Hydrogel–electrospun fiber mat composite coatings for neural prostheses
- 1 William G. Lowrie Department of Chemical and Biomolecular Engineering, The Ohio State University, Columbus, OH, USA
- 2 Department of Materials and Science Engineering, The Ohio State University, Columbus, OH, USA
- 3 Department of Biomedical Engineering, The Ohio State University, Columbus, OH, USA
Achieving stable, long-term performance of implanted neural prosthetic devices has been challenging because of implantation related neuron loss and a foreign body response that results in encapsulating glial scar formation. To improve neuron–prosthesis integration and form chronic, stable interfaces, we investigated the potential of neurotrophin-eluting hydrogel–electrospun fiber mat (EFM) composite coatings. In particular, poly(ethylene glycol)-poly(ε-caprolactone) (PEGPCL) hydrogel–poly(ε-caprolactone) EFM composites were applied as coatings for multielectrode arrays. Coatings were stable and persisted on electrode surfaces for over 1 month under an agarose gel tissue phantom and over 9 months in a PBS immersion bath. To demonstrate drug release, a neurotrophin, nerve growth factor (NGF), was loaded in the PEGPCL hydrogel layer, and coating cytotoxicity and sustained NGF release were evaluated using a PC12 cell culture model. Quantitative MTT assays showed that these coatings had no significant toxicity toward PC12 cells, and neurite extension at day 7 and 14 confirmed sustained release of NGF at biologically significant concentrations for at least 2 weeks. Our results demonstrate that hydrogel–EFM composite materials can be applied to neural prostheses to improve neuron–electrode proximity and enhance long-term device performance and function.
Introduction
Neural prostheses have been explored for over two decades for their potential to restore lost function in the central nervous system (CNS; Grill et al., 2001; Loewenstein et al., 2004; Schwartz, 2004; Schwartz et al., 2006). However, clinical applications of these devices have been limited by the long-term instability of the CNS tissue–device interface (Butson and McIntyre, 2005; Polikov et al., 2005). There are two primary factors that compromise electrode efficiency: (1) local tissue anatomy, which frequently prevents proximal device implantation and results in a significant separation of electrodes from target neurons (Lawrence et al., 2003; Loewenstein et al., 2004) and (2) the formation of an electrically insulating glial sheath composed of compact activated astrocytes and microglia around the device as a result of cellular and tissue responses to the sustained presence of the device (Heiduschka and Thanos, 1998; Turner et al., 1999; Frampton et al., 2010). These limitations ultimately lead to an increase in electrode impedance, which in turn affects device performance. Thus, to reduce the reactive response to implanted devices, considerable efforts have been made to improve the biocompatibility of neural prostheses, including modifications of the electrode material, electrode shape, and implantation techniques (Lawrence et al., 2003; Szarowski et al., 2003; George et al., 2005), application of drugs to minimize reactive gliosis (Retterer et al., 2004; Winter et al., 2007; Jhaveri et al., 2009), and application of electrode coatings such as hydrogels (Winter et al., 2007; Jun et al., 2008; Jhaveri et al., 2009), layer-by-layer films (He and Bellamkonda, 2005; He et al., 2006; Wu et al., 2007), and conducting polymers (Cui et al., 2001, 2003; Abidian and Martin, 2009).
Neurotrophins are a family of proteins widely used in nerve regeneration for their ability to promote neuronal survival and growth (Greene, 1978; Huang and Reichardt, 2001). By incorporating neurotrophins (e.g., nerve growth factor, NGF) into electrode coatings for controlled release to surrounding tissue, target neurons can be stimulated to survive and extend neurites toward electrode sites. This should reduce the separation distance between target neurons and electrodes. Unfortunately, the extremely short in vivo half-lives of neurotrophins make their sustained release challenging (Maysinger et al., 1996; Kishino et al., 2001). Previously, we demonstrated the potential of neurotrophin-eluting poly(ethylene glycol)-poly(lactic acid) (PEGPLA) hydrogels as coatings for planar microelectrode arrays (MEAs; Winter et al., 2007). Photo-polymerized, biodegradable, PEG-based hydrogels were selected for their biocompatibility (Yowell and Blackwell, 2002; Eugene, 2004), ability to gel in situ (Anseth et al., 2002; Jain et al., 2006), and facile tailoring of release characteristics by changing polymer chain length, degradable repeat ratio, and cross-linking density (Sawhney et al., 1993). We showed that PEGPLA hydrogel coatings persisted on electrode surfaces for ∼7 days in PBS immersion bath and ∼11 days under an agarose gel, and released bioactive NGF for at least 9 days (Winter et al., 2007). Although these results were promising in terms of reducing acute immune response caused by the trauma of electrode implantation (∼1.5–3 weeks post-implantation; Turner et al., 1999); coatings capable of longer electrode adhesion and neurotrophin elution are required to address the chronic immune response, which is believed to occur for at least 2 months post-implantation (Turner et al., 1999).
Here, we present hydrogel–electrospun fiber mat (EFM) composite electrode coatings, which adhered to electrode surfaces for over 9 months and eluted neurotrophins for at least 25 days. Since the duration of drug release and coating adhesion on electrodes can be increased by reducing polymer degradation rate (Sawhney et al., 1993; West and Hubbell, 1995), poly(ethylene glycol)-poly(ε-caprolactone) (PEGPCL) hydrogels were used in the composite coating instead of comparative molecular weight PEGPLA because of their slower degradation rate (Gadzinowski et al., 2000; Sun et al., 2003). EFMs have been extensively studied as tissue engineering constructs for their potential to topographically mimic the extracellular matrix (ECM), which is important in controlling cell adhesion, cell morphology, and tissue architecture (Matthews et al., 2002; Luu et al., 2003; Boudriot et al., 2006). Also, EFMs with high porosity and surface area are attractive candidates in controlled drug delivery (Kim et al., 2004) and are capable of maintaining the bioactivity of agents incorporated into fibers by electrospinning (Chew et al., 2005). Poly(ε-caprolactone) (PCL) hydrophobic EFMs were used to construct hydrogel–EFM composite coatings because of their excellent biocompatibility and low biodegradation rate (Huatan et al., 1995). These novel hydrogel–EFM composites combine the appealing features of both hydrogel and EFM systems, and hold great potential to improve the biocompatibility of implanted devices, thereby enhancing chronic electrode performance.
Materials and Methods
PEGPCL Hydrogel–PCL EFM Composite Formation
All chemicals were used as received from Sigma-Aldrich, Inc., unless otherwise stated. Acryl-PEGPCL copolymers were synthesized using a modification of the procedure (Rao et al., 2011) of Hubell and colleagues (Sawhney et al., 1993). Briefly, ring- opening polymerization of ε-caprolactone on the terminal hydroxyl of poly(ethylene glycol) (PEG; Mw: 950–1,050) was initiated by stannous octanoate [Tin(II)2-ethylhexanoate], and completed after 24 h at 130°C under an argon atmosphere. Acryloyl chloride in anhydrous dichloromethane (4 M excess) was added to a solution of purified PEGPCL intermediate and triethylamine (TEA) in anhydrous dichloromethane at a rate of ∼0.20 mL/min in an ∼ −30°C dry ice-acetone bath over 8 h. A thermocouple probe was used to monitor the bath temperature, which was controlled within a range of ±5°C. Acrylation was completed after continuous stirring for 60 h. Byproduct TEA–HCl salt was removed by alternate repeated solvent evaporation and Buchner funnel filtration. After a series of purification steps described in (Rao et al., 2011), the collected diacryl-PEGPCL copolymers were characterized by Fourier transform infrared spectroscopy (Nicolet 6700 FT-IR spectrometer, Thermo Scientific) and nuclear magnetic resonance spectroscopy (1H NMR Bruker DPX400). The 1H NMR and FT-IR spectra confirmed successful synthesis of diacryl-PEGPCL copolymer with a yield of 85.2%.
Poly(ε-caprolactone) mats were fabricated by electrospinning. A 12-wt% solution of PCL (Mw: 65,000) in dichloromethane (Mallinckrodt Chemicals) was dispensed at a flow rate of 3 mL/h, electrospun under a voltage of +30 kV, and deposited onto an aluminum foil target at a 20-cm tip-to-substrate distance (Gaumer et al., 2009). PCL mats were dried in a vacuum oven overnight (Nam et al., 2008) to remove residual dichloromethane. Morphology of PCL EFMs was characterized by scanning electron microscopy (SEM, Quanta 200, FEI). The average fiber diameter was 0.56 ± 0.084 μm (mean ± SD), and the range of fiber diameter distribution was approximately 0.24–8.5 μm. Circular discs (∼1.5 mm dia.) were then cut from EFMs using dermal punches (Acuderm) for composite formation.
Poly(ethylene glycol)-poly(ε-caprolactone) hydrogel–PCL EFM composites were created via photo-polymerization, following the method of Hubbell and colleagues (Sawhney et al., 1993). Precursor solution was prepared by adding initiators (Irgacure 2959, 0.1 wt%, Ciba, Tarrytown, NY, USA, and 1-vinyl-2-pyrrolidone, 0.5 wt%) into a solution of 22 wt% diacryl-PEGPCL polymer in Dulbecco’s phosphate buffered saline (D-PBS). 2 μL of precursor solution was placed on the desired substrate (i.e., Teflon™ for Eluted NGF measurement exp., electrode arrays for electrode coating adhesion, biocompatibility, and eluted NGF bioactivity tests), and exposed to UV illumination (100 W, 365 nm wavelength, Blak-Ray, B-100-AP, UVP) for 2 min to form PEGPCL hydrogel boluses. MEAs for coating adhesion were generously provided by Dr. Stuart Cogan (EIC Laboratories, Norwood, MA, USA), as shown in Figure 1. The arrays are composed of 15 (3 × 5), 400 μm circular, gold electrodes covered by a sputtered 300 nm film of iridium oxide (SIROF) and a 50-nm titanium film between the electrode sites and SIROF, fabricated on a ∼10 mm thick polyimide substrate (Klein et al., 1989). For composites formed on Teflon™ (shown in Figure 2A), PCL mats (D = 1.5 mm) were placed on Teflon™, followed by the deposition of 2 μL PEGPCL precursor solution on top of each mat, and exposed to UV illumination for 90 s. A second PCL mat was then placed on top of each bolus, and exposed to UV illumination for an additional 45 s. For composite electrode coating formation (shown in Figure 2B), 2 mL of PEGPCL precursor solution was dispensed over the distal end of a 3 × 5 electrode array, exposed to UV illumination for 90 s, followed by addition of a PCL mat (D = 1.5 mm) to the top of the bolus and continuing UV exposure for another 45 s.

Figure 1. Optical micrograph of the distal tip of the MEA array.
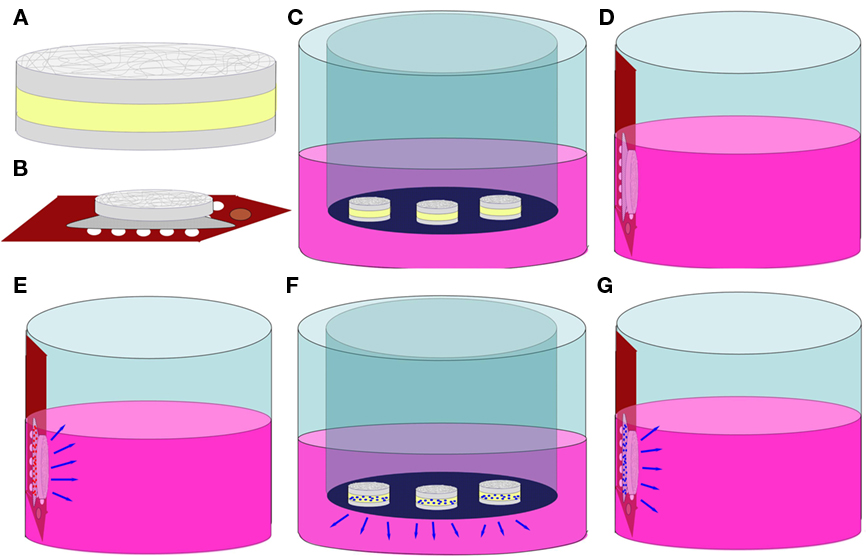
Figure 2. Schematics of biocompatibility, NGF elution, and bioactivity tests. (A) hydrogel–EFM composite material. Gray = EFM, Yellow = Hydrogel; (B) composite coating on a MEA; (C) sample C (hydrogel–EFM composite); (D) sample EC (electrode with composite coating); (E) sample ECB (electrode with BSA-eluting composite coating); (F) NGF elution; (G) NGF bioactivity test.
Characterization of Composite Electrode Coating Adhesion
Poly(ethylene glycol)-poly(ε-caprolactone) hydrogel–PCL EFM composites were deposited as electrode array coatings through UV illumination as described above. The coated electrode array was then inserted into an agarose gel (1% wt/v in D-PBS) and removed immediately. The integrity of coating was visually evaluated by phase contrast microscopy at 10× and 20× magnification. Long-term composite coating adhesion (up to 9 months in D-PBS solution; N = 2 coated electrodes) was assessed by reflected differential interference contrast (Khademhosseini et al., 2004) optical microscopy (Olympus BX41) and SEM (Quanta 200, FEI). Additionally, coated electrode arrays were placed under an agarose gel phantom (1% wt/v in D-PBS), which mimics the tissue that would constrain composite coatings under in vivo conditions. All images were converted to grayscale using Adobe Photoshop (Version 10.0).
Biocompatibility of Composite Electrode Coatings
Before utilization in cell culture and NGF elution experiments, PEGPCL precursor solution was sterile-filtered through a 0.22 μm syringe filter with a 0.8 μm pre-filter (Millipore). Biocompatibility of composite electrode coatings was evaluated by culturing PC12 cells (ATCC, CRL-1721, Manassas, VA, USA) at 1 × 104 cells/cm2 in collagen-coated 24-well tissue-culture plates. Cells were incubated at 37°C, 5% CO2 overnight in 0.5 mL standard culture medium [Ham’s F12K Medium with 2 mM L-glutamine, 1.5 g/L sodium bicarbonate, 2.5% fetal bovine serum, 15% horse serum (all from Sigma-Aldrich), and 1% penicillin–streptomycin (Gibco, Carlsbad, CA, USA)]. Three different samples were evaluated in addition to a control sample (PC12 cells in culture wells). Sample C: Hydrogel–EFM composites (N = 3) of 2 mL hydrogel phase were created on Teflon™ and placed in a transwell insert (Figure 2C). Sample EC: a composite-coated electrode array was attached to the side wall of a well using adhesive carbon tape to avoid direct contact with PC12 cells at the bottom of the well (Figure 2D). Sample ECB: an electrode array coated by composite containing 5 wt% bovine serum albumin (BSA, model drug) in the hydrogel phase was attached to the side wall of a well (Figure 2E). Cells were cultured in the presence of samples (N = 3 for each sample) for 2 weeks and then analyzed by the MTT cell viability assay following the manufacturer’s instructions. Data was collected using a Versamax UV-visible micro-plate reader with measurements performed in triplicate. Pooled data from each repetition was analyzed using one way analysis of variance (ANOVA; α = 0.01). Pair-wise comparisons were performed using the Holm–Sidak method (α = 0.05).
Measurement of Eluted NGF Using Elisa
For NGF elution experiments, sterile PEGPCL precursor solution was added to lyophilized NGF (2.5 S, Promega, Madison, WI, USA) at a concentration of 1 μg/μL. Hydrogel–EFM composites (N = 3) with 2 μL NGF-containing hydrogel phase were created on Teflon™ as described in Section “PEGPCL Hydrogel–PCL EFM Composite Formation,” and placed on the upper surface of 0.4 μm membrane transwell inserts (Corning; Figure 2F). The insert was then placed in a 24-well tissue-culture plate (BD Biosciences) filled with 0.5 mL D-PBS placed in a 37°C, 5% CO2 incubator. To determine the amount of eluted NGF, 450 mL samples (N = 3) were collected at each time point (up to 25 days) and measured using the NGF EMax ELISA assay (Promega), following the manufacturer’s instructions for each sample. Fresh D-PBS (450 μL) was then used to replace the sample collected.
Characterization of Eluted NGF Bioactivity Through PC12 Cell Neurite Extension
To evaluate bioactivity of eluted NGF, PC12 cells were used because of their ability to exhibit a neuronal phenotype including neurite extensions upon exposure to NGF, and retract neurites after NGF is withdrawn from the culture media. In the absence of NGF, PC12 cells maintain a rounded morphology with few neurite extensions. Cells were cultured at 1 × 104 cells/cm2 in 24-well plates overnight. Electrode arrays coated with composites containing 5 wt% BSA or 1 μg NGF/μL in the hydrogel phase were attached to the side walls of wells (one electrode array/well, triplicate for each sample; Note that cells do not contact the composite coatings directly; Figure 2G). Cells were cultured for 2 weeks with medium exchanged every 2–3 days. Samples were compared to two controls, a positive control (N = 3) which received 50 ng/mL NGF added directly to the medium and a negative control (N = 3) which received no NGF. Neurite extension was assessed using phase contrast microscopy (Olympus IX71) at 10× and 20× magnifications at day 7 and day 14 for all samples. Five pictures were randomly taken for each well. Neurite length (from the tip of each neurite branch to the soma) was measured using Image J software. Kruskal–Wallis one way ANOVA by ranks (α = 0.01, for non-parametric data) was applied to analyze pooled data from each repetition, whereas the Tukey test (α = 0.05) was performed for pair-wise comparisons.
Results
Characterization of Composite Coating Electrode Adhesion
Poly(ethylene glycol)-poly(ε-caprolactone) hydrogels and PEGPCL hydrogel–PCL EFM composites adhere to electrode arrays over the short-term optical micrographs (Figures 3A,B) and SEM (Figure 3C). The interaction of PCL EFMs with PEGPCL hydrogels can be seen by the roughness of the coating surface near the PCL EFM in Figure 3C, which probably resulted from the non-uniform shrinkage of the hydrogel as it was constrained by the EFM during dehydration. Coatings demonstrate both short and long-term adhesion on electrode surfaces in the presence of PBS (Figures 3D,E, 45 days and Figure 3F, 9 months). In Figures 3D,E, irregular margin outside the original hydrogel–EFM composite interface indicates the spread of degraded hydrogel. In contrast with Figure 3D, the degraded hydrogel film in Figure 3F has broken into small pieces covering individual electrode sites. Although degradation of the PEGPCL hydrogel component is evidently advancing in Figures 3D–F as seen by the receding edge of hydrogels, the presence of PCL EFMs reduces degradation of the hydrogel component by physically constraining hydrogels to electrode surfaces and reducing diffusion to the hydrogel surface. Additionally, some wrinkling of the EFM is evident in Figure 3F, likely driven by shrinkage of the hydrogel layer beneath it. Composite coatings demonstrated adhesion for at least 1 month in a tissue phantom test, in which a coated electrode array was placed under an agarose gel mimicking the effect of tissue (Figure 3G).

Figure 3. Micrographs of PEGPCL hydrogel–PCL EFM adhesion to electrode array surfaces. (A) Optical micrographs of the distal tip of an MEA array coated with PEGPCL hydrogel and (B) coated with PEGPCL hydrogel–PCL EFM composite at day 0. (C) SEM micrograph of the distal tip of an MEA coated with PEGPCL hydrogel–PCL EFM composite at day 0. (D,E) Optical micrographs of composite coating after soaking in PBS buffer for 45 days. (F) SEM micrograph of composite coating after soaking in PBS solution for 9 months. (G) Optical micrograph of composite coating at day 32 under an agarose tissue phantom. (H,I) Optical micrographs of composite + electrode array after insertion into and removal from an agarose tissue phantom. [Black arrows indicate the edge of the PEGPCL hydrogel; white arrows indicate the edge of PCL EFMs. Scale bar: 1 mm for (A–D,F,H); 500 mm for (E,G,I)].
We also investigated the potential of coatings to withstand implantation into the brain. Each of the three composite electrode coatings was inserted into a 1% wt/v agarose gel (brain tissue has similar mechanical properties to softer 0.6% wt/v agarose gels; Chen et al., 2004). No delamination was observed for the coatings, demonstrating their potential to withstand the physical stresses associated with in vivo implantation (Figures 3H,I). These results suggest that the constraint created by PCL EFMs increases the persistence of the composite coating on the electrode surface and provide a first examination of the potential for clinical application. However, since the arachnoidea, which is substantially tougher than 1% wt/v agarose gel, provides the first barrier in real brain; more tests (i.e., in vivo composite-coated electrode implantation studies) are required to validate coating resistance to implantation forces.
Biocompatibility of Composite Electrode Coating
Poly(ethylene glycol)-poly(ε-caprolactone) hydrogel–PCL EFM composite electrode coatings did not demonstrate evident toxicity in a PC12 cell culture model. An MTT assay (Figure 4) indicated that negative control samples (PC12 cells cultured without exposure to composites or composite electrode coatings) demonstrate no significant difference in cell number compared with samples exposed to composites (C), composite electrode coatings (EC), and composite electrode coatings releasing BSA (ECB; p = 0.81, N = 3, average of three replicates). Also, in phase contrast optical micrographs, cells responded to C, EC, and ECB by maintaining their undifferentiated, rounded morphology (data not shown). No qualitative morphology change was observed in all samples.
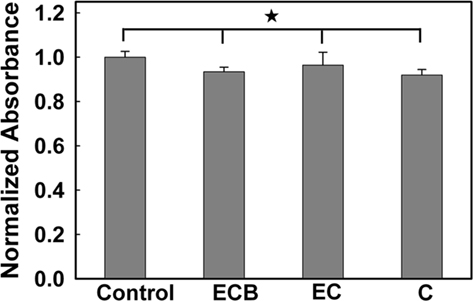
Figure 4. Normalized absorbance from an MTT cell viability test. (The asterisk indicates no significant difference; p < 0.05).
Measurement of Eluted NGF
The NGF Emax ELISA assay was used to determine the quantity of NGF eluted from PEGPCL hydrogels and PEGPCL hydrogel–PCL EFM composites. NGF was released from all samples over a period of 25 days (Figure 5). The concentration of NGF released from composite materials was higher than that of PEGPCL hydrogels at each time point, despite equal initial NGF loading. After 25 days exposure, composite materials showed significantly extended release of NGF vs. PEGPCL hydrogel controls, which may result from the diffusion barrier provided by the EFM layers. Also, it is possible that the fabrication method used for composite materials might better preserve NGF conformation than that used for PEGPCL hydrogels.
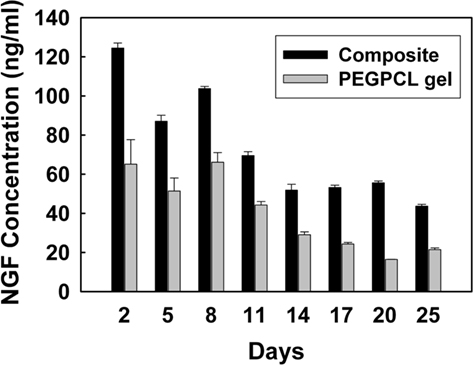
Figure 5. Concentration of eluted NGF as measured by ELISA.
Characterization of Eluted NGF Bioactivity Through PC12 Cell Neurite Extension
The quality of NGF eluted from hydrogel–EFM composite electrode coatings was evaluated using PC12 cell neurite extension. Composite electrode coatings delivered the targeted amount of bioactive NGF for over 2 weeks. Neurite extension of PC12 cells exposed to samples was characterized at day 7 and 14. At day 14, neurite extension was still evident (Figure 6A, NGF positive control and Figure 6B, coating), indicating that sufficient, bioactive NGF was released from composite electrode coatings to elicit a neuronal phenotype. The neurite number and length were similar to that of a positive control (which received 50 ng/mL NGF every other day for 14 days). Also, PC12 cells exposed to a negative control (no NGF) and a sham (BSA-releasing coatings) displayed rounded, undifferentiated morphologies at day 14 (Figures 6C,D), consistent with the expected morphology of PC12 cells not exposed to NGF.
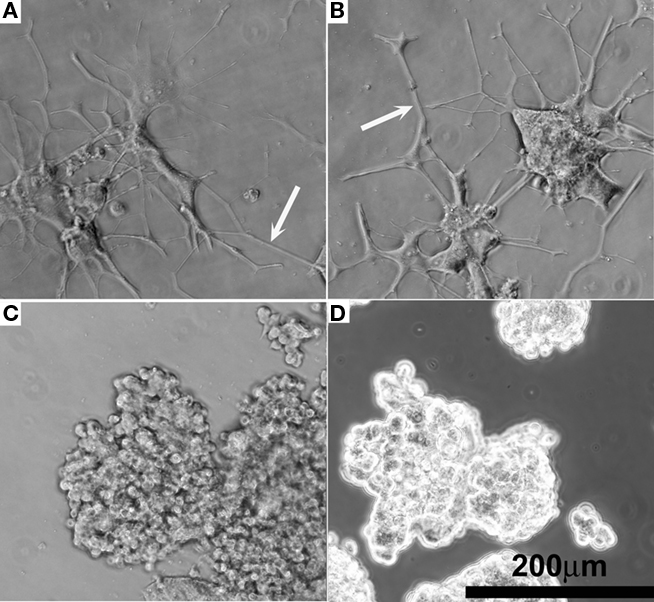
Figure 6. Representative phase contrast optical micrographs of PC12 cells after 14 days cell culture. (A) Positive control receiving 50 ng/mL NGF in the media. (B) NGF-eluting composite coating as shown in Figure 2G. (C) Negative control receiving 0 ng/mL NGF. (D) Sham, BSA-releasing composite coating.
For PEGPCL hydrogel–PCL EFM composite electrode coatings, neurite length distributions at day 7 and day 14 were statistically insignificant (p > 0.05) from NGF positive control samples (Figures 7A,B), and statistically significant (p < 0.05) from negative control and sham (BSA-releasing coatings) samples (data not shown). These results were consistent with the observations in Figure 5. Also, for both composite electrode coatings and NGF+ control samples, neurite length distributions at day 14 were statistically different (p < 0.05) from those at day 7 (Figure 8), which reflected the sustained release of NGF from electrode coatings for over 14 days at a concentration sufficient to induce neurite extension. It should be noted that cells continued to extend neurites beyond the 14 day period investigated, but neurite length measurements beyond 14 days became difficult as a result of neurite branching and network formation in both NGF+ control and electrode coating samples. Qualitatively, neurite extension of composite electrode coating samples remained visibly similar to NGF+ control samples and distinct from that of negative control and sham samples beyond 14 days of cell culture.
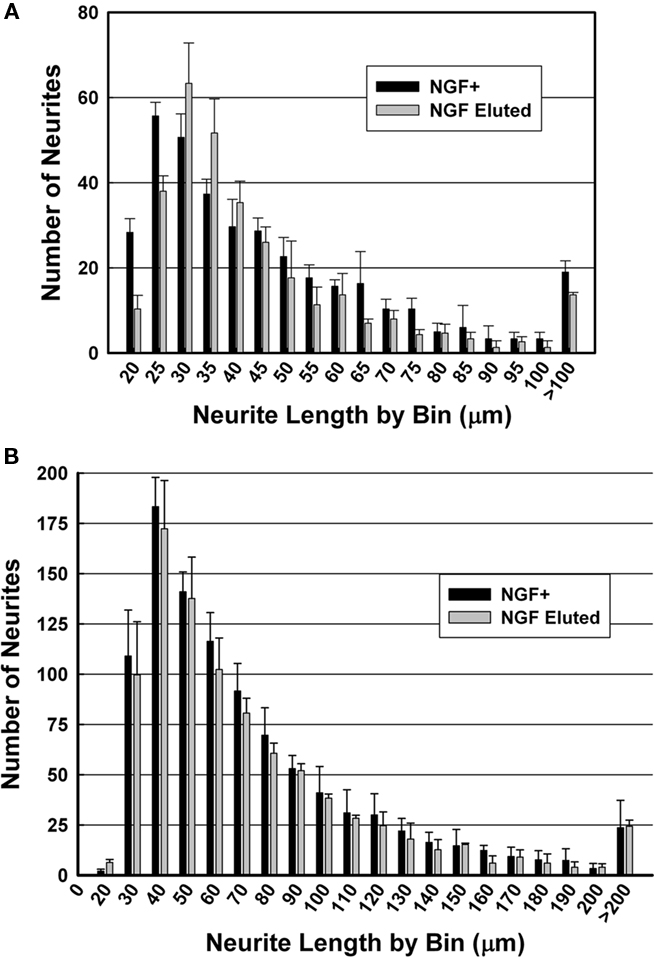
Figure 7. Histograms of neurite length after culture for (A) 7 days and (B) 14 days. Black: NGF positive control; gray: NGF-eluting composite electrode coatings.
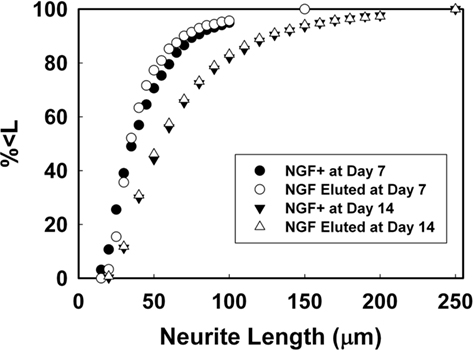
Figure 8. Percent of neurites possessing length < L at day 7 and 14.
Discussion
We have preliminarily demonstrated the potential of PEGPCL hydrogel–PCL EFM composites as electrode coatings to improve the biocompatibility of implanted devices. However, additional, in vivo composite coating studies are needed to confirm these results. Compared to the PEGPCL (Rao et al., 2011) and PEGPLA (Winter et al., 2007) hydrogel coatings investigated previously, which adhere to the electrode array surface for at least 28 and 10 days, respectively, PEGPCL hydrogel–PCL EFM composite coatings displayed significantly longer electrode adhesion [over 9 months in PBS bath immersion tests (Figure 3F) and over 1 month in tissue phantom tests (Figure 3G)]. We also observed that coating regions not covered by EFMs (i.e., PEGPCL hydrogel only) degraded much more rapidly than regions covered by PCL EFMs (Figures 3E,F). These results suggest that PCL EFMs physically constrain the hydrogel bolus and increase the water diffusion barrier, thereby significantly reducing hydrogel degradation and improving coating adhesion duration. It is thus possible that adhesion duration of PEGPCL hydrogel–PCL EFM composite electrode coatings could be controlled by applying EFMs of different shapes and sizes. Apart from adhesion, composite coatings were also capable of resisting implantation forces, as evidenced by insertion and removal tests in tissue phantoms (Figure 3I). These results are similar to those for PEGPCL hydrogel coatings observed previously (Rao et al., 2011). Thus composite coatings were able to withstand compression or shearing forces encountered during implantation. An interesting possibility is that the stability imparted by EFMs to the coatings may provide additional resistance to device movement associated with use, a target for future research.
To reduce device-related tissue damage, it is crucial to minimize the total size of neural prostheses, including coatings. The thickness of hydrogel–EFM composites is easily controlled. By changing the weight percentage of polymer solution and the volume of the hydrogel bolus, we can modify the thickness of the PEGPCL hydrogel layer. The application of PCL EFMs on top of the hydrogel layer can improve the uniformity of the hydrogel coating, and the thicknesses of PCL EFMs can be easily modified by changing electrospinning fabrication parameters, i.e., polymer solution flow rate, deposition time, and tip-to-substrate distance.
Although electrochemical properties of composite electrode coatings were not characterized through cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS), and potential transient measurements, the results of previous electrochemical characterization (Winter et al., 2007) showed very little effect of PEGPLA hydrogel coatings on the charge-injection properties of coated electrodes, and no significant difference of the maximum electrochemical potential excursions and access voltage between coated and uncoated electrodes. Since PEGPCL hydrogels have similar physical and electrochemical properties to PEGPLA hydrogels, we believe it is very unlikely that a significant difference in electrochemical properties would be observed. Also, the effect of the coating on the electrode electrical properties would be mitigated by continued polymer degradation. Furthermore, to fully expose the electrode sites to surrounding tissue without any compromise of their electrical properties, patterning techniques such as photolithography (Whitesides et al., 2001) or microfluidic channels (Khademhosseini et al., 2004) can be applied to control the spatial properties of the hydrogel coatings. For example, electrode sites could be exposed while the rest of the MEA could be covered by coatings. Similarly, to avoid the potential influence of composite EFMs on electrode electrochemical properties, the shape of EFMs can be tailored to the pattern of electrode array, exposing the electrode sites to surrounding tissue.
These results have also shown that composite electrode coatings are biocompatible and can be used as controlled neurotrophin delivery vehicles. Hydrogel–EFM composites are comprised of biocompatible materials, PEGPCL, and PCL, which have been extensively used in the clinic without evident cytotoxicity (Holland et al., 1986). Also, these biomaterials displayed NGF release durations for over 25 days (Figure 5), near our target of 4 weeks, demonstrating their potential for future in vivo applications. Since NGF release profiles from composite materials are closely related to the area of PEGPCL hydrogel covered by EFMs; longer release duration can easily be obtained by changing the size and thickness of EFMs or by applying patterning techniques (e.g., photolithography) to reduce the exposed hydrogel area. NGF was eluted from composite materials at a higher concentration than from PEGPCL hydrogel boluses at each time point, especially in the first week. This indicates slower diffusion rates and degradation profiles, most likely resulting from the presence of EFMs.
We have also demonstrated that neurotrophin-eluting composite electrode coatings have the potential to enhance neuron proximity to devices. Coatings can induce neurite extension in PC12 cells for over 2 weeks (Figure 6B). Thus, composite electrode coatings could not only improve interaction between adjacent neurons and the device, but could also potentially impact cells several hundred microns from the electrode surface. Apart from neurotrophin releasing systems, other methods have been proposed to enhance tissue–device contact, including the employment of passive surface coatings (Moxon et al., 2004; Nam et al., 2004; He and Bellamkonda, 2005), which promote neurite extension and cell adhesion through direct contact to modified or patterned surfaces. However, with the potential to release soluble factors, PEGPCL hydrogel–PCL EFM composites offer the substantial advantage of influencing cells distant from the implant site. Additionally, the potential of EFMs to guide cell adhesion has been extensively explored in the past decade (Chua et al., 2006; Chew et al., 2008); thus the outer EFM layer of composite electrode coatings may also enhance neuronal adhesion. To further enhance adhesion, adhesion molecules (e.g., polylysine, collagen, laminin) could be used to modify EFM surfaces through standard bioconjugation methods. Thus, composite coatings, such as those presented here, that provide sustained, multiple (e.g., soluble and adhesive) cues hold tremendous promise in improving the neuronal tissue–electrode interface.
Conclusion
These results demonstrate that PEGPCL hydrogel–PCL EFM composite coatings have the potential to improve neural prosthesis biocompatibility by delivering neurotrophins at biologically significant concentrations in a controlled and sustained manner to stimulate neural survival and differentiation. In vivo composite coating studies (e.g., implantation, electrochemical properties, biocompatibility, and neurotrophin-eluting) will be performed to further validate that potential. The incorporation of PCL EFMs in hydrogel-based coatings can constrain the hydrogel bolus, providing an additional diffusion barrier and therefore extending both coating adhesion to electrode surfaces and neurotrophin release. These hydrogel–EFM composite coatings thus provide a promising approach to encourage neuronal sprouting toward device surface and enhance neural prosthesis performance over the long-term, which will be further evaluated through in vivo tests.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge financial support from The Ohio State University, the National Science Foundation (CBET 0854015, EEC-0425626 and CMMI-0928315) and the H.C. “Slip” Slider Professorship (to Jessica O. Winter).
References
Abidian, M. R., and Martin, D. C. (2009). Multifunctional nanobiomaterials for neural interfaces. Adv. Funct. Mater. 19, 573–585.
Anseth, K. S., Metters, A. T., Bryant, S. J., Martens, P. J., Elisseeff, J. H., and Bowman, C. N. (2002). In situ forming degradable networks and their application in tissue engineering and drug delivery. J. Control Release 78, 199–209.
Boudriot, U., Dersch, R., Greiner, A., and Wendorff, J. H. (2006). Electrospinning approaches toward scaffold engineering – a brief overview. Artif. Organs 30, 785–792.
Butson, C. R., and McIntyre, C. C. (2005). Tissue and electrode capacitance reduce neural activation volumes during deep brain stimulation. Clin. Neurophysiol. 116, 2490–2500.
Chen, Z. J., Gillies, G. T., Broaddus, W. C., Prabhu, S. S., Fillmore, H., Mitchell, R. M., Corwin, F. D., and Fatouros, P. P. (2004). A realistic brain tissue phantom for intraparenchymal infusion studies. J. Neurosurg. 101, 314–322.
Chew, S. Y., Mi, R., Hoke, A., and Leong, K. W. (2008). The effect of the alignment of electrospun fibrous scaffolds on Schwann cell maturation. Biomaterials 29, 653–661.
Chew, S. Y., Wen, J., Yim, E. K., and Leong, K. W. (2005). Sustained release of proteins from electrospun biodegradable fibers. Biomacromolecules 6, 2017–2024.
Chua, K. N., Chai, C., Lee, P. C., Tang, Y. N., Ramakrishna, S., Leong, K. W., and Mao, H. Q. (2006). Surface-aminated electrospun nanofibers enhance adhesion and expansion of human umbilical cord blood hematopoietic stem/progenitor cells. Biomaterials 27, 6043–6051.
Cui, X., Lee, V. A., Raphael, Y., Wiler, J. A., Hetke, J. F., Anderson, D. J., and Martin, D. C. (2001). Surface modification of neural recording electrodes with conducting polymer/biomolecule blends. J. Biomed. Mater. Res. 56, 261–272.
Cui, X., Wiler, J., Dzaman, M., Altschuler, R. A., and Martin, D. C. (2003). In vivo studies of polypyrrole/peptide coated neural probes. Biomaterials 24, 777–787.
Eugene, M. (2004). Polyethyleneglycols and immunocamouflage of the cells tissues and organs for transplantation. Cell. Mol. Biol. (Noisy-le-grand) 50, 209–215.
Frampton, J. P., Hynd, M. R., Shuler, M. L., and Shain, W. (2010). Effects of glial cells on electrode impedance recorded from neuralprosthetic devices in vitro. Ann. Biomed. Eng. 38, 1031–1047.
Gadzinowski, M., Slomkowski, S., Elaissari, A., and Pichot, C. (2000). Phase transfer and characterization of poly(epsilon-caprolactone) and poly(L-lactide) microspheres. J. Biomater. Sci. Polym. Ed. 11, 459–480.
Gaumer, J., Prasad, A., Lee, D., and Lannutti, J. (2009). Source-to-ground distance and the mechanical properties of electrospun fiber. Acta Biomater. 2009, 1552–1561.
George, P. M., Lyckman, A. W., LaVan, D. A., Hegde, A., Leung, Y., Avasare, R., Testa, C., Alexander, P. M., Langer, R., and Sur, M. (2005). Fabrication and biocompatibility of polypyrrole implants suitable for neural prosthetics. Biomaterials 26, 3511–3519.
Greene, L. A. (1978). Nerve growth factor prevents the death and stimulates the neuronal differentiation of clonal PC12 pheochromocytoma cells in serum-free medium. J. Cell Biol. 78, 747–755.
Grill, W. M., McDonald, J. W., Peckham, P. H., Heetderks, W., Kocsis, J., and Weinrich, M. (2001). At the interface: convergence of neural regeneration and neural prostheses for restoration of function. J. Rehabil. Res. Dev. 38, 633–639.
He, W., and Bellamkonda, R. V. (2005). Nanoscale neuro-integrative coatings for neural implants. Biomaterials 26, 2983–2990.
He, W., McConnell, G. C., and Bellamkonda, R. V. (2006). Nanoscale laminin coating modulates cortical scarring response around implanted silicon microelectrode arrays. J. Neural Eng. 3, 316–326.
Heiduschka, P., and Thanos, S. (1998). Implantable bioelectric interfaces for lost nerve functions. Prog. Neurobiol. 55, 433–461.
Holland, S. J., Tighe, B. J., and Gould, P. L. (1986). Polymers for biodegradable medical devices. 1. The potential of polyesters as controlled macromolecular release systems. J. Control Release 4, 155–180.
Huang, E. J., and Reichardt, L. F. (2001). Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24, 677–736.
Huatan, H., Collett, J. H., Attwood, D., and Booth, C. (1995). Preparation and characterization of poly(epsilon-caprolactone) polymer blends for the delivery of proteins. Biomaterials 16, 1297–1303.
Jain, A., Kim, Y. T., McKeon, R. J., and Bellamkonda, R. V. (2006). In situ gelling hydrogels for conformal repair of spinal cord defects, and local delivery of BDNF after spinal cord injury. Biomaterials 27, 497–504.
Jhaveri, S. J., Hynd, M. R., Dowell-Mesfin, N., Turner, J. N., Shain, W., and Ober, C. K. (2009). Release of nerve growth factor from HEMA hydrogel-coated substrates and its effect on the differentiation of neural cells. Biomacromolecules 10, 174–183.
Jun, S. B., Hynd, M. R., Dowell-Mesfin, N. M., Al-Kofahi, Y., Roysam, B., Shain, W., and Kim, S. J. (2008). Modulation of cultured neural networks using neurotrophin release from hydrogel-coated microelectrode arrays. J. Neural Eng. 5, 203–213.
Khademhosseini, A., Yeh, J., Jon, S., Eng, G., Suh, K. Y., Burdick, J. A., and Langer, R (2004). Molded polyethylene glycol microstructures for capturing cells within microfluidic channels. Lab Chip 4, 425–430.
Kim, K., Luu, Y. K., Chang, C., Fang, D., Hsiao, B. S., Chu, B., and Hadjiargyrou, M. (2004). Incorporation and controlled release of a hydrophilic antibiotic using poly(lactide-co-glycolide)-based electrospun nanofibrous scaffolds. J. Control Release 98, 47–56.
Kishino, A., Katayama, N., Ishige, Y., Yamamoto, Y., Ogo, H., Tatsuno, T., Mine, T., Noguchi, H., and Nakayama, C. (2001). Analysis of effects and pharmacokinetics of subcutaneously administered BDNF. Neuroreport 12, 1067–1072.
Klein, J. D., Clauson, S. L., and Cogan, S. F. (1989). Morphology and charge capacity of sputtered iridium oxide-films. J. Vac. Sci. Technol. A 7, 3043–3047.
Lawrence, S. M., Dhillon, G. S., and Horch, K. W. (2003). Fabrication and characteristics of an implantable, polymer-based, intrafascicular electrode. J. Neurosci. Methods 131, 9–26.
Loewenstein, J. I., Montezuma, S. R., and Rizzo, J. F. III (2004). Outer retinal degeneration: an electronic retinal prosthesis as a treatment strategy. Arch. Ophthalmol. 122, 587–596.
Luu, Y. K., Kim, K., Hsiao, B. S., Chu, B., and Hadjiargyrou, M. (2003). Development of a nanostructured DNA delivery scaffold via electrospinning of PLGA and PLA-PEG block copolymers. J. Control Release 89, 341–353.
Matthews, J. A., Wnek, G. E., Simpson, D. G., and Bowlin, G. L. (2002). Electrospinning of collagen nanofibers. Biomacromolecules 3, 232–238.
Maysinger, D., Krieglstein, K., Filipovic-Grcic, J., Sendtner, M., Unsicker, K., and Richardson, P. (1996). Microencapsulated ciliary neurotrophic factor: physical properties and biological activities. Exp. Neurol. 138, 177–188.
Moxon, K. A., Kalkhoran, N. M., Markert, M., Sambito, M. A., McKenzie, J. L., and Webster, J. T. (2004). Nanostructured surface modification of ceramic-based microelectrodes to enhance biocompatibility for a direct brain-machine interface. IEEE Trans. Biomed. Eng. 51, 881–889.
Nam, J., Huang, Y., Agarwal, S., and Lannutti, J. (2008). Materials selection and residual solvent retention in biodegradable electrospun fibers. J. Appl. Polym. Sci. 107, 1547–1554.
Nam, Y., Chang, J. C., Wheeler, B. C., and Brewer, G. J. (2004). Gold-coated microelectrode array with thiol linked self-assembled monolayers for engineering neuronal cultures. IEEE Trans. Biomed. Eng. 51, 158–165.
Polikov, V. S., Tresco, P. A., and Reichert, W. M. (2005). Response of brain tissue to chronically implanted neural electrodes. J. Neurosci. Methods 148, 1–18.
Rao, S. S., Han, N., and Winter, J. O. (2011). Polylysine-modified PEG-based hydrogels to enhance the neuro-electrode interface. J. Biomater. Sci. Polym. Ed. 22, 611–625.
Retterer, S. T., Smith, K. L., Bjornsson, C. S., Neeves, K. B., Spence, A. J., Turner, J. N., Shain, W., and Isaacson, M. S. (2004). Model neural prostheses with integrated microfluidics: a potential intervention strategy for controlling reactive cell and tissue responses. IEEE Trans. Biomed. Eng. 51, 2063–2073.
Sawhney, A. S., Pathak, C. P., and Hubbell, J. A. (1993). Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(alpha-hydroxy acid) diacrylate macromers. Macromolecules 26, 581–587.
Schwartz, A. B., Cui, X. T., Weber, D. J., and Moran, D. W. (2006). Brain-controlled interfaces: movement restoration with neural prosthetics. Neuron 52, 205–220.
Sun, S. W., Jeong, Y. I., and Kim, S. H. (2003). Surfactant-free microspheres of poly(epsilon-caprolactone)/poly (ethylene glycol)/poly(epsilon-caprolactone) triblock copolymers as a protein carrier. Arch. Pharm. Res. 26, 504–510.
Szarowski, D. H., Andersen, M. D., Retterer, S., Spence, A. J., Isaacson, M., Craighead, H. G., Turner, J. N., and Shain, W. (2003). Brain responses to micro-machined silicon devices. Brain Res. 983, 23–35.
Turner, J. N., Shain, W., Szarowski, D. H., Andersen, M., Martins, S., Isaacson, M., and Craighead, H. (1999). Cerebral astrocyte response to micromachined silicon implants. Exp. Neurol. 156, 33–49.
West, J. L., and Hubbell, J. A. (1995). Photopolymerized hydrogel materials for drug-delivery applications. React. Polym. 25, 139–147.
Whitesides, G. M., Ostuni, E., Takayama, S., Jiang, X., and Ingber, D. E. (2001). Soft lithography in biology and biochemistry. Annu. Rev. Biomed. Eng. 3, 335–373.
Winter, J. O., Cogan, S. F., Rizzo, J. F. III (2007). Neurotrophin-eluting hydrogel coatings for neural stimulating electrodes. J. Biomed. Mater. Res. B Appl. Biomater. 81, 551–563.
Wu, Z. R., Ma, J., Liu, B. F., Xu, Q. Y., and Cui, F. Z. (2007). Layer-by-layer assembly of polyelectrolyte films improving cytocompatibility to neural cells. J. Biomed. Mater. Res. A. 81, 355–362.
Keywords: neural prostheses, hydrogels, electrospun fibers, coatings, drug release
Citation: Han N, Rao SS, Johnson J, Parikh KS, Bradley PA, Lannutti JJ and Winter JO (2011) Hydrogel–electrospun fiber mat composite coatings for neural prostheses. Front. Neuroeng. 4:2. doi: 10.3389/fneng.2011.00002
Received: 16 August 2010;
Accepted: 21 February 2011;
Published online: 11 March 2011.
Edited by:
Laura Ballerini, University of Trieste, ItalyReviewed by:
Andre Poot, University of Twente, NetherlandsJens Schouenborg, Neuronano Research Center, Sweden
Copyright: © 2011 Han, Rao, Johnson, Parikh, Bradley, Lannutti and Winter. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Jessica O. Winter, William G. Lowrie Department of Chemical and Biomolecular Engineering, Department of Biomedical Engineering, The Ohio State University, 125A Koffolt Laboratories, 140 West 19th Avenue, Columbus, OH 43210, USA. e-mail: winter.63@osu.edu