 Yair M. Gozal1,2,3 Eric B. Dammer2,3,4,5 Duc M. Duong2,3,4,5 Dongmei Cheng2,3,4,5 Marla Gearing1,2,3 Howard D. Rees1,2,3 Junmin Peng2,3,4,5 James J. Lah1,2,3* Allan I. Levey1,2,3*
Yair M. Gozal1,2,3 Eric B. Dammer2,3,4,5 Duc M. Duong2,3,4,5 Dongmei Cheng2,3,4,5 Marla Gearing1,2,3 Howard D. Rees1,2,3 Junmin Peng2,3,4,5 James J. Lah1,2,3* Allan I. Levey1,2,3*- 1 Department of Neurology, School of Medicine, Emory University, Atlanta, GA, USA
- 2 Center for Neurodegenerative Diseases, School of Medicine, Emory University, Atlanta, GA, USA
- 3 Alzheimer’s Disease Research Center, School of Medicine, Emory University, Atlanta, GA, USA
- 4 Department of Human Genetics, School of Medicine, Emory University, Atlanta, GA, USA
- 5 Emory Proteomics Service Center, School of Medicine, Emory University, Atlanta, GA, USA
Frontotemporal lobar degeneration (FTLD) is the most common cause of dementia with pre-senile onset, accounting for as many as 20% of cases. A common subset of FTLD cases is characterized by the presence of ubiquitinated inclusions in vulnerable neurons (FTLD-U). While the pathophysiological mechanisms underlying neurodegeneration in FTLD-U have not yet been elucidated, the presence of inclusions in this disease indicates enhanced aggregation of one or several proteins. Moreover, these inclusions suggest altered expression, processing, or degradation of proteins during FTLD-U pathogenesis. Thus, one approach to understanding disease mechanisms is to delineate the molecular changes in protein composition in FTLD-U brain. Using a combined approach consisting of laser capture microdissection (LCM) and high-resolution liquid chromatography-tandem mass spectrometry (LC–MS/MS), we identified 1252 proteins in hippocampal dentate granule cells excised from three post-mortem FTLD-U and three unaffected control cases processed in parallel. Additionally, we employed a labeling-free quantification technique to compare the abundance of the identified proteins between FTLD-U and control cases. Quantification revealed 54 proteins with selective enrichment in FTLD-U, including TAR–DNA binding protein 43 (TDP-43), a recently identified component of ubiquitinated inclusions. Moreover, 19 proteins were selectively decreased in FTLD-U. Subsequent immunohistochemical analysis of TDP-43 and three additional protein candidates suggests that our proteomic profiling of FTLD-U dentate granule cells reveals both inclusion-associated proteins and non-aggregated disease-specific proteins. Application of LCM is a valuable tool in the molecular analysis of complex tissues, and its application in the proteomic characterization of neurodegenerative disorders such as FTLD-U may be used to identify proteins altered in disease.
Introduction
Frontotemporal lobar degeneration (FTLD) is a progressive neurologic disorder that manifests profound behavioral, personality, and language symptoms (Neary et al., 2005; Kumar-Singh and Van Broeckhoven, 2007). In the past few decades, the pathologic classification of FTLD has been primarily accomplished using biochemical and immunohistochemical approaches to differentiate the composition of proteinaceous aggregates found in the brain. Historically, three major neuropathological subtypes have been identified in FTLD, including tauopathies, FTLD cases lacking distinctive histopathology, and FTLD-ubiquitinated (FTLD-U) type (Woulfe et al., 2001). Recent studies have suggested that FTLD-U is the most common of these subtypes, accounting for approximately half of all FTLD cases (Graff-Radford and Woodruff, 2007; Snowden et al., 2007). The histopathologic hallmark of FTLD-U is the presence of ubiquitin-positive, tau and α-synuclein-negative intraneuronal inclusions primarily in the hippocampal dentate gyrus and frontotemporal cortex (Kovari et al., 2004). Interestingly, a similar pathology is also seen in cases of FTLD-U with concomitant motor neuron disease (MND), which suggests that FTLD-U and MND share a common pathogenesis (Forman et al., 2006).
Pathological lesions in many neurodegenerative diseases exhibit ubiquitin immunoreactivity, reflecting common downstream cellular responses to protein complexes. However, the identities of the inciting proteins that aggregate differ depending on the particular disease. In FTLD-U inclusions, the primary protein constituents have proven difficult to characterize, hampering understanding of key early events in the disease pathogenesis. This obstacle has been primarily technical in nature, as the small size and distribution of the inclusions precluded the application of biochemical purification and identification approaches common in the study of other pathologic lesions. In fact, it was only recently that the first non-ubiquitin components of FTLD-U inclusions were identified as TAR–DNA-Binding Protein 43 (TDP-43; Neumann et al., 2006) and Fused in Sarcoma (FUS; Kwiatkowski et al., 2009; Vance et al., 2009), both nuclear RNA binding proteins involved in RNA splicing and transcriptional regulation (Yang et al., 1998; Buratti et al., 2001; Tan and Manley, 2010).
The identification of TDP-43 as a primary aggregating protein in FTLD-U has fueled efforts to further define the mechanisms leading to protein aggregation, inclusion formation, and neurodegeneration in this disorder. To date, however, limited progress has been made in the characterization of additional functionally relevant proteins in sporadic FTLD-U. In contrast, several genes have been associated with the pathogenesis of familial disease forms (Rollinson et al., 2009), including the discovery of pathogenic mutations in the progranulin gene (GRN; Baker et al., 2006; Cruts et al., 2006), valosin-containing protein (VCP) on chromosome 9p21 (Watts et al., 2004), and charged multivesicular body protein 2B (CHMP2B) on chromosome 3p11 (Skibinski et al., 2005). GRN mutations cause a loss-of-function (haploinsufficiency) by introducing premature termination codons or missense mutations that result in rapid mRNA degradation or non-functional protein expression (Baker et al., 2006; Eriksen and Mackenzie, 2008). Thus, unlike TDP-43, mutations in GRN do not result in the accumulation of aggregated progranulin. Similarly, neither VCP nor CHMP2B have been shown to systematically accumulate in the ubiquitin-immunoreactive neuropathology. Notably, extensive histopathological characterization of familial and sporadic FTLD-U cases reveals differences in aggregate distribution, density, and morphology, suggesting that they may not share a common molecular basis (Mackenzie and Rademakers, 2007). As such, further molecular analysis of sporadic FTLD-U tissues is warranted.
The development of laser capture microdissection (LCM) technology over the past 15 years (Emmert-Buck et al., 1996) has given investigators a new method to isolate and study neurodegenerative disease tissues. LCM is a rapid, reliable method for the isolation of specific cells, or small biologically relevant areas, from complex tissues (Emmert-Buck et al., 1996). Using a low-power laser to melt a thermoplastic film onto a tissue section, a target of interest as small as 3–5 μm in diameter can be isolated (Bonner et al., 1997). Multiple laser shots can be combined on the same film in order to procure cell clusters or more complicated tissue structures (Simone et al., 1998). Importantly, the remarkable precision demonstrated in the laser capture process, coupled with minimal direct handling and processing of the captured material, reduces contamination in collected samples and minimizes the impact on downstream analyses (Ornstein et al., 2000). However, the process of LCM allows the recovery of only a minimal amount of protein from captured tissues, a limitation that may be largely addressed by the application of high-sensitivity proteomics platforms such as liquid chromatography – tandem mass spectrometry (LC–MS/MS).
The combination of LCM and LC–MS/MS offers a unique opportunity to study neurodegenerative disorders because these diseases are characterized by the presence of selectively vulnerable populations of neurons (Morrison et al., 1998) and by distinct neuropathological lesions that can be microdissected and analyzed. For example, we have previously applied this combined approach in the characterization of senile plaques from post-mortem Alzheimer’s disease (AD) brain tissues (Liao et al., 2004; Gozal et al., 2006). Specifically, we demonstrated the co-isolation of 488 proteins with the plaques, including more than 80% of the previously documented plaque proteins. More significantly, quantitative comparison of plaques and non-plaque tissues revealed at least 2-fold enrichment of 26 proteins in the plaque regions, an indication of the complexity and diversity of cellular processes involved in the formation of plaques. Thus, in this study, we coupled LCM and LC–MS/MS to identify and quantitate proteins isolated from neurons containing ubiquitinated inclusions in the hippocampal dentate gyrus of FTLD-U patients. We reveal significant changes in 73 proteins in FTLD-U compared with unaffected controls, and evaluate the potential of this approach for profiling protein expression in cells that specifically accumulate ubiquitin-immunoreactive inclusions and other complex aggregates in neurodegeneration.
Materials and Methods
Case Material
All cases used in these studies were obtained from the Alzheimer’s Disease Research Center (ADRC) and Center for Neurodegenerative Disease (CND) Brain Bank at Emory University School of Medicine. The inclusion of FTLD-U cases was based on extensive neuropathologic characterization required for diagnosis based on consensus criteria (McKhann et al., 2001; Trojanowski and Dickson, 2001; Cairns et al., 2007). All cases exhibited small, ubiquitin-positive, tau and α-synuclein-negative neuronal cytoplasmic inclusions in the hippocampal dentate gyrus. Additionally, these cases did not meet criteria for neuropathological diagnosis of AD (Mirra et al., 1991; The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease, 1997), Lewy body disease (McKeith et al., 2005), or tau pathology consistent with a tauopathy (Litvan et al., 1996; Dickson, 1998). TDP-43 immunoreactivity was histochemically confirmed in all FTLD-U cases. Control cases had neither a clinical history nor a neuropathologic diagnosis of neurologic disease, and were selected to match FTLD-U cases in post-mortem interval (PMI) to prevent significant variability in protein quality. Samples were separated into three independent FTLD-U/Control comparisons for processing and analysis.
Antibodies
Commercially available primary antibodies used in these studies were to the proteins TDP-43 (rabbit polyclonal; Protein Tech Group, Chicago, IL, USA), septin 3 (goat polyclonal; Abcam, Cambridge, MA, USA), septin 7 (rabbit polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA, USA), glial fibrillary acidic protein (GFAP; mouse monoclonal; DAKO, Carpinteria, CA, USA), and ubiquitin (rabbit polyclonal; DAKO).
Preparation of Tissues for LCM
Isolation of hippocampal dentate gyrus granule cells by LCM was performed based on previously developed protocols (Emmert-Buck et al., 1996; Goldsworthy et al., 1999). Ethanol-fixed, paraffin-embedded 10 μm-thick hippocampal sections mounted on uncoated and uncharged glass slides were deparaffinized in xylene and rehydrated. Sections were subsequently stained for 1 min in Histogene Staining Solution (Arcturus, Mountain View, CA, USA), differentiated in 75% ethanol for 1 min, subjected to dehydration in graded alcohols, cleared for 5 min in fresh xylene, and air-dried for 5 min. Finally, sections were desiccated prior to LCM (Emmert-Buck et al., 1996).
Laser Capture Microdissection
Laser capture microdissection was performed using a Pixcell II laser capture system (Arcturus). Hippocampal dentate granule cells were visualized and captured with short-duration pulses (1 ms) of an infrared laser (laser spot size: 7.5 μm) using a laser power setting of 80–90 mW. Typically, granule cells of the dentate gyrus from three to four consecutive sections (10 μm thick) were captured on a single CapSure Macro LCM cap (Arcturus), and 50–60 sections were processed from each individual.
Protein Extraction from LCM Caps
Protein extractions were performed as previously described (Gozal et al., 2006). Briefly, caps were extracted for 15 min at 65°C with 20 μl of lysis buffer composed of 2% sodium dodecyl sulfate (SDS), 10% glycerol, 10 mM dithiothreitol (DTT), 1 mM ethylenediaminetetraacetic acid (EDTA), and protease inhibitor cocktail (PIC; Roche Applied Science) in phosphate-buffered saline, pH 7.2. The caps were then re-extracted for 15 min at 65°C with a fresh 20 μl aliquot of lysis buffer. The lysis buffer was reused in the sequential extractions of up to four caps containing captured hippocampal dentate gyri from the same case. The procedure was repeated until all caps corresponding to a particular case were extracted, and the extracts were then pooled to produce one extracted sample for each case. To normalize total proteins submitted for proteomic analysis, protein concentration was estimated from a modified silver staining (Shevchenko et al., 1996) of 5% of the sample as previously described (Gozal et al., 2006).
Analysis and Protein Identification by Mass Spectrometry
Mass spectrometric analysis and protein identification was performed as described previously (Liao et al., 2004; Gozal et al., 2009). Briefly, samples were first separated on a 10% SDS gel (0.75 mm thickness), and visualized with Coomassie Blue G-250. Sample lanes were cut into 6 gel bands and digested with trypsin. The resulting digested peptides were extracted from each of the gel pieces and analyzed by LC–MS/MS using a hybrid LTQ–FT linear ion trap/7-T Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (Thermo Electron, San Jose, CA, USA). The acquired MS/MS spectra were searched against the human reference database (29,575 proteins) of the National Center for Biotechnology Information (January 2007) using the SEQUEST-Sorcerer algorithm version 3.11 r11 (Sage-N-Research, San Jose, CA, USA; Eng et al., 1994). A target-decoy filtering strategy was subsequently used to evaluate the peptide identification false discovery rate (Peng et al., 2003). False positive matches were thus filtered out first by mass accuracy (15 ppm) and then by dynamically increasing XCorr (minimal 1.8) and ΔCn (minimal 0.05) values such that the false discovery rate was less than 0.2%. Generally, following manual validation of the spectra (Peng et al., 2003), we accepted proteins identified by at least one unique peptide.
Label-free Quantification: Extracted Ion Current
Quantification of proteins in the LCM extracts was based on the comparison of paired peptides within each of the three independent FTLD-U/Control comparisons as previously described (Gozal et al., 2009). Ion current intensities for identified peptides were extracted in MS survey scans, and a ratio of the peak intensities for the peptide precursor ion was calculated. The resultant ratio was used as a measure of the relative abundance of the peptide within each FTLD-U/control comparison (Wang et al., 2003). Abundance ratios for all peptides of a particular protein were averaged to determine the protein abundance ratio. To evaluate the statistical significance of the protein changes, a histogram of all protein abundance ratios after logarithmic transformation (log2) was fitted to a Gaussian distribution according to the null hypothesis. Proteins identified and consistently reduced or enriched in at least two of the independent FTLD-U/Control comparisons with log2 ratios falling more than two SD from the mean of the Gaussian distribution were marked as significantly altered.
Immunohistochemistry
Free-floating 50 μm-thick frontal cortex and hippocampus sections from 6 FTLD-U and six control cases were incubated with 3% hydrogen peroxide (H2O2) to quench endogenous peroxidase activity, blocked in normal serum, and incubated overnight at 4°C with primary antibody. Sections were then incubated with a biotinylated secondary antibody and signal was visualized using an avidin–biotin–peroxidase complex method (Vector Elite ABC Kit, Vector Laboratories, Burlingame, CA, USA) with 3,3′-diaminobenzidine (DAB) as a chromogen.
Western Blotting
Post-mortem frontal cortex from AD, FTLD-U, or control cases was extracted at 5 mL/g (volume/weight) with ice-cold low salt (LS) buffer (10 mM Tris, pH 7.5, 5 mM EDTA, 1 mM DTT, 10% sucrose, 10 mM β-glycerophosphate, 10 mM sodium orthovanadate, 10 mM tetrasodium pyrophosphate, 50 mM sodium fluoride, 1× Roche complete PIC). The resulting homogenate was diluted in an equal volume of 2× RIPA buffer (100 mM Tris·Cl pH 7.3, 300 mM sodium chloride, 2 mM EDTA, 2% Triton-X-100, 2% deoxycholate, and 0.2% SDS) with 1× Roche complete PIC and 10 mM iodoacetic acid (IAA). Samples were sonicated on ice and centrifuged at 25,000 × g for 15 min at 4°C. The RIPA soluble extract (supernatant) was collected and the pellet (insoluble fraction) was subsequently incubated in 20 μL of 8 M urea in SDS loading buffer (63 mM Tris HCl, 10% Glycerol, 2% SDS, 0.0025% Bromophenol Blue, 5% 2-Mercaptoethanol, 1× PIC, and 10 mM IAA) prior to trituration and sonication. Immunoblotting was performed according to standard procedures as described previously (Seyfried et al., 2010) with either 25 μg of insoluble fraction or 60 μg of RIPA soluble fraction.
Results
Identification of proteins in FTLD-U dentate granule cells by LC–MS/MS
Using LCM, we selectively dissected the granule cell layer of the dentate gyrus, a site of abundant pathologic inclusions in FTLD-U, from three clinically and pathologically confirmed FTLD-U cases and three unaffected controls for proteomic analysis (Table 1). Each FTLD-U case was paired with a control case after matching for gender and PMI (±6 h) in order to minimize variation due to other variables. To collect sufficient material for proteomics, the maximum number of granule cells from each dentate gyrus were captured (Figures 1A,B) from 50 to 60 ethanol-fixed, paraffin-embedded, post-mortem hippocampal sections per case, requiring 3000–5000 laser pulses per section. Protein captured from each case was subsequently extracted with SDS, separated by mass using SDS gel electrophoresis, and quantified to estimate total protein yield by silver stain (Figure 1C). For each case, ∼5 μg of total protein was collected by LCM of hippocampal sections, thus requiring that the samples be analyzed by MS in their entirety and precluding the processing of a replicate sample. Moreover, while the total protein analyzed was controlled for each FTLD-U and control pair, significant variations in the amount of analyzed protein may have been present between paired comparisons in order to maximize the number of proteins identified in each case. Following the division of the samples into three independent FTLD-U/control comparisons, the extracted proteins were separated on a second SDS gel. Each gel lane was subsequently cut into six pieces by molecular weight, and exposed to in-gel digestion with trypsin. The resultant tryptic peptides were analyzed by high-resolution LC–MS/MS, the method of choice for large-scale proteomics. The spectra acquired for each sample were searched against a human protein database, and further filtered by mass accuracy and matching scores. We identified 6694 peptides corresponding to 1252 proteins across the three paired case/control comparisons. Of these, 218 proteins were found in all three paired comparisons (Figure 2).
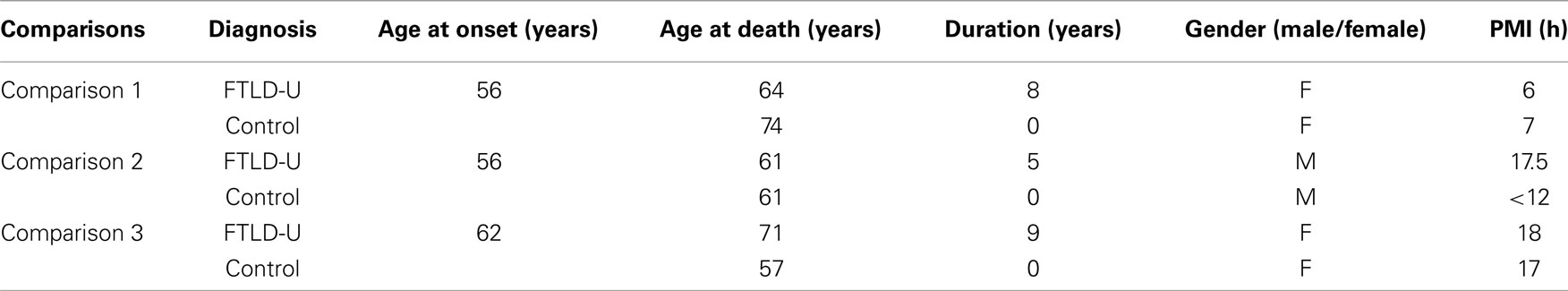
Table 1. Demographic information.
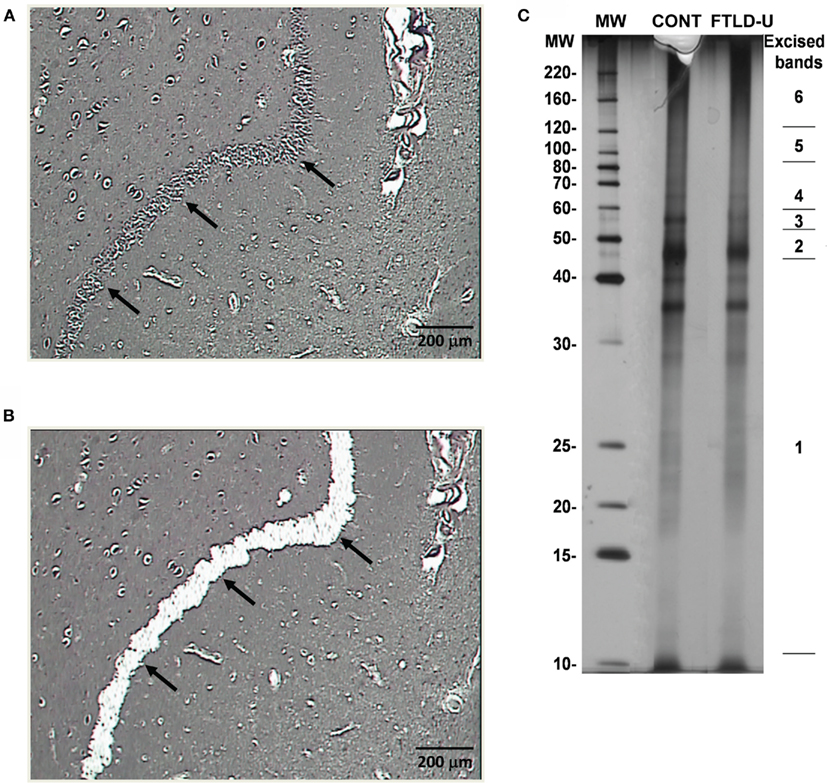
Figure 1. Dissection and preparation of LCM samples for proteomics. (A) Before and (B) after images depicting efficient removal of the hippocampal dentate gyrus via LCM. (C) Representative silver stain of extracted proteins captured from one FTLD-U case and one unaffected control. Proteins were extracted using SDS-containing lysis buffer, and a small fraction of each sample (∼5%) was separated by SDS-PAGE and examined by silver staining. The remainder of each sample (∼95%) was resolved on a separate SDS gel and stained with Coomassie Blue G-250. Each sample lane was then cut into six pieces according to the molecular weight marker as is shown on the right.
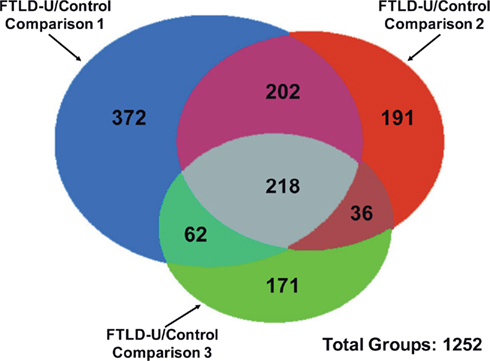
Figure 2. Identification of proteins in independent FTLD-U/Control comparisons. Proteins identified in each comparison are represented by colored circles. The number of overlapped proteins between comparisons is indicated in the relevant areas. A total of 1252 proteins were identified across all three FTLD-U/Control comparisons. Of these, 218 were independently identified in each of the three comparisons.
Relative Quantification of Identified Proteins by a Label-free Strategy
Quantitation of protein changes between FTLD-U and control dentate granule cell proteomes was based on the ratio of the extracted ion currents of peptides identified in each case. Abundance ratios (FTLD-U/Control) corresponding to the relative protein abundance between the FTLD-U and control proteomes were calculated for all of the peptides obtained in this experiment. To evaluate the significance of the protein changes and to correct the quantitative errors resulting from sample handling and/or ionization instability, the abundance ratios for all 1252 proteins identified in this experiment were averaged over the three comparisons, transformed into log2 (ratio), and plotted as shown in Figure 3 (Li et al., 2003; Cheng et al., 2006). Because the majority of proteins tend to display similar abundance in diseased and control tissues, the resultant experimental distribution was fitted to a normal distribution on the basis of the central limit theorem. Using the normal distribution parameters, we identified significant enrichment (≥2 SD from the mean) of 54 proteins in FTLD-U dentate granule cells compared with control. Moreover, 19 proteins were found to be significantly reduced in FTLD-U cases. In order to increase the reliability of the dataset, we considered only proteins detected in at least two out of three FTLD-U/Control comparisons (518 proteins). Finally, only the proteins demonstrating consistency in the direction of the measured change (i.e., enriched in all comparisons) were included in this final list (Tables 2 and 3).
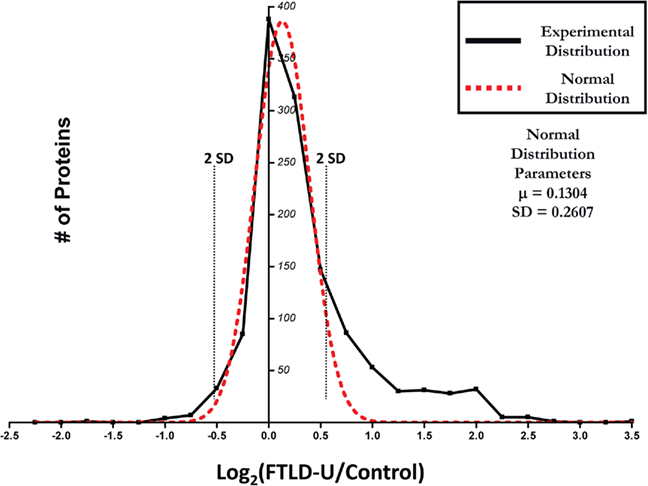
Figure 3. Distribution of abundance ratios reveals a subset of altered FTLD-U proteins. Logarithmic (base 2) transformation of protein ratios from FTLD-U/control comparisons was performed and plotted with each point corresponding to the number of proteins in each 0.25 unit window (solid black line). A normal distribution was subsequently fitted to the data (red line) and used to determine the two SD threshold used in filtering (dotted black line). Total proteins = 1252.
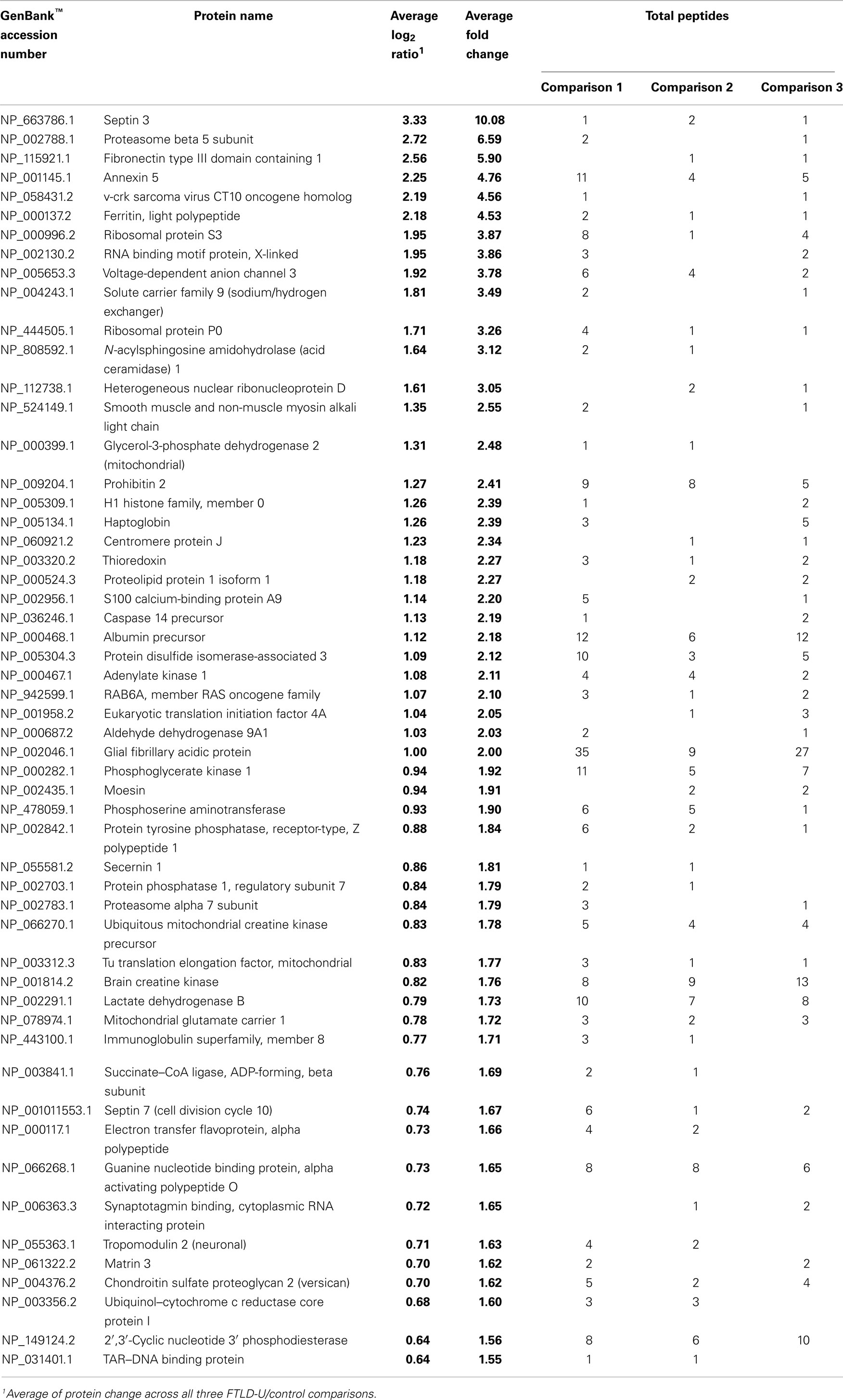
Table 2. Proteins enriched in FTLD-U dentate granule cells.
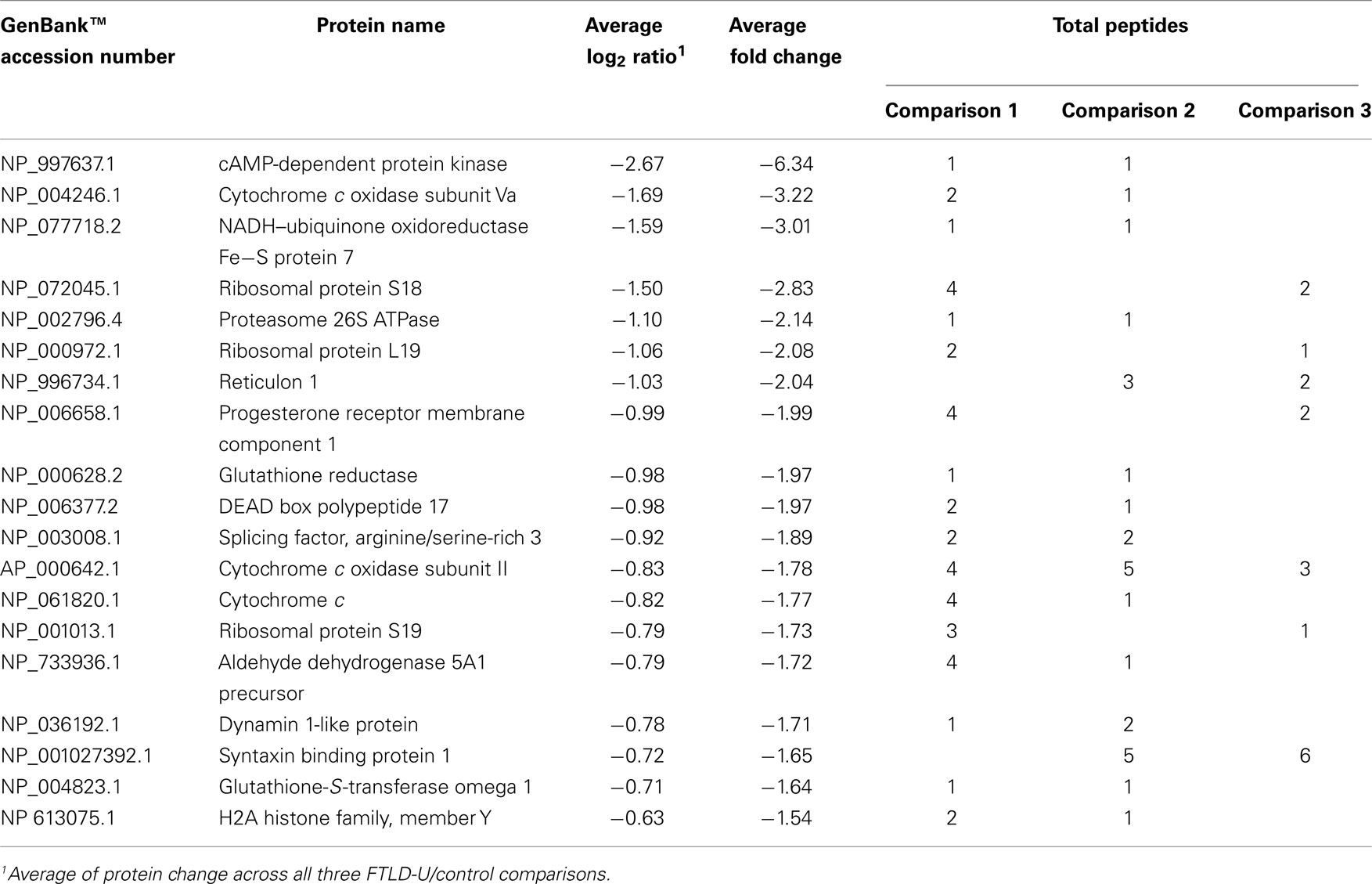
Table 3. Proteins decreased in FTLD-U dentate granule cells.
Analysis of selected FTLD-U enriched components
Our quantitative comparison of proteins captured by LCM identifies a list of candidate molecules that might play a significant role in FTLD-U pathogenesis. Immunohistochemical staining for TDP-43 revealed abundant labeled inclusions in the FTLD-U dentate granule cells (Figure 4A) while the controls showed no labeling. The identification of TDP-43, a well-established disease protein known to associate with FTLD-U pathological lesions, substantiates the sensitivity of our proteomic strategy and validates our use of LCM samples in proteomics analyses. However, while TDP-43 was among the proteins meeting these filtering restrictions, it showed only a moderate ∼1.5 fold increase in FTLD-U. Several additional candidate proteins were selected for immunohistochemical (IHC) analysis based on the availability of specific antibodies and the magnitude of enrichment in FTLD-U. IHC was performed in frontal cortex and hippocampal sections from six additional post-mortem FTLD-U cases and six unaffected controls using antibodies to ubiquitin (Figure 4B), septin 3 (Figure 4C), septin 7 (Figure 4D), and GFAP (data not shown). These antibodies did not reveal associations with inclusions or staining overlap with ubiquitin immunoreactivity in the cases studied, suggesting that many of the enriched proteins identified through our LCM and LC–MS/MS approach represent soluble proteins rather than components of insoluble aggregates. To further examine the relationship of septin 3 and septin 7 with insoluble aggregates in FTLD-U, we performed immunoblot analysis on biochemically extracted FTLD-U, AD, and control frontal cortex (Figure 5). Interestingly, while detergent-insoluble urea samples did not reveal enrichment of either protein, septin 3 was noted to be up-regulated in detergent-soluble fractions. Thus, while enriched in FTLD-U tissues, neither protein appears to comprise the pathologic aggregates in this disorder.
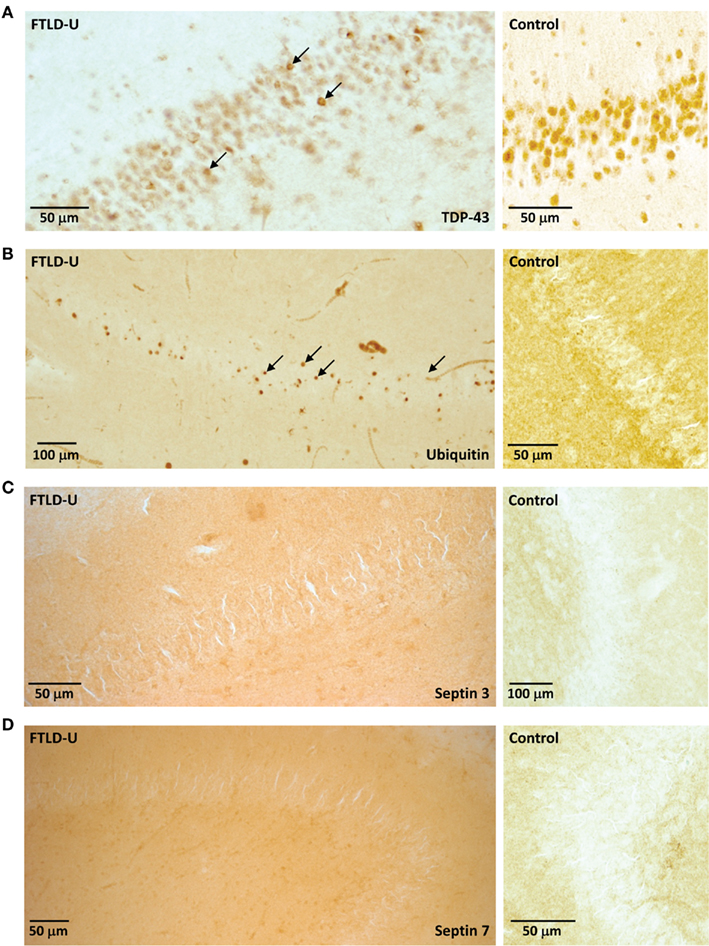
Figure 4. Examination of LCM proteomic candidates by immunohistochemistry. Representative staining of (A) TDP-43, (B) Ubiquitin, (C) Septin 3, and (D) Septin 7 in hippocampal sections from FTLD-U (left) or control (right). While inclusions are apparent with both TDP-43 and ubiquitin immunohistochemistry (arrows), these bodies are not identified by Septin 3 and Septin 7.
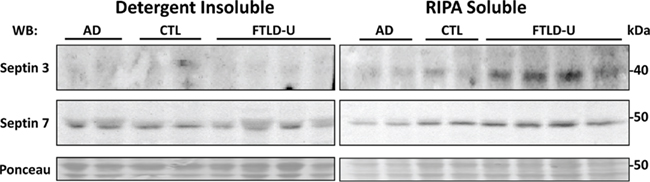
Figure 5. Immunoblots of septin 3 and septin 7 confirm proteomic findings. Frontal cortex samples from two AD, two control, and four FTLD-U cases were sequentially extracted with RIPA buffer (RIPA soluble) and 8 M urea (detergent-insoluble). Both soluble and insoluble fractions were immunoblotted with antibodies to septin 3 (top panel) or septin 7 (middle panel). Notably, septin 3 is enriched in the soluble but not the insoluble fractions. To ensure equal loading and complete transfer of proteins from the gel, membranes were reversibly stained with Ponceau S (bottom panel).
Discussion
The complete composition of inclusions and the mechanisms underlying their formation in neurodegeneration are currently unclear. The evaluation of pathological inclusions in FTLD-U and other diseases has, to date, been based on conventional neuropathologic approaches aimed at differentiating these lesions from other abnormal protein aggregates. As a result, immunohistochemical identification has been limited to a subset of proteins classically associated with neurodegeneration, including β-amyloid, α-synuclein, tau, and more recently, TDP-43 and FUS (Okamoto et al., 1991; Mackenzie et al., 2009). The combination of LCM and LC–MS/MS has been successfully applied in the proteomic characterization of multiple neurodegenerative disease aggregates, including amyloid plaques (Liao et al., 2004), neurofibrillary tangles (Wang et al., 2005), and Lewy bodies (Leverenz et al., 2007). However, in contrast with the capture of amyloid plaques (Liao et al., 2004), where 2000 aggregates (50–100 μm in diameter) were specifically microdissected from four cortical sections, each FTLD-U section contained <50 much smaller inclusions. The small size of these inclusions, typically 1–8 μm in diameter (Okamoto et al., 1991), and their relatively sparse distribution rendered their direct capture and analysis impractical.
Since direct comparison of inclusion-containing dentate granule cells with adjacent unaffected granule cells was technically impossible, we performed more global comparisons between independent pairs of FTLD-U and control cases. We employed a label-free quantitative proteomics approach to characterize dentate granule cells microdissected from FTLD-U hippocampal sections. Only a small proportion of the final lysate extracted from the LCM caps consisted of aggregating proteins, and our approach identifies mainly non-aggregated proteins whose expression is altered in FTLD-U dentate granule cells. Analysis of three independent FTLD-U and control sample comparisons resulted in the identification of 1252 proteins, of which 73 were significantly altered in FTLD-U. Several of the identified proteins have been previously implicated in known pathogenic pathways in FTLD-U or ALS, including VCP (Gitcho et al., 2009) which demonstrated enrichment of ∼1.3-fold. Moreover, TDP-43, a known component of ubiquitin-immunoreactive inclusions, was significantly enriched in FTLD-U samples compared with controls. Identification of these established proteins serves to validate our experimental strategy.
Mass spectrometry is a rapid and sensitive method for the identification of critical proteins in neurodegenerative diseases. Our MS findings are highly consistent with global alterations in protein levels in FTLD-U, but the initial proteins targeted for additional immunohistochemical analysis did not localize to ubiquitin-immunoreactive inclusions. While specific association of a protein with pathological lesions offers the most straightforward evidence for involvement in disease, this approach is inherently limited by the fact that accumulated inclusions in autopsy specimens may only reveal end-stage pathology resulting from many years of disease. Several proteins have been shown to play important roles in the pathogenesis of FTLD-U despite failing to localize within inclusion bodies. These include progranulin (Baker et al., 2006; Cruts et al., 2006) and CHMP2B (Skibinski et al., 2005), both of which were shown to play critical genetic roles in the disease process. To our knowledge, this study provides the first unbiased proteomic analysis identifying a list of candidate proteins with increased expression in the FTLD-U hippocampus. The altered level of expression in FTLD-U suggests that some of our 73 candidate proteins contribute to disease pathogenesis, and additional case–control comparisons may help further delineate those proteins with consistent change in disease tissues. Moreover, the identification of proteins in several pathways previously implicated in other neurodegenerative diseases, including metabolism and oxidative stress, protein degradation, and components of the cytoskeleton, suggest that additional validation of proteins in our list may be warranted. Their role in FTLD-U and their potential utility as biomarkers of the disease deserves further study.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Michael Iuvone for technical help with laser capture microdissection and Craig J. Heilman for help in the selection of antibodies. This work was supported by NIH grant P50 AG025688, P30 NS055077, and NIH training grants F30 NS057902 to Yair M. Gozal and T32 NS007480 to Eric B. Dammer. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
Baker, M., Mackenzie, I. R., Pickering-Brown, S. M., Gass, J., Rademakers, R., Lindholm, C., Snowden, J., Adamson, J., Sadovnick, A. D., Rollinson, S., Cannon, A., Dwosh, E., Neary, D., Melquist, S., Richardson, A., Dickson, D., Berger, Z., Eriksen, J., Robinson, T., Zehr, C., Dickey, C. A., Crook, R., McGowan, E., Mann, D., Boeve, B., Feldman, H., and Hutton, M. (2006). Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919.
Bonner, R. F., Emmert-Buck, M., Cole, K., Pohida, T., Chuaqui, R., Goldstein, S., and Liotta, L. A. (1997). Laser capture microdissection: molecular analysis of tissue. Science 278, 1481,1483.
Buratti, E., Dork, T., Zuccato, E., Pagani, F., Romano, M., and Baralle, F. E. (2001). Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 20, 1774–1784.
Cairns, N. J., Bigio, E. H., Mackenzie, I. R., Neumann, M., Lee, V. M., Hatanpaa, K. J., White, C. L. III, Schneider, J. A., Grinberg, L. T., Halliday, G., Duyckaerts, C., Lowe, J. S., Holm, I. E., Tolnay, M., Okamoto, K., Yokoo, H., Murayama, S., Woulfe, J., Munoz, D. G., Dickson, D. W., Ince, P. G., Trojanowski, J. Q., and Mann, D. M. (2007). Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 114, 5–22.
Cheng, D., Hoogenraad, C. C., Rush, J., Ramm, E., Schlager, M. A., Duong, D. M., Xu, P., Rukshan, S., Hanfelt, J., Nakagawa, T., Sheng, M., and Peng, J. (2006). Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell. Proteomics 5, 1158–1170.
Cruts, M., Gijselinck, I., van der Zee, J., Engelborghs, S., Wils, H., Pirici, D., Rademakers, R., Vandenberghe, R., Dermaut, B., Martin, J. J., van Duijn, C., Peeters, K., Sciot, R., Santens, P., De Pooter, T., Mattheijssens, M., Van den Broeck, M., Cuijt, I., Vennekens, K., De Deyn, P. P., Kumar-Singh, S., and Van Broeckhoven, C. (2006). Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924.
Emmert-Buck, M. R., Bonner, R. F., Smith, P. D., Chuaqui, R. F., Zhuang, Z., Goldstein, S. R., Weiss, R. A., and Liotta, L. A. (1996). Laser capture microdissection. Science 274, 998–1001.
Eng, J., McCormack, A. L., and Yates, J. R. III. (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989.
Eriksen, J. L., and Mackenzie, I. R. (2008). Progranulin: normal function and role in neurodegeneration. J. Neurochem. 104, 287–297.
Forman, M. S., Farmer, J., Johnson, J. K., Clark, C. M., Arnold, S. E., Coslett, H. B., Chatterjee, A., Hurtig, H. I., Karlawish, J. H., Rosen, H. J., Van Deerlin, V., Lee, V. M., Miller, B. L., Trojanowski, J. Q., and Grossman, M. (2006). Frontotemporal dementia: clinicopathological correlations. Ann. Neurol. 59, 952–962.
Gitcho, M. A., Strider, J., Carter, D., Taylor-Reinwald, L., Forman, M. S., Goate, A. M., and Cairns, N. J. (2009). VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J. Biol. Chem. 284, 12384–12398.
Goldsworthy, S. M., Stockton, P. S., Trempus, C. S., Foley, J. F., and Maronpot, R. R. (1999). Effects of fixation on RNA extraction and amplification from laser capture microdissected tissue. Mol. Carcinog. 25, 86–91.
Gozal, Y. M., Cheng, D., Duong, D. M., Lah, J. J., Levey, A. I., and Peng, J. (2006). Merger of laser capture microdissection and mass spectrometry: a window into the amyloid plaque proteome. Meth. Enzymol. 412, 77–93.
Gozal, Y. M., Duong, D. M., Gearing, M., Cheng, D., Hanfelt, J. J., Funderburk, C., Peng, J., Lah, J. J., and Levey, A. I. (2009). Proteomics analysis reveals novel components in the detergent-insoluble subproteome in Alzheimer’s disease. J. Proteome Res. 8, 5069–5079.
Graff-Radford, N. R., and Woodruff, B. K. (2007). Frontotemporal dementia. Semin. Neurol. 27, 48–57.
Kovari, E., Gold, G., Giannakopoulos, P., and Bouras, C. (2004). Cortical ubiquitin-positive inclusions in frontotemporal dementia without motor neuron disease: a quantitative immunocytochemical study. Acta Neuropathol. 108, 207–212.
Kumar-Singh, S., and Van Broeckhoven, C. (2007). Frontotemporal lobar degeneration: current concepts in the light of recent advances. Brain Pathol. 17, 104–114.
Kwiatkowski, T. J. Jr., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., Davis, A., Gilchrist, J., Kasarskis, E. J., Munsat, T., Valdmanis, P., Rouleau, G. A., Hosler, B. A., Cortelli, P., de Jong, P. J., Yoshinaga, Y., Haines, J. L., Pericak-Vance, M. A., Yan, J., Ticozzi, N., Siddique, T., McKenna-Yasek, D., Sapp, P. C., Horvitz, H. R., Landers, J. E., and Brown, R. H. Jr. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208.
Leverenz, J. B., Umar, I., Wang, Q., Montine, T. J., McMillan, P. J., Tsuang, D. W., Jin, J., Pan, C., Shin, J., Zhu, D., and Zhang, J. (2007). Proteomic identification of novel proteins in cortical lewy bodies. Brain Pathol. 17, 139–145.
Li, X. J., Zhang, H., Ranish, J. A., and Aebersold, R. (2003). Automated statistical analysis of protein abundance ratios from data generated by stable-isotope dilution and tandem mass spectrometry. Anal. Chem. 75, 6648–6657.
Liao, L., Cheng, D., Wang, J., Duong, D. M., Losik, T. G., Gearing, M., Rees, H. D., Lah, J. J., Levey, A. I., and Peng, J. (2004). Proteomic characterization of postmortem amyloid plaques isolated by laser capture microdissection. J. Biol. Chem. 279, 37061–37068.
Litvan, I., Hauw, J. J., Bartko, J. J., Lantos, P. L., Daniel, S. E., Horoupian, D. S., McKee, A., Dickson, D., Bancher, C., Tabaton, M., Jellinger, K., and Anderson, D. W. (1996). Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J. Neuropathol. Exp. Neurol. 55, 97–105.
Mackenzie, I. R., Neumann, M., Bigio, E. H., Cairns, N. J., Alafuzoff, I., Kril, J., Kovacs, G. G., Ghetti, B., Halliday, G., Holm, I. E., Ince, P. G., Kamphorst, W., Revesz, T., Rozemuller, A. J., Kumar-Singh, S., Akiyama, H., Baborie, A., Spina, S., Dickson, D. W., Trojanowski, J. Q., and Mann, D. M. (2009). Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 117, 15–18.
Mackenzie, I. R., and Rademakers, R. (2007). The molecular genetics and neuropathology of frontotemporal lobar degeneration: recent developments. Neurogenetics 8, 237–248.
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’Brien, J. T., Feldman, H., Cummings, J., Duda, J. E., Lippa, C., Perry, E. K., Aarsland, D., Arai, H., Ballard, C. G., Boeve, B., Burn, D. J., Costa, D., Del Ser, T., Dubois, B., Galasko, D., Gauthier, S., Goetz, C. G., Gomez-Tortosa, E., Halliday, G., Hansen, L. A., Hardy, J., Iwatsubo, T., Kalaria, R. N., Kaufer, D., Kenny, R. A., Korczyn, A., Kosaka, K., Lee, V. M., Lees, A., Litvan, I., Londos, E., Lopez, O. L., Minoshima, S., Mizuno, Y., Molina, J. A., Mukaetova-Ladinska, E. B., Pasquier, F., Perry, R. H., Schulz, J. B., Trojanowski, J. Q., and Yamada, M. (2005). Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65, 1863–1872.
McKhann, G. M., Albert, M. S., Grossman, M., Miller, B., Dickson, D., and Trojanowski, J. Q. (2001). Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch. Neurol. 58, 1803–1809.
Mirra, S. S., Heyman, A., McKeel, D., Sumi, S. M., Crain, B. J., Brownlee, L. M., Vogel, F. S., Hughes, J. P., van Belle, G., and Berg, L. (1991). The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486.
Morrison, B. M., Hof, P. R., and Morrison, J. H. (1998). Determinants of neuronal vulnerability in neurodegenerative diseases. Ann. Neurol. 44, S32–S44.
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., Bruce, J., Schuck, T., Grossman, M., Clark, C. M., McCluskey, L. F., Miller, B. L., Masliah, E., Mackenzie, I. R., Feldman, H., Feiden, W., Kretzschmar, H. A., Trojanowski, J. Q., and Lee, V. M. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133.
Okamoto, K., Hirai, S., Yamazaki, T., Sun, X. Y., and Nakazato, Y. (1991). New ubiquitin-positive intraneuronal inclusions in the extra-motor cortices in patients with amyotrophic lateral sclerosis. Neurosci. Lett. 129, 233–236.
Ornstein, D. K., Gillespie, J. W., Paweletz, C. P., Duray, P. H., Herring, J., Vocke, C. D., Topalian, S. L., Bostwick, D. G., Linehan, W. M., Petricoin, E. F. III, and Emmert-Buck, M. R. (2000). Proteomic analysis of laser capture microdissected human prostate cancer and in vitro prostate cell lines. Electrophoresis 21, 2235–2242.
Peng, J., Elias, J. E., Thoreen, C. C., Licklider, L. J., and Gygi, S. P. (2003). Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J. Proteome Res. 2, 43–50.
Rollinson, S., Rizzu, P., Sikkink, S., Baker, M., Halliwell, N., Snowden, J., Traynor, B. J., Ruano, D., Cairns, N., Rohrer, J. D., Mead, S., Collinge, J., Rossor, M., Akay, E., Guerreiro, R., Rademakers, R., Morrison, K. E., Pastor, P., Alonso, E., Martinez-Lage, P., Graff-Radford, N., Neary, D., Heutink, P., Mann, D. M., Van Swieten, J., and Pickering-Brown, S. M. (2009). Ubiquitin associated protein 1 is a risk factor for frontotemporal lobar degeneration. Neurobiol. Aging 30, 656–665.
Seyfried, N. T., Gozal, Y. M., Dammer, E. B., Xia, Q., Duong, D. M., Cheng, D., Lah, J. J., Levey, A. I., and Peng, J. (2010). Multiplex SILAC analysis of a cellular TDP-43 proteinopathy model reveals protein inclusions associated with SUMOylation and diverse polyubiquitin chains. Mol. Cell. Proteomics 9, 705–718.
Shevchenko, A., Wilm, M., Vorm, O., and Mann, M. (1996). Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68, 850–858.
Simone, N. L., Bonner, R. F., Gillespie, J. W., Emmert-Buck, M. R., and Liotta, L. A. (1998). Laser-capture microdissection: opening the microscopic frontier to molecular analysis. Trends Genet. 14, 272–276.
Skibinski, G., Parkinson, N. J., Brown, J. M., Chakrabarti, L., Lloyd, S. L., Hummerich, H., Nielsen, J. E., Hodges, J. R., Spillantini, M. G., Thusgaard, T., Brandner, S., Brun, A., Rossor, M. N., Gade, A., Johannsen, P., Sorensen, S. A., Gydesen, S., Fisher, E. M., and Collinge, J. (2005). Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 37, 806–808.
Snowden, J., Neary, D., and Mann, D. (2007). Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol. 114, 31–38.
Tan, A. Y., and Manley, J. L. (2010). TLS inhibits RNA polymerase III transcription. Mol. Cell. Biol. 30, 186–196.
The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease (1997). Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. Neurobiol. Aging 18, S1–S2.
Trojanowski, J. Q., and Dickson, D. (2001). Update on the neuropathological diagnosis of frontotemporal dementias. J. Neuropathol. Exp. Neurol. 60, 1123–1126.
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., Hu, X., Smith, B., Ruddy, D., Wright, P., Ganesalingam, J., Williams, K. L., Tripathi, V., Al-Saraj, S., Al-Chalabi, A., Leigh, P. N., Blair, I. P., Nicholson, G., de Belleroche, J., Gallo, J. M., Miller, C. C., and Shaw, C. E. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211.
Wang, Q., Woltjer, R. L., Cimino, P. J., Pan, C., Montine, K. S., Zhang, J., and Montine, T. J. (2005). Proteomic analysis of neurofibrillary tangles in Alzheimer disease identifies GAPDH as a detergent-insoluble paired helical filament tau binding protein. FASEB J. 19, 869–871.
Wang, W., Zhou, H., Lin, H., Roy, S., Shaler, T. A., Hill, L. R., Norton, S., Kumar, P., Anderle, M., and Becker, C. H. (2003). Quantification of proteins and metabolites by mass spectrometry without isotopic labeling or spiked standards. Anal. Chem. 75, 4818–4826.
Watts, G. D., Wymer, J., Kovach, M. J., Mehta, S. G., Mumm, S., Darvish, D., Pestronk, A., Whyte, M. P., and Kimonis, V. E. (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381.
Woulfe, J., Kertesz, A., and Munoz, D. G. (2001). Frontotemporal dementia with ubiquitinated cytoplasmic and intranuclear inclusions. Acta Neuropathol. 102, 94–102.
Keywords: neurodegeneration, dementia, laser capture microdissection, mass spectrometry, ubiquitin
Citation: Gozal YM, Dammer EB, Duong DM, Cheng D, Gearing M, Rees HD, Peng J, Lah JJ and Levey AI (2011) Proteomic analysis of hippocampal dentate granule cells in frontotemporal lobar degeneration: application of laser capture technology. Front. Neur. 2:24. doi: 10.3389/fneur.2011.00024
Received: 02 September 2010;
Accepted: 01 April 2011;
Published online: 25 April 2011.
Edited by:
Patrick A. Lewis, University College London, UKReviewed by:
Elisa Greggio, University of Padova, ItalyGaia Skibinski, The J. David Gladstone Institutes, USA
Shushant Jain, Vu Medical Center, Netherlands
Copyright: © 2011 Gozal, Dammer, Duong, Cheng, Gearing, Rees, Peng, Lah and Levey. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: James J. Lah, Department of Neurology, Emory University School of Medicine, Whitehead Biomedical Research Building, 615 Michael Street, Room 505C, Atlanta, GA 30322, USA. e-mail: jlah@emory.edu Allan I. Levey, Department of Neurology, Emory University School of Medicine, Woodruff Memorial Research Building, 101 Woodruff Circle, Suite 6000, Atlanta, GA 30322. USA. e-mail: alevey@emory.edu