 David Z. Rose1 Scott E. Kasner2*
David Z. Rose1 Scott E. Kasner2*- 1 Stroke Division, Department of Neurology, University of South Florida, Tampa General Hospital, Tampa, FL, USA
- 2 Stroke Division, Department of Neurology, University of Pennsylvania, Philadelphia, PA, USA
Successful implementation of a randomized clinical trial (RCT) for neuro-vascular emergencies such as cerebral infarction, intracerebral hemorrhage, or subarachnoid hemorrhage is extraordinarily challenging. Besides establishing an accurate, hyper-expedited diagnosis among many mimics in a person with acute neurological deficits, informed consent must be obtained from this vulnerable group of patients who may be unable to convey their own wishes, grasp the gravity of their situation, or give a complete history or examination. We review the influences, barriers, and factors investigators encounter when providing established and putative stroke therapies, and focus on informed consent, the most important research protector of human subjects, as the rate-limiting step for enrollment into acute stroke RCTs. The informed consent process has received relatively little attention in the stroke literature, but is especially important for stroke victims with acute cognitive, aural, lingual, motor, or visual impairments. Consent by a surrogate may not accurately reflect the patient’s wishes. Further, confusion about trial methodology, negative opinions of placebo-controlled studies, and therapeutic misconception by patients or surrogates may impede trial enrollment and requires further study. Exception from informed consent offers an opportunity that is rarely if ever utilized for stroke RCTs. Ultimately, advancing the knowledge base and treatment paradigms for acute stroke is essential but autonomy, beneficence (non-malfeasance), and justice must also be carefully interwoven into any well-designed RCT.
Neuro-vascular emergencies, including cerebral infarction, intracerebral hemorrhage (ICH), and subarachnoid hemorrhage (SAH), are arguably the most difficult medical conditions in which to successfully construct and implement a randomized clinical trial (RCT). First, an accurate, hyper-expedited stroke diagnosis must be established among a large array of potential mimics in a patient with acute neurological deficits, based on an incomplete history, limited examination, and imperfect neuroimaging. Second, informed consent must be obtained from potentially vulnerable patients who are often suddenly unable to convey their own wishes, communicate with physicians and family, or comprehend the gravity of their situation. Third, both established and putative acute therapies must be administered rapidly to maximize their potential benefits. Extensive research has focused on improving stroke diagnosis and on developing systems to optimize the timely delivery of treatment to stroke patients. However, the issue of informed consent in stroke trials has received relatively little attention, yet it is often the rate-limiting step in RCTs of acute stroke therapy. Clinical trials involving potential treatments for stroke, as well as many other neurological emergencies, face a cluster of unique challenges to informed consent that likely contribute to the consistently slow recruitment pace of these trials (The National Institute of Neurological Disorders and Stroke Rt-PA Stroke Study Group, 1995; Alexandrov, 2006; Elkins et al., 2006).
Voluntary informed consent, authorized by a competent, alert, informed individual, has been the ethical foundation of human research for decades in the Belmont Report, published in 1979 (National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research, 1979; Beauchamp, 2004). Informed consent has been labeled the “most important protector” of human subjects in research (Prentice, 1999). The process of informed consent must include a description of the precise objectives of the RCT, its methods and procedures, its risks and discomforts (known and unknown), liability in case of injury, alternatives to participation, plans for use of personal and private information, discontinuation options (for the subject and for the sponsor or investigators), and the promise of possible benefit. To give informed consent, a person must at the time of consent be 18 years of age or older, possess adequate reasoning faculties, and clearly understand the facts, implications, and future consequences of their decision (Food and Drug Administration, 1996; Marwick, 1997; Adams and Wegener, 1999; Biros et al., 1999; Prentice, 1999; Morris et al., 2004; Kasner et al., 2009, 2010). Subjects who lose their autonomy due to acute stroke-related cognitive impairment require special consideration and additional protections. Without autonomy, a subject cannot understand the risks and benefits of a RCT nor provide informed consent for trial enrollment. Notably, patients with the most severe ischemic strokes are the least likely to benefit from currently available thrombolytic therapy and are also the least likely to be able to provide their own consent to trial participation (Flaherty et al., 2008).
In addition to autonomy, beneficence/non-malfeasance, and justice together triangulate ethical trial design (Burrows and Hodgson, 1997). Beneficence in clinical trials refers to the goal of promoting the best interest and health of study subjects. This principle may conflict with autonomy, as may occur when a patient has no viable standard treatment options yet is incapacitated from making a decision about an experimental intervention. Justice requires that research subjects are treated fairly and not placed at a disadvantage or faced with excessive risk. Patients with severe stroke must not be given more or less opportunity to participate in RCTs than those with minor strokes. Again, this may be a challenge when there is loss of autonomy. Justice also refers to a social justice for the community of subjects with the medical problem, for whom the research must be timely and relevant. Ultimately, autonomy, beneficence/non-malfeasance, and justice in a well-designed RCT make the RCT itself potentially highly valuable both to the individual and to society (Burrows and Hodgson, 1997). Exclusion of patients who are unable to provide their own consent for stroke RCTs undoubtedly impacts both validity and generalizability of the results and thereby limits the value of such a RCT, not to mention its progress. Thus, to overcome this obstacle, and advance the knowledge base and treatment paradigms for acute stroke, improved and alternative approaches to the consent process must be sought (Table 1).
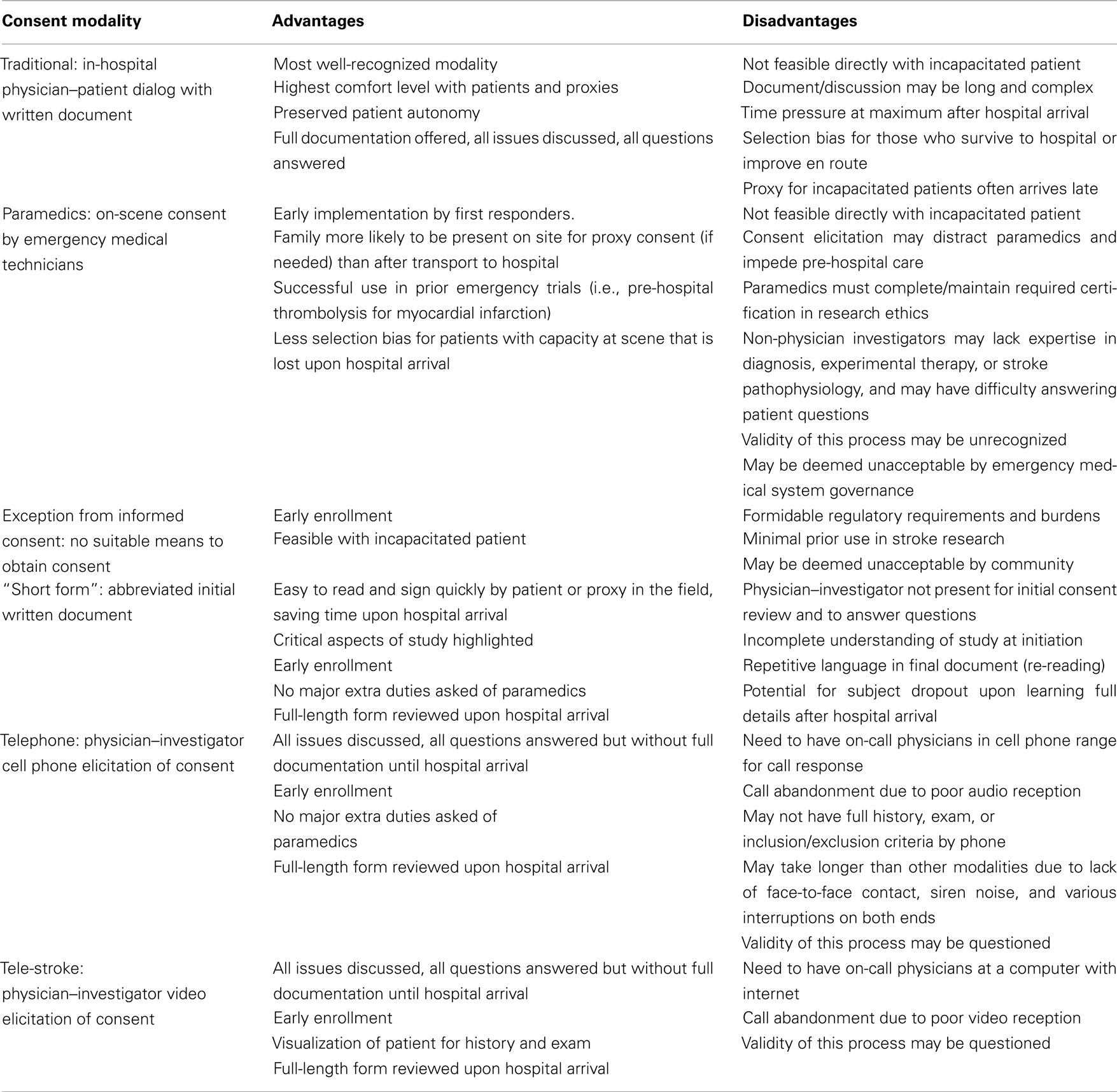
Table 1. Advantages and disadvantages of various modalities of informed consent for stroke trials.
Time Pressure
Acute neuro-vascular brain injury will lead to irreversible damage without rapid intervention. As with standard therapy such as thrombolysis for ischemic stroke, there is also extraordinary time pressure for trial enrollment, which requires an accelerated approach to the process of informed consent itself. For the acute stroke patient, arrival in the emergency department is followed quickly by questions from healthcare professionals, physical and neurological examinations and evaluations, blood draws, and urgent neuroimaging. The patient will be offered standard acute therapies if appropriate and learn rapidly about their potential benefits and risks. Amid all of this rampage, the patient may be verbally offered participation in a trial but given mere minutes to read the trial’s informed consent document, understand it, mull it over, and make a decision about participation. The narrow time windows for standard therapy, informed consent, decision-making, randomization, and study drug administration, are often prohibitive. As there are many personal, medical, sociocultural, emotional, spiritual, and legal factors involved in giving informed consent for a RCT, patients should be able consider their options thoughtfully. When asked about the time allotted to make such a decision for a stroke trial, 30 min was deemed sufficient for only 56% of respondents (Del Giudice et al., 2009). Subjects were more likely to participate if they felt that they had enough time, though many wished they had more time and opportunity to discuss their choice with others. In order to augment enrollment, investigators should make every effort to start the consent process as early as possible, in order to allocate more than just a few pressurized moments for the potential subject to make what could be the biggest decision of his or her life.
Research Insight and Literacy
Potential study subjects may have an incomplete, imperfect understanding of the proposed trial and its key concepts, even if cognitively normal (Kasner et al., 2009, 2010). The typical informed consent form for a stroke trial ranges from 8 to 16 pages, depending on numerous factors but most prominently the risks and invasiveness of the intervention and the policies of the institution. While attempts are reliably made to use 10th grade or lower reading levels, consent forms are nevertheless laden with medical jargon and other information that is often quite new for the reader. Illiteracy in general remains a problem in the U.S. but is widespread in developing countries, and medical illiteracy is a major global challenge for clinical research.(Lynoe et al., 1998; Kilmarx et al., 2001; Marshall, 2006) Patients may conflate their standard therapy with the study intervention leading to further difficulty with the overall process.(Lynoe et al., 2004) Thus, any information given to patients to review for entry into acute RCTs must be clear, concise, and intended for subjects with little medical knowledge (Grotta and Bratina, 1995). In one survey of 685 stroke patients in the post-acute phase (and their proxies) in 12 hospitals across Italy, education was found to be the primary sociodemographic determinant of independent clinical decision-making (Ciccone et al., 2001). The preference for thrombolytic therapy, for example, was higher among more educated people (Ciccone et al., 2001). Nevertheless, the role of education plays a somewhat questionable role in research contexts.
In less urgent settings, educational level as well as age, sex, race/ethnicity, and numerous sociocultural factors have been reported as probable factors associated with RCT participation (Marwick, 1997; Adams and Wegener, 1999; Biros et al., 1999; Prentice, 1999; Lynoe et al., 2004; Morris et al., 2004; Marshall, 2006; Kasner et al., 2009, 2010). However, none of these factors appeared to be significantly associated with the decision to participate in RCTs involving cerebral infarction, ICH, or SAH (Del Giudice et al., 2009; Kasner et al., 2009). This suggests that these factors may not be directly relevant to giving informed consent in these clinical situations.
Critical research concepts such as randomization and clinical equipoise are complex and may not be easily interpreted or appropriately incorporated into the research decision-making process, particularly when pressed for time. Confusion about the overall trial design and therapeutic misconception may result. Potential subjects believe they will get a new medication that would likely help them, without full realization that they ultimately may be better off in the placebo or standard therapy arm of the trial. In both structured interviews and in focus groups involving people either at risk for or with prior stroke or traumatic brain injury, participants expressed that in the face of an acute neurological catastrophe, “experimental treatment must be better than nothing” (Kasner et al., 2010). When confronted with the possibility of not receiving the investigational agent (i.e., given placebo) one said, “I think everyone should get the treatment, I mean that’s not fair. You’re allowing people to be experimented upon and not giving them a fair shot of taking a risk” (Kasner et al., 2010). Participants consistently had negative opinions of placebo-controlled studies and described feelings of being “left out” if not given experimental treatment (Kasner et al., 2010). This therapeutic misconception may have been cultivated by a misunderstanding of supportive care, in that it is still the standard of care. Conversely, when considering an alternative RCT design comparing two active treatments, subjects then said they did not feel “deprived of treatment” (Kasner et al., 2010).
Thus, concerted public education as well as rapid counseling in the acute setting may improve comprehension of these key concepts and thereby improve research participation. Community outreach campaigns also may encourage more people to consider participating in a stroke RCT, should they or their loved ones suffer from this malady. Naturally, any stroke awareness information sessions must be developed in a straightforward, culturally sensitive way, without undue influence, financially, socially, or otherwise. The aim is not to deemphasize the role of informed consent or the physician–patient interaction, but instead to enhance the decision-making process. This is accomplished by having potential enrollees and their families preemptively identify with the concept of a RCT, its generation, function, goal, and significance. Such a campaign could help demystify the prospects of a RCT before a stroke ever happens.
Loss of Decision-Making Capacity
Stroke victims’ acute cognitive, aural, lingual, motor, or visual impairments can partially or completely render them unable to take part in the informed consent process. This issue is fairly unique to neuro-vascular trials, as RCTs involving other types of disorders, even in an emergency situation, seldom encounter acute brain dysfunction that removes subjects’ capacity for informed consent. Indeed, loss of autonomy is often the rule than the exception, with only 30% of subjects in some stroke RCTs being able provide their own consent prior to enrollment (Flaherty et al., 2008). Some researchers seriously doubt the validity of acute stroke patients’ informed consent when at the nadir of their illness because even stroke victims who recover rapidly have a foggy recollection of their event (Grotta and Bratina, 1995).
Loss of capacity to give informed consent has also been linked to increasing age and baseline neurological deficit, according to an analysis of patients enrolled in four urgent therapeutic trials (Demarquay et al., 2005). Aphasia was the main reason for inability to self-consent in 67% of patients. In the third International stroke trial (IST-3), patients with more severe stroke as well as those with non-lacunar hemispheric stroke syndromes also were less likely to have been recruited by their own consent (Kane et al., 2006). It would be helpful to have a standardized tool to assess acute stroke patients’ capacity to consent for RCT enrollment, but currently no such validated or appropriately rapid test exists (Bateman et al., 2003). So for the time being, researchers continue to subjectively estimate each patient’s capacity to give informed consent prior to RCT enrollment and then decide on their own if surrogate consent is needed.
Surrogate Informed Consent
As long as the interests of patients are generally best served by participation, research involving incapacitated individuals is ethically permissible by surrogate informed consent (World Medical Association, 2000, 2004). According to the Declaration of Helsinki, a document initially adopted in 1964 defining essential ethical principles guiding clinical research, subjects without capacity to give informed consent “should not be included in research unless research is necessary to promote the health of the population represented and research cannot instead be performed on legally competent persons” (World Medical Association, 2000, 2004). This language differs from the 1947 Nuremberg Code on human experimentation which made Informed Consent an “absolutely essential” requirement (Simmons, 1964; Faden et al., 1996; Dickens, 1999). Article II of the Declaration of Helsinki stipulates that doctors should try to acquire consent “if at all possible” but if this is impossible and a surrogate such as a legal guardian is available, then research is allowed without the patient’s own explicit consent.
In the NINDS trial investigating tissue plasminogen activator (t-PA) for acute ischemic stroke, surrogate consent was used to enroll 439 of 624 (70%) of subjects (The National Institute of Neurological Disorders and Stroke Rt-PA Stroke Study Group, 1995; Flaherty et al., 2008). Compared to subjects who provided their own consent, those enrolled by surrogate consent generally were about 5 years older, with higher NIH stroke scale (NIHSS) scores (median 17 versus 9, p < 0.001), and less likely to have a good recovery (26 versus 53% had a modified Rankin score of 0–1 at 90 days, p < 0.001; Flaherty et al., 2008). Over a decade later, in a RCT of pre-hospital acute stroke therapy, (D:SaverandTheFAST-MAGInvestigators:2011]) surrogate-consented subjects were again found to be older by about 7 years and with higher NIHSS scores (median 16 versus 6, p < 0.001). A small study from Scotland also demonstrated that infarct lesion volume on MRI, in addition to stroke severity, was associated with capacity to consent for acute stroke trials. (Dani et al., 2008) Stroke lesion burden was greater for incapable of consent compared to those enrolled by their own consent. In a registry involving 20 major stroke centers across Canada, stroke victims with cognitive impairment and/or altered mental status were excluded and the in-hospital mortality rate was 6.9% for those who participated and 22% for those who did not (p < 0.001; Tu et al., 2004). This strongly suggests that those included in the registry were not representative of the typical stroke population. Together, these studies demonstrate that subjects who provide their own consent differ substantially from those requiring surrogate consent (Figures 1 and 2). Further, in SAH trials, only 28% of patients believed they had the capacity to make their own decision at the time of enrollment (Schats et al., 2003). The inclusion of subjects with stroke via surrogate consent therefore dramatically expands the diversity of the study population and enhances the generalizability of the research.
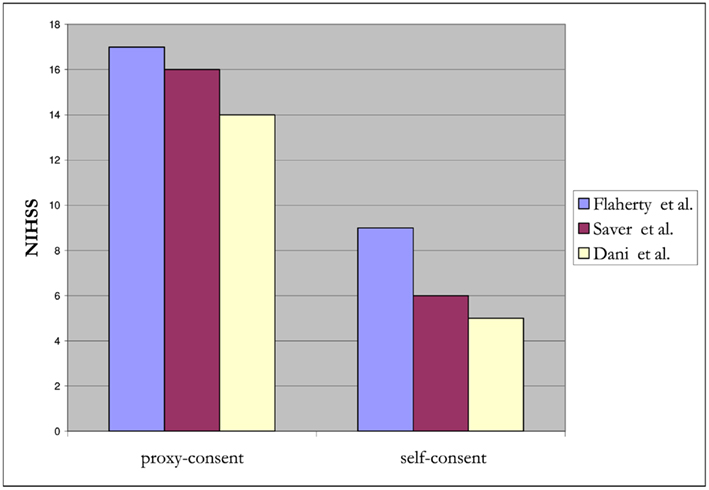
Figure 1. Relationship between NIHSS scores and proxy- versus self-consent for acute stroke trials.
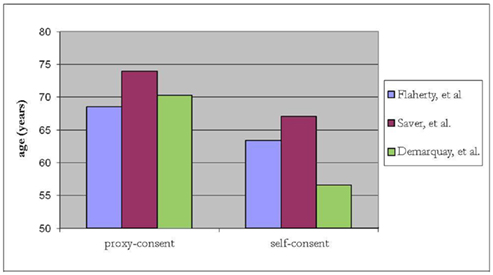
Figure 2. Relationship between age and proxy- versus self-consent in acute stroke trials.
In the U.S., regulations for surrogate consent in medical research vary from state to state. In some areas, there are no formal guidelines at all, forcing Institutional Review Boards (IRBs) and investigators to grapple with this issue themselves (Kim and Karlawish, 2003). Interpretations by IRBs in several states and countries prohibit surrogate informed consent for research participation unless durable power of attorney is furnished (Saver, 1996; Flaherty et al., 2008). However, IRBs in some regions have been reported to temporarily suspend enrollment by surrogate informed consent, demanding legislative clarification before resuming such research entry (Flaherty et al., 2008). Reluctance to allow surrogate informed consent may be particularly prominent with research that carries substantial risk as opposed to investigations of more benign interventions (Flaherty et al., 2008). Some IRBs insist that prior to collecting data, researchers obtain informed consent from the patient – even though the well-known, oft-cited, federal privacy law, health insurance portability and accountability act (HIPAA), permits informed consent waivers (Kulynych and Korn, 2002). Nevertheless, this and other U.S. federal statutes do not delineate criteria for someone to be considered “a legally authorized representative,” so investigators and IRBs continue to deal with it themselves or seek guidance from state law (Bateman et al., 2003). On a global scale, there is even greater variability in the law and its interpretation, and increasingly complex sociocultural issues regarding who can make decisions on behalf of a patient (Marshall, 2006). While not studied specifically for stroke trials, there are numerous examples where consent appeared to be more acceptable by a husband or an elder from the husband’s family than by a wife (Kilmarx et al., 2001; Decosta et al., 2004). Yet with the silent or ambiguous laws and sociocultural idiosyncrasies that pepper each individual region, a circle of chaos emerges about who is authorized to make medical decisions for the incapacitated especially in the acute setting (Saver, 1996; Bateman et al., 2003). Such medico-legal uncertainty can decelerate the development of effective stroke therapy. The RCTs that only enroll patients who can definitely give informed consent will analyze healthier subjects who do not adequately resemble the clinical spectrum of stroke patients, and thus any conclusions drawn from such research may be deemed underpowered, unreliable, and consequently invalidated (Dani et al., 2008; Flaherty et al., 2008).
Even when legally authorized representatives are acceptable and available for decision-making, enrollment into acute stroke RCTs is still low. When offered a hypothetical stroke trial, only 40% of surrogates agreed to participation compared to 59% of stroke patients (Kasner et al., 2009). One explanation may be that there is more uncertainty or anxiety about RCT participation among surrogate decision-makers than patients, especially when decisions are made for a relative (41% of proxies could not reach a decision versus 17% for self-indecision, p < 0.001) according to one review (Ciccone et al., 2001). Another study found that zero of 10 patients (0%) declared feeling uncomfortable when giving self-consent, but seven out of 13 relatives (54%) said the “psychological stress induced by urgent decision-making” made them uneasy when providing consent for their family members (Demarquay et al., 2005). Categorically, stroke patients are much more inclined to participate in a RCT than their proxy decision-makers.
When surrogate decision-makers are not immediately available, extensive efforts may be required to find them, but this can be tedious. One individual suggested: “Go into my wallet and maybe I have a phone number in there (or) maybe I have a cell phone and see if you can call someone” (Kasner et al., 2010). However, there is typically minimal time to do a thorough search in the setting of an acute stroke. Further, there is substantial variability in regional legal statutes and precedents about who may serve as a surrogate in this context (Saver, 1996; Kulynych and Korn, 2002; Bateman et al., 2003; Kim and Karlawish, 2003; Flaherty et al., 2008). Even if someone suitable is found, he or she may not be able to get to the hospital in time to read, decide, and sign the consent document. If a surrogate can only be reached by telephone, many IRBs reject telephone consent for RCT participation. In one study, only 18% of IRBs allowed surrogate consent to be obtained over the phone (Leira et al., 2010). However, physician phone elicitation of informed consent from legally authorized representatives has been successfully utilized in the pre-hospital setting in the Field administration of stroke therapy – magnesium (FAST-MAG) trial, permitting rapid patient study entry, without delaying transport times (Saver et al., 2006; Sanossian et al., 2010). Further, a suitable surrogate decision-maker could be identified but he or she may not be comfortable making any decision for the patient.
In the end, a surrogate may not necessarily make a decision that is consistent with the stroke patient’s own preferences (Kasner et al., 2009, 2010). Whether or not is it fair to tell surrogates that they may underestimate the patient’s wishes to participate remains to be put to an ethical test.
Exception from Informed Consent
Under certain circumstances, such as in emergency situations that involve acutely neurologically impaired patients, research participation is possible without explicit informed consent (Food and Drug Administration, 1996). The U.S. Food and Drug Administration (FDA) permits enrollment into trials under their Exception from informed consent (EFIC) directive, FDA Rule 21 CFR 50.24, which parallels the Department of health and human services (DHHS) waiver of Informed Consent Rule 45 CFR 46.101.(Biros et al., 1999) To qualify a study for EFIC, the following criteria must be met:
(i) the subject must be in a life-threatening situation,
(ii) existing therapies are unproven or unsatisfactory,
(iii) unable to obtain informed consent due to the medical condition, and
(iv) unable to use surrogate consent due to the acuity of the treatment.
The pathology of the condition need not be immediately life-threatening, but the condition of the subject must be life-threatening during the therapeutic window of the investigational agent, with a high likelihood of patient death or disability unless the course of the pathology is interrupted (Food and Drug Administration, 1996; Marwick, 1997; Adams and Wegener, 1999; Biros et al., 1999; Prentice, 1999; Morris et al., 2004; Kasner et al., 2009, 2010). There must be no reasonable way, prospectively, to identify subjects eligible for participation in the RCT. According to the DHHS rule, current treatments must be declared “unproven or unsatisfactory,” preclinical studies and related evidence must “support the potential for the intervention,” and risks associated with the investigation deemed “reasonable” compared to standard therapy. The FDA also requires that an independent data safety monitoring board (DSMB) provide ongoing oversight of all trials invoking EFIC, regardless of size. Investigators must demonstrate that their RCT could not practicably be carried out without EFIC. Good-faith, appropriately documented attempts to locate a proxy (to avoid proceeding without consent) should be monitored by an IRB. Lastly, for each EFIC protocol using a drug, device, or biologic agent, an application for investigational new drug (IND) or investigational device exemption (IDE) must be submitted. Although these FDA requisites may appear onerous, the intention of the EFIC is to protect subjects whom, without a waiver, would not participate in any research at all and hence forgo the potential benefit offered by scientific advancement.
Before a trial is conducted under EFIC, additional requirements include community consultation and public disclosure in accord with 21 CFR 50.24 (Food and Drug Administration, 1996). Community consultation is not well defined, but should “involve and engage members of the community in which the research will take place and members of the community from which prospective study subjects may be drawn (if these two communities are not the same). The proposed trial must therefore be openly discussed with community members who are representative of the potential study population and the study community” (Food and Drug Administration, 1996; Kasner et al., 2010). This consultation fosters a spirit of inclusion for the community, prompting its members to learn about local research and voice concerns about real or imagined abuses by investigators (Leira et al., 2010). Community consultation may be reasonably achieved by individual interviews, focus groups, town hall meetings, and other forums. A series of focus groups involving stroke survivors and family members of stroke victims acknowledged that if there were no other treatment options available, or if experimental treatment were the last resort, then their willingness to participate would be high and they would accept enrollment into a RCT without consent under the EFIC regulations (Blixen and Agich, 2005; Kasner et al., 2009). In particular, stroke survivors were noted to be well aware of the long-term sequelae of stroke, the limitations of current therapies, and the need for more research in acute therapies, which likely made them more open to EFIC than the general public (Blixen and Agich, 2005). Focus group participants were generally content having the decision made by their physician or, if necessary, an independent committee of physicians and medical ethics experts with the patient’s best interests in mind. The inclusion of a layperson with personal experience with stroke may also be helpful, as stroke survivors have been reported to “exchange information, ideas, and encouragement … my online (stroke survivor) community might be closer to the mark for me because I share things with them that my family does not know about because they understand things that my family does not” (Kasner et al., 2010). One participant summed it up by saying, “If my situation were so severe that I really had no other option, like I had nothing to lose, then I would say yes”’ (Kasner et al., 2010). Thus, the perceptions of beneficence for the patient prevailed over autonomy. Of note, focus group participants were less trusting of emergency medical technicians (EMTs) than physicians, more leery of an out-of-hospital RCT than a RCT set up in an emergency room, and thoroughly skeptical that the general public has enough education or relevant experience to represent their views on informed consent in these circumstances. Unequivocally, they did not endorse any other political, religious, or professional organization as suitable for consultation at all.
The past two decades have seen an upsurge in the interest and dedication to the study of stroke. However, no RCTs for acute stroke therapies have utilized EFIC. Practically, EFIC has been difficult to implement in the U.S. overall, as many people view EFIC as more ethically problematic than surrogate informed consent (Salzman et al., 2007). EFIC has been used successfully in other neuroemergency trials, including the North American Brain Injury Study-Hypothermia, the Progesterone for Traumatic brain injury Experimental Clinical Treatment, and the Rapid Anticonvulsant Medications Prior to Arrival Trial for seizures. Although EFIC remains unused for stroke RCTs, it appears to be an acceptable option to stroke survivors and their families. Additional research will further elucidate the utility, efficiency, and satisfaction of community consultation and public disclosure activities for stroke and other neuroemergency trials.
Nevertheless, for stroke victims, EFIC may represent their best chance to participate in research. In a emergency department-based survey involving a hypothetical stroke trial, 55% of eligible respondents were willing to be enrolled into a RCT without informed consent, and an independent predictor to participate was if a greater than 50% chance of full recovery was deemed possible (Goldstein et al., 2010).
Willingness to Participate
Few studies have attempted to quantitatively assess the proportion of people who would be willing to participate in acute stroke trials. Not surprisingly, the results have been strongly related to the severity of the stroke and the risk of the proposed intervention. For a hypothetical trial involving acute SAH, which poses an enormous threat of death and disability, a survey of emergency department patients (who did not SAH) found that 76% were likely to participate (Del Giudice et al., 2009). A survey of patients and their surrogates with acute ischemic stroke or ICH found that 57% were likely to participate in the proposed RCT, though only 50% agreed if the treatment was potentially dangerous while 90% agreed when the risk was relatively low (Kasner et al., 2009). Willingness to participate in a hypothetical trial has been shown to be a valid predictor of actual participation, and assessment of enrollment preferences in hypothetical situations rather than in dire emergencies might better reflect patients’ genuine interests (Halpern et al., 2001). Another hypothetical RCT, exploring motivations for participation, found that personal health benefit, helping others, and contributing to scientific knowledge were the top three reasons subjects were willing to enroll, while the most common concerns were the need for medication adjustments (56%), inconvenience of being in a trial (38%), anxiety of study drug side effects (35%), and placebo futility (24%; Halpern et al., 2003).
Factors such as age, sex, race, and educational level have not been shown to bear significantly on the decision to participate in stroke trials (Kasner et al., 2009), although such issues have not been well explored on an international scale. Notably, prior exposure to a neurological injury or illness was prominently associated with the desire to participate in a RCT, likely due to the impact of the prior event and the potential subject’s opinion that a trial intervention could only help them (Kasner et al., 2009, 2010). Stroke survivors “have a very devil-may-care attitude…after we’ve gone through this” and are often quite eager to try something new that may not have been an option at the time of the first stroke (Blixen and Agich, 2005; Kasner et al., 2010).
Public outreach campaigns have educated people about stroke and thereby accelerated hospital arrival times and acute treatments (Morgenstern et al., 2003). However, attitudes toward and expectations of clinical research are mixed and could be targeted for similar educational efforts. In one study, acute stroke RCTs were rated as very positive by 5%, positive by 45%, neutral by 44%, negative by 5%, and very negative by 2% (Kasner et al., 2009). One virtually unchangeable obstacle to successful enrollment is a person’s fixed unassailable concerns about research (i.e., rigid pre-conceived notions and fears about experimentation, or explicit demand for non-enrollment). Prior negative attitudes about RCTs have been shown to markedly impede trial enrollment, and for some people, this mind-set may not be changed by educational efforts (Flaherty et al., 2008). However, it remains unknown if this perspective changes after one experiences the chronic aftermath of a devastating neurological injury (Marwick, 1997; Adams and Wegener, 1999; Biros et al., 1999; Prentice, 1999; Morris et al., 2004; Kasner et al., 2009, 2010).
Willingness to Enroll
In a European study examining physicians’ views of emergency trials, 218 questionnaires were completed, and attitudes were predominantly positive (Iwanowski et al., 2008). For the most part, physicians felt that patients’ trust was not compromised during emergency research involving stroke or acute coronary syndrome. Less than 15% of respondents believed that patients fully read written information anyway such as an Informed Consent document, with 19% hardly reading anything at all (Iwanowski et al., 2008).
Nevertheless, many stroke researchers have called for an international ethical consensus to mediate consent issues fairly for both investigators and stroke patients from diverse cultural backgrounds and various belief systems (Leira et al., 2010). In a survey about their experience with their local IRBs (or equivalent), 90 stroke researchers from 15 different countries compared their personal beliefs about the ethics of acute stroke trials with those of their IRB members. Substantial diversity existed between the ethical priorities of the researchers and the IRBs with respect to informed consent standards. For example, using EFIC for enrolling a patient with impaired consciousness was acceptable to 41% of investigators but only 33% of IRB members (Leira et al., 2010). Similarly, only 36% of IRB members but 62% of investigators felt that NIHSS score was sufficient to characterize decisional capacity. Perhaps a collaborative effort to attain a standardized, global informed consent practice could bridge this controversy. Working toward common ethical standards for acute stroke research may more rigorously and equally protect subjects across the globe (Leira et al. 2010).
Conclusion and the Future
The rate-limiting step in acute stroke trials is the process of informed consent. The majority of people are inclined to participate once they have weighed their options, but individuals with stroke seldom make that choice for themselves. Family members are more hesitant than patients to consent to trial participation, and they are often not available to make a well-timed decision. Despite reasonable laws to waive informed consent, EFIC remains underutilized for stroke RCTs, though it is deemed an acceptable option to many stroke survivors and their families. EFIC seems to be an inevitable part of the solution to expand the population of stroke patients able to participate and to improve the timeliness of stroke clinical research. Educational outreach programs are also needed to reduce confusion about trial methods, negative opinions, and therapeutic misconception. Additional research will further elucidate the utility, efficiency, and satisfaction of community consultation and public disclosure activities for stroke and other neuroemergency trials. Perhaps the ultimate message for clinician–investigators designing an acute stroke RCT is to motivate the public and potential subjects with the hope of improving outcomes of stroke in their community and throughout the world.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adams, J. G., and Wegener, J. (1999). Acting without asking: an ethical analysis of the Food and Drug Administration waiver of informed consent for emergency research. Ann. Emerg. Med. 33, 218–223.
Alexandrov, A. V. (2006). Slow recruitment in clinical trials: failure is not an option! Int. J. Stroke 1, 160.
Bateman, B. T., Meyers, P. M., Schumacher, H. C., Mangla, S., and Pile-Spellman, J. (2003). Conducting stroke research with an exception from the requirement for informed consent. Stroke 34, 1317–1323.
Beauchamp, T. (2004). The legacy and the future. 30 years after the Belmont report, Beauchamp sets the record straight. Prot. Hum. Subj. 1–3.
Biros, M. H., Fish, S. S., and Lewis, R. J. (1999). Implementing the Food and Drug Administration’s final rule for waiver of informed consent in certain emergency research circumstances. Acad. Emerg. Med. 6, 1272–1282.
Blixen, C. E., and Agich, G. J. (2005). Stroke patients’ preferences and values about emergency research. J. Med. Ethics 31, 608–611.
Burrows, R. C., and Hodgson, R. E. (1997). De facto gatekeeping and informed consent in intensive care. Med. Law 16, 17–27.
Ciccone, A., Sterzi, R., Crespi, V., Defanti, C. A., and Pasetti, C. (2001). Thrombolysis for acute ischemic stroke: the patient’s point of view. Cerebrovasc. Dis. 12, 335–340.
Dani, K. A., Mccormick, M. T., and Muir, K. W. (2008). Brain lesion volume and capacity for consent in stroke trials: potential regulatory barriers to the use of surrogate markers. Stroke 39, 2336–2340.
Decosta, A., D’Souza, N., Krishnan, S., Chhabra, M. S., Shihaam, I., and Goswami, K. (2004). Community based trials and informed consent in rural north India. J. Med. Ethics 30, 318–323.
Del Giudice, A., Plaum, J., Maloney, E., Kasner, S. E., Le Roux, P. D., and Baren, J. M. (2009). Who will consent to emergency treatment trials for subarachnoid hemorrhage? Acad. Emerg. Med. 16, 309–315.
Demarquay, G., Derex, L., Nighoghossian, N., Adeleine, P., Philippeau, F., Honnorat, J., and Trouillas, P. (2005). Ethical issues of informed consent in acute stroke. Analysis of the modalities of consent in 56 patients enrolled in urgent therapeutic trials. Cerebrovasc. Dis. 19, 65–68.
Dickens, B. M. (1999). Vulnerable persons in biomedical research: 50 years after the Nuremberg Code. J. Int. Bioethique 10, 13–23.
Elkins, J. S., Khatabi, T., Fung, L., Rootenberg, J., and Johnston, S. C. (2006). Recruiting subjects for acute stroke trials: a meta-analysis. Stroke 37, 123–128.
Faden, R. R., Lederer, S. E., and Moreno, J. D. (1996). US medical researchers, the Nuremberg doctors trial, and the Nuremberg Code. A review of findings of the advisory committee on human radiation experiments. JAMA 276, 1667–1671.
Flaherty, M. L., Karlawish, J., Khoury, J. C., Kleindorfer, D., Woo, D., and Broderick, J. P. (2008). How important is surrogate consent for stroke research? Neurology 71, 1566–1571.
Food and Drug Administration. (1996). Protection of human subjects; informed consent–FDA. Final rule. Fed. Regist. 61, 51498–51533.
Goldstein, J. N., Espinola, J. A., Fisher, J., Pallin, D. J., and Camargo, C. A. (2010). Public opinion of a stroke clinical trial using exception from informed consent. Int. J. Emerg. Med. 3, 385–389.
Grotta, J., and Bratina, P. (1995). Subjective experiences of 24 patients dramatically recovering from stroke. Stroke 26, 1285–1288.
Halpern, S. D., Karlawish, J. H., Casarett, D., Berlin, J. A., Townsend, R. R., and Asch, D. A. (2003). Hypertensive patients’ willingness to participate in placebo-controlled trials: implications for recruitment efficiency. Am. Heart J. 146, 985–992.
Halpern, S. D., Metzger, D. S., Berlin, J. A., and Ubel, P. A. (2001). Who will enroll? Predicting participation in a phase II AIDS vaccine trial. J. Acquir. Immune Defic. Syndr. 27, 281–288.
Iwanowski, P., Budaj, A., Czlonkowska, A., Wasek, W., Kozlowska-Boszko, B., Oledzka, U., and Maselbas, W. (2008). Informed consent for clinical trials in acute coronary syndromes and stroke following the European clinical trials directive: investigators’ experiences and attitudes. Trials 9, 45.
Kane, I., Lindley, R., Lewis, S., and Sandercock, P. (2006). Impact of stroke syndrome and stroke severity on the process of consent in the third international stroke trial. Cerebrovasc. Dis. 21, 348–352.
Kasner, S. E., Baren, J. M., Le Roux, P. D., Nathanson, P. G., Lamond, K., Rosenberg, S. L., and Karlawish, J. (2010). Community views on neurologic emergency treatment trials. Ann. Emerg. Med. 57, 346–354.
Kasner, S. E., Del Giudice, A., Rosenberg, S., Sheen, M., Luciano, J. M., Cucchiara, B. L., Messe, S. R., Sansing, L. H., and Baren, J. M. (2009). Who will participate in acute stroke trials? Neurology 72, 1682–1688.
Kilmarx, P. H., Ramjee, G., Kitayaporn, D., and Kunasol, P. (2001). Protection of human subjects’ rights in HIV-preventive clinical trials in Africa and Asia: experiences and recommendations. AIDS 15(Suppl. 5), S73–S79.
Kim, S. Y., and Karlawish, J. H. (2003). Ethics and politics of research involving subjects with impaired decision-making abilities. Neurology 61, 1645–1646.
Kulynych, J., and Korn, D. (2002). The effect of the new federal medical-privacy rule on research. N. Engl. J. Med. 346, 201–204.
Leira, E. C., Kaldjian, L. C., Ludwig, B. R., Torner, J. C., Olalde, H. M., Hacke, W., and Adams, H. P. Jr. (2010). Lack of international consensus on ethical aspects of acute stroke trials. J. Stroke Cerebrovasc. Dis. doi:10.1016/j.jstrokecerebrovasdis.2010.06.008.[Epub ahead of print].
Lynoe, N., Sandlund, M., Jacobsson, L., Nordberg, G., and Jin, T. (2004). Informed consent in China: quality of information provided to participants in a research project. Scand. J. Public Health 32, 472–475.
Lynoe, N., Sandlund, M., Westberg, K., and Duchek, M. (1998). Informed consent in clinical training – patient experiences and motives for participating. Med. Educ. 32, 465–471.
Marshall, P. A. (2006). Informed consent in international health research. J. Empir. Res. Hum. Res. Ethics 1, 25–42.
Morgenstern, L. B., Bartholomew, L. K., Grotta, J. C., Staub, L., King, M., and Chan, W. (2003). Sustained benefit of a community and professional intervention to increase acute stroke therapy. Arch. Intern. Med. 163, 2198–2202.
Morris, M. C., Nadkarni, V. M., Ward, F. R., and Nelson, R. M. (2004). Exception from informed consent for pediatric resuscitation research: community consultation for a trial of brain cooling after in-hospital cardiac arrest. Pediatrics 114, 776–781.
National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research. (1979). Protection of human subjects; Belmont report: notice of report for public comment. Fed. Regist. 44, 23191–23197.
Prentice, E. D. (1999). Informed consent: the most important protector. Acad. Emerg. Med. 6, 774–775.
Salzman, J. G., Frascone, R. J., Godding, B. K., Provo, T. A., and Gertner, E. (2007). Implementing emergency research requiring exception from informed consent, community consultation, and public disclosure. Ann. Emerg. Med. 50, 448–455; 455 e441–444.
Sanossian, N., Starkman, S., Eckstein, M., Stratton, S., Pratt, F., Conwit, R., and Saver, J. L. (2010). Intercontinental elicitation of informed consent for enrollment in stroke research. Cerebrovasc. Dis. 30, 323–324.
Saver, J. L., Kidwell, C., Eckstein, M., Ovbiagele, B., and Starkman, S. (2006). Physician-investigator phone elicitation of consent in the field: a novel method to obtain explicit informed consent for prehospital clinical research. Prehosp. Emerg. Care 10, 182–185.
Saver, J. L.The FAST-MAG Investigators. (2011). Patient versus proxy explicit informed consent for enrollment in a prehospital stroke treatment trial, International Stroke Conference Oral Presentations. Stroke 42, e58.
Saver, R. S. (1996). Critical care research and informed consent. North Carol. Law Rev. 75, 205–271.
Schats, R., Brilstra, E. H., Rinkel, G. J., Algra, A., and Van Gijn, J. (2003). Informed consent in trials for neurological emergencies: the example of subarachnoid haemorrhage. J. Neurol. Neurosurg. Psychiatr. 74, 988–991.
The National Institute of Neurological Disorders and Stroke Rt-PA Stroke Study Group. (1995). Tissue plasminogen activator for acute ischemic stroke. N. Engl. J. Med. 333, 1581–1587.
Tu, J. V., Willison, D. J., Silver, F. L., Fang, J., Richards, J. A., Laupacis, A., and Kapral, M. K. (2004). Impracticability of informed consent in the registry of the Canadian stroke network. N. Engl. J. Med. 350, 1414–1421.
World Medical Association. (2000). Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 284, 3043–3045.
Keywords: informed consent, acute stroke, cerebral infarction, clinical trial
Citation: Rose DZ and Kasner SE (2011) Informed consent: the rate-limiting step in acute stroke trials. Front. Neur. 2:65. doi: 10.3389/fneur.2011.00065
Received: 16 August 2011; Paper pending published: 08 September 2011;
Accepted: 21 September 2011; Published online: 17 October 2011.
Edited by:
Yuko Y. Palesch, Medical University of South Carolina, USAReviewed by:
Ru-Lan Hsieh, Shin Kong Wu Ho-Su Memorial Hospital, TaiwanNerses Sanossian, University of Southern California, USA
Copyright: © 2011 Rose and Kasner. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Scott E. Kasner, Stroke Division, Department of Neurology, University of Pennsylvania, 3 West Gates Building, 3400 Spruce Street, Philadelphia, PA 19104, USA. e-mail: kasner@mail.med.upenn.edu