The brain's supply and demand in obesity
- 1Medical Clinic 1, University of Lübeck, Lübeck, Germany
- 2Department of Neuroendocrinology, University of Lübeck, Lübeck, Germany
- 3Department of Psychiatry and Human Behavior, University of California, Irvine, CA, USA
- 4Institute of Technical Computational Mathematics, University of Braunschweig, Braunschweig, Germany
During psychosocial stress, the brain demands extra energy from the body to satisfy its increased needs. For that purpose it uses a mechanism referred to as “cerebral insulin suppression” (CIS). Specifically, activation of the stress system suppresses insulin secretion from pancreatic beta-cells, and in this way energy—particularly glucose—is allocated to the brain rather than the periphery. It is unknown, however, how the brain of obese humans organizes its supply and demand during psychosocial stress. To answer this question, we examined 20 obese and 20 normal weight men in two sessions (Trier Social Stress Test and non-stress control condition followed by either a rich buffet or a meager salad). Blood samples were continuously taken and subjects rated their vigilance and mood by standard questionnaires. First, we found a low reactive stress system in obesity. While obese subjects showed a marked hormonal response to the psychosocial challenge, the cortisol response to the subsequent meal was absent. Whereas the brains of normal weight subjects demanded for extra energy from the body by using CIS, CIS was not detectable in obese subjects. Our findings suggest that the absence of CIS in obese subjects is due to the absence of their meal-related cortisol peak. Second, normal weight men were high reactive during psychosocial stress in changing their vigilance, thereby increasing their cerebral energy need, whereas obese men were low reactive in this respect. Third, normal weight subjects preferred carbohydrates after stress to supply their brain, while obese men preferred fat and protein instead. We conclude that the brain of obese people organizes its need, supply, and demand in a low reactive manner.
Introduction
Continuous supply of cerebral energy is mandatory for survival. The selection of substrates by the brain is specific. While the brain essentially demands for glucose as “brain fuel,” peripheral organs (muscle) can also metabolize fat or proteins as “body fuels.” The brain takes up approximately 65% of the circulating glucose (Reinmuth et al., 1965). Only under certain conditions, such as starvation, exercise, or stress, the organism produces significant amounts of alternative substrates such as ketones or lactate which may take over a role in the energy supply of the brain. The flux of glucose through the blood-brain barrier varies with different behavioral states: deep sleep decreases the global cerebral energy supply by 40% (Boyle et al., 1994) and a mental challenge increases it by 12% (Madsen et al., 1995).
How can the brain adjust its energy supply to its varying energy needs? Energy fluxes to the brain have been described by a “cerebral supply chain,” ranging from the remote to the near environment, to the body and toward the brain (Peters and Langemann, 2009). According to that concept, the brain regulates cerebral energy homeostasis via “brain-pull mechanisms,” i.e., by actively demanding energy from the body. Of central relevance for brain-pull function is our stress system with its principle components the sympathetic nervous system (SNS) and the hypothalamus-pituitary adrenal axis (HPA). The stress system functions to procure fuel for the brain when needed. In a recent study in normal weight men, evidence was provided for a brain-pull mechanism referred to as “cerebral insulin suppression” (CIS) (Hitze et al., 2010; Peters et al., 2011b).
Several molecular and cellular mechanisms have already been discovered, which may fulfill the physiological function of CIS (Figure 1): both the medial amygdalar nucleus and the ventromedial hypothalamus contain glucose-responsive neurons, which control the activity of the SNS and the HPA (Miki et al., 2001; Zhou et al., 2010). The medial amygdalar nucleus sends heavy input to the ventromedial hypothalamus and thereby exerts control over its activity (Petrovich et al., 2001). In the ventromedial hypothalamus, ATP-sensitive potassium (KATP) channels have been shown to monitor intracellular ATP (Spanswick et al., 1997). If the cerebral intracellular ATP concentrations fall, ventromedial hypothalamus neurons depolarize due to GABAergic disinhibition (Chan et al., 2007). Via glutamatergic mechanisms they activate the sympatho-adrenal system (Tong et al., 2007). Specific sympathetic efferences project to pancreatic beta-cells and in so doing inhibit insulin secretion (Ahren, 2000). Ventromedial hypothalamus-controlled sympathetic efferences also project to muscle and fat tissue, where they inhibit insulin-dependent glucose transport via GLUT 4 (Mulder et al., 2005). Catecholamines released at nerve endings adjacent to adipocytes interfere with trafficking of GLUT 4 molecules, i.e., they block the translocation of GLUT 4 from the inside of the cell to its cell membrane. Thus, the ventromedial hypothalamus neurons are capable of limiting the flux of glucose from the blood into the peripheral energy stores (muscle and fat tissue). As a consequence, glucose is now available via insulin-independent GLUT1-transport across the blood-brain barrier (Hasselbalch et al., 1995; Seaquist et al., 2001). In this way, CIS can be interpreted as a brain-pull mechanism that functions to demand energy from the body to cover the increased cerebral need during stress.
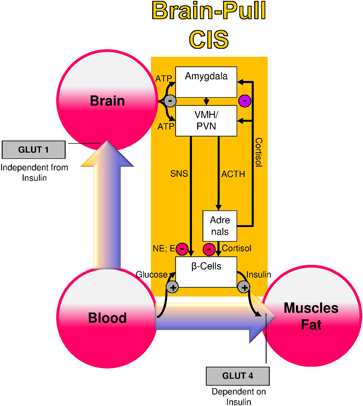
Figure 1. A conceptual diagram that shows key physiological mechanisms, which fulfill the brain-pull function of cerebral insulin suppression (CIS). The flux of glucose is either directed to the brain or to muscle and fat. If neuronal ATP concentrations fall, activation of neurons in the amygdala, in the ventromedial hypothalamus (VMH), and in the paraventricular nucleus (PVN) occurs, which in turn activates the sympathetic nervous system and the hypothalamus pituitary adrenal system (SNS and HPA). Both the SNS and the HPA system suppress insulin release from pancreatic beta cells, thereby decreasing glucose transporter 4 (GLUT-4) mediated glucose uptake into muscle and fat. In this way the brain limits glucose uptake in peripheral tissues and procures itself with glucose on demand. Finally, the CIS-brain-pull system is hierarchically organized and adaptive. The strength of the central glucocorticoid feedback determines whether the CIS-brain-pull system reacts in a high or low reactive manner. ACTH, Adrenocorticotropic hormone; E, epinephrine; NE, norepinephrine.
Cerebral energy demand mechanisms like CIS are not only present during psychosocial stress in normal weight humans. The concept of the neural control of the endocrine pancreas has already been described in detail by Steve Woods and Daniel Porte in the 1970s (Woods and Porte, 1974). CIS is known to occur in various kinds of stress states including caloric restriction (Peters et al., 2011a), sleep deprivation (Spiegel et al., 1999), myocardial infarction (Taylor et al., 1969), cerebral ischemia (Harada et al., 2009; McPherson et al., 2009), burns (Allison et al., 1968), hemorrhagic shock (Cerchio et al., 1971), subarachnoid hemorrhage (Kruyt et al., 2011), hypoxia (Chen et al., 2007), and chronic obstructive pulmonary disease in the pink puffer type (Franssen et al., 2011). In all these states of metabolic challenges, the brain can activate CIS as a demand mechanism to furnish extra-glucose fuel, which finally serves to prevent cerebral energy depletion.
There are still open questions with respect to the cerebral energy supply and demand in obesity. It is doubtful that obese subjects display high reactivity of their stress system—as normal weight subjects do—to exert the CIS-brain-pull mechanism during a psychosocial challenge. This doubt arises because of both experimental and theoretical reasons. There is experimental evidence that obese subjects exhibit low stress reactivity. First, a low epinephrine reactivity to a mental challenge was the strongest predictor for body mass gain in an 18-years-lasting Norwegian cohort study (Flaa et al., 2008). Second, obese subjects have consistently been shown to exhibit a low sympatho-adrenal system reactivity to divers challenges such as exercise (Eliakim et al., 2006; Azarbayjani et al., 2011), cold (Nielsen et al., 1993), hypoglycemia (Vea et al., 1994), and oral glucose load (Spraul et al., 1994). Similarly, obesity prone rats are known to exhibit a low sympatho-adrenal system reactivity to hypoglycemia (Tkacs and Levin, 2004), overfeeding (Shin et al., 2010), and chronic stress (Levin et al., 2000). Moreover, it has been shown theoretically, that there is an inherent property in the cerebral supply chain, indicating that a low reactive (“incompetent”) brain-pull (i.e., the force with which the brain actively demands energy from the body) results in accumulation of energy in the body storages—culminating in obesity and diabetes type 2 (Peters and Langemann, 2009).
On this background, we address here the following questions: (1) do obese subjects display a high reactivity of their stress system—as normal weight subjects do—to exert CIS following acute psychosocial stress in order to demand for cerebral energy? and (2) what do obese subjects eat after stress to replenish the brain and the body with energy?
Methods
Study Population
Twenty obese men of Caucasian descent with a Body-Mass-Index ≥ 30 kg/m2 were recruited by notice board postings. In a previous study, we already investigated normal weight men of Caucasian descent in the same study design (Hitze et al., 2010). From that study, normal weight subjects randomly assigned to either the rich buffet or the meager salad experimental groups are used here for comparison. Participants met the following inclusion criteria: normal physical examination and routine laboratory tests, no physical, or mental disease, no abuse of nicotine, alcohol or drugs, no nightshifts, no disturbed sleep or exceptional stress during the past two weeks and no blood donation during the past four weeks prior to the study. In normal weight men, abnormalities in eating behavior and chronic stress were excluded by the German version of the Three-Factor-Eating-Questionnaire (Pudel and Westenhöfer, 1989) and by the Trier Inventory for Chronic Stress (Schulz et al., 2004), respectively. Educational level was assessed for classification of socio-economic status (low vs. high).
The study was approved by the local medical ethics committee of Lübeck University. All subjects provided their fully informed and written consent before participation.
Study Protocol
Subjects were randomly assigned to two different experimental groups according to the energy provided (group 1: rich buffet + i.v. control (i.e., Ringer-infusion), group 2: meager salad + i.v. control). Each subject participated in two sessions (stress intervention and non-stress control condition) with an interval of 7–14 days between these two sessions. The order of sessions was balanced across subjects.
As described previously, experiments took place in a sound attenuated room with the subjects resting on a bed. One venous catheter was placed in each arm. With one cannula the infusion was applied; the other cannula was connected to a long thin tube that enabled blood sampling from an adjacent room without awareness of the subject. A microphone and video camera were installed for communication and observation purposes (Hitze et al., 2010).
After a fasting period of 2½ h, participants arrived at the medical research unit at 12:30. Adjacent to a short medical exploration and interview including study relevant medical history, each subject received a 250 ml Ringer-infusion (isomolar, consisting of sodium chloride, potassium chloride, calcium chloride, and water) to adjust the fluid balance and to compensate for the following blood loss. Weight was assessed to the nearest gram by an electronic scale. Height was measured to the nearest 0.5 cm by stadiometer. BMI was calculated as weight (kg) / height (m2). Afterwards, a standardized lunch was offered consisting of potatoes, mixed vegetables, butter, chicken breast, margarine, gravy, tomatoes, and yoghurt dressing. Between 15:00 and 16:00 blood samples were taken every 15 min. At 16:00, a second Ringer-infusion was applied with an infusion rate of 7.5 ml/h/kg body mass (duration: 40 min).
At 16:00, the Trier Social Stress Test began. Directly after stress intervention (at 16:25 and 16:30), a blood sample was taken and a meal (rich buffet or meager salad) was offered at 16:30. Experimental group 1 was offered a rich buffet, from which they could choose food for 1 h [for composition of free-choice rich buffet see (Hitze et al., 2010)]. Consumed food was assessed and analyzed for its amounts of energy and macro-nutrients by a dietician. Experimental group 2 received a meager salad only, from which they could eat for 1 h. During and after meal ingestion nine blood samples were taken according to the following schedule: 16:40; between 17:00 and 18:00 every 15 min and between 18:00 and 19:30 every 30 min. The procedure in the non-stress control condition was the same, except that the stress test was omitted.
Psychosocial Stress Test Intervention
At 16:00, the subjects were introduced to the task they would have to perform, i.e., a free speech in front of an audience and a mental arithmetic task (Kirschbaum et al., 1993). The subjects were taken to another room, where an audience already sat at a table, and a microphone as well as video camera was installed. It was announced that a video analysis of the subject's performance would be performed. The audience consisted of a man and a woman in white lab coats, who deprived the subjects of social support, i.e., they did not encourage the subjects. After a brief preparation period (3 min), the subjects were asked to stand at the microphone and to deliver a free speech as a job applicant who was invited for a personal interview with the company manager (5 min). If the subject finished in less than 5 min, the jury members told the subjects that he had still some time left. If the subject stopped a second time, the audience was quiet for some seconds and then started to ask prepared questions. Afterwards, subjects had to perform a mental arithmetic task consisting of serial subtractions (5 min). On a failure, subjects had to restart.
Assessment of Symptoms, Mood, and Vigilance
Subjects were asked to rate neuroglycopenic and autonomic symptoms at baseline (14:30), immediately after social stress intervention (16:25) and 1½ h after meal ingestion in the post-stress replenishment phase (18:00). Subjects rated the following symptoms from 0 (not at all) to 9 (severely): anxiety, sweating, tremor, palpitation, and nervousness (autonomic symptoms) as well as tingling, blurred vision, difficulty to concentrate, dizziness, and faintness (neuroglycopenic symptoms). Ratings were averaged for analysis. Likewise, fatigue was assessed. Fatigue ratings were reversed into vigilance and also averaged for analysis.
Together with the symptom scales (at 14:30, 16:25, and 18:00), a short form of the MDBF [Multidimensional Mood State Questionnaire; (Steyer et al., 1997)] was delivered to assess current mood. Subjects rated their mood from 0 (not at all) to 5 (severely) by the following adjectives: satisfied, bad, good, unwell. Ratings were averaged for analysis.
Depression index was assessed by Beck-Depression-Inventory (Lustman et al., 1997).
Laboratory Methods
All blood samples were immediately centrifuged, and the supernatants were stored at −60°C till analysis. Plasma glucose was measured by hexokinase method (Abbott Clinical Chemistry, IL, USA, intra-assay and inter-assay CV: <5%). Using the Immulite 2000 system (Siemens, Los Angeles, USA), serum insulin, plasma ACTH, and serum cortisol were determined by immunometric assay (insulin: intra-assay CV: 3.3–5.5%; inter-assay CV: 4.1–7.3%; ACTH: intra-assay CV: 6.7–9.5%; inter-assay CV: 6.1–10.0%; cortisol: intra-assay CV: 5.2–7.4%, inter-assay CV: 7.2–9.4%). Plasma epinephrine and norepinephrine were analyzed by HPLC (Chromsystems Diagnostics by HPLC, Munich, Germany, intra-assay CV 1.7–11.4%, inter-assay CV 3.7–12.7%).
Statistics
Data analysis was performed using SPSS statistical software (SPSS 12.0, Inc., Chicago, USA). Descriptive statistics are given as mean ± SEM. Within-group differences were calculated by paired t-test. χ2-Test was applied to analyze differences in frequency distributions. ANOVA for repeated measures was used to test differences in the variation of time between stress intervention and non-stress control condition. In this approach, time, and stress intervention were entered as within-subject factors. Where appropriate, experimental groups (rich buffet vs. meager salad) and weight groups (normal weight vs. obesity) were entered as between-subject-factors. Two-level hierarchical linear modeling (HLM) growth curve analyses (Raudenbush and Bryk, 2002; Singer et al., 2003) were conducted using the HLM 6.01 for windows software package to predict changes in glucose and insulin concentrations in the post-stress replenishment phase by meal-related cortisol peak. Level 1 captured the post-stress replenishment phase as random predictors of glucose and insulin concentrations, respectively. Meal-related cortisol increase was calculated for each individual and entered at level 2. A P-value (two-sided) of 0.05 was considered significant.
Results
Characterization of the Study Population
Participant characteristics are presented in Table 1. Obese men had a higher BMI than normal weight subjects and were 4 years older. They displayed marked hyperinsulinemia despite normoglycemia. On average, obese men had a lower socio-economic status. Furthermore, obese men showed social overload, more depression symptoms, and higher values in disturbances in eating behavior than normal weight men.
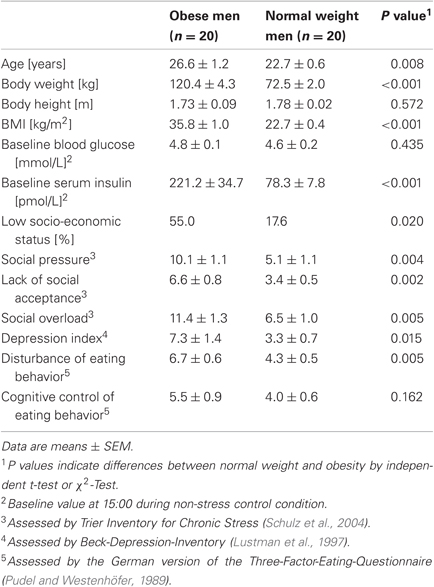
Table 1. Characterization of the study population.
Low HPA Reactivity in Obesity
We first investigated how psychosocial stress activates the stress system. Normal weight men showed high hormonal and autonomic symptomatic stress responses to psychosocial stress and a subsequent meal (Hitze et al., 2010). While the obese subjects studied here also showed a marked response to psychosocial stress (Figure 2) (which did not differ from that in normal weight subjects, all P > 0.05), they did not display any ACTH- and cortisol response to the subsequent meal. In detail, cortisol concentrations increased by 23.5 ± 18.9% in response to meal in normal weight subjects, while cortisol concentrations decreased by 31.9 ± 2.6% in obese men (P < 0.01). Thus, normal weight subjects showed more pronounced meal-related cortisol peaks after stress intervention than obese subjects (Figure 2) [interaction time (17:00–17:30) × weight group F = 10.0, P = 0.005; main effect time F = 0.2, P = 0.682; main effect weight group F = 0.4, P = 0.570; effect covariate age F = 0.2, P = 0.629]. These results demonstrate that the stress system is low reactive in obesity.
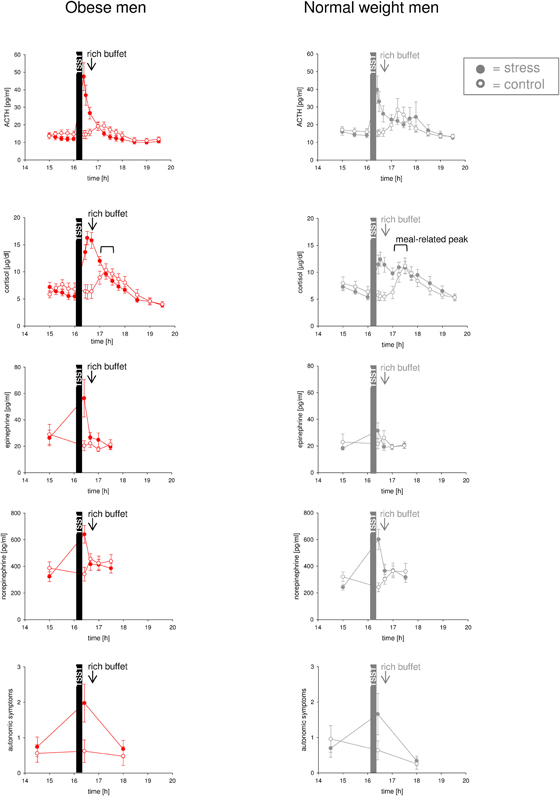
Figure 2. Hormonal and autonomic symptomatic reactivity during stress intervention and non-stress control condition in obese men (colored symbols) and normal weight men (gray symbols). All subjects were offered a rich buffet at the same time (see arrow). Noteworthy, the meal-related cortisol peak was absent in obese subjects during stress intervention (in contrast to normal weight subjects and the non-stress control conditions). Values are means ± SEM; closed symbols, stress intervention and open symbols, non-stress control condition. For comparison, data of normal weight subjects are taken from [Hitze et al. (2010)].
Cerebral Insulin Suppression was not Evident in Obesity
In order to test whether CIS was operative during psychosocial stress we compared the stress with the non-stress condition. Normal weight men exert CIS in the post-stress replenishment phase (Hitze et al., 2010). That is, blood glucose concentrations increased, but were not accompanied by corresponding increases in serum insulin concentrations. As we know that cortisol suppresses pancreatic insulin secretion (Billaudel and Sutter, 1982) and thus is able to exert CIS, we next tested in a HLM whether the meal-related cortisol peak was linked to CIS. In normal weight men, a steeper meal-related cortisol increase (time: 17:00 till 17:30) was associated with a more pronounced suppression of insulin concentrations in the post-stress replenishment phase (16:00 and 16:40–17:30) (coefficient for interaction time slope insulin and meal-related cortisol increase = −0.131246, SE = 0.035799, P = 0.001, for stress intervention only; HLM), whereas the meal-related cortisol increase (time: 17:00 till 17:30) was not associated with glucose concentrations in the post-stress replenishment phase (16:00 and 16:40–17:30) (coefficient = −0.001658, SE = 0.002909, P = 0.571 for stress intervention only; HLM). These results support the notion that CIS was operative in our normal weight subjects.
In the obese men, however, stress did not lead to an observable increase in glucose concentrations in the post-stress replenishment phase (Figure 3) (interaction time (16:00 and 16:40 till 17:30) × stress F = 0.6, P = 0.554; main effect time F = 16.2, P = 0.001; main effect stress F = 6.1, P = 0.035). Likewise, stress did not result in a detectable increase in insulin concentrations in the post-stress replenishment phase (Figure 3) (interaction time (16:00 and 16:40 till 17:30) × stress F = 0.4, P = 0.771; main effect time F = 9.7, P = 0.001; main effect stress F = 3.2, P = 0.111). Thus, CIS was not evident in obesity.
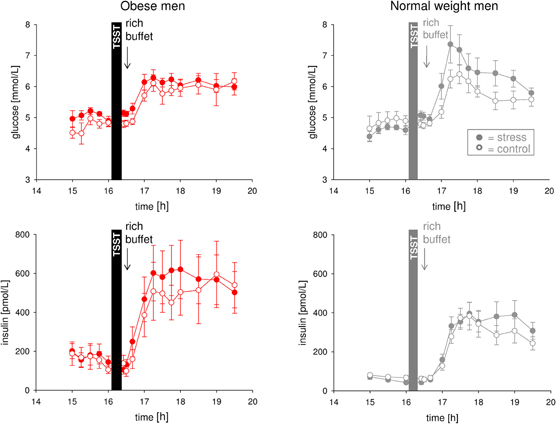
Figure 3. Glucose and insulin concentrations during stress intervention and the non-stress control condition in obese men (red symbols) and normal weight men (gray symbols). All subjects were offered a rich buffet at the same time (see arrow). While normal weight men displayed suppression of insulin (CIS) under stress conditions (that is blood glucose concentrations increased, but were not accompanied by corresponding increases in serum insulin concentrations), dynamical differences in glucose and insulin concentrations between stress intervention and the non-stress control condition were not observable in obese subjects. Legends as in Figure 2.
The absence of CIS in obese men may be due to the absence of their meal-related cortisol peak, as in the normal weight group there was a link between the meal-related cortisol peak and CIS. These findings in obese subjects suggest low reactivity of brain-pull function leading to an absence of CIS.
The Hyperinsulinemia was not Driven by Blood Glucose
When comparing the obese and normal weight subjects, we found the post-stress insulin concentrations markedly higher in the obese men (main effect weight group F = 7.0, P = 0.016; main effect time (16:25 till 17:15) F = 21.9, P < 0.001; interaction time × weight group F = 2.4, P = 0.103) (Figure 3). By contrast, the post-stress blood glucose profiles did not show any difference between obese and normal weight subjects (main effect weight group F = 0.1, P = 0.740; main effect time F = 25.2, P < 0.001; interaction time (16:25 till 17:15) × weight group F = 3.3, P = 0.065). Thus, it is unlikely that differences in glucose concentrations between normal weight and obese men have caused a hyperinsulinaemic state. This glucose-insulin pattern (hyperinsulinemia at normal blood glucose) was not only observed in the post-stress phase, but also under baseline conditions (Table 1). Thus, obese subjects displayed marked hyperinsulinemia despite normoglycemia before and after stress. In all, our results indicate that in the obese subjects glucose concentrations did not drive (i.e., were not the cause of) hyperinsulinemia.
Hypervigilance was not Evident in Obese Men
We hypothesized that social stress elicits hypervigilance in obese men. Normal weight men switched into a higher state of vigilance during stress (interaction time (14:30–16:25) × stress F = 8.0, P = 0.011; main effect time F = 1.5, P = 0.234; main effect stress F = 0.0, P = 0.856) (Figure 4). By contrast, stressed obese subjects remained in their prevailing state of vigilance (interaction time (14:30–16:25) × stress F = 0.4, P = 0.555; main effect time F = 0.0, P = 0.915; main effect stress F = 4.1, P = 0.057) (Figure 4). Thus, psychosocial stress elicited hypervigilance in normal weight men, but that was not evident in obese subjects.
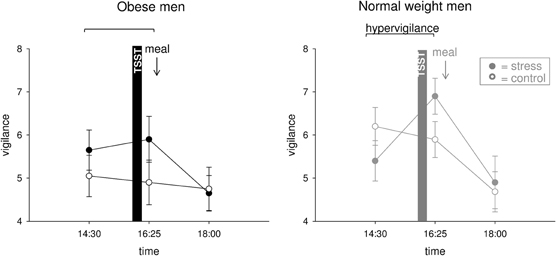
Figure 4. Vigilance ratings during stress intervention and non-stress control condition in obese (black symbols) and normal weight men (gray symbols). Social stress elicited hypervigilance in normal weight men, but that was not observable in obesity. Original scale of vigilance ratings ranges from 0 to 9. Legends as in Figure 2.
Post-Stress Neuroglycopenia in Normal Weight, but not in Obese Subjects
The observed stress-induced hypervigilance in normal weight men was accompanied by the development of a neuroglycopenic state (Hitze et al., 2010) (Figure 5). Obese subjects neither showed evidence of hypervigilance, nor of a neuroglycopenic state after stress (Figure 5) (interaction time (14:30–16:25) × stress F = 1.5, P = 0.241; main effect time F = 5.0, P = 0.037; main effect stress F = 0.02, P = 0.899).
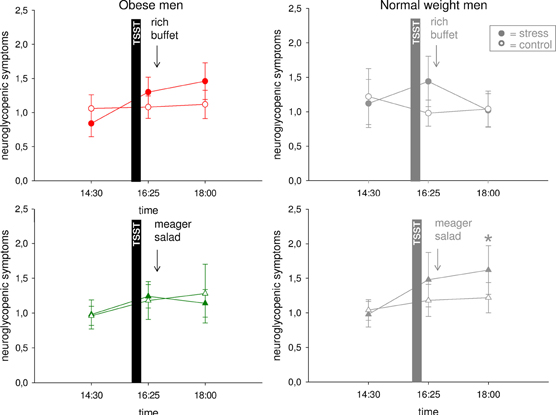
Figure 5. Neuroglycopenic symptoms during stress intervention and non-stress control condition in obese men (colored symbols) and normal weight men (gray symbols). Social stress induced a neuroglycopenic state in normal weight, but that was not detectable in obesity. Original scale of neuroglycopenic symptoms ranges from 0 to 9. Legends as in Figure 2. *P < 0.05, significantly different from non-stress control condition, by dependent t-test.
In normal weight subjects, high energy supplementation resolved the post-stress neuroglycopenic state, whereas in the meager salad experimental group neuroglycopenic symptoms persisted at a high-level (Hitze et al., 2010). To address the question whether low energy supplementation also affected neuroglycopenic symptoms in obese subjects we investigated another experimental group, who were offered a meager salad only. In obese subjects, energy supplementation (that is rich buffet vs. meager salad) did not influence post-stress neuroglycopenic symptoms (Figure 5) [interaction time (16:25–18:00) × stress F = 0.03, P = 0.869; interaction time × stress × experimental group F = 0.5; P = 0.511; main effect time F = 0.2, P = 0.682; main effect stress F = 0.7, P = 0.421; main effect experimental group F = 0.01, P = 0.915].
Thus, in obese subjects neither signs of hypervigilance nor signs of a neuroglycopenic state were detectable.
Obese Men did not use Carbohydrates for Recovery from Stress
We then tested the hypothesis that social stress alters food choice. Total energy intake from the rich buffet of normal weight and obese men was equal both during stress intervention and non-stress control condition (Figure 6). This finding was independent of both cognitive control in eating behavior and disturbances in eating behavior. Food choice from the rich buffet, however, differed between normal weight and obese men. Carbohydrate intake was increased after stress in normal weight subjects by 34 g and was larger than in the obese men (183.3 ± 15.6 g vs. 120.5 ± 23.5 g; P = 0.039). By contrast, fat and protein intake was increased after stress in obese men in total by 26 g (fat: + 11 g and protein: + 15 g) (Figure 6). Detailed analysis of specific buffet components did not reveal any differences in the ingestion of the respective components such as salmon, potato chips, salami, gummy bears, chocolate, bap, sweet food, and sweet drinks when comparing the stress intervention and the non-stress control condition. These results demonstrate that food preferences were altered after stress: normal weight men preferred carbohydrates, obese men fat and protein.
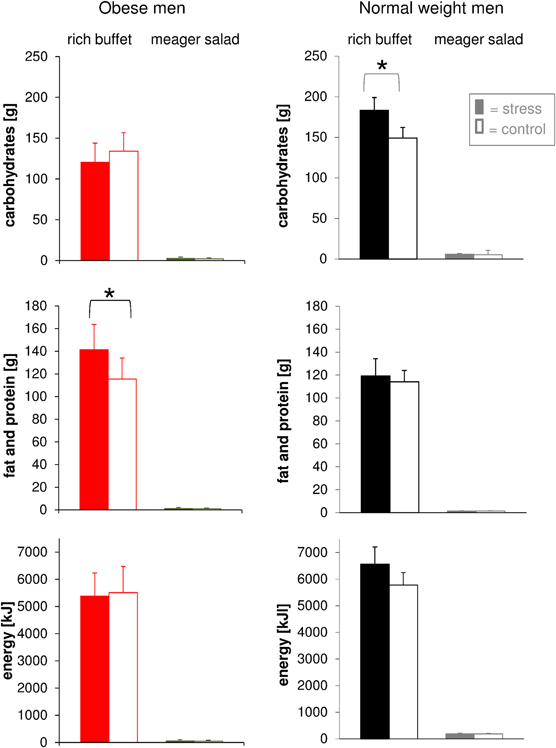
Figure 6. Post-stress macro-nutrient and energy intake in obese men (colored bars) and normal weight men (gray bars) offered a rich buffet or a meager salad; non-stress control condition (white bars). Normal weight men increased their carbohydrate intake, whereas obese men increased fat and protein intake. Legend as in Figure 2. *P < 0.05, significantly different from non-stress control condition, by dependent t-test.
Good Mood Requires Recovery from HPA Overactivation and Neuroglycopenia
We finally asked whether mood is impaired by psychosocial stress. In normal weight men, mood was rated to worsen immediately after stress. In the post-stress replenishment phase, mood recovered when high-energy was provided, but when a meager salad was offered mood remained bad (Hitze et al., 2010) (Figure 7).
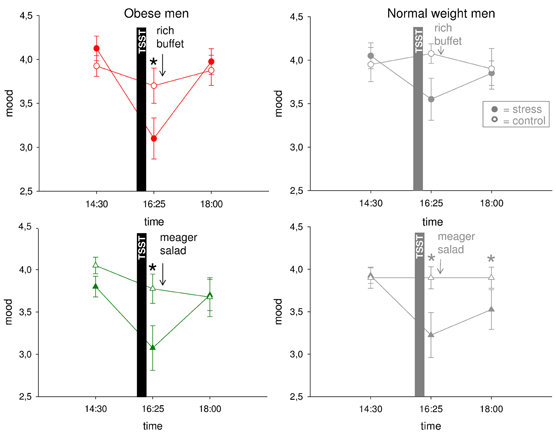
Figure 7. Mood ratings during stress intervention and non-stress control condition in obese men (colored symbols) and normal weight men (gray symbols). Mood recovered with resolving HPA overactivation and neuroglycopenia. Original scale of mood ratings ranges from 0 to 9. Legends as in Figure 2. *P < 0.05, significantly different from non-stress control condition, by dependent t-test.
In the obese subjects studied here, mood (Figure 7) was also deteriorated by social stress [interaction time (14:30–16:25) × stress F = 8.5, P = 0.009; main effect time F = 23.7, P < 0.001; main effect stress F = 8.2, P = 0.010]. In the post-stress replenishment phase, mood ratings were restored [interaction time (16:25–18:00) × stress F = 22.1, P < 0.001; main effect time F = 27.4, P < 0.001; main effect stress F = 3.8, P = 0.070]. In the obese subjects, recovery from bad mood occurred even with ingestion of the meager salad. There was no difference between both experimental groups (i.e., rich buffet vs. meager salad) (interaction time × stress × group F = 0.0, P = 0.935; main effect group F = 0.2, P = 0.637). Note: A neuroglycopenic state was not evident in obese subjects.
In all, in the stress phase, mood deteriorated both in normal weight and obese men. In the post-stress replenishment phase, however, obese subjects recovered their mood in parallel with decreasing cortisol concentrations. In the normal weight subjects, however, the decrease in cortisol concentrations was not sufficient—recovery from neuroglycopenia was also necessary to restore good mood.
Discussion
The experiments presented here show that CIS is not evident in obese men, i.e., insulin concentrations were not suppressed by our stress intervention. Neither were there any signs of hypervigilance during psychosocial stress in obese subjects. Further, obese men increased their fat and protein intake for replenishment after stress, while normal weight subjects increased their carbohydrate intake to supply their brain and body with energy (Table 2).
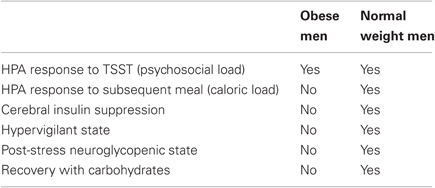
Table 2. Summary of results.
Our first main finding was that obese subjects displayed a much lower HPA reactivity than the normal controls during the psychosocial stress experiment (Figure 2). While obese subjects showed a marked immediate sympatho-adrenal response to the psychosocial load, they did not respond to the subsequent meal (caloric load). In this respect, obese subjects showed a sort of stiffness or low reactivity in their stress system. This finding is in line with several previous studies mentioned in the introduction showing that on the one hand low stress reactivity is predictive of weight gain (Flaa et al., 2008) and on the other hand obese subjects and animals exhibit low stress reactivity to various challenges (Nielsen et al., 1993; Spraul et al., 1994; Vea et al., 1994; Levin et al., 2000; Tkacs and Levin, 2004; Eliakim et al., 2006; Shin et al., 2010; Azarbayjani et al., 2011). Moreover, heart rate variability, a measure of the autonomic-visceromotor system's reactivity, is known to be reduced in obese subjects, which indicates the stress system's low reactivity (Felber et al., 2006). Reduced heart rate variability has also been shown to predict the development of hyperglycemia and diabetes (Carnethon et al., 2003; Hemingway et al., 2005). All these findings support the importance of the stress system's reactivity in the development and progression of obesity and type 2 diabetes.
A limitation of our study was that the psychosocial load imposed to our obese and normal weight subjects in the Trier Social Stress Test was not necessarily equal. It is known, that obese subjects severely suffer from weight discrimination (Puhl and Heuer, 2009). In fact, our obese subjects showed greater signs of social pressure and feelings of social non-acceptance, signs of social overload, and a high number of depressive symptoms (Table 1). Moreover, obese men had a lower socio-economic status, which is not only associated with increased social stress but also is a strong predictor of weight gain (Martikainen and Marmot, 1999). It is possible that our normal weight TSST jury members might have been perceived as more stressful by obese than by normal weight subjects. Since we cannot rule out that the psychosocial load was stronger for obese than for normal weight men, our observation of an apparently equal stress-hormone response to psychosocial load must be interpreted with caution.
The second main finding was that CIS was not detectable in our obese subjects (Figure 3). In other words, insulin concentrations were not suppressed by the stress-system. That means that in obese men brain-pull function was not highly activated to demand for cerebral energy. Our results indicate that in normal weight subjects, who highly activated their CIS-brain-pull, the energy flux in the post-stress-replenishment phase was rather directed to the brain, and that in obese subjects, who did not activate their CIS-brain-pull, the flux was rather directed to the peripheral energy stores. The absence of CIS in obesity was likely due to the low reactivity of their stress system evidenced by the absence of a meal-related cortisol peak (Figure 2). We found that in normal weight subjects the reactivity of the stress system assessed by HPA-activity was clearly linked to CIS. In contrast, our obese subjects neither showed a meal-related cortisol peak, nor CIS during the meal. Similarly, it was found in the ARIC-study that the reactivity of the stress system—assessed by heart rate variability—was associated with CIS; in detail, those subjects who displayed low heart rate variability (i.e., low reactivity) did not suppress their insulin concentrations, no matter how their levels of glycemia were (Schroeder et al., 2005). In this way, the ARIC-data support our first and second main finding. Our finding, that obese people do not activate CIS, has already been forecasted by the “Selfish Brain” theory postulating that in the cerebral supply chain a low reactive brain-pull constitutes the underlying cause of obesity (Peters and Langemann, 2009).
What could be on the other hand the underlying causes of a low reactive brain-pull? Various causes of low reactive brain-pull have been described in the medical literature ranging from chronic stress, appetitive conditioning, perinatal programming, psychotropic, and metabotrobe drugs, brain tumors, genetic, and epigenetic abnormalities, all being associated with weight gain (Peters and Langemann, 2009). However, chronic stress in adult life may constitute the most frequent cause of low reactive brain-pull. Evidence exists that habituation (i.e., reduction of reactivity) of the stress system to a persistent stressor represents a “metabolic coping strategy” of the brain, which on the one hand allows to relieve the overloaded stress system and to improve mood, but on the other hand makes it necessary to increase eating behavior in order to supply the brain (Peters et al., 2011b). Specifically, it has been shown that several domains of chronic psychosocial stress were associated with weight gain over a 9-year period (Block et al., 2009).
Does a low activity of CIS result in elevated serum insulin concentrations? We found that the hyperinsulinemia in the obese subjects was not driven by blood glucose (Figure 3). It is known that hyperinsulinemia can be driven by blood glucose or other factors, e.g., autonomic or neuroendocrine input to the ß-cells (Ahren, 2000). Blood glucose abnormalities are known to occur in the “insulin resistant state.” Experiments in animals with targeted disruption of the GLUT4 selectively in muscle showed that not only an insulin resistant state developed, but also glucose abnormalities and hyperinsulinemia (Zisman et al., 2000). In humans, the development of glucose abnormalities precede the onset of type 2 diabetes mellitus by at least 13 years, and hyperinsulinemia is already manifest at this earlier stage of the metabolic disease (Tabak et al., 2009). The widely held view that in the insulin resistant state hyperinsulinemia develops in order to compensate blood glucose concentration does hold true only, if blood glucose is actually elevated. The validity of this common view is limited because of the following reason: In a closed-loop-control system, which consists of one controlling variable (x) and one controlled variable (y), with the latter feeding back on the former, changes in the one variable (e.g., insulin) do definitely not occur without changes in the other (e.g., glucose), unless there are other external factors acting (e.g., autonomic input) (DiStefano et al., 1967). As glucose abnormalities were not observed in our obese subjects, we conclude that their hyperinsulinemia was not driven by glucose. By showing that our obese subjects were unable to suppress their insulin concentration by their stress-system, we provide clear evidence that low activity of CIS was a cause of their hyperinsulinemia.
Our third main finding was that hypervigilance was induced by stress in normal weight, but not in obese men (Figure 4). The mechanisms, which underlie that observation, cannot be identified by the present data. However, the results of other neurobiological studies suggest how habituation of the stress reactivity in obese subjects might have led to a low reactivity during psychosocial stress in changing their vigilance.
The locus coeruleus norepinephrine system—located in the brainstem—plays a major role in regulating the behavioral state during stress (Berridge and Waterhouse, 2003; Aston-Jones and Cohen, 2005). Locus coeruleus neurons exhibit two modes of activity, phasic, and tonic. In the phasic mode, locus coeruleus cells exhibit phasic activation in response to the processing of task-relevant stimuli but display only a moderate level of tonic discharge. This mode of function is consistently associated with high levels of task performance. In this way, the locus coeruleus neurons, which send diffuse projections throughout the brain, focus ongoing neuronal activity in a limited number of brain regions. In the tonic mode, locus coeruleus cells fail to respond phasically to task events but exhibit higher levels of ongoing tonic activity. This mode is associated with poor performance on tasks that require focused attention and corresponds to apparent increases in distractibility, alternative behavioral strategies are searched for instead (exploration). In this case, the locus coeruleus allows neuronal activity in a larger number of brain regions. How the neuronal activity and energy consumption change due to the regulation of the locus coeruleus norepinephrine system can be estimated from experiments assessing global brain energy supply. Using invasive measurement techniques, it has been found that different behavioral states differ in cerebral energy supply: deep sleep decreases global cerebral glucose uptake by 40% (Boyle et al., 1994), while acute mental stress increases it by 12% (Madsen et al., 1995).
During psychosocial stress, several brain areas are involved in stress response, e.g., the lateral and central amygdala, the bed nucleus of the stria terminalis provide inputs to the SNS, HPA, and the locus coeruleus norepinephrine system (Dong et al., 2001; Valentino and Van Bockstaele, 2008; Reyes et al., 2011). Corticotropin-releasing factor is the crucial mediator of stress-elicited locus coeruleus activation (Valentino and Van Bockstaele, 2008). Corticotropin-releasing factor elicits an increase in tonic and decrease in phasic discharge of locus coeruleus neurons (Valentino and Van Bockstaele, 2008). These changes of locus coeruleus activity may promote a state of hypervigilance during psychosocial stress (Valentino and Van Bockstaele, 2008).
As shown in our present study, obese subjects displayed a lower HPA reactivity, which can be regarded as a form of habituation in their HPA-system. Such a habituation might occur at the level of the lateral amygdala, the central amygdala, and the bed nucleus of the stria terminalis, which are all glucocorticoid-sensitive (Lechner and Valentino, 1999) and provide heavy input into the ventromedial hypothalamus and the paraventricular nucleus. In addition, the central amygdala and the bed nucleus of the stria terminalis send inputs into the locus coeruleus norepinephrine system. In case of a habituation, the bed nucleus neurons may sustain locus coeruleus neurons in their phasic mode—thus preventing a switch to a tonic mode, which can typically be observed during stress. In this way, a habituation at the striato-pallidal level of the stress system could be responsible for our observation that obese subjects were also low reactive during psychosocial stress in changing their state of vigilance.
The stress-induced hypervigilant state in our normal weight subjects was accompanied by the development of a neuroglycopenic state. Neither hypervigilance nor neuroglycopenia were detectable in our obese subjects (Figure 5). Neuroglycopenic symptoms, which refer to a “shortage of cerebral glucose,” are indicative of an active brain process which serves neuroprotection (Peters et al., 2007). In order to maintain cerebral energy homeostasis during an energy crisis (e.g., hypoglycemia), the brain switches to an energy sparing mode and shuts down certain brain functions. The clinical symptoms indicate ubiquitous cerebral non-functioning, as for example tingling, blurred vision, difficulty to concentrate, etc. It is known that cerebral energy concentration only decreases when the brain's need exceeds the supply. As brain's supply increases during mental stress (Madsen et al., 1995), neuroglycopenia is indicative of an increased cerebral need. The exact origin for neuroglycopenia in normal weight men cannot be identified by the present data. It is likely, however, that the hypervigilance during stress in normal weight subjects has induced such a state of cerebral exhaustion. Thus, in obese men, a neuroglycopenic state may not occur, as psychosocial stress did not elicit hypervigilance. This may also explain, why the brain of normal weight men—in contrast to that of obese men—demands for extra cerebral energy by activating the CIS-brain-pull mechanism, to match the brain's energy supply with the increased cerebral energy needs.
Our fourth main finding was that normal weight subjects chose carbohydrates to recover from stress, whereas obese men chose fat and protein (Figure 6). Thus, it appears that normal weight subjects preferred brain fuel, while obese subjects preferred body fuel. Other researchers also found the carbohydrate intake enhanced after an acute mental challenge (Wardle et al., 2000; Rutters et al., 2009). Taking up food replenishes the energy that the brain has extracted from the body stores during the stressful episode. Stress and eating behavior are closely related (Peters et al., 2011b). Many neuroendocrine mechanisms underlying the regulation of energy replenishment have been discovered recently. For example, neurons of the lateral hypothalamus respond to changes in extracellular glucose concentrations by modulating their tandem-pore K+ channels (Burdakov et al., 2006; Gonzalez et al., 2008). Upon a fall in extracellular glucose, these neurons depolarize. With activation of the lateral hypothalamus neurons, eating behavior is initiated to replenish energy in blood and peripheral buffer stores. In our normal weight men, social stress increased carbohydrate intake from a rich buffet. As CIS was operative in these normal weight men, our results support the view that these stress extra carbohydrates were allocated to the brain to resolve the above mentioned neuroglycopenic state, i.e., to cover the increased cerebral need during stress. For that reason, normal weight men might have chosen carbohydrates when stressed. By contrast, obese men increased their fat and protein intake after stress (Figure 6). As CIS was not detectable in these obese men, our results support the notion that the extra fat and protein was allocated to the open body stores. For that reason, obese men might have chosen body fuel when stressed.
Our fifth main finding was that good mood was re-established with resolving HPA overactivation and neuroglycopenia (Figure 7). How stress and the stress response influence our emotions is a central question in psychology and medicine. Previous studies have already shown that stress deteriorates mood (Kuhlmann et al., 2005; Het and Wolf, 2007; Hitze et al., 2010). We can now specify that changes in the HPA system are parallel to the changes in mood (Table 3). Our results add new aspects to the “comfort food” hypothesis (Dallman et al., 2003), which associated decreased HPA-system activity with increased ingestion of energy-rich food. Our results now indicate that the re-establishment of cerebral energy homeostasis is also necessary to regain a balanced emotional state. Good mood was only achieved when both cortisol concentrations were in a normal range and cerebral energy homeostasis (i.e., absence of neuroglycopenia) was achieved (Table 3). In all, a balanced neuroenergetic homeostasis and the stress system being in a state of rest is a necessary requirement for feeling good.
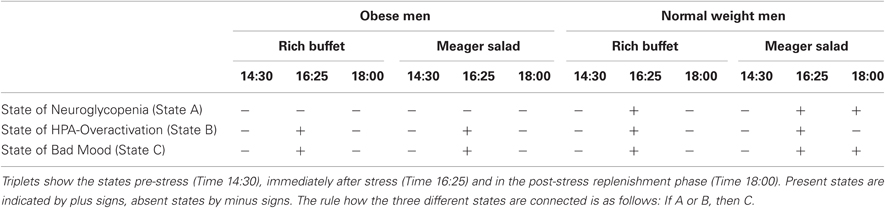
Table 3. Overview of the interrelation between the states of neuroglycopenia, HPA-overactivation and bad mood.
Are the changes we found in the reactivity in obese people due to default or adaptation? Medical reasoning always refers to categories like health and disease, competence or incompetence, efficiency or inefficiency. In our first paper describing the supply chain of the brain (Peters and Langemann, 2009) we used such medical reasoning for interpretation and regarded a low reactivity of the stress system (brain-pull) as an “incompetence.” From an evolutionary prospective, however, high and low reactivity of the stress system (brain-pull) can also be regarded as a phenotypic trait of an individual belonging to a certain population, which might be favorable or unfavorable under respective environmental conditions. We are inclined now to prefer the evolutionary prospective using the concept of “weight diversity,” particularly because recent data—which report on the so-called “obesity paradox”—provide evidence that the changes in physiologic variables in obese people are indeed adaptive. The obesity paradox relies on data sets showing that in the general population a higher body weight (e.g., BMI 30) is associated with increased mortality (Whitlock et al., 2009); paradoxically in an population which is burdened by diseases like type 2 diabetes and cardiovascular disease, a lower body weight (BMI 25) has been found to be associated with an even more increased mortality (Doehner et al., 2011). The same paradox has been shown for people who carry the load of diseases like myocardial infarction, stroke, cerebral hemorrhage, heart and kidney disease, chronic obstructive pulmonary disease, and sepsis (Buettner et al., 2007; Hallin et al., 2007; Elsayed et al., 2008; Sakr et al., 2008; Kim et al., 2011; Lavie et al., 2011; Vemmos et al., 2011): under all these stressful circumstances obese people display better survival rates than normal weight people. How can these “counterintuitive” data be explained?
Recently, a solution of the “obesity paradox” has been proposed (Peters and McEwen, 2012): In short, key to understanding is the reactivity of the stress system (brain-pull). On the one hand the reactivity of the stress system (brain-pull) is positively related to mortality; on the other hand it is inversely related to body weight. In detail, the lower the reactivity of the stress system (brain-pull) is, the lower is the increase of cortisol concentrations in people who are burdened by chronic psychosocial stress or by diseases (McEwen, 1998); and if the cortisol concentrations are lower, then also the peoples' all-cause and cardiovascular mortality is lower (Schoorlemmer et al., 2009; Vogelzangs et al., 2010; Kumari et al., 2011). In contrast, the lower the reactivity of the stress system (brain-pull) is, the higher is the body weight of people who have access to a sufficiently large food offer (Peters and Langemann, 2009). Thus, people who habituate in their stress response, when they are exposed to persistent environmental stressors and when food is available, on the one hand display decreased mortality, but on the other hand display increased body weight. All these data support the notion, that phenotypic plasticity of the stress system may serve to reduce the allostatic load, e.g., the sequels of long-term overactivation of the stress system: muscle loss, osteoporosis, disruption of growth and reproduction, memory impairment, depression, and mortality. Thus, it appears as if the phenotypic plasticity of the stress system of people who are chronically stressed stabilize their organism in a new equilibrium at higher body mass values allowing better survival.
The changes we found in the reactivity of vigilance in obese people are of crucial benefit too. During acute psychosocial stress, we found that normal weight subjects increased both their vigilance and their cerebral need (Hitze et al., 2010). More need usually prompts more demand, but a higher cerebral demand would burden the stress-brain-pull system. During chronic stress, switching into a higher state of vigilance would add to the allostatic load. Thus, a lower reactivity of vigilance, as we observed in obese people, may be regarded as protective against the various sequels of long-term overactivation of the stress system [(McEwen and Peters (eds.), 2012)]—both in favor of the brain and the body.
In conclusion, our results indicate that the brain of obese people organizes its need, supply and demand in a low reactive manner.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Ann-Sophie Lindenberg, Kristin Schlichting, Irena Zeiß, Saskia Otte, Kay Duysen, Kirstin Nordhausen, and Jutta Schwanbom for carrying out the experiments in normal weight men and for their expert administrative and laboratory assistance.
Disclosure
This work was supported by a grant from the German Research Foundation (Clinical Research Group KFO-126).
References
Ahren, B. (2000). Autonomic regulation of islet hormone secretion–implications for health and disease. Diabetologia 43, 393–410.
Allison, S. P., Hinton, P., and Chamberlain, M. J. (1968). Intravenous glucose-tolerance, insulin, and free-fatty-acid levels in burned patients. Lancet 2, 1113–1116.
Aston-Jones, G., and Cohen, J. D. (2005). An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu. Rev. Neurosci. 28, 403–450.
Azarbayjani, M. A., Vaezepor, F., Rasaee, M. J., Tojaril, F., Pournemati, P., Jourkesh, M., Ostojic, S. M., and Stannard, S. R. (2011). Daily timing of salivary cortisol responses and aerobic performance in lean and obese active females. Bratisl. Lek. Listy 112, 213–217.
Berridge, C. W., and Waterhouse, B. D. (2003). The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res. Brain Res. Rev. 42, 33–84.
Billaudel, B., and Sutter, B. C. (1982). Immediate in-vivo effect of corticosterone on glucose-induced insulin secretion in the rat. J. Endocrinol. 95, 315–320.
Block, J. P., He, Y., Zaslavsky, A. M., Ding, L., and Ayanian, J. Z. (2009). Psychosocial stress and change in weight among US adults. Am. J. Epidemiol. 170, 181–192.
Boyle, P. J., Scott, J. C., Krentz, A. J., Nagy, R. J., Comstock, E., and Hoffman, C. (1994). Diminished brain glucose metabolism is a significant determinant for falling rates of systemic glucose utilization during sleep in normal humans. J. Clin. Invest. 93, 529–535.
Buettner, H. J., Mueller, C., Gick, M., Ferenc, M., Allgeier, J., Comberg, T., Werner, K. D., Schindler, C., and Neumann, F. J. (2007). The impact of obesity on mortality in UA/non-ST-segment elevation myocardial infarction. Eur. Heart J. 28, 1694–1701.
Burdakov, D., Jensen, L. T., Alexopoulos, H., Williams, R. H., Fearon, I. M., O'Kelly, I., Gerasimenko, O., Fugger, L., and Verkhratsky, A. (2006). Tandem-pore K+ channels mediate inhibition of orexin neurons by glucose. Neuron 50, 711–722.
Carnethon, M. R., Golden, S. H., Folsom, A. R., Haskell, W., and Liao, D. (2003). Prospective investigation of autonomic nervous system function and the development of type 2 diabetes: the Atherosclerosis Risk in Communities study, 1987–1998. Circulation 107, 2190–2195.
Cerchio, G. M., Moss, G. S., Popovich, P. A., Butler, E., and Siegel, D. C. (1971). Serum insulin and growth hormone response to hemorrhagic shock. Endocrinology 88, 138–143.
Chan, O., Lawson, M., Zhu, W., Beverly, J. L., and Sherwin, R. S. (2007). ATP-sensitive K(+) channels regulate the release of GABA in the ventromedial hypothalamus during hypoglycemia. Diabetes 56, 1120–1126.
Chen, X. Q., Dong, J., Niu, C. Y., Fan, J. M., and Du, J. Z. (2007). Effects of hypoxia on glucose, insulin, glucagon, and modulation by corticotropin-releasing factor receptor type 1 in the rat. Endocrinology 148, 3271–3278.
Dallman, M. F., Pecoraro, N., Akana, S. F., La Fleur, S. E., Gomez, F., Houshyar, H., Bell, M. E., Bhatnagar, S., Laugero, K. D., and Manalo, S. (2003). Chronic stress and obesity: a new view of “comfort food”. Proc. Natl. Acad. Sci. U.S.A. 100, 11696–11701.
DiStefano, J. J., Stubberud, A. R., and Williams, I. J. (1967). Theory and Problems of Feedback and Control Systems. New York, NY: McGraw-Hill.
Doehner, W., Erdmann, E., Cairns, R., Clark, A. L., Dormandy, J. A., Ferrannini, E., and Anker, S. D. (2011). Inverse relation of body weight and weight change with mortality and morbidity in patients with type 2 diabetes and cardiovascular co-morbidity: an analysis of the PROactive study population. Int. J. Cardiol. (Epub ahead of print).
Dong, H. W., Petrovich, G. D., and Swanson, L. W. (2001). Topography of projections from amygdala to bed nuclei of the stria terminalis. Brain Res. Brain Res. Rev. 38, 192–246.
Eliakim, A., Nemet, D., Zaldivar, F., McMurray, R. G., Culler, F. L., Galassetti, P., and Cooper, D. M. (2006). Reduced exercise-associated response of the GH-IGF-I axis and catecholamines in obese children and adolescents. J. Appl. Physiol. 100, 1630–1637.
Elsayed, E. F., Sarnak, M. J., Tighiouart, H., Griffith, J. L., Kurth, T., Salem, D. N., Levey, A. S., and Weiner, D. E. (2008). Waist-to-hip ratio, body mass index, and subsequent kidney disease and death. Am. J. Kidney Dis. 52, 29–38.
Felber, D. D., Schindler, C., Schwartz, J., Barthelemy, J. C., Tschopp, J. M., Roche, F., von Eckardstein, A., Brandli, O., Leuenberger, P., Gold, D. R., Gaspoz, J. M., and Ackermann-Liebrich, U. (2006). Heart rate variability in an ageing population and its association with lifestyle and cardiovascular risk factors: results of the SAPALDIA study. Europace 8, 521–529.
Flaa, A., Sandvik, L., Kjeldsen, S. E., Eide, I. K., and Rostrup, M. (2008). Does sympathoadrenal activity predict changes in body fat? An 18-y follow-up study. Am. J. Clin. Nutr. 87, 1596–1601.
Franssen, F. M., Sauerwein, H. P., Ackermans, M. T., Rutten, E. P., Wouters, E. F., and Schols, A. M. (2011). Increased postabsorptive and exercise-induced whole-body glucose production in patients with chronic obstructive pulmonary disease. Metabolism 60, 957–964.
Gonzalez, J. A., Jensen, L. T., Fugger, L., and Burdakov, D. (2008). Metabolism-independent sugar sensing in central orexin neurons. Diabetes 57, 2569–2576.
Hallin, R., Gudmundsson, G., Suppli, U. C., Nieminen, M. M., Gislason, T., Lindberg, E., Brondum, E., Aine, T., Bakke, P., and Janson, C. (2007). Nutritional status and long-term mortality in hospitalised patients with chronic obstructive pulmonary disease (COPD). Respir. Med. 101, 1954–1960.
Harada, S., Fujita, W. H., Shichi, K., and Tokuyama, S. (2009). The development of glucose intolerance after focal cerebral ischemia participates in subsequent neuronal damage. Brain Res. 1279, 174–181.
Hasselbalch, S. G., Knudsen, G. M., Jakobsen, J., Hageman, L. P., Holm, S., and Paulson, O. B. (1995). Blood-brain barrier permeability of glucose and ketone bodies during short-term starvation in humans. Am. J. Physiol. 268, E1161–E1166.
Hemingway, H., Shipley, M., Brunner, E., Britton, A., Malik, M., and Marmot, M. (2005). Does autonomic function link social position to coronary risk? The Whitehall II study. Circulation 111, 3071–3077.
Het, S., and Wolf, O. T. (2007). Mood changes in response to psychosocial stress in healthy young women: effects of pretreatment with cortisol. Behav. Neurosci. 121, 11–20.
Hitze, B., Hubold, C., van Dyken, R., Schlichting, K., Lehnert, H., Entringer, S., and Peters, A. (2010). How the selfish brain organizes its “supply and demand”. Front. Neuroenerg. 2:7. doi: 10.3389/fnene.2010.00007
Kim, B. J., Lee, S. H., Ryu, W. S., Kim, C. K., Lee, J., and Yoon, B. W. (2011). Paradoxical longevity in obese patients with intracerebral hemorrhage. Neurology 76, 567–573.
Kirschbaum, C., Pirke, K. M., and Hellhammer, D. H. (1993). The “Trier Social Stress Test”-a tool for investigating psychobiological stress responses in a laboratory setting. Neuropsychobiology 28, 76–81.
Kruyt, N. D., Musters, A., Biessels, G. J., Devries, J. H., Coert, B. A., Vergouwen, M. D., Horn, J., and Roos, Y. B. (2011). Beta-cell dysfunction and insulin resistance after subarachnoid haemorrhage. Neuroendocrinology 93, 126–132.
Kuhlmann, S., Piel, M., and Wolf, O. T. (2005). Impaired memory retrieval after psychosocial stress in healthy young men. J. Neurosci. 25, 2977–2982.
Kumari, M., Shipley, M., Stafford, M., and Kivimaki, M. (2011). Association of diurnal patterns in salivary cortisol with all-cause and cardiovascular mortality: findings from the Whitehall II study. J. Clin. Endocrinol. Metab. 96, 1478–1485.
Lavie, C. J., De, S. A., Patel, D., Artham, S. M., and Milani, R. V. (2011). Body composition and coronary heart disease mortality-an obesity or a lean paradox? Mayo Clin. Proc. 86, 857–864.
Lechner, S. M., and Valentino, R. J. (1999). Glucocorticoid receptor-immunoreactivity in corticotrophin-releasing factor afferents to the locus coeruleus. Brain Res. 816, 17–28.
Levin, B. E., Richard, D., Michel, C., and Servatius, R. (2000). Differential stress responsivity in diet-induced obese and resistant rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R1357–R1364.
Lustman, P. J., Clouse, R. E., Griffith, L. S., Carney, R. M., and Freedland, K. E. (1997). Screening for depression in diabetes using the Beck Depression Inventory. Psychosom. Med. 59, 24–31.
Madsen, P. L., Hasselbalch, S. G., Hagemann, L. P., Olsen, K. S., Bulow, J., Holm, S., Wildschiodtz, G., Paulson, O. B., and Lassen, N. A. (1995). Persistent resetting of the cerebral oxygen/glucose uptake ratio by brain activation: evidence obtained with the Kety-Schmidt technique. J. Cereb. Blood Flow Metab. 15, 485–491.
Martikainen, P. T., and Marmot, M. G. (1999). Socioeconomic differences in weight gain and determinants and consequences of coronary risk factors. Am. J. Clin. Nutr. 69, 719–726.
McEwen, B. S. (1998). Protective and damaging effects of stress mediators. N. Engl. J. Med. 338, 171–179.
McPherson, R. J., Mascher-Denen, M., and Juul, S. E. (2009). Postnatal stress produces hyperglycemia in adult rats exposed to hypoxia-ischemia. Pediatr. Res. 66, 278–282.
Miki, T., Liss, B., Minami, K., Shiuchi, T., Saraya, A., Kashima, Y., Horiuchi, M., Ashcroft, F., Minokoshi, Y., Roeper, J., and Seino, S. (2001). ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat. Neurosci. 4, 507–512.
Mulder, A. H., Tack, C. J., Olthaar, A. J., Smits, P., Sweep, F. C., and Bosch, R. R. (2005). Adrenergic receptor stimulation attenuates insulin-stimulated glucose uptake in 3T3-L1 adipocytes by inhibiting GLUT4 translocation. Am. J. Physiol. Endocrinol. Metab. 289, E627–E633.
Nielsen, B., Astrup, A., Samuelsen, P., Wengholt, H., and Christensen, N. J. (1993). Effect of physical training on thermogenic responses to cold and ephedrine in obesity. Int. J. Obes. Relat. Metab. Disord. 17, 383–390.
Peters, A., Bosy-Westphal, A., Kubera, B., Langemann, D., Goele, K., Later, W., Heller, M., Hubold, C., and Müller, M. J. (2011a). Why doesn't the brain lose weight, when obese people diet? Obes. Facts 4, 151–157.
Peters, A., Kubera, B., Hubold, C., and Langemann, D. (2011b). The selfish brain: stress and eating behavior. Front. Neurosci. 5:74. doi: 10.3389/fnins.2011.00074
Peters, A., and Langemann, D. (2009). Build-ups in the supply chain of the brain: on the neuroenergetic cause of obesity and type 2 diabetes mellitus. Front. Neuroenergetics 1:2. doi: 10.3389/neuro.14.002.2009
Peters, A., and McEwen, B. S. (2012). Editorial introduction to the allostatic load special issue. Physiol. Behav. 106, 1–4.
Peters, A., Pellerin, L., Dallman, M. F., Oltmanns, K. M., Schweiger, U., Born, J., and Fehm, H. L. (2007). Causes of obesity: looking beyond the hypothalamus. Prog. Neurobiol. 81, 61–88.
Petrovich, G. D., Canteras, N. S., and Swanson, L. W. (2001). Combinatorial amygdalar inputs to hippocampal domains and hypothalamic behavior systems. Brain Res. Brain Res. Rev. 38, 247–289.
Pudel, V., and Westenhöfer, J. (1989). Fragebogen zum Eßverhalten (FEV) Handanweisung. Göttingen: Verlag für Psychologie Dr. C. J. Hogrefe.
Puhl, R. M., and Heuer, C. A. (2009). The stigma of obesity: a review and update. Obesity (Silver Spring) 17, 941–964.
Raudenbush, S. W., and Bryk, A. S. (2002). Hierarchical Linear Models: Applications and Data Analysis Methods. Thousand Oaks, CA: Sage Publications.
Reinmuth, O. M., Scheinberg, P., and Bourne, B. (1965). Total cerebral blood flow and metabolism. Arch. Neurol. 12, 49–66.
Reyes, B. A., Carvalho, A. F., Vakharia, K., and Van Bockstaele, E. J. (2011). Amygdalar peptidergic circuits regulating noradrenergic locus coeruleus neurons: linking limbic and arousal centers. Exp. Neurol. 230, 96–105.
Rutters, F., Nieuwenhuizen, A. G., Lemmens, S. G., Born, J. M., and Westerterp-Plantenga, M. S. (2009). Acute stress-related changes in eating in the absence of hunger. Obesity (Silver Spring) 17, 72–77.
Sakr, Y., Madl, C., Filipescu, D., Moreno, R., Groeneveld, J., Artigas, A., Reinhart, K., and Vincent, J. L. (2008). Obesity is associated with increased morbidity but not mortality in critically ill patients. Intensive Care Med. 34, 1999–2009.
Schoorlemmer, R. M., Peeters, G. M., van Schoor, N. M., and Lips, P. (2009). Relationships between cortisol level, mortality and chronic diseases in older persons. Clin. Endocrinol. (Oxf.) 71, 779–786.
Schroeder, E. B., Chambless, L. E., Liao, D., Prineas, R. J., Evans, G. W., Rosamond, W. D., and Heiss, G. (2005). Diabetes, glucose, insulin, and heart rate variability: the Atherosclerosis Risk in Communities (ARIC) study. Diabetes Care 28, 668–674.
Schulz, P., Schlotz, W., and Becker, P. (2004). Trier Inventar zum chronischen Stress (TICS). Göttingen: Hogrefe.
Seaquist, E. R., Damberg, G. S., Tkac, I., and Gruetter, R. (2001). The effect of insulin on in vivo cerebral glucose concentrations and rates of glucose transport/metabolism in humans. Diabetes 50, 2203–2209.
Shin, A. C., MohanKumar, S. M., Sirivelu, M. P., Claycombe, K. J., Haywood, J. R., Fink, G. D., and MohanKumar, P. S. (2010). Chronic exposure to a high-fat diet affects stress axis function differentially in diet-induced obese and diet-resistant rats. Int. J. Obes. (Lond.) 37, 1218–1226.
Singer, J. D., and Willett, J. B. (2003). Applied Longitudinal Data Analysis: Modeling Change and Event Occurence. New York, NY: Oxford University Press.
Spanswick, D., Smith, M. A., Groppi, V. E., Logan, S. D., and Ashford, M. L. (1997). Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature 390, 521–525.
Spiegel, K., Leproult, R., and Van Cauter, E. (1999). Impact of sleep debt on metabolic and endocrine function. Lancet 354, 1435–1439.
Spraul, M., Anderson, E. A., Bogardus, C., and Ravussin, E. (1994). Muscle sympathetic nerve activity in response to glucose ingestion. Impact of plasma insulin and body fat. Diabetes 43, 191–196.
Steyer, R., Schwenkmezger, O., Notz, P., and Eid, M. (1997). Der Mehrdimensionale Befindlichkeits-fragebogen (MDBF). Göttingen: Hogrefe.
Tabak, A. G., Jokela, M., Akbaraly, T. N., Brunner, E. J., Kivimaki, M., and Witte, D. R. (2009). Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: an analysis from the Whitehall II study. Lancet 373, 2215–2221.
Taylor, S. H., Saxton, C., Majid, P. A., Dykes, J. R., Ghosh, P., and Stoker, J. B. (1969). Insulin secretion following myocardial infarction with particular respect to the pathogenesis of cardiogenic shock. Lancet 2, 1373–1378.
Tkacs, N. C., and Levin, B. E. (2004). Obesity-prone rats have preexisting defects in their counterregulatory response to insulin-induced hypoglycemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R1110–R1115.
Tong, Q., Ye, C., McCrimmon, R. J., Dhillon, H., Choi, B., Kramer, M. D., Yu, J., Yang, Z., Christiansen, L. M., Lee, C. E., Choi, C. S., Zigman, J. M., Shulman, G. I., Sherwin, R. S., Elmquist, J. K., and Lowell, B. B. (2007). Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab. 5, 383–393.
Valentino, R. J., and Van Bockstaele, E. (2008). Convergent regulation of locus coeruleus activity as an adaptive response to stress. Eur. J. Pharmacol. 583, 194–203.
Vea, H., Jorde, R., Sager, G., Vaaler, S., and Sundsfjord, J. (1994). Glycemic thresholds for hypoglycemic responses in obese subjects. Int. J. Obes. Relat. Metab. Disord. 18, 111–116.
Vemmos, K., Ntaios, G., Spengos, K., Savvari, P., Vemmou, A., Pappa, T., Manios, E., Georgiopoulos, G., and Alevizaki, M. (2011). Association between obesity and mortality after acute first-ever stroke: the obesity-stroke paradox. Stroke 42, 30–36.
Vogelzangs, N., Beekman, A. T., Milaneschi, Y., Bandinelli, S., Ferrucci, L., and Penninx, B. W. (2010). Urinary cortisol and six-year risk of all-cause and cardiovascular mortality. J. Clin. Endocrinol. Metab. 95, 4959–4964.
Wardle, J., Steptoe, A., Oliver, G., and Lipsey, Z. (2000). Stress, dietary restraint and food intake. J. Psychosom. Res. 48, 195–202.
Whitlock, G., Lewington, S., Sherliker, P., Clarke, R., Emberson, J., Halsey, J., Qizilbash, N., Collins, R., and Peto, R. (2009). Body-mass index and cause-specific mortality in 900 000 adults: collaborative analyses of 57 prospective studies. Lancet 373, 1083–1096.
Woods, S. C., and Porte, D. Jr. (1974). Neural control of the endocrine pancreas. Physiol. Rev. 54, 596–619.
Zhou, L., Podolsky, N., Sang, Z., Ding, Y., Fan, X., Tong, Q., Levin, B. E., and McCrimmon, R. J. (2010). The medial amygdalar nucleus: a novel glucose-sensing region that modulates the counterregulatory response to hypoglycemia. Diabetes 59, 2646–2652.
Zisman, A., Peroni, O. D., Abel, E. D., Michael, M. D., Mauvais-Jarvis, F., Lowell, B. B., Wojtaszewski, J. F., Hirshman, M. F., Virkamaki, A., Goodyear, L. J., Kahn, C. R., and Kahn, B. B. (2000). Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat. Med. 6, 924–928.
Keywords: brain metabolism, brain-pull, cerebral insulin suppression, experimental human study, obesity, selfish brain theory, stress
Citation: Kubera B, Hubold C, Zug S, Wischnath H, Wilhelm I, Hallschmid M, Entringer S, Langemann D and Peters A (2012) The brain's supply and demand in obesity. Front. Neuroenerg. 4:4. doi: 10.3389/fnene.2012.00004
Received: 05 December 2011; Paper pending published: 10 January 2012;
Accepted: 24 February 2012; Published online: 08 March 2012.
Edited by:
Sabine Bahn, University of Cambridge, UKReviewed by:
Hirac Gurden, CNRS and University Paris Sud, FranceGianfrancesco Goracci, University of Perugia, Italy
Susan Ozanne, University of Cambridge, UK
Paul Guest, University of Cambridge, UK
Copyright: © 2012 Kubera, Hubold, Zug, Wischnath, Wilhelm, Hallschmid, Entringer, Langemann and Peters. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Achim Peters, Head of the Clinical Research Group SELFISH BRAIN, Brain Metabolism, Neuroenergetics, Obesity and Diabetes, Medical Clinic I, University of Lübeck, Ratzeburger Allee 160, 23538 Lübeck, Germany. e-mail: achim.peters@uksh.de
† These authors contributed equally to this work.