
- McGill Parkinson Program, Department of Neurology and Neurosurgery, Montreal Neurological Institute, McGill University, Montreal, QC, Canada
Parkinson’s disease (PD) is characterized by massive degeneration of dopaminergic neurons in the substantia nigra. Whereas the majority of PD cases are sporadic, about 5–10% of cases are familial and associated with genetic factors. The loss of parkin or PINK1, two such factors, leads to an early onset form of PD. Importantly, recent studies have shown that parkin functions downstream of PINK1 in a common genetic pathway affecting mitochondrial homeostasis. More precisely, parkin has been shown to mediate the autophagy of damaged mitochondria (mitophagy) in a PINK1-dependent manner. However, much of the work characterizing this pathway has been carried out in immortalized cell lines overexpressing high levels of parkin. In contrast, whether or how endogenous parkin and PINK1 contribute to mitophagy in neurons is much less clear. Here we review recent work addressing the role of parkin/PINK1-dependent mitophagy in neurons. Clearly, it appears that mitophagy pathways differ spatially and kinetically in neurons and immortalized cells, and therefore might diverge in their ultimate outcome and function. While evidence suggests that parkin can translocate to mitochondria in neurons, the function and mechanism of mitophagy downstream of parkin recruitment in neurons remains to be clarified. Moreover, it is noteworthy that most work has focused on the downstream signaling events in parkin/PINK1 mitophagy, whereas the upstream signaling pathways remain comparatively poorly characterized. Identifying the upstream signaling mechanisms that trigger parkin/PINK1 mitophagy will help to explain the nature of the insults affecting mitochondrial function in PD, and a better understanding of these pathways in neurons will be the key in identifying new therapeutic targets in PD.
Introduction
Over the past two decades, the identification of genes responsible for complex neurodegenerative disorders has profoundly changed our understanding of pathogenic mechanisms leading to neuronal cell death. Whereas previous research focused on post-mortem studies, analysis of the physiological function of causative genes now allows us to focus directly on key cellular pathways involved in pathology (1).
Among these pathways, dysregulation of mitochondrial quality control has emerged as a common theme for many neurological diseases and in particular for Parkinson’s disease (PD) (2). Mitochondrial dysfunction has been a longstanding theme in PD following observations that mitochondrial toxins such as MPTP and rotenone could induce acute parkinsonism (3, 4) and that mitochondrial respiration was defective in the substantia nigra pars compacta (SNpc) of post-mortem PD patients (5). Furthermore, the SNpc of patient brains has been shown to have a higher occurrence of mtDNA deletions compared to aged-matched controls (6, 7).
More recently, two PD-linked genes – parkin and PINK1 – have been implicated in mitochondrial quality control, via the degradation of dysfunctional mitochondria by autophagy (a process termed mitophagy). This suggests that the mitochondrial dysfunction observed in PD may be the result of compromised mitochondrial quality control mechanisms. Most studies examining this process, however, have employed immortalized cell lines in place of primary cell cultures, and few have studied it in neurons. In this review, we summarize the evidence for a physiological role for mitophagy in neurons, discussing the possible role of parkin and PINK1 in such a pathway and its relevance to PD.
Mitochondrial Dynamics and Quality Control in Health and Disease
Mitochondria – double membrane-bound organelles originating from the symbiosis between an early eukaryotic cell and a prokaryotic cell – are essential for generating energy through the process of oxidative phosphorylation (OXPHOS) and also play important roles in fatty acid metabolism, apoptosis, and calcium-buffering (8).
Long regarded as individual, “bean-shaped” organelles, mitochondria are now understood as a dynamic, inter-connected network, linked to other organelles and important players in a myriad of cellular signaling pathways (9). By regulating the connectivity and the size of the mitochondrial network, the cell can regulate energy production and most other mitochondrial processes (9). While the shape of the mitochondrial network is controlled by fusion- and fission-specific GTPases, the size of the network is controlled by de novo mitochondrial biogenesis and macroautophagy.
Mitochondria are the cellular site of OXPHOS, as well as many other biosynthetic reactions. These essential processes generate, as by-products, reactive intermediates, and oxidizing agents, which in turn damage mitochondrial proteins and lipids (10). To this end, distinct mitochondrial quality control mechanisms – the degradation of unfolded mitochondrial proteins by mitochondrial proteases, the elimination of selective cargo by mitochondria-derived vesicles and the elimination of whole mitochondria by mitophagy – function in response to the degree of mitochondrial damage present (10–14).
Nowhere is the requirement for effective mitochondrial quality control systems more important than in neurons, where high energetic demands and need for high calcium-buffering capacity due to action potential-driven calcium influxes rely heavily on proper mitochondrial function (15). This mitochondrial dependence renders neurons especially vulnerable to mitochondrial damage, and, in turn, efficient and properly functioning mitochondrial quality control pathways are paramount to neuronal survival. Highlighting this are genetic studies demonstrating the involvement of genes regulating mitochondrial morphology – MFN2 and GDAP-1 in Charcot-Marie-Tooth type2A, as well as OPA1in Optic Atrophy – and mitochondrial quality control – AFG3L2 in Spinocerebellar Ataxia type 28, parkin and PINK1 in PD – in neurodegenerative disease (16–23).
The PINK1/Parkin Pathway: A Link between Mitochondrial Quality Control and Parkinson’s Disease
It has been hypothesized recently that the decline in mitochondrial function observed in PD may stem from the rapid deregulation of mitochondrial quality control mechanisms in patients affected by the disease (10, 24). Importantly, recent studies have implicated two genes linked to autosomal-recessive juvenile parkinsonism (AR-JP) in humans – PINK1, a mitochondrially targeted serine/threonine kinase, and parkin, an E3 ubiquitin ligase – in a mitochondrial quality control pathway involving the degradation of damaged mitochondria by autophagy.
Initial genetic evidence from Drosophila had suggested that both parkin and PINK1 function in a common pathway regulating mitochondrial morphology by promoting mitochondrial fission – either by inhibiting the pro-fusion protein Fzo1 (the major Drosophila mitofusin homolog) or by activating the pro-fission protein Drp1 (25, 26). However, a clear consensus of how these genes affect morphology in mammalian cells has yet to be established (27–29). In regulating mitochondrial function, however, PINK1 has been shown to promote mitochondrial respiration and increase mitochondrial membrane potential (Δψm) (30–32), with a specific link to complex I (33, 34), as well as proper calcium homeostasis in mammalian cell lines (35–38).
Overwhelming evidence in mammalian cell lines has implicated parkin and PINK1 in the mitophagic degradation of dysfunctional, depolarized mitochondria. Upon ablation of Δψm by the chemical uncoupler CCCP, the Δψm-dependent mitochondrial import of PINK1 – a polypeptide that, basally, is rapidly turned over by proteases once inside mitochondria – is halted, allowing PINK1, bound to the TOM complex, to build up on the outer mitochondrial membrane (39–42). Here, PINK1 recruits parkin from the cytosol, in a manner dependent on functional PINK1 kinase activity, and promotes parkin’s E3 ubiquitin ligase activity, possibly through direct phosphorylation of parkin by PINK1 (43–47). Once recruited to depolarized mitochondria, parkin-dependent ubiquitination and proteasomal degradation of outer membrane proteins – notably the mitofusins, VDACs, and Miro – ultimately lead to autophagy, a step that possibly involves the rupture of the outer mitochondrial membrane (43, 48–55). PINK1-/parkin-dependent mitophagy enlists the canonical ATG (autophagy-related gene) pathway, originally identified in yeast (56). The ubiquitination of mitochondrial proteins by parkin has been suggested to recruit ubiquitin-binding adaptor proteins, such as p62/SQSTM1, to depolarized mitochondria (43, 57, 58). This in turn was shown to induce mitochondrial clustering around the nucleus (43, 57, 58), possibly facilitating the autophagy of mitochondria by increasing their proximity to the endoplasmic reticulum, a possible source of autophagic membranes. Although PINK1/parkin mitophagy has not been fully characterized with respect to the canonical ATG pathway, the requirement for LC3, p62, and ATG5 suggests that depolarization-induced, PINK1-/parkin-dependent mitophagy indeed makes use of the conserved ATG pathway to remove damaged mitochondria.
Clearly, the ability to pharmacologically disrupt Δψm has enabled the study of the PINK1/parkin pathway using a robust and effective paradigm, although parkin-dependent mitophagy has also been observed under less severe conditions. For example, in fusion-deficient cells, parkin recruitment to depolarized mitochondria (arising from uneven fission) has been demonstrated at the steady-state (50). Furthermore, in cells harboring severe mtDNA mutations, parkin has been shown to selectively remove dysfunctional mitochondria over time (59). However, a truly robust, physiological assay with which to determine the effectiveness of PINK1- and parkin-dependent mitophagy has remained elusive.
Types of Mitophagy Under Physiological Conditions
Selective mitophagy (depicted in Figure 1) is critical during the development of cells that specifically degrade their mitochondria as they mature. The most-studied example concerns red blood cells (RBCs), which lose their mitochondria in order to transport oxygen instead of consuming it (60). While it was long known that RBCs are devoid of nuclei and organelles such as mitochondria and Golgi apparatus (61), only recently have studies identified mitophagy as the mechanism by which mitochondria are removed (62). Mitophagy in RBCs occurs canonically, according to the conserved ATG protein pathway (56), as well as through a redundant ATG7-independent mechanism involving NIX/BNIP3L, a protein related to Bcl-2 (63–65). Moreover, NIX/BNIP3L has been shown to be an essential mediator of mitochondrial depolarization prior to autophagy (66, 67).
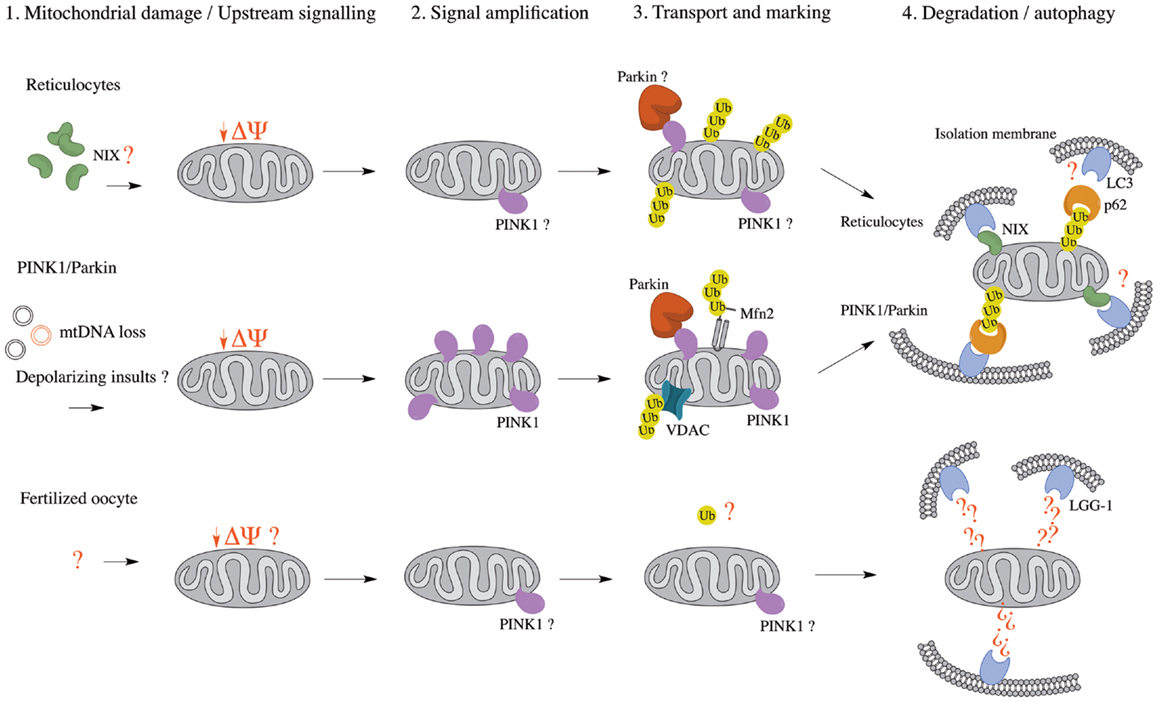
Figure 1. Comparison of known mitophagy pathways (Reticulocytes, PINK1/Parkin, and Fertilized oocyte) in four major steps. (1) Upstream signaling or mitochondrial damage activates mitophagy pathways. (2) The initial mitochondrial signaling or mitochondrial damage converges at a flagship protein, which amplifies the signal. (3) Mitochondria are transported to isolation membrane-rich sites and marked for either proteasomal or autophagic degradation. (4) Mitochondria are enveloped by isolation membranes and delivered to autophagosomes. Δψ, mitochondrial membrane potential; LC3, microtubule-associated protein 1 light chain 3; LGG-1, LC3, GABARAP, and GATE16 family 1; Mfn2, mitofusin 2; mtDNA, mitochondrial DNA; NIX, NIP3-like protein X; PINK1, PTEN-induced putative kinase 1; Ub, ubiquitin; VDAC, voltage-dependent anion channel.
The observation that both NIX- and parkin-/PINK1-dependent mitophagy seem to rely on mitochondrial depolarization as an upstream mechanism prior to autophagy suggests a conserved mechanism among pathways (Table 1), whether they are programed (reticulocyte differentiation) or reactionary (parkin/PINK1). Indeed, NIX was found to be necessary for LC3 activation following Δψm loss and parkin recruitment to the mitochondria, suggesting it might be essential to the parkin/PINK1 mitophagy pathway (63). This also raises the possibility that parkin and PINK1 function in a programed mitophagy pathway, although it would be unlikely to involve RBCs, as one would then expect parkin- or PINK1-associated PD patients to present with anemia (68).

Table 1. Comparison of mitophagy pathways.
A second type of programed mitophagy consists of the elimination of paternal mitochondria in the fertilized oocyte (69). While the notion that mitochondrial DNA is inherited uniquely from the mother has been long established, only recently have studies in C. elegans found that the degradation of paternal mitochondria, as well as its mtDNA, occurs through mitophagy (70–72). Whereas autophagy of paternal mitochondria was demonstrated to rely on the ATG-associated machinery for mitochondrial degradation, it was also shown that mitochondrial ubiquitination does not appear prior to engulfment, as opposed to mitophagy occurring during reticulocyte maturation and the parkin/PINK1 pathway (Table 1). However, further work on autophagy of paternal mitochondria, clarifying whether mitochondria are depolarized prior to engulfment, remains to be done; it would be interesting to see if parkin and PINK1 are implicated in this pathway, possibly by examining the occurrence of autophagy of paternal mitochondria in fertilized ova of parkin- or PINK1-null mice. Interestingly, parkin- and PINK1-null flies both show mitochondrial abnormalities in embryonic development, characterized by swollen or vacuolated nebenkern – spheres encompassing two giant mitochondria in the developing fly spermatid – which result in male sterility (73–75). These findings suggest that, in spermatogenesis, parkin and PINK1 may have a role in mitophagy-related events.
In essence, it seems that many mitophagic pathways utilize the canonical ATG-associated machinery, yet differ both in the manner through which mitochondria are signaled for autophagosomal engulfment and in upstream signaling mechanisms. In the case of parkin-/PINK1-dependent mitophagy, it is the latter that is poorly defined, and it is plausible that a programed mitophagic pathway, utilizing parkin and PINK1, exists in mammals and contributes to PD pathogenesis. In addition, we have already described that PINK1 can accumulate on mitochondria upon disruption of mitochondrial import in the absence of depolarization (39), suggesting that several upstream mechanisms may impinge on a canonical, programed parkin/PINK1 mitophagic pathway.
Mitophagy in Parkinson’s Disease
Although studies examining parkin-/PINK1-dependent mitophagy as a quality control mechanism have relied heavily on the use of chemical uncouplers in heterologous cell culture (42, 43, 45, 47, 50), the existence and relevance of such a pathway in neurons has remained elusive, based on a handful of studies relying predominantly on parkin overexpression (76–78). Alarmingly, other groups were unable to show recruitment of overexpressed parkin in neurons following mitochondrial depolarization (79), or showed that endogenous parkin failed to mediate mitophagy in neurons and cultured cells (80).
As described previously, complete autophagy of the mitochondrial network can occur in many cell types in response to intrinsic or extrinsic signals. Neurons, however, cannot switch to glycolytic metabolism (as an ATP-generating mechanism) during acute mitochondrial stress, and hence are utterly dependent on mitochondria for energy production (81, 82). Therefore, it is unlikely that molecular pathways have evolved to remove the whole mitochondrial network following mitochondrial damage in neurons. Moreover, it has been shown that neurons divert glucose away from glycolysis to the pentose phosphate pathway – in order to maintain a high level of reduced glutathione – and instead generate ATP predominantly through OXPHOS (83, 84). As such, disruption of the OXPHOS process by uncouplers or other mitochondrial toxins results in a bioenergetic crisis inherent to neurons, and may contribute to the ambiguity surrounding findings concerning the PINK1/parkin pathway obtained from this cell type.
Thus, two important questions arise from the controversy surrounding the relevance of the PINK1/parkin pathway in neurons: (1) whether or not parkin is recruited to depolarized mitochondria in these cells and, if so, (2) what is the physiological role of this recruitment in neurons? While parkin recruitment was shown to be robust and reproducible in immortalized cells, the data indicate that this is more variable in neurons. This is not so surprising since neuronal culture protocols carry many more variables than those for immortalized cells. When analyzing data from the five studies on parkin-/PINK1-dependent mitophagy in neurons (Table 2), we find that many components of neuronal media could influence parkin translocation. Cai and colleagues used inhibitors of apoptosis in their neuronal culture (the caspase inhibitor Z-VAD-FMK) to counter the effects of high doses of chemical uncouplers triggering apoptosis in an environment devoid of protective glia. While these conditions do lead to parkin translocation in neurons, they may also mask the normal physiological reaction of neurons to gross depolarization of the mitochondrial network. It is unlikely that neurons have evolved to adapt to this type of insult, and apoptosis may be the physiological response. Interestingly, in the study (77) that showed the highest percentage of parkin translocation (about 70%), it was found that recruitment was dependent on the absence of antioxidants (in the form of the B-27 supplement) in the media. Taken together, these factors might explain why some groups (79) were not able to detect significant parkin translocation upon mitochondrial depolarization in neurons. Importantly, parkin translocation was also much slower (12–24 h) in the study that used B-27 than in studies without it (4 h). This indicates that neuronal cultures containing antioxidant supplements may counteract the action of chemical uncouplers. It would however be important to determine if, under growth conditions lacking antioxidants, neurons can survive over long time periods following mitochondrial depolarization. In light of these recent studies, we conclude that parkin can translocate to mitochondria in neurons following depolarization, given the proper culture conditions. However, most of these studies did not quantify mitophagy following parkin translocation (76–78). While Seibler and colleagues found that parkin-positive cells have reduced mitochondrial DNA copy numbers after exposure to CCCP, they did not rule out decreased mitochondrial biogenesis as a possible mechanism. This raises the question of whether parkin translocation proceeds to mitophagy in neurons, or plays another role. Rakovic and colleagues addressed this question by looking at the degradation of a number of mitochondrial proteins both at the outer membrane, the inner membrane, and the matrix. Surprisingly, they found that even when overexpressing parkin in induced pluripotent stem (iPS) cell-derived dopaminergic neurons, parkin does not promote mitophagy upon mitochondria depolarization. However, given that the kinetics of parkin recruitment to mitochondria seem considerably slower in neurons (70% recruitment at 4 h) than in immortalized cells (100% at 2 h) (Table 3), it is plausible that mitophagy may proceed more slowly and may not be detectable at 16 h with the 1 μM valinomycin used by Rakovic and colleagues. Again, one obvious issue may be that incubating neuronal cultures with chemical uncouplers for an extended period may induce apoptosis. Adding apoptotic inhibitors may circumvent this limitation, allowing for the study of parkin-dependent mitophagy in neurons on a longer time scale, as shown by Cai and colleagues. Whereas this allowed them to show colocalization between autophagic markers and mitochondria in isolated events, Cai and colleagues did not quantify them in parkin-overexpressing versus mock-transfected neurons.
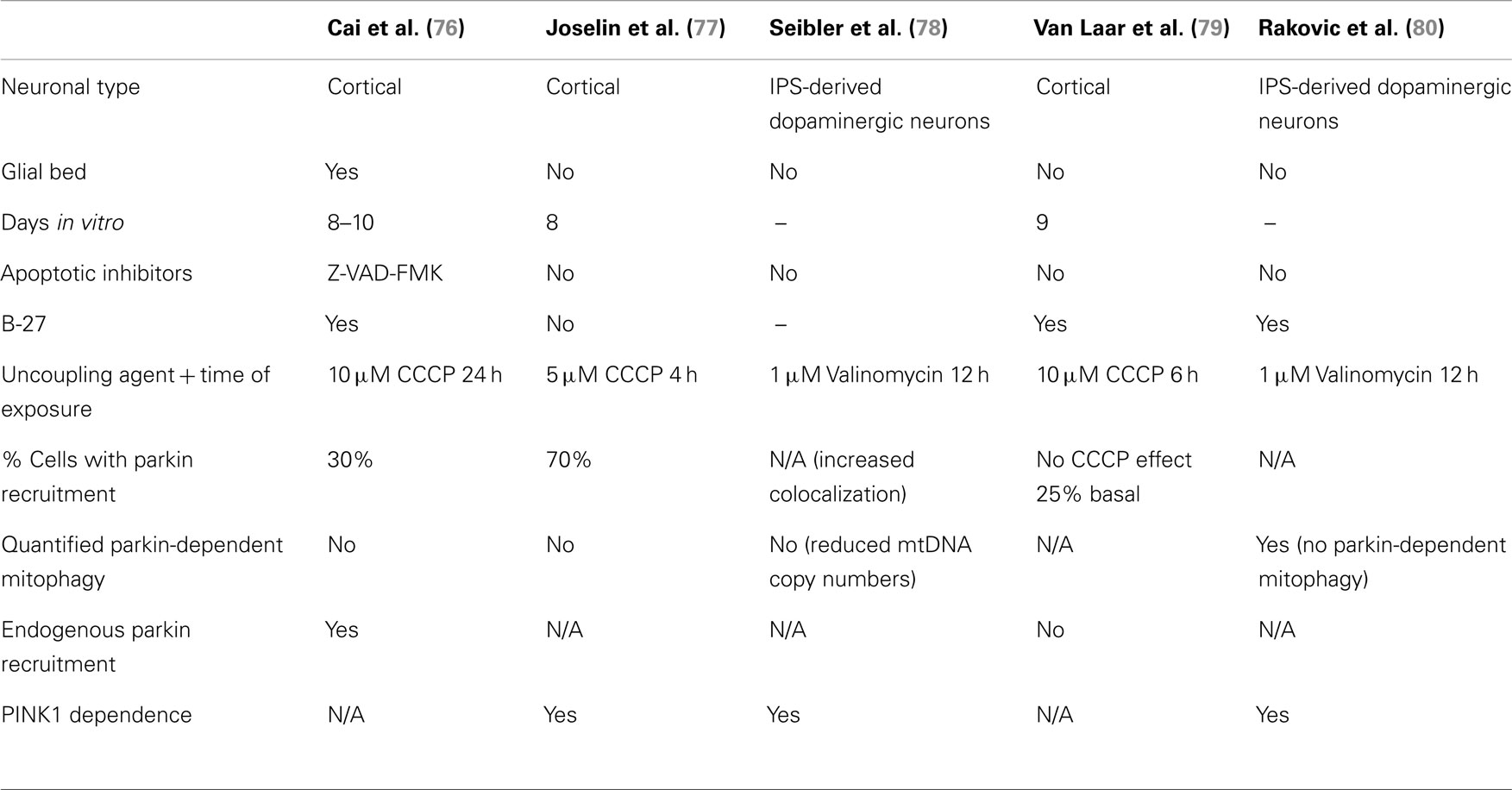
Table 2. Comparison of data on Parkin/PINK1-dependent mitophagy in neurons.
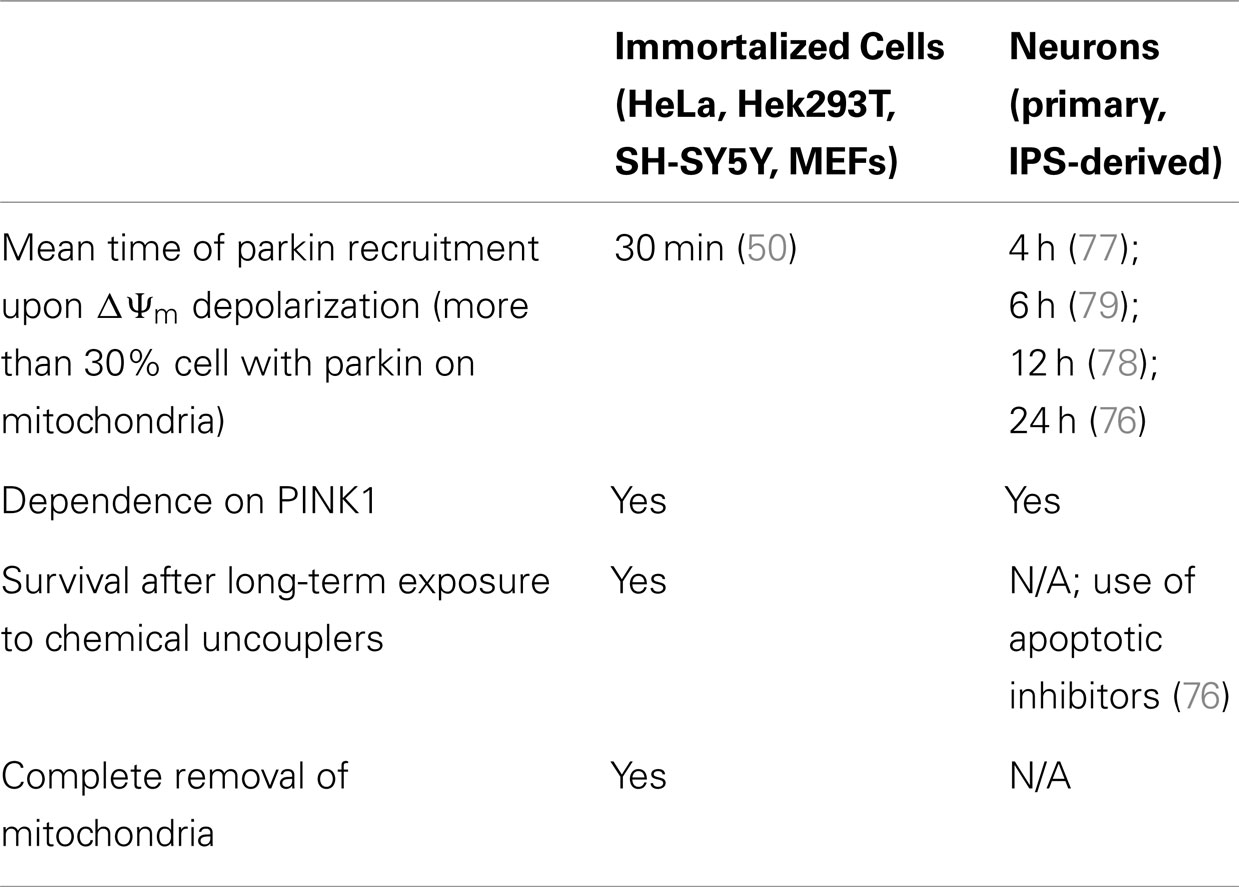
Table 3. Comparison of parkin recruitment in immortalized cells versus neurons.
To overcome the limitations of in vitro neuronal cultures, Sterky and colleagues crossed MitoPark mice – which develop progressive parkinsonism and mitochondrial abnormalities stemming from the ablated expression of mitochondrial transcription factor A in dopaminergic neurons (85) – with parkin knockout mice. These mice did not show increased neurodegeneration or accumulation of damaged mitochondria, suggesting that parkin had no role in degrading damaged mitochondria (86). Moreover, they overexpressed parkin in the MitoPark mouse and found that parkin was not recruited to mitochondria at the steady-state. While these data suggest that parkin may not be involved in mitophagy in the brain in vivo, it is noteworthy that the mitochondrial defects of MitoPark mice have not been fully characterized, and, as such, the mitochondria of these mice may not be sufficiently depolarized to stabilize PINK1 levels and trigger parkin recruitment. Interestingly, a recent study by Vincow and colleagues demonstrated that parkin null flies exhibit a slower turnover of mitochondrial proteins (87). Moreover, they showed that electron transport chain (ETC) protein turnover is especially affected in both parkin and PINK1 single-null flies, suggesting that, under physiological conditions, parkin and PINK1 might have a specific role in regulating the levels of ETC proteins, as opposed to the complete removal of mitochondria following depolarization in cell lines. In light of these results, it is clear that further studies will be required to test whether parkin promotes mitophagy in neurons and what are its consequences in vivo.
Conclusion
Parkin and PINK1 are the first two PD-associated genes to be implicated in a common genetic pathway. More specifically, the association of parkin and PINK1 in a common mitochondrial quality control pathway has consolidated the hypothesis that mitochondrial defects are central to PD pathogenesis. However, the physiological relevance of such a pathway in neurons requires further investigation. Moreover, neuronal parkin and PINK1 may play roles in mitochondrial homeostasis other than degrading damaged mitochondria, such as regulating ETC protein turnover. Upon review of the few, pioneering studies that have aimed to clarify these questions, we conclude that, although it is robust and implicates many other players subsequent to parkin recruitment, the parkin/PINK1 mitophagy pathway still lacks proper upstream signaling characterization. This is reflected in the inability to find a consensus on the proper conditions with which to study this pathway in a more disease-relevant cell type. We also conclude that, while redistribution of parkin to depolarized mitochondria has now been shown in neurons, the physiological role of such recruitment – specifically, whether or not this proceeds to mitophagy – remains elusive. By understanding the physiological function of parkin and PINK1 in neurons, future studies will undoubtedly reveal key molecular mechanisms underlying neurodegeneration and hence novel therapeutic targets for the treatment of PD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Lill CM, Bertram L. Towards unveiling the genetics of neurodegenerative diseases. Semin Neurol (2011) 31:531–41. doi:10.1055/s-0031-1299791
2. Karbowski M, Neutzner A. Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol (2012) 123:157–71. doi:10.1007/s00401-011-0921-0
3. Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science (1983) 219:979–80. doi:10.1126/science.6823561
4. Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol (2008) 7:97–109. doi:10.1016/S1474-4422(07)70327-7
5. Mann VM, Cooper JM, Krige D, Daniel SE, Schapira AH, Marsden CD. Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson’s disease. Brain (1992) 115(Pt 2):333–42. doi:10.1093/brain/115.2.333
6. Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet (2006) 38:515–7. doi:10.1038/ng1769
7. Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet (2006) 38:518–20. doi:10.1038/ng1778
8. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell (2012) 148:1145–59. doi:10.1016/j.cell.2012.02.035
9. Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol (2010) 11:872–84. doi:10.1038/nrm3013
10. Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J (2008) 27:306–14. doi:10.1038/sj.emboj.7601972
11. Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ (2013) 20:31–42. doi:10.1038/cdd.2012.81
12. Shutt TE, McBride HM. Staying cool in difficult times: mitochondrial dynamics, quality control and the stress response. Biochim Biophys Acta (2013) 1833:417–24. doi:10.1016/j.bbamcr.2012.05.024
13. Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol (2012) 22:135–41. doi:10.1016/j.cub.2011.11.057
14. Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, McBride HM. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS ONE (2012) 7:e52830. doi:10.1371/journal.pone.0052830
15. Chan CS, Gertler TS, Surmeier DJ. Calcium homeostasis, selective vulnerability and Parkinson’s disease. Trends Neurosci (2009) 32:249–56. doi:10.1016/j.tins.2009.01.006
16. Baxter RV, Ben Othmane K, Rochelle JM, Stajich JE, Hulette C, Dew-Knight S, et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet (2002) 30:21–2. doi:10.1038/ng796
17. Cuesta A, Pedrola L, Sevilla T, Garcia-Planells J, Chumillas MJ, Mayordomo F, et al. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat Genet (2002) 30:22–5. doi:10.1038/ng798
18. Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet (2000) 26:207–10. doi:10.1038/79936
19. Di Bella D, Lazzaro F, Brusco A, Plumari M, Battaglia G, Pastore A, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet (2010) 42:313–21. doi:10.1038/ng.544
20. Edener U, Wollner J, Hehr U, Kohl Z, Schilling S, Kreuz F, et al. Early onset and slow progression of SCA28, a rare dominant ataxia in a large four-generation family with a novel AFG3L2 mutation. Eur J Hum Genet (2010) 18:965–8. doi:10.1038/ejhg.2010.40
21. Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet (2000) 25:302–5. doi:10.1038/77060
22. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science (2004) 304:1158–60. doi:10.1126/science.1096284
23. Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet (2004) 36:449–51. doi:10.1038/ng1341
24. Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron (2011) 70:1033–53. doi:10.1016/j
25. Deng H, Dodson MW, Huang H, Guo M. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A (2008) 105:14503–8. doi:10.1073/pnas.0803998105
26. Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A (2008) 105:1638–43. doi:10.1073/pnas.0709336105
27. Exner N, Treske B, Paquet D, Holmstrom K, Schiesling C, Gispert S, et al. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci (2007) 27:12413–8. doi:10.1523/JNEUROSCI.0719-07.2007
28. Lutz AK, Exner N, Fett ME, Schlehe JS, Kloos K, Lammermann K, et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J Biol Chem (2009) 284:22938–51. doi:10.1074/jbc.M109.035774
29. Mortiboys H, Thomas KJ, Koopman WJ, Klaffke S, Abou-Sleiman P, Olpin S, et al. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann Neurol (2008) 64:555–65. doi:10.1002/ana.21492
30. Amo T, Sato S, Saiki S, Wolf AM, Toyomizu M, Gautier CA, et al. Mitochondrial membrane potential decrease caused by loss of PINK1 is not due to proton leak, but to respiratory chain defects. Neurobiol Dis (2011) 41:111–8. doi:10.1016/j.nbd.2010.08.027
31. Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A (2008) 105:11364–9. doi:10.1073/pnas.0802076105
32. Gegg ME, Cooper JM, Schapira AH, Taanman JW. Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS ONE (2009) 4:e4756. doi:10.1371/journal.pone.0004756
33. Morais VA, Verstreken P, Roethig A, Smet J, Snellinx A, Vanbrabant M, et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol Med (2009) 1:99–111. doi:10.1002/emmm.200900006
34. Vilain S, Esposito G, Haddad D, Schaap O, Dobreva MP, Vos M, et al. The yeast complex I equivalent NADH dehydrogenase rescues pink1 mutants. PLoS Genet (2012) 8:e1002456. doi:10.1371/journal.pgen.1002456
35. Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell (2009) 33:627–38. doi:10.1016/j.molcel.2009.02.013
36. Heeman B, Van den Haute C, Aelvoet SA, Valsecchi F, Rodenburg RJ, Reumers V, et al. Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J Cell Sci (2011) 124:1115–25. doi:10.1242/jcs.078303
37. Marongiu R, Spencer B, Crews L, Adame A, Patrick C, Trejo M, et al. Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux. J Neurochem (2009) 108:1561–74. doi:10.1111/j.1471-4159.2009.05932.x
38. Sandebring A, Thomas KJ, Beilina A, van der Brug M, Cleland MM, Ahmad R, et al. Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS ONE (2009) 4:e5701. doi:10.1371/journal.pone.0005701
39. Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, et al. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep (2012) 13:378–85. doi:10.1038/embor.2012.14
40. Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol (2010) 191:933–42. doi:10.1083/jcb.201008084
41. Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell (2012) 22:320–33. doi:10.1016/j.devcel.2011.12.014
42. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol (2010) 8:e1000298. doi:10.1371/journal.pbio.1000298
43. Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol (2010) 12:119–31. doi:10.1038/ncb2012
44. Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ. PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol (2013) 200:163–72. doi:10.1083/jcb.201210111
45. Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol (2010) 189:211–21. doi:10.1083/jcb.200910140
46. Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, et al. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep (2012) 2:1002. doi:10.1038/srep01002
47. Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A (2010) 107:378–83. doi:10.1073/pnas.0911187107
48. Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet (2011) 20:1726–37. doi:10.1093/hmg/ddr048
49. Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet (2010) 19:4861–70. doi:10.1093/hmg/ddq419
50. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol (2008) 183:795–803. doi:10.1083/jcb.200809125
51. Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE (2010) 5:e10054. doi:10.1371/journal.pone.0010054
52. Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol (2010) 191:1367–80. doi:10.1083/jcb.201007013
53. Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell (2011) 147:893–906. doi:10.1016/j.cell.2011.10.018
54. Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem (2011) 286:19630–40. doi:10.1074/jbc.M110.209338
55. Ziviani E, Tao RN, Whitworth AJ. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci U S A (2010) 107:5018–23. doi:10.1073/pnas.0913485107
56. Zhang J, Ney PA. Reticulocyte mitophagy: monitoring mitochondrial clearance in a mammalian model. Autophagy (2010) 6:405–8. doi:10.4161/auto.6.3.11245
57. Narendra D, Kane LA, Hauser DN I, Fearnley M, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy (2010) 6:1090–106. doi:10.4161/auto.6.8.13426
58. Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells (2010) 15:887–900. doi:10.1111/j.1365-2443.2010.01426.x
59. Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci U S A (2010) 107:11835–40. doi:10.1073/pnas.0914569107
60. Ney PA. Normal and disordered reticulocyte maturation. Curr Opin Hematol (2011) 18:152–7. doi:10.1097/MOH.0b013e328345213e
61. Heynen MJ, Verwilghen RL. A quantitative ultrastructural study of normal rat erythroblasts and reticulocytes. Cell Tissue Res (1982) 224:397–408. doi:10.1007/BF00216882
62. Fader CM, Colombo MI. Multivesicular bodies and autophagy in erythrocyte maturation. Autophagy (2006) 2:122–5.
63. Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, et al. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem (2010) 285:27879–90. doi:10.1074/jbc.M110.119537
64. Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep (2010) 11:45–51. doi:10.1038/embor.2009.256
65. Zhang J, Randall MS, Loyd MR, Dorsey FC, Kundu M, Cleveland JL, et al. Mitochondrial clearance is regulated by Atg7-dependent and -independent mechanisms during reticulocyte maturation. Blood (2009) 114:157–64. doi:10.1182/blood-2008-04-151639
66. Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature (2008) 454:232–5. doi:10.1038/nature07006
67. Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A (2007) 104:19500–5. doi:10.1073/pnas.0708818104
68. Mortensen M, Ferguson DJ, Edelmann M, Kessler B, Morten KJ, Komatsu M, et al. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci USA (2010) 107:832–7. doi:10.1073/pnas.0913170107
69. Dumollard R, Duchen M, Carroll J. The role of mitochondrial function in the oocyte and embryo. Curr Top Dev Biol (2007) 77:21–49. doi:10.1016/S0070-2153(06)77002-8
70. Al Rawi S, Louvet-Vallee S, Djeddi A, Sachse M, Culetto E, Hajjar C, et al. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science (2011) 334:1144–7. doi:10.1126/science.1211878
71. Cummins JM. Fertilization and elimination of the paternal mitochondrial genome. Hum Reprod (2000) 15(Suppl 2):92–101. doi:10.1093/humrep/15.suppl_2.92
72. Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science (2011) 334:1141–4. doi:10.1126/science.1210333
73. Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature (2006) 441:1162–6. doi:10.1038/nature04779
74. Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A (2003) 100:4078–83. doi:10.1073/pnas.0737556100
75. Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature (2006) 441:1157–61. doi:10.1038/nature04788
76. Cai Q, Zakaria HM, Simone A, Sheng ZH. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol (2012) 22:545–52. doi:10.1016/j.cub.2012.02.005
77. Joselin AP, Hewitt SJ, Callaghan SM, Kim RH, Chung YH, Mak TW, et al. ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum Mol Genet (2012) 21:4888–903. doi:10.1093/hmg/dds325
78. Seibler P, Graziotto J, Jeong H, Simunovic F, Klein C, Krainc D. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J Neurosci (2011) 31:5970–6. doi:10.1523/JNEUROSCI.4441-10.2011
79. Van Laar VS, Arnold B, Cassady SJ, Chu CT, Burton EA, Berman SB. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum Mol Genet (2011) 20:927–40. doi:10.1093/hmg/ddq531
80. Rakovic A, Shurkewitsch K, Seibler P, Grunewald A, Zanon A, Hagenah J, et al. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J Biol Chem (2013) 288:2223–37. doi:10.1074/jbc.M112.391680
81. Almeida A, Almeida J, Bolanos JP, Moncada S. Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically generated ATP in astrocyte protection. Proc Natl Acad Sci U S A (2001) 98:15294–9. doi:10.1073/pnas.261560998
82. Almeida A, Moncada S, Bolanos JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol (2004) 6:45–51. doi:10.1038/ncb1080
83. Bolanos JP, Almeida A, Moncada S. Glycolysis: a bioenergetic or a survival pathway? Trends Biochem Sci (2010) 35:145–9. doi:10.1016/j.tibs.2009.10.006
84. Herrero-Mendez A, Almeida A, Fernandez E, Maestre C, Moncada S, Bolanos JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol (2009) 11:747–52. doi:10.1038/ncb1881
85. Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A (2007) 104:1325–30. doi:10.1073/pnas.0605208103
86. Sterky FH, Lee S, Wibom R, Olson L, Larsson NG. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci USA (2011) 108:12937–42. doi:10.1073/pnas.1103295108
87. Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, Maccoss MJ, et al. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci U S A (2013) 110:6400–5. doi:10.1073/pnas.1221132110
88. Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol (2010) 189:671–9. doi:10.1083/jcb.201001039
89. Sun Y, Vashisht AA, Tchieu J, Wohlschlegel JA, Dreier L. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J Biol Chem (2012) 287:40652–60. doi:10.1074/jbc.M112.419721
Keywords: autophagy, Mitochondria, mitophagy, neurons, parkin, Parkinson’s disease, PINK1
Citation: Grenier K, McLelland G-L and Fon EA (2013) Parkin- and PINK1-dependent mitophagy in neurons: will the real pathway please stand up? Front. Neurol. 4:100. doi: 10.3389/fneur.2013.00100
Received: 31 March 2013; Paper pending published: 03 June 2013;
Accepted: 08 July 2013; Published online: 19 July 2013.
Edited by:
Heather Lynn Montie, Philadelphia College of Osteopathic Medicine, USAReviewed by:
Denis Soulet, Laval University, CanadaKah-Leong Lim, National University of Singapore, Singapore
Copyright: © 2013 Grenier, McLelland and Fon. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Edward A. Fon, Montreal Neurological Institute, 3801 University Street, Montreal, QC H3A 2B4, Canada e-mail: ted.fon@mcgill.ca