post-inspiratory/expiratory phase in preBötC neurons (Del Negro et al., 2009). The tonic discharge seen in XII motor output after 5 min of exposure to strophanthidin may result from a large number of preBötC neurons depolarizing collectively. It may also be partially attributable to depolarization of XII motoneurons by Na/K ATPase blockade. Pathological depolarization due to loss of the tonic component of Ipump, combined with loss of the transient component of Ipump may give rise to a population of highly depolarized preBötC neurons that can no longer participate in rhythmogenesis. In the experiments where rhythmogenesis persisted in strophanthidin (e.g., Figure 1), we speculate that fewer preBötC neurons are involved in rhythm generation since the network output, as assessed by XII output, was greatly diminished.
Role of IK–Na and IK–ATP
IK–Na and IK–ATP function like the transient component of Ipump. Removing IK–Na and IK–ATP also caused depolarization, depolarization block, and eventually rhythm cessation. Removal of IK–Na (Figures 4 and 5) enhanced inspiratory bursts transiently, consistent with model predictions (as we described above). The inspiratory burst showed an incrementing pattern, in contrast to the decremental pattern observed before blocking IK–Na. At this early stage of the experiment, it is unlikely that quinidine affected the whole rhythmogenic network, so its effects on the burst-terminating mechanisms of the recorded neuron were more apparent. Figure 7 showed qualitatively similar data for IK–ATP blockade, in which the first 2 min showed a transient enhancement of burst magnitude probably due to selective removal of the activity dependent outward current before the drug affected the entire rhythm-generating network. A prior report showed that 50 μM glibenclamide enhanced inspiratory burst magnitude and duration, which is also consistent with a role for IK–ATP in burst termination (Haller et al., 2001).
Physiological Significance
Simulated bursts allowed us to measure post-burst hyperpolarization responses that reflect Ipump, IK–Na, and IK–ATP. IK–Na may play the largest role in burst termination because it caused −7.7 mV of post-burst hyperpolarization lasting 0.50–0.75 s. This is consistent with the almost universally overlooked role of IK–Na in causing delayed outward currents in a wide range of mammalian neurons (Budelli et al., 2009). In the preBötC specifically, Ipump may also play a significant role. It generated −6.4 mV of post-burst hyperpolarization lasting 1.6 s. The present results are consistent with the possible role of Ipump suggested by preliminary experiments and modeling studies (Del Negro et al., 2009; Rubin et al., 2009). Since burst generation and termination appear linked to Na+, it is logical that both of these Na+-activated currents play a larger role in termination. IK–ATP may be the least significant in terms of magnitude; it caused −3 mV of hyperpolarization lasting 0.4 s, and in some cases may not be involved at all, since in one out of three neurons tested showed no evidence of IK–ATP.
Most in vitro experiments that permit detailed cellular and systems level experiments are performed on neonatal and sometimes embryonic tissues. The cellular mechanisms of burst generation and termination may continue to change during post-natal development and into adulthood. In particular, the role of synaptic inhibition may become more important to promote inspiratory–expiratory phase transitions (Manzke et al., 2010).
In neonatal (Del Negro et al., 2009) and adult (Richter, 1982, among others) rodents, burst termination is accomplished within ∼1 s in inspiratory neurons. The mechanism may depend on pre-synaptic factors (Rubin et al., 2009) as well as outward currents not necessarily coupled to Na+ accumulation. Nevertheless, the present data indicate that inspiratory burst termination involves active outward currents such as Ipump and IK–Na, as well as IK–ATP to a lesser extent, which should be considered when studying and modeling the preBötC.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by NIH 1R01HL104127-01 (PI: Christopher A. Del Negro). Authors Rebecca A. Krey, Adam M. Goodreau, and Thomas B. Arnold received support by from the Biological Sciences Research and Training program for undergraduates, funded by a grant to The College of William and Mary from the Howard Hughes Medical Institute. The authors thank John A. Hayes, Jens C. Rekling, and Gilles Fortin for critical discussions and editing.
References
Ballerini, L., Bracci, E., and Nistri, A. (1997). Pharmacological block of the electrogenic sodium pump disrupts rhythmic bursting induced by strychnine and bicuculline in the neonatal rat spinal cord. J. Neurophysiol. 77, 17–23.
Bhattacharjee, A., Joiner, W. J., Wu, M., Yang, Y., Sigworth, F. J., and Kaczmarek, L. K. (2003). Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J. Neurosci. 23, 11681–11691.
Bouvier, J., Thoby-Brisson, M., Renier, N., Dubreuil, V., Ericson, J., Champagnat, J., Pierani, A., Chedotal, A., and Fortin, G. (2010). Hindbrain interneurons and axon guidance signaling critical for breathing. Nat. Neurosci. 13, 1066–1074.
Budelli, G., Hage, T. A., Wei, A., Rojas, P., Jong, Y. J., O’Malley, K., and Salkoff, L. (2009). Na+-activated K+ channels express a large delayed outward current in neurons during normal physiology. Nat. Neurosci. 12, 745–750.
Büsselberg, D., Bischoff, A. M., and Richter, D. W. (2003). A combined blockade of glycine and calcium-dependent potassium channels abolishes the respiratory rhythm. Neuroscience 122, 831–841.
Crowder, E. A., Saha, M. S., Pace, R. W., Zhang, H., Prestwich, G. D., and Del Negro, C. A. (2007). Phosphatidylinositol 4,5-bisphosphate regulates inspiratory burst activity in the neonatal mouse preBotzinger complex. J. Physiol. 582, 1047–1058.
Darbon, P., Tscherter, A., Yvon, C., and Streit, J. (2003). The role of the electrogenic Na/K pump in disinhibition-induced bursting in cultured spinal networks. J. Neurophysiol. 90, 3119–3129.
Del Negro, C. A., Hsiao, C. F., and Chandler, S. H. (1999). Outward currents influencing bursting dynamics in guinea pig trigeminal motoneurons. J. Neurophysiol. 81, 1478–1485.
Del Negro, C. A., Kam, K., Hayes, J. A., and Feldman, J. L. (2009). Asymmetric control of inspiratory and expiratory phases by excitability in the respiratory network of neonatal mice in vitro. J. Physiol. 587, 1217–1231.
Del Negro, C. A., Morgado-Valle, C., Hayes, J. A., Mackay, D. D., Pace, R. W., Crowder, E. A., and Feldman, J. L. (2005). Sodium and calcium current-mediated pacemaker neurons and respiratory rhythm generation. J. Neurosci. 25, 446–453.
Feldman, J. L., and Del Negro, C. A. (2006). Looking for inspiration: new perspectives on respiratory rhythm. Nat. Rev. Neurosci. 7, 232–242.
Gray, P. A., Hayes, J. A., Ling, G., Llona, I., Tupai, S., Picardo, M. C., Ross, S., Hirata, T., Corbin, J. G., Eugenin, J., and Del Negro, C. A. (2010). Developmental origin of preBötzinger complex respiratory neurons. J. Neurosci. 30, 14883–14895.
Grillner, S. (2003). The motor infrastructure: from ion channels to neuronal networks. Nat. Rev. Neurosci. 4, 573–586.
Grillner, S. (2006). Biological pattern generation: the cellular and computational logic of networks in motion. Neuron 52, 751–766.
Haller, M., Mironov, S. L., Karschin, A., and Richter, D. W. (2001). Dynamic activation of K(ATP) channels in rhythmically active neurons. J. Physiol. 537, 69–81.
Johnson, S. W., Seutin, V., and North, R. A. (1992). Burst firing in dopamine neurons induced by N-methyl-d-aspartate: role of electrogenic sodium pump. Science 258, 665–667.
Koizumi, H., and Smith, J. C. (2008). Persistent Na+ and K+-dominated leak currents contribute to respiratory rhythm generation in the pre-Bötzinger complex in vitro. J. Neurosci. 28, 1773–1785.
Koizumi, H., Wilson, C. G., Wong, S., Yamanishi, T., Koshiya, N., and Smith, J. C. (2008). Functional imaging, spatial reconstruction, and biophysical analysis of a respiratory motor circuit isolated in vitro. J. Neurosci. 28, 2353–2365.
Manzke, T., Niebert, M., Koch, U. R., Caley, A., Vogelgesang, S., Hulsmann, S., Ponimaskin, E., Muller, U., Smart, T. G., Harvey, R. J., and Richter, D. W. (2010). Serotonin receptor 1A-modulated phosphorylation of glycine receptor alpha3 controls breathing in mice. J. Clin. Invest. 120, 4118–4128.
Mironov, S. (2009). Respiratory circuits: function, mechanisms, topology, and pathology. Neuroscientist 15, 194–208.
Mironov, S. L. (2008). Metabotropic glutamate receptors activate dendritic calcium waves and TRPM channels which drive rhythmic respiratory patterns in mice. J. Physiol. 586, 2277–2291.
Mironov, S. L., Langohr, K., Haller, M., and Richter, D. W. (1998). Hypoxia activates ATP-dependent potassium channels in inspiratory neurones of neonatal mice. J. Physiol. 509(Pt 3), 755–766.
Onimaru, H., Ballanyi, K., and Homma, I. (2003). Contribution of Ca2+-dependent conductances to membrane potential fluctuations of medullary respiratory neurons of newborn rats in vitro. J. Physiol. 552, 727–741.
Pace, R. W., Mackay, D. D., Feldman, J. L., and Del Negro, C. A. (2007). Inspiratory bursts in the preBötzinger complex depend on a calcium-activated nonspecific cationic current linked to glutamate receptors. J. Physiol. 582, 113–125.
Pierrefiche, O., Bischoff, A. M., and Richter, D. W. (1996). ATP-sensitive K+ channels are functional in expiratory neurones of normoxic cats. J. Physiol. 494 (Pt 2), 399–409.
Ptak, K., Yamanishi, T., Aungst, J., Milescu, L. S., Zhang, R., Richerson, G. B., and Smith, J. C. (2009). Raphe neurons stimulate respiratory circuit activity by multiple mechanisms via endogenously released serotonin and substance P. J. Neurosci. 29, 3720–3737.
Rekling, J. C., Champagnat, J., and Denavit-Saubie, M. (1996). Electroresponsive properties and membrane potential trajectories of three types of inspiratory neurons in the newborn mouse brain stem in vitro. J. Neurophysiol. 75, 795–810.
Rekling, J. C., and Feldman, J. L. (1998). PreBötzinger complex and pacemaker neurons: hypothesized site and kernel for respiratory rhythm generation. Annu. Rev. Physiol. 60, 385–405.
Richter, D. W. (1982). Generation and maintenance of the respiratory rhythm. J. Exp. Biol. 100, 93–107.
Ruangkittisakul, A., Panaitescu, B., and Ballanyi, K. (2010). K+ and Ca2+ dependence of inspiratory-related rhythm in novel “calibrated” mouse brainstem slices. Respir. Physiol. Neurobiol. (in press).
Rubin, J. E., Hayes, J. A., Mendenhall, J. L., and Del Negro, C. A. (2009). Calcium-activated nonspecific cation current and synaptic depression promote network-dependent burst oscillations. Proc. Natl. Acad. Sci. U.S.A. 106, 2939–2944.
Tan, W., Janczewski, W. A., Yang, P., Shao, X. M., Callaway, E. M., and Feldman, J. L. (2008). Silencing preBotzinger complex somatostatin-expressing neurons induces persistent apnea in awake rat. Nat. Neurosci. 11, 538–540.
Wallen, P., Robertson, B., Cangiano, L., Low, P., Bhattacharjee, A., Kaczmarek, L. K., and Grillner, S. (2007). Sodium-dependent potassium channels of a Slack-like subtype contribute to the slow afterhyperpolarization in lamprey spinal neurons. J. Physiol. 585, 75–90.
Yuan, A., Santi, C. M., Wei, A., Wang, Z. W., Pollak, K., Nonet, M., Kaczmarek, L., Crowder, C. M., and Salkoff, L. (2003). The sodium-activated potassium channel is encoded by a member of the Slo gene family. Neuron 37, 765–773.
Keywords: central pattern generation, breathing, respiration, brainstem, rhythmic networks, Na/K ATPase, sodium-dependent potassium current, ATP-dependent potassium current
Citation: Krey RA, Goodreau AM, Arnold TB and Del Negro CA (2010) Outward currents contributing to inspiratory burst termination in preBötzinger Complex neurons of neonatal mice studied in vitro. Front. Neural Circuits 4:124. doi: 10.3389/fncir.2010.00124
Received: 29 September 2010;
Paper pending published: 20 October 2010;
Accepted: 11 November 2010;
Published online: 29 November 2010.
Edited by:
Paul S. Katz, Georgia State University, USAReviewed by:
Stephen M. Johnson, University of Wisconsin, USAGregory D. Funk, University of Alberta, Canada
Copyright: © 2010 Krey, Goodreau, Arnold and Del Negro. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Christopher A. Del Negro, Department of Applied Science, The College of William and Mary, McGlothlin-Street Hall, Room 318, Williamsburg, VA 23187-8795, USA. e-mail: cadeln@wm.edu