Pericyte-mediated regulation of capillary diameter: a component of neurovascular coupling in health and disease
- Department of Neuroscience, Physiology and Pharmacology, University College London, London, UK
Because regional blood flow increases in association with the increased metabolic demand generated by localized increases in neural activity, functional imaging researchers often assume that changes in blood flow are an accurate read-out of changes in underlying neural activity. An understanding of the mechanisms that link changes in neural activity to changes in blood flow is crucial for assessing the validity of this assumption, and for understanding the processes that can go wrong during disease states such as ischaemic stroke. Many studies have investigated the mechanisms of neurovascular regulation in arterioles but other evidence suggests that blood flow regulation can also occur in capillaries, because of the presence of contractile cells, pericytes, on the capillary wall. Here we review the evidence that pericytes can modulate capillary diameter in response to neuronal activity and assess the likely importance of neurovascular regulation at the capillary level for functional imaging experiments. We also discuss evidence suggesting that pericytes are particularly sensitive to damage during pathological insults such as ischaemia, Alzheimer’s disease and diabetic retinopathy, and consider the potential impact that pericyte dysfunction might have on the development of therapeutic interventions and on the interpretation of functional imaging data in these disorders.
Pericytes as Contractile Cells
Pericytes are found on almost all capillaries, as well as on small arterioles and venules. They are small cells located on the outside of the vessel, between the endothelial cell layer and the parenchyma. They are separated from the parenchyma by the basal lamina, a thin layer of which also interposes between the pericyte and endothelial cells (red layer in Figure 1 B; Frank et al., 1987 ). Pericytes vary morphologically depending on the vascular bed and position along the arterio-venous axis, but in general send out primary projections along the vessel in each direction from the soma, from which secondary and even tertiary processes project around the vessel (Figure 1 A; Shepro and Morel, 1993 ). They seem to have important roles in a range of functions, including angiogenesis, vessel stabilization, endothelial cell regulation and maintenance of the blood-brain barrier (Hirschi and D’Amore, 1996 ; Lai and Kuo, 2005 ). Pericytes also exhibit macrophage-like activity (Thomas, 1999 ) and may be multipotent precursors for several different cell types, forming neurons and glia in the CNS and forming liver and skin cells in those tissues (Dore-Duffy et al., 2006 , 2008 ). Long ago, however, the perivascular location and morphology of pericytes led to the suggestion that they might be contractile cells, involved in regulation of capillary blood flow in response to vasoactive agents and neural activity (Dore, 1923 ). It has taken over 100 years to show that this is indeed the case, and important questions still remain as to the precise role of these cells in the co-ordination of changes in blood flow in response to altered neural activity.
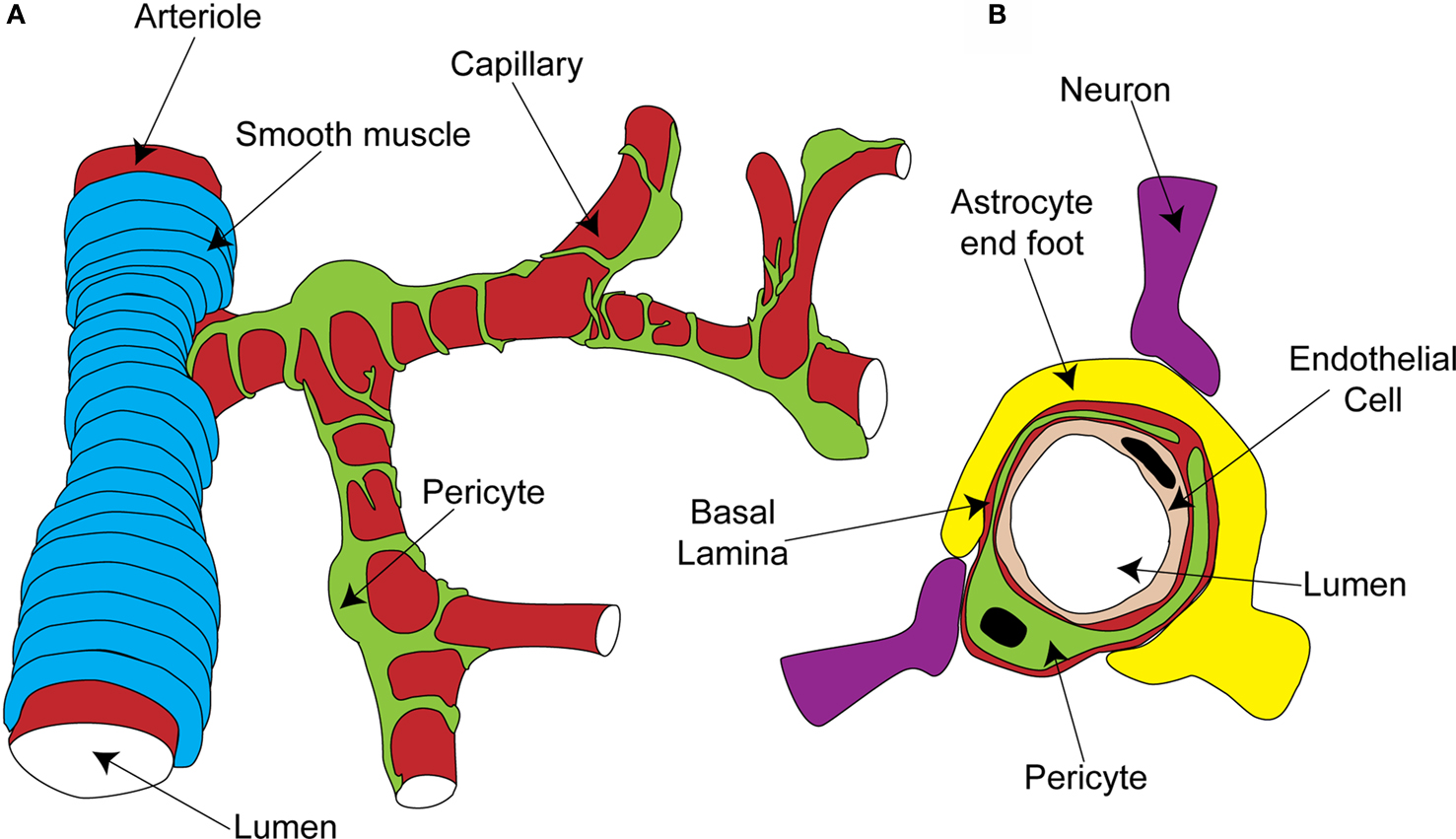
Figure 1. Organisation of the capillary neurovascular unit. (A) Rings of smooth muscle encircle arterioles, while pericytes send processes along and around capillaries, without fully covering the vessel. (B) Pericytes are located outside the endothelial cells and are separated from them and the parenchyma by a layer of basal lamina. In the parenchyma, astrocyte end-feet and neuronal terminals are closely associated with the capillary.
The morphology and location of pericytes provided the first clues that they might have a role in blood flow regulation. Other aspects of CNS pericyte anatomy lend further support to this idea. In the central nervous system, there are more pericytes per endothelial cell than in other areas of the body (Frank et al., 1987 ; Shepro and Morel, 1993 ), suggesting that pericytes might play a particularly important role in the nervous system, where regional blood flow control is particularly important. Neuronal terminals which contain dopamine (Favard et al., 1990 ; Krimer et al., 1998 ), GABA (Gragera et al., 1993 ), vasoactive intestinal peptide (VIP; Benagiano et al., 1996 ), acetylcholine (Arneric et al., 1988 ) and nitric oxide (NO; Roufail et al., 1995 ) are closely apposed to capillaries and pericytes suggesting that, as for arterioles (Sato and Sato, 1990 ; Krimer et al., 1998 ; Cauli et al., 2004 ), capillary blood flow could be modulated by neuronal input (Figure 1 B). For arterioles, glutamate released during neuronal activity plays a critical role in signaling to the blood vessel a requirement for increased local blood flow (Faraci and Breese, 1993 ; Mathiesen et al., 1998 ). Astrocytes are a major target for this glutamate signal, metabotropic glutamate receptor activation producing Ca2+ rises in their end-feet, which surround arterioles. This stimulates the production of vasoactive metabolites of arachidonic acid [prostaglandin E2 (PGE2) and epoxyeicosotrienoic acids (EETs)] which diffuse to the arteriolar smooth muscle and produce vasodilation (Zonta et al., 2003 ; Metea and Newman, 2006 ; Gordon et al., 2008 ). Astrocyte end-feet surround capillaries to a similar extent as they do arterioles (perhaps even more densely; Maynard et al., 1957 ) and so are in a prime position to transmit signals from neurons to capillaries (Figure 1 B).
Anatomical evidence also supports the view that pericytes are contractile. Localized constrictions can be observed in corrosion casts of cat spinal cord capillaries, where pericytes express the smooth muscle-specific isoform of actin (αSMA: Toribatake et al., 1997 ). A subpopulation of retinal and brain pericytes also express αSMA, with stronger expression in pericytes on capillaries which branch directly from arterioles and venules, than in pericytes on mid-capillary regions (Nehls and Drenckhahn, 1991 ), though αSMA is expressed in some of these mid-capillary cells (Bandopadhyay et al., 2001 ). In peripheral tissues, other contractile proteins such as tropomyosin have been found to follow the same pattern of expression as αSMA (Joyce et al., 1985 ), suggesting that this will be the case in the brain as well. Electron micrographs of brain pericytes reveal that they contain microfilaments resembling actin- and myosin-containing muscle fibers (Le Beux and Willemot, 1978 ; Ho, 1985 ), and immunohistochemistry shows that activation of endothelin-1 (ET-1) receptors to raise the calcium concentration in cultured pericytes produces alignment of F-actin and intermediate filaments, which coincides with constriction (Dehouck et al., 1997 ). Pericytes do not express exactly the same contractile machinery as smooth muscle, however. For example, they lack the calcium-binding protein calponin, which regulates contractile function in smooth muscle cells (Bandopadhyay et al., 2001 ).
Much evidence that these pericyte muscle fibers are functional comes from studies of cultured retinal pericytes, in which pericyte surface area or the degree of wrinkling of the silicone surface on which the cells are grown serve as an indication of pericyte tone. Cultured pericytes constrict (or increase wrinkling) in response to some endothelial-derived vasoactive agents, such as ET-1, thromboxane A2 and angiotensin II (Dodge et al., 1991 ; Matsugi et al., 1997a ), but dilate in response to others, such as prostacyclin (PGI2; Dodge et al., 1991 ). Consistent with potential neural, as well as endothelial, control of pericyte tone, catecholamines (which are present in neurons innervating capillaries) can modulate cultured pericyte tone, with serotonin, histamine and noradrenaline all constricting cultured pericytes (Kelley et al., 1988 ; Markhotina et al., 2007 ). Other vasoactive molecules which are produced in neurons dilate cultured pericytes, such as VIP (Markhotina et al., 2007 ), NO (Haefliger et al., 1994 ), which can be produced by neurons or the endothelium, or adenosine (Matsugi et al., 1997b ), which is a breakdown product of ATP and is produced during conditions of high metabolic demand. Adenosine dilates arterioles as well and has been proposed as a messenger by which increased blood flow could be directed to areas of increased metabolic demand (Dirnagl et al., 1994 ). Cultured pericytes also dilate in response to a more acid extracellular pH (Chen and Anderson, 1997 ), which is another consequence of increased energy use (Chesler and Kaila, 1992 ), making the pH sensitivity of pericyte tone a further potential mechanism for balancing capillary blood flow with metabolic demand.
Studies on cultured pericytes therefore support a contractile role for these cells, but it seems that expression of contractile proteins may be increased by culturing as in situ only a subpopulation of pericytes express αSMA (Nehls and Drenckhahn, 1991 ; Bandopadhyay et al., 2001 ), while in culture all pericytes can stain positive for muscle-specific actins (Herman and D’Amore, 1985 ). Such differences in αSMA expression might be paralleled by changes in expression levels of other proteins, reducing the potential physiological relevance of these studies. However, many of the findings from cultured CNS pericytes have been replicated using acute ex vivo preparations (e.g. tissue slices, freshly isolated microvessels or whole mount retinae). For example, angiotensin II and endothelin I both constrict retinal pericytes in situ (Schonfelder et al., 1998 ) and extracellular decreases in pH dilate them (Reber et al., 2003 ), while noradrenaline constricts pericytes in cerebellar slices (Peppiatt et al., 2006 ). In addition NO generates hyperpolarizing currents that would be expected to relax pericytes on freshly isolated retinal capillaries (Sakagami et al., 2001a ). Some differences with cultured cells have been observed, however, as histamine dilates rather than constricts retinal pericytes in situ (Schonfelder et al., 1998 ). This study also found that bradykinin and cholinergic agonists dilated retinal pericytes. Other new discoveries from using acute tissue preparations include the constriction of retinal pericytes to GABA antagonists (Peppiatt et al., 2006 ) and, particularly interestingly, the dilation of cerebellar pericytes to glutamate (Peppiatt et al., 2006 ). This finding suggests that active neurons could signal to capillaries in the same manner as to arterioles, with glutamate generating vasoactive molecules such as NO, PGE2 or EETs in neurons or astrocytes which instruct pericytes to dilate capillaries and increase blood flow.
The suggestion from cultured pericytes that the metabolic state of the tissue might be important in their response (Chen and Anderson, 1997 ; Matsugi et al., 1997b ) is backed up by acute preparations, as adenosine, a product of ATP breakdown, generates hyperpolarizing currents that would be expected to relax pericytes (Li and Puro, 2001 ). Additionally, lactate, the production of which is increased by neuronal activity, produces a constriction of retinal pericytes on isolated capillaries in high oxygen (when presumably there is no need for an increased oxygen supply) but a dilation in low oxygen (Yamanishi et al., 2006 ), when an increased oxygen supply may be required. Similarly, PDGF-B, the release of which from endothelial cells is critical for pericyte function (Bjarnegard et al., 2004 ), produces a constriction of retinal pericytes in control conditions but a dilation during ischaemia (Sakagami et al., 2001b ). In conditions of metabolic demand, ATP is released from neurons both physiologically and when the energy supply is disrupted during ischaemia (Fields and Burnstock, 2006 ). In high oxygen, extracellular ATP constricts pericytes on retinal capillaries when in situ (Peppiatt et al., 2006 ) or when vessels are isolated from surrounding neural tissue (Kawamura et al., 2003 ), but it is not known whether this is switched to a dilation at low oxygen concentrations (as occurs for the effect of glutamate on arterioles: Gordon et al., 2008 ).
Potentially therefore, pericytes could couple neural activity to increased capillary blood flow via two broad mechanisms: either neural activity could directly produce vasoactive molecules (e.g. NO, VIP, PGE2,, noradrenaline) to modulate pericyte tone, or the decrease in oxygen level and increase in lactate and adenosine levels produced by the increased energy use by active neurons could directly dilate pericytes and increase nutrient supply. A recent in vivo study suggests, however, that at least changes in oxygen level in the tissue do not directly regulate neurovascular coupling, because when cortex was kept at a high oxygen concentration by perfusion with hyperbaric oxygen, blood flow responses to forepaw stimulation or cortical spreading depression were not affected (Lindauer et al., 2010 ). If glucose rather than oxygen is limiting for ATP production, however, the metabolic state of the tissue could still feed back to regulate blood flow via changes in the levels of metabolites such as lactate or adenosine. Furthermore, oxygen-dependent regulation may still occur in conditions of ischaemia or hypoxia.
Mechanisms of Regulation of Pericyte Tone
What are the pathways by which pericytes can respond to neurotransmitters and vasoactive agents with an alteration in their contractile tone?
In general, smooth muscle contraction can be triggered by an elevated intracellular calcium concentration, leading to calcium- and calmodulin-dependent activation of myosin light chain kinase (MLCK, Figure 2 A). This generates contraction by phosphorylating myosin light chain (MLC) and prompting its interaction with αSMA (Webb, 2003 ). Conversely, relaxation of smooth muscle is favoured by low calcium levels and decreased MLCK activity, and by the action of myosin light chain phosphatase (MLCP; in its active, unphosphorylated form), which decreases the phosphorylation of MLC and therefore inhibits its interaction with αSMA (Webb, 2003 ).
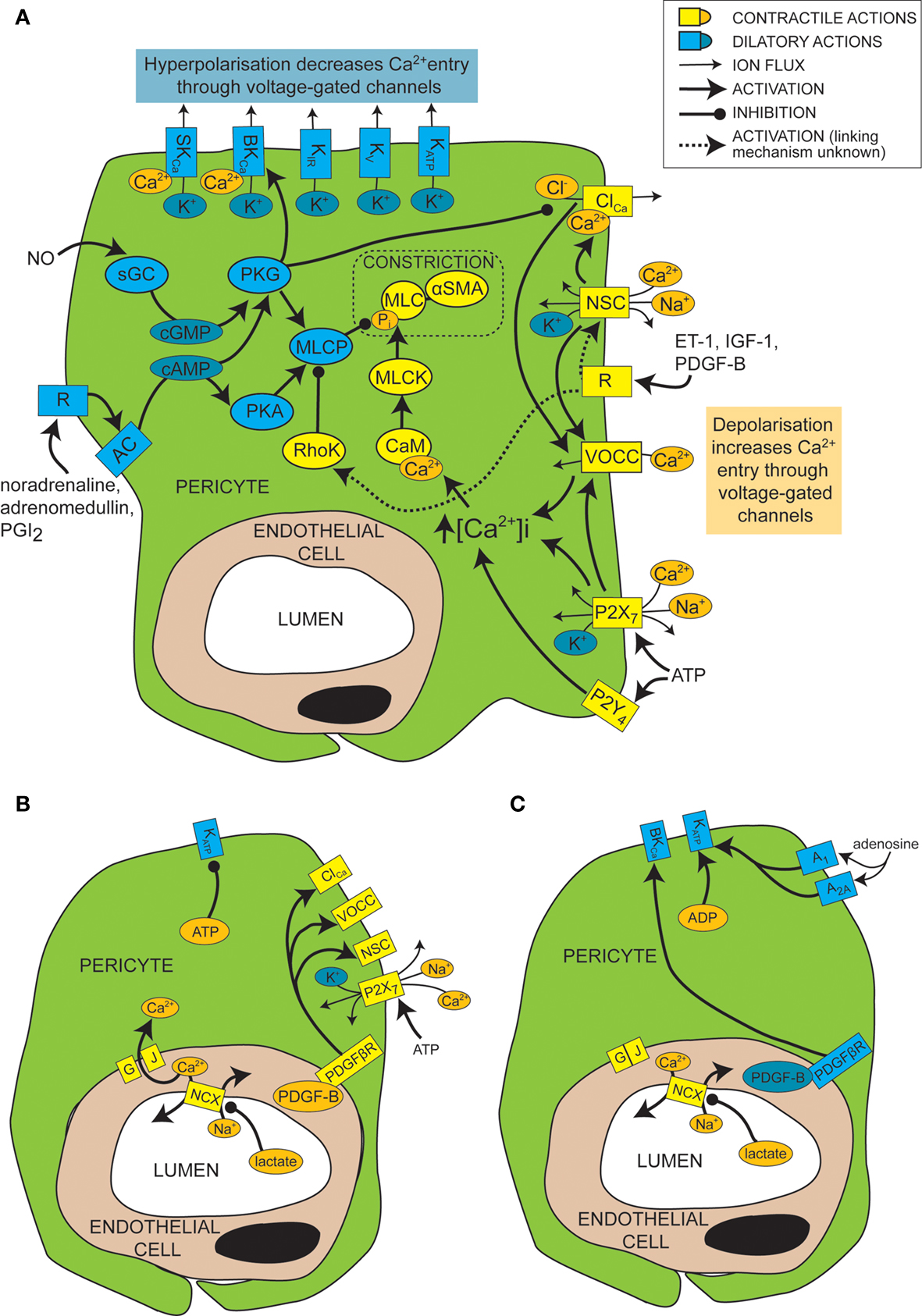
Figure 2. Mechanisms of control of capillary tone. (A) Molecular mechanisms that link vasoactive molecules acting on receptors (labelled “R”) to pericyte constriction (yellow and orange shapes) or dilation (blue shapes). Rectangles are membrane proteins, while ovals are cytosolic species. SKCa, small conductance, calcium-activated potassium channel; BKCa, large conductance, calcium-activated potassium channel; KIR, inwardly rectifying potassium channel; KV, voltage-gated potassium channel; KATP, ATP-sensitive potassium channel; ClCa, calcium-activated chloride channel; NSC, non-specific cation channels; R, ligand-binding receptor; VOCC, voltage-operated calcium channel; MLC, myosin light chain; αSMA, alpha smooth muscle actin; MLCK, myosin light chain kinase; CaM, calmodulin; RhoK, Rho kinase; sGC, soluble guanylyl cyclase; PKG, protein kinase G; AC, adenylyl cyclase; PKA, protein kinase A; MLCP, myosin light chain phosphatase; ET-1, endothelin-1; IGF-1, insulin-like growth factor 1; PDGF-B, platelet-derived growth factor-B; PGI2, prostacyclin. (B) When there is a plentiful supply of O2 and ATP, stimuli such as lactate, PDGF-B and intracellular ATP favour pericyte constriction. GJ, gap junction; PDGFβR, PDGF-B receptor. (C) When O2 and/or ATP are low, lactate, PDGF-B and high intracellular ADP levels favour dilation.
Pericyte Constriction
Like for vascular smooth muscle cells, pericyte contraction is controlled by the intracellular calcium concentration (Sakagami et al., 1999 , 2001a ; Sugiyama et al., 2005 ). For example, the constriction that is produced by electrically stimulating pericytes is blocked by removing extracellular calcium (Peppiatt et al., 2006 ). Vasoconstrictors and vasodilators such as ET-1 and NO have been used to elucidate, by patch clamp experiments, the electrophysiological changes that control intracellular calcium levels in cultured or freshly isolated retinal pericytes. Some vasoconstrictors are thought to bring about an intracellular calcium rise by depolarizing the cell membrane from the resting voltage, usually between −35 and −60 mV, to a voltage which activates L-type voltage-operated calcium channels (VOCCs; see Figure 2 A; Sakagami et al., 1999 , 2001a ). Binding of several vasoconstrictors, such as ET-1 (Kawamura et al., 2002 ), insulin-like growth factor-1 (IGF-1; Sakagami et al., 1999 and PDGF-B; Sakagami et al., 2001b ), to their receptors leads to activation of non-specific cation channels (NSCs) that depolarize the cells and trigger calcium entry through VOCCs (Figure 2 A). Calcium entry also opens calcium-activated chloride (ClCa) channels, which, due to the high intracellular chloride concentration of pericytes, allow chloride to leave the cell, producing a further depolarization allowing further activation of VOCCs (Kawamura et al., 2002 ). Voltage clamp recordings show that ClCa channels in pericytes open transiently and repetitively (Sakagami et al., 1999 ). This characteristic may help to maximise the calcium influx through VOCCs, as desensitization of the VOCCs is minimized by reducing the time each channel is open (Sakagami et al., 1999 ). VOCCs are also thought to be activated by the depolarization caused by ATP binding to P2X7 receptors, which are themselves calcium-permeable (Figure 2 A; Sugiyama et al., 2005 ). ATP also increases the intracellular calcium concentration via P2Y4 receptor activation (Kawamura et al., 2003 ).
In smooth muscle, vasoactive stimuli (e.g. ET-1) can increase constriction by modifying the phosphorylation state of MLCP, via pathways that can include Rho kinase (Figure 2 A; Hilgers and Webb, 2005 ). On phosphorylation, MLCP becomes inactive and is unable to remove the phosphate from the MLC (Webb, 2003 ). This pathway also affects pericyte function as over-expression of Rho-GTPase in pericytes leads to increased constriction (shown by wrinkling of the silicon upon which they were cultured), while inhibition of Rho kinase reduces constriction (Kutcher et al., 2007 ).
As well as the transient events discussed above, neurons also induce more long-term changes in pericytes by release of glutamate, subsequent activation of astrocyte metabotropic glutamate receptors and an increase of NFATc3 nuclear accumulation in pericytes (Filosa et al., 2007 ). NFAT is a calcium-regulated transcription factor associated with the control of vascular cell contractility (Gonzalez Bosc et al., 2004 ; Filosa et al., 2007 ).
Pericyte Dilation
Just as depolarization and an increase in intracellular calcium level favors constriction of pericytes, so hyperpolarization and a decrease of intracellular calcium level favors dilation. A major pathway which hyperpolarizes and relaxes vascular smooth muscle cells is activation of potassium channels (Jackson, 2005 ). It seems that this is also the case for pericytes, as several vasodilators (e.g. PGI2, adenosine and β-adrenoceptor agonists) produce their actions via potassium channel activation (Li and Puro, 2001 ; Quignard et al., 2003 ; Hirao et al., 2004 ; Burnette and White, 2006 ). Pericytes express potassium channels that are voltage-sensitive, including inward rectifying (KIR) channels (von Beckerath et al., 2000 ; Cao et al., 2006 ), voltage-gated potassium channels (von Beckerath et al., 2000 ) and big and small conductance calcium-activated potassium channels (BKCa and SKCa; Wiederholt et al., 1995 ; Quignard et al., 2003 ), as well as ATP-sensitive potassium channels (KATP; Li and Puro, 2001 ; Hirao et al., 2004 ), which are not voltage-gated but do exhibit inward rectification at positive potentials (Li and Puro, 2001 ). Activation of these channels by vasodilators produces vessel relaxation by hyperpolarizing the cell and therefore reducing VOCC activation (Figure 2 A). An increase in extracellular potassium concentration can also produce dilation by promoting an efflux of potassium at the resting membrane potential, as a result of a raised [K+]o increasing the conductance of KIR channels (as shown previously for smooth muscle; Filosa et al., 2006 ). As well as inducing vasodilation, potassium channel activation in pericytes may also serve to modulate and limit constriction, since serum-derived molecules which constrict pericytes (e.g. IGF-1) also open voltage-dependent potassium channels (Sakagami et al., 1999 ). Conversely, some vasoconstrictors inhibit potassium channels to increase contractile tone, e.g. ET-1 decreases activity of KATP channels (Kawamura et al., 2002 ). Not all vasodilators target potassium channels, however. NO has been found to dilate retinal pericytes at least in part by inhibiting VOCCs and ClCa channels, thereby reducing depolarization and calcium entry into the cell and favoring dilation over constriction (Figure 2 A; Sakagami et al., 2001a ).
Cyclic nucleotides are frequently involved in the pathways by which receptor activation on pericytes leads to regulation of ion channels or MLCP (Figure 2 A). Activation of PGI2 receptors or β-adrenoceptors, for example, elicits opening of BKCa channels via activation of adenylyl cyclase and production of cAMP (Quignard et al., 2003 ; Burnette and White, 2006 ) which, rather than activating protein kinase A (PKA), in this case produces activation of protein kinase G (PKG; Burnette and White, 2006 ). Even though PKG is more sensitive to cGMP than cAMP, cross-activation can occur as intracellular cAMP levels are generally much higher than cGMP levels, and PKG can become more sensitive to cAMP following autophosphorylation (Smith et al., 1996 ). Alternatively, cAMP might decrease degradation of cGMP by phosphodiesterase V, boosting cGMP levels and thus PKG activity (Pelligrino and Wang, 1998 ). In other cell types, PKG activation also occurs in response to NO, because NO activates soluble guanylyl cyclase (sGC) to generate cGMP, which then activates PKG (Garthwaite, 2000 ). This seems to be the way in which pericytes respond to NO, as pericytes express both sGC (Fessenden and Schacht, 1997 ) and PKG (Tian et al., 1999 ) and the dilating effect of NO donors is mimicked by 8-Br-cGMP, a membrane permeable cGMP analog (Sakagami et al., 2001a ), and prevented by inhibition of sGC (Haefliger et al., 1994 ).
Ion channels are not the only target of cyclic nucleotide pathways which evoke relaxation. Adrenomedullin, a vasoactive peptide, which is expressed in CNS endothelial cells and neurons, and is known to increase cerebral blood flow (Serrano et al., 2002 ) produces an increase in cAMP level in pericytes, activating PKA and decreasing the phosphorylation of MLC, leading to relaxation (Takata et al., 2009 ). In vascular smooth muscle cells, PKG activation is known to increase the activity of MLCP (Lincoln et al., 2001 ), which would also produce this effect, so it is possible that the same mechanism is also active in pericytes.
Mechanisms of Metabolic Regulation of Pericyte Tone
As mentioned above, the metabolic state of the tissue can regulate pericyte tone (Figures 2 B,C). KATP channels are inherently metabolically regulated, increasing in activity when the intracellular level of ATP falls and that of ADP increases, hyperpolarizing the cell and thus favoring dilation. This effect can be enhanced, as the hyperpolarization closes channels that are activated by depolarization, such as VOCCs and NSCs. This increases the membrane resistance so further current flow through KATP channels may produce a larger hyperpolarization. Conversely, as the ATP level rises and that of ADP falls, KATP channels decrease in activity, favoring constriction. An increase in adenosine level, which can occur during hypoxia, activates A1 and A2A adenosine receptors which then also increase KATP channel activity (Li and Puro, 2001 ) and increase capillary blood flow (Hirao et al., 2004 ). KATP currents can exert different levels of control over the contractile tone of pericytes, depending on the metabolic state of the tissue. In normoxic conditions, for example, PDGF-B causes a depolarization and constriction of retinal pericytes through activation of NSCs, ClCa channels and VOCCs, but in simulated ischaemia, PDGF-B causes dilation by activation of KATP channels and a subsequent decrease of intracellular calcium concentration (Sakagami et al., 2001b ).
Cell-cell communication can also be modulated by the metabolic state of the tissue. Lactate is another bidirectional vasoactive signal, which constricts pericytes in conditions of high oxygen level, but relaxes them when oxygen is scarce (Yamanishi et al., 2006 ). Constriction in this instance is thought to involve communication with endothelial cells (Figure 2 B), which express sodium-calcium exchangers that are inhibited by lactate, following a complicated cascade involving activation of monocarboxylate transporters, sodium-hydrogen exchangers and the sodium-potassium ATPase. This results in a decrease in calcium extrusion on lactate application and thus a rise in intracellular calcium concentration that is transmitted through gap junctions to pericytes, producing constriction. Hypoxia apparently closed gap junctions between pericytes and endothelial cells, preventing any calcium rise in endothelial cells from reaching the pericytes, and thus preventing constriction (Yamanishi et al., 2006 ). The mechanism by which hypoxia alters gap junction coupling is unclear, though agonist induced closure can occur via activation of protein kinase C (PKC), for example following prolonged stimulation with ET-1 (Kawamura et al., 2002 ). It is interesting to note that the closure of gap junctions by hypoxia will increase the membrane resistance of pericytes, so that opening of pericyte ion channels (e.g. KATP channels as discussed above) will be more able to influence the membrane potential.
Pericyte Regulation of Capillary Diameter: In Vivo Evidence and Implications for Neuroimaging
Pericytes can modulate capillary diameters in vitro, suggesting that they may play a role in the in vivo regulation of blood flow. Capillaries are more numerous and densely spaced than arterioles: in the hippocampus, for example, the average distance between a neuron and a capillary is 8–23 µm, depending on the cell layer, while the average distance between a neuron and an arteriole is 70–160 µm (Lovick et al., 1999 ). Capillaries therefore seem better placed than arterioles to rapidly detect any increase in neuronal activity and to respond to the requirement for increased blood flow. Of particular relevance to neuroimaging studies, the greater density of capillaries also means that capillary-level regulation of blood flow could potentially allow more spatially-restricted changes in blood flow on occurrence of a localized increase in neuronal activity. Technological advances allowing the use of smaller voxel sizes in imaging techniques that employ blood oxygenation or blood volume as an indicator of neural activation (e.g. the blood-oxygen level dependent (BOLD) signal in functional magnetic resonance imaging, fMRI) may therefore reveal a finer level of spatial detail in the underlying neuronal activity than has previously been observed.
The in vivo evidence for regulation of capillary diameter by pericytes is somewhat patchy, however. Optical recordings have allowed comparison of the relative spatial scale of passive vascular changes (i.e. the areas of increased oxygen extraction and deoxyhemoglobin formation due to increased neuronal activity) and active vascular changes, whereby changes in vessel resistance lead to increases in cerebral blood flow and volume. While immediate increases in deoxyhemoglobin level localise well to regions of neuronal activity, suggestive of passive changes in blood oxygenation at the capillary level, later increases in blood flow and volume were originally found to cover much larger areas, suggesting that these occur at the level of arterioles and arteries (Iadecola et al., 1997 ; Vanzetta and Grinvald, 2008 ). More recently, however, evidence has been found that changes in cerebral blood flow and volume do occur on a more spatially-restricted scale, consistent with regulation of capillary blood flow. These studies find that when the interfering contribution of large surface vessels is removed, either at the analysis stage (Vanzetta et al., 2004 ), or by considering only the first (Frostig et al., 1990 ; Sheth et al., 2004 ) or last (Berwick et al., 2008 ) few seconds of the signal, localized increases of cerebral blood volume do occur in the parenchyma (assumed to represent the capillary network), with a spatial resolution of 100–400 µm, corresponding to the areas of cortex activated by whisker stimulation or by a single ocular dominance column in visual cortex (Frostig et al., 1990 ; Sheth et al., 2004 ; Vanzetta et al., 2004 ). From the relative spacing of capillaries and arterioles (see above), each capillary perfuses a tissue volume with a diameter of 15–50 µm, while arterioles perfuse a volume of diameter 140–320 µm. Arterioles could therefore only provide spatially localized perfusion to a single activated column if the arteriole was located directly in the center of a given cortical column, but if blood flow were regulated at the capillary level as well, the capillary network could easily provide this degree of spatial resolution. Indeed, dye injection of penetrating arterioles in rat barrel cortex found that although the area of tissue perfused by a single penetrating arteriole was roughly the same size as a cortical barrel, arteriolar domains did not match barrel domains, so spatial fine tuning by the microvessel network must occur in order to account for the specific change of perfusion in a single cortical barrel (Woolsey et al., 1996 ).
It is possible that there is a contribution of small arterioles to some of the parenchymal changes that have been assumed to reflect changes in blood volume in capillaries, since the use of increased magnification has shown that some small arterioles may be present, and activity in these vessels may contribute to the localized changes which were earlier attributed to capillaries (Frostig et al., 1990 ; Berwick et al., 2008 ). There is, however, further support for a capillary basis for at least some parenchymal blood volume changes, as dilations and constrictions of cerebral capillaries have been observed in rat somatosensory cortex on forepaw stimulation (Stefanovic et al., 2008 ) and in rat cortex in response to hypercapnia (Hutchinson et al., 2006 ). Changes in blood volume or diameter in capillaries do not, in themselves, demonstrate active regulation of cerebral blood flow at the capillary level, as upstream arteriolar changes could also produce these effects in capillaries via an alteration in perfusion pressure. Nevertheless, tentative evidence for active control of capillary diameter was found by Charpak’s group (Chaigneau et al., 2003 ). If no active regulation occurs at the capillary level, all capillaries in an interconnected network should either dilate or constrict passively in response to an upstream increase or decrease of perfusion pressure. In rat olfactory bulb, however, odour stimulation only produced dilations in those capillaries within an activated olfactory glomerulus (approximate diameter: 200 µm), while capillaries just outside this glomerulus, though closely interconnected, did not respond (Chaigneau et al., 2003 ). This suggests that active regulation of capillary flow can occur and that changes in capillary flow across the brain can occur on a spatial scale similar to that observed using optical imaging. Furthermore, a simple calculation suggests that, if the change in capillary diameter produced by pericytes in cerebellar slices also occurs in vivo, then capillary-level regulation could reduce vascular resistance sufficiently to contribute significantly to the blood flow increase evoked by neural activity (Peppiatt et al., 2006 ).
Such estimates of the magnitude of the effect on blood flow of altering capillary diameter depend on knowing the relative contributions of arterioles, capillaries and venules to the total vascular resistance, so that the effect of a decrease in resistance in one of these compartments can be assessed. Several models of blood flow through the cerebral vasculature have been formulated which address this issue, which yield estimates for the percentage of vascular resistance at the capillary level ranging from 16%, in a simple multicompartmental model (Lu et al., 2004 ), to 70% in a more complicated model incorporating branching, parallel vessels, (which assumes that a constant arterial pressure is provided at the upstream end of the penetrating arterioles and a constant venous pressure is maintained downstream of the venules; Boas et al., 2008 ). Poiseuille’s law predicts that flow depends on the fourth power of the vessel radius, and can be used to estimate changes in vessel resistance following diameter changes (though as blood has cells suspended in it that are comparable in size to the capillary diameter, Poiseuille’s law is not strictly valid and will actually underestimate the effect of a change in capillary diameter as it neglects the energy required to deform blood cells at very narrow vessel diameters; Secomb, 1987 ). This means that a 2.1-fold increase in capillary diameter, as is seen after glutamate application to capillaries in vitro (Peppiatt et al., 2006 ), would decrease capillary resistance by (at least) 20-fold, decrease total vascular resistance by between 15 and 67% and therefore increase blood flow by a factor of 1.18 to 2.98 (for calculation see Peppiatt et al., 2006 , Supplementary Material). Clearly experimental evidence is required to assess which of these values is more accurate, but adaptations of these models could be valuable to assess the effect of localized changes in capillary diameter on the spatiotemporal pattern of flow through upstream arterioles, parallel capillaries and downstream venules. Indeed, a recent model simulated capillary dilation over a relatively wide region and predicted spatially-restricted increases in flow that were, nevertheless, not confined to the area of dilation (Reichold et al., 2009 ). Further modeling using more spatially limited regions of capillary dilation and more anatomically accurate representations of the capillaries will be valuable for generating predictions as to the likely impact of pericyte-mediated capillary diameter regulation.
As well as looking at the spatial patterns of changes in vessel diameters, the relative time courses of haemodynamic changes in parenchymal and arteriolar compartments can also be examined to understand the mechanisms by which changes in vessel resistance are co-ordinated. If capillaries dilate before arterioles, it is likely that they, rather than arterioles, “sense” the requirement for increased blood flow. If, however, capillaries respond later than arterioles, then arterioles are presumably the initial sensor, with capillary dilation occurring as a passive response to increased pressure or via an independent (slower) active dilation pathway. Two studies have found the early parenchymal increase in deoxyhemoglobin concentration in visual cortex, observed after visual stimulation, to be accompanied by a local increase in parenchymal total hemoglobin that is well localized to activated cortical columns (Malonek and Grinvald, 1996, indicating capillary-level regulation) and which occurred before blood flow to the region increased (Malonek et al., 1997 ). This was interpreted as representing a small degree of capillary dilation, possibly recruiting blood cells from other capillaries, before later upstream increases in blood flow (Malonek and Grinvald, 1996 ; Malonek et al., 1997 ). Most studies, however, do not observe this and find that arteriolar flow changes following sensory or electrical stimulation occur at the same time (Matsuura et al., 1999 ; Li et al., 2003 ; Hillman et al., 2007 ), or just before (Vanzetta et al., 2005 ) changes in capillary perfusion, suggesting that at least the predominant hemodynamic changes originate upstream of capillaries, rather than dilation by capillaries being retrogradely conducted to arterioles.
If retrograde transmission of dilation does not occur, then an important question arises. At what level is increased neuronal activity communicated to the vasculature? Is a local increase in neuronal activity signaled directly to a distant arteriole, e.g. via the astrocytic syncytium (Gordon et al., 2007 ) or does the signal reach the vasculature more directly, at the (nearer) capillary, without necessarily producing a diameter change? In skeletal muscle, local muscle fibre stimulation produces a current and calcium change in local capillary endothelial cells and elicits upstream arteriole dilation (without local capillary dilation; Sarelius et al., 2000 ; Murrant et al., 2004 ). It seems this signal is communicated in part, but not exclusively, by electrotonic current spread through endothelial gap junctions (Sarelius et al., 2000 ). Propagated dilation has been observed from cerebral arterioles to arteries (Iadecola et al., 1997 ) but has not been observed in cerebral capillaries, though propagated constriction has been reported (Peppiatt et al., 2006 ) and hyperpolarizations have been shown to propagate via gap junctionally coupled pericytes and endothelial cells in isolated retinal capillaries (Wu et al., 2006 ), suggesting propagated dilation between pericytes is plausible. As seen above, pericyte hyperpolarization is associated with dilation (Puro, 2007 ) but often only a fraction of pericytes demonstrate a visible alteration in tone, even when they are all electrophysiologically responsive (e.g. Yamanishi et al., 2006 ). It may therefore be possible for a neuronal or astrocytic signal to reach capillaries and be signaled upstream to arterioles, as a hyperpolarization of the endothelium and pericytes, without an immediate alteration in capillary diameter. If so, pericytes across the capillary network may exist in different states of rigidity, depending on the vasoactive signals they have received. Their relative tone could therefore dictate which capillaries become most perfused when increased flow arises from the arteriolar level. This would be consistent with increases in red blood cell content in specific capillaries (functional recruitment) which have been observed on electrical stimulation of rat cerebellar and somatosensory cortex (Akgoren and Lauritzen, 1999 ; Schulte et al., 2003 ).
In summary, the spatial resolution of changes in cerebral blood volume following sensory stimulation is suggestive of regulation of flow at the capillary level, and dilations and constrictions of capillaries have been observed in vivo. Evidence for active regulation of capillary tone in vivo is more limited, however, though differential responses in interconnected capillaries suggest that it does occur. Although the limited evidence available points to the initiation of flow increases occurring at the arteriolar level, it remains to be seen whether or not arterioles are the primary site of the neuronal or astrocytic vasoactive signals reaching the vasculature. Further work will doubtless address whether capillaries contribute to upstream transmission of vasodilatory (or vasoconstrictory) signals and how these signals fine tune blood flow through the capillary network.
What are the implications of a possible capillary-level regulation of blood flow for functional imaging studies? The most direct read-out of neuronal activity from the BOLD signal is the increase in deoxyhemoglobin level in capillaries due to increased oxygen extraction by active regions of cortex, but this “initial dip” is not easily detected (Vanzetta and Grinvald, 2008 ). Indeed it is possible that active capillary-level blood flow regulation could serve to decrease the initial dip, by increasing the amount of oxygenated blood present. As discussed above, blood volume changes with high spatial resolution have been detected optically in primary sensory cortices and the resolution limits are probably set by responses in the capillary network. These spatial resolution limits of 100–400 µm have not been possible previously using magnetic resonance imaging (MRI), but as magnetic field strengths increase (Mangia et al., 2009 ), and by orienting MRI slices to avoid large blood vessels (Vanzetta and Grinvald, 2008 ), resolution using the positive phase of the BOLD signal has improved to allow the detection of cortical laminae (Goense and Logothetis, 2006 ), orientation columns (Yacoub et al., 2008 ) and olfactory glomeruli (Kida et al., 2002 ), with voxel diameters reaching 500 µm in humans and 200 µm in animals (Mangia et al., 2009 ), thus approaching the limits observed using optical techniques. What limits could capillary-level alterations in blood flow therefore impose on the potentially achievable spatial resolution using MRI? In primary sensory cortices, the resolution attainable with optical imaging methods corresponds to a functional cortical column, and where responses are bigger than a single column, this has been suggested to reflect the underlying spread of neuronal activity, rather than a poorly localized blood flow response (Berwick et al., 2008 ). In other words, the potential resolution of the vascular response may be significantly greater than is apparent when imaging primary sensory cortices. It would therefore be valuable to compare vascular responses in regions where more spatially localized neuronal firing occurs in response to physiological stimulation, in order to assess whether, in these regions, different stimuli or tasks generate spatially different vascular responses, hopefully reflecting differences in the underlying pattern of neuronal firing. It is conceivable that different vascular responses could occur with a much finer spatial resolution, approximating the spatial density of capillaries (20–40 µm).
Pericytes and Pathophysiology
Pericyte damage has been linked with several different pathological conditions. For example, changes in pericyte morphology at the electron microscope level, suggestive of decreased function and degeneration, have been reported in brain tissue from epilepsy (Liwnicz et al., 1990 ) and multiple sclerosis (Claudio et al., 1995 ) patients, while pericyte coverage of capillaries from aged rats is less than in younger controls (Heinsen and Heinsen, 1983 ). In spontaneously hypertensive rats, conversely, more αSMA-expressing pericytes are found on brain capillaries (Herman and Jacobson, 1988 ), suggesting that pericytes may be more able to modulate capillary diameter in hypertension, an effect which may either contribute to cardiovascular disease or be adaptive. There is more substantial evidence, however, showing changes in CNS pericytes in three major conditions: Alzheimer’s disease, diabetic retinopathy and cerebral ischaemia.
Alzheimer’s Disease
Amyloid deposits within degenerating pericytes have been observed in the brains of patients with early onset familial (Wisniewski et al., 1992 ) and normal sporadic Alzheimer’s disease (Szpak et al., 2007 ), and exposure to and internalisation of β-amyloid is toxic to pericytes (Verbeek et al., 1997 ; Wilhelmus et al., 2007 ). Accumulation of β-amyloid around capillaries and larger vessels of Alzheimer’s disease patients is thought to occur because the perivascular space represents an important clearance pathway for β-amyloid from the interstitial fluid surrounding brain cells (Preston et al., 2003 ; Carare et al., 2008 ). Proteins are normally removed from the perivascular space via the lymphatic drainage pathways or by transport across the blood-brain barrier (Weller et al., 2008 ). Indeed, pericytes may be directly involved in β-amyloid clearance across the blood-brain barrier as they express proteins that can act as β-amyloid receptors and can internalise β-amyloid (Wilhelmus et al., 2007 ). In Alzheimer’s disease β-amyloid clearance is insufficient, either due to overproduction of β-amyloid or, as now seems more likely (Zlokovic et al., 2005 ), due to decreased functioning of the clearance pathways (Weller et al., 2008 ), so β-amyloid accumulates, forming aggregates or plaques. The presence of β-amyloid produces vasoconstriction, endothelial cell damage and a decrease in cerebral blood flow in larger blood vessels (Thomas et al., 1996 ; Niwa et al., 2001 ), and also reduces the reactivity of cerebral blood vessels to neuronal activity or to endothelium-dependent vasodilators (Iadecola et al., 1999 ; Niwa et al., 2000 ). The mechanism for these changes seems to involve generation of superoxide by endothelial cells, and, at least in part, a decrease of NO bioavailability (Thomas et al., 1996 ; Iadecola et al., 1999 ; Niwa et al., 2001 ). As pericytes are likely to also be involved in neurovascular coupling it is probable that β-amyloid will have similar effects on the regulation of capillary diameter. Thus, initially low levels of β-amyloid may produce free radicals and hence endothelial damage, capillary constriction and impaired neurovascular coupling (Figure 3 A). This reduction of the energy supply to the brain may increase neuronal damage, and the decrease in flow may also further impair β-amyloid clearance causing amyloid to accumulate further in a vicious cycle, since blood flow supports protein clearance via the perivascular space and across the blood-brain barrier (Ball et al., 2010 ). Capillaries may, in fact, be particularly sensitive to β-amyloid-mediated damage, as β-amyloid is more toxic to cultured pericytes than to vascular smooth muscle cells (Verbeek et al., 1997 ), so alterations in β-amyloid clearance and neurovascular coupling may occur even earlier in capillaries than in larger blood vessels. It is therefore of importance to establish the role of capillaries in impaired vascular function in order to fully understand the pathogenesis of Alzheimer’s disease and, hopefully, to identify potential novel drug targets to treat the disease.
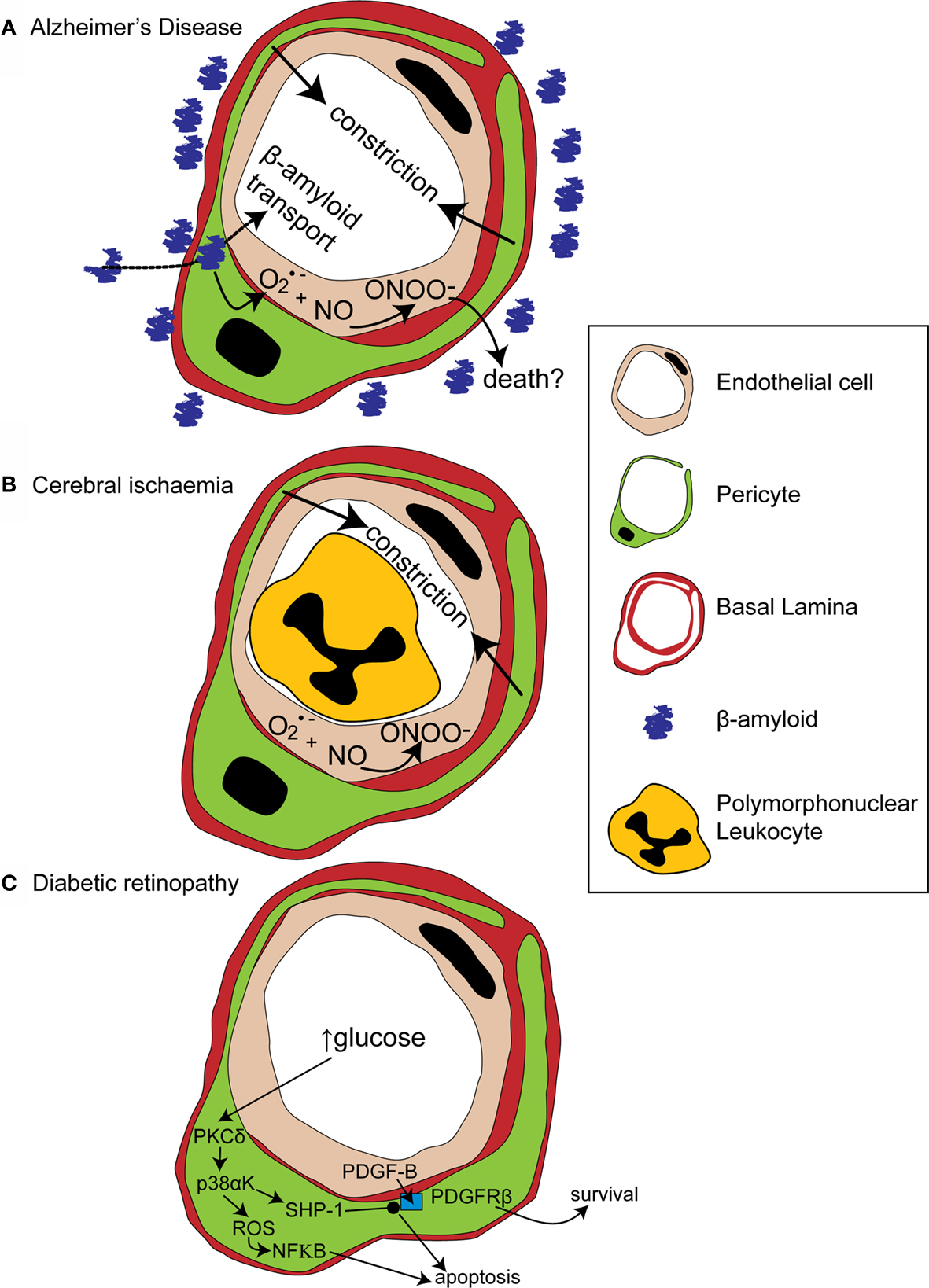
Figure 3. Alterations in pericyte function in disease states. (A) In Alzheimer’s disease, β-amyloid removal across the blood-brain barrier slows so β-amyloid aggregates are formed around blood vessels. β-amyloid is toxic to pericytes and produces superoxide (O2•−), which scavenges NO, forming peroxynitrite (ONOO−) and constricting vessels. (B) After cerebral ischaemia, the energy supply to an ischaemic region is not fully restored as many capillaries fail to reperfuse due to blockade by polymorphonuclear leukocytes, generation of reactive oxygen species and capillary constriction. (C) In diabetic retinopathy, high glucose levels trigger pericyte apoptosis and capillary malfunction via activation of protein kinase C δ (PKCδ) and p38α mitogen activated protein kinase (p38αK). Reactive oxygen species (ROS) are generated, producing apoptosis via nuclear factor κB (NF-κB) activation and Src homology-2 domain-containing phosphatase-1 (SHP-1) is activated, which inhibits PDGFR-β, decreasing activity of pro-survival pathways and further promoting apoptosis.
Of relevance to functional imaging studies, low levels of β-amyloid disrupt basal CBF and neurovascular coupling before affecting neuronal activity (as assessed by rates of glucose utilisation; Niwa et al., 2000 , 2002 ). Early changes in the BOLD signal related to vascular malfunction are therefore likely to occur in people who will go on to develop Alzheimer’s disease, before the onset of neuronal death or cognitive decline. Importantly, as these changes are due to vascular abnormalities, not altered cortical processing, they could confound the interpretation of functional imaging studies on subjects with premorbid Alzheimer’s disease. It is therefore important to consider the potential effects of altered neurovascular coupling due to Alzheimer’s disease, or other age-related conditions, in the design of studies using older people (D’Esposito et al., 2003 ).
Cerebral Ischaemia
Cerebral ischaemia has long been known to disrupt cerebral blood flow at the capillary level. Following transient focal ischaemia, after a brief period of reactive hyperaemia, cerebral blood flow is reduced, even after removal of arterial occlusion (Leffler et al., 1989 ), and many cerebral capillaries show poor reperfusion of blood cells (del Zoppo et al., 1991 ; Mori et al., 1992 ). This low reflow phenomenon occurs, in part, due to blockade of capillaries by activated polymorphonuclear leukocytes (Mori et al., 1992 ) and of post-capillaries by activated aggregating platelets and fibrin deposition (del Zoppo, 2008 ). Activation and increased adherence of these species occurs as a consequence of blood-brain barrier breakdown (del Zoppo, 2008 ) and is detrimental to the brain, since preventing loss of vessel patency by inhibiting platelet binding to fibrin decreases both microvessel occlusions and infarct volume (Choudhri et al., 1998 ; Abumiya et al., 2000 ). Recently, however, pericytes have also been shown to constrict capillaries soon after the onset of simulated retinal ischaemia, and after transient cerebral ischaemia followed by reperfusion (Peppiatt et al., 2006 ; Yemisci et al., 2009 ). In the latter study, capillary patency and infarct size was improved by blocking NO synthesis or by scavenging superoxide, and labeling for 3- nitrotyrosine (a product of peroxynitrite-mediated reactions) was found on capillaries, suggesting both that peroxynitrite formation might underlie the constriction of capillaries by pericytes (Yemisci et al., 2009 ) and that this constriction reduces energy supply to the brain sufficiently to exacerbate neuronal damage. Thus capillary blockade both by activated platelets and leukocytes, and by pericyte constriction, can decrease cerebral blood flow and increase neuronal damage following transient ischaemia (Figure 3 B).
Interestingly, several electron microscopic studies have observed a migration of pericytes away from the vessel wall following global ischaemia and reperfusion (Takahashi et al., 1997 ; Gonul et al., 2002 ; Melgar et al., 2005 ). This migration began at around the same time as the period of secondary hypoperfusion and continued for at least 12–24 h, by which time pericytes could be observed several microns away from the capillary in the neuropil (Melgar et al., 2005 ). The function of pericyte migration is not clear, but in a model of traumatic brain injury, in which pericyte migration was also observed, those pericytes which did not migrate showed cytoplasmic changes consistent with degeneration, suggesting that migration may protect the pericyte from death (Dore-Duffy et al., 2000 ) (or that pericytes which died rapidly could not migrate). Alternatively, pericytes could be acting as macrophages (Thomas, 1999 ), to clear up debris from degenerating neurons, or may even be primed to replace dying cells as, in culture at least, pericytes can differentiate into neuronal, astroglial and oligodendrocytic phenotypes (Dore-Duffy et al., 2006 ). Further study is therefore required to investigate whether pericyte migration is beneficial or damaging to functional recovery from ischaemia, which pathways control this phenomenon and whether any elements of these pathways could potentially be manipulated by therapeutic interventions.
Diabetic Retinopathy
Pericytes are more numerous on retinal capillaries than in any other vascular bed in the body (Frank et al., 1987 ; Shepro and Morel, 1993 ). Their loss in the retina occurs at the earliest stages of diabetic retinopathy (Cogan et al., 1961 ) and is associated with subsequent microaneurysms, haemorrhages, blocked capillaries and changes to the basal lamina. Later, in some patients, blindness can occur as the disease progresses to the proliferative stage, characterized by oedema over the macular region and angiogenesis, which obscures vision and threatens sight (Motiejunaite and Kazlauskas, 2008 ; Antonetti, 2009 ). Communication between pericytes and endothelial cells seems to be critically important for maintaining pericytes’ presence on retinal capillaries. Platelet-derived growth factor-B (PDGF-B) is produced by endothelial cells, while its receptor, PDGFR-β is expressed on pericytes (Figure 3 C). Mice in which PDGF-B is knocked out are not viable and die as embryos with an absence of brain pericytes and evidence of lethal haemorrhages (Lindahl et al., 1997 ). Heterozygotes survive, though with a 30% reduction in the number of retinal pericytes (Hammes et al., 2002 ). These mice also display retinal microaneurysms and an increased number of acellular capillaries (consisting of tubes of basal lamina empty of endothelial cells and pericytes). The number of pericytes decreases further (in comparison to both diabetic controls and nondiabetic PDGF-B+/− mice) if the mice are made diabetic, and the retinopathy worsens. In addition, in response to hypoxia, they show more de novo angiogenesis than do controls (Hammes et al., 2002 ). Mice with selective endothelial ablation of the PDGF-B gene are also viable and show a wide variation in the number of pericytes present on vessels (Enge et al., 2002 ). The pericyte density on CNS capillaries turns out to be negatively correlated with the degree of retinopathy, including regressed capillaries and microaneurysms, and proliferative retinopathy was found in all animals where pericyte density on CNS capillaries was decreased by more than 50% (Enge et al., 2002 ). Together these studies suggest that PDGF-B signaling from endothelial cells to pericytes is important for their association with capillaries and that this association is important for preserving capillary stability, maintaining vessel wall tone (to prevent microaneurysm formation) and preventing both vessel regression and excessive angiogenesis.
A recent paper has cast light on how diabetes may disrupt PDGF-B signaling and thus disrupt pericyte association with retinal capillaries (Geraldes et al., 2009 ). PKC activity has been previously linked with hyperglycaemia-mediated vascular dysfunction (Koya and King, 1998 ). Geraldes et al. (2009) showed that levels of PKCδ, a novel PKC isoform (Sampson and Cooper, 2006 ) which is expressed in retinal pericytes, are increased by high glucose concentrations or by an animal model of diabetes (Figure 3 C). Conversely, knockout of PKCδ prevents diabetes from inducing pericyte loss and prevents the development of retinopathic features such as acellular capillaries and increased vascular leakage. Using knockout mice and cultured retinal pericytes, PKCδ activation was found to increase p38α MAPK phosphorylation and activation, the effects of which were to increase both reactive oxygen species-mediated activation of NF-κB and expression of Src homology-2 domain–containing phosphatase-1 (SHP-1), a novel target for p38 α MAPK (Geraldes et al., 2009 ). SHP-1, which inhibits the PDGFR-β (Yu et al., 1998 ), prevented phosphorylation of the pro-survival targets Akt and ERK in response to endothelial PDGF-B (Geraldes et al., 2009 ). Both NF-κB activation (Romeo et al., 2002 ) and decreased phosphorylation of Akt and ERK trigger apoptosis in pericytes and other cell types (Liu et al., 2000 ; Geraldes et al., 2009 ). Endothelial cell-pericyte communication via PDGF-B is therefore critical for maintaining phosphorylation of pro-survival targets in pericytes and disruption of this pathway by increased activity of PKCδ and its downstream targets, in response to high glucose levels, leads to pericyte loss and the development of retinopathy (Figure 3 C).
The manner in which pericytes malfunction in disease highlights their importance in roles from controlling vessel tone and capillary perfusion, to maintenance of the blood-brain barrier and stabilising capillary structure. Further understanding of the pathways that are compromised and lead to pericyte death and malfunction in different pathological conditions is likely to be important for the development of new pharmaceutical targets. Equally, the concept that conditions such as stroke and Alzheimer’s disease, in addition to producing cognitive impairments, can also alter neurovascular coupling at all levels of the cerebrovascular system, is important not only for understanding disease progression but also when interpreting the BOLD signal in studies using these patient groups.
Overall Conclusions
Pericytes have convincingly been demonstrated to constrict or dilate in response to many different neurotransmitters and vasoactive molecules, and many of the second messenger pathways that lead to constriction or dilation have been elucidated. As yet, however, their behavior following physiological neuronal activity has not been conclusively revealed in vivo. It remains to be seen whether pericyte-mediated neurovascular coupling at the capillary level plays an important role in generating the blood flow response to localized neuronal activity, and what the role of pericytes and capillaries is in signaling a requirement for increased flow to upstream arterioles and arteries. As altered pericyte function is associated with several pathological conditions, in terms of their contractile state and other roles such as maintenance of the blood-brain barrier and provision of macrophages or precursors to the neuropil, increased knowledge of pericyte physiology and pathophysiology will be critical for further understanding disease processes, for developing new treatments, and for understanding the contribution of pericytes to functional imaging signals.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by the Fondation Leducq, Wellcome Trust, Medical Research Council UK and the European Research Council.
References
Abumiya, T., Fitridge, R., Mazur, C., Copeland, B. R., Koziol, J. A., Tschopp, J. F., Pierschbacher, M. D., and del Zoppo, G. J. (2000). Integrin alpha(IIb)beta(3) inhibitor preserves microvascular patency in experimental acute focal cerebral ischemia. Stroke 31, 1402–1409.
Akgoren, N., and Lauritzen, M. (1999). Functional recruitment of red blood cells to rat brain microcirculation accompanying increased neuronal activity in cerebellar cortex. Neuroreport 10, 3257–3263.
Antonetti, D. (2009). Eye vessels saved by rescuing their pericyte partners. Nat. Med. 15, 1248–1249.
Arneric, S. P., Honig, M. A., Milner, T. A., Greco, S., Iadecola, C., and Reis, D. J. (1988). Neuronal and endothelial sites of acetylcholine synthesis and release associated with microvessels in rat cerebral cortex: ultrastructural and neurochemical studies. Brain Res. 454, 11–30.
Ball, K. K., Cruz, N. F., Mrak, R. E., and Dienel, G. A. (2010). Trafficking of glucose, lactate, and amyloid-beta from the inferior colliculus through perivascular routes. J. Cereb. Blood Flow Metab. 30, 162–176.
Bandopadhyay, R., Orte, C., Lawrenson, J. G., Reid, A. R., De, S. S., and Allt, G. (2001). Contractile proteins in pericytes at the blood-brain and blood-retinal barriers. J. Neurocytol. 30, 35–44.
Benagiano, V., Virgintino, D., Maiorano, E., Rizzi, A., Palombo, S., Roncali, L., and Ambrosi, G. (1996). VIP-like immunoreactivity within neurons and perivascular neuronal processes of the human cerebral cortex. Eur. J. Histochem. 40, 53–56.
Berwick, J., Johnston, D., Jones, M., Martindale, J., Martin, C., Kennerley, A. J., Redgrave, P., and Mayhew, J. E. (2008). Fine detail of neurovascular coupling revealed by spatiotemporal analysis of the hemodynamic response to single whisker stimulation in rat barrel cortex. J. Neurophysiol. 99, 787–798.
Bjarnegard, M., Enge, M., Norlin, J., Gustafsdottir, S., Fredriksson, S., Abramsson, A., Takemoto, M., Gustafsson, E., Fassler, R., and Betsholtz, C. (2004). Endothelium-specific ablation of PDGFB leads to pericyte loss and glomerular, cardiac and placental abnormalities. Development 131, 1847–1857.
Boas, D. A., Jones, S. R., Devor, A., Huppert, T. J., and Dale, A. M. (2008). A vascular anatomical network model of the spatio-temporal response to brain activation. Neuroimage 40, 1116–1129.
Burnette, J. O., and White, R. E. (2006). PGI2 opens potassium channels in retinal pericytes by cyclic AMP-stimulated, cross-activation of PKG. Exp. Eye Res. 83. 1359–1365.
Cao, C., Goo, J. H., Lee-Kwon, W., and Pallone, T. L. (2006). Vasa recta pericytes express a strong inward rectifier K+ conductance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R1601–R1607.
Carare, R. O., Bernardes-Silva, M., Newman, T. A., Page, A. M., Nicoll, J. A., Perry, V. H., and Weller, R. O. (2008). Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol. Appl. Neurobiol. 34, 131–144.
Cauli, B., Tong, X. K., Rancillac, A., Serluca, N., Lambolez, B., Rossier, J., and Hamel, E. (2004). Cortical GABA interneurons in neurovascular coupling: relays for subcortical vasoactive pathways. J. Neurosci. 24, 8940–8949.
Chaigneau, E., Oheim, M., Audinat, E., and Charpak, S. (2003). Two-photon imaging of capillary blood flow in olfactory bulb glomeruli. Proc. Natl. Acad. Sci. U.S.A. 100, 13081–13086.
Chen, Q., and Anderson, D. R. (1997). Effect of CO2 on intracellular pH and contraction of retinal capillary pericytes. Invest Ophthalmol. Vis. Sci. 38, 643–651.
Chesler, M., and Kaila, K. (1992). Modulation of pH by neuronal activity. Trends Neurosci. 15, 396–402.
Choudhri, T. F., Hoh, B. L., Zerwes, H. G., Prestigiacomo, C. J., Kim, S. C., Connolly, E. S. Jr., Kottirsch, G., and Pinsky, D. J. (1998). Reduced microvascular thrombosis and improved outcome in acute murine stroke by inhibiting GP IIb/IIIa receptor- mediated platelet aggregation. J. Clin. Invest 102, 1301–1310.
Claudio, L., Raine, C. S., and Brosnan, C. F. (1995). Evidence of persistent blood-brain barrier abnormalities in chronic-progressive multiple sclerosis. Acta Neuropathol. 90, 228–238.
Cogan, D. G., Toussaint, D., and Kuwabara, T. (1961). Retinal vascular patterns. IV. Diabetic retinopathy. Arch. Ophthalmol. 66, 366–378.
Dehouck, M. P., Vigne, P., Torpier, G., Breittmayer, J. P., Cecchelli, R., and Frelin, C. (1997). Endothelin-1 as a mediator of endothelial cell-pericyte interactions in bovine brain capillaries. J. Cereb. Blood Flow Metab. 17, 464–469.
del Zoppo, G. J. (2008). Virchow’s triad: the vascular basis of cerebral injury. Rev. Neurol. Dis. 5(Suppl. 1), S12–S21.
del Zoppo, G. J., Schmid-Schonbein, G. W., Mori, E., Copeland, B. R., and Chang, C. M. (1991). Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke 22, 1276–1283.
D’Esposito, M., Deouell, L. Y., and Gazzaley, A. (2003). Alterations in the BOLD fMRI signal with ageing and disease: a challenge for neuroimaging. Nat. Rev. Neurosci. 4, 863–872.
Dirnagl, U., Niwa, K., Lindauer, U., and Villringer, A. (1994). Coupling of cerebral blood flow to neuronal activation: role of adenosine and nitric oxide. Am. J. Physiol. 267, H296–H301.
Dodge, A. B., Hechtman, H. B., and Shepro, D. (1991). Microvascular endothelial-derived autacoids regulate pericyte contractility. Cell Motil. Cytoskeleton 18, 180–188.
Dore, S. E. (1923). On the contractility of and nervous supply of capillaries. Br. J. Dermatol. 35, 398–404.
Dore-Duffy, P. (2008). Pericytes: pluripotent cells of the blood brain barrier. Curr. Pharm. Des. 14, 1581–1593.
Dore-Duffy, P., Katychev, A., Wang, X., and Van, B. E. (2006). CNS microvascular pericytes exhibit multipotential stem cell activity. J. Cereb. Blood Flow Metab. 26, 613–624.
Dore-Duffy, P., Owen, C., Balabanov, R., Murphy, S., Beaumont, T., and Rafols, J. A. (2000). Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc. Res. 60, 55–69.
Enge, M., Bjarnegard, M., Gerhardt, H., Gustafsson, E., Kalen, M., Asker, N., Hammes, H. P., Shani, M., Fassler, R., and Betsholtz, C. (2002). Endothelium-specific platelet-derived growth factor-B ablation mimics diabetic retinopathy. EMBO J. 21, 4307–4316.
Faraci, F. M., and Breese, K. R. (1993). Nitric oxide mediates vasodilatation in response to activation of N-methyl-D-aspartate receptors in brain. Circ. Res. 72, 476–480.
Favard, C., Simon, A., Vigny, A., and Nguyen-Legros, J. (1990). Ultrastructural evidence for a close relationship between dopamine cell processes and blood capillary walls in Macaca monkey and rat retina. Brain Res. 523, 127–133.
Fessenden, J. D., and Schacht, J. (1997). Localization of soluble guanylate cyclase activity in the guinea pig cochlea suggests involvement in regulation of blood flow and supporting cell physiology. J. Histochem. Cytochem. 45, 1401–1408.
Fields, R. D., and Burnstock, G. (2006). Purinergic signalling in neuron-glia interactions. Nat. Rev. Neurosci. 7, 423–436.
Filosa, J. A., Bonev, A. D., Straub, S. V., Meredith, A. L., Wilkerson, M. K., Aldrich, R. W., and Nelson, M. T. (2006). Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat. Neurosci. 9, 1397–1403.
Filosa, J. A., Nelson, M. T., and Gonzalez Bosc, L. V. (2007). Activity-dependent NFATc3 nuclear accumulation in pericytes from cortical parenchymal microvessels. Am. J. Physiol., Cell Physiol. 293, C1797–C1805.
Frank, R. N., Dutta, S., and Mancini, M. A. (1987). Pericyte coverage is greater in the retinal than in the cerebral capillaries of the rat. Invest Ophthalmol. Vis. Sci. 28, 1086–1091.
Frostig, R. D., Lieke, E. E., Ts’o, D. Y., and Grinvald, A. (1990). Cortical functional architecture and local coupling between neuronal activity and the microcirculation revealed by in vivo high-resolution optical imaging of intrinsic signals. Proc. Natl. Acad. Sci. U.S.A. 87, 6082–6086.
Garthwaite, J. (2000). “The physiological roles of nitric oxide in the central nervous system,” in Nitric Oxide, ed. B. Mayer (Berlin: Springer-Verlag), 259–275.
Geraldes, P., Hiraoka-Yamamoto, J., Matsumoto, M., Clermont, A., Leitges, M., Marette, A., Aiello, L. P., Kern, T. S., and King, G. L. (2009). Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat. Med. 15, 1298–1306.
Goense, J. B., and Logothetis, N. K. (2006). Laminar specificity in monkey V1 using high-resolution SE-fMRI. Magn. Reson. Imaging 24, 381–392.
Gonul, E., Duz, B., Kahraman, S., Kayali, H., Kubar, A., and Timurkaynak, E. (2002). Early pericyte response to brain hypoxia in cats: an ultrastructural study. Microvasc. Res. 64, 116–119.
Gonzalez Bosc, L. V., Wilkerson, M. K., Bradley, K. N., Eckman, D. M., Hill-Eubanks, D. C., and Nelson, M. T. (2004). Intraluminal pressure is a stimulus for NFATc3 nuclear accumulation: role of calcium, endothelium-derived nitric oxide, and cGMP-dependent protein kinase. J. Biol. Chem. 279, 10702–10709.
Gordon, G. R., Choi, H. B., Rungta, R. L., Ellis-Davies, G. C., and MacVicar, B. A. (2008). Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature 456, 745–749.
Gordon, G. R., Mulligan, S. J., and MacVicar, B. A. (2007). Astrocyte control of the cerebrovasculature. Glia 55, 1214–1221.
Gragera, R. R., Muniz, E., and Martinez-Rodriguez, R. (1993). Electron microscopic immunolocalization of GABA and glutamic acid decarboxylase (GAD) in cerebellar capillaries and their microenvironment. Cell Mol. Biol. (Noisy-le-grand) 39, 809–817.
Haefliger, I. O., Zschauer, A., and Anderson, D. R. (1994). Relaxation of retinal pericyte contractile tone through the nitric oxide-cyclic guanosine monophosphate pathway. Invest Ophthalmol. Vis. Sci. 35, 991–997.
Hammes, H. P., Lin, J., Renner, O., Shani, M., Lundqvist, A., Betsholtz, C., Brownlee, M., and Deutsch, U. (2002). Pericytes and the pathogenesis of diabetic retinopathy. Diabetes 51, 3107–3112.
Heinsen, H., and Heinsen, Y. L. (1983). Cerebellar capillaries. Qualitative and quantitative observations in young and senile rats. Anat. Embryol. 168, 101–116.
Herman, I. M., and D’Amore, P. A. (1985). Microvascular pericytes contain muscle and nonmuscle actins. J. Cell Biol. 101, 43–52.
Herman, I. M., and Jacobson, S. (1988). In situ analysis of microvascular pericytes in hypertensive rat brains. Tissue Cell 20, 1–12.
Hilgers, R. H., and Webb, R. C. (2005). Molecular aspects of arterial smooth muscle contraction: focus on Rho. Exp. Biol. Med. (Maywood) 230, 829–835.
Hillman, E. M., Devor, A., Bouchard, M. B., Dunn, A. K., Krauss, G. W., Skoch, J., Bacskai, B. J., Dale, A. M., and Boas, D. A. (2007). Depth-resolved optical imaging and microscopy of vascular compartment dynamics during somatosensory stimulation. Neuroimage 35, 89–104.
Hirao, M., Oku, H., Goto, W., Sugiyama, T., Kobayashi, T., and Ikeda, T. (2004). Effects of adenosine on optic nerve head circulation in rabbits. Exp. Eye Res. 79, 729–735.
Hirschi, K. K., and D’Amore, P. A. (1996). Pericytes in the microvasculature. Cardiovasc. Res. 32, 687–698.
Ho, K. L. (1985). Ultrastructure of cerebellar capillary hemangioblastoma. IV. Pericytes and their relationship to endothelial cells. Acta Neuropathol. 67, 254–264.
Hutchinson, E. B., Stefanovic, B., Koretsky, A. P., and Silva, A. C. (2006). Spatial flow-volume dissociation of the cerebral microcirculatory response to mild hypercapnia. Neuroimage 32, 520–530.
Iadecola, C., Yang, G., Ebner, T. J., and Chen, G. (1997). Local and propagated vascular responses evoked by focal synaptic activity in cerebellar cortex. J. Neurophysiol. 78, 651–659.
Iadecola, C., Zhang, F. Y., Niwa, K., Eckman, C., Turner, S. K., Fischer, E., Younkin, S., Borchelt, D. R., Hsiao, K. K., and Carlson, G. A. (1999). SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat. Neurosci. 2, 157–161.
Jackson, W. F. (2005). Potassium channels in the peripheral microcirculation. Microcirculation 12, 113–127.
Joyce, N. C., Haire, M. F., and Palade, G. E. (1985). Contractile proteins in pericytes. I. Immunoperoxidase localization of tropomyosin. J. Cell Biol. 100, 1379–1386.
Kawamura, H., Oku, H., Li, Q., Sakagami, K., and Puro, D. G. (2002). Endothelin-induced changes in the physiology of retinal pericytes. Invest Ophthalmol. Vis. Sci. 43, 882–888.
Kawamura, H., Sugiyama, T., Wu, D. M., Kobayashi, M., Yamanishi, S., Katsumura, K., and Puro, D. G. (2003). ATP: a vasoactive signal in the pericyte-containing microvasculature of the rat retina. J. Physiol. 551, 787–799.
Kelley, C., D’Amore, P., Hechtman, H. B., and Shepro, D. (1988). Vasoactive hormones and cAMP affect pericyte contraction and stress fibres in vitro. J. Muscle Res. Cell Motil. 9, 184–194.
Kida, I., Xu, F., Shulman, R. G., and Hyder, F. (2002). Mapping at glomerular resolution: fMRI of rat olfactory bulb. Magn Reson. Med. 48, 570–576.
Koya, D., and King, G. L. (1998). Protein kinase C activation and the development of diabetic complications. Diabetes 47, 859–866.
Krimer, L. S., Muly, E. C. III, Williams, G. V., and Goldman-Rakic, P. S. (1998). Dopaminergic regulation of cerebral cortical microcirculation. Nat. Neurosci. 1, 286–289.
Kutcher, M. E., Kolyada, A. Y., Surks, H. K., and Herman, I. M. (2007). Pericyte Rho GTPase mediates both pericyte contractile phenotype and capillary endothelial growth state. Am. J. Pathol. 171, 693–701.
Lai, C. H., and Kuo, K. H. (2005). The critical component to establish in vitro BBB model: Pericyte. Brain Res. Brain Res. Rev. 50, 258–265.
Le Beux, Y. J., and Willemot, J. (1978). Actin- and myosin-like filaments in rat brain pericytes. Anat. Rec. 190, 811–826.
Leffler, C. W., Busija, D. W., Mirro, R., Armstead, W. M., and Beasley, D. G. (1989). Effects of ischemia on brain blood flow and oxygen consumption of newborn pigs. Am. J. Physiol. 257, H1917–H1926.
Li, P., Luo, Q., Luo, W., Chen, S., Cheng, H., and Zeng, S. (2003). Spatiotemporal characteristics of cerebral blood volume changes in rat somatosensory cortex evoked by sciatic nerve stimulation and obtained by optical imaging. J. Biomed. Opt. 8, 629–635.
Li, Q., and Puro, D. G. (2001). Adenosine activates ATP-sensitive K+ currents in pericytes of rat retinal microvessels: role of A1 and A2a receptors. Brain Res. 907, 93–99.
Lincoln, T. M., Dey, N., and Sellak, H. (2001). Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: from the regulation of tone to gene expression. J. Appl. Physiol. 91, 1421–1430.
Lindahl, P., Johansson, B. R., Leveen, P., and Betsholtz, C. (1997). Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 277, 242–245.
Lindauer, U., Leithner, C., Kaasch, H., Rohrer, B., Foddis, M., Fuchtemeier, M., Offenhauser, N., Steinbrink, J., Royl, G., Kohl-Bareis, M., and Dirnagl, U. (2010). Neurovascular coupling in rat brain operates independent of hemoglobin deoxygenation. J. Cereb. Blood Flow Metab. 30, 757–768.
Liu, W., Ahmad, S. A., Reinmuth, N., Shaheen, R. M., Jung, Y. D., Fan, F., and Ellis, L. M. (2000). Endothelial cell survival and apoptosis in the tumor vasculature. Apoptosis 5, 323–328.
Liwnicz, B. H., Leach, J. L., Yeh, H. S., and Privitera, M. (1990). Pericyte degeneration and thickening of basement membranes of cerebral microvessels in complex partial seizures: electron microscopic study of surgically removed tissue. Neurosurgery 26, 409–420.
Lovick, T. A., Brown, L. A., and Key, B. J. (1999). Neurovascular relationships in hippocampal slices: physiological and anatomical studies of mechanisms underlying flow-metabolism coupling in intraparenchymal microvessels. Neuroscience 92, 47–60.
Lu, K., Clark, J. W. Jr., Ghorbel, F. H., Robertson, C. S., Ware, D. L., Zwischenberger, J. B., and Bidani, A. (2004). Cerebral autoregulation and gas exchange studied using a human cardiopulmonary model. Am. J. Physiol. Heart Circ. Physiol. 286, H584–H601.
Malonek, D., Dirnagl, U., Lindauer, U., Yamada, K., Kanno, I., and Grinvald, A. (1997). Vascular imprints of neuronal activity: relationships between the dynamics of cortical blood flow, oxygenation, and volume changes following sensory stimulation. Proc. Natl. Acad. Sci. U.S.A. 94, 14826–14831.
Malonek, D., and Grinvald, A. (1996). Interactions between electrical activity and cortical microcirculation revealed by imaging spectroscopy: implications for functional brain mapping. Science 272, 551–554.
Mangia, S., Giove, F., Tkac, I., Logothetis, N. K., Henry, P. G., Olman, C. A., Maraviglia, B., Di, S. F., and Ugurbil, K. (2009). Metabolic and hemodynamic events after changes in neuronal activity: current hypotheses, theoretical predictions and in vivo NMR experimental findings. J. Cereb. Blood Flow Metab. 29, 441–463.
Markhotina, N., Liu, G. J., and Martin, D. K. (2007). Contractility of retinal pericytes grown on silicone elastomer substrates is through a protein kinase A-mediated intracellular pathway in response to vasoactive peptides. IET Nanobiotechnol. 1, 44–51.
Mathiesen, C., Caesar, K., Akgoren, N., and Lauritzen, M. (1998). Modification of activity-dependent increases of cerebral blood flow by excitatory synaptic activity and spikes in rat cerebellar cortex. J. Physiol. 512(Pt 2), 555–566.
Matsugi, T., Chen, Q., and Anderson, D. R. (1997a). Contractile responses of cultured bovine retinal pericytes to angiotensin II. Arch. Ophthalmol. 115, 1281–1285.
Matsugi, T., Chen, Q., and Anderson, D. R. (1997b). Adenosine-induced relaxation of cultured bovine retinal pericytes. Invest. Ophthalmol. Vis. Sci. 38, 2695–2701.
Matsuura, T., Fujita, H., Seki, C., Kashikura, K., and Kanno, I. (1999). Hemodynamics evoked by microelectrical direct stimulation in rat somatosensory cortex. Comp. Biochem. Physiol., Part A Mol. Integr. Physiol. 124, 47–52.
Maynard, E. A., Schultz, R. L., and Pease, D. C. (1957). Electron microscopy of the vascular bed of rat cerebral cortex. Am. J. Anat. 100, 409–433.
Melgar, M. A., Rafols, J., Gloss, D., and Diaz, F. G. (2005). Postischemic reperfusion: ultrastructural blood-brain barrier and hemodynamic correlative changes in an awake model of transient forebrain ischemia. Neurosurgery 56, 571–581.
Metea, M. R., and Newman, E. A. (2006). Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J. Neurosci. 26, 2862–2870.
Mori, E., del Zoppo, G. J., Chambers, J. D., Copeland, B. R., and Arfors, K. E. (1992). Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischemia in baboons. Stroke 23, 712–718.
Motiejunaite, R., and Kazlauskas, A. (2008). Pericytes and ocular diseases. Exp. Eye Res. 86, 171–177.
Murrant, C. L., Duza, T., Kim, M. B., Cohen, K. D., and Sarelius, I. H. (2004). Arteriolar dilations induced by contraction of hamster cremaster muscle are dependent on changes in endothelial cell calcium. Acta Physiol. Scand. 180, 231–238.
Nehls, V., and Drenckhahn, D. (1991). Heterogeneity of microvascular pericytes for smooth muscle type alpha-actin. J. Cell Biol. 113, 147–154.
Niwa, K., Kazama, K., Younkin, S. G., Carlson, G. A., and Iadecola, C. (2002). Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol. Dis. 9, 61–68.
Niwa, K., Porter, V. A., Kazama, K., Cornfield, D., Carlson, G. A., and Iadecola, C. (2001). A beta-peptides enhance vasoconstriction in cerebral circulation. Am. J. Physiol. Heart Circ. Physiol. 281, H2417–H2424.
Niwa, K., Younkin, L., Ebeling, C., Turner, S. K., Westaway, D., Younkin, S., Ashe, K. H., Carlson, G. A., and Iadecola, C. (2000). Abeta 1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc. Natl. Acad. Sci. U.S.A. 97, 9735–9740.
Pelligrino, D. A., and Wang, Q. (1998). Cyclic nucleotide crosstalk and the regulation of cerebral vasodilation. Prog. Neurobiol. 56, 1–18.
Peppiatt, C. M., Howarth, C., Mobbs, P., and Attwell, D. (2006). Bidirectional control of CNS capillary diameter by pericytes. Nature 443, 700–704.
Preston, S. D., Steart, P. V., Wilkinson, A., Nicoll, J. A., and Weller, R. O. (2003). Capillary and arterial cerebral amyloid angiopathy in Alzheimer’s disease: defining the perivascular route for the elimination of amyloid beta from the human brain. Neuropathol. Appl. Neurobiol. 29, 106–117.
Puro, D. G. (2007). Physiology and pathobiology of the pericyte-containing retinal microvasculature: new developments. Microcirculation 14, 1–10.
Quignard, J. F., Harley, E. A., Duhault, J., Vanhoutte, P. M., and Feletou, M. (2003) K+ channels in cultured bovine retinal pericytes: effects of beta-adrenergic stimulation. J. Cardiovasc. Pharmacol. 42, 379–388.
Reber, F., Gersch, U., and Funk, R. W. (2003). Blockers of carbonic anhydrase can cause increase of retinal capillary diameter, decrease of extracellular and increase of intracellular pH in rat retinal organ culture. Graefes Arch. Clin. Exp. Ophthalmol. 241, 140–148.
Reichold, J., Stampanoni, M., Lena, K. A., Buck, A., Jenny, P., and Weber, B. (2009). Vascular graph model to simulate the cerebral blood flow in realistic vascular networks. J. Cereb. Blood Flow Metab. 29, 1429–1443.
Romeo, G., Liu, W. H., Asnaghi, V., Kern, T. S., and Lorenzi, M. (2002). Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes 51, 2241–2248.
Roufail, E., Stringer, M., and Rees, S. (1995). Nitric oxide synthase immunoreactivity and NADPH diaphorase staining are co-localised in neurons closely associated with the vasculature in rat and human retina. Brain Res. 684, 36–46.
Sakagami, K., Kawamura, H., Wu, D. M., and Puro, D. G. (2001a). Nitric oxide/cGMP-induced inhibition of calcium and chloride currents in retinal pericytes. Microvasc. Res. 62, 196–203.
Sakagami, K., Kodama, T., and Puro, D. G. (2001b). PDGF-induced coupling of function with metabolism in microvascular pericytes of the retina. Invest. Ophthalmol. Vis. Sci. 42, 1939–1944.
Sakagami, K., Wu, D. M., and Puro, D. G. (1999). Physiology of rat retinal pericytes: modulation of ion channel activity by serum-derived molecules. J. Physiol. 521(Pt 3), 637–650.
Sampson, S. R., and Cooper, D. R. (2006). Specific protein kinase C isoforms as transducers and modulators of insulin signaling. Mol. Genet. Metab. 89, 32–47.
Sarelius, I. H., Cohen, K. D., and Murrant, C. L. (2000). Role for capillaries in coupling blood flow with metabolism. Clin. Exp. Pharmacol. Physiol. 27, 826–829.
Sato, A., and Sato, Y. (1990). Cerebral cortical vasodilatation in response to stimulation of cholinergic fibres originating in the nucleus basalis of Meynert. J. Auton. Nerv. Syst. 30(Suppl) S137–S140.
Schonfelder, U., Hofer, A., Paul, M., and Funk, R. H. (1998). In situ observation of living pericytes in rat retinal capillaries. Microvasc. Res. 56, 22–29.
Schulte, M. L., Wood, J. D., and Hudetz, A. G. (2003). Cortical electrical stimulation alters erythrocyte perfusion pattern in the cerebral capillary network of the rat. Brain Res. 963, 81–92.
Secomb, T. W. (1987). Flow-dependent rheological properties of blood in capillaries. Microvasc. Res. 34, 46–58.
Serrano, J., Alonso, D., Fernandez, A. P., Encinas, J. M., Lopez, J. C., Castro-Blanco, S., Fernandez-Vizarra, P., Richart, A., Santacana, M., Uttenthal, L. O., Bentura, M. L., Martinez-Murillo, R., Martinez, A., Cuttitta, F., and Rodrigo, J. (2002). Adrenomedullin in the central nervous system. Microsc. Res. Tech. 57, 76–90.
Sheth, S. A., Nemoto, M., Guiou, M., Walker, M., Pouratian, N., Hageman, N., and Toga, A. W. (2004). Columnar specificity of microvascular oxygenation and volume responses: implications for functional brain mapping. J. Neurosci. 24, 634–641.
Smith, J. A., Francis, S. H., Walsh, K. A., Kumar, S., and Corbin, J. D. (1996). Autophosphorylation of type Ibeta cGMP-dependent protein kinase increases basal catalytic activity and enhances allosteric activation by cGMP or cAMP. J. Biol. Chem. 271, 20756–20762.
Stefanovic, B., Hutchinson, E., Yakovleva, V., Schram, V., Russell, J. T., Belluscio, L., Koretsky, A. P., and Silva, A. C. (2008). Functional reactivity of cerebral capillaries. J. Cereb. Blood Flow Metab. 28, 961–972.
Sugiyama, T., Kawamura, H., Yamanishi, S., Kobayashi, M., Katsumura, K., and Puro, D. G. (2005). Regulation of P2X7-induced pore formation and cell death in pericyte-containing retinal microvessels. Am. J. Physiol. Cell Physiol. 288, C568–C576.
Szpak, G. M., Lewandowska, E., Wierzba-Bobrowicz, T., Bertrand, E., Pasennik, E., Mendel, T., Stepien, T., Leszczynska, A., and Rafalowska, J. (2007). Small cerebral vessel disease in familial amyloid and non-amyloid angiopathies: FAD-PS-1 (P117L) mutation and CADASIL. Immunohistochemical and ultrastructural studies. Folia Neuropathol. 45, 192–204.
Takahashi, A., Park, H. K., Melgar, M. A., Alcocer, L., Pinto, J., Lenzi, T., Diaz, F. G., and Rafols, J. A. (1997). Cerebral cortex blood flow and vascular smooth muscle contractility in a rat model of ischemia: a correlative laser Doppler flowmetric and scanning electron microscopic study. Acta Neuropathol. 93, 354–368.
Takata, F., Dohgu, S., Nishioku, T., Takahashi, H., Harada, E., Makino, I., Nakashima, M., Yamauchi, A., and Kataoka, Y. (2009). Adrenomedullin-induced relaxation of rat brain pericytes is related to the reduced phosphorylation of myosin light chain through the cAMP/PKA signaling pathway. Neurosci. Lett. 449, 71–75.
Thomas, T., Thomas, G., McLendon, C., Sutton, T., and Mullan, M. (1996). beta-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature 380, 168–171.
Thomas, W. E. (1999). Brain macrophages: on the role of pericytes and perivascular cells. Brain Res. Brain Res. Rev. 31, 42–57.
Tian, F., Fessenden, J. D., and Schacht, J. (1999). Cyclic GMP-dependent protein kinase-I in the guinea pig cochlea. Hear. Res. 131. 63–70.
Toribatake, Y., Tomita, K., Kawahara, N., Baba, H., Ohnari, H., and Tanaka, S. (1997). Regulation of vasomotion of arterioles and capillaries in the cat spinal cord: role of alpha actin and endothelin-1. Spinal Cord 35, 26–32.
Vanzetta, I., and Grinvald, A. (2008). Coupling between neuronal activity and microcirculation: implications for functional brain imaging. HFSP J. 2, 79–98.
Vanzetta, I., Hildesheim, R., and Grinvald, A. (2005). Compartment-resolved imaging of activity-dependent dynamics of cortical blood volume and oximetry. J. Neurosci. 25, 2233–2244.
Vanzetta, I., Slovin, H., Omer, D. B., and Grinvald, A. (2004). Columnar resolution of blood volume and oximetry functional maps in the behaving monkey; implications for FMRI. Neuron 42, 843–854.
Verbeek, M. M., de Waal, R. M., Schipper, J. J., and Van Nostrand, W. E. (1997). Rapid degeneration of cultured human brain pericytes by amyloid beta protein. J. Neurochem. 68, 1135–1141.
von Beckerath, N., Nees, S., Neumann, F. J., Krebs, B., Juchem, G., and Schomig, A. (2000). An inward rectifier and a voltage-dependent K+ current in single, cultured pericytes from bovine heart. Cardiovasc. Res. 46, 569–578.
Weller, R. O., Subash, M., Preston, S. D., Mazanti, I., and Carare, R. O. (2008). Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 18, 253–266.
Wiederholt, M., Berweck, S., and Helbig, H. (1995). Electrophysiological properties of cultured retinal capillary pericytes. Prog. Retin. Eye Res. 14, 437–451.
Wilhelmus, M. M., Otte-Holler, I., van Triel, J. J., Veerhuis, R., Maat-Schieman, M. L., Bu, G., de Waal, R. M., and Verbeek, M. M. (2007). Lipoprotein receptor-related protein-1 mediates amyloid-beta-mediated cell death of cerebrovascular cells. Am. J. Pathol. 171, 1989–1999.
Wisniewski, H. M., Wegiel, J., Wang, K. C., and Lach, B. (1992). Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer’s disease. Acta Neuropathol. 84, 117–127.
Woolsey, T. A., Rovainen, C. M., Cox, S. B., Henegar, M. H., Liang, G. E., Liu, D., Moskalenko, Y. E., Sui, J., and Wei, L. (1996). Neuronal units linked to microvascular modules in cerebral cortex: response elements for imaging the brain. Cereb. Cortex 6, 647–660.
Wu, D. M., Minami, M., Kawamura, H., and Puro, D. G. (2006). Electrotonic transmission within pericyte-containing retinal microvessels. Microcirculation 13, 353–363.
Yacoub, E., Harel, N., and Ugurbil, K. (2008). High-field fMRI unveils orientation columns in humans. Proc. Natl. Acad. Sci. U.S.A. 105, 10607–10612.
Yamanishi, S., Katsumura, K., Kobayashi, T., and Puro, D. G. (2006). Extracellular lactate as a dynamic vasoactive signal in the rat retinal microvasculature. Am. J. Physiol. Heart Circ. Physiol. 290: H925–H934.
Yemisci, M., Gursoy-Ozdemir, Y., Vural, A., Can, A., Topalkara, K., and Dalkara, T. (2009). Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med. 15, 1031–1037.
Yu, Z., Su, L., Hoglinger, O., Jaramillo, M. L., Banville, D., and Shen, S. H. (1998). SHP-1 associates with both platelet-derived growth factor receptor and the p85 subunit of phosphatidylinositol 3-kinase. J. Biol. Chem. 273, 2694–3687.
Zlokovic, B. V., Deane, R., Sallstrom, J., Chow, N., and Miano, J. M. (2005). Neurovascular pathways and Alzheimer amyloid beta-peptide. Brain Pathol. 15, 78–83.
Keywords: pericyte, capillary, neurovascular coupling, brain
Citation: Hamilton NB, Attwell D and Hall CN (2010) Pericyte-mediated regulation of capillary diameter: a component of neurovascular coupling in health and disease. Front. Neuroenerg. 2:5. doi: 10.3389/fnene.2010.00005
Received: 11 February 2010;
Paper pending published: 25 March 2010;
Accepted: 28 April 2010;
Published online: 21 May 2010
Edited by:
Anna Devor, University of California San Diego, USAReviewed by:
Eric Newman, University of Minnesota, USARay Koehler, Johns Hopkins University, USA
Copyright: © 2010 Hamilton, Attwell and Hall. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Catherine N. Hall, Department of Neuroscience, Physiology and Pharmacology, University College London, Gower St, London, WC1E 6BT, UK. e-mail: catherine.hall@ucl.ac.uk