
- 1 Division of Pulmonary and Critical Care Medicine, Department of Internal Medicine, University of Texas Medical Branch, Galveston, TX, USA
- 2 Division of Cognitive and Behavioral Neurology, Department of Neurology, University of Texas Medical Branch, Galveston, TX, USA
- 3 Encephalogistics, Inc., Galveston, TX, USA
- 4 Brain-Mind Project, Inc., Galveston, TX, USA
- 5 Brown Foundation, Institute of Molecular Medicine, University of Texas Health Science Center, Houston, TX, USA
Lifestyle factors such as intellectual stimulation, cognitive and social engagement, nutrition, and various types of exercise appear to reduce the risk for common age-associated disorders such as Alzheimer’s disease (AD) and vascular dementia. In fact, many studies have suggested that promoting physical activity can have a protective effect against cognitive deterioration later in life. Slowing or a deterioration of walking speed is associated with a poor performance in tests assessing psychomotor speed and verbal fluency in elderly individuals. Fitness training influences a wide range of cognitive processes, and the largest positive impact observed is for executive (a.k.a. frontal lobe) functions. Studies show that exercise improves additional cognitive functions such as tasks mediated by the hippocampus, and result in major changes in plasticity in the hippocampus. Interestingly, this exercise-induced plasticity is also pronounced in APOE ε4 carriers who express a risk factor for late-onset AD that may modulate the effect of treatments. Based on AD staging by Braak and Braak (1991) and Braak et al. (1993) we propose that the effects of exercise occur in two temporo-spatial continua of events. The “inward” continuum from isocortex (neocortex) to entorhinal cortex/hippocampus for amyloidosis and a reciprocal “outward” continuum for neurofibrillary alterations. The exercise-induced hypertrophy of the hippocampus at the core of these continua is evaluated in terms of potential for prevention to stave off neuronal degeneration. Exercise-induced production of growth factors such as the brain-derived neurotrophic factor (BDNF) has been shown to enhance neurogenesis and to play a key role in positive cognitive effects. Insulin-like growth factor (IGF-1) may mediate the exercise-induced response to exercise on BDNF, neurogenesis, and cognitive performance. It is also postulated to regulate brain amyloid β (Aβ) levels by increased clearance via the choroid plexus. Growth factors, specifically fibroblast growth factor and IGF-1 receptors and/or their downstream signaling pathways may interact with the Klotho gene which functions as an aging suppressor gene. Neurons may not be the only cells affected by exercise. Glia (astrocytes and microglia), neurovascular units and the Fourth Element may also be affected in a differential fashion by the AD process. Analyses of these factors, as suggested by the multi-dimensional matrix approach, are needed to improve our understanding of this complex multi-factorial process, which is increasingly relevant to conquering the escalating and intersecting world-wide epidemics of dementia, diabetes, and sarcopenia that threaten the global healthcare system. Physical activity and interventions aimed at enhancing and/or mimicking the effects of exercise are likely to play a significant role in mitigating these epidemics, together with the embryonic efforts to develop cognitive rehabilitation for neurodegenerative disorders.
Introduction
Plasticity can be broadly defined as the property of the nervous system to adapt to changes in the external environment and/or its integrity (i.e., lesions) in order to maintain or recover and optimize its functions. From a morphological standpoint, brain plasticity includes the potential of neurons to change their synaptic connections (Ashford and Jarvik, 1985). The elongation of axons, growth of collateral ramifications, and remodeling allow establishment of new synapses and new cognitive and behavioral operations, that can be adaptive or maladaptive to the situation that triggered the phenomenon. Exercise-induced plasticity is but one such situation. Exercise-enhanced neuronal plasticity might help neural circuits spared, or less affected by a disease to compensate for deteriorated circuits and improve other network performance and overall neurological function (Palop et al., 2006). We focus this review in many aspects by which skeletal muscle exercise affects brain plasticity and may arrest, slow down or even reverse the pathophysiological evolution to mild cognitive impairment (MCI) and possibly to Alzheimer’s disease (AD). During aging, all physiological functions gradually decline (Lamberts et al., 1997). There is a diminished capacity for cellular protein synthesis, a decline in immune function, an increase in fat mass, a loss of muscle mass and strength, and a decrease in bone mineral density (Lamberts et al., 1997).
Sarcopenia
In an increasing number of elderly patients, loss of muscle strength and mass (sarcopenia) is the main limiting factor that determines their chances of living an independent life until death. The lack of sufficient physical exercise has been linked to immune system dysfunction, metabolic disease, musculoskeletal disorders, accidents and injuries, pulmonary diseases, and certain types of cancer and neurological disorder (Booth et al., 2002). A sedentary lifestyle is therefore a major risk factor for many chronic pathologies, compounding the effect of aging alone. This is also relevant to the world-wide intersection of the epidemics of diabetes and dementia (Kuljiš and Šalkovic-Petrišic, 2011).
Physical inactivity has also been shown unequivocally to increase the rates of morbidity and mortality (Erikssen et al., 1998; Hu et al., 2004). Therefore, the capacity to exercise is a strong predictor of overall mortality rates, regardless of health status (Handschin and Spiegelman, 2008). Indeed, the devastating effects of a sedentary lifestyle have been repeatedly observed in the elderly (Hollmann et al., 2007). A decrease in muscle function in the elderly population is directly linked to sarcopenia and diminished aerobic capacity. In such patients, a loss of strength affects the ability to carry out daily tasks and social interactions, and to preserve adequate mobility, cognitive performance, and life expectancy (Hollmann et al., 2007). Loss of muscle strength is also an important determinant in the development of frailty throughout the lifespan. Even in the early years, changes in physical activity have deleterious consequences for health status and life expectancy. For example, men 70 years of age or older who engaged in exercise increased significantly their probability of living until age 90 (Yates et al., 2008).
Lifestyle
There has also been a steadily increasing focus on the impact of lifestyle factors, including intellectual involvement, cognitive vitality, social exchanges, nutrition, and physical activity on cognition in old age. Studies have thus examined whether lifestyle factors reduce the risk of age-associated diseases such as AD or vascular dementia. These studies have suggested that physical activity can have a protective effect on cognition later in life (Podewils et al., 2005; Larson et al., 2006; Anderson et al., 2010a,b; Kattenstroth et al., 2010; Ang et al., 2010; Kim et al., 2011). Furthermore, a slowing or a deterioration of the walking speed was associated with a poor performance in tests assessing psychomotor speed and verbal fluency in the elderly (Soumare et al., 2009). Fitness training widely influences numerous cognitive processes, and the largest positive impact observed was for executive control (Colcombe and Kramer, 2003). Executive control processes encompass planning, scheduling, working memory, inhibitory processes, and multitasking. Fitness training was more efficient when programs of aerobic training were associated with strength and flexibility training. Aerobic training induces an increased activity in the frontal and parietal regions of the brain, which are considered to be involved in efficient attentional control and performance on a focused attention task (Colcombe et al., 2004). Those effects in frontal, parietal, and temporal regions were expressed also as a significant increase in gray matter volume (Colcombe et al., 2003; Kramer et al., 2006), which, although relatively well known nowadays, could hardly have been predicted merely a decade ago. Strikingly also, losses of brain volume may be restored by short exercise interventions (Colcombe et al., 2003, 2006; Kramer et al., 2006) raising the intriguing possibility that these rather macroscopic changes may be implemented into regimented strategies for both palliation and therapy.
Lifestyle in Predisposed Individuals
The above macroscopic exercise-induced changes foster hope that predisposed individuals having potential risk factors for dementia may also be sensitive to physical activity and exercise. For example, Larson et al. (2006) showed that the association of regular exercise with a reduced risk for dementia and AD is not influenced by a genetic predisposition for AD such as one or even two ε4 alleles on the APOE gene. Another study showed that exercise improves cognitive functions such as tasks dependent on the hippocampus and resulted in major changes in plasticity in the hippocampus of mice that are APOE ε4 carriers (Nichol et al., 2009). Furthermore, improvements with exercise on a hippocampus-dependent task were specific to ε4 carriers (Nichol et al., 2009). These findings are in line with findings in humans indicating that sedentary APOE ε4 carriers share an increased risk of cognitive decline, and, as such, may exhibit a greater response from physical activity than non-carriers, perhaps in a way similar to mice expressing APOE ε4.
The Entorhinal and Hippocampal Regions: A Target for Exercise-Induced Changes?
The chronology of exercise-induced mitigation may be related to the progression of physiopathological events. Exercise has been shown to suppress liposaccharide (LPS)-inhibited neurogenesis and to enhance the proliferation of multipotent and neural stem cells and their differentiation into neurons (Wu et al., 2007) that appear responsible for significant improvement in learning and memory performance (Wu et al., 2007; Hoveida et al., 2011). Exercise has also been shown to deplete beta-amyloid load in a transgenic mouse model of AD (Adlard et al., 2005). In humans, a population-based, prospective study of a cohort of patients (MCI, AD, and cognitively stable) by magnetic resonance imaging (MRI) and cognitive testing suggested that physical activity (evaluation based on a questionnaire) had not been associated with a reduced rate of white matter lesion progression (Podewils et al., 2007). However, in multiple sclerosis, cardiorespiratory fitness was associated with a sparing of gray matter volume and white matter tract integrity usually undermined by the disease (Prakash et al., 2010).
Skeletal muscle exercise increases neurotrophic factors (Neeper et al., 1995, 1996), β-endorphin production (Koehl et al., 2008), and cell proliferation in the hippocampus and dentate gyrus (Trejo et al., 2001; Glasper et al., 2010; Llorens-Martin et al., 2010a,b,c). In cognitively normal older adults, a 10-year history of physical training (walking, running, and jogging) which met the recommendations of the American Heart Association (AHA) of 7.5 metabolic equivalent (MET)-hours/week (30 min of moderate exercise 5 days/week) had a distribution of biomarkers significantly associated with a lower risk of AD (Liang et al., 2010). The distribution associated with a greater exercise training (AHA) was a low mean cortical binding potential (MCBP) as evaluated by positron emission tomography (PET) which detects the amount of Aβ deposition by a radiotracer, the Pittsburgh compound-B (PIB). In AD, Aβ aggregation leads to plaque formation and thus to a higher uptake of PIB while in trained elderly individuals the PIB uptake is low (Liang et al., 2010). Therefore, CSF levels of Aβ1–42 by higher clearance were also higher in those trained subjects. The levels of biomarkers, tau and phosphorylated tau p(tau)181, reflecting indirectly AD axonal death or neuronal degeneration in brain regions as the hippocampus (De Meyer et al., 2010), were low in the CSF of physically trained elders (Liang et al., 2010). The distribution of biomarkers in sedentary subjects with high PIB was reversed, i.e., low CSF Aβ1–42, high tau and phosphorylated tau p(tau)181. Cognitively normal adult APOE ε4 carriers expressed elevated PIB uptake, lower Aβ1–42, but no difference in tau and p(tau)181 was observed. The overarching question is, therefore, not whether these components of the limbic loop express plastic properties. Rather, why is exercise stopping the evolution of neuropathological lesions? Why is this unknown underlying mechanism associated with the remarkable plasticity of the hippocampus, entorhinal cortex, and dentate gyrus?
Exercise-Induced Entorhinal and Hippocampal Plasticity
The mechanisms that mediate the therapeutic effects of exercise and the pathological changes elicited by a sedentary lifestyle in humans remain largely unknown. Very little is known about the precise structural brain changes resulting from lack of exercise, and the connection between skeletal muscle, cardiovascular exercise leading to those changes, albeit the association has been demonstrated consistently.
MRI Studies and Exercise-Induced Modifications
Magnetic resonance imaging (MRI) has increasingly served to evaluate the effects of skeletal muscle exercise on hippocampal size. Visualization (ultra-high field 7-T MRI) of the hippocampal CA1 apical neuropil layer thinning in subjects with mild AD suggests a greater role for synaptic loss than neuronal loss (Kerchner et al., 2010). Postmortem studies have demonstrated that the hippocampal CA1 apical neuropil is one of the initial sites of pathology, with tau aggregation and atrophy occurring before there is significant loss of the CA1 pyramidal neurons (Braak and Braak, 1991; Braak et al., 1994, 2000). Age-related reduced levels of growth factors such as brain-derived neurotrophic factor (BDNF) correlate with the decline in hippocampal volume and elevated memory deficits (Erickson et al., 2010). There is also a growing body of evidence that aerobic exercise training in older humans increases (serum) BDNF levels, the size of the (anterior) hippocampus, improves spatial memory (Erickson et al., 2011) as well as the plasticity of brain networks (frontal executive, fronto-parietal, primary motor cortex, and primary auditory cortex; Voss et al., 2010). Such effects on hippocampal volume and memory have also been shown in preadolescent children (Chaddock et al., 2010) and to increase the volume of the cerebral gray matter in multiple sclerosis patients (Prakash et al., 2007, 2010). Aging is associated with impaired spatial memory and reduced resting cerebral blood flow (CBF), whereas increased blood flow to the hippocampus is associated with greater memory performance in older adults (Heo et al., 2010). However, the effect on hippocampal–entorhinal cortices of an intervention such as skeletal muscle exercise is difficult to evaluate, especially in advanced AD because of the potential lack of patients’ compliance with the intervention. Indeed, exercise may also mediate its effects on cognition via movement as seen in other neurophysiopathological processes, e.g., proprioception (Bak, 2011).
Animal Studies
Previous studies with animals have shown that aerobic exercise can produce growth of new capillaries in the brain (Black et al., 1990), increase the length and number of dendritic connections between neurons (Cotman and Berchtold, 2002), and also increase cell production in the hippocampus (Van et al., 1999a). Repetitive running exercise promotes angiogenesis in the motor cortex of the adult mammalian brain (Swain et al., 2003). Capillary growth was observed within 30 days from the onset of exercise regimen. It has been shown that more neurons were generated post-exercise in the dentate gyrus of young and aged animals (Van et al., 1999b; Kim et al., 2004). Farmer showed that BDNF mRNA levels were elevated in the dentate gyrus of running animals (Farmer et al., 2004). The enhancement of neurogenesis and learning in exercising animals may be related to increased levels of BDNF (Berchtold et al., 2010; Adlard et al., 2011). Indeed, it has been shown that exercise increases BDNF levels in the hippocampus in both young and aged brains (Neeper et al., 1995, 1996; Cotman and Berchtold, 2002). However, further research is required to understand the relationship between BDNF and age-related neuronal loss as well as the role of exercise in mitigating changes in BDNF. BDNF also regulates multiple neurotransmitters, including the dopaminergic, cholinergic, and GABAergic systems (Knusel et al., 1991). Exercise may also be a potential intervention in reducing the onset or the rate of progression in Parkinson’s disease (Smith and Zigmond, 2003). Therefore, BDNF is believed to play a key role in exercise-induced positive cognitive effects although the underlying mechanisms remain to be elucidated. The greatest effects of exercise on BDNF seem to materialize in highly plastic, or transformable areas, responsive to environmental stimuli (Volkmar and Greenough, 1972; Castren et al., 1992; Neeper et al., 1995).
Systemic Growth Factors and the Limbic Loop
Blood levels and brain uptake of insulin-like growth factor (IGF-1) are elevated with exercise (Carro et al., 2001; Trejo et al., 2001). Historically, the first biochemical pathway shown to influence brain aging was the insulin–IGF-1 pathway (Kenyon, 2010). Blocking the entry of IGF-1 into the brain resulted in the blockage of the exercise-induced neuron proliferation in the dentate gyrus, suggesting that IGF-1 is also a key factor in neurogenesis (Trejo et al., 2001). Therefore, IGF-1 may mediates the response to exercise on BDNF, neurogenesis, and cognitive performance.
Treatment with systemic slow-release of IGF-1 reverts the impairment of spatial learning, memory, and decreases the total brain Aβ load (Carro et al., 2006a). Serum IGF-1 regulates brain Aβ levels by increased clearance via the choroid plexus (Carro et al., 2002, 2006a). Serum IGF-1 controls brain Aβ levels by facilitating the passage of Aβ protein transport such as albumin and/or apolipoprotein J (Trejo et al., 2004) into the brain through the choroid plexus and therefore may obliterate an “amyloid sink” whereby brain Aβ can be rapidly eliminated from the brain. A deficient clearance of Aβ in the brain via the choroid plexus may thus contribute to AD-related amyloidosis. Blockage of IGF-1 receptors in the choroid plexus triggers a cascade of brain disturbances in rodents that are felt to resemble those found in AD (Carro et al., 2006b). The latter includes amyloidosis, cognitive impairment, synaptic vesicle protein loss, gliosis, and abnormally phosphorylated forms of tau. Insights into mechanisms of reduced sensitivity or blockage of IGF-1 receptors at the choroids plexus may thus shed the light on the pathogenesis of sporadic AD. In humans, a large prospective multi-center study revealed an increased blood Aβ1–42/Aβ1–40 ratio, which is strongly associated with a lower risk of sporadic dementia (AD and mixed/vascular dementia; Lambert et al., 2009). However, based on these results the clearance of overall Aβ cannot be determined. It is also unclear whether the “amyloid sink” is overwhelmed, or whether Aβ accumulates within brain plaques as the production is increased. The reduction in plasma Aβ may reflect a decrease of soluble Aβ in the systemic circulation, as it accumulates within insoluble brain plaques (Mayeux et al., 2003). Low CSF Aβ1–42 levels appear to reflect plaque formation and may be considered as an in vivo biomarker of AD pathology (De Meyer et al., 2010; Prins and van Swieten, 2010). The production of Aβ1–42 and Aβ1–40 was found to be similar in AD patients and controls, whereas clearance of Aβ1–42 and Aβ1–40 significantly decreased in AD patients vs. cognitively normal controls (Mawuenyega et al., 2010). However, a direct cause-effect relationship between decreased CSF Aβ1–42 levels and CNS clearance of Aβ1–42 remains to be determined. Therefore, experimental analysis of whether similar or different effects on the parameters relevant to modulating the risk for dementia occur as the result of different types of exercise is needed.
Beta-Endorphins
A deficiency of beta-endorphins totally blocks the exercise-induced increase in hippocampal cell proliferation in vivo in non-sedentary mice (Koehl et al., 2008). Therefore, β-endorphins are necessary and perhaps even critical to hippocampal neurogenesis during induced by exercise. Beta-endorphin is synthesized mainly in the pituitary gland. It is released into the circulation and is transported into other areas of the brain through nerve fibers, where it acts preferentially on μ opioid receptors (MOR) distributed throughout the brain, including the hippocampus (Koehl et al., 2008). Under non-stress conditions, the circulating levels of β-endorphin are extremely low. In contrast, exercise at an intensity greater than 50% of 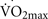 significantly increases the plasma levels of β-endorphins in normal brains (Taylor et al., 1994; Pedersen and Hoffman-Goetz, 2000). Therefore, presumably low levels of β-endorphins in aging brains in sedentary elders may negatively affect the plasticity of the hippocampal and entorhinal regions. This hypothesis provides yet another target for experimental assessment and therapeutic intervention.
significantly increases the plasma levels of β-endorphins in normal brains (Taylor et al., 1994; Pedersen and Hoffman-Goetz, 2000). Therefore, presumably low levels of β-endorphins in aging brains in sedentary elders may negatively affect the plasticity of the hippocampal and entorhinal regions. This hypothesis provides yet another target for experimental assessment and therapeutic intervention.
Biological Mediators of Physical Activity
How may exercise-induced systemic mediators affect the brain? Strength and endurance exercises activate many distinct signal-transduction pathways and result in specific beneficial adaptations of skeletal muscle. Physical activity is closely linked to inflammation and immunity in a complex way (Febbraio, 2007). Regular, moderate exercise reduces the level of systemic inflammation (Gleeson, 2007). The entire array of the mediators of this beneficial effect are unclear, but at least a few candidate mechanisms have been identified. Exercise increases the release of adrenaline, cortisol, growth hormone, dehydroepiandrosterone (DHEA), prolactin, and other factors that have immunomodulatory effects (Nieman, 2003). Myokines, cytokines that are produced and secreted by skeletal muscle cells provide further insight on the association between exercise and inflammation (Febbraio, 2007) and may provide markers of the beneficial systemic effects of exercise on non-muscle tissues (Handschin and Spiegelman, 2008). Contraction of muscle fibers produce IL-6, IL-8, and IL-15 (Pedersen et al., 2007). Exercise induces the appearance of several other cytokines into the circulation such as IL-1, IL-10, and tumor necrosis factor-α (TNF-α; Pedersen et al., 2007; Handschin and Spiegelman, 2008). In elderly humans, higher levels of circulating IL-6 are inversely correlated with levels of physical activity, with muscle mass, and with the anabolic mediator IGF-1 (Zaldivar et al., 2006). Strenuous exercise leads to increased levels of pro-inflammatory cytokines, but simultaneously cytokine inhibitors and anti-inflammatory cytokines restrict the magnitude and duration of the inflammatory response to exercise. Brief exercise sessions lead to seemingly paradoxical changes in the pattern of intracellular cytokines and growth factor expression in leukocytes, reflecting pro-inflammatory and anti-inflammatory function and growth mediators. The release of cytokines such as vascular endothelial growth factor (VEGF) and Il-6 is associated with angiogenesis and muscle growth (Egginton, 2009). Microcirculatory endothelial growth is a key player during exercise, and vascular remodeling follows dynamically the biochemical changes induced by the mechanically powerful stimuli of exercise. It is necessary to understand this growth regulation by transcription and stability of mRNA, production of proteins and interactions with other growth factors. Pituitary (basic) β-FGF, an endothelial cell mitogen, is active ubiquitously on various tissues such as capillary endothelial cells with powerful angiogenic activity in vivo, vascular smooth cells and adrenal cortex (Esch et al., 1985; Montesano et al., 1986). Since β-FGF has been isolated from the brain (Gospodarowicz et al., 1985, 1986), it may also likely involve neurotrophic activity of hippocampal neurons (Walicke et al., 1986). Basic fibroblast growth factor (FGF) promotes neuronal survival, rapid neuronal growth, elaboration of neurites, and is not produced merely by astrocyte proliferation (Morrison et al., 1986; Walicke et al., 1986). However, there is also evidence that β-FGF may have a mitogenic effect on astrocytes and oligodendrocytes (Morrison and de Vellis, 1981). In contrast, the AD phenotype is a consequence of convergence of multiple risk factors or dysregulations (Fernández et al., 2008). Neuroinflammation is a key component that activates microglia, produces nuclear factor (NF)-κB, and induces multiple cell apoptosis-promoting signals, such as TNF-α, IL-1β, and IL-6, that lead to the production of pathological isoforms of the tau protein (Fernández et al., 2008; Maccioni et al., 2009a, Maccioni et al., 2009a,b). These abnormal signals result in over-activation of some cell cycle enzymes, such as cdk5 and the neuronal glycogen synthase kinase, with the consequent tau hyperphosphorylation (Fernández et al., 2008; Maccioni et al., 2009a,b). Overexpression of IL-1, other pro-inflammatory cytokines, and glial activation have been associated in the pathogenesis of both AD and Parkinson’s disease (Griffin et al., 2006). Although several lines of evidence indicate a role for inflammatory mediators in the neurodegenerative cascade, they may explain only a portion of a more complex pathophysiological process, yet at the outset it seems that this phenomenon may be strongly linked to the strong epidemiological evidence for a beneficial effect of exercise on neurodegeneration.
Growth Factors, Neural Stem Cells and Klotho
What other age- or exercise-related events are triggered by changes in the levels of regulatory molecules? And what, in turn, modulates the regulators? Can the levels of growth factors change with exercise training because of alterations in the regulators?
On one hand, exercise results in an increase in p38 MAP-kinase (p38 MAPK) phosphorylation that may last for hours after mild/moderate exercise (Geiger et al., 2005). There is a growing line of evidence suggesting that activation of p38 is in the pathway of the exercise-induced stimulation of the glucose transport by insulin and skeletal muscle contractions (Geiger et al., 2005, 2006). Phosphorylation of p38 MAPK is significantly increased (2 to 16-fold) by exercise (Hulmi et al., 2010). Exercise induces a lower basal p38 MAPK activity (Chen et al., 2010). The p38 MAPK/PGC-1alpha regulatory axis participates in the control of contractile protein expression, angiogenesis, mitochondrial biogenesis, and other adaptations of the skeletal muscle (Pogozelski et al., 2009; Lira et al., 2010). In human skeletal muscle, activation of cytosolic p38 MAPK in this axis may be a signal to increase nuclear concentration and activate of PGC-1alpha in response to endurance exercise (Little et al., 2010). On the other hand, p38 activity is significantly increased in AD (Muñoz et al., 2007; Chang et al., 2010; Muñoz and Ammit, 2010). Glutathione (GSH) synthesis induces a neuroinflammatory response in human microglia and astrocytes associated with activation of p38 MAP-kinase (Lee et al., 2010, 2011). This is a new model for inducing neuroinflammation and suggests that increasing GSH levels in glial cells may confer neuroprotection in neurodegenerative diseases, such as Alzheimer disease, which have a prominent neuroinflammatory component. p38 is a key downstream effector of Aβ-induced neurodegeneration (Zhu et al., 2001, 2005).
An interesting mouse model has a bearing on p38 signaling in the brain. Mice carrying a loss-of-function mutation in the Klotho gene develop a syndrome resembling human aging, including shortened life span, skin atrophy, muscle atrophy, early sterility, osteoporosis, arteriosclerosis, cognitive impairment, neurodegeneration, hearing loss and pulmonary emphysema (Kuro-o et al., 1997). Conversely, the overexpression of the Klotho gene significantly extends the life span of mice (Kuro-o et al., 1997; Kuro-o, 2008). These observations suggest that the Klotho gene functions as an aging suppressor gene (Kurosu et al., 2005) and that this effect is exerted, at least in part, through its interactions with the growth factor FGF23 (Kurosu et al., 2006) and its effect on several growth factor receptors, such as EGFR, IGF1R, frizzled, and TGFβ, and their downstream signaling pathways. Therefore, the Klotho protein may respond or may be affected by physical activity since Klotho interactions are mediated via growth factors such as IGF-1. Moreover, we have recently shown that Klotho has a significant inhibitory effect on p38 activity and signaling in the liver (Hsieh et al., 2010) and our latest data demonstrates that this effect is preserved in the brain (Brobey et al., 2011). The effect of Klotho in the brains of the overexpression models appears to be neuro-protective and prevents the loss of dopaminergic neurons within the substantia nigra after exposure to the neurotoxin MPTP. p38 levels were very low in the whole brains and within the nigra of the overexpression transgenic animals, but the converse was true in the brains of the Klotho knockout animals. The pathway depends on an inhibitory action of Klotho on the ASK1 signalosome, which is a major regulator of p38 activity downstream. Several neurotrophic and growth factors have a neuro-protective capacity on hippocampal and cortical cell structures (Mark et al., 1997). One of the most effective growth factors is the basic (β-FGF) which protects hippocampal and cortical neurons against glutamate toxicity (Mattson et al., 1989), glucose depletion, hypoxia (Cheng and Mattson, 1991; Mattson et al., 1995), oxidative stress (Zhang et al., 1993), Aβ amyloid toxicity (Mark et al., 1997) and lesion-induced cell death (Anderson et al., 1988).
Moreover, the Klotho protein increases resistance to oxidative stress at the cellular and organismal level in mammals through its effects on various anti-oxidant and stress resistant proteins (Yamamoto et al., 2005; Kuro-o, 2008) which may be affected by physical activity. Recent results have revealed that Klotho overexpression in mice may be neuro-protective in mouse models of Parkinson’s disease, indicating that the Klotho signaling networks may provide a fertile ground for the development of novel therapeutic targets in neurodegenerative diseases. The α-Klotho (a-kl) gene encodes a type I membrane protein that is predominantly expressed in tissues that are involved in calcium homeostasis in the choroid plexus, kidneys, and parathyroid glands among other tissue loci (Kuro-o et al., 1997; Kurosu et al., 2005; Imura et al., 2007). Furthermore, cleaved 139-kD extracellular domain of (a-kl) is secreted into the blood and cerebrospinal fluid (CSF; Kurosu et al., 2005; Imura et al., 2007). We are also noticing in our on-going experiments a neuro-protective effect of the Klotho protein especially on human neural stem cells (Figure 1, Foster and Rosenblatt, unpublished).
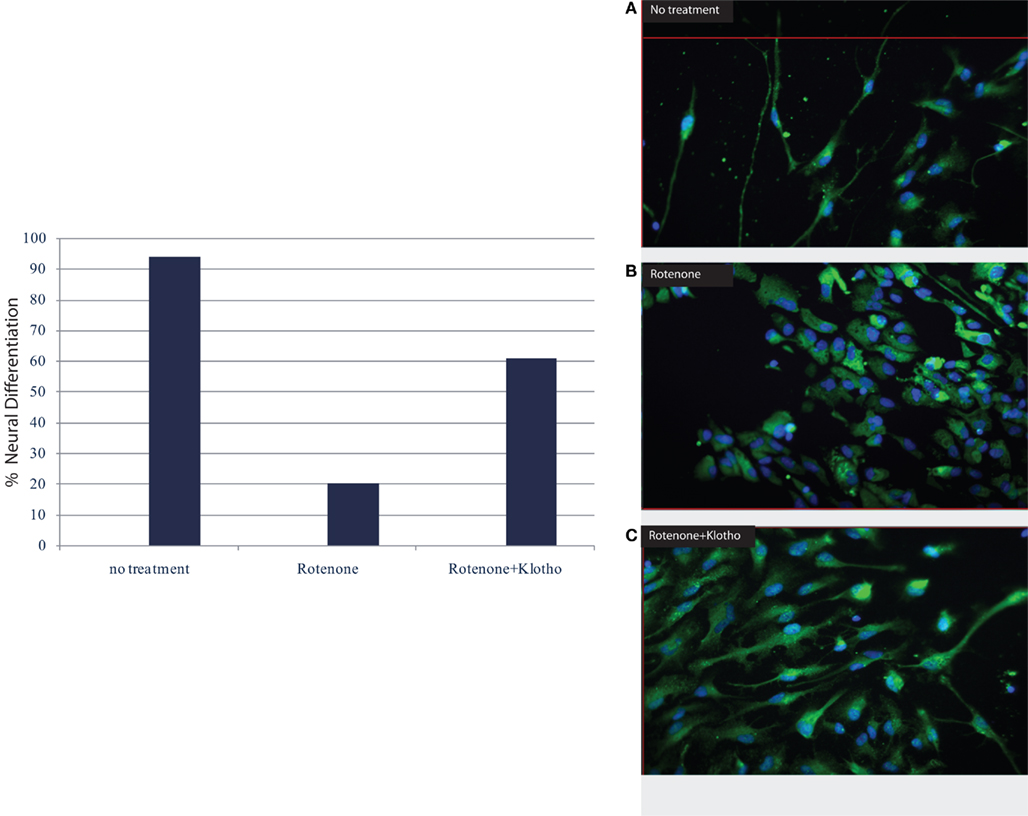
Figure 1. Klotho protein and neuronal differentiation ratio of human neural stem cells (hNSC): NF–M (medium neurofilament protein) vs. astrocyte/GFAP (glial fibrillary acidic protein). (A) no treatment, (B) in the presence of rotenone; apoptosis with destruction of cell membrane and nucleus, loss of neuronal differentiation; (C) the addition of Klotho protein to rotenone increases survival, induces stem cell proliferation and neuronal differentiation (Rosenblatt and Foster, unpublished data).
Potential Markers of Exercise-Induced Efficacy
It is likely that the effect of skeletal muscle exercise produces a maximal efficiency at the pre-symptomatic and prodromal stages rather than at AD dementia stage. Various markers may allow the evaluation of the effect of exercise toward a future diagnostic algorithm. The diagnosis of AD is expected to rely on clinical evidence of the disease phenotype and potential in vivo biological cues of Alzheimer’s pathology (Dubois et al., 2007, 2010). Specific pattern of cognitive changes such as episodic impairment and in vivo structural/biological manifestations of the disease can now be integrated into an algorithm defining the diagnosis of AD (Dubois et al., 2010). Laboratory and neuroimaging biomarkers are very highly correlated with the neuropathological lesions of AD (Buerger et al., 2009; Tapiola et al., 2009; Ewers et al., 2011) as defined in the section below (Braak and Braak, 1991). Hypothetical pathophysiological markers reflect the two cardinal degenerative processes that characterize Alzheimer’s pathology: amyloidosis (SP) and tauopathy (NFT). The International Working Group (Dubois et al., 2010) distinguishes (in vivo) pathophysiological markers which closely correlate with their respective neuropathological lesions, including CSF Aβ (Buerger et al., 2009; (Jack et al., 2010) and PIB–PET with SP (Rinne et al., 2010), and total tau and phospho-tau with NFT (Ewers et al., 2011). These pathophysiological markers are proposed to be predictive of eventual cognitive decline and the development of clinical signs of AD in cognitively normal elderly individuals (Dubois et al., 2010). In line with the pathophysiological findings from MRI and animals studies, topographical markers assessing regional distribution of Alzheimer’s pathology include MRI medial temporal lobe atrophy (Vemuri et al., 2010). Apart from their diagnostic benefits, those markers may also serve as indices of exercise effect and other treatment efficacy in clinical trials. The former topographical marker may also prove particularly useful given the direct relationship between exercise and hippocampal size (Chaddock et al., 2010; Erickson et al., 2010, 2011; Heo et al., 2010; Voss et al., 2010).
Exercise-Induced Low Cholesterol, Impact on Aβ Oligomers and Neurodegeneration
In a large multi-center cohort (3C), the metabolic syndrome had a negative impact on cognitive function tested using standardized clinical protocols (Raffaitin et al., 2011). The link between Aβ and cholesterol metabolism and atherogenesis suggests a plausible mechanism for increased atherosclerosis in patients with AD (Roher et al., 2004; Gamba et al., 2011; Sierra et al., 2011). Statins prevented declining cognitive function by mitigating the amyloidogenic process (Kurata et al., 2011), changes in permeability of the blood–brain barrier and protection against tau hyperphosphorylation (Sierra et al., 2011). Secretase activation in macrophages of atherosclerotic plaques cleaves Aβ from platelet-derived APP (De Meyer et al., 2002, 2010). Aβ appears to produce atherosclerosis by enhancing local inflammatory reactions through activation of scavenger receptors. Clearly, intra-vascular presence of Aβ or Aβ oligomers enhances atherosclerosis in AD patients (Roher et al., 2004). Although regular exercise is related to a higher ratio HDL cholesterol/total cholesterol (Williams et al., 1979; Berg et al., 1996) which would lower the risk of AD via the aforementioned pathway, there are probably other direct or indirect mechanisms that mediate the palliative effects of exercise. Aβ also has powerful effects on neurodegeneration, but there is increasing evidence that Aβ also has major influence on systemic and cerebral blood vessels. Non-deposited Aβ, especially the predominant short form (Niwa et al., 2002; Iadecola, 2004), or soluble Aβ forms “oligomers” (Kayed et al., 2003; Lansbury and Lashuel, 2006) in the cerebral circulation are critical factors in cerebrovascular dysfunction. Aβ accumulation and synaptic loss produces further vascular dysregulation and hypoperfusion which, in turn, facilitates Aβ formation and enhances neurodegeneration.
Potential Inference of Exercise-Induced Myokines on Circulatory Networks
Many of the neuronal and vascular lesions of AD were first been described by Alois Alzheimer in 1907 (Alzheimer et al., 1995; An English translation of Alzheimer’s original 1907 paper). However, it is unclear how these lesions relate to each other and how they contribute to a chronic neurodegenerative and clinically heterogenous disorder. Several lines of evidence converge toward a cerebral circulatory network alteration compounding the glial and neuronal dysfunction in AD. This participation is highlighted by the potential association between AD and the following: (1) cerebrovascular disease (de la Torre, 2004; Gorelick, 2004; Greenberg et al., 2004; Giannakopoulos et al., 2007, 2009; Stopa et al., 2008; Kalaria, 2009; Knopman and Roberts, 2010; Salmina et al., 2010); (2) atherosclerosis (Casserly and Topol, 2004; Roher et al., 2004; Honig et al., 2005; Utter et al., 2008); (3) angiogenesis deficiency (Paris et al., 2004; Wu et al., 2005), and (4) deficiency of Aβ clearance across the blood–brain barrier (Shibata et al., 2000, 2008; Carro et al., 2002, 2003, 2005, 2006b; Trejo et al., 2004). These converging mechanisms indicate that dysfunction of cerebral circulatory networks is probably a critical feature of AD that may play a major role in the pathogenesis of this chronic neurodegenerative condition (de la Torre, 2004). Altered brain capillary circulatory networks physiology, vascular neuroinflammatory response, and disruption of brain activity–mediated CBF regulation are key factors as well for the pathogenesis of cognitive decline in the Alzheimer’s process (Wu et al., 2005). In AD, down-regulation of certain genes (MEOX2 or GAX), exclusively profiling human brain endothelial cells, appear important in the mediation of aberrant angiogenesis (Wu et al., 2005). These are yet additional putative targets for the beneficial effects of exercise in AD.
Early in the twentieth century, Ramón y Cajal (1909) describes a Third Element, which he perceived correctly as different from neurons and astrocytes. Over a decade later, del Rio Hortega et al. (1919) discovered microglial cells among the components of the former Third Element. More recently, a growing body of evidence has shown that immune exercise-induced mechanisms may positively regulate the remodeling of neural circuits, promoting neurogenesis, memory consolidation, hippocampal function (Yirmiya and Goshen, 2011). These beneficial effects of the immune system are mediated by complex interactions cells with immune functions such as microglia and astrocytes, neurons, and neural precursor cells involving the response of neurons and glia to low concentrations of inflammatory cytokines, such as interleukin IL-1, IL-6, and TNF-α, and other mediators, such as prostaglandins and neurotrophins plasticity is considered as crucial for learning and memory encoding. Glia are known to modulate synaptic scaling, homeostatic plasticity and metaplasticity via changes in synaptic coverage, release of chemokines, cytokines and astrocytes, or microglial transmitters release (Ben and Pascual, 2010). However, glial hypertrophy correlates with synaptic numbers via synaptogenesis following motor-skill learning but not with increased capillary density through angiogenesis post-exercise (Anderson et al., 1994). Therefore, in theory, repetitive routine exercise alone may not be sufficient to induce a glial hypertrophy. The fourth element cell (4EC) is a newly identified large population of “unconventional” cells (Reyners et al., 1982, 1986; Bergles et al., 2000) which might also be a target for chemokines and thus exercise. They are antigenically different from astrocytes, oligodendrocytes, and microglia. A mechanism of interest in the pathophysiological AD process is that the 4EC receive glutaminergic synaptic signaling from projection neurons. These 4EC also express all characteristics of neuron-to-neuron synapses (Bergles et al., 2000; Nishiyama et al., 2009; Kuljiš, 2010c). Because of the plasticity of 4EC and its potential involvement in the AD process, these cells may also be a target for exercise-induced effects. Virtually, nothing is known about them, however, making this an essentially virgin territory for research (Kuljiš, 2010c).
Virtual Reality Running or Moving and Spatial Cognition Networks
Cognition has multiple scales and dimensions and includes recently formulated notion of the “connectome” which hopes to attain a complete connection mapping of the brain (Connectome Project; Kuljiš, 2010b). Exercise-induced effects may affect brain neural networks. The hippocampus, entorhinal, and transentorhinal cortices are proposed to constitute the starting point of neurodegeneration in AD (Braak and Braak, 1991; Braak et al., 1993). The hippocampus has been implicated in spatial learning (Hafting et al., 2005), episodic, declarative, or autobiographical memory (Doeller et al., 2010). Therefore, the neural representation of this exercise-induced effect resides in a network of regions which supports spatial cognition. Memory loss and disorientation in space are among the first behavioral manifestations of the condition. Exercise-induced (or movement-) hippocampal and entorhinal volume appears to be associated with improved performance in spatial memory (Chaddock et al., 2010; Heo et al., 2010; Erickson et al., 2011). Path-integration maintains permanent tracking of the direction and distance from some reference point (landmark) during navigation in the environment (Hafting et al., 2005) and involves the activation of the hippocampal and entorhinal networks.
The mechanisms underlying spatial memory – which is targeted early in AD – have begun to be elucidated recently. Evidence for certain specific cells supporting spatial cognition in those hippocampal and entorhinal networks has been shown in animals. Hippocampal place cells fire in response to several spatial inputs such as extrinsic landmarks and translational or directional movement inputs (O’Keefe and Burgess, 1996). Place cells fire in response to a unique, specified position in the environment (O’Keefe and Burgess, 1996; Doeller et al., 2010), whereas grid cells, located in the entorhinal cortex, are firing in multiple locations of the environment geometrically defined in a Euclidean plane (spatial autocorrelogram; Hafting et al., 2005). This geometrical superimposition onto a map of the environment is repeating a pattern of equilateral triangles which “tiles” the space (Hafting et al., 2005). Therefore, grid cells may be an element of a global path-integration-based map of the spatial environment. Their response is modulated by direction and speed of motion (Doeller et al., 2010). The basic unit of the map is the grid cell, whose multiple discrete firing fields invariantly form a stable, regularly tessellating structure of equilateral triangles (Hafting et al., 2005).
The recently acquired knowledge may be helpful to elucidate how exercise produces its effects in the brain. Stimulation provided by external landmarks may be a major player. Persistence of firing structure after sensory input suggests that operation of the grids may be based on network mechanisms (Hafting et al., 2005). The maintenance of grid structure during visual deprivation targets path-integration as a probable key to the spatial periodicity (Hafting et al., 2005). In humans, functional MRI suggests that the signal is modulated by direction and running speed (Doeller et al., 2010). The grid orientation appears to vary randomly across individuals, suggesting that the activity of grid cells is independent of landmarks in the surroundings (Doeller et al., 2010). However, path-integration is an independent navigational system which may be functioning without reference to landmarks. Although the map is anchored to external landmarks (i.e., visual), it persists without them once acquired (Doeller et al., 2010). Therefore, patients in whom exercise or sustained physical activity is not possible may benefit of the input/stimulation by the exploration of a virtual reality environment. Depending on the importance of the virtual navigation paradigm in the stimulation process, a virtual training might enhance hippocampal and entorhinal volume and improve spatial cognition performance. These effects would presumably be independent from additional effects brought about by cytokines and other modulators produced also by exercise, as discussed in the preceding sections.
Exercise and Kinetics of Neuronal Degeneration
How does cognitive performance devolve with normal aging? What is the transition state from normal aging to dementia? Is there a neurodegenerative continuum, and, if so, what is its profile? Can exercise intervention stop or reverse this continuum to dementia? It has been thought that extending the lifespan by slowing down aging may also be associated with an increase in the rate or likelihood of neurodegenerative processes, e.g., AD (Kenyon, 2010). However, mutations such as down-regulation of the insulin/IGF-1 signaling pathway, inhibition of the TOR pathway, and dietary restriction that slow aging may also postpone age-related disease (Kenyon, 2010). However, slowing a degenerative process might seem like an overwhelming challenge, as the decline is so pervasive and polyfacetic. Neuron degeneration in patients with Parkinson’s disease, Huntington’s disease and in cultured hippocampal neurons is best described by a constant or exponentially decreasing risk of cell death (Clarke et al., 2000). The time of death (or apoptosis) of any neuron is a random process. The kinetics of cell loss may be described by the differential equation

where X is a measured variable reflecting the cell damage or loss, τ(t) designates the risk of neuron death at time age t, and can be estimated (Clarke et al., 2000), if the risk is exponentially decreasing as

where τ0(t) designates the initial probability of cell death, a is a constant, and D is the delay or time before death begins, or, if the risk is constant, it may be expressed as

An exponentially decreasing risk indicates that the likelihood of cell death decreases in direct proportion to the number of the remaining cells. This type of kinetics may result from an increase in the concentration of neurotrophic factor, such as β-FGF (Gospodarowicz et al., 1985, 1986; Montesano et al., 1986; Morrison et al., 1986) or from a decrease of the amount of a toxic factor as the population of cells declines. Interestingly, on Figure 2, modified from various authors (Fearnley and Lees, 1990, 1991; Clarke et al., 2000), the initial rapid decline of the remaining cells represents the critical portion of the curve. The neurodegenerative process is rapid and the largest amount of neurons is lost during the initial phase. It is a critical phase in which a mitigation process will have more impact in preventing the neurodegeneration and the clinical outcome (Figure 3). Therefore, if the hypothesis is true, and exercise plays a powerful enough mitigating/palliative role, it should be performed very early and aggressively, and ideally prophylactic years before the onset of this process. We can only speculate when associating the initial portion of the curve with MCI. Indeed, MCI may only occur when a significant portion of neurons has already been lost. However, an early mitigation is likely to have greater impact on the neurodegenerative process and the symptoms, a consideration that must be addressed aggressively given the rapidly increasing numbers of patients with MCI and AD (Kuljiš, 2010a, Kuljiš, 2010c).
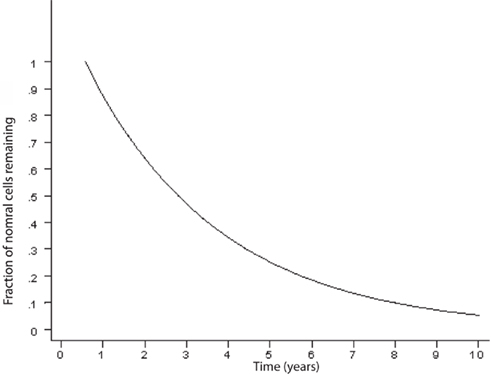
Figure 2. Curve describing the kinetics of neuronal death in neurodegenerative diseases based on an animal model (adapted; Clarke et al., 2000). There is an exponential decline of neuronal number in time.
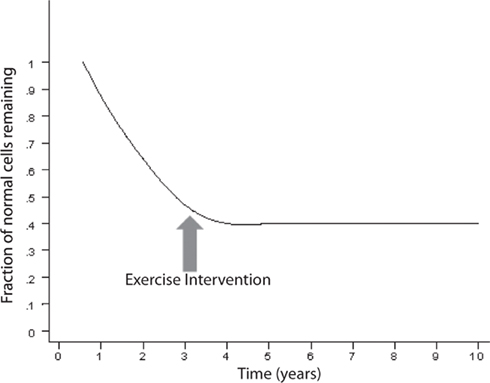
Figure 3. The exercise may halt the neurodegenerative process depending on the time of intervention.
Impairment in cognitive performance as a result of normal aging evolves along a continuum (Small et al., 2008). The objective cognitive deficits are only minimal during normal aging and if the patient enters a state of MCI, the slope becomes more pronounced (Small et al., 2008) a illustrated in Figure 4. MCI often reflects a transitional state between normal aging cognitive decline and dementia (Petersen, 2004; Winblad et al., 2004). Insoluble proteins with amyloid properties start to aggregate in plaques Aβ and neurofibrillary tangles (tau; Glenner, 1980a,b). They have been observed in autopsy studies of young and middle-aged adults and they accumulate in a predictable spatial cerebral pattern in normal aging, MCI and AD albeit rarer in normal aging (Braak and Braak, 1991; Price and Morris, 1999; Petersen et al., 2006). Neurofibrillary tangles may be observed in the hippocampus in MCI, and spread to parietal and frontal neocortical areas as MCI advances to AD. When daily activities of living are altered and the decline includes other cognitive spheres, dementia is usually diagnosed (Small et al., 2008). The evolution to AD, as seen in Figure 4, expresses a progressive continuum. The curve depicting the progressive loss of cognitive performance ψ(t) as a function of time, in Figure 4, may be expressed by the following logarithmic expression

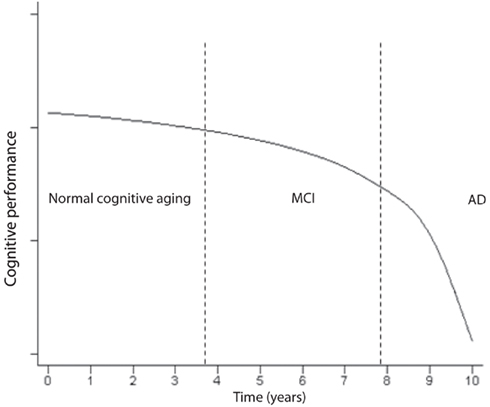
Figure 4. Time course of the impairment continuum of cognition (adapted; Small et al., 2008).
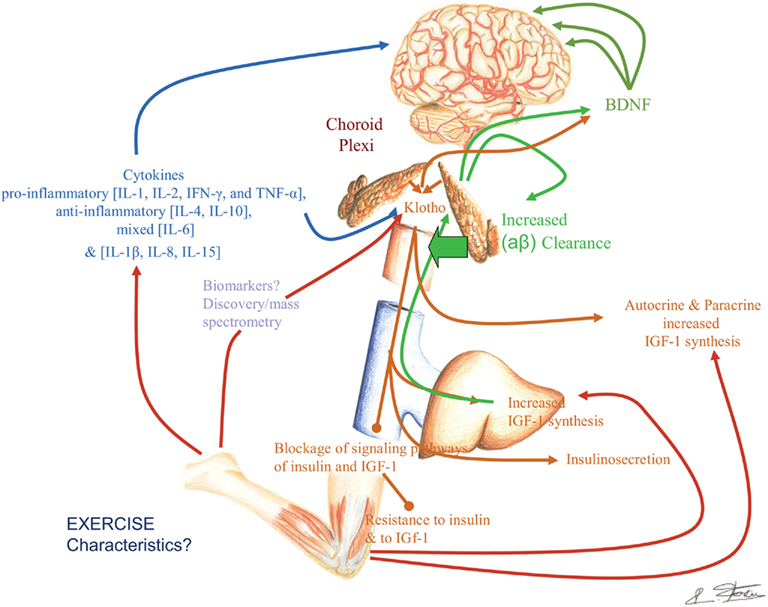
Figure 5. Exercise, growth factors, myokines, Klotho, and potential effects on the brain.
where B and c are constants. The natural logarithm and exponential functions are inverse functions albeit Eqs 2 and 4 are not strictly inverse functions. As logical as it may seem, the decline in cognitive performance may not be proportional to the fraction of cells remaining or to τ(t), e.g., in the hippocampus. Rather, ψ(t) may almost be inversely related to τ(t) which means that the performance may still be acceptable while a significant amount of neuron destruction has already occurred. However, it may not be legitimate or accurate to equate epidemiological studies in humans to animal or cell culture experiments. The kinetics of decline in cognitive performance and neuronal degeneration may only apply to AD, the most common dementia which expresses an insidious onset and a progressive course of decline. In contrast, vascular dementia is often diagnosed by abrupt onset, and superimposed cognitive deficits may progress in a stepwise manner as structural damage due to cerebrovascular disease accumulates (Gascon-Bayarri et al., 2007; Meguro et al., 2007). Because the transition from MCI to AD requires a significant amount of time and patent neuronal degeneration might already be at an advanced stage (Figure 4), the postulate is to start as early as possible the exercise training when there is a suspicion of MCI, or when much hoped for pre-symptomatic markers become positive. This early exercise intervention may prevent further rapid neuronal degeneration at the first portion of the curve in Figure 4, and potentially slow down or, ideally, reverse the process.
The situation is certainly more complex since, apart from mere “cell loss,” the clinical manifestations of neurodegenerative disorders are caused also by less well characterized and even less understood phenomena. These include dysfunction of the cells without known anatomical abnormality (e.g., synaptic dysfunction without synapse loss), synaptic degeneration without neuronal loss, and a likely continuum of neuronal dysfunction that precedes neuronal death.
Conclusion
However, both for simplicity and to illustrate the relatively rigorous predictions that can be made on the basis of cell death alone – for which reasonably well-agreed upon figures exist – we have provided theoretical calculations that can demonstrate potentially fruitful experimental algorithms that can help answer the key questions addressed.
Readers are likely to feel, like us, bewildered by the growing complexity of this challenging topic, and especially by the dilemma of how to factor together and prioritize the information relevant to physical exercise, its role in maintaining and enhancing cognitive and behavioral abilities, and perhaps even more so, in palliating and preventing the devastating effects of aging. The rapidly increasing amount of putative molecular mechanisms (Figure 5) relevant to this problem is perhaps the most challenging aspect of this dilemma, but it is by no means the only essential aspect. The reward for accomplishing an eventual new synthesis will indeed be vast, but necessitate addressing unresolved biotechnology and conceptual challenges posed by the Damage Signals hypothesis of AD pathogenesis (Fernández et al., 2008), the multi-dimensional formulation of both normal and disordered cognition (Kuljiš, 2009) and other rapidly redeveloping reformulations on the pathogenesis of dementia, especially on AD, as decades-old dogmas crumble under the weight of on-going experimental lack of confirmation (Herrup, 2010; Kuljiš, 2010a). In a nutshell, as more putative mechanisms are proposed, there is no agreed upon conceptual framework to assemble them together into a coherent framework that can be implemented into an experimental testing algorithm that is capable of rigorously refuting this apparent network of mechanisms. This is all made even more complex by the fact that such mechanisms span multiple spatio-temporal and conceptual levels, at least from the molecular through the evolutionary, passing through the behavioral and the societal levels (Kuljiš, 2009, 2010a,b). The clearly established but poorly understood interconnections between physical exercise and cognition/behavior are no exception to this dilemma, but their study – like the interrelation between dementia and diabetes (Kuljiš and Šalkovic-Petrišic, 2011) – is quite likely to provide key conceptual and empirical tools to eventually overcome the problem, and thus obtain practical solutions to increasingly common, diseases that compound and mutually worsen each other and afflict us world-wide (Kuljiš, 2010c).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adlard, P. A., Engesser-Cesar, C., and Cotman, C. W. (2011). Mild stress facilitates learning and exercise improves retention in aged mice 1. Exp. Gerontol. 46, 53–59.
Adlard, P. A., Perreau, V. M., Pop, V., and Cotman, C. W. (2005). Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer’s disease 1. J. Neurosci. 25, 4217–4221.
Alzheimer, A., Schnitzlein, H. N., and Murtagh, F. R. (1995). An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde” 1. Clin. Anat. 8, 429–431.
Anderson, B. J., Greenwood, S. J., and McCloskey, D. (2010a). Exercise as an intervention for the age-related decline in neural metabolic support. Front. Aging Neurosci. 4:30. doi: 10.3389/fnagi.2010.00030
Anderson, E. S., Winett, R. A., Wojcik, J. R., and Williams, D. M. (2010b). Social cognitive mediators of change in a group randomized nutrition and physical activity intervention: social support, self-efficacy, outcome expectations and self-regulation in the guide-to-health trial. J. Health Psychol. 15, 21–32.
Anderson, B. J., Li, X., Alcantara, A. A., Isaacs, K. R., Black, J. E., and Greenough, W. T. (1994). Glial hypertrophy is associated with synaptogenesis following motor-skill learning, but not with angiogenesis following exercise. Glia 11, 73–80.
Anderson, K. J., Dam, D., Lee, S., and Cotman, C. W. (1988). Basic fibroblast growth factor prevents death of lesioned cholinergic neurons in vivo. Nature 332, 360–361.
Ang, E. T., Tai, Y. K., Lo, S. Q., Seet, R., and Soong, T. W. (2010). Neurodegenerative diseases: exercising toward neurogenesis and neuroregeneration. Front. Aging Neurosci. 2:25. doi: 10.3389/fnagi.2010.00025
Ashford, J. W., and Jarvik, L. (1985). Alzheimer’s disease: does neuron plasticity predispose to axonal neurofibrillary degeneration? N. Engl. J. Med. 313, 388–389.
Bak, T. H. (2011). Movement disorders: why movement and cognition belong together 5. Nat. Rev. Neurol. 7, 10–12.
Ben, A. S., and Pascual, O. (2010). Glia: the many ways to modulate synaptic plasticity. Neurochem. Int. 57, 440–445.
Berchtold, N. C., Castello, N., and Cotman, C. W. (2010). Exercise and time-dependent benefits to learning and memory 8. Neuroscience 167, 588–597.
Berg, A., Halle, M., Baumstark, M., Keul, J., and Northoff, H. (1996). Spontaneously low LDL cholesterol and reaction to exercise-induced stress. Lancet 347, 405.
Bergles, D. E., Roberts, J. D., Somogyi, P., and Jahr, C. E. (2000). Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus 33. Nature 405, 187–191.
Black, J. E., Isaacs, K. R., Anderson, B. J., Alcantara, A. A., and Greenough, W. T. (1990). Learning causes synaptogenesis, whereas motor activity causes angiogenesis, in cerebellar cortex of adult rats. Proc. Natl. Acad. Sci. U.S.A. 87, 5568–5572.
Booth, F. W., Chakravarthy, M. V., Gordon, S. E., and Spangenburg, E. E. (2002). Waging war on physical inactivity: using modern molecular ammunition against an ancient enemy. J. Appl. Physiol. 93, 3–30.
Braak, E., Braak, H., and Mandelkow, E. M. (1994). A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 87, 554–567.
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259.
Braak, H., Braak, E., and Bohl, J. (1993). Staging of Alzheimer-related cortical destruction. Eur. Neurol. 33, 403–408.
Braak, H., Del, T. K., Schultz, C., and Braak, E. (2000). Vulnerability of select neuronal types to Alzheimer’s disease. Ann. N. Y. Acad. Sci. 924, 53–61.
Brobey, R., German, D., Gurnani, P., Pastor, J., Hsieh, C.-C., Papaconstantinou, J., Kuro-o, M., and Rosenblatt, K. P. (2011). “Klotho activation is neuroprotective against oxidative stress through antioxidant and apoptotic signaling pathways,” in Conference Proceeding, Houston, TX.
Buerger, K., Frisoni, G., Uspenskaya, O., Ewers, M., Zetterberg, H., Geroldi, C., Binetti, G., Johannsen, P., Rossini, P. M., Wahlund, L. O., Vellas, B., Blennow, K., and Hampel, H. (2009). Validation of Alzheimer’s disease CSF and plasma biological markers: the multicentre reliability study of the pilot European Alzheimer’s Disease Neuroimaging Initiative (E-ADNI) 2. Exp. Gerontol. 44, 579–585.
Carro, E., Trejo, J. L., Nunez, A., and Torres-Aleman, I. (2003). Brain repair and neuroprotection by serum insulin-like growth factor I. Mol. Neurobiol. 27, 153–162.
Carro, E., Trejo, J. L., Busiguina, S., and Torres-Aleman, I. (2001). Circulating insulin-like growth factor I mediates the protective effects of physical exercise against brain insults of different etiology and anatomy. J. Neurosci. 21, 5678–5684.
Carro, E., Trejo, J. L., Gerber, A., Loetscher, H., Torrado, J., Metzger, F., and Torres-Aleman, I. (2006a). Therapeutic actions of insulin-like growth factor I on APP/PS2 mice with severe brain amyloidosis. Neurobiol. Aging 27, 1250–1257.
Carro, E., Trejo, J. L., Spuch, C., Bohl, D., Heard, J. M., and Torres-Aleman, I. (2006b). Blockade of the insulin-like growth factor I receptor in the choroid plexus originates Alzheimer’s-like neuropathology in rodents: new cues into the human disease? Neurobiol. Aging 27, 1618–1631.
Carro, E., Trejo, J. L., Gomez-Isla, T., LeRoith, D., and Torres-Aleman, I. (2002). Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat. Med. 8, 1390–1397.
Carro, E., Trejo, J. L., Nunez, A., and Torres-Aleman, I. (2003). Brain repair and neuroprotection by serum insulin-like growth factor I. Mol. Neurobiol. 27, 153–162.
Casserly, I., and Topol, E. (2004). Convergence of atherosclerosis and Alzheimer’s disease: inflammation, cholesterol, and misfolded proteins 3. Lancet 363, 1139–1146.
Castren, E., Zafra, F., Thoenen, H., and Lindholm, D. (1992). Light regulates expression of brain-derived neurotrophic factor mRNA in rat visual cortex. Proc. Natl. Acad. Sci. U.S.A. 89, 9444–9448.
Chaddock, L., Erickson, K. I., Prakash, R. S., Kim, J. S., Voss, M. W., Vanpatter, M., Pontifex, M. B., Raine, L. B., Konkel, A., Hillman, C. H., Cohen, N. J., and Kramer, A. F. (2010). A neuroimaging investigation of the association between aerobic fitness, hippocampal volume, and memory performance in preadolescent children 1. Brain Res. 1358, 172–183.
Chang, K. H., de, P. Y., Lee, H. P., Lee, H. G., Smith, M. A., and Shah, K. (2010). Cdk5 is a major regulator of p38 cascade: relevance to neurotoxicity in Alzheimer’s disease. J. Neurochem. 113, 1221–1229.
Chen, M. F., Chen, H. I., and Jen, C. J. (2010). Exercise training upregulates macrophage MKP-1 and affects immune responses in mice 5. Med. Sci. Sports Exerc. 42, 2173–2179.
Cheng, B., and Mattson, M. P. (1991). NGF and bFGF protect rat hippocampal and human cortical neurons against hypoglycemic damage by stabilizing calcium homeostasis. Neuron 7, 1031–1041.
Clarke, G., Collins, R. A., Leavitt, B. R., Andrews, D. F., Hayden, M. R., Lumsden, C. J., and McInnes, R. R. (2000). A one-hit model of cell death in inherited neuronal degenerations. Nature 406, 195–199.
Colcombe, S., and Kramer, A. F. (2003). Fitness effects on the cognitive function of older adults: a meta-analytic study. Psychol. Sci. 14, 125–130.
Colcombe, S. J., Erickson, K. I., Raz, N., Webb, A. G., Cohen, N. J., McAuley, E., and Kramer, A. F. (2003). Aerobic fitness reduces brain tissue loss in aging humans. J. Gerontol. A Biol. Sci. Med. Sci. 58, 176–180.
Colcombe, S. J., Erickson, K. I., Scalf, P. E., Kim, J. S., Prakash, R., McAuley, E., Elavsky, S., Marquez, D. X., Hu, L., and Kramer, A. F. (2006). Aerobic exercise training increases brain volume in aging humans. J. Gerontol. A Biol. Sci. Med. Sci. 61, 1166–1170.
Colcombe, S. J., Kramer, A. F., Erickson, K. I., Scalf, P., McAuley, E., Cohen, N. J., Webb, A., Jerome, G. J., Marquez, D. X., and Elavsky, S. (2004). Cardiovascular fitness, cortical plasticity, and aging. Proc. Natl. Acad. Sci. U.S.A. 101, 3316–3321.
Cotman, C. W., and Berchtold, N. C. (2002). Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 25, 295–301.
de la Torre, J. C. (2004). Is Alzheimer’s disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics 11. Lancet Neurol. 3, 184–190.
De Meyer, G., Shapiro, F., Vanderstichele, H., Vanmechelen, E., Engelborghs, S., De Deyn, P. P., Coart, E., Hansson, O., Minthon, L., Zetterberg, H., Blennow, K., Shaw, L., and Trojanowski, J. Q. (2010). Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people 3. Arch. Neurol. 67, 949–956.
De Meyer, G. R., De Cleen, D. M., Cooper, S., Knaapen, M. W., Jans, D. M., Martinet, W., Herman, A. G., Bult, H., and Kockx, M. M. (2002). Platelet phagocytosis and processing of beta-amyloid precursor protein as a mechanism of macrophage activation in atherosclerosis. Circ. Res. 90, 1197–1204.
del Rio Hortega, P. (1919). El “tercer elemento” de los centros nerviosos. Poder fagocitario y movilidad de la microglía. Bol. R. Soc. Esp. Hist. Nat. Secc. Biol. 8, 62–82.
Doeller, C. F., Barry, C., and Burgess, N. (2010). Evidence for grid cells in a human memory network 1. Nature 463, 657–661.
Dubois, B., Feldman, H. H., Jacova, C., Cummings, J. L., Dekosky, S. T., Barberger-Gateau, P., Delacourte, A., Frisoni, G., Fox, N. C., Galasko, D., Gauthier, S., Hampel, H., Jicha, G. A., Meguro, K., O’Brien, J., Pasquier, F., Robert, P., Rossor, M., Salloway, S., Sarazin, M., de Souza, L. C., Stern, Y., Visser, P. J., and Scheltens, P. (2010). Revising the definition of Alzheimer’s disease: a new lexicon 1. Lancet Neurol. 9, 1118–1127.
Dubois, B., Feldman, H. H., Jacova, C., Dekosky, S. T., Barberger-Gateau, P., Cummings, J., Delacourte, A., Galasko, D., Gauthier, S., Jicha, G., Meguro, K., O’brien, J., Pasquier, F., Robert, P., Rossor, M., Salloway, S., Stern, Y., Visser, P. J., and Scheltens, P. (2007). Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria 2. Lancet Neurol. 6, 734–746.
Erickson, K. I., Prakash, R. S., Voss, M. W., Chaddock, L., Heo, S., McLaren, M., Pence, B. D., Martin, S. A., Vieira, V. J., Woods, J. A., McAuley, E., and Kramer, A. F. (2010). Brain-derived neurotrophic factor is associated with age-related decline in hippocampal volume 10. J. Neurosci. 30, 5368–5375.
Erickson, K. I., Voss, M. W., Prakash, R. S., Basak, C., Szabo, A., Chaddock, L., Kim, J. S., Heo, S., Alves, H., White, S. M., Wojcicki, T. R., Mailey, E., Vieira, V. J., Martin, S. A., Pence, B. D., Woods, J. A., McAuley, E., and Kramer, A. F. (2011). Exercise training increases size of hippocampus and improves memory 1. Proc. Natl. Acad. Sci. U.S.A. 108, 3017–3022.
Erikssen, G., Liestol, K., Bjornholt, J., Thaulow, E., Sandvik, L., and Erikssen, J. (1998). Changes in physical fitness and changes in mortality. Lancet 352, 759–762.
Esch, F., Baird, A., Ling, N., Ueno, N., Hill, F., Denoroy, L., Klepper, R., Gospodarowicz, D., Bohlen, P., and Guillemin, R. (1985). Primary structure of bovine pituitary basic fibroblast growth factor (FGF) and comparison with the amino-terminal sequence of bovine brain acidic FGF. Proc. Natl. Acad. Sci. U.S.A. 82, 6507–6511.
Ewers, M., Cheng, X., Nural, H. F., Walsh, C., Meindl, T., Teipel, S. J., Buerger, K., Shen, Y., and Hampel, H. (2011). Increased CSF-BACE1 activity associated with decreased hippocampus volume in Alzheimer’s disease 1. J. Alzheimers Dis. PMID: 21460439. [Epub ahead of print]
Farmer, J., Zhao, X., Van, P. H., Wodtke, K., Gage, F. H., and Christie, B. R. (2004). Effects of voluntary exercise on synaptic plasticity and gene expression in the dentate gyrus of adult male Sprague-Dawley rats in vivo. Neuroscience 124, 71–79.
Fearnley, J. M., and Lees, A. J. (1990). Striatonigral degeneration. A clinicopathological study. Brain 113(Pt 6), 1823–1842.
Fearnley, J. M., and Lees, A. J. (1991). Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114(Pt 5), 2283–2301.
Fernández, Rojo, L., Kuljiš, R. O., and Maccioni, R. B. (2008). The damage signals hypothesis of Alzheimer’s disease pathogenesis 6. J. Alzheimers Dis. 14, 329–333.
Gamba, P., Leonarduzzi, G., Tamagno, E., Guglielmotto, M., Testa, G., Sottero, B., Gargiulo, S., Biasi, F., Mauro, A., Viña, J., and Poli, G. (2011). Interaction between 24-hydroxycholesterol, oxidative stress and amyloid-beta in amplifying neuronal damage in Alzheimer’s disease: three partners in crime. Aging Cell. doi: 10.1111/j.1474-9726.2011.00681.x. [Epub ahead of print]
Gascon-Bayarri, J., Reñé, R., Del Barrio, J. L., De Pedro-Cuesta, J., Ramón, J. M., Manubens, J. M., Sánchez, C., Hernández, M., Estela, J., Juncadella, M., and Rubio, F. R. (2007). Prevalence of dementia subtypes in El Prat de Llobregat, Catalonia, Spain: the PRATICON study. Neuroepidemiology 28, 224–234.
Geiger, P. C., Han, D. H., Wright, D. C., and Holloszy, J. O. (2006). How muscle insulin sensitivity is regulated: testing of a hypothesis. Am. J. Physiol. Endocrinol. Metab. 291, E1258–E1263.
Geiger, P. C., Wright, D. C., Han, D. H., and Holloszy, J. O. (2005). Activation of p38 MAP kinase enhances sensitivity of muscle glucose transport to insulin 76. Am. J. Physiol. Endocrinol. Metab. 288, E782–E788.
Giannakopoulos, P., Gold, G., Kovari, E., von, G. A., Imhof, A., Bouras, C., and Hof, P. R. (2007). Assessing the cognitive impact of Alzheimer disease pathology and vascular burden in the aging brain: the Geneva experience 29. Acta Neuropathol. 113, 1–12.
Giannakopoulos, P., Kovari, E., Herrmann, F. R., Hof, P. R., and Bouras, C. (2009). Interhemispheric distribution of Alzheimer disease and vascular pathology in brain aging 18. Stroke 40, 983–986.
Glasper, E. R., Llorens-Martin, M. V., Leuner, B., Gould, E., and Trejo, J. L. (2010). Blockade of insulin-like growth factor-I has complex effects on structural plasticity in the hippocampus. Hippocampus 20, 706–712.
Glenner, G. G. (1980a). Amyloid deposits and amyloidosis. The beta-fibrilloses (first of two parts). N. Engl. J. Med. 302, 1283–1292.
Glenner, G. G. (1980b). Amyloid deposits and amyloidosis: the beta-fibrilloses (second of two parts). N. Engl. J. Med. 302, 1333–1343.
Gorelick, P. B. (2004). Risk factors for vascular dementia and Alzheimer disease 4. Stroke 35, 2620–2622.
Gospodarowicz, D., Baird, A., Cheng, J., Lui, G. M., Esch, F., and Bohlen, P. (1986). Isolation of fibroblast growth factor from bovine adrenal gland: physicochemical and biological characterization. Endocrinology 118, 82–90.
Gospodarowicz, D., Cheng, J., Lui, G. M., Baird, A., Esch, F., and Bohlen, P. (1985). Corpus luteum angiogenic factor is related to fibroblast growth factor. Endocrinology 117, 2383–2391.
Greenberg, S. M., Gurol, M. E., Rosand, J., and Smith, E. E. (2004). Amyloid angiopathy-related vascular cognitive impairment 3. Stroke 35, 2616–2619.
Griffin, W. S., Liu, L., Li, Y., Mrak, R. E., and Barger, S. W. (2006). Interleukin-1 mediates Alzheimer and Lewy body pathologies. J. Neuroinflammation 3, 5.
Hafting, T., Fyhn, M., Molden, S., Moser, M. B., and Moser, E. I. (2005). Microstructure of a spatial map in the entorhinal cortex 1. Nature 436, 801–806.
Handschin, C., and Spiegelman, B. M. (2008). The role of exercise and PGC1alpha in inflammation and chronic disease. Nature 454, 463–469.
Heo, S., Prakash, R. S., Voss, M. W., Erickson, K. I., Ouyang, C., Sutton, B. P., and Kramer, A. F. (2010). Resting hippocampal blood flow, spatial memory and aging 18. Brain Res. 1315, 119–127.
Herrup, K. (2010). Reimagining Alzheimer’s disease–an age-based hypothesis 3. J. Neurosci. 30, 16755–16762.
Hollmann, W., Struder, H. K., Tagarakis, C. V., and King, G. (2007). Physical activity and the elderly. Eur. J. Cardiovasc. Prev. Rehabil. 14, 730–739.
Honig, L. S., Kukull, W., and Mayeux, R. (2005). Atherosclerosis and AD: analysis of data from the US National Alzheimer’s Coordinating Center 62. Neurology 64, 494–500.
Hoveida, R., Alaei, H., Oryan, S., Parivar, K., and Reisi, P. (2011). Treadmill running improves spatial memory in an animal model of Alzheimer’s disease 1. Behav. Brain Res. 216, 270–274.
Hsieh, C. C., Kuro-o, M., Rosenblatt, K. P., Brobey, R., and Papaconstantinou, J. (2010). The ASK1-Signalosome regulates p38 MAPK activity in response to levels of endogenous oxidative stress in the Klotho mouse models of aging. Aging (Albany NY) 2, 597–611.
Hu, F. B., Willett, W. C., Li, T., Stampfer, M. J., Colditz, G. A., and Manson, J. E. (2004). Adiposity as compared with physical activity in predicting mortality among women. N. Engl. J. Med. 351, 2694–2703.
Hulmi, J. J., Walker, S., Ahtiainen, J. P., Nyman, K., Kraemer, W. J., and Hakkinen, K. (2010). Molecular signaling in muscle is affected by the specificity of resistance exercise protocol 1. Scand. J. Med. Sci. Sports. doi: 10.1111/j.1600-0838.2010.01198.x. [Epub ahead of print]
Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 5, 347–360.
Imura, A., Tsuji, Y., Murata, M., Maeda, R., Kubota, K., Iwano, A., Obuse, C., Togashi, K., Tominaga, M., Kita, N., Tomiyama, K., Iijima, J., Nabeshima, Y., Fujioka, M., Asato, R., Tanaka, S., Kojima, K., Ito, J., Nozaki, K., Hashimoto, N., Ito, T., Nishio, T., Uchiyama, T., Fujimori, T., and Nabeshima, Y. (2007). Alpha-Klotho as a regulator of calcium homeostasis. Science 316, 1615–1618.
Jack, C. R. Jr., Knopman, D. S., Jagust, W. J., Shaw, L. M., Aisen, P. S., Weiner, M. W., Petersen, R. C., and Trojanowski, J. Q. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128.
Kalaria, R. N. (2009). Neurodegenerative disease: diabetes, microvascular pathology and Alzheimer disease 15. Nat. Rev. Neurol. 5, 305–306.
Kattenstroth, J. C., Kolankowska, I., Kalisch, T., and Dinse, H. R. (2010). Superior sensory, motor, and cognitive performance in elderly individuals with multi-year dancing activities. Front. Aging Neurosci. 2:31. doi: 10.3389/fnagi.2010.00031
Kayed, R., Head, E., Thompson, J. L., McIntire, T. M., Milton, S. C., Cotman, C. W., and Glabe, C. G. (2003). Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489.
Kerchner, G. A., Hess, C. P., Hammond-Rosenbluth, K. E., Xu, D., Rabinovici, G. D., Kelley, D. A., Vigneron, D. B., Nelson, S. J., and Miller, B. L. (2010). Hippocampal CA1 apical neuropil atrophy in mild Alzheimer disease visualized with 7-T MRI 1. Neurology 75, 1381–1387.
Kim, K. W., Park, J. H., Kim, M. H., Kim, M. D., Kim, B. J., Kim, S. K., Kim, J. L., Moon, S. W., Bae, J. N., Woo, J. I., Ryu, S. H., Yoon, J. C., Lee, N. J., Lee, D. Y., Lee, D. W., Lee, S. B., Lee, J. J., Lee, J. Y., Lee, C. U., Chang, S. M., Jhoo, J. H., and Cho, M. J. (2011). A nationwide survey on the prevalence of dementia and mild cognitive impairment in South Korea. J. Alzheimers Dis. 23, 281–291.
Kim, Y. P., Kim, H., Shin, M. S., Chang, H. K., Jang, M. H., Shin, M. C., Lee, S. J., Lee, H. H., Yoon, J. H., Jeong, I. G., and Kim, C. J. (2004). Age-dependence of the effect of treadmill exercise on cell proliferation in the dentate gyrus of rats. Neurosci. Lett. 355, 152–154.
Knopman, D. S., and Roberts, R. (2010). Vascular risk factors: imaging and neuropathologic correlates 12. J. Alzheimers Dis. 20, 699–709.
Knusel, B., Winslow, J. W., Rosenthal, A., Burton, L. E., Seid, D. P., Nikolics, K., and Hefti, F. (1991). Promotion of central cholinergic and dopaminergic neuron differentiation by brain-derived neurotrophic factor but not neurotrophin 3. Proc. Natl. Acad. Sci. U.S.A. 88, 961–965.
Koehl, M., Meerlo, P., Gonzales, D., Rontal, A., Turek, F. W., and Abrous, D. N. (2008). Exercise-induced promotion of hippocampal cell proliferation requires beta-endorphin. FASEB J. 22, 2253–2262.
Kramer, A. F., Erickson, K. I., and Colcombe, S. J. (2006). Exercise, cognition, and the aging brain. J. Appl. Physiol. 101, 1237–1242.
Kuljiš, R. O. (2009). Toward a multi-dimensional formulation of the pathogenesis and pathophysiology of the Alzheimer dementia-like syndrome applicable to a variety of degenerative disorders and normal cognition 4. Med. Hypotheses 73, 315–318.
Kuljiš, R. O. (2010a). Grand challenges in dementia 2010. Front. Neurol. 1:4. doi: 10.3389/fneur.2010.00004
Kuljiš, R. O. (2010b). Integrative understanding of emergent brain properties, quantum brain hypotheses, and connectome alterations in dementia are key challenges to conquer Alzheimer’s disease 2. Front. Neurol. 1:15. doi: 10.3389/fneur.2010.00015
Kuljiš, R. O. (2010c). The fourth element targeting hypothesis of Alzheimer’s disease pathogenesis and pathophysiology 1. Front. Neurol. 1:144. doi: 10.3389/fneur.2010.00144
Kuljiš, R. O., and Šalkovic-Petrišic, M. (2011). Dementia, diabetes, Alzheimer’s disease, and insulin resistance in the brain: progress, dilemmas, new opportunities, and a hypothesis to tackle intersecting epidemics. J. Alzheimers Dis. PMID: 21335659 [Epub ahead of print]
Kurata, T., Miyazaki, K., Kozuki, M., Panin, V. L., Morimoto, N., Ohta, Y., Nagai, M., Ikeda, Y., Matsuura, T., and Abe, K. (2011). Atorvastatin and pitavastatin improve cognitive function and reduce senile plaque and phosphorylated tau in aged APP mice. Brain Res. 1371, 161–170.
Kuro-o, M. (2008). Klotho as a regulator of oxidative stress and senescence. Biol. Chem. 389, 233–241.
Kuro-o, M., Matsumura, Y., Aizawa, H., Kawaguchi, H., Suga, T., Utsugi, T., Ohyama, Y., Kurabayashi, M., Kaname, T., Kume, E., Iwasaki, H., Iida, A., Shiraki-Iida, T., Nishikawa, S., Nagai, R., and Nabeshima, Y. I. (1997). Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390, 45–51.
Kurosu, H., Ogawa, Y., Miyoshi, M., Yamamoto, M., Nandi, A., Rosenblatt, K. P., Baum, M. G., Schiavi, S., Hu, M. C., Moe, O. W., and Kuro-o, M. (2006). Regulation of fibroblast growth factor-23 signaling by klotho. J. Biol. Chem. 281, 6120–6123.
Kurosu, H., Yamamoto, M., Clark, J. D., Pastor, J. V., Nandi, A., Gurnani, P., McGuinness, O. P., Chikuda, H., Yamaguchi, M., Kawaguchi, H., Shimomura, I., Takayama, Y., Herz, J., Kahn, C. R., Rosenblatt, K. P., and Kuro-o, M. (2005). Suppression of aging in mice by the hormone Klotho. Science 309, 1829–1833.
Lambert, J. C., Schraen-Maschke, S., Richard, F., Fievet, N., Rouaud, O., Berr, C., Dartigues, J. F., Tzourio, C., Alpérovitch, A., Buée, L., and Amouyel, P. (2009). Association of plasma amyloid beta with risk of dementia: the prospective three-city study. Neurology 73, 847–853.
Lamberts, S. W., van den Beld, A. W., and van der Lely, A. J. (1997). The endocrinology of aging. Science 278, 419–424.
Lansbury, P. T., and Lashuel, H. A. (2006). A century-old debate on protein aggregation and neurodegeneration enters the clinic 4. Nature 443, 774–779.
Larson, E. B., Wang, L., Bowen, J. D., McCormick, W. C., Teri, L., Crane, P., and Kukull, W. (2006). Exercise is associated with reduced risk for incident dementia among persons 65 years of age and older. Ann. Intern. Med. 144, 73–81.
Lee, M., Cho, T., Jantaratnotai, N., Wang, Y. T., McGeer, E., and McGeer, P. L. (2010). Depletion of GSH in glial cells induces neurotoxicity: relevance to aging and degenerative neurological diseases. FASEB J. 24, 2533–2545.
Lee, M., Jantaratnotai, N., McGeer, E., McLarnon, J. G., and McGeer, P. L. (2011). Mg2+ ions reduce microglial and THP-1 cell neurotoxicity by inhibiting Ca2+ entry through purinergic channels. Brain Res. 1369, 21–35.
Liang, K. Y., Mintun, M. A., Fagan, A. M., Goate, A. M., Bugg, J. M., Holtzman, D. M., Morris, J. C., and Head, D. (2010). Exercise and Alzheimer’s disease biomarkers in cognitively normal older adults. Ann. Neurol. 68, 311–318.
Lira, V. A., Benton, C. R., Yan, Z., and Bonen, A. (2010). PGC-1alpha regulation by exercise training and its influences on muscle function and insulin sensitivity 7. Am. J. Physiol. Endocrinol. Metab. 299, E145–E161.
Little, J. P., Safdar, A., Cermak, N., Tarnopolsky, M. A., and Gibala, M. J. (2010). Acute endurance exercise increases the nuclear abundance of PGC-1alpha in trained human skeletal muscle 10. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R912–R917.
Llorens-Martin, M., Tejeda, G. S., and Trejo, J. L. (2010a). Differential regulation of the variations induced by environmental richness in adult neurogenesis as a function of time: a dual birthdating analysis. PLoS ONE 5, e12188. doi: 10.1371/journal.pone.0012188
Llorens-Martin, M., Torres-Aleman, I., and Trejo, J. L. (2010b). Exercise modulates insulin-like growth factor 1-dependent and -independent effects on adult hippocampal neurogenesis and behaviour. Mol. Cell. Neurosci. 44, 109–117.
Llorens-Martin, M. V., Rueda, N., Tejeda, G. S., Florez, J., Trejo, J. L., and Martinez-Cue, C. (2010c). Effects of voluntary physical exercise on adult hippocampal neurogenesis and behavior of Ts65Dn mice, a model of Down syndrome. Neuroscience, 171, 1228–1240.
Maccioni, R. B., Farías, G. A., Rojo, L. E., Sekler, M. A., and Kuljiš, R. O. (2009a). “What have we learned from the tau hypothesis?” in: Current Hypotheses and Research Milestones in Alzheimer’s Disease, ed. R. B. P. G. Maccioni (New York: Springer Science & Business Media), 49–62.
Maccioni, R. B., Rojo, L. E., Fernandez, J. A., and Kuljiš, R. O. (2009b). The role of neuroimmunomodulation in Alzheimer’s disease 5. Ann. N. Y. Acad. Sci. 1153, 240–246.
Mark, R. J., Keller, J. N., Kruman, I., and Mattson, M. P. (1997). Basic FGF attenuates amyloid beta-peptide-induced oxidative stress, mitochondrial dysfunction, and impairment of Na + /K + -ATPase activity in hippocampal neurons. Brain Res. 756, 205–214.
Mattson, M. P., Lovell, M. A., Furukawa, K., and Markesbery, W. R. (1995). Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of intracellular Ca2+ concentration, and neurotoxicity and increase antioxidant enzyme activities in hippocampal neurons. J. Neurochem. 65, 1740–1751.
Mattson, M. P., Murrain, M., Guthrie, P. B., and Kater, S. B. (1989). Fibroblast growth factor and glutamate: opposing roles in the generation and degeneration of hippocampal neuroarchitecture. J. Neurosci. 9, 3728–3740.
Mawuenyega, K. G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., Yarasheski, K. E., and Bateman, R. J. (2010). Decreased clearance of CNS beta-amyloid in Alzheimer’s disease 1. Science 330, 1774.
Mayeux, R., Honig, L. S., Tang, M. X., Manly, J., Stern, Y., Schupf, N., and Mehta, P. D. (2003). Plasma A[beta]40 and A[beta]42 and Alzheimer’s disease: relation to age, mortality, and risk. Neurology 61, 1185–1190.
Meguro, K., Ishii, H., Kasuya, M., Akanuma, K., Meguro, M., Kasai, M., Lee, E., Hashimoto, R., Yamaguchi, S., and Asada, T. (2007). Incidence of dementia and associated risk factors in Japan: The Osaki-Tajiri Project. J. Neurol. Sci. 260, 175–182.
Montesano, R., Vassalli, J. D., Baird, A., Guillemin, R., and Orci, L. (1986). Basic fibroblast growth factor induces angiogenesis in vitro. Proc. Natl. Acad. Sci. U.S.A. 83, 7297–7301.
Morrison, R. S., and de Vellis, J. (1981). Growth of purified astrocytes in a chemically defined medium. Proc. Natl. Acad. Sci. U.S.A. 78, 7205–7209.
Morrison, R. S., Sharma, A., de, V. J., and Bradshaw, R. A. (1986). Basic fibroblast growth factor supports the survival of cerebral cortical neurons in primary culture. Proc. Natl. Acad. Sci. U.S.A. 83, 7537–7541.
Muñoz, L., and Ammit, A. J. (2010). Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 58, 561–568.
Muñoz, L., Ralay, R. H., Roy, S. M., Hu, W., Craft, J. M., McNamara, L. K., Chico, L. W., Van Eldik, L. J., and Watterson, D. M. (2007). A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer’s disease mouse model. J. Neuroinflammation 4, 21.
Neeper, S. A., Gomez-Pinilla, F., Choi, J., and Cotman, C. (1995). Exercise and brain neurotrophins. Nature 373, 109.
Neeper, S. A., Gomez-Pinilla, F., Choi, J., and Cotman, C. W. (1996). Physical activity increases mRNA for brain-derived neurotrophic factor and nerve growth factor in rat brain. Brain Res. 726, 49–56.
Nichol, K., Deeny, S. P., Seif, J., Camaclang, K., and Cotman, C. W. (2009). Exercise improves cognition and hippocampal plasticity in APOE epsilon4 mice. Alzheimers Dement. 5, 287–294.
Nieman, D. C. (2003). Current perspective on exercise immunology. Curr. Sports Med. Rep. 2, 239–242.
Nishiyama, A., Komitova, M., Suzuki, R., and Zhu, X. (2009). Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity 3. Nat. Rev. Neurosci. 10, 9–22.
Niwa, K., Kazama, K., Younkin, L., Younkin, S. G., Carlson, G. A., and Iadecola, C. (2002). Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am. J. Physiol. Heart Circ. Physiol. 283, H315–H323.
O’Keefe, J., and Burgess, N. (1996). Geometric determinants of the place fields of hippocampal neurons 35. Nature 381, 425–428.
Palop, J. J., Chin, J., and Mucke, L. (2006). A network dysfunction perspective on neurodegenerative diseases 5. Nature 443, 768–773.
Paris, D., Patel, N., DelleDonne, A., Quadros, A., Smeed, R., and Mullan, M. (2004). Impaired angiogenesis in a transgenic mouse model of cerebral amyloidosis 5. Neurosci. Lett. 366, 80–85.
Pedersen, B. K., Akerstrom, T. C., Nielsen, A. R., and Fischer, C. P. (2007). Role of myokines in exercise and metabolism. J. Appl. Physiol. 103, 1093–1098.
Pedersen, B. K., and Hoffman-Goetz, L. (2000). Exercise and the immune system: regulation, integration, and adaptation. Physiol. Rev. 80, 1055–1081.
Petersen, R. C. (2004). Mild cognitive impairment as a diagnostic entity. J. Intern. Med. 256, 183–194.
Petersen, R. C., Parisi, J. E., Dickson, D. W., Johnson, K. A., Knopman, D. S., Boeve, B. F., Jicha, G. A., Ivnik, R. J., Smith, G. E., Tangalos, E. G., Braak, H., and Kokmen, E. (2006). Neuropathologic features of amnestic mild cognitive impairment. Arch. Neurol. 63, 665–672.
Podewils, L. J., Guallar, E., Beauchamp, N., Lyketsos, C. G., Kuller, L. H., and Scheltens, P. (2007). Physical activity and white matter lesion progression: assessment using MRI 1. Neurology 68, 1223–1226.
Petersen, R. C. (2004). Mild cognitive impairment as a diagnostic entity. J. Intern. Med. 256, 183–194.
Petersen, R. C., Parisi, J. E., Dickson, D. W., Johnson, K. A., Knopman, D. S., Boeve, B. F., Jicha, G. A., Ivnik, R. J., Smith, G. E., Tangalos, E. G., Braak, H., and Kokmen, E. (2006). Neuropathologic features of amnestic mild cognitive impairment. Arch. Neurol. 63, 665–672.
Podewils, L. J., Guallar, E., Beauchamp, N., Lyketsos, C. G., Kuller, L. H., and Scheltens, P. (2007). Physical activity and white matter lesion progression: assessment using MRI 1. Neurology 68, 1223–1226.
Podewils, L. J., Guallar, E., Kuller, L. H., Fried, L. P., Lopez, O. L., Carlson, M., and Lyketsos, C. G. (2005). Physical activity, APOE genotype, and dementia risk: findings from the cardiovascular health cognition study. Am. J. Epidemiol. 161, 639–651.
Pogozelski, A. R., Geng, T., Li, P., Yin, X., Lira, V. A., Zhang, M., Chi, J. T., and Yan, Z. (2009). p38gamma mitogen-activated protein kinase is a key regulator in skeletal muscle metabolic adaptation in mice. PLoS ONE 4, e7934. doi: 10.1371/journal.pone.0007934
Prakash, R. S., Snook, E. M., Erickson, K. I., Colcombe, S. J., Voss, M. W., Motl, R. W., and Kramer, A. F. (2007). Cardiorespiratory fitness: a predictor of cortical plasticity in multiple sclerosis 472. Neuroimage 34, 1238–1244.
Prakash, R. S., Snook, E. M., Motl, R. W., and Kramer, A. F. (2010). Aerobic fitness is associated with gray matter volume and white matter integrity in multiple sclerosis 19. Brain Res. 1341, 41–51.
Price, J. L., and Morris, J. C. (1999). Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann. Neurol. 45, 358–368.
Prins, N. D., and van Swieten, J. C. (2010). Alzheimer disease: MRI and CSF biomarkers in AD-accuracy and temporal change 1. Nat. Rev. Neurol. 6, 650–651.
Raffaitin, C., Feart, C., Le, G. M., Amieva, H., Helmer, C., Akbaraly, T. N., Tzourio, C., Gin, H., and Barberger-Gateau, P. (2011). Metabolic syndrome and cognitive decline in French elders: the three-city study 1. Neurology 76, 518–525.
Ramón y Cajal, S. (1909). Histologie Du Système Nerveux De L’Homme Et Des Vertébrés. Paris: A. Maloine.
Reyners, H., Gianfelici de, R. E., and Maisin, J. R. (1982). The beta astrocyte: a newly recognized radiosensitive glial cell type in the cerebral cortex 5. J. Neurocytol. 11, 967–983.
Reyners, H., Gianfelici de, R. E., Regniers, L., and Maisin, J. R. (1986). A glial progenitor cell in the cerebral cortex of the adult rat 2. J. Neurocytol. 15, 53–61.
Rinne, J. O., Brooks, D. J., Rossor, M. N., Fox, N. C., Bullock, R., Klunk, W. E., Mathis, C. A., Blennow, K., Barakos, J., Okello, A. A., Rodriguez Martinez de Liano, S., Liu, E., Koller, M., Gregg, K. M., Schenk, D., Black, R., and Grundman, M. (2010). 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 9, 363–372.
Roher, A. E., Esh, C., Rahman, A., Kokjohn, T. A., and Beach, T. G. (2004). Atherosclerosis of cerebral arteries in Alzheimer disease 1. Stroke 35, 2623–2627.
Salmina, A. B., Inzhutova, A. I., Malinovskaya, N. A., and Petrova, M. M. (2010). Endothelial dysfunction and repair in Alzheimer-type neurodegeneration: neuronal and glial control 6. J. Alzheimers Dis. 22, 17–36.
Shibata, M., Yamada, S., Kumar, S. R., Calero, M., Bading, J., Frangione, B., Holtzman, D. M., Miller, C. A., Strickland, D. K., Ghiso, J., and Zlokovic, B. V. (2000). Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier 1. J. Clin. Invest. 106, 1489–1499.
Shibata, S., Hastings, J. L., Prasad, A., Fu, Q., Okazaki, K., Palmer, M. D., Zhang, R., and Levine, B. D. (2008). “Dynamic” Starling mechanism: effects of ageing and physical fitness on ventricular-arterial coupling. J. Physiol. 586, 1951–1962.
Sierra, S., Ramos, M. C., Molina, P., Esteo, C., Vazquez, J. A., and Burgos, J. S. (2011). Statins as neuroprotectants: a comparative in vitro study of lipophilicity, blood-brain-barrier penetration, lowering of brain cholesterol, and decrease of neuron cell death. J. Alzheimers Dis. 23, 307–318.
Small, G. W., Bookheimer, S. Y., Thompson, P. M., Cole, G. M., Huang, S. C., Kepe, V., and Barrio, J. R. (2008). Current and future uses of neuroimaging for cognitively impaired patients. Lancet Neurol. 7, 161–172.
Smith, A. D., and Zigmond, M. J. (2003). Can the brain be protected through exercise? Lessons from an animal model of parkinsonism. Exp. Neurol. 184, 31–39.
Soumare, A., Tavernier, B., Alperovitch, A., Tzourio, C., and Elbaz, A. (2009). A cross-sectional and longitudinal study of the relationship between walking speed and cognitive function in community-dwelling elderly people. J. Gerontol. A Biol. Sci. Med. Sci. 64, 1058–1065.
Stopa, E. G., Butala, P., Salloway, S., Johanson, C. E., Gonzalez, L., Tavares, R., Hovanesian, V., Hulette, C. M., Vitek, M. P., and Cohen, R. A. (2008). Cerebral cortical arteriolar angiopathy, vascular beta-amyloid, smooth muscle actin, Braak stage, and APOE genotype. Stroke 39, 814–821.
Swain, R. A., Harris, A. B., Wiener, E. C., Dutka, M. V., Morris, H. D., Theien, B. E., Konda, S., Engberg, K., Lauterbur, P. C., and Greenough, W. T. (2003). Prolonged exercise induces angiogenesis and increases cerebral blood volume in primary motor cortex of the rat. Neuroscience 117, 1037–1046.
Tapiola, T., Alafuzoff, I., Herukka, S. K., Parkkinen, L., Hartikainen, P., Soininen, H., and Pirttila, T. (2009). Cerebrospinal fluid beta-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain 1. Arch. Neurol. 66, 382–389.
Taylor, D. V., Boyajian, J. G., James, N., Woods, D., Chicz-Demet, A., Wilson, A. F., and Sandman, C. A. (1994). Acidosis stimulates beta-endorphin release during exercise. J. Appl. Physiol. 77, 1913–1918.
Trejo, J. L., Carro, E., Garcia-Galloway, E., and Torres-Aleman, I. (2004). Role of insulin-like growth factor I signaling in neurodegenerative diseases. J. Mol. Med. 82, 156–162.
Trejo, J. L., Carro, E., and Torres-Aleman, I. (2001). Circulating insulin-like growth factor I mediates exercise-induced increases in the number of new neurons in the adult hippocampus. J. Neurosci. 21, 1628–1634.
Utter, S., Tamboli, I. Y., Walter, J., Upadhaya, A. R., Birkenmeier, G., Pietrzik, C. U., Ghebremedhin, E., and Thal, D. R. (2008). Cerebral small vessel disease-induced apolipoprotein E leakage is associated with Alzheimer disease and the accumulation of amyloid beta-protein in perivascular astrocytes 19. J. Neuropathol. Exp. Neurol. 67, 842–856.
Van, P. H., Christie, B. R., Sejnowski, T. J., and Gage, F. H. (1999a). Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc. Natl. Acad. Sci. U.S.A. 96, 13427–13431.
Van, P. H., Kempermann, G., and Gage, F. H. (1999b). Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat. Neurosci. 2, 266–270.
Vemuri, P., Simon, G., Kantarci, K., Whitwell, J. L., Senjem, M. L., Przybelski, S. A., Gunter, J. L., Josephs, K. A., Knopman, D. S., Boeve, B. F., Ferman, T. J., Dickson, D. W., Parisi, J. E., Petersen, R. C., and Jack, C. R. Jr. (2010). Antemortem differential diagnosis of dementia pathology using structural MRI: differential-STAND 1. Neuroimage. 55, 522–531.
Volkmar, F. R., and Greenough, W. T. (1972). Rearing complexity affects branching of dendrites in the visual cortex of the rat. Science 176, 1445–1447.
Voss, M. W., Prakash, R. S., Erickson, K. I., Basak, C., Chaddock, L., Kim, J. S., Alves, H., Heo, S., Szabo, A. N., White, S. M., Wójcicki, T. R., Mailey, E. L., Gothe, N., Olson, E. A., McAuley, E., and Kramer, A. F. (2010). Plasticity of brain networks in a randomized intervention trial of exercise training in older adults 4. Front. Aging Neurosci. 2:32. doi: 10.3389/fnagi.2010.00032
Walicke, P., Cowan, W. M., Ueno, N., Baird, A., and Guillemin, R. (1986). Fibroblast growth factor promotes survival of dissociated hippocampal neurons and enhances neurite extension. Proc. Natl. Acad. Sci. U.S.A. 83, 3012–3016.
Williams, P., Robinson, D., and Bailey, A. (1979). High-density lipoprotein and coronary risk factors in normal men. Lancet 1, 72–75.
Winblad, B., Palmer, K., Kivipelto, M., Jelic, V., Fratiglioni, L., Wahlund, L. O., Nordberg, A., Bäckman, L., Albert, M., Almkvist, O., Arai, H., Basun, H., Blennow, K., de Leon, M., DeCarli, C., Erkinjuntti, T., Giacobini, E., Graff, C., Hardy, J., Jack, C., Jorm, A., Ritchie, K., van Duijn, C., Visser, P., and Petersen, R. C. (2004). Mild cognitive impairment–beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J. Intern. Med. 256, 240–246.
Wu, C. W., Chen, Y. C., Yu, L., Chen, H. I., Jen, C. J., Huang, A. M., Tsai, H. J., Chang, Y. T., and Kuo, Y. M. (2007). Treadmill exercise counteracts the suppressive effects of peripheral lipopolysaccharide on hippocampal neurogenesis and learning and memory 2. J. Neurochem. 103, 2471–2481.
Wu, Z., Guo, H., Chow, N., Sallstrom, J., Bell, R. D., Deane, R., Brooks, A. I., Kanagala, S., Rubio, A., Sagare, A., Liu, D., Li, F., Armstrong, D., Gasiewicz, T., Zidovetzki, R., Song, X., Hofman, F., and Zlokovic, B. V. (2005). Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease 1. Nat. Med. 11, 959–965.
Yamamoto, M., Clark, J. D., Pastor, J. V., Gurnani, P., Nandi, A., Kurosu, H., Miyoshi, M., Ogawa, Y., Castrillon, D. H., Rosenblatt, K. P., and Kuro-o, M. (2005). Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 280, 38029–38034.
Yates, L. B., Djousse, L., Kurth, T., Buring, J. E., and Gaziano, J. M. (2008). Exceptional longevity in men: modifiable factors associated with survival and function to age 90 years. Arch. Intern. Med. 168, 284–290.
Yirmiya, R., and Goshen, I. (2011). Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 25, 181–213.
Zaldivar, F., Wang-Rodriguez, J., Nemet, D., Schwindt, C., Galassetti, P., Mills, P. J., Wilson, L. D., and Cooper, D. M. (2006). Constitutive pro- and anti-inflammatory cytokine and growth factor response to exercise in leukocytes. J. Appl. Physiol. 100, 1124–1133.
Zhang, Y., Tatsuno, T., Carney, J. M., and Mattson, M. P. (1993). Basic FGF, NGF, and IGFs protect hippocampal and cortical neurons against iron-induced degeneration. J. Cereb. Blood Flow Metab. 13, 378–388.
Zhu, X., Mei, M., Lee, H. G., Wang, Y., Han, J., Perry, G., and Smith, M. A. (2005). P38 activation mediates amyloid-beta cytotoxicity. Neurochem. Res. 30, 791–796.
Keywords: hippocampus, entorhinal cortex, insulin-like growth factor, reduction of systemic inflammation, p38 effector of Aβ-induced neurodegeneration, virtual reality environment, exponentially decreasing risk of cell death, loss of cognitive performance
Citation: Foster PP, Rosenblatt KP and Kuljiš RO (2011) Exercise-induced cognitive plasticity, implications for mild cognitive impairment and Alzheimer’s disease. Front. Neur. 2:28. doi: 10.3389/fneur.2011.00028
Received: 11 April 2011; Accepted: 18 April 2011;
Published online: 06 May 2011.
Edited by:
Cristian Lasagna Reeves, University of Texas Medical Branch, USAReviewed by:
Craig Atwood, University of Wisconsin, USACristian Lasagna Reeves, University of Texas Medical Branch, USA
Diana Laura Castillo-Carranza, University of Texas Medical Branch, USA
Marcos Jair Guerrero-Munoz, University of Texas Medical Branch, USA
Copyright: © 2011 Foster, Rosenblatt and Kuljiš. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Philip P. Foster, Department of Internal Medicine, University of Texas Medical Branch, 301 University Boulevard, Galveston, TX 77555-0561, USA. e-mail: ppfoster@utmb.edu