
- Department of Integrative Physiology and Neuroscience, Sleep and Performance Research Center, Washington State University, Spokane, WA, USA
Modafinil, in its two clinical formulations (Provigil® and Nuvigil®), is a widely prescribed wake-promoting therapeutic agent. It binds competitively to the cell-membrane dopamine (DA) transporter and is dependent on catecholaminergic (dopaminergic and adrenergic) signaling for its wake-promoting effects. The clinical spectrum of effects for modafinil is distinct from the effects seen with other catecholaminergic agents. Relative to other commonly used agents that act through catecholaminergic mechanisms, modafinil has a relatively low abuse potential, produces wakefulness with an attenuated compensatory sleep recovery thereafter, and does not ameliorate cataplexy in narcolepsy. These clinically relevant phenomenological differences between modafinil and agents such as amphetamines and cocaine do not eliminate catecholaminergic effects as a possible mediator of its wake-promoting action; they merely reflect its unique pharmacological profile. Modafinil is an exceptionally weak, but apparently very selective, DA transporter inhibitor. The pharmacodynamic response to modafinil, as measured by DA levels in brain microdialyzate, is protracted relative to other agents that act via catecholaminergic mechanisms. The conformational constraints on the interaction of modafinil with the DA transporter – and probably, as a consequence, its effects on trace amine receptor signaling in the catecholaminergic cell – are unique among catecholaminergic agents. These unique pharmacological properties of modafinil should be considered both in seeking to thoroughly understand its putatively elusive mechanism of action and in the design of novel therapeutic agents.
Introduction
Modafinil was originally introduced in the clinical literature as a wake-promoting agent in 1988 (1). Modafinil was first approved by the US Food and Drug Administration (FDA) in 1998 and marketed as the racemic mixture of R- and S-enantiomers (2) and later as a formulation containing only the R-enantiomer, which is pharmacokinetically distinct from the S-enantiomer in humans (3) as described later (4). It has been viewed throughout its history (5–7) as a “novel” wake-promoting therapeutic, and apparently is still viewed in the same manner to this day (8). The fact that this mainstream therapeutic agent is still thought of as novel presumably stems from some unique pharmacological and clinically relevant properties of modafinil relative to other wake-promoting agents. The purpose of this review is to summarize the known pharmacological properties of modafinil and to explain the unique clinical responses to modafinil in the context of these properties. In so doing, this review will expose some unanswered questions regarding the mechanism of action of modafinil and will offer insights relevant to the discovery and preclinical development of other novel (in a stricter sense) wake-promoting agents.
Modafinil Binds to the Dopamine Transporter and is Dependent on Catecholaminergic Signaling for Its Wake-Promoting Effects
Like many compounds, modafinil was found to be clinically useful long before its pharmacological target was known. Still, as with any new wake-promoting agent, a number of potential targets came to mind in the search for its mechanism of action. Among the potential targets for modafinil were the cell-membrane monoamine transporters. These monoamine-selective transporters serve, in a non-selective fashion, to clear the monoamines dopamine (DA), noradrenaline (NE), and serotonin (5-HT) from the extracellular space surrounding the neurons that release them. The transporters are named for the neurochemical identity of the cells that express them at the highest levels – the dopamine transporter (DAT), the noradrenaline transporter (NET), and the serotonin transporter (SERT). Despite this nomenclature, they are not truly selective for their namesake neuromodulators. For instance, the Michaelis constant (Km) for DA uptake by the NET is lower than the Km for NE uptake by the NET [at least in genetically engineered cells expressing NET under an exogenous construct (9)], indicating a higher affinity for DA. This fact, and the promiscuity among catecholaminergic (dopaminergic and adrenergic) receptors in responding to both DA and NE, may lead to some confusion with regard to modafinil’s mechanism of action (see below).
At the time when modafinil’s wake-promoting effect was discovered, various agents that bind to and inhibit the activity of monoamine transporters, such as cocaine and amphetamines, were known to also promote wakefulness. Thus, it was reasonable to hypothesize that modafinil might act through monoamine transporter inhibition to produce wakefulness. The first indication that modafinil binds competitively to a monoamine transporter, specifically DAT, came in 1994 (10). In that study, modafinil competitively displaced the binding of radiolabeled (−)-2-β-Carbomethoxy-3-β-(4-fluorophenyl)tropane (abbreviated β-CFT, also known as WIN 35,428), a known DAT ligand, in extracts of a tissue enriched for DAT, the striatum, with a concentration that inhibits 50% (IC50) value of 3.19 μM. The ability of modafinil to displace SERT and NET ligands was investigated in the same study and no effect was detected.
Competitive binding of modafinil to the DAT has been replicated in human embryonic kidney HEK293 cells transfected with DAT-encoding genetic constructs. In these studies, racemic modafinil displaced WIN 35,428 with inhibition constant [Ki] values of 2.1 μM (11) and in a separate study 2.3 μM (12). The latter study additionally reported enantiomer-specific Ki values, which were threefold higher for S-modafinil (2.5 μM) than R-modafinil (0.78 μM). These data are summarized in Table 1.
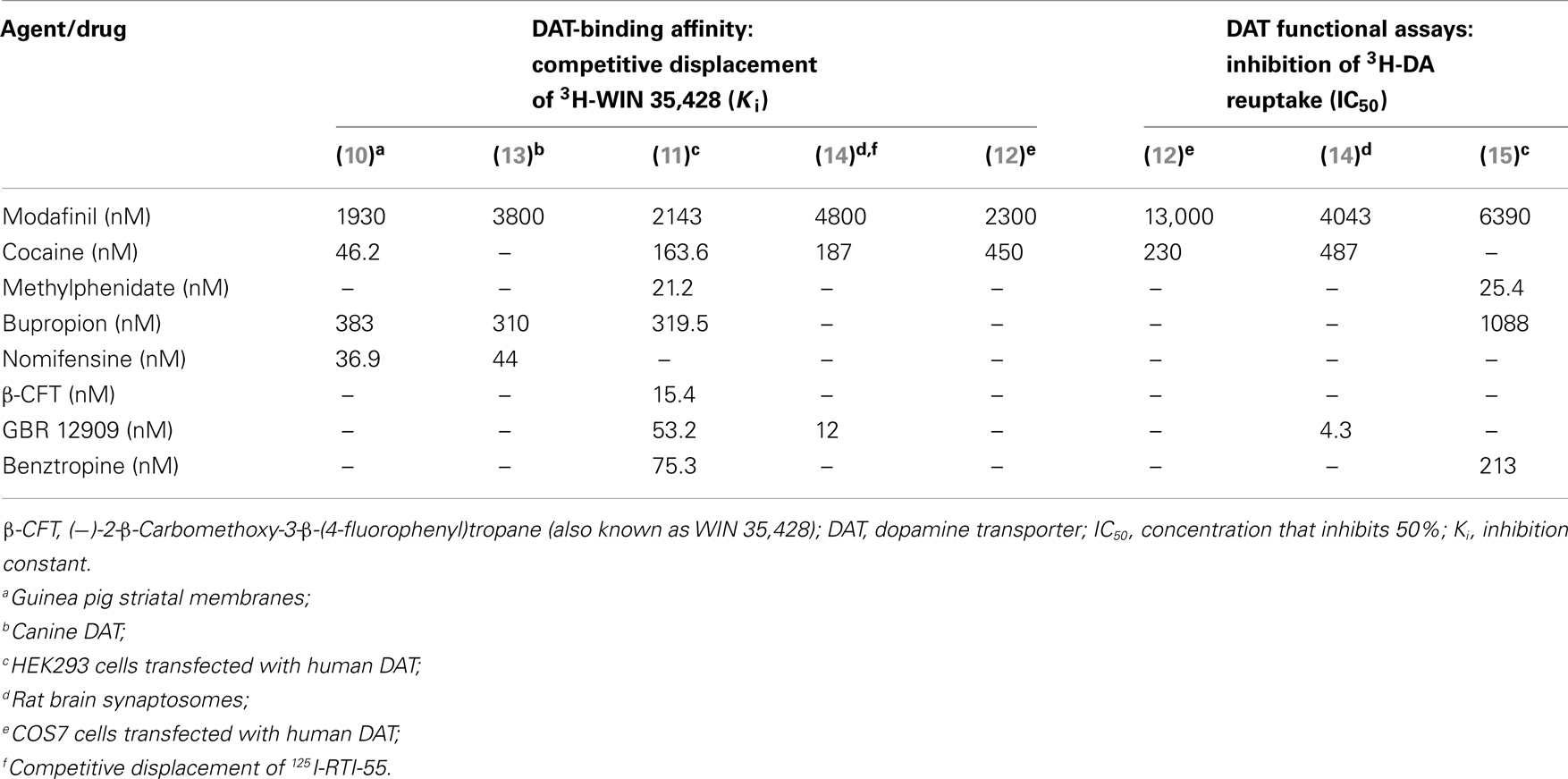
Table 1. Affinity of modafinil compared to other dopamine reuptake inhibitors.
Other studies showed that high concentrations of modafinil (relative to known DAT inhibitors) block DA uptake by cell lines stably transfected with the DAT. IC50 values for modafinil in these in vitro assays range from 4.0 to 13 μM (12, 14, 15). IC50 values for DA uptake inhibition are enantiomer-specific and twofold higher for S-modafinil (8.7 μM) relative to R-modafinil [4.0 μM; (12)]. Additionally, it has been demonstrated in positron emission tomography (PET) studies that modafinil causes the displacement of the DA-receptor ligand raclopride and the DAT ligand cocaine in the human brain (16). Similarly, modafinil displaces WIN 35,428 in the non-human primate brain (15). Displacement of a DA-receptor PET ligand is not necessarily evidence of direct binding of modafinil to the receptor. The displacement of a DA-receptor ligand by modafinil is likely to be a consequence of elevated extracellular DA concentrations, a known consequence of modafinil administration (12, 14, 17), rather than binding of modafinil to the DA receptor.
There is some evidence that modafinil binds to the NET in addition to the DAT. In PET studies, modafinil displaced the binding of radiolabeled NET ligand [11C]MeNER in the monkey thalamus (15). In cultured HEK293 cells transfected with human NET, modafinil inhibited NE uptake with an IC50 value of 35.6 μM (15). However, in other studies where NET binding or effects on NET activity were assessed in vitro (10, 12–14, 18), modafinil was found to be devoid of interactions with NET. Furthermore, the absence of therapeutic efficacy for modafinil in treating cataplexy in narcoleptic humans (19) or animal models (20, 21) makes it unlikely that modafinil is a functional NET inhibitor in vivo. NET inhibitors are very effective as anti-cataplectic agents (22), whereas DAT inhibitors are not (13).
If modafinil is a DAT inhibitor, and the blockade of DATs by modafinil is central to its wake-promoting effects, several predictions can be made and tested experimentally. First, one would expect that genetic ablation of the DAT would nullify the wake-promoting effect of modafinil if indeed this is the site of action. In fact, the wake-promoting effect of modafinil is abolished in mice genetically deficient for DAT (17). Second, one would expect modafinil administration to elevate extracellular DA concentrations in vivo, and it does. Modafinil administration increased extracellular DA concentrations in the caudate nuclei of narcoleptic dogs by twofold relative to baseline (17) and in the nucleus accumbens of mice (12) and rats (14, 23) by approximately two to threefold relative to baseline. Despite enantiomeric differences in DAT ligand displacement and DA uptake inhibition mentioned previously, R-modafinil and S-modafinil were equally effective in elevating microdialyzate DA concentration in the mouse nucleus accumbens at systemic doses of 30–300 mg/kg (12).
If elevation of dopaminergic tone underlies the wake-promoting effect of modafinil, one would expect therapeutic responses to modafinil to be dependent on the activation of DA receptors. Either a D1 or D2 DA antagonist is sufficient to block the wake-promoting effects of low wake-promoting doses (≤45 mg/kg) of modafinil in wild-type mice. In D2-deficient mice, the arousal response to high-dose (90–180 mg/kg) modafinil is attenuated relative to the wild-type response in the absence of pharmacological receptor blockade and is abolished by D1-antagonist application (24). Are these DA receptor-dependent effects of modafinil necessarily secondary to DAT blockade, or could they be indicative of agonist activity at D1 or D2 receptors? A single publication reported, in native rat striatal homogenates, that R-modafinil, but not S-modafinil, displaces the D2 receptor ligand domperidone with nanomolar potency (25). This result contrasts the work of others showing no displacement of D2 ligands by racemic modafinil at concentrations less than 10 mM [sulpiride in Ref. (10); N-methylspiperone in Ref. (14)]. Further work, including measuring the effects of R-modafinil and S-modafinil separately in D2-deficient mice, may help to clarify whether binding to the D2 receptor contributes to the wake-promoting effects of the R-enantiomer, specifically.
Collectively, these data make a compelling case for the concept that the wake-promoting effects of modafinil are mediated by its interaction with the DAT and elevation of dopaminergic tone.
Putative Non-Dopaminergic Effects of Modafinil In vivo may be Secondary to DAT Binding
Neural signaling systems other than DA and its receptors have been implicated in the brain’s response to modafinil, but the other responses could be triggered secondary to elevated concentrations of brain DA. For instance, the alpha-1 adrenergic antagonist prazosin prevented modafinil-induced, behaviorally defined nocturnal awakenings in monkeys (26) and electroencephalogram (EEG)-defined wakefulness in cats (27). Two DA-dependent mechanisms might explain this linkage of modafinil’s wake-promoting effect to adrenergic receptors. As a ligand for alpha-1 adrenergic receptors, DA is very nearly equipotent with NE (28). So the elevation of extracellular DA concentrations by modafinil should be expected to directly activate adrenergic receptors wherever they lie in close proximity to DAT-bearing dopaminergic terminals in the brain.
Additionally, modafinil elevates NE concentrations in both the prefrontal cortex and the hypothalamus (29). This response can be explained by a D1 receptor-mediated effect, as DA infusion into the prefrontal cortex elevates extracellular NE concentrations in a D1 receptor-dependent manner (30). Whether the adrenergic component of the response to modafinil is a direct effect of DA binding to adrenergic receptors or secondary to D1 receptor-induced elevation of NE, the role for alpha-1 adrenergic receptors does not violate the conceptual framework of modafinil as a DAT blocker.
Similar logic applies to other neurotransmitter responses to modafinil. Modafinil precipitates a decrease in concentrations of gamma-aminobutyric acid (GABA) in microdialyzates from various brain areas (31–33). This effect, at least in the cerebral cortex, is dependent on catecholaminergic signaling, as it is attenuated by the catecholaminergic toxin 6-hydroxy-DA (32). Furthermore, D1 agonists precipitate a reduction in GABA concentrations in cortical microdialyzates (34).
A similar line of reasoning applies to glutamate. Modafinil promotes an increase in extracellular glutamate concentrations in the striatum (31) and the hippocampus (35). The DA agonist apomorphine promotes an increase in extracellular glutamate concentrations in the striatum (36, 37), although the interactions of DA in this region are admittedly complex and not entirely consistent across experiments (38). DA itself promotes an increase in glutamate release in the hippocampus (39). The increase in glutamate release in these areas after modafinil administration may, thus, be secondary to elevated extracellular DA. This logic could be applied to the other transmitter systems known to be affected by modafinil (40).
So yes, modafinil has effects on adrenergic, GABAergic, and glutamatergic transmission, but all of these effects can be explained by its known pharmacology as a DAT blocker. Still, though it is one thing to argue that these responses are secondary to elevated dopaminergic tone, it is another to ascertain that they are. To do so, one would have to show that each of these hypothesized secondary responses is abolished in DAT-deficient animals and in wild-type animals treated with a panel of DA-receptor antagonists. Given the preponderance of evidence for a dopaminergic mechanism, these experiments should be a top priority for anyone seeking to document any putative non-dopaminergic mechanism of action.
Why Do Modafinil and Other Commonly Used (and Abused) DAT Inhibitors have Distinct Effects on Clinically Relevant Measures?
The effects of modafinil on sleep and sleep disorders are distinct from those of methamphetamines. Sleep loss induced by sleep deprivation is followed by a change in EEG parameters, including increased time spent asleep, increased duration of individual sleep episodes, elevated slow-wave activity in the EEG, and decreased numbers of awakenings. This constellation of changes, sometimes referred to as hypersomnolence or sleep rebound, has been observed in experimental rodents (41) and humans (42, 43) alike.
The occurrence of hypersomnolence after sleep loss is taken as evidence that sleep is a homeostatic process, and that homeostatic sleep need builds as a function of time spent awake (44). In this context, the effects of modafinil on sleep homeostasis differed from those of methamphetamine when the two were compared. Whereas hypersomnolence occurred after methamphetamine-induced wakefulness in rats, it was not detected after an equivalent wake-promoting dose of modafinil (6, 45). One interpretation for this difference is that the two compounds have distinct effects on the biological substrates for homeostatic sleep need. Specifically, modafinil might decelerate, or methamphetamine might accelerate, the rate at which sleep need accumulates during wakefulness. Regarding the first of these two possibilities, a head-to-head comparison of the severity of hypersomnolence subsequent to modafinil-induced wakefulness and sleep deprivation-induced wakefulness in mice found no difference between conditions (46). Similarly, in human subjects, administration of modafinil during enforced wakefulness did not, relative to placebo, attenuate the increase in slow-wave activity that occurred in subsequent sleep (47, 48). Therefore, modafinil does not decelerate the rate at which sleep need accumulates during wakefulness.
It is possible that methamphetamine accelerates the accumulation of sleep need; this effect may be due to its activity as a disruptor of NET and SERT. One might hypothesize that the direct action of methamphetamine on serotonergic and noradrenergic terminals (49) contributes to methamphetamine-induced hypersomnolence. While the activity of 5-HT-producing cells of the raphe- and NE-producing cells of the locus ceruleus is greatest in wakefulness and strongly reduced during sleep, both NE (50) and 5-HT (51–53) promote homeostatic sleep drive. The direct perturbation of noradrenergic and serotonergic terminals by amphetamines may contribute to the hypersomnolence that they cause. Assessment of the effects of amphetamines on sleep in NET- and SERT-null mutants might address this possibility. Attenuation of amphetamine-induced hypersomnolence by knockout of either NET or SERT would confirm that they contribute to amphetamine-induced hypersomnolence.
Measures of gross locomotor behavior have long been applied to measure the psychostimulant effects of cocaine, amphetamines, and other DAT-binding agents. Locomotor effects of modafinil have been compared and contrasted to those of cocaine and amphetamines and the data are not consistent across studies. Acute administration of modafinil increases locomotor activity in rodents (23, 54, 55), much like cocaine and amphetamines. However, electroencephalographic studies in rats demonstrated that the intensity of locomotor activity (amount of locomotor activity per hour of wakefulness) in a home cage environment was not increased by modafinil relative to vehicle controls, in contrast to d-methamphetamine which elevated the amount of locomotor activity per hour of wakefulness (6). Similarly, video monitoring of Syrian hamsters revealed that modafinil increased time spent in quiet wakefulness (“arousal without ambulation, head up, eyes open”), but not wake with ambulation, relative to a vehicle injection (56). Cocaine and methamphetamine effects on behavior were not measured in the latter study. Hence, the results cannot be taken as evidence for a unique effect of modafinil; there may be a species-specific response to DAT inhibitors, for instance. One study reported a “non-amphetaminic mechanism” for modafinil based on the absence of stereotyped climbing (“repetitive locomotor activity”) in modafinil-treated rodents (57). However, at least two other studies did report stereotypy after modafinil administration [“repetitive movements” in Ref. (14) and “repetitive oral movements, such as gnawing, biting, and sniffing” in Ref. (58)]. Inconsistency across studies aside, none of these behavioral studies demonstrated a pharmacological mechanism of action for modafinil.
Both amphetamines and cocaine produce locomotor sensitization, in which the amount of induced locomotor activity increases with repeated daily administration over time. Rats exhibit locomotor sensitization to repeated daily modafinil administration at 64 mg/kg (23, 58). Mice subjected to modafinil at 75 mg/kg do not undergo locomotor sensitization with repeated doses (55), whereas mice subjected to modafinil at 150 mg/kg do. Cross-sensitization (wherein repeated administration of one agent potentiates the locomotor response to acute administration of another), is taken as indirect evidence that two agents act on a similar neurobiological substrate. Mice subjected to repeated administration of modafinil exhibit potentiated locomotor responses to acutely administered methamphetamine (59). Likewise, mice subjected to repeated administration of methamphetamine exhibit potentiated locomotor responses to acutely administered modafinil (59). While not mechanistic, these cross-sensitization studies suggest that modafinil and amphetamines share a common, or at least overlapping, neurobiological substrate. Repeated administration of the D1/D2 receptor agonist apomorphine cross-sensitizes rats to modafinil (58); this fact provides further support for the notion that the relevant substrate is dopaminergic transmission.
The concept that modafinil acts via DAT inhibition might be regarded as controversial because of inconsistencies in the preclinical and clinical literature on the potential for abuse and addiction. The purpose of this article is not to review exhaustively the addictive potential of modafinil; this topic is covered elsewhere from a clinical perspective (60, 61). Rather, a brief survey of the pertinent literature serves to illustrate why differences between modafinil and other DAT-binding agents in putative measures of abuse potential does not nullify DAT binding as the mechanism of action for modafinil.
There is some evidence, from preclinical models purported to measure the potential for abuse and addiction, that modafinil has rewarding properties. For example, modafinil has discriminative stimulus effects in animals trained to engage in operant behavior when exposed to cocaine. This effect of modafinil has been documented in rodents (12, 54, 62), rhesus monkeys (63, 64), and humans (60). In mice, R-modafinil and S-modafinil were equipotent in discriminative stimulus assays (12), which, like microdialysis data mentioned above, leaves in question the significance of the enantiomeric differences in DAT-binding pharmacology. Modafinil also has a modest (relative to methylphenidate) discriminative stimulus effect in rats trained to engage in operant behavior when exposed to d-amphetamine (65). Drug-naïve mice exposed to modafinil at 75 (55) or 125 mg/kg (66) exhibited conditioned-place preference (CPP), a behavioral gage of reward that also has been found to be induced by cocaine [for instance, (55)]. However, this finding was not replicated in rats. Rats exhibited either no place preference [64 mg/kg; (67)] or a significant aversion to the environment in which they had previously been exposed to modafinil [32 or 64 mg/kg; (23)]. Furthermore, modafinil does not promote the reinstatement of cocaine self-administration (68) or methamphetamine-seeking (69–71) in rats.
Data from human subjects discriminate modafinil from cocaine in terms of abuse potential. Cocaine users do not report a high when exposed to modafinil (72, 73); rather, they report that modafinil blunts the subjective effect of cocaine when the two drugs are administered simultaneously (74, 75). One clinical trial reported that modafinil increased the maximum number of consecutive days of cocaine abstinence across a 12-week clinical trial. However, at the end of that 12-week trial there was no evidence of a decrease in total days of abstinence (76). And other clinical trials in humans, in which modafinil has failed to significantly improve abstinence rates during methamphetamine withdrawal (77, 78) or cocaine withdrawal (79), are revealing from both a conceptual standpoint and a practical one. If modafinil were a pharmacological mimetic of either of these agents, presumably it would substitute more effectively and promote sustained abstinence from the original agent. Therefore, although there are conflicting data in the literature, the majority of both preclinical and clinical data suggest that modafinil is pharmacologically distinct from both cocaine and amphetamines in the context of abuse and addiction.
Thus, ambiguities in the literature on drug abuse and sleep contribute to the concept that modafinil is somehow novel and distinct from amphetamines and cocaine. Notwithstanding the fact that modafinil, cocaine, and amphetamines all interact with DAT, the pharmacology of modafinil is distinct from that of cocaine and amphetamines. Whereas cocaine (80) and amphetamines (49) bind to the DA transporter with nanomolar affinity, modafinil acts as a DAT ligand at micromolar concentrations (10, 14). This low affinity may contribute to the slow kinetics of its effect on extracellular DA concentration (discussed below). Considering the monoamine transporters DAT, NET, and SERT, modafinil is highly selective as a DAT ligand (10, 12, 13, 18), albeit at micromolar concentrations. Cocaine (80) and amphetamines (49) disrupt the function of all three of the cell-membrane monoamine transporters. Additionally, both cocaine and amphetamines disrupt the vesicular monoamine transporter that packages monoamines into vesicles within the cell (81). Whether modafinil does so remains an open question.
The physiological relevance of the multi-transporter mechanism in responses to cocaine is illustrated by the failure of genetic inactivation of any one transporter to nullify the behavioral effects of cocaine. Neither DAT knockout (82) nor dual NET/SERT knockout (83) eliminated cocaine-induced CPP. A 90% reduction of DAT expression, induced by modifications of promoter sequences in the gene, was also not sufficient to disrupt cocaine-induced locomotion or CPP (84). These observation were taken as support for the concept that the rewarding effects of cocaine are mediated by multiple transporters. However, subsequent analyses with more sophisticated models led to a more complicated story. DAT knockout mice exhibited self-administration of cocaine only transiently and at reduced frequency relative to wild-type controls (85). A mouse genetic model was engineered in which DAT is expressed at normal levels but is modified in sequence such that it 89-fold less sensitive to inhibition by cocaine (86). These mice failed to exhibit cocaine-induced operant behavior (conditioned-place preference) and exhibited a decrease in locomotor activity in response to cocaine. These data indicate that (a) DAT mediates rewarding effects of cocaine and (b) while cocaine has pharmacological effects on other monoamine transporters, it does not promote reward or locomotor activity through those transporters unless DAT is genetically inactivated. The effects of cocaine on multiple transporters are in direct contrast to modafinil, the wake-promoting effect of which is abolished in DAT-deficient mice (17). The selectivity of modafinil as a DAT inhibitor is pertinent to the treatment of narcolepsy. Modafinil is distinguished from amphetamines by its lack of efficacy in treating cataplexy in narcolepsy (19). Amphetamines and other agents that block uptake by NET are effective anti-cataplectic agents (21).
Modafinil is further distinguished from amphetamines and cocaine by virtue of the physical nature of its interactions with DAT. As a neurotransmitter sodium symporter, DAT undergoes a sequence of conformational changes in the process of transporting its ligand into the cell (87). The sequence begins when extracellular sodium promotes the assumption of an open-to-out (also known as outward-facing) conformation, which primes the transporter for ligand binding. Ligand binding causes a shift to the closed-to-out (also known as occluded) conformation. The presence of additional ligand molecules in the extracellular milieu promotes a shift from the closed-to-out conformation to the open-to-in (also known as inward-facing) conformation, which releases the bound ligand into the cytoplasm and frees the transporter to repeat this sequence of changes.
In the context of this sequence of conformational changes, DAT inhibitors can behave very differently. Those that exhibit abuse potential, such as cocaine, facilitate the open-to-out conformation. By contrast, those that do not exhibit abuse potential, such as GBR12909, facilitate the closed-to-out or open-to-in conformation. The classification of compounds into these two categories (“cocaine-like” vs. “atypical” inhibitors) can be ascertained in site-directed mutagenesis studies: point mutations that cause the transporter to preferentially adopt an open-to-out conformation increase the IC50 value of cocaine at DAT, but not that of atypical inhibitors, by 200-fold. According to this type of analysis, modafinil is an atypical DAT-binding agent (11), and this distinction from cocaine is true for both R- and S-modafinil assayed independently (12). Therefore, the relatively low abuse potential attributed to modafinil may reflect the nature of its interaction with DAT, not the absence of an interaction with DAT. The relationship between DAT conformation and abuse potential is admittedly a relatively new concept. Why the physicochemical nature of binding influences abuse potential is uncertain, but this emerging line of work offers a potential explanation for the relatively low abuse potential associated with modafinil without requiring some putative unknown mechanism.
Amphetamines are rather complex modulators of monoaminergic function. Consideration of their effects on dopaminergic cells distinguishes them from modafinil. Amphetamines are a substrate for monoamine transporters (88) and are imported into the cell (14). In so doing, they promote the reverse transport of other DAT substrates [3H]1-methyl-4-phenylpyridinium (14) and DA itself (89) out of the cell via the DAT. The distinct mechanisms of modafinil and amphetamines in the cell are illustrated by their pharmacological interactions: amphetamine-induced [3H]1-methyl-4-phenylpyridinium efflux is abolished by co-administration of modafinil with amphetamines (14). Amphetamines are further distinguished from modafinil by the fact that they act as agonists for the trace amine-associated receptor 1 (TAAR1) (89). Modafinil exhibits no activity as a TAAR1 agonist at physiologically relevant concentrations [below 100 micromolar; (15)]. TAAR1 activation promotes wakefulness (90) and simultaneously increases protein kinase C (PKC) activity in vitro (91). One effect of amphetamine exposure in vivo is the phosphorylation of known PKC targets (92), although this effect has yet to be linked to TAAR1 activation, specifically. PKC activation via direct TAAR1 stimulation may therefore contribute to methamphetamine-induced hypersomnolence. It is possible that this unique property of amphetamines relative to modafinil underlies, at least in part, their distinct effects on sleep-wake cycles. In this context, it would be informative to measure the impact of wakefulness induced by selective TAAR1 receptor ligands on subsequent sleep and to determine whether TAAR1 knockout alters the course of sleep-wake cycles subsequent to methamphetamine-induced wakefulness.
Finally, there are pharmacokinetic differences between modafinil and other DAT-binding agents. The timing of peak plasma levels and the plasma half-life of orally administered racemic modafinil in humans are twice those of methylphenidate (93). Half-life is enantiomer-specific: the half-life of R-modafinil [15 h at an oral dose of 50–400 mg; (94)] is threefold greater than that of S-modafinil (3). Peak plasma levels are achieved much more rapidly with nasally administered cocaine (95), smoked cocaine (96), or orally administered methylphenidate (93). These agents yield peak plasma concentrations within an hour of administration. The pharmacokinetic difference between modafinil and cocaine is accompanied by a difference in the temporal profile of the effects of these compounds in vivo on extracellular DA in the nucleus accumbens in mice. Whereas cocaine caused extracellular DA concentrations to increase to peak levels by 30 min and decrease to less than half of peak values within an hour, modafinil-induced elevation of extracellular DA did not peak until approximately 1 h after administration and remained at peak values until the experiment was terminated at 6 h (12). Table 2 summarizes the similarities and differences between modafinil and classical stimulants discussed in the preceding section.
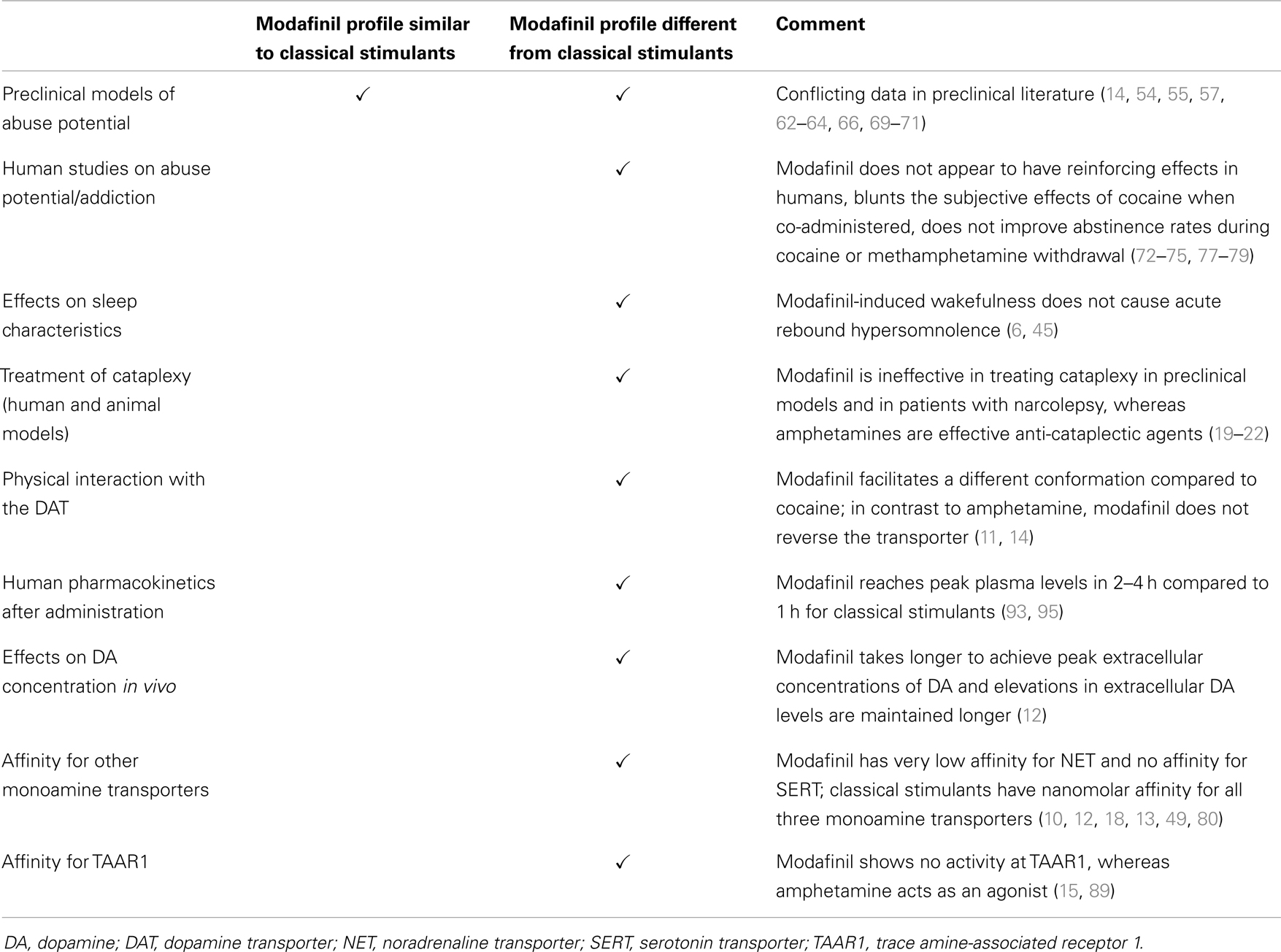
Table 2. Effects of modafinil compared to classical stimulants.
The relative reinforcing efficacy of DAT-binding agents is inversely proportional to their pharmacokinetic profiles: those compounds that are rapidly absorbed into and rapidly cleared from the system exhibit more addictive potential than those with slower kinetics (97–99). Therefore, putative differences in addictive potential and perceived rewarding effects between modafinil and DAT-binding agents such as cocaine, methamphetamine, and methylphenidate do not require the invocation of distinct sites of action.
Conclusion
Parsimony dictates that distinct effects on clinically relevant measures be attributed to the known pharmacological differences between modafinil and other DAT-binding compounds, rather than to occult, unknown effects of modafinil at sites other than DAT. Modafinil elevates extracellular DA concentrations by binding to and disrupting the activity of the cell-membrane DAT. The resulting elevation of extracellular DA, through increased stimulation of dopaminergic and adrenergic receptors, results in wakefulness. Other signaling mechanisms that have been implicated in the response to modafinil are likely to be secondary to its catecholaminergic effects. While modafinil can be distinguished from other catecholaminergic agents in terms of clinical endpoints such as post-treatment hypersomnolence and its potential for addiction and abuse, these differences may be due to differences in selectivity for DAT, pharmacokinetics, or distinctions in the physicochemical nature of their interactions with the DAT. Additional work might clarify the exact basis for these subtle distinctions among wake-promoting agents. Still, the preponderance of information on the mechanism of action of modafinil points us in the direction of the DAT.
Future efforts to understand and therapeutically exploit wake-promoting mechanisms might better be directed toward more innovative strategies. The use of ligands for histamine H3 receptors (100), hypocretin receptors (101), or TAAR1 receptors (90) for instance, to ameliorate potential cognitive deficits secondary to sleep loss, is a relatively novel and potentially impactful strategy. The application of non-pharmacological approaches to treat performance deficits secondary to sleep insufficiency also has potential. In the context of narcolepsy, regenerative therapies for the hypocretin system, which undergoes degeneration in narcolepsy, are worthy of attention and effort (102, 103). In addition, transcranial manipulation of the electrical activity of the cerebral cortex (ultimately the seat of fatigue-related deficits) for therapeutic intervention is fast becoming a reality (104, 105).
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Assistance with the initial literature search for this article was provided by Nancy Holland, Ph.D., of Synchrony Medical Communications, West Chester, PA, USA under the direction of the author. Funding for this support was provided by Teva Pharmaceuticals, North Wales, PA, USA.
References
1. Bastuji H, Jouvet M. Successful treatment of idiopathic hypersomnia and narcolepsy with modafinil. Prog Neuropsychopharmacol Biol Psychiatry (1988) 12(5):695–700. doi:10.1016/0278-5846(88)90014-0
3. Wong YN, Simcoe D, Hartman LN, Laughton WB, King SP, McCormick GC, et al. A double-blind, placebo-controlled, ascending-dose evaluation of the pharmacokinetics and tolerability of modafinil tablets in healthy male volunteers. J Clin Pharmacol (1999) 39:30–40. doi:10.1177/00912709922007534
5. Ballas CA, Kim D, Baldassano CF, Hoeh N. Modafinil: past, present and future. Expert Rev Neurother (2002) 2(4):449–57. doi:10.1586/14737175.2.4.449
6. Edgar DM, Seidel WF. Modafinil induces wakefulness without intensifying motor activity or subsequent rebound hypersomnolence in the rat. J Pharmacol Exp Ther (1997) 283(2):757–69.
7. Hendricks JC, Kirk D, Panckeri K, Miller MS, Pack AI. Modafinil maintains waking in the fruit fly Drosophila melanogaster. Sleep (2003) 26(2):139–46.
8. Kim D. Practical use and risk of modafinil, a novel waking drug. Environ Health Toxicol (2012) 27:e2012007. doi:10.5620/eht.2012.27.e2012007
9. Giros B, Wang YM, Suter S, McLeskey SB, Pifl C, Caron MG. Delineation of discrete domains for substrate, cocaine, and tricyclic antidepressant interactions using chimeric dopamine-norepinephrine transporters. J Biol Chem (1994) 269(23):15985–8.
10. Mignot E, Nishino S, Guilleminault C, Dement WC. Modafinil binds to the dopamine uptake carrier site with low affinity. Sleep (1994) 17(5):436–7.
11. Schmitt KC, Reith ME. The atypical stimulant and nootropic modafinil interacts with the dopamine transporter in a different manner than classical cocaine-like inhibitors. PLoS ONE (2011) 6(10):e25790. doi:10.1371/journal.pone.0025790
12. Loland CJ, Mereu M, Okunola OM, Cao J, Prisinzano TE, Mazier S, et al. R-modafinil (armodafinil): a unique dopamine uptake inhibitor and potential medication for psychostimulant abuse. Biol Psychiatry (2012) 72(5):405–13. doi:10.1016/j.biopsych.2012.03.022
13. Nishino S, Mao J, Sampathkumaran R, Shelton J. Increased dopaminergic transmission mediates the wake-promoting effects of CNS stimulants. Sleep Res Online (1998) 1(1):49–61.
14. Zolkowska D, Jain R, Rothman RB, Partilla JS, Roth BL, Setola V, et al. Evidence for the involvement of dopamine transporters in behavioral stimulant effects of modafinil. J Pharmacol Exp Ther (2009) 329(2):738–46. doi:10.1124/jpet.108.146142
15. Madras BK, Xie Z, Lin Z, Jassen A, Panas H, Lynch L, et al. Modafinil occupies dopamine and norepinephrine transporters in vivo and modulates the transporters and trace amine activity in vitro. J Pharmacol Exp Ther (2006) 319(2):561–9. doi:10.1124/jpet.106.106583
16. Volkow ND, Fowler JS, Logan J, Alexoff D, Zhu W, Telang F, et al. Effects of modafinil on dopamine and dopamine transporters in the male human brain: clinical implications. JAMA (2009) 301(11):1148–54. doi:10.1001/jama.2009.351
17. Wisor JP, Nishino S, Sora I, Uhl GH, Mignot E, Edgar DM. Dopaminergic role in stimulant-induced wakefulness. J Neurosci (2001) 21(5):1787–94.
18. Cao J, Prisinzano TE, Okunola OM, Kopajtic T, Shook M, Katz JL, et al. Structure-activity relationships at the monoamine transporters for a novel series of modafinil (2-[(diphenylmethyl)sulfinyl]acetamide) analogues. ACS Med Chem Lett (2010) 2(1):48–52. doi:10.1021/ml1002025
19. Billiard M, Besset A, Montplaisir J, Laffont F, Goldenberg F, Weill JS, et al. Modafinil: a double-blind multicentric study. Sleep (1994) 17(8 Suppl):S107–12.
20. Fujiki N, Cheng T, Yoshino F, Nishino S. Specificity of direct transition from wake to REM sleep in orexin/ataxin-3 transgenic narcoleptic mice. Exp Neurol (2009) 217(1):46–54. doi:10.1016/j.expneurol.2009.01.015
21. Shelton J, Nishino S, Vaught J, Dement WC, Mignot E. Comparative effects of modafinil and amphetamine on daytime sleepiness and cataplexy of narcoleptic dogs. Sleep (1995) 18(10):817–26.
22. Mignot E, Renaud A, Nishino S, Arrigoni J, Guilleminault C, Dement WC. Canine cataplexy is preferentially controlled by adrenergic mechanisms: evidence using monoamine selective uptake inhibitors and release enhancers. Psychopharmacology (Berl) (1993) 113(1):76–82. doi:10.1007/BF02244337
23. Uguen M, Perrin D, Belliard S, Ligneau X, Beardsley PM, Lecomte JM, et al. Preclinical evaluation of the abuse potential of Pitolisant, a histamine H(3) receptor inverse agonist/antagonist compared with Modafinil. Br J Pharmacol (2013) 169:632–44. doi:10.1111/bph.12149
24. Qu WM, Huang ZL, Xu XH, Matsumoto N, Urade Y. Dopaminergic D1 and D2 receptors are essential for the arousal effect of modafinil. J Neurosci (2008) 28(34):8462–9. doi:10.1523/JNEUROSCI.1819-08.2008
25. Seeman P, Guan HC, Hirbec H. Dopamine D2High receptors stimulated by phencyclidines, lysergic acid diethylamide, salvinorin A, and modafinil. Synapse (2009) 63:698–704. doi:10.1002/syn.20647
26. Duteil J, Rambert FA, Pessonnier J, Hermant JF, Gombert R, Assous E. Central alpha 1-adrenergic stimulation in relation to the behaviour stimulating effect of modafinil; studies with experimental animals. Eur J Pharmacol (1990) 180(1):49–58. doi:10.1016/0014-2999(90)90591-S
27. Lin JS, Roussel B, Akaoka H, Fort P, Debilly G, Jouvet M. Role of catecholamines in the modafinil and amphetamine induced wakefulness, a comparative pharmacological study in the cat. Brain Res (1992) 591(2):319–26. doi:10.1016/0006-8993(92)91713-O
28. Zhang WP, Ouyang M, Thomas SA. Potency of catecholamines and other L-tyrosine derivatives at the cloned mouse adrenergic receptors. Neuropharmacology (2004) 47(3):438–49. doi:10.1016/j.neuropharm.2004.04.017
29. De Saint Hilaire Z, Orosco M, Rouch C, Blanc G, Nicolaidis S. Variations in extracellular monoamines in the prefrontal cortex and medial hypothalamus after modafinil administration: a microdialysis study in rats. Neuroreport (2001) 12(16):3533–7. doi:10.1097/00001756-200111160-00032
30. Pan WH, Yang SY, Lin SK. Neurochemical interaction between dopaminergic and noradrenergic neurons in the medial prefrontal cortex. Synapse (2004) 53(1):44–52. doi:10.1002/syn.20034
31. Ferraro L, Antonelli T, O’Connor WT, Tanganelli S, Rambert FA, Fuxe K. The effects of modafinil on striatal, pallidal and nigral GABA and glutamate release in the conscious rat: evidence for a preferential inhibition of striato-pallidal GABA transmission. Neurosci Lett (1998) 253(2):135–8. doi:10.1016/S0304-3940(98)00629-6
32. Tanganelli S, Ferraro L, Bianchi C, Fuxe K. 6-Hydroxy-dopamine treatment counteracts the reduction of cortical GABA release produced by the vigilance promoting drug modafinil in the awake freely moving guinea-pig. Neurosci Lett (1994) 171(1–2):201–4. doi:10.1016/0304-3940(94)90639-4
33. Tanganelli S, Fuxe K, Ferraro L, Janson AM, Bianchi C. Inhibitory effects of the psychoactive drug modafinil on gamma-aminobutyric acid outflow from the cerebral cortex of the awake freely moving guinea-pig. Possible involvement of 5-hydroxytryptamine mechanisms. Naunyn Schmiedebergs Arch Pharmacol (1992) 345(4):461–5. doi:10.1007/BF00176625
34. Abekawa T, Ohmori T, Ito K, Koyama T. D1 dopamine receptor activation reduces extracellular glutamate and GABA concentrations in the medial prefrontal cortex. Brain Res (2000) 867(1–2):250–4. doi:10.1016/S0006-8993(00)02298-8
35. Ferraro L, Antonelli T, O’Connor WT, Tanganelli S, Rambert F, Fuxe K. The antinarcoleptic drug modafinil increases glutamate release in thalamic areas and hippocampus. Neuroreport (1997) 8(13):2883–7. doi:10.1097/00001756-199709080-00016
36. Exposito I, Sanz B, Porras A, Mora F. Effects of apomorphine and L-methionine sulphoximine on the release of excitatory amino acid neurotransmitters and glutamine in the striatum of the conscious rat. Eur J Neurosci (1994) 6(2):287–91. doi:10.1111/j.1460-9568.1994.tb00271.x
37. Porras A, Mora F. Dopamine – glutamate – GABA interactions and ageing: studies in the striatum of the conscious rat. Eur J Neurosci (1995) 7(11):2183–8. doi:10.1111/j.1460-9568.1995.tb00640.x
38. David HN, Ansseau M, Abraini JH. Dopamine-glutamate reciprocal modulation of release and motor responses in the rat caudate-putamen and nucleus accumbens of “intact” animals. Brain Res Brain Res Rev (2005) 50(2):336–60. doi:10.1016/j.brainresrev.2005.09.002
39. Clinckers R, Smolders I, Meurs A, Ebinger G, Michotte Y. Anticonvulsant action of hippocampal dopamine and serotonin is independently mediated by D and 5-HT receptors. J Neurochem (2004) 89(4):834–43. doi:10.1111/j.1471-4159.2004.02355.x
40. Minzenberg MJ, Carter CS. Modafinil: a review of neurochemical actions and effects on cognition. Neuropsychopharmacology (2008) 33(7):1477–502. doi:10.1038/sj.npp.1301534
41. Wisor JP, O’Hara BF, Terao A, Selby CP, Kilduff TS, Sancar A, et al. A role for cryptochromes in sleep regulation. BMC Neurosci (2002) 3:20. doi:10.1186/1471-2202-3-20
42. Drapeau C, Carrier J. Fluctuation of waking electroencephalogram and subjective alertness during a 25-hour sleep-deprivation episode in young and middle-aged subjects. Sleep (2004) 27(1):55–60.
43. Spiegel K, Leproult R, Van CE. Impact of sleep debt on metabolic and endocrine function. Lancet (1999) 354(9188):1435–9. doi:10.1016/S0140-6736(99)01376-8
44. Borbély AA, Achermann P. Sleep homeostasis and models of sleep regulation. 3rd ed. In: Kryger MH, Roth T, Dement WC, editors. Principles and Practice of Sleep Medicine. Philadelphia: Saunders (2000). p. 377–90.
45. Wisor JP, Dement WC, Aimone L, Williams M, Bozyczko-Coyne D. Armodafinil, the R-enantiomer of modafinil: wake-promoting effects and pharmacokinetic profile in the rat. Pharmacol Biochem Behav (2006) 85(3):492–9. doi:10.1016/j.pbb.2006.09.018
46. Kopp C, Petit JM, Magistretti P, Borbely AA, Tobler I. Comparison of the effects of modafinil and sleep deprivation on sleep and cortical EEG spectra in mice. Neuropharmacology (2002) 43(1):110–8. doi:10.1016/S0028-3908(02)00070-9
47. Bodenmann S, Hohoff C, Freitag C, Deckert J, Retey JV, Bachmann V, et al. Polymorphisms of ADORA2A modulate psychomotor vigilance and the effects of caffeine on neurobehavioural performance and sleep EEG after sleep deprivation. Br J Pharmacol (2012) 165(6):1904–13. doi:10.1111/j.1476-5381.2011.01689.x
48. Bodenmann S, Xu S, Luhmann UF, Arand M, Berger W, Jung HH, et al. Pharmacogenetics of modafinil after sleep loss: catechol-O-methyltransferase genotype modulates waking functions but not recovery sleep. Clin Pharmacol Ther (2009) 85(3):296–304. doi:10.1038/clpt.2008.222
49. Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, et al. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse (2001) 39(1):32–41. doi:10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3
50. Cirelli C, Huber R, Gopalakrishnan A, Southard TL, Tononi G. Locus ceruleus control of slow-wave homeostasis. J Neurosci (2005) 25(18):4503–11. doi:10.1523/JNEUROSCI.4845-04.2005
51. Imeri L, Opp MR. How (and why) the immune system makes us sleep. Nat Rev Neurosci (2009) 10(3):199–210. doi:10.1038/nrn2576
52. Jouvet M, Bobillier P, Pujol JF, Renault J. Permanent insomnia and diminution of cerebral serotonin due to lesion of the raphe system in cats. J Physiol Paris (1967) 59(1 Suppl):248.
53. Morrow JD, Vikraman S, Imeri L, Opp MR. Effects of serotonergic activation by 5-hydroxytryptophan on sleep and body temperature of C57BL/6J and interleukin-6-deficient mice are dose and time related. Sleep (2008) 31(1):21–33.
54. Paterson NE, Fedolak A, Olivier B, Hanania T, Ghavami A, Caldarone B. Psychostimulant-like discriminative stimulus and locomotor sensitization properties of the wake-promoting agent modafinil in rodents. Pharmacol Biochem Behav (2010) 95(4):449–56. doi:10.1016/j.pbb.2010.03.006
55. Shuman T, Cai DJ, Sage JR, Anagnostaras SG. Interactions between modafinil and cocaine during the induction of conditioned place preference and locomotor sensitization in mice: implications for addiction. Behav Brain Res (2012) 235(2):105–12. doi:10.1016/j.bbr.2012.07.039
56. Webb IC, Pollock MS, Mistlberger RE. Modafinil [2-[(diphenylmethyl) sulfinyl]acetamide] and circadian rhythms in syrian hamsters: assessment of the chronobiotic potential of a novel alerting compound. J Pharmacol Exp Ther (2006) 317:882–9. doi:10.1124/jpet.105.099010
57. Simon P, Hemet C, Ramassamy C, Costentin J. Non-amphetaminic mechanism of stimulant locomotor effect of modafinil in mice. Eur Neuropsychopharmacol (1995) 5(4):509–14. doi:10.1016/0924-977X(95)80011-P
58. Chang ST, Tung CS, Lin YL, Chuang CH, Lee AR, Liu YP. Behavioral and cross sensitization after repeated exposure to modafinil and apomorphine in rats. Chin J Physiol (2010) 53:318–27. doi:10.4077/CJP.2010.AMK067
59. da Costa CS, Moreira KD, Abrahao KP, Quadros IM, Oliveira MG. Individual differences are critical in determining modafinil-induced behavioral sensitization and cross-sensitization with methamphetamine in mice. Behav Brain Res (2012) 233:367–74. doi:10.1016/j.bbr.2012.05.023
60. Jasinski DR. An evaluation of the abuse potential of modafinil using methylphenidate as a reference. J Psychopharmacol (2000) 14(1):53–60. doi:10.1177/026988110001400107
61. Myrick H, Malcolm R, Taylor B, LaRowe S. Modafinil: preclinical, clinical, and post-marketing surveillance – a review of abuse liability issues. Ann Clin Psychiatry (2004) 16(2):101–9. doi:10.1080/10401230490453743
62. Dopheide MM, Morgan RE, Rodvelt KR, Schachtman TR, Miller DK. Modafinil evokes striatal [(3)H]dopamine release and alters the subjective properties of stimulants. Eur J Pharmacol (2007) 568(1–3):112–23. doi:10.1016/j.ejphar.2007.03.044
63. Gold LH, Balster RL. Evaluation of the cocaine-like discriminative stimulus effects and reinforcing effects of modafinil. Psychopharmacology (Berl) (1996) 126(4):286–92. doi:10.1007/BF02247379
64. Newman JL, Negus SS, Lozama A, Prisinzano TE, Mello NK. Behavioral evaluation of modafinil and the abuse-related effects of cocaine in rhesus monkeys. Exp Clin Psychopharmacol (2010) 18(5):395–408. doi:10.1037/a0021042
65. Heal DJ, Buckley NW, Gosden J, Slater N, France CP, Hackett D. A preclinical evaluation of the discriminative and reinforcing properties of lisdexamfetamine in comparison to d-amfetamine, methylphenidate and modafinil. Neuropharmacology (2013) 73C:348–58. doi:10.1016/j.neuropharm.2013.05.021
66. Nguyen TL, Tian YH, You IJ, Lee SY, Jang CG. Modafinil-induced conditioned place preference via dopaminergic system in mice. Synapse (2011) 65(8):733–41. doi:10.1002/syn.20892
67. Quisenberry AJ, Prisinzano TE, Baker LE. Modafinil alone and in combination with low dose amphetamine does not establish conditioned place preference in male Sprague-Dawley rats. Exp Clin Psychopharmacol (2013) 21:252–8. doi:10.1037/a0031832
68. Mahler SV, Hensley-Simon M, Tahsili-Fahadan P, Lalumiere RT, Thomas C, Fallon RV, et al. Modafinil attenuates reinstatement of cocaine seeking: role for cystine-glutamate exchange and metabotropic glutamate receptors. Addict Biol (2013). doi:10.1111/j.1369-1600.2012.00506.x
69. Holtz NA, Lozama A, Prisinzano TE, Carroll ME. Reinstatement of methamphetamine seeking in male and female rats treated with modafinil and allopregnanolone. Drug Alcohol Depend (2012) 120(1–3):233–7. doi:10.1016/j.drugalcdep.2011.07.010
70. Reichel CM, See RE. Modafinil effects on reinstatement of methamphetamine seeking in a rat model of relapse. Psychopharmacology (Berl) (2010) 210(3):337–46. doi:10.1007/s00213-010-1828-5
71. Reichel CM, See RE. Chronic modafinil effects on drug-seeking following methamphetamine self-administration in rats. Int J Neuropsychopharmacol (2012) 15(7):919–29. doi:10.1017/S1461145711000988
72. Rush CR, Kelly TH, Hays LR, Baker RW, Wooten AF. Acute behavioral and physiological effects of modafinil in drug abusers. Behav Pharmacol (2002) 13(2):105–15. doi:10.1097/00008877-200203000-00002
73. Rush CR, Kelly TH, Hays LR, Wooten AF. Discriminative-stimulus effects of modafinil in cocaine-trained humans. Drug Alcohol Depend (2002) 67(3):311–22. doi:10.1016/S0376-8716(02)00082-0
74. Hart CL, Haney M, Vosburg SK, Rubin E, Foltin RW. Smoked cocaine self-administration is decreased by modafinil. Neuropsychopharmacology (2008) 33(4):761–8. doi:10.1038/sj.npp.1301472
75. Malcolm R, Swayngim K, Donovan JL, DeVane CL, Elkashef A, Chiang N, et al. Modafinil and cocaine interactions. Am J Drug Alcohol Abuse (2006) 32(4):577–87. doi:10.1080/00952990600920425
76. Anderson AL, Reid MS, Li SH, Holmes T, Shemanski L, Slee A, et al. Modafinil for the treatment of cocaine dependence. Drug Alcohol Depend (2009) 104:133–9. doi:10.1016/j.drugalcdep.2009.04.015
77. Anderson AL, Li SH, Biswas K, McSherry F, Holmes T, Iturriaga E, et al. Modafinil for the treatment of methamphetamine dependence. Drug Alcohol Depend (2012) 120(1–3):135–41. doi:10.1016/j.drugalcdep.2011.07.007
78. Shearer J, Darke S, Rodgers C, Slade T, van BI, Lewis J, et al. A double-blind, placebo-controlled trial of modafinil (200 mg/day) for methamphetamine dependence. Addiction (2009) 104(2):224–33. doi:10.1111/j.1360-0443.2008.02437.x
79. Dackis CA, Kampman KM, Lynch KG, Plebani JG, Pettinati HM, Sparkman T, et al. A double-blind, placebo-controlled trial of modafinil for cocaine dependence. J Subst Abuse Treat (2012) 43(3):303–12. doi:10.1016/j.jsat.2011.12.014
80. Kozikowski AP, Zhao L, Zhang A, Wang CZ, Flippen-Anderson J, Johnson KM. Structural remodeling of cocaine: design and synthesis of trisubstituted cyclopropanes as selective serotonin reuptake inhibitors. ChemMedChem (2006) 1(1):58–65. doi:10.1002/cmdc.200500016
81. Brown JM, Hanson GR, Fleckenstein AE. Regulation of the vesicular monoamine transporter-2: a novel mechanism for cocaine and other psychostimulants. J Pharmacol Exp Ther (2001) 296(3):762–7.
82. Sora I, Wichems C, Takahashi N, Li XF, Zeng Z, Revay R, et al. Cocaine reward models: conditioned place preference can be established in dopamine- and in serotonin-transporter knockout mice. Proc Natl Acad Sci U S A (1998) 95:7699–704. doi:10.1073/pnas.95.13.7699
83. Hall FS, Li XF, Sora I, Xu F, Caron M, Lesch KP, et al. Cocaine mechanisms: enhanced cocaine, fluoxetine and nisoxetine place preferences following monoamine transporter deletions. Neuroscience (2002) 115(1):153–61. doi:10.1016/S0306-4522(02)00379-2
84. Tilley MR, Cagniard B, Zhuang X, Han DD, Tiao N, Gu HH. Cocaine reward and locomotion stimulation in mice with reduced dopamine transporter expression. BMC Neurosci (2007) 8:42. doi:10.1186/1471-2202-8-42
85. Thomsen M, Han DD, Gu HH, Caine SB. Lack of cocaine self-administration in mice expressing a cocaine-insensitive dopamine transporter. J Pharmacol Exp Ther (2009) 331:204–11. doi:10.1124/jpet.109.156265
86. Chen R, Tilley MR, Wei H, Zhou F, Zhou FM, Ching S, et al. Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter. Proc Natl Acad Sci U S A (2006) 103:9333–8. doi:10.1073/pnas.0600905103
87. Schmitt K, Rothman RB, Reith M. Non-classical pharmacology of the dopamine transporter: atypical inhibitors, allosteric modulators and partial substrates. J Pharmacol Exp Ther (2013) 346(1):2–10. doi:10.1124/jpet.111.191056
88. Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, et al. The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat Neurosci (2008) 11(7):780–9. doi:10.1038/nn.2146
89. Xie Z, Miller GM. A receptor mechanism for methamphetamine action in dopamine transporter regulation in brain. J Pharmacol Exp Ther (2009) 330(1):316–25. doi:10.1124/jpet.109.153775
90. Revel FG, Moreau JL, Pouzet B, Mory R, Bradaia A, Buchy D, et al. A new perspective for schizophrenia: TAAR1 agonists reveal antipsychotic- and antidepressant-like activity, improve cognition and control body weight. Mol Psychiatry (2013) 18(5):543–56. doi:10.1038/mp.2012.57
91. Xie Z, Miller GM. Trace amine-associated receptor 1 is a modulator of the dopamine transporter. J Pharmacol Exp Ther (2007) 321(1):128–36. doi:10.1124/jpet.106.117382
92. Szabo ST, Machado-Vieira R, Yuan P, Wang Y, Wei Y, Falke C, et al. Glutamate receptors as targets of protein kinase C in the pathophysiology and treatment of animal models of mania. Neuropharmacology (2009) 56(1):47–55. doi:10.1016/j.neuropharm.2008.08.015
93. Wong YN, King SP, Laughton WB, McCormick GC, Grebow PE. Single-dose pharmacokinetics of modafinil and methylphenidate given alone or in combination in healthy male volunteers. J Clin Pharmacol (1998) 38(3):276–82. doi:10.1002/j.1552-4604.1998.tb04395.x
94. Darwish M, Kirby M, Hellriegel ET, Yang R, Robertson P. Pharmacokinetic profile of armodafinil in healthy subjects: pooled analysis of data from three randomized studies. Clin Drug Investig (2009) 29:87–100. doi:10.2165/0044011-200929020-00003
95. Lukas SE, Sholar M, Kouri E, Fukuzako H, Mendelson JH. Marijuana smoking increases plasma cocaine levels and subjective reports of euphoria in male volunteers. Pharmacol Biochem Behav (1994) 48(3):715–21. doi:10.1016/0091-3057(94)90338-7
96. Jenkins AJ, Keenan RM, Henningfield JE, Cone EJ. Correlation between pharmacological effects and plasma cocaine concentrations after smoked administration. J Anal Toxicol (2002) 26(7):382–92. doi:10.1093/jat/26.7.382
97. Kimmel HL, Negus SS, Wilcox KM, Ewing SB, Stehouwer J, Goodman MM, et al. Relationship between rate of drug uptake in brain and behavioral pharmacology of monoamine transporter inhibitors in rhesus monkeys. Pharmacol Biochem Behav (2008) 90(3):453–62. doi:10.1016/j.pbb.2008.03.032
98. Wee S, Carroll FI, Woolverton WL. A reduced rate of in vivo dopamine transporter binding is associated with lower relative reinforcing efficacy of stimulants. Neuropsychopharmacology (2006) 31(2):351–62. doi:10.1038/sj.npp.1300795
99. Woolverton WL, Wang Z. Relationship between injection duration, transporter occupancy and reinforcing strength of cocaine. Eur J Pharmacol (2004) 486(3):251–7. doi:10.1016/j.ejphar.2004.01.003
100. Barbier AJ, Berridge C, Dugovic C, Laposky AD, Wilson SJ, Boggs J, et al. Acute wake-promoting actions of JNJ-5207852, a novel, diamine-based H3 antagonist. Br J Pharmacol (2004) 143(5):649–61. doi:10.1038/sj.bjp.0705964
101. Dietrich H, Jenck F. Intact learning and memory in rats following treatment with the dual orexin receptor antagonist almorexant. Psychopharmacology (Berl) (2010) 212(2):145–54. doi:10.1007/s00213-010-1933-5
102. Blanco-Centurion C, Liu M, Konadhode R, Pelluru D, Shiromani PJ. Effects of orexin gene transfer in the dorsolateral pons in orexin knockout mice. Sleep (2013) 36(1):31–40. doi:10.5665/sleep.2296
103. Liu M, Blanco-Centurion C, Konadhode R, Begum S, Pelluru D, Gerashchenko D, et al. Orexin gene transfer into zona incerta neurons suppresses muscle paralysis in narcoleptic mice. J Neurosci (2011) 31(16):6028–40. doi:10.1523/JNEUROSCI.6069-10.2011
104. Cohen DA, Freitas C, Tormos JM, Oberman L, Eldaief M, Pascual-Leone A. Enhancing plasticity through repeated rTMS sessions: the benefits of a night of sleep. Clin Neurophysiol (2010) 121(12):2159–64. doi:10.1016/j.clinph.2010.05.019
Keywords: modafinil, amphetamines, cocaine, dopamine transporter, monoamines, sleep homeostasis, addiction, wake-promoting therapeutics
Citation: Wisor J (2013) Modafinil as a catecholaminergic agent: empirical evidence and unanswered questions. Front. Neurol. 4:139. doi: 10.3389/fneur.2013.00139
Received: 28 June 2013; Paper pending published: 15 July 2013;
Accepted: 06 September 2013; Published online: 07 October 2013.
Edited by:
Ralph E. Mistlberger, Simon Fraser University, CanadaReviewed by:
J. M. Monti, Clinics Hospital, UruguayIan C. Webb, University of Mississippi Medical Center, USA
Maarten E. Reith, New York University School of Medicine, USA
Copyright: © 2013 Wisor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan Wisor, WWAMI Medical Education Program, Department of Integrative Physiology and Neuroscience, Sleep and Performance Research Center, Washington State University, 280K Health Sciences Building, Spokane, WA 99210-1945, USA e-mail: jonathan.wisor@wsu.edu