
- 1C. Eugene Bennett Department of Chemistry, West Virginia University, Morgantown, WV, USA
- 2NanoSAFE, West Virginia University, Morgantown, WV, USA
- 3The Center for Neurosciences, West Virginia University, Morgantown, WV, USA
There are a vast number of neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD), associated with the rearrangement of specific proteins to non-native conformations that promotes aggregation and deposition within tissues and/or cellular compartments. These diseases are commonly classified as protein-misfolding or amyloid diseases. The interaction of these proteins with liquid/surface interfaces is a fundamental phenomenon with potential implications for protein-misfolding diseases. Kinetic and thermodynamic studies indicate that significant conformational changes can be induced in proteins encountering surfaces, which can play a critical role in nucleating aggregate formation or stabilizing specific aggregation states. Surfaces of particular interest in neurodegenerative diseases are cellular and subcellular membranes that are predominately comprised of lipid components. The two-dimensional liquid environments provided by lipid bilayers can profoundly alter protein structure and dynamics by both specific and non-specific interactions. Importantly for misfolding diseases, these bilayer properties can not only modulate protein conformation, but also exert influence on aggregation state. A detailed understanding of the influence of (sub)cellular surfaces in driving protein aggregation and/or stabilizing specific aggregate forms could provide new insights into toxic mechanisms associated with these diseases. Here, we review the influence of surfaces in driving and stabilizing protein aggregation with a specific emphasis on lipid membranes.
Introduction
A common motif of several neurodegenerative diseases is the ordered aggregation of specific proteins, leading to their deposition in tissues or cellular compartments (Chiti and Dobson, 2006). Often referred to as protein conformational or misfolding disorders, such diseases include Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyloidoses, α1-antitrypsin deficiency, and the prion encephalopathies to name a few. The common structural motif of protein aggregates associated with these diseases is the formation of extended, β-sheet rich fibrils, referred to as amyloid. Despite no apparent correlation between aggregating proteins in size or primary amino acid sequence, the characteristic lesions of each disease typically contain fibrillar structures with common biochemical characteristics (Dobson, 2003; Chiti and Dobson, 2006), indicating the potential for a conserved mechanism of pathogenesis linking these phenotypically diverse diseases. The earliest potential event in the disease process may be the conversion of a protein to a critical abnormal conformation, resulting in toxic gain of function for the monomer, and/or the formation of toxic nanoscale aggregates (Figure 1; Naeem and Fazili, 2011). The elusive toxic species, whether monomeric or higher-order, may subsequently initiate a cascade of pathogenic protein–protein interactions that culminate in neuronal dysfunction. The precise timing of such interactions and the mechanisms by which altered protein conformations or aggregates trigger neuronal dysfunction are unclear.
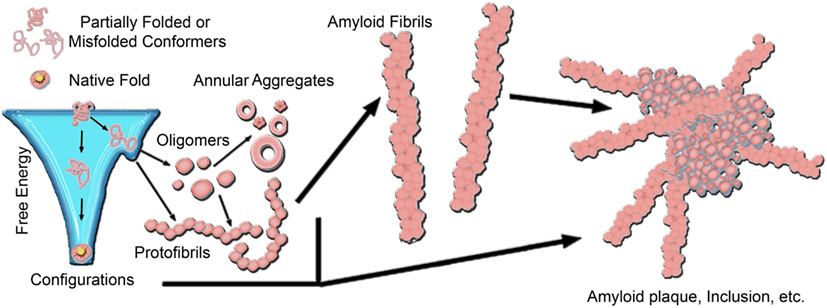
Figure 1. A generic aggregation scheme for amyloid-forming proteins. Proteins fold into their native structure, which is typically a low free energy configuration. However, the energy landscape for protein folding often can have localized minima in which a protein can become trapped into a misfolded conformation, which can lead to aggregation into β-sheet rich amyloid fibrils. The formation of fibrils often proceeds through a heterogeneous mixture of intermediate species, including oligmers and protofibrils. Off-pathway aggregates can also form, such as annular aggregates. These aggregates accumulate into amyloid plaques or inclusions in the diseased brain. The aggregation pathway for any given amyloid-forming protein can vary considerably depending on the protein and its folding environment.
The formation of fibrils often proceeds via a heterogeneous mixture of intermediate aggregate structures, including a variety of protofibrils and oligomers (Figure 1). Amyloid formation typically occurs via a nucleation-growth mechanisms that features an initial lag-phase due to a thermodynamically unfavorable nucleation event (Lomakin et al., 1996; Murphy, 2002; Chiti and Dobson, 2006). Once nucleation occurs, aggregation proceeds via an exponential growth phase associated with the addition of monomers into aggregate forms. The initial lag-phase can be circumvented by the presence of pre-existing aggregates that can act as seeds for amyloid formation (Lansbury, 1997; Hu et al., 2009; Langer et al., 2011; Hamaguchi et al., 2012). To further complicate the issue, several aggregates have been identified that may be off-pathway to fibril formation, such as annular structures (Wetzel, 1994; Wacker et al., 2004). While there are often specific mutations or dysfunctional processing that can be directly linked to aggregation, the nature, and location of protein aggregates in vivo depends on the specific protein associated with disease. The specific protein involved also influences the specific form of the critical aggregation nucleus. For example, synthetic polyglutamine (polyQ) peptides are thought to have a monomeric critical nucleus (Chen et al., 2002a,b; Wetzel, 2012); however the addition of flanking sequences associated with the first exon of the huntingtin (htt) protein can change the size of the critical nucleus to a tetramer (Jayaraman et al., 2012; Wetzel, 2012). This can be further modulated by the addition of β-hairpin motifs within the polyQ domain (Kar et al., 2013). The extent of the lag-phase, and subsequent aggregation of polyQ peptides and htt proteins is dependent on the size of the polyQ domain (Legleiter et al., 2010; Kar et al., 2011). As protein aggregation often progresses from misfolded monomers to oligomeric precursors and finally mature fibrils, intensive research activity has been devoted to determining the most toxically relevant aggregate species in many of these diseases. This is particularly important, as for the vast majority of these diseases, there are no widely effective preventative measures or therapeutic treatments.
Fibril structures associated with several different amyloid-forming proteins have been experimentally resolved, and a common motif of fibrillar aggregates is a cross-β structure (Eanes and Glenner, 1968; Glenner et al., 1971; Kirschner et al., 1986; Sunde et al., 1997; Berriman et al., 2003; Tycko and Ishii, 2003; Tycko, 2004, 2006; Nelson et al., 2005; Fandrich, 2007). While the structural spine of fibrils share this common intermolecular β-sheet structure, a variety of possibilities are available for the packing of protofilaments into the fibril structure, even for the same protein/peptide. This variability can lead to distinct amyloid fibril morphologies. Such variable protofilament arrangements give rise to distinct fibril morphologies, often termed polymorphisms (Kodali and Wetzel, 2007). For example, Aβ has been shown to form a variety of fibril structures in vitro dependent on the peptide preparation and aggregation conditions (Kodali et al., 2010). Furthermore, fibril polymorphs have been observed for several other amyloid-forming proteins, such as calcitonin (Bauer et al., 1995), amylin (Goldsbury et al., 1997), glucagon (Pedersen et al., 2006), the SH3 domain of phosphatidylinositol-3′-kinase (Jimenez et al., 1999; Chamberlain et al., 2000; Pedersen et al., 2006), insulin (Bouchard et al., 2000; Jimenez et al., 2002; Dzwolak et al., 2004), and lysozyme (Chamberlain et al., 2000). Polymorphic fibrils can differ in the cross-sectional thickness or helical pitch of the fibril, which can be observed via high resolution imaging techniques like transmission electron microscopy (TEM) and atomic force microscopy (AFM) or distinguished with spectroscopic techniques like circular dichroism (CD; Petkova et al., 2005; Kurouski et al., 2010, 2012; Mossuto et al., 2010; Norlin et al., 2012). While polymorphs are often observed for various in vitro aggregation reactions, polymorphs have been observed in amyloid fibrils extracted from tissue as well (Crowther and Goedert, 2000; Jimenez et al., 2001), affirming that in vivo aggregation can be heterogeneous and complex. Furthermore, it has been proposed that polymorphic fibrils may result in distinct biological activities and variable toxicity related to the different aggregate structures (Seilheimer et al., 1997; Petkova et al., 2005). These distinct fibril morphologies may also have distinct aggregate intermediates associated with their formation, adding to the heterogeneity of potential protein aggregates and further complicating efforts aimed at elucidating the relative role of discrete aggregates in disease-related toxicity.
While protein preparation and environment influence the structural polymorphs of protein aggregates in vitro, determining what environmental factors influence aggregation in vivo remains difficult. However, the interaction of proteins at solid interfaces, including cellular membranes comprised of lipid bilayers, may prove to be a fundamental phenomenon with potential implications for protein-misfolding diseases. Solid surfaces, such as mica, graphite, gold, and Teflon, have been shown to heavily influence aggregation kinetics and the resulting aggregate morphology for a variety of amyloid-forming proteins (Goldsbury et al., 1999; Hoyer et al., 2004; Morriss-Andrews and Shea, 2012). A variety of kinetic and thermodynamic studies point to significant conformational changes being induced in proteins encountering surfaces (Gray, 2004). These surface induced conformational changes in proteins could play a critical role in nucleating amyloid formation or altering aggregate morphology to specific toxic species. Such phenomenon are well demonstrated by a study of immunoglobulin light-chain aggregation on mica (Zhu et al., 2002). Small pieces of mica were incubated in solutions containing a recombinant amyloidogenic light-chain variable domain of smooth muscle actin (SMA) antibody, under conditions in which fibrils normally do not form (i.e., low concentration and no agitation). At short times, amorphous aggregates appeared on mica, and fibrils were observed within 10 h and fibrils were not formed in the solution within the same time frame. The fibrils on the surface of mica grew from the amorphous aggregates and the assemblies of oligomers present on mica. The use of such solid surfaces as model systems provides the opportunity to elucidate how specific surface environment influence protein aggregation.
In regards to disease-related protein aggregation, surfaces of more physiological relevance are cellular and subcellular membranes that are predominately comprised of lipid bilayers. Like solid surfaces, the presence of lipid membranes can alter the aggregation of disease-related proteins by increasing aggregation rates, nucleating aggregation, promoting specific polymorphs, or even stabilizing potentially toxic, transient aggregate intermediates. A significant question remains regarding why amyloid fibrils form in vivo at concentrations that are orders of magnitude lower (Seubert et al., 1992) than the critical nucleation concentrations required in vitro (Lomakin et al., 1996; Sabate and Estelrich, 2005). A possible answer is the ability to create local concentrations of protein adsorbed onto molecular surfaces, such as cellular and subcellular membranes (Kim et al., 2006; Aisenbrey et al., 2008). Lipid interaction appears to be a common modulator in fibril formation, as studies of α-synuclein (α-syn; Jo et al., 2000, 2004; Necula et al., 2003), islet amyloid polypeptide (IAPP; Knight and Miranker, 2004), and β-amyloid (Aβ; McLaurin and Chakrabartty, 1996, 1997; Choo-Smith et al., 1997; Yip and McLaurin, 2001; Yip et al., 2002) all demonstrate accelerated fibril formation in a membrane environment in comparison to bulk solution. General physicochemical properties of lipid membrane, including phase state, bilayer curvature, elasticity, and modulus, surface charge, and degree of hydration, modulate protein aggregation (Gorbenko and Kinnunen, 2006). The exact chemical composition and lipid constituents of a lipid bilayer can also influence the aggregation process (Evangelisti et al., 2012). Potentially important chemical properties of membrane components include the extent of acyl chain unsaturation, conformation and dynamics of lipid headgroups and acyl chains, and protein–lipid selectivity arising from factors such as the hydrophobic matching at the protein–lipid interface (Jensen and Mouritsen, 2004). Although lipid bilayers may act catalytically to induce aggregation by providing environments that promote protein conformation and orientation conducive to fibril assembly (Thirumalai et al., 2003; Sparr et al., 2004; Zhao et al., 2004), cell membranes may also be targeted by protein aggregates to induce physical changes in the membrane, leading to dysfunction and cell death. This may be due to the ability of amyloid-forming peptides to induce membrane permeabilization by altering bilayer structure via the sequestration of membrane components into fibrils (Michikawa et al., 2001; Lins et al., 2002; Sparr et al., 2004; Zhao et al., 2004; Valincius et al., 2008) or by forming unregulated pore-like structures (Jang et al., 2007). A variety of amyloid-forming proteins, including Aβ, IAPP, and htt, have been show to locally change the rigidity of model lipid bilayers in a generic manner (Burke et al., 2013). Furthermore, the presence of lipid membranes can also influence the ability of small molecules to prevent or destabilize protein aggregates, having a major impact on several therapeutic strategies. Such a scenario has been demonstrated experimentally as (-)-epigallocatechin gallate (EGCG), which has been shown to inhibit the aggregation of several amyloid-forming proteins in the absence of surfaces (Bieschke et al., 2010; Popovych et al., 2012), was less effective at inhibiting aggregation of human IAPP at a phospholipid interface (Engel et al., 2012).
Here, we review the influence of surfaces in driving and stabilizing protein aggregation with a specific emphasis on lipid membranes. We will initially focus on Aβ as an illustrative example, and then quickly review some interesting features of the interaction of other select amyloid-forming proteins with surface interfaces.
The Aggregation of Aβ on Solid Surfaces
The ordered aggregation of Aβ into neuritic plaques is one of the major hallmarks of AD. Aβ is a secreted peptide derived from the endoproteolysis of the amyloid precursor protein (APP), a receptor-like transmembrane protein, and is ubiquitously expressed in neural and non-neural cells. Successive cleavage of APP by β-secretase and γ-secretase results in the release of an intact Aβ peptide. Aβ contains a portion of APP’s transmembrane domain, as well as an extracellular portion, resulting in an amphiphilic peptide ∼39–43 residues in length. The Aβ component of amyloid plaques found in the diseased brain consist primarily of two versions of the peptide, which are 40 and 42 amino acids long [Aβ(1–40) and Aβ(1–42) respectively]. Aβ(1–42) aggregates more quickly than Aβ(1–40) and is thought to play a major role in AD (Jarrett and Lansbury, 1993). The amphiphilic nature of Aβ is thought to drive its aggregation and may play an important role in its interaction with solid surfaces and ability to insert and/or penetrate lipid membranes (Lansbury and Lashuel, 2006; Williams and Serpell, 2011). The extra addition of two hydrophobic residues in Aβ(1–42) may also lead to variations in the interaction of this peptide with surfaces compared to Aβ(1–40).
Hydrophobic Teflon surfaces can be considered mimics of the non-polar plane of lipid membranes. While both Teflon and Aβ carry a negative charge at physiological pH, protein dehydration effects lead to substantial adsorption of Aβ at pH 7(Giacomelli and Norde, 2003). Aβ(1–40) and Aβ(1–42) adsorption to Teflon particles increased aggregation and fibrillogenesis (Linse et al., 2007). Adsorption of Aβ to another hydrophobic surface, highly ordered pyrolytic graphite, results in extended aggregate formation in a nucleation dependent manner (Kowalewski and Holtzman, 1999). Using a variety of surfaces with tunable hydrophobicity or hydrophilicity (as well as supported lipid bilayers), weakly adsorbed peptides with two-dimensional diffusivity were found to be critical precursors to surface growth of Aβ(1–42) fibrils (Shen et al., 2012). As the adsorption of Aβ on highly hydrophilic surfaces was negligible, fibril growth was inhibited on such surfaces. On highly hydrophobic surfaces, the two-dimensional diffusion of Aβ along the surface was too low, also inhibiting fibril formation. It appears that surface properties that promote weak adsorption of Aβ to the surface and maintain translational mobility result in local concentrations of Aβ due to confinement within the plane of the surface, allowing for fibril formation at a concentration far below the critical concentration observed in bulk solutions. The adsorption of Aβ to hydrophilic silica surfaces is pH dependent, occurring at pH 4 and 7 when Aβ has an overall positive charge (Giacomelli and Norde, 2005), suggesting a vital role of electrostatics on Aβ‘s adsorption to surfaces.
Due to the ability of AFM to be operated in solution and track the formation and fate of individual aggregates with time on surfaces (Goldsbury et al., 1999), the impact of surface chemistry on the morphology of Aβ aggregates has been extensively studied with this technique. On mica, a hydrophilic surface, Aβ(1–40) (Blackley et al., 2000) and Aβ(1–42) (Kowalewski and Holtzman, 1999) form small, highly mobile oligomeric aggregates that organize into extended pre-fibrillar aggregates that continually elongate with time (Figure 2A). These aggregate structures are similar in morphology to those formed in bulk solution from similarly prepped Aβ stocks (Kowalewski and Holtzman, 1999; Legleiter and Kowalewski, 2004). However, Aβ(1–42) aggregates into morphologically distinct structures on a graphite surface (Figure 2B), forming extended nanoribbons with heights of ∼1–1.2 nm and widths of ∼18 nm (Kowalewski and Holtzman, 1999). These dimensions suggest that Aβ adopts a fully extended β-sheet conformation perpendicular to the long axis of the nanoribbons. These nanoribbons elongated with time, organize themselves into parallel, raft-like structures with a preferential alignment along the graphite lattice. Aβ adsorbs to and aggregates on surfaces functionalized with methyl, carboxyl, or amine groups; however, aggregate morphology and surface affinity is dependent on the specific surface chemistries (Moores et al., 2011). Hydrophobic surfaces promote formation of spherical amorphous clusters; charged surfaces promote the formation of protofibrils (Moores et al., 2011). Studies of the aggregation of Aβ peptides containing single point mutations on mica further support the notion that electrostatics play an important role in Aβ adsorption and aggregation on surfaces (Yates et al., 2011). These mutations are clustered around the central hydrophobic core of Aβ (E22G Arctic mutation, E22K Italian mutation, D23N Iowa mutation, and A21G Flemish mutation) and are associated with familial forms of AD. In bulk solution and under identical preparatory conditions, these Aβ mutants form aggregated species that were morphologically similar to those of Wild Type Aβ; however, on a mica surface the aggregates differ in morphology (Figure 3). While Wild Type Aβ forms oligomers and putative protofibrils on mica similar to other previously described studies, Arctic Aβ aggregate into extended, fibrillar aggregates on mica that orient on the surface similar to the previously described Wild Type Aβ aggregates on graphite. However, the dimensions of the Arctic Aβ aggregates on mica indicate they most likely contain a β-turn as opposed to the fully elongated Wild Type Aβ nanoribbons on graphite. Italian Aβ, which replaces a negatively charged residue with a positive one, adsorbs quickly to mica and predominantly forms oligomeric aggregates reminiscent of those formed by wild type Aβ on mica. However, there was a small percentage of Italian Aβ aggregates similar in morphology to those formed by Arctic Aβ on mica.
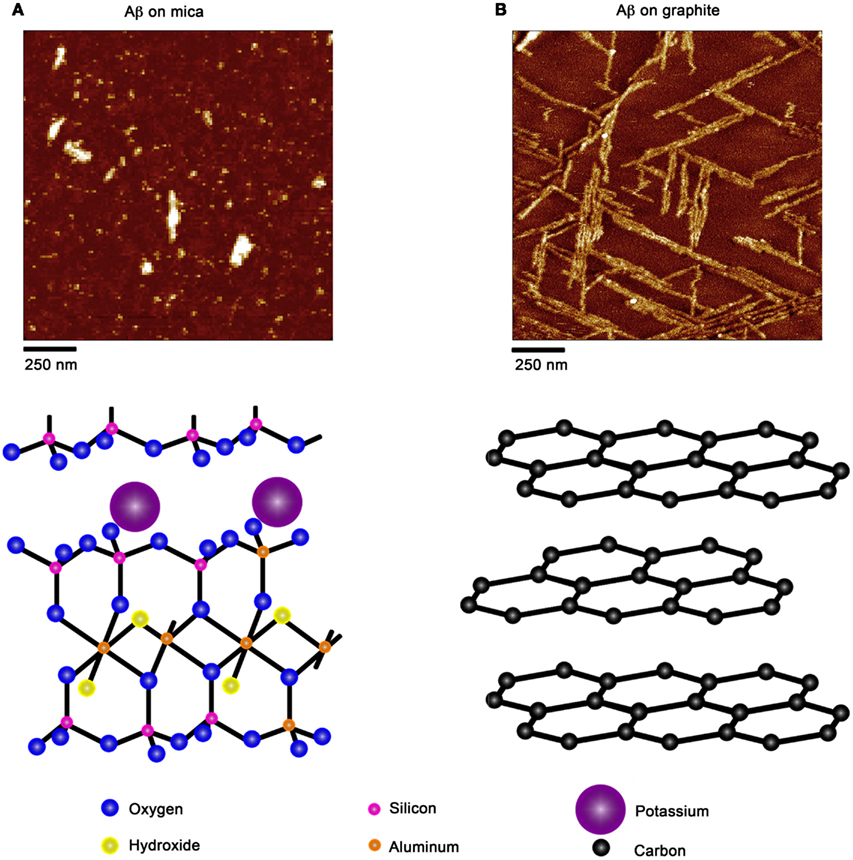
Figure 2. Aβ aggregation is modulated by the presence of chemically distinct solid surfaces. (A) On highly ordered pyrolytic graphite, Aβ aggregates into extended nanoribbons that are epitaxially ordered on the surface. The distinct orientation of Aβ aggregates on graphite is attributed to the optimization of the contact between the peptide and underlying hydrophobic carbon lattice. (B) On a negatively charged, hydrophilic mica surface, Aβ forms discrete oligomers that maintained some lateral mobility along the plane of the surface. These oligomers could organize into elongated protofibrillar structures. Schematic representations of the structure of each surface (graphite and mica) are provided under each image.
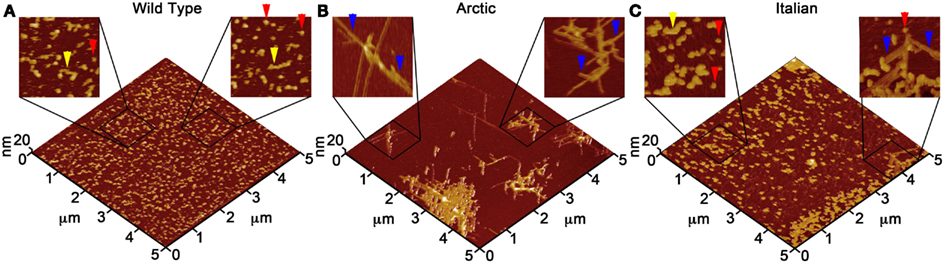
Figure 3. Point mutations in Aβ(1–40) modulate aggregate morphology in the presence of a mica surface. Using solution AFM, the aggregation of Wild Type, Arctic (E22G), and Italian (E22K) Aβ was monitored on a mica surface (Aβ concentration was 20 μM for all experiments). 5 μm × 5 μm images are presented in 3D with indicated zoomed in areas of 1 μm × 1 μm shown in 2D. (A) Wild Type Aβ formed a large population of oligomers (red arrows) and highly curved, elongated protofibrils (yellow arrows) with aggregate heights of ∼3–5 nm similar to presented in Figure 2. (B) Arctic Aβ formed rigid, branched, and highly ordered fibrillar aggregates (blue arrows) along the crystallographic lattice of mica with aggregate heights of ∼2–5 nm along the contour. These Arctic Aβ aggregates morphologically distinct from those formed by Wild Type Aβ. (C) Italian Aβ predominately aggregated into small oligomers (2–3 nm tall, red arrows) that coalesced into larger protofibrils (yellow arrows), in a similar fashion to Wild Type Aβ; however, a small number of rigid, elongated “Arctic-like” fibrillar aggregates of Italian Aβ also formed (blue arrow).
The Interaction of Aβ with Lipid Surfaces
While studies on model surfaces can provide mechanistic detail on how solid interfaces alter and/or promote Aβ aggregation, ultimately, pathological protein aggregation occurs in a cellular environment, dictating the need to study protein-misfolding and aggregation on more physiologically relevant surfaces. This is not to say that studies on solid surfaces are irrelevant. For example, the aforementioned dependence on lateral mobility of Aβ on a surface being critical in fibril formation was directly extended to lipid surfaces as well (Shen et al., 2012). This phenomenon could be important in light of single molecule studies demonstrating that Aβ inserted into anionic lipid membranes demonstrate high lateral mobility until aggregating into oligomers (King et al., 2012).
It has been hypothesized that a potential pathway for Aβ toxicity may lie in its ability to modulate lipid membrane function. This hypothesis is based on the observation that Aβ bears a portion of the APP transmembrane domain. Thus, elucidating the interaction between Aβ and membrane lipids could be critical in understanding potential pathways of Aβ toxicity, especially given the results of studies that demonstrate that changes in membrane composition occur in AD along with the association with plaques, tangles, and neuritic dystrophy. Importantly, it has often been observed that exogenously added Aβ will selectively bind a subset of cells in an apparent homogenous population of cells in culture (Lacor et al., 2004; De Felice et al., 2008). Such an initial cellular binding event may play a critical role in toxic mechanisms and cell to cell propagation of disease. This cell selectivity may be influenced by the presence of specific lipid components or membrane properties (Okada et al., 2007; Wakabayashi and Matsuzaki, 2007; Lin et al., 2008). Once Aβ aggregation begins in or near a membrane, the potential toxic mechanism include disruption of the bilayer structure, changes in bilayer curvature, and/or the creation of membrane pores or channels (Arispe et al., 1993a,b; McLaurin and Chakrabartty, 1996, 1997; Mirzabekov et al., 1996; Gorbenko and Kinnunen, 2006; Figure 4). The majority of studies on membrane-mediated fibrillogenesis have been undertaken with model systems including amyloidogenic peptides or proteins and lipid vesicles or supported bilayers of varying composition (Terzi et al., 1997; Lindstrom et al., 2002; Bokvist et al., 2004; Sparr et al., 2004). These studies often point to the importance of the chemical nature of membrane lipids and the mode of protein–lipid interactions in determining fibrillogenic properties of membrane bound Aβ. Lipids can also stabilize toxic protofibrils and even revert mature fibrils into such toxic species (Martins et al., 2008), providing another potential role for lipid surfaces in toxicity.
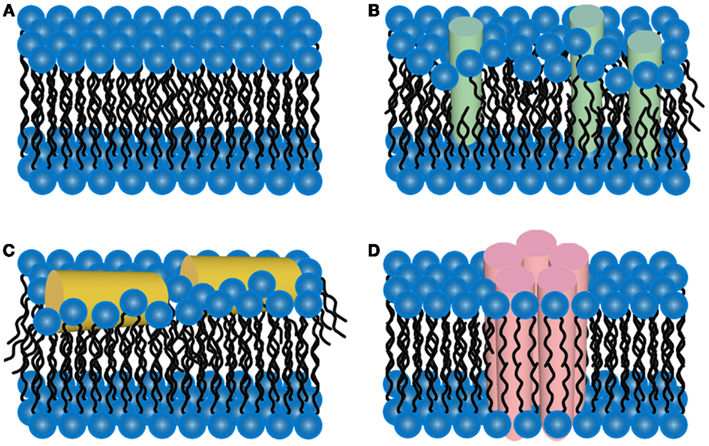
Figure 4. Schematic representations of potential mechanisms of amyloid/lipid association. (A) A schematic representation of simplified, undisrupted bilayer is presented. This bilayer structure can be perturbed by (B) amyloid-protein insertion or (C) association of amphiphilic α-helices lipid-binding domains. Such scenarios could lead to membrane thinning and non-specific membrane leakage. (D) Many amyloid-forming proteins have been shown to form pore-like structures that can act as unregulated ion-selective channels.
A large number of biophysical techniques have been applied in understanding the specific interactions between lipid membranes and Aβ. Due to the ability to control bilayer composition, biomimetic unilamellar vesicles have been extensively used to elucidate the interaction between Aβ and membranes (Williams et al., 2010). Simple vesicles comprised of a single lipid component, soybean PC, have been used to demonstrate that the presence of neutral PC delays the characteristic lag time to initiate Aβ aggregation in a lipid concentration-dependent manner (Sabate et al., 2005). Lipids can also induce changes in the secondary structure of Aβ, as CD studies demonstrated that a variety of lipids induce a transition from an α-helical to β-sheet structure in Aβ(McLaurin and Chakrabartty, 1997). As with solid surfaces, the charge of the lipid membrane surfaces, determined by the headgroups of phospholipids, dictate the extent of Aβ/membrane association due to electrostatic considerations. For example, similarly prepared Aβ(1-40) displays a stronger affinity to liposomes comprised of POPG compared to those comprised of POPC, with only the association with POPG enhancing the rate of Aβ aggregation (Kremer and Murphy, 2003). Freshly prepared Aβ(1–40) preferentially binds negatively charged PG membranes and composite membranes containing negatively charged lipids in comparison to neutral membranes; however, the relative affinity for fibril aggregates of Aβ with these lipid membranes is altered (Lin et al., 2007). Allowing Aβ(1–40) to form fibrils causes the affinity for negatively charged membranes to be smaller compared to the affinity for neutral membranes, suggesting that Aβ aggregation state can further modulate the interaction with lipid surfaces.
A potential mechanism for amyloid-forming proteins, such as Aβ, is their ability to alter membrane structure and integrity, leading to permeation of cellular membranes (Figure 4). Detergent-like effects arise from the amphiphilic nature of Aβ, leading to reduced membrane surface tension leading to membrane thinning and hole formation (Hebda and Miranker, 2009). Several AFM studies performed in solution have provided valuable insight into the aggregation of Aβ on a variety of model lipid membranes, leading to altered membrane morphology. The interaction of Aβ(1–40) with bilayers formed from total brain lipid extract (TBLE) revealed that Aβ(1–40) will partially insert into bilayers, growing into small fibers (Yip and McLaurin, 2001). In the same study, larger fiber-like structures associated with disruption of the bilayer morphology and integrity were observed as measured by increased surface roughness and formation of holes, respectively. Large fibrils were often highly branched and associated with edges of disrupted bilayer. The TBLE bilayers also aided in nucleation and enhancement of fibril growth. Interestingly, preformed fibrils were not capable of disrupting the TBLE bilayers, which may indicate that the act of aggregation, that is pre-fibrillar aggregates, may be key in Aβ-induced membrane disruption. Similar experiments exposing DMPC bilayers to Aβ(1–40) resulted in the formation of globular aggregates that were associated with small holes in the bilayer, whereas, fibril growth and/or extensive bilayer disruption was not observed. Aβ(1–42) demonstrated a different interaction/aggregation pattern on TBLE bilayers (Yip et al., 2002). Discrete molecules of Aβ(1–42) could be detected on the surface that were replaced by distinctly larger aggregates with time. However, bilayer defects were rarely detected upon exposure to Aβ(1–42). Point mutations in Aβ(1–40) also altered the aggregation on and ability to disrupt TBLE bilayers (Figure 5; Pifer et al., 2011). These same point mutations were shown to cause polymorphic aggregation of Aβ on mica. Aggregation in the presence of TBLE bilayers resulted in a variety of polymorphic aggregates in a mutation dependent manner and a variable ability to disrupt bilayer morphology/integrity. Such results highlight the potential role electrostatic and hydrophobic properties of Aβ play in its ability to bind, insert, and potentially disrupt lipid membranes.

Figure 5. Point mutations in Aβ influence peptide aggregation in the presence of total brain lipid bilayers. Using solution AFM, aggregation of Wild Type, Arctic (E22G), or Italian (E22K) Aβ in the presence of supported TBLE bilayers was monitored (Aβ concentration was 20 μM for all experiments). 3D images are presented (4 μm × 4 μm and 6 μm × 6 μm) with indicated zoomed in areas of 1 μm × 1 μm and 2 μm × 2 μm shown in 2D. (A) With time, Wild Type Aβ aggregated into discrete oligomers and fibrils that were associated with regions of the bilayer with perturbed morphology (an increase in surface roughness). (B) While many small oligomers of Arctic Aβ were observed on the bilayer, highly curved fibrils that were associated with membrane disruption were the dominant aggregate species. These Arctic Aβ fibrils were morphologically distinct from fibrils observed for Wild Type Aβ. (C) While Italian Aβ also formed similar oligomers compared Wild Type and Arctic Aβ, large patches of disrupted bilayer morphology developed that may be associated with distinct fibril aggregates.
Another proposed toxic mechanism points to Aβ‘s ability to alter cellular ion concentrations, calcium in particular, through the formation of membrane pores (Figure 4D). Initial evidence for this scenario came from the observation that PS bilayers that had Aβ(1–40) directly incorporated into them displayed linear current/voltage relationships in symmetrical solutions (Arispe et al., 1993a). Further evidence for this scenario was provided by studies on phospholipid vesicles that had either Aβ(1–42) (Rhee et al., 1998) or Aβ(1–40) (Lin et al., 1999) directly incorporated into them. In both cases, these vesicles stiffened in the presence of calcium, due to calcium ion-induced charge–charge repulsion inside the vesicles, binding of calcium to lipids and proteins, and an enhanced efficiency of lipid–protein interactions. This increased stiffness of the vesicles could be blocked by pretreatment with anti-Aβ antibodies, Tris, or zinc, all of which would block putative calcium channels. Reconstituting Aβ(1–42) with a planar lipid bilayer resulted in the formation of multimeric channel-like structures with symmetries suggesting tetramer or hexamer pore-like structures of Aβ(Lin et al., 2001; Quist et al., 2005). The formation of a variety of similar aggregate structures in lipid membranes have also been demonstrated computationally (Capone et al., 2012; Tofoleanu and Buchete, 2012).
Similar impacts on membranes due to exposure to Aβ have been detected in cellular models. Cells exposed to Aβ(1–40), Aβ(1–42), and Aβ(25–35) on endothelial cells undergo morphological changes and cell disruption, with the highest sensitivity to Aβ(1–42) (Zhu et al., 2000). While cell disruption was induced by nanomolar concentrations of Aβ(1–42), micromolar concentrations of Aβ(1–40) were required to trigger similar effects. Similar observations were reported for fibroblasts in the presence of nanomolar Aβ(1–42), as morphological changes along the periphery of the cell were observed that could be blocked by anti-Aβ antibodies, zinc, and the removal of calcium (Zhu et al., 2000). Protofibrils and low molecular weight oligomers of Aβ can alter the electrical activity of neurons and reproducibly induced toxicity in mixed brain cultures in a time- and concentration-dependent manner, suggesting changes in membrane integrity and depolarization (Hartley et al., 1999). Aβ peptides induce ion channel-like ion flux in model lipid membranes and neuronal membranes independent from the ability of Aβ to modulate intrinsic cellular ion channels or transporter proteins (Capone et al., 2009) Even intracellular forms of Aβ can alter the electrophysiological properties of cultured human primary neurons (Hou et al., 2009). Aβ(1–42) oligomers form single ion channel permeable to Ca2+ in oocytes are highly toxic and not attributable to endogenous oocyte channels (Demuro et al., 2011).
The discussed potential mechanism of Aβ toxicity associated with lipid membranes are not exhaustive, nor are they mutually exclusive. Aβ-induced membrane disruption of POPC, POPOC/POPS/gangliosides, and TBLE systems occurred in a two-step process (Sciacca et al., 2012). The initial step involved the formation of ion-selective pores, followed by non-specific fragmentation of the lipid membrane due to fibrillization. This demonstrates that different mechanism of membrane disruption may be associated with specific stages of aggregation. Large unilamellar lipid vesicles (LUVs) encapsulating self-quenching fluorescent dyes can be used as reporters of membrane disruption and leakage. Such systems have been used to elucidate the ability of Aβ to disrupt membrane integrity. Upon exposure of LUVs comprised of DMPC and containing calcein to Aβ(1–42) oligomers, the LUV structure is disrupted, allowing leakage of the dye, but preformed fibrils have a decreased ability to disrupt LUVs (Williams et al., 2010). However, the oligomers that interacted with the DMPC LUVs formed fibrils, suggesting that the aggregation process may actually play a role in membrane disruption.
A variety of studies indicate the ability of Aβ to bind membranes is highly dependent on the presence of specific lipid components, i.e., cholesterol (Yip et al., 2001; Reiss et al., 2004; Yu and Zheng, 2012), sphingolipids (van Echten-Deckert and Walter, 2012), gangliosides (McLaurin and Chakrabartty, 1996), and neutral or charged phospholipids (McLaurin and Chakrabartty, 1997; Sabate et al., 2005, 2012). This may be due to specific chemical/electrostatic interactions between membrane components and Aβ and/or the mechanical properties of the bilayer associated with their specific composition. For example, altering the cholesterol content of supported TBLE bilayers changes Aβ aggregation on membranes. Aβ(1–40) induced bilayer disruption and its ability to form fibrils on the bilayer was strongly dependent on cholesterol content of the supported bilayers (Yip et al., 2001). Cholesterol depletion of bilayers inhibited the ability of Aβ(1–40) to perturb bilayer structure. When Aβ(1–40) was added to TBLE bilayers that had been enriched with 10% exogenous cholesterol, discrete Aβ(1–40) peptides appeared on the bilayer within ∼30 min. Eventually, ring-like Aβ(1–40) structures with diameters of 55–80 nm as well as short fibrils and small aggregates were observed on the cholesterol enriched bilayer, but no membrane disruption was observed. At higher cholesterol content (30% of the total lipid), these Aβ(1–40) aggregates were not observed. The ability of Aβ(1–40) to disrupt the TBLE bilayers with varying amounts of cholesterol correlated with bilayer fluidity, indicating that decreased fluidity (modulated by cholesterol content) of the membrane somehow enhanced the interaction between the bilayer and Aβ. Simulation of POPC bilayers containing different mole fractions of cholesterol demonstrate that cholesterol induces changes in bilayer properties, i.e., membrane structure, dynamics, and surface chemistry, that cause increased bilayer thickness, ordering of hydrophobic chains, surface hydrophobicity, and decreased lipid mobility (Yu and Zheng, 2012). These effects promoted the binding of Aβ(1–42) to the model POPC lipid bilayers.
Cholesterol is also critical in the insertion of oligomeric forms of Aβ(1–42) into POPC membranes (Ashley et al., 2006). With DOPC model bilayers, the addition of cholesterol acts as a target for the binding of Aβ to the membrane (Drolle et al., 2012). AFM studies further illustrate that the alteration of bilayer mechanical properties induced by lipid composition impact the ability of Aβ to bind membranes, by demonstrating that astrocyte secreted lipoprotein particles containing different isoforms of apolipoprotein E (apoE), of which the apoE4 allele is a major risk factor for the development of AD, protect TBLE bilayers from Aβ(1–40) induced disruption (Legleiter et al., 2011). The apoE4 allele was less effective in protecting these bilayers from Aβ(1–40) compared with their apoE3 counterparts, and further analysis revealed that this was due to the varying ability of the lipoprotein particles containing different alleles of apoE to modulate the fluidity of bilayers by acquiring bilayer components (most likely cholesterol and/or oxidatively damaged lipids). There is evidence that peptide/membrane affinity in vascular cells can also be related to the ability of cholesterol to modulate membrane fluidity and structure (Subasinghe et al., 2003). Other cell culture assays, using PC-12 and SH-SY5Y cells, demonstrated that depleting cells of cholesterol increased the cellular binding of Aβ(1–40) (Yip et al., 2001).
While the mechanical properties of bilayers can influence their susceptibility to Aβ binding, once Aβ binds a membrane, this association may also alter the mechanical properties of the membrane, leading to dysfunction. Such a scenario is plausible considering the observed morphological changes associated with lipid membranes exposed to Aβ(Yip et al., 2002; Legleiter et al., 2011; Pifer et al., 2011). Anisotropy studies with POPC and POPG lipid membranes demonstrated that monomeric Aβ had initially little impact on bilayer fluidity; however, oligomers were able to decrease bilayer fluidity (Kremer et al., 2000). Furthermore, oligomers prepared at pH 6 had a larger impact on bilayer fluidity compared to oligomers that formed at a neutral pH, suggesting that distinct, polymorphic oligomers were formed under the different conditions (Kremer et al., 2000). Studies performed on supported phospholipid membranes revealed that exposure to Aβ modifies morphology and local mechanical properties of bilayers, reducing the force required to break through the membrane with an AFM probe (Dante et al., 2011). The lysis tension of unilamellar vesicles containing oxysterols are altered by exposure to nanomolar concentration of Aβ peptides (Kim and Frangos, 2008). Collectively, these results suggest Aβ can negatively impact the mechanical integrity of lipid membranes.
The major risk factor associated with AD is age. Age-related changes in membrane composition and/or physical properties may facilitate an increased cellular susceptibility to Aβ cytotoxicity. For example, both enhanced cellular cholesterol content (Wood et al., 2002; Cutler et al., 2004; Panchal et al., 2010) and oxidative damage (Chen and Yu, 1994; Choe et al., 1995) are associated with aging, decreased fluidity of membranes, and AD. Oxidative damage of polyunsaturated fatty acids, in general, increase lipid bilayer rigidity as a result of increased steric hindrance restricting the movement of lipid acyl chains (Choe et al., 1995; Choi and Yu, 1995). Furthermore, Aβ oligomers display preferential accumulate at oxidatively damaged plasma membranes of cells (Cecchi et al., 2007), and there is evidence of enhanced oxidative damage in AD brains (Williamson et al., 2008; Ansari and Scheff, 2010). Such studies suggest that altered membrane mechanics play a role in facilitating Aβ/lipid interactions.
Surface Aggregation of Other Amyloid-Forming Proteins
Surfaces can also modulate the aggregation of other amyloid-forming proteins associated with neurodegenerative diseases. Specifically, these proteins may also alter membrane homeostasis, presumably via similar mechanisms as described for Aβ. Here we will briefly discuss some features of the interaction of other amyloid-forming proteins: htt, α-syn, apolipoprotein C-II, and prions. Many studies of the aggregation of these two proteins have focused on lipid vesicles or organelles, which have membranes comprised predominately of lipids, and these studies further highlight how aggregation can be modulated by membrane composition.
The Interaction of α-syn with Surfaces
Parkinson’s disease is a neurodegenerative disease caused by the sporadic misfolding and aggregation of the protein α-synuclein (α-syn) leading to the appearance of inclusions termed Lewy bodies. Electron microscopy (EM) and ex situ AFM have shown that, while a heterogeneous population of oligomers, protofibrils, and annular aggregates exists (Conway et al., 2000; Apetri et al., 2006), over time the predominant aggregate species are fibrillar (Conway et al., 1998; Narhi et al., 1999; Apetri et al., 2006). While Lewy bodies have long been known to be comprised of fibrils (Duffy and Tennyson, 1965) it is now widely believed that pre-fibrillar and pre-Lewy body inclusions aggregates are responsible for disease. The fragmentation of the Golgi apparatus, for example, corresponds to the appearance of protofibrils, rather than fibrils (Gosavi et al., 2002). This notion that pre-fibrillar aggregates are the cause of disease is supported by dementia with Lewy bodies patient brains lysates containing elevated levels of α-syn oligomers (Paleologou et al., 2009) compared to control and AD patient brains. Toxicity in cell models is usually displayed without fibrillar or protofibrillar species, but rather a 54–83 kDa aggregate, perhaps comprised of 17 kDa oligomers, that appears to mediate neurotoxicity (Xu et al., 2002). Transgenic mice, unlike human patients, exhibited neurodegeneration and inclusions comprised of fine granular material and clear vacuoles, not fibrils (Masliah et al., 2000). Furthermore, appearance of Thioflavin T (ThT) reactive aggregates have been shown to correspond with decreased fluidity of lipid acyl chains in membranes (Smith et al., 2008).
Surface stabilized α-syn aggregates have been observed by in solution AFM studies, where fibrillar sheets grew in length along two directions 120° from each other reflecting the pseudo-hexagonal surface geometry of muscovite mica. Altering the surface substrate from mica, a hydrophilic surface, to highly order pyrolytic graphite, a hydrophobic surface, impeded sheet formation, demonstrating a specific surface dependent growth mechanism (Hoyer et al., 2004). This is contrary to what has been seen in EM and ex situ AFM studies in which α-syn is aggregated in bulk solution. In these studies, α-syn forms oligomers and fibrils without any discernible directionality (Conway et al., 2000).
In pre-synaptic termini, α-syn exists in both free and plasma membrane or vesicle bound states (McLean et al., 2000). Densitometric analysis of rat brain fractionation demonstrated that ∼15% α-syn in the supernatant is membrane bound (Lee et al., 2002). Homozygous deletions of α-syn in mouse models and overexpression of α-syn in a neuronal cell line corresponded with changes in membrane fluidity and cellular fatty acid uptake and metabolism (Sharon et al., 2003; Castagnet et al., 2005; Golovko et al., 2005). Similarly, α-syn has been shown to have a strong interaction with synthetic anionic phospholipid vesicles (Davidson et al., 1998; Jo et al., 2000; Ramakrishnan et al., 2003), crude brain vesicles, cellular membranes, lipid rafts, and lipid droplets (Jensen et al., 1998; McLean et al., 2000; Cole et al., 2002; Fortin et al., 2004). EPR studies have demonstrated that the α-syn helix extends parallel to the curved lipid (Jao et al., 2008), while electron microscopy experiments note α-syn’s ability to tubulate vesicles (Varkey et al., 2010). Ex situ AFM studies of PG vesicles exposed to α-syn lead to membrane fragmentation (Volles et al., 2001) and in solution AFM experiments of α-syn aggregation on mica supported lipid bilayers demonstrate that α-syn association leads to bilayer disruption and eventual fibril formation on the exposed mica surface (Jo et al., 2000). This interaction with lipid structures is believed to be directed by the first N-terminal 60 amino acids of α-syn, which contains an amphipathic α-helix structurally similar to apolipoproteins-binding domains (Clayton and George, 1998).
Thus, the first 60 amino acids of α-syn causing subcellular localization may lead to 1) an increase in local α-syn concentration and nucleation sites or 2) the α-helical structure of the membrane bound α-syn might impede misfolding into high-ordered aggregates. Supporting the first hypothesis, FTIR and far-UV CD studies demonstrate that aggregation of α-syn depends on the proximity of the membrane; amorphous aggregates were formed on or close to membranes whereas fibrillar aggregates were formed distant to membranes (Munishkina et al., 2003). Fluorescence and AFM experiments with polytetrafluroethylene balls and α-syn also highlight that aggregate formation is dominated by reactions at hydrophobic interfaces, like lipid membranes (Pronchik et al., 2010). Similarly, fluorescence studies on supported lipid bilayers demonstrate that α-syn clustering on membranes is a function of anionic lipid and/or protein concentration (Pandey et al., 2009). Double electron–electron resonance studies reveal well-defined α-syn aggregates with lipids that could form part of larger aggregates and serve as nucleation sites (Drescher et al., 2010). The second hypothesis that α-helical membrane bound α-syn impedes aggregation into higher ordered aggregates, is supported a fluorescence resonance energy transfer study, where membrane binding alters the tertiary conformation of α-syn such that oligomerization is inhibited (Narayanan and Scarlata, 2001). However, it is important to note that the two hypotheses may not be mutually exclusive. It is possible that α-syn binding to a membrane stabilizes and nucleates a toxic aggregate specie.
Circular dichroism studies have also hinted at surface altered aggregate species as α-syn in PBS is in a random coiled secondary structure, whereas α-syn in the presence of POPC/POPS small unilamellar vesicles (SUVs) formed an α-helical structure. These studies further demonstrated that α-syn aggregation was not an effect of surface curvature as POPC/POPS multilamellar vesicles (MLVs), POPC/POPI, and POPC/POPA SUVs do not result in α-syn α-helical structure, whereas with the addition of PE to POPC/PI and POPC/POPA SUVs α-syn’s α-helical content increased (Jo et al., 2000). These studies suggest that surface membrane composition plays a role in stabilizing aggregates. Stabilized annular aggregates have been found in in vitro studies and human brain samples. This stabilized pore-like structure is hypothesized to lead to membrane ion leakage (Lashuel et al., 2002; Pountney et al., 2004). Thus, surface stabilized aggregates, such the membrane stabilized annular aggregates, may be one key toward understanding the mechanism of toxicity in PD.
The Interaction of Huntingtin with Surfaces
Huntington’s disease is another neurodegenerative disease caused by a polyQ expansion within exon1 htt. The length of the polyQ domain is intimately correlated to age of onset and severity of disease (Snell et al., 1993; Penney et al., 1997; Tobin and Signer, 2000). Inclusion bodies, the hallmark of disease, once thought to be the toxic species, have been shown by a survival analysis to potentially have a beneficial rather than pathogenic response to htt aggregation (Arrasate et al., 2004). AFM experiments with both GST-fusion htt exon1 proteins and synthetic polyQ peptides demonstrate a heterogeneous and complex aggregation mechanism, including oligomers, fibrils, annular aggregates, and inclusions, in which antibodies detect numerous different conformations of these aggregates (Legleiter et al., 2009, 2010). Analytical size exclusion chromatography experiments have demonstrated that flanking sequences of the polyQ domain alter aggregation rates considerably. Specifically, in bulk solution the first 17 N-terminal amino acids accelerate aggregation while the a C-terminal polyproline (polyP) domain retards aggregation rates (Thakur et al., 2009). Different types of HD models have shown that in neurons, both normal and mutant htt proteins localize to several subcellular compartments, such as endosomes, pre-synaptic, and clathrin-coated vesicles, and dendritic plasma membrane (Harjes and Wanker, 2003). Furthermore, htt inclusion bodies developed in cell lines expressing large N-terminal htt fragments incorporate multi-vesicular membranes, autophagosomes, and mitochondria into their surfaces (Kegel et al., 2000; Qin et al., 2004).
Immunohistochemical studies and subcellular fractions have also highlighted the fact the htt is enriched in membrane-containing fractions (Gutekunst et al., 1995). In fact, ∼50% of endogenous htt distributes with membranes after subcellular fractionation of neuron-like clonal striatal cells (Kegel et al., 2005). Thus, the wide subcellular localization and membrane-incorporated aggregates suggest that there is a strong htt interaction with lipid bilayers, which may be directed by the first 17 amino acids on the N-terminus of htt exon1. This domain appears to adopt a highly conserved amphipathic α-helix with membrane binding properties (Atwal et al., 2007), which may be facilitated by the polyP domain on the C-terminal side of the polyQ domain (Qin et al., 2004). Similar to PD, subcellular localization of htt may lead to a local increase in htt concentration creating aggregation nucleation sites or stabilization of the α-helical conformation may actually stabilize specific aggregate species that are transiently formed in bulk solution. It is possible that htt association to membranes nucleates some types of aggregation while potentially stabilizing specific intermediates along that aggregation pathway.
Surface stabilized aggregates of simple polyQ peptides have been observed via in solution AFM studies that demonstrated that, while the majority of peptide formed extensive fibrillar networks, discrete oligomers formed on a mica surface (Legleiter et al., 2010; Burke et al., 2011). These studies are contrary to previous assumptions based on bulk solution experiments that aggregation of pure polyQ peptides proceeded directly from monomer to fibril without oligomeric intermediates (Chen et al., 2002a,b). Similar to α-syn, htt has also been observed by CD to alter its structure in the presence of POPC and POPS:POPC SUVs, both compositions of endoplasmic reticulum (ER) and ER derived vesicles (Atwal et al., 2007). These studies were able to show that while htt does have α-helical content in free solution, α-helical content is altered in the presence of SUVs. Interestingly, although no structural data was provided, densitometry data from Western blots were able to demonstrate that htt/lipid interaction is modulated by membrane composition and polyQ length (Kegel et al., 2009). Here, increased polyQ length had a preferential association with multivalent phospholipids. Stabilized oligomers have been identified to be associated with mitochondrial structural proteins in HD patient brains. Here, it is believed that the oligomeric species lead to mitochondrial fragmentation, abnormal mitochondrial dynamics, and oxidative DNA damage (Shirendeb et al., 2011). Surface stabilized htt aggregates, such as mitochondrial stabilized oligomeric species, may lead to understanding potential toxic mechanisms and therefore therapeutic targets.
Posttranslational modifications of htt further modify its trafficking and interaction with membranous cellular surfaces. Sumoylation of the first 17 N-terminal amino acids in htt exon1 leads to its being trafficked to the nucleus (Steffan et al., 2004). This sumoylation of mutant htt also increases soluble diffuse aggregates that elicit greater cytotoxicity and neurotoxicity in HD Drosphila models (Steffan et al., 2004). More specifically, when Rhes, a protein selectively localized in the striatum that increases sumoylation in transgenic mice, is overexpressed in mutant htt knock-in striatal cells, cell survival is reduced by 60% whereas there is no effect with wild type htt (Subramaniam et al., 2009). Similarly, phosphomimetic mutations at serine 13 and 16 have been shown to alter the kinetics of aggregation by reducing fibrillization while accumulating alternative aggregates (Gu et al., 2009). YAC128 mouse models demonstrated that ganglioside GM1 treatment induced phosphorylation at serines 13 and 16 resulting in a restoration of normal motor behavior (Di Pardo et al., 2012). Furthermore, structural studies have determined that phosphorylation of serines 13 and 16 inhibit the first 17 N-terminal amino acids’ amphipathic α-helix, altering the localization of htt within cells (Atwal et al., 2011). Collectively, these posttranslational modifications of the N-terminal domain modulate its lipid-binding properties and the cellular trafficking of htt.
The Interaction of apoC-II with Surfaces
Not all amyloid diseases are neurodegenerative in nature, and insights into the ability of lipid association to promote specific aggregation pathways and structure can be gleaned from these systems. One such illustrative system is amyloid deposition associated with aortic atherosclerotic lesions (Westermark et al., 1995; Mucchiano et al., 2001; Rocken et al., 2006), which contain numerous plasma apolipoproteins, such as apolipoprotein C-II (apoC-II; Medeiros et al., 2004). Lipid stabilized conformations of apoC-II have long been observed. CD studies have shown apoC-II exists in a highly disordered conformation in bulk solution (Tajima et al., 1982), whereas, in the presence of sodium dodecylsulfate, trifluoroethanol, and phosphatidylcholine vesicles apoC-II adopts a helical structure (Tajima et al., 1982). Furthermore, in the absence of lipid, TEM, and AFM studies have revealed that apoC-II forms stable fibrillar ribbons (Hatters et al., 2000; Teoh et al., 2011) with increased β-sheet content as measured by CD (Hatters et al., 2000), whereas TEM and turbidity assays have demonstrated that DHPC micelles inhibit amyloid formation while inducing α-helical formation believed to be amphipathic (Hatters et al., 2001). ThT fluorescence assays have even demonstrated that a 1:4 apoC-II60–70 peptide to D5PC lipid ratio is sufficient to inhibit fibril formation up to 24 h (Hung et al., 2008). In the presence of sub-micellar phospholipid concentrations, apoC-II forms a tetrameric structure that when seeded forms apoC-II fibrils, thus indicating that the tetramer specie is on-pathway to fibril formation (Ryan et al., 2008).
Intriguingly, TEM and CD studies have observed apoC-II polymorphisms by altering the lipid environment present during fibrillization. Under low-lipid concentrations, two apoC-II populations are observed in solution, which are believed to have competing fibril assembly pathways resulting in two distinct fibril structures. One fibril structure is believed to occur via the same pathway as “lipid-free” conditions, resulting in the rapid formation of ribbonlike fibrils. The second fibril structure results in a slower development of straight fibrils and is believed to form from the remaining population of lipid-associated apoC-II. Furthermore, the population of ribbonlike fibrils appears to decline as the straight fibrils are assembling, thus it is believed that apoC-II is able to transition from a mature ribbonlike fibril into the straight fibrillar assembly pathway (Griffin et al., 2008). Therefore, the lipid stabilized straight fibrils may be key toward understanding the toxic mechanism associated with atherosclerosis. The mechanisms by which lipids trigger specific aggregate forms of apoC-II may inform us concerning similar phenomena in neurodegenerative diseases.
The Interaction of Prions with Surfaces and Parallels with Cell to Cell Transport of Amyloid-Forming Proteins
Of all of the neurodegenerative diseases, prion diseases (or transmissible spongiform encephalopathies) have long been considered unique due to their infectious nature (Prusiner and Hsiao, 1994; Prusiner, 1998; Aguzzi and Calella, 2009). Prion diseases are caused by the posttranslational misfolding of the benign, α-helical prion protein cellular isoform (PrPC) into an infectious disease-related, β-sheet rich form (PrPSc; Caughey et al., 1991; Pan et al., 1993). AFM and EM experiments using PrP proteins and fragments demonstrate a complex aggregation mechanism involving oligomers (Serio et al., 2000), polymorphic fibrils (Anderson et al., 2006), and amorphous aggregates (Pan et al., 1993). Prions replicate by forcing PrPC of the host animal to adopt the PrPSc form, and this infectious, protein-only mechanism is now widely accepted (Soto, 2011).
As exposure to the PrPSc form occurs extracellular, the interaction of prions with the exterior surface of cells may play an important role in a variety of toxic or infectious mechanisms. Several studies point to a role for lipid membranes in the conversion of PrPC to PrPSc that leads to aggregation (Stahl et al., 1990; Sanghera and Pinheiro, 2002; Robinson and Pinheiro, 2010). The model prion protein fragment (PrP118-135) undergoes conformational and orientational changes in model POPG lipid bilayers (Li et al., 2012). Furthermore, the interaction between prions and cellular membranes lead directly to liposome fusion and apoptotic cell death (Pillot et al., 1997, 2000). SDS-PAGE and subcellular fractionation studies demonstrated that PrPC is a glycosyl-phosphatidylinositol (GPI)-anchored cell surface protein (Oesch et al., 1985; Meyer et al., 1986), and fluorescence studies have indicated that membrane environment alters the conformation of recombinant PrP lacking a GPI anchor (Morillas et al., 1999), playing an important role in the initial formation of the PrPSc form. Furthermore, lipid rafts or caveola-like domains are believed to be involved in the conformational transition of PrP (Gorodinsky and Harris, 1995; Vey et al., 1996). FTIR studies of PrPC binding to lipid membranes composed of DMPC, sphingomyelin, cerebroside, and cholesterol observed PrPC forming β-sheets at the membrane interface as the concentration of PrPC reached a concentration threshold (Elfrink et al., 2008). Studies using CD spectroscopy also demonstrated that β-sheet formation in PrP106-126 fragments is induced by the clustered negative surface charges on a lipid membrane surface (Miura et al., 2007). Recently, an immunofluorescence analysis of MYC-tagged PrPSc exposed to Rocky Mountain Laboratory mouse prions was able to demonstrate that the infectious isoform PrPSc was present primarily located at the plasma membrane within 1 min of exposure (Goold et al., 2011). Collectively, these studies suggest that the exterior surfaces of cell may play a role in the initial formation of PrPSc and its subsequent propagation.
Recently, several other amyloid-forming proteins have been shown to have prion-like infectious properties. The ability to circumvent the lag-phase of amyloid formation by adding preformed aggregates in a process called seeding (Jarrett and Lansbury, 1993; Lansbury, 1997; Paravastu et al., 2006; Nonaka et al., 2010; Jucker and Walker, 2011; Serem et al., 2011), demonstrating that such seeds can impose aberrant structure on other proteins. Such a phenomenon appears to play a role in the cell to cell translation of the disease state across specific regions of the brain (Vonsattel and DiFiglia, 1998; Braak et al., 2003; Ravits et al., 2007; Braak and Del Tredici, 2011), and this is reminiscent of the infectious nature of prions. Acceleration of AD has been observed in several transgenic mouse studies by the injection of preformed Aβ aggregates, suggesting that Aβ may have self-propagating conformations that can seed aggregation in vivo (Kane et al., 2000; Meyer-Luehmann et al., 2006; Stohr et al., 2012). A similar phenomenon has been observed for tau (Clavaguera et al., 2009) and α-syn (Mougenot et al., 2012). While cellular membranes may still represent a target in many toxic mechanisms, for predominately extracellular Aβ, the ability of misfolded conformers to induce/seed aggregation does not necessarily depend on cellular uptake. However, for seeding of protein aggregation associated in neurodegenerative diseases associated with intracellular inclusions/deposits, uptake of the self-propagating conformers is necessary, and this may be facilitated by the interaction with the cell membrane or other lipid-containing structures. Cellular uptake of aggregates of several amyloid-forming proteins has been demonstrated. Aggregates superoxide dismutase-1 associated with ALS can penetrate cells by macropinocytosis and seed further aggregation (Munch and Bertolotti, 2012). Pure polyQ and htt exon1 aggregates have both been shown to penetrate mammalian cells, inducing aggregation (Ren et al., 2009; Trevino et al., 2012). Experiments with cultured cells have demonstrated that extracellular aggregates of tau are endocytosed by cells, inducing the aggregation of intracellular tau (Frost et al., 2009; Nonaka et al., 2010; Guo and Lee, 2011), and the propagation of tau aggregates within the brain of a mouse model via a prion-like mechanism has been demonstrated (de Calignon et al., 2012). The ability of α-syn aggregates to seed intracellular aggregation in a variety of cellular systems (Danzer et al., 2009; Hansen et al., 2011) has also been demonstrated and mouse models (Mougenot et al., 2012) has been demonstrated. The interaction of amyloid-forming proteins with cellular surfaces may also stabilize aggregates with seeding capabilities. Such a scenario has been demonstrated for α-syn (Lee et al., 2002).
Conclusion
While the aggregation of amyloid-forming proteins in bulk solution has been extensively studied, there is still much to understand at the molecular level about protein aggregation associated with surfaces. Of particular interest are lipid membrane surfaces, which cannot only mediate and influence protein aggregation, but also may be directly targeted by toxic protein aggregates. Due to the transient nature of several aggregate species and the continuing debate concerning specific toxic species, the mechanisms associated with the ability of surfaces, like lipid membranes, to potentially stabilize (Drescher et al., 2010) or promote (Martins et al., 2008) specific aggregates need to be further elucidated. Understanding these phenomenon may prove crucial in the effectiveness of therapeutic strategies based on manipulating the aggregation pathways of amyloid-forming proteins, as has been demonstrated for EGCG (Engel et al., 2012). The exact mechanisms associated with amyloid-forming proteins leading to cellular dysfunction and death have not fully been elucidated. The ability of such proteins to perturb membrane integrity via a variety of scenarios could directly lead to membrane dysfunction, disrupting organelles, or cellular homeostasis. Still, the specific aggregate species that cause membrane destabilization are not entirely clear, and it could be that the aggregation process itself occurring at lipid surfaces may play a critical role in damaging membranes. It is an intriguing possibility that induced changes in lipid membranes may represent a common toxic motif. Continued research into the mechanism of interaction between specific conformers capable of seeding aggregation with cellular membranes is needed to fully understand how amyloid propagates from cell to cell (Munch and Bertolotti, 2012). How specific changes in cellular properties, such as membrane mechanics, influence the susceptibility of specific cells to the prion-like propagation of these protein aggregates remain unclear (Cecchi et al., 2007). Here, we highlighted some specific features of amyloid aggregation at model surfaces and lipid membranes. While the studies reviewed here are not exhaustive, we hope that collectively they offer a compelling argument that such surface induced aggregation may play a role in a variety of toxic mechanisms associated with these diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Support from the National Science Foundation (NSF#1054211), and the Alzheimer’s Association (NIRG-11-203834) is gratefully acknowledged.
References
Aguzzi, A., and Calella, A. M. (2009). Prions: protein aggregation and infectious diseases. Physiol. Rev. 89, 1105–1152.
Aisenbrey, C., Borowik, T., Bystrom, R., Bokvist, M., Lindstrom, F., Misiak, H., et al. (2008). How is protein aggregation in amyloidogenic diseases modulated by biological membranes? Eur. Biophys. J. 37, 247–255.
Anderson, M., Bocharova, O. V., Makarava, N., Breydo, L., Salnikov, V. V., and Baskakov, I. V. (2006). Polymorphism and ultrastructural organization of prion protein amyloid fibrils: an insight from high resolution atomic force microscopy. J. Mol. Biol. 358, 580–596.
Ansari, M. A., and Scheff, S. W. (2010). Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 69, 155–167.
Apetri, M. M., Maiti, N. C., Zagorski, M. G., Carey, P. R., and Anderson, V. E. (2006). Secondary structure of alpha-synuclein oligomers: characterization by Raman and atomic force microscopy. J. Mol. Biol. 355, 63–71.
Arispe, N., Pollard, H. B., and Rojas, E. (1993a). Giant multilevel cation channels formed by alzheimer-disease amyloid beta-protein A-beta-P-(1-40) in bilayer-membranes. Proc. Natl. Acad. Sci. U.S.A. 90, 10573–10577.
Arispe, N., Rojas, E., and Pollard, H. B. (1993b). Alzheimer disease amyloid β protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. U.S.A. 90, 567–571.
Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., and Finkbeiner, S. (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810.
Ashley, R. H., Harroun, T. A., Hauss, T., Breen, K. C., and Bradshaw, J. P. (2006). Autoinsertion of soluble oligomers of Alzheimer’s A beta(1-42) peptide into cholesterol-containing membranes is accompanied by relocation of the sterol towards the bilayer surface. BMC Struct. Biol. 6:21. doi:10.1186/1472-6807-6-21
Atwal, R. S., Desmond, C. R., Caron, N., Maiuri, T., Xia, J., Sipione, S., et al. (2011). Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat. Chem. Biol. 7, 453–460.
Atwal, R. S., Xia, J., Pinchev, D., Taylor, J., Epand, R. M., and Truant, R. (2007). Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum. Mol. Genet. 16, 2600–2615.
Bauer, H. H., Aebi, U., Haner, M., Hermann, R., Muller, M., Arvinte, T., et al. (1995). Architecture and polymorphism of fibrillar supramolecular assemblies produced by in-vitro aggregation of human calcitonin. J. Struct. Biol. 115, 1–15.
Berriman, J., Serpell, L. C., Oberg, K. A., Fink, A. L., Goedert, M., and Crowther, R. A. (2003). Tau filaments from human brain and from in vitro assembly of recombinant protein show cross-beta structure. Proc. Natl. Acad. Sci. U.S.A. 100, 9034–9038.
Bieschke, J., Russ, J., Friedrich, R. P., Ehrnhoefer, D. E., Wobst, H., Neugebauer, K., et al. (2010). EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. U.S.A. 107, 7710–7715.
Blackley, H. K. L., Sanders, G. H. W., Davies, M. C., Roberts, C. J., Tendler, S. J. B., and Wilkinson, M. J. (2000). In-situ atomic force microscopy study of β-amyloid fibrillization. J. Mol. Biol. 298, 833–840.
Bokvist, M., Lindstrom, F., Watts, A., and Grobner, G. (2004). Two types of Alzheimer’s β-amyloid (1-40) peptide membrane interactions: aggregation preventing transmembrane anchoring Versus accelerated surface fibril formation. J. Mol. Biol. 335, 1039–1049.
Bouchard, M., Zurdo, J., Nettleton, E. J., Dobson, C. M., and Robinson, C. V. (2000). Formation of insulin amyloid fibrils followed by FTIR simultaneously with CD and electron microscopy. Protein Sci. 9, 1960–1967.
Braak, H., and Del Tredici, K. (2011). Alzheimer’s pathogenesis: is there neuron-to-neuron propagation? Acta Neuropathol. 121, 589–595.
Braak, H., Del Tredici, K., Rub, U., De Vos, R. A. I., Steur, E., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211.
Burke, K. A., Godbey, J., and Legleiter, J. (2011). Assessing mutant huntingtin fragment and polyglutamine aggregation by atomic force microscopy. Methods 53, 275–284.
Burke, K. A., Yates, E. A., and Legleiter, J. (2013). Amyloid-forming proteins alter the local mechanical properties of lipid membranes. Biochemistry 52, 808–817.
Capone, R., Jang, H., Kotler, S. A., Kagan, B. L., Nussinov, R., and Lal, R. (2012). Probing structural features of Alzheimer’s amyloid-β pores in bilayers using site-specific amino acid substitutions. Biochemistry 51, 776–785.
Capone, R., Quiroz, F. G., Prangkio, P., Saluja, I., Sauer, A. M., Bautista, M. R., et al. (2009). Amyloid-β-induced ion flux in artificial lipid bilayers and neuronal cells: resolving a controversy. Neurotox. Res. 16, 1–13.
Castagnet, P. I., Golovko, M. Y., Barcelo-Coblijn, G. C., Nussbaum, R. L., and Murphy, E. J. (2005). Fatty acid incorporation is decreased in astrocytes cultured from α-synuclein gene-ablated mice. J. Neurochem. 94, 839–849.
Caughey, B. W., Dong, A., Bhat, K. S., Ernst, D., Hayes, S. F., and Caughey, W. S. (1991). Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry 30, 7672–7680.
Cecchi, C., Fiorillo, C., Baglioni, S., Pensalfini, A., Bagnoli, S., Nacmias, B., et al. (2007). Increased susceptibility to amyloid toxicity in familial Alzheimer’s fibroblasts. Neurobiol. Aging 28, 863–876.
Chamberlain, A. K., Macphee, C. E., Zurdo, J., Morozova-Roche, L. A., Hill, H. A. O., Dobson, C. M., et al. (2000). Ultrastructural organization of amyloid fibrils by atomic force microscopy. Biophys. J. 79, 3282–3293.
Chen, J. J., and Yu, B. P. (1994). Alterations in mitochondrial membrane fluidity by lipid peroxidation products. Radic. Biol. Med. 17, 411–418.
Chen, S. M., Berthelier, V., Hamilton, J. B., O’Nuallain, B., and Wetzel, R. (2002a). Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry 41, 7391–7399.
Chen, S. M., Ferrone, F. A., and Wetzel, R. (2002b). Huntington’s disease age-of-onset linked to polyglutamine aggregation nucleation. Proc. Natl. Acad. Sci. U.S.A. 99, 11884–11889.
Chiti, F., and Dobson, C. M. (2006). Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366.
Choe, M., Jackson, C., and Yu, B. P. (1995). Lipid-peroxidation contributes to age-related membrane rigidity. Radic. Biol. Med. 18, 977–984.
Choi, J. H., and Yu, B. P. (1995). Brain synaptosomal aging: free radicals and membrane fluidity. Radic. Biol. Med. 18, 133–139.
Choo-Smith, L. P., Garzon-Rodriguez, W., Glabe, C. G., and Surewicz, W. K. (1997). Acceleration of amyloid fibril formation by specific binding of Aβ-(1-40) peptide to ganglioside-containing membrane vesicles. J. Biol. Chem. 272, 22987–22990.
Clavaguera, F., Bolmont, T., Crowther, R. A., Abramowski, D., Frank, S., Probst, A., et al. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–U325.
Clayton, D. F., and George, J. M. (1998). The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 21, 249–254.
Cole, N. B., Murphy, D. D., Grider, T., Rueter, S., Brasaemle, D., and Nussbaum, R. L. (2002). Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein α-synuclein. J. Biol. Chem. 277, 6344–6352.
Conway, K. A., Harper, J. D., and Lansbury, P. T. (1998). Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 4, 1318–1320.
Conway, K. A., Lee, S. J., Rochet, J. C., Ding, T. T., Williamson, R. E., and Lansbury, P. T. (2000). Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. U.S.A. 97, 571–576.
Crowther, R. A., and Goedert, M. (2000). Abnormal tau-containing filaments in neurodegenerative diseases. J. Struct. Biol. 130, 271–279.
Cutler, R. G., Kelly, J., Storie, K., Pedersen, W. A., Tammara, A., Hatanpaa, K., et al. (2004). Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 2070–2075.
Dante, S., Hauss, T., Steitz, R., Canale, C., and Dencher, N. A. (2011). Nanoscale structural and mechanical effects of beta-amyloid (1-42) on polymer cushioned membranes: a combined study by neutron reflectometry and AFM Force Spectroscopy. Biochim. Biophys. Acta 1808, 2646–2655.
Danzer, K. M., Krebs, S. K., Wolff, M., Birk, G., and Hengerer, B. (2009). Seeding induced by α-synuclein oligomers provides evidence for spreading of α-synuclein pathology. J. Neurochem. 111, 192–203.
Davidson, W. S., Jonas, A., Clayton, D. F., and George, J. M. (1998). Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449.
de Calignon, A., Polydoro, M., Suarez-Calvet, M., William, C., Adamowicz, D. H., Kopeikina, K. J., et al. (2012). Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73, 685–697.
De Felice, F. G., Wu, D., Lambert, M. P., Fernandez, S. J., Velasco, P. T., Lacor, P. N., et al. (2008). Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by Aβ oligomers. Neurobiol. Aging 29, 1334–1347.
Demuro, A., Smith, M., and Parker, I. (2011). Single-channel Ca2+ imaging implicates Aβ1-42 amyloid pores in Alzheimer’s disease pathology. J. Cell Biol. 195, 515–524.
Di Pardo, A., Maglione, V., Alpaugh, M., Horkey, M., Atwal, R. S., Sassone, J., et al. (2012). Ganglioside GM1 induces phosphorylation of mutant huntingtin and restores normal motor behavior in Huntington disease mice. Proc. Natl. Acad. Sci. U.S.A. 109, 3528–3533.
Drescher, M., Van Rooijen, B. D., Veldhuis, G., Subramaniam, V., and Huber, M. (2010). A stable lipid-induced aggregate of α-synuclein. J. Am. Chem. Soc. 132, 4080–4082.
Drolle, E., Gaikwad, R. M., and Leonenko, Z. (2012). Nanoscale electrostatic domains in cholesterol-laden lipid membranes create a target for amyloid binding. Biophys. J. 103, L27–L29.
Duffy, P. E., and Tennyson, V. M. (1965). Phase and electron microscopic observations of Lewy bodies and melanin granules in substantia nigra and locus caeruleus in Parkinsons disease. J. Neuropathol. Exp. Neurol. 24, 398–414.
Dzwolak, W., Smirnovas, V., Jansen, R., and Winter, R. (2004). Insulin forms amyloid in a strain-dependent manner: an FT-IR spectroscopic study. Protein Sci. 13, 1927–1932.
Eanes, E. D., and Glenner, G. G. (1968). X-ray diffraction studies on amyloid filaments. J. Histochem. Cytochem. 16, 673–677.
Elfrink, K., Ollesch, J., Stohr, J., Willbold, D., Riesner, D., and Gerwert, K. (2008). Structural changes of membrane-anchored native PrP(C). Proc. Natl. Acad. Sci. U.S.A. 105, 10815–10819.
Engel, M. F. M., Vandenakker, C. C., Schleeger, M., Velikov, K. P., Koenderink, G. H., and Bonn, M. (2012). The polyphenol EGCG inhibits amyloid formation less efficiently at phospholipid interfaces than in bulk solution. J. Am. Chem. Soc. 134, 14781–14788.
Evangelisti, E., Cecchi, C., Cascella, R., Sgromo, C., Becatti, M., Dobson, C. M., et al. (2012). Membrane lipid composition and its physicochemical properties define cell vulnerability to aberrant protein oligomers. J. Cell. Sci. 125, 2416–2427.
Fandrich, M. (2007). On the structural definition of amyloid fibrils and other polypeptide aggregates. Cell. Mol. Life Sci. 64, 2066–2078.
Fortin, D. L., Troyer, M. D., Nakamura, K., Kubo, S., Anthony, M. D., and Edwards, R. H. (2004). Lipid rafts mediate the synaptic localization of α-synuclein. J. Neurosci. 24, 6715–6723.
Frost, B., Jacks, R. L., and Diamond, M. I. (2009). Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 284, 12845–12852.
Giacomelli, C. E., and Norde, W. (2003). Influence of hydrophobic Teflon particles on the structure of amyloid β-peptide. Biomacromolecules 4, 1719–1726.
Giacomelli, C. E., and Norde, W. (2005). Conformational changes of the amyloid beta-peptide (1-40) adsorbed on solid surfaces. Macromol. Biosci. 5, 401–407.
Glenner, G. G., Ein, D., Eanes, E. D., Bladen, H. A., Terry, W., and Page, D. L. (1971). Creation of amyloid fibrils from Bence Jones proteins in-vitro. Science 174, 712–714.
Goldsbury, C., Kistler, J., Aebi, U., Arvinte, T., and Cooper, G. J. S. (1999). Watching amyloid fibrils grow by time-lapse atomic force microscopy. J. Mol. Biol. 285, 33–39.
Goldsbury, C. S., Cooper, G. J. S., Goldie, K. N., Muller, S. A., Saafi, E. L., Gruijters, W. T. M., et al. (1997). Polymorphic fibrillar assembly of human amylin. J. Struct. Biol. 119, 17–27.
Golovko, M. Y., Faergeman, N. J., Cole, N. B., Castagnet, P. I., Nussbaum, R. L., and Murphy, E. J. (2005). α-synuclein gene deletion decreases brain palmitate uptake and alters the palmitate metabolism in the absence of α-synuclein palmitate binding. Biochemistry 44, 8251–8259.
Goold, R., Rabbanian, S., Sutton, L., Andre, R., Arora, P., Moonga, J., et al. (2011). Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat. Commun. 2, 281.
Gorbenko, G. P., and Kinnunen, P. K. J. (2006). The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem. Phys. Lipids 141, 72–82.
Gorodinsky, A., and Harris, D. A. (1995). Glycolipid-anchored proteins in neuroblastoma cells form detergent-resistant complexes without caveolin. J. Cell Biol. 129, 619–627.
Gosavi, N., Lee, H. J., Lee, J. S., Patel, S., and Lee, S. J. (2002). Golgi fragmentation occurs in the cells with prefibrillar α-synuclein aggregates and precedes the formation of fibrillar inclusion. J. Biol. Chem. 277, 48984–48992.
Gray, J. J. (2004). The interaction of proteins with solid surfaces. Curr. Opin. Struct. Biol. 14, 110–115.
Griffin, M. D., Mok, M. L., Wilson, L. M., Pham, C. L., Waddington, L. J., Perugini, M. A., et al. (2008). Phospholipid interaction induces molecular-level polymorphism in apolipoprotein C-II amyloid fibrils via alternative assembly pathways. J. Mol. Biol. 375, 240–256.
Gu, X., Greiner, E. R., Mishra, R., Kodali, R., Osmand, A., Finkbeiner, S., et al. (2009). Serines 13 and 16 are critical determinants of full-length human mutant Huntingtin induced disease pathogenesis in HD mice. Neuron 64, 828–840.
Guo, J. L., and Lee, V. M. Y. (2011). Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 286, 15317–15331.
Gutekunst, C. A., Levey, A. I., Heilman, C. J., Whaley, W. L., Yi, H., Nash, N. R., et al. (1995). Identification and localization of Huntingtin in brain and human lymphoblastoid cell-lines with anti-fusion antibodies. Proc. Natl. Acad. Sci. U.S.A. 92, 8710–8714.
Hamaguchi, T., Eisele, Y. S., Varvel, N. H., Lamb, B. T., Walker, L. C., and Jucker, M. (2012). The presence of Aβ seeds, and not age per se, is critical to the initiation of Aβ deposition in the brain. Acta Neuropathol. 123, 31–37.
Hansen, C., Angot, E., Bergstrom, A.-L., Steiner, J. A., Pieri, L., Paul, G., et al. (2011). α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Ivest. 121, 715–725.
Harjes, P., and Wanker, E. E. (2003). The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem. Sci. 28, 425–433.
Hartley, D. M., Walsh, D. M., Ye, C. P. P., Diehl, T., Vasquez, S., Vassilev, P. M., et al. (1999). Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J. Neurosci. 19, 8876–8884.
Hatters, D. M., Lawrence, L. J., and Howlett, G. J. (2001). Sub-micellar phospholipid accelerates amyloid formation by apolipoprotein C-II. FEBS Lett. 494, 220–224.
Hatters, D. M., Macphee, C. E., Lawrence, L. J., Sawyer, W. H., and Howlett, G. J. (2000). Human apolipoprotein C-II forms twisted amyloid ribbons and closed loops. Biochemistry 39, 8276–8283.
Hebda, J. A., and Miranker, A. D. (2009). The interplay of catalysis and toxicity by amyloid intermediates on lipid bilayers: insights from Type II diabetes. Ann. Rev. Biophys. 38, 125–152.
Hou, J. F., Cui, J., Yu, L. C., and Zhang, Y. (2009). Intracellular amyloid induces impairments on electrophysiological properties of cultured human neurons. Neurosci. Lett. 462, 294–299.
Hoyer, W. G., Cherny, D., Subramaniam, V., and Jovin, T. M. (2004). Rapid self-assembly of α-synuclein observed by in situ atomic force microscopy. J. Mol. Biol. 340, 127–139.
Hu, X., Crick, S. L., Bu, G., Frieden, C., Pappu, R. V., and Lee, J.-M. (2009). Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc. Natl. Acad. Sci. U.S.A. 106, 20324–20329.
Hung, A., Griffin, M. D., Howlett, G. J., and Yarovsky, I. (2008). Effects of oxidation, pH and lipids on amyloidogenic peptide structure: implications for fibril formation? Eur. Biophys. J. 38, 99–110.
Jang, H., Zheng, J., and Nussinov, R. (2007). Models of β-amyloid ion channels in the membrane suggest that channel formation in the bilayer is a dynamic process. Biophys. J. 93, 1938–1949.
Jao, C. C., Hegde, B. G., Chen, J., Haworth, I. S., and Langen, R. (2008). Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. U.S.A. 105, 19666–19671.
Jarrett, J. T., and Lansbury, P. T. (1993). Seeding one-dimensional crystallization of amyloid – a pathogenic mechanism in Alzheimers disease and scrapie. Cell 73, 1055–1058.
Jayaraman, M., Mishra, R., Kodali, R., Thakur, A. K., Koharudin, L. M. I., Gronenborn, A. M., et al. (2012). Kinetically competing huntingtin aggregation pathways control amyloid polymorphism and properties. Biochemistry 51, 2706–2716.
Jensen, M. O., and Mouritsen, O. G. (2004). Lipids do influence protein function – the hydrophobic matching hypothesis revisited. Biochim. Biophys. Acta 1666, 205–226.
Jensen, P. H., Nielsen, M. S., Jakes, R., Dotti, G., and Goedert, M. (1998). Binding of α-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem. 273, 26292–26294.
Jimenez, J. L., Guijarro, J. L., Orlova, E., Zurdo, J., Dobson, C. M., Sunde, M., et al. (1999). Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. EMBO J. 18, 815–821.
Jimenez, J. L., Nettleton, E. J., Bouchard, M., Robinson, C. V., Dobson, C. M., and Saibil, H. R. (2002). The protofilament structure of insulin amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 99, 9196–9201.
Jimenez, J. L., Tennent, G., Pepys, M., and Saibil, H. R. (2001). Structural diversity of ex vivo amyloid fibrils studied by cryo-electron microscopy. J. Mol. Biol. 311, 241–247.
Jo, E., Darabie, A. A., Han, K., Tandon, A., Fraser, P. E., and Mclaurin, J. (2004). α-synuclein-synaptosomal membrane interactions – implications for fibrillogenesis. Eur. J. Biochem. 271, 3180–3189.
Jo, E. J., Mclaurin, J., Yip, C. M., St George-Hyslop, P., and Fraser, P. E. (2000). α-synuclein membrane interactions and lipid specificity. J. Biol. Chem. 275, 34328–34334.
Jucker, M., and Walker, L. C. (2011). Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann. Neurol. 70, 532–540.
Kane, M. D., Lipinski, W. J., Callahan, M. J., Bian, F., Durham, R. A., Schwarz, R. D., et al. (2000). Evidence for seeding of β-amyloid by intracerebral infusion of Alzheimer brain extracts in β-amyloid precursor protein-transgenic mice. J. Neurosci. 20, 3606–3611.
Kar, K., Hoop, C. L., Drombosky, K. W., Baker, M. A., Kodali, R., Arduini, I., et al. (2013). β-Hairpin-mediated nucleation of polyglutamine amyloid formation. J. Mol. Biol. doi:10.1016/j.jmb.2013.01.016
Kar, K., Jayaraman, M., Sahoo, B., Kodali, R., and Wetzel, R. (2011). Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat. Struct. Mol. Biol. 18, 328.
Kegel, K. B., Kim, M., Sapp, E., Mcintyre, C., Castano, J. G., Aronin, N., et al. (2000). Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J. Neurosci. 20, 7268–7278.
Kegel, K. B., Sapp, E., Alexander, J., Valencia, A., Reeves, P., Li, X., et al. (2009). Polyglutamine expansion in huntingtin alters its interaction with phospholipids. J. Neurochem. 110, 1585–1597.
Kegel, K. B., Sapp, E., Yoder, J., Cuiffo, B., Sobin, L., Kim, Y. J., et al. (2005). Huntingtin associates with acidic phospholipids at the plasma membrane. J. Biol. Chem. 280, 36464–36473.
Kim, D. H., and Frangos, J. A. (2008). Effects of amyloid β-peptides on the lysis tension of lipid bilayer vesicles containing oxysterols. Biophys. J. 95, 620–628.
Kim, S.-I., Yi, J.-S., and Ko, Y.-G. (2006). Amyloid β oligomerization is induced by brain lipid rafts. J. Cell. Biochem. 99, 878–889.
King, O. D., Gitler, A. D., and Shorter, J. (2012). The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 1462, 61–80.
Kirschner, D. A., Abraham, C., and Selkoe, D. J. (1986). X-ray-diffraction from intraneuronal paired helical filaments and extraneuronal amyloid fibers in Alzheimers disease indcates cross-β conformation. Proc. Natl. Acad. Sci. U.S.A. 83, 503–507.
Knight, J. D., and Miranker, A. D. (2004). Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol. 341, 1175–1187.
Kodali, R., and Wetzel, R. (2007). Polymorphism in the intermediates and products of amyloid assembly. Curr. Opin. Struct. Biol. 17, 48–57.
Kodali, R., Williams, A. D., Chemuru, S., and Wetzel, R. (2010). Aβ(1-40) forms five distinct amyloid structures whose beta-sheet contents and fibril stabilities are correlated. J. Mol. Biol. 401, 503–517.
Kowalewski, T., and Holtzman, D. M. (1999). In situ atomic force microscopy study of Alzheimer’s β-amyloid peptide on different substrates: new insights into mechanism of β-sheet formation. Proc. Natl. Acad. Sci. U.S.A. 96, 3688–3693.
Kremer, J. J., and Murphy, R. M. (2003). Kinetics of adsorption of β-amyloid peptide Aβ(1-40) to lipid bilayers. J. Biochem. Biophys. Methods 57, 159–169.
Kremer, J. J., Pallitto, M. M., Sklansky, D. J., and Murphy, R. M. (2000). Correlation of β-amyloid aggregate size and hydrophobicity with decreased bilayer fluidity of model membranes. Biochemistry 39, 10309–10318.
Kurouski, D., Dukor, R. K., Lu, X., Nafie, L. A., and Lednev, I. K. (2012). Spontaneous inter-conversion of insulin fibril chirality. Chem. Commun. 48, 2837–2839.
Kurouski, D., Lombardi, R. A., Dukor, R. K., Lednev, I. K., and Nafie, L. A. (2010). Direct observation and pH control of reversed supramolecular chirality in insulin fibrils by vibrational circular dichroism. Chem. Commun. 46, 7154–7156.
Lacor, P. N., Buniel, M. C., Chang, L., Fernandez, S. J., Gong, Y. S., Viola, K. L., et al. (2004). Synaptic targeting by Alzheimer’s-related amyloid β oligomers. J. Neurosci. 24, 10191–10200.
Langer, F., Eisele, Y. S., Fritschi, S. K., Staufenbiel, M., Walker, L. C., and Jucker, M. (2011). Soluble Aβ seeds are potent inducers of cerebral β-amyloid deposition. J. Neurosci. 31, 14488–14495.
Lansbury, P. T. (1997). Structural neurology: are seeds at the root of neuronal degeneration? Neuron 19, 1151–1154.
Lansbury, P. T., and Lashuel, H. A. (2006). A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature 443, 774–779.
Lashuel, H. A., Petre, B. M., Wall, J., Simon, M., Nowak, R. J., Walz, T., et al. (2002). α-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 322, 1089–1102.
Lee, H. J., Choi, C., and Lee, S. J. (2002). Membrane-bound α-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J. Biol. Chem. 277, 671–678.
Legleiter, J., Fryer, J. D., Holtzman, D. M., and Kowalewski, A. (2011). The modulating effect of mechanical changes in lipid bilayers caused by apoE-containing lipoproteins on Aβ induced membrane disruption. ACS Chem. Neurosci. 2, 588–599.
Legleiter, J., and Kowalewski, T. (2004). Atomic force microscopy of β-amyloid: static and dynamic studies of nanostructure and its formation. Methods Mol. Biol. 242, 349–364.
Legleiter, J., Lotz, G. P., Miller, J., Ko, J., Ng, C., Williams, G. L., et al. (2009). Monoclonal antibodies recognize distinct conformational epitopes formed by polyglutamine in a mutant Huntingtin fragment. J. Biol. Chem. 284, 21647–21658.
Legleiter, J., Mitchell, E., Lotz, G. P., Sapp, E., Ng, C., Difiglia, M., et al. (2010). Mutant Huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. J. Biol. Chem. 285, 14777–14790.
Li, H., Ye, S., Wei, F., Ma, S., and Luo, Y. (2012). In situ molecular-level insights into the interfacial structure changes of membrane-associated prion protein fragment [118–135] investigated by sum frequency generation vibrational spectroscopy. Langmuir 28, 16979–16988.
Lin, H., Bhatia, R., and Lal, R. (2001). Amyloid β protein forms ion channels: implications for Alzheimer’s disease pathophysiology. FASEB J. 15, 2433–2444.
Lin, H., Zhu, Y. W. J., and Lal, R. (1999). Amyloid βprotein (1-40) forms calcium-permeable, Zn2+-sensitive channel in reconstituted lipid vesicles. Biochemistry 38, 11189–11196.
Lin, M.-S., Chen, L.-Y., Wang, S. S. S., Chang, Y., and Chen, W.-Y. (2008). Examining the levels of ganglioside and cholesterol in cell membrane on attenuation the cytotoxicity of beta-amyloid peptide. Colloids Surf. B Biointerfaces 65, 172–177.
Lin, M.-S., Chiu, H.-M., Fan, F.-J., Tsai, H.-T., Wang, S. S. S., Chang, Y., et al. (2007). Kinetics and enthalpy measurements of interaction between β-amyloid and liposomes by surface plasmon resonance and isothermal titration microcalorimetry. Colloids Surf. B Biointerfaces 58, 231–236.
Lindstrom, F., Bokvist, M., Sparrman, T., and Grobner, G. (2002). Association of amyloid-β peptide with membrane surfaces monitored by solid state NMR. Phys. Chem. Chem. Phys. 4, 5524–5530.
Lins, L., Flore, C., Chapelle, L., Talmud, P. J., Thomas, A., and Brasseur, R. (2002). Lipid-interacting properties of the N-terminal domain of human apolipoprotein C-III. Protein Eng. 15, 513–520.
Linse, S., Cabaleiro-Lago, C., Xue, W.-F., Lynch, I., Lindman, S., Thulin, E., et al. (2007). Nucleation of protein fibrillation by nanoparticles. Proc. Natl. Acad. Sci. U.S.A. 104, 8691–8696.
Lomakin, A., Chung, D. S., Benedek, G. B., Kirschner, D. A., and Teplow, D. B. (1996). On the nucleation and growth of amyloid beta-protein fibrils: detection of nuclei and quantitation of rate constants. Proc. Natl. Acad. Sci. U.S.A. 93, 1125–1129.
Martins, I. C., Kuperstein, I., Wilkinson, H., Maes, E., Vanbrabant, M., Jonckheere, W., et al. (2008). Lipids revert inert Aβ amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J. 27, 224–233.
Masliah, E., Rockenstein, E., Veinbergs, I., Mallory, M., Hashimoto, M., Takeda, A., et al. (2000). Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science 287, 1265–1269.
McLaurin, J., and Chakrabartty, A. (1996). Membrane disruption by Alzheimer β-amyloid peptides mediated through specific finding to either phospholipids or gangliosides – implications for neurotoxicity. J. Biol. Chem. 271, 26482–26489.
McLaurin, J., and Chakrabartty, A. (1997). Characterization of the interactions of Alzheimer β-amyloid peptides with phospholipid membranes. Eur. J. Biochem. 245, 355–363.
McLean, P. J., Kawamata, H., Ribich, S., and Hyman, B. T. (2000). Membrane association and protein conformation of α-synuclein in intact neurons – effect of Parkinson’s disease-linked mutations. J. Biol. Chem. 275, 8812–8816.
Medeiros, L. A., Khan, T., El Khoury, J. B., Pham, C. L., Hatters, D. M., Howlett, G. J., et al. (2004). Fibrillar amyloid protein present in atheroma activates CD36 signal transduction. J. Biol. Chem. 279, 10643–10648.
Meyer, R. K., Mckinley, M. P., Bowman, K. A., Braunfeld, M. B., Barry, R. A., and Prusiner, S. B. (1986). Separation and properties of cellular and scrapie prion proteins. Proc. Natl. Acad. Sci. U.S.A. 83, 2310–2314.
Meyer-Luehmann, M., Coomaraswamy, J., Bolmont, T., Kaeser, S., Schaefer, C., Kilger, E., et al. (2006). Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 313, 1781–1784.
Michikawa, M., Gong, J. S., Fan, Q. W., Sawamura, N., and Yanagisawa, K. (2001). A novel action of Alzheimer’s amyloid β-protein (Aβ): Oligomeric Aβ promotes lipid release. J. Neurosci. 21, 7226–7235.
Mirzabekov, T. A., Lin, M. C., and Kagan, B. L. (1996). Pore formation by the cytotoxic islet amyloid peptide amylin. J. Biol. Chem. 271, 1988–1992.
Miura, T., Yoda, M., Takaku, N., Hirose, T., and Takeuchi, H. (2007). Clustered negative charges on the lipid membrane surface induce β-sheet formation of prion protein fragment 106-126. Biochemistry 46, 11589–11597.
Moores, B., Drolle, E., Attwood, S. J., Simons, J., and Leonenko, Z. (2011). Effect of surfaces on amyloid fibril formation. PLoS ONE 6:e25954. doi:10.1371/journal.pone.0025954
Morillas, M., Swietnicki, W., Gambetti, P., and Surewicz, W. K. (1999). Membrane environment alters the conformational structure of the recombinant human prion protein. J. Biol. Chem. 274, 36859–36865.
Morriss-Andrews, A., and Shea, J.-E. (2012). Kinetic pathways to peptide aggregation on surfaces: the effects of β-sheet propensity and surface attraction. J. Chem. Phys. 136:065103. doi:10.1063/1.3682986
Mossuto, M. F., Dhulesia, A., Devlin, G., Frare, E., Kumita, J. R., De Laureto, P. P., et al. (2010). The non-core regions of human lysozyme amyloid fibrils influence cytotoxicity. J. Mol. Biol. 402, 783–796.
Mougenot, A.-L., Nicot, S., Bencsik, A., Morignat, E., Verchere, J., Lakhdar, L., et al. (2012). Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol. Aging 33, 2225–2228.
Mucchiano, G. I., Haggqvist, B., Sletten, K., and Westermark, P. (2001). Apolipoprotein A-1-derived amyloid in atherosclerotic plaques of the human aorta. J. Pathol. 193, 270–275.
Munch, C., and Bertolotti, A. (2012). Propagation of the prion phenomenon: beyond the seeding principle. J. Mol. Biol. 421, 491–498.
Munishkina, L. A., Phelan, C., Uversky, V. N., and Fink, A. L. (2003). Conformational behavior and aggregation of α-synuclein in organic solvents: modeling the effects of membranes. Biochemistry 42, 2720–2730.
Murphy, R. M. (2002). Peptide aggregation in neurodegenerative disease. Annu. Rev. Biomed. Eng. 4, 155–174.
Naeem, A., and Fazili, N. (2011). Defective protein folding and aggregation as the basis of neurodegenerative diseases: the darker aspect of proteins. Cell Biochem. Biophys. 61, 237–250.
Narayanan, V., and Scarlata, S. (2001). Membrane binding and self-association of α-synucleins. Biochemistry 40, 9927–9934.
Narhi, L., Wood, S. J., Steavenson, S., Jiang, Y. J., Wu, G. M., Anafi, D., et al. (1999). Both familial Parkinson’s disease mutations accelerate α-synuclein aggregation. J. Biol. Chem. 274, 9843–9846.
Necula, M., Chirita, C. N., and Kuret, J. (2003). Rapid anionic micelle-mediated α-synuclein fibrillization in vitro. J. Biol. Chem. 278, 46674–46680.
Nelson, R., Sawaya, M. R., Balbirnie, M., Madsen, A. O., Riekel, C., Grothe, R., et al. (2005). Structure of the cross-βspine of amyloid-like fibrils. Nature 435, 773–778.
Nonaka, T., Watanabe, S. T., Iwatsubo, T., and Hasegawa, M. (2010). Seeded aggregation and toxicity of α-synuclein and tau: cellular moels of neurodegeneative disease. J. Biol. Chem. 285, 34885–34898.
Norlin, N., Hellberg, M., Filippov, A., Sousa, A. A., Gröbner, G., Leapman, R. D., et al. (2012). Aggregation and fibril morphology of the Arctic mutation of Alzheimer’s Aβ peptide by CD, TEM, STEM and in situ AFM. J. Struct. Biol. 180, 174–189.
Oesch, B., Westaway, D., Walchli, M., Mckinley, M. P., Kent, S. B., Aebersold, R., et al. (1985). A cellular gene encodes scrapie PrP 27-30 protein. Cell 40, 735–746.
Okada, T., Wakabayashi, M., Ikeda, K., and Matsuzaki, K. (2007). Formation of toxic fibrils of Alzheimer’s amyloid β-protein-(1-40) by monosialoganglioside GM1, a neuronal membrane component. J. Mol. Biol. 371, 481–489.
Paleologou, K. E., Kragh, C. L., Mann, D. M. A., Salem, S. A., Al-Shami, R., Allsop, D., et al. (2009). Detection of elevated levels of soluble-synuclein oligomers in post-mortem brain extracts from patients with dementia with Lewy bodies. Brain 132, 1093–1101.
Pan, K.-M., Baldwin, M., Nguyen, J., Gasset, M., Serban, A., Groth, D., et al. (1993). Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. U.S.A. 90, 10962–10966.
Panchal, M., Loeper, J., Cossec, J.-C., Perruchini, C., Lazar, A., Pompon, D., et al. (2010). Enrichment of cholesterol in microdissected Alzheimer’s disease senile plaques as assessed by mass spectrometry. J. Lipid Res. 51, 598–605.
Pandey, A. P., Haque, F., Rochet, J.-C., and Hovis, J. S. (2009). Clustering of α-synuclein on supported lipid bilayers: role of anionic lipid, protein, and divalent ion concentration. Biophys. J. 96, 540–551.
Paravastu, A. K., Petkova, A. T., and Tycko, R. (2006). Polymorphic fibril formation by residues 10-40 of the Alzheimer’s β-amyloid peptide. Biophys. J. 90, 4618–4629.
Pedersen, J. S., Dikov, D., Flink, J. L., Hjuler, H. A., Christiansen, G., and Otzen, D. E. (2006). The changing face of glucagon fibrillation: structural polymorphism and conformational imprinting. J. Mol. Biol. 355, 501–523.
Penney, J. B., Vonsattel, J. P., Macdonald, M. E., Gusella, J. F., and Myers, R. H. (1997). CAG repeat number governs the development rate of pathology in Huntington’s disease. Ann. Neurol. 41, 689–692.
Petkova, A. T., Leapman, R. D., Guo, Z. H., Yau, W. M., Mattson, M. P., and Tycko, R. (2005). Self-propagating, molecular-level polymorphism in Alzheimer’s β-amyloid fibrils. Science 307, 262–265.
Pifer, P. M., Yates, E. A., and Legleiter, J. (2011). Point mutations in Aβ result in the formation of distinct polymorphic aggregates in the presence of lipid bilayers. PLoS ONE 6:e16248. doi:10.1371/journal.pone.0016248
Pillot, T., Drouet, B., Pincon-Raymond, R. L., Vandekerckhove, J., Rosseneu, T., and Chambaz, J. (2000). A nonfibrillar form of the fusogenic prion protein fragment 118-135 induces apoptotic cell death in rat cortical neurons. J. Neurochem. 75, 2298–2308.
Pillot, T., Lins, L., Goethals, M., Vanloo, B., Baert, J., Vandekerckhove, J., et al. (1997). The 118-135 peptide lot the human prion protein forms amyloid fibrils and induces liposome fusion. J. Mol. Biol. 274, 381–393.
Popovych, N., Brender, J. R., Soong, R., Vivekanandan, S., Hartman, K., Basrur, V., et al. (2012). Site specific interaction of the polyphenol EGCG with the SEVI amyloid precursor peptide PAP(248-286). J. Phys. Chem. B 116, 3650–3658.
Pountney, D. L., Lowe, R., Quilty, M., Vickers, J. C., Voelcker, N. H., and Gai, W. P. (2004). Annular α-synuclein species from purified multiple system atrophy inclusions. J. Neurochem. 90, 502–512.
Pronchik, J., He, X., Giurleo, J. T., and Talaga, D. S. (2010). In vitro formation of amyloid from α-synuclein is dominated by reactions at hydrophobic interfaces. J. Am. Chem. Soc. 132, 9797–9803.
Qin, Z. H., Wang, Y. M., Sapp, E., Cuiffo, B., Wanker, E., Hayden, M. R., et al. (2004). Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J. Neurosci. 24, 269–281.
Quist, A., Doudevski, I., Lin, H., Azimova, R., Ng, D., Frangione, B., et al. (2005). Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. U.S.A. 102, 10427–10432.
Ramakrishnan, M., Jensen, P. H., and Marsh, D. (2003). α-Synuclein association with phosphatidylglycerol probed by lipid spin labels. Biochemistry 42, 12919–12926.
Ravits, J., Paul, P., and Jorg, C. (2007). Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 68, 1571–1575.
Reiss, A. B., Siller, K. A., Rahman, M. M., Chan, E. S. L., Ghiso, J., and De Leon, M. J. (2004). Cholesterol in neurologic disorders of the elderly: stroke and Alzheimer’s disease. Neurobiol. Aging 25, 977–989.
Ren, P.-H., Lauckner, J. E., Kachirskaia, I., Heuser, J. E., Melki, R., and Kopito, R. R. (2009). Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 11, 219–225.
Rhee, S. K., Quist, A. P., and Lal, R. (1998). Amyloid β protein-(1-42) forms calcium-permeable, Zn2+-sensitive channel. J. Biol. Chem. 273, 13379–13382.
Robinson, P. J., and Pinheiro, T. J. (2010). Phospholipid composition of membranes directs prions down alternative aggregation pathways. Biophys. J. 98, 1520–1528.
Rocken, C., Tautenhahn, J., Buhling, F., Sachwitz, D., Vockler, S., Goette, A., et al. (2006). Prevalence and pathology of amyloid in atherosclerotic arteries. Arterioscler. Thromb. Vasc. Biol. 26, 676–677.
Ryan, T. M., Howlett, G. J., and Bailey, M. F. (2008). Fluorescence detection of a lipid-induced tetrameric intermediate in amyloid fibril formation by apolipoprotein C-II. J. Biol. Chem. 283, 35118–35128.
Sabate, R., Espargaro, A., Barbosa-Barros, L., Ventura, S., and Estelrich, J. (2012). Effect of the surface charge of artificial model membranes on the aggregation of amyloid β-peptide. Biochimie 94, 1730–1738.
Sabate, R., and Estelrich, J. (2005). Evidence of the existence of micelles in the fibrillogenesis of β-amyloid peptide. J. Phys. Chem. B 109, 11027–11032.
Sabate, R., Gallardo, M., and Estelrich, J. (2005). Spontaneous incorporation of beta-amyloid peptide into neutral liposomes. Colloids Surf. A Physicochem. Eng. Asp. 270, 13–17.
Sanghera, N., and Pinheiro, T. J. T. (2002). Binding of prion protein to lipid membranes and implications for prion conversion. J. Mol. Biol. 315, 1241–1256.
Sciacca, M. F. M., Kotler, S. A., Brender, J. R., Chen, J., Lee, D.-K., and Ramamoorthy, A. (2012). Two-step mechanism of membrane disruption by Aβ through membrane fragmentation and pore formation. Biophys. J. 103, 702–710.
Seilheimer, B., Bohrmann, B., Bondolfi, L., Muller, F., Stuber, D., and Dobeli, H. (1997). The toxicity of the Alzheimer’s β-amyloid peptide correlates with a distinct fiber morphology. J. Struct. Biol. 119, 59–71.
Serem, W. K., Bett, C. K., Ngunjiri, J. N., and Garno, J. C. (2011). Studies of the growth, evolution, and self-aggregation of β-amyloid fibrils using tapping-mode atomic force microscopy. Microsc. Res. Tech. 74, 699–708.
Serio, T. R., Cashikar, A. G., Kowal, A. S., Sawicki, G. J., Moslehi, J. J., Serpell, L., et al. (2000). Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 289, 1317–1321.
Seubert, P., Vigopelfrey, C., Esch, F., Lee, M., Dovey, H., Davis, D., et al. (1992). Isolation and quantification of soluble Alzheimers β-peptide from biological fluids. Nature 359, 325–327.
Sharon, R., Bar-Joseph, I., Mirick, G. E., Serhan, C. N., and Selkoe, D. J. (2003). Altered fatty acid composition of dopaminergic neurons expressing α-synuclein and human brains with α-synucleinopathies. J. Biol. Chem. 278, 49874–49881.
Shen, L., Adachi, T., Vanden Bout, D., and Zhu, X. Y. (2012). A mobile precursor determines amyloid-β Peptide Fibril Formation at Interfaces. J. Am. Chem. Soc. 134, 14172–14178.
Shirendeb, U., Reddy, A. P., Manczak, M., Calkins, M. J., Mao, P., Tagle, D. A., et al. (2011). Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum. Mol. Genet. 20, 1438–1455.
Smith, D. P., Tew, D. J., Hill, A. F., Bottomley, S. P., Masters, C. L., Barnham, K. J., et al. (2008). Formation of a high affinity lipid-binding intermediate during the early aggregation phase of α-synuclein. Biochemistry 47, 1425–1434.
Snell, R. G., Macmillan, J. C., Cheadle, J. P., Fenton, I., Lazarou, L. P., Davies, P., et al. (1993). Relationship between trinucelotide repeat expansion and phenotypic variation in Huntington’s disease. Nat. Genet. 4, 393–397.
Sparr, E., Engel, M. F. M., Sakharov, D. V., Sprong, M., Jacobs, J., De Kruijff, B., et al. (2004). Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett. 577, 117–120.
Stahl, N., Borchelt, D. R., and Prusiner, S. B. (1990). Differential release of cellular and scrapie prion proteins from cellular membranes by phosphatidylinositol-specific phospholipase-C. Biochemistry 29, 5405–5412.
Steffan, J. S., Agrawal, N., Pallos, J., Rockabrand, E., Trotman, L. C., Slepko, N., et al. (2004). SUMO modification of Huntingtin and Huntington’s disease pathology. Science 304, 100–104.
Stohr, J., Watts, J. C., Mensinger, Z. L., Oehler, A., Grillo, S. K., Dearmond, S. J., et al. (2012). Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc. Natl. Acad. Sci. U.S.A. 109, 11025–11030.
Subasinghe, S., Unabia, S., Barrow, C. J., Mok, S. S., Aguilar, M. I., and Small, D. H. (2003). Cholesterol is necessary both for the toxic effect of Aβ peptides on vascular smooth muscle cells and for Aβ binding to vascular smooth muscle cell membranes. J. Neurochem. 84, 471–479.
Subramaniam, S., Sixt, K. M., Barrow, R., and Snyder, S. H. (2009). Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 324, 1327–1330.
Sunde, M., Serpell, L. C., Bartlam, M., Fraser, P. E., Pepys, M. B., and Blake, C. C. F. (1997). Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 273, 729–739.
Tajima, S., Yokoyama, S., Kawai, Y., and Yamamoto, A. (1982). Behavior of apolipoprotein C-II in an aqueous solution. J. Biochem. 91, 1273–1279.
Teoh, C. L., Pham, C. L., Todorova, N., Hung, A., Lincoln, C. N., Lees, E., et al. (2011). A structural model for apolipoprotein C-II amyloid fibrils: experimental characterization and molecular dynamics simulations. J. Mol. Biol. 405, 1246–1266.
Terzi, E., Holzemann, G., and Seelig, J. (1997). Interaction of Alzheimer β-amyloid peptide(1-40) with lipid membranes. Biochemistry 36, 14845–14852.
Thakur, A. K., Jayaraman, M., Mishra, R., Thakur, M., Chellgren, V. M., Byeon, I.-J. L., et al. (2009). Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 16, 380–389.
Thirumalai, D., Klimov, D. K., and Dima, R. I. (2003). Emerging ideas on the molecular basis of protein and peptide aggregation. Curr. Opin. Struct. Biol. 13, 146–159.
Tobin, A. J., and Signer, E. R. (2000). Huntington’s disease: the challenge for cell biologists. Trends Cell Biol. 10, 531–536.
Tofoleanu, F., and Buchete, N.-V. (2012). Alzheimer’s Aβ beta peptide interactions with lipid membranes fibrils, oligomers and polymorphic amyloid channels. Prion 6, 339–345.
Trevino, R. S., Lauckner, J. E., Sourigues, Y., Pearce, M. M., Bousset, L., Melki, R., et al. (2012). Fibrillar structure and charge determine the interaction of polyglutamine protein aggregates with the cell surface. J. Biol. Chem. 287, 29722–29728.
Tycko, R. (2004). Progress towards a molecular-level structural understanding of amyloid fibrils. Curr. Opin. Struct. Biol. 14, 96–103.
Tycko, R., and Ishii, Y. (2003). Constraints on supramolecular structure in amyloid fibrils from two-dimensional solid-state NMR spectroscopy with uniform isotopic labeling. J. Am. Chem. Soc. 125, 6606–6607.
Valincius, G., Heinrich, F., Budvytyte, R., Vanderah, D. J., Mcgillivray, D. J., Sokolov, Y., et al. (2008). Soluble amyloid β-oligomers affect dielectric membrane properties by bilayer insertion and domain formation: implications for cell toxicity. Biophys. J. 95, 4845–4861.
van Echten-Deckert, G., and Walter, J. (2012). Sphingolipids: critical players in Alzheimer’s disease. Prog. Lipid Res. 51, 378–393.
Varkey, J., Isas, J. M., Mizuno, N., Jensen, M. B., Bhatia, V. K., Jao, C. C., et al. (2010). Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J. Biol. Chem. 285, 32486–32493.
Vey, M., Pilkuhn, S., Wille, H., Nixon, R., Dearmond, S. J., Smart, E. J., et al. (1996). Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc. Natl. Acad. Sci. U.S.A. 93, 14945–14949.
Volles, M. J., Lee, S. J., Rochet, J. C., Shtilerman, M. D., Ding, T. T., Kessler, J. C., et al. (2001). Vesicle permeabilization by protofibrillar α-synuclein: implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry 40, 7812–7819.
Vonsattel, J. P. G., and DiFiglia, M. (1998). Huntington disease. J. Neuropathol. Exp. Neurol. 57, 369–384.
Wacker, J. L., Zareie, M. H., Fong, H., Sarikaya, M., and Muchowski, P. J. (2004). Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat. Struct. Mol. Biol. 11, 1215–1222.
Wakabayashi, M., and Matsuzaki, K. (2007). Formation of amyloids by Aβ-(1-42) on NGF-differentiated PC12 cells: roles of gangliosides and cholesterol. J. Mol. Biol. 371, 924–933.
Westermark, P., Mucchiano, G., Marthin, T., Johnson, K. H., and Sletten, K. (1995). Apolipoprotein A1-derived amyloid in human aortic atherosclerotic plaques. Am. J. Pathol. 147, 1186–1192.
Wetzel, R. (1994). Mutations and off-pathway aggregation of proteins. Trends Biotechnol. 12, 193–198.
Wetzel, R. (2012). Physical chemistry of polyglutamine: intriguing tales of a monotonous sequence. J. Mol. Biol. 421, 466–490.
Williams, T. L., Day, I. J., and Serpell, L. C. (2010). The effect of Alzheimer’s Aβ aggregation state on the permeation of biomimetic lipid vesicles. Langmuir 26, 17260–17268.
Williams, T. L., and Serpell, L. C. (2011). Membrane and surface interactions of Alzheimer’s Aβ peptide – insights into the mechanism of cytotoxicity. FEBS J. 278, 3905–3917.
Williamson, R., Usardi, A., Hanger, D. P., and Anderton, B. H. (2008). Membrane-bound β-amyloid oligomers are recruited into lipid rafts by a fyn-dependent mechanism. FASEB J. 22, 1552–1559.
Wood, W. G., Schroeder, F., Igbavboa, U., Avdulov, N. A., and Chochina, V. V. (2002). Brain membrane cholesterol domains, aging and amyloid beta-peptides. Neurobiol. Aging 23, 685–694.
Xu, J., Kao, S. Y., Lee, F. J. S., Song, W. H., Jin, L. W., and Yankner, B. A. (2002). Dopamine-dependent neurotoxicity of α-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat. Med. 8, 600–606.
Yates, E. A., Cucco, E. M., and Legleiter, J. (2011). Point Mutations in Aβ induce polymorphic aggregates at liquid/solid interfaces. ACS Chem. Neurosci. 2, 294–307.
Yip, C. M., Darabie, A. A., and Mclaurin, J. (2002). A(42-Peptide assembly on lipid bilayers. J. Mol. Biol. 318, 97–107.
Yip, C. M., Elton, E. A., Darabie, A. A., Morrison, M. R., and Mclaurin, J. (2001). Cholesterol, a modulator of membrane-associated Aβ-fibrillogenesis and neurotoxicity. J. Mol. Biol. 311, 723–734.
Yip, C. M., and McLaurin, J. (2001). Amyloid-β peptide assembly: a critical step in fibrillogenesis and membrane disruption. Biophys. J. 80, 1359–1371.
Yu, X., and Zheng, J. (2012). Cholesterol promotes the interaction of Alzheimer β-amyloid monomer with lipid bilayer. J. Mol. Biol. 421, 561–571.
Zhao, H. X., Tuominen, E. K. J., and Kinnunen, P. K. J. (2004). Formation of amyloid fibers triggered by phosphatidylserine-containing membranes. Biochemistry 43, 10302–10307.
Zhu, M., Souillac, P. O., Ionescu-Zanetti, C., Carter, S. A., and Fink, A. L. (2002). Surface-catalyzed amyloid fibril formation. J. Biol. Chem. 277, 50914–50922.
Keywords: amyloid disease, lipid membranes, protein aggregation, Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, prion disease
Citation: Burke KA, Yates EA and Legleiter J (2013) Biophysical insights into how surfaces, including lipid membranes, modulate protein aggregation related to neurodegeneration. Front. Neurol. 4:17. doi: 10.3389/fneur.2013.00017
Received: 29 November 2012; Accepted: 09 February 2013;
Published online: 01 March 2013.
Edited by:
Heather L. Montie, Thomas Jefferson University, USAReviewed by:
Denis Soulet, Laval University, CanadaDanny Hatters, The University of Melbourne, Australia
Copyright: © 2013 Burke, Yates and Legleiter. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Justin Legleiter, C. Eugene Bennett Department of Chemistry, West Virginia University, 217 Clark Hall, PO Box 6045, Morgantown, WV 26506, USA. e-mail: justin.legleiter@mail.wvu.edu