 Carmela Giordano1
Carmela Giordano1 Maddalena Marchiò
Maddalena Marchiò Elena Timofeeva
Elena Timofeeva Giuseppe Biagini
Giuseppe Biagini- 1Laboratory of Experimental Epileptology, Department of Biomedical, Metabolic and Neural Sciences, University of Modena and Reggio Emilia, Modena, Italy
- 2Neuropediatric Unit, Department of Medical and Surgical Sciences for Children and Adults, Policlinico Hospital, University of Modena and Reggio Emilia, Modena, Italy
- 3Department of Neurosciences, NOCSAE Hospital, Modena, Italy
- 4Département Psychiatrie et Neurosciences, Faculté de Médecine, Centre de Recherche de l’Institut Universitaire de Cardiologie et de Pneumologie de Québec, Université Laval, Québec, QC, Canada
Various ketogenic diet (KD) therapies, including classic KD, medium chain triglyceride administration, low glycemic index treatment, and a modified Atkins diet, have been suggested as useful in patients affected by pharmacoresistant epilepsy. A common goal of these approaches is to achieve an adequate decrease in the plasma glucose level combined with ketogenesis, in order to mimic the metabolic state of fasting. Although several metabolic hypotheses have been advanced to explain the anticonvulsant effect of KDs, including changes in the plasma levels of ketone bodies, polyunsaturated fatty acids, and brain pH, direct modulation of neurotransmitter release, especially purinergic (i.e., adenosine) and γ-aminobutyric acidergic neurotransmission, was also postulated. Neuropeptides and peptide hormones are potent modulators of synaptic activity, and their levels are regulated by metabolic states. This is the case for neuroactive peptides such as neuropeptide Y, galanin, cholecystokinin, and peptide hormones such as leptin, adiponectin, and growth hormone-releasing peptides (GHRPs). In particular, the GHRP ghrelin and its related peptide des-acyl ghrelin are well-known controllers of energy homeostasis, food intake, and lipid metabolism. Notably, ghrelin has also been shown to regulate the neuronal excitability and epileptic activation of neuronal networks. Several lines of evidence suggest that GHRPs are upregulated in response to starvation and, particularly, in patients affected by anorexia and cachexia, all conditions in which also ketone bodies are upregulated. Moreover, starvation and anorexia nervosa are accompanied by changes in other peptide hormones such as adiponectin, which has received less attention. Adipocytokines such as adiponectin have also been involved in modulating epileptic activity. Thus, neuroactive peptides whose plasma levels and activity change in the presence of ketogenesis might be potential candidates for elucidating the neurohormonal mechanisms involved in the beneficial effects of KDs. In this review, we summarize the current evidence for altered regulation of the synthesis of neuropeptides and peripheral hormones in response to KDs, and we try to define a possible role for specific neuroactive peptides in mediating the antiepileptic properties of diet-induced ketogenesis.
The lack of a satisfactory response to pharmacological therapy in patients affected by epilepsy is a major neurological problem. In addition, in patients affected by epilepsy, antiepileptic drugs (AEDs) may induce serious side effects. Finding a solution to pharmacoresistance has been recently recognized as a priority by clinicians, parents, and stakeholders (1). Although various new AEDs are available to address refractory epilepsy, no improvements in treating drug resistance have been obtained, even with the release of additional new drugs (2). Alternatively to drug therapy, pharmacoresistant patients could be eligible for treatments such as: (i) neurosurgical resection of epileptic foci, (ii) intracranial or extracranial neurostimulation, and (iii) ketogenic diets (KDs).
Surgical resection appears to be effective in a high percentage (up to 73% are seizure free after 1 year post-surgery) of patients (3, 4). However, surgical intervention could also result in impairment of cognitive functions, such as verbal and visuospatial memory (5). In any case, the surgical approach to pharmacoresistance is a valuable alternative that is warranted when an epileptic focus is clearly identified, whereas in the opposite case alternative treatments, such as intracranial and extracranial neurostimulation and KDs, have to be considered.
Extracranial or intracranial stimulation has been proposed as a possible therapy for refractory epilepsy. In the first case, stimulation is applied to the vagal nerve (6). Recent meta-analyses of the results obtained with vagal stimulation show the limited efficacy of this approach (7, 8), limitations confirmed by a recent randomized active controlled trial in children (9). Other authors recommended the use of vagal nerve stimulation in children and adults, although the researchers concluded that more information about this technique is required to better define its impact on lifestyle and adverse effects (10). In addition to this extracranial neurostimulation technique, electrodes can be inserted directly into the brain to address specific cerebral regions, such as the cerebellum, centromedian thalamus, subthalamus, neostriatum, hippocampus, and brainstem. A few small trials have addressed the efficacy of intracranial stimulation procedures (11); however, these studies did not address the efficacy or safety of long term stimulation. Low- and high-frequency stimulation protocols have been evaluated, but the studies were inconclusive (12–14).
An effective approach for controlling pharmacoresistant seizures is represented by the nutritional induction of ketogenesis. Until now, the therapeutic use of KDs has been mainly limited to children (15), but recent studies suggest that KDs may be effectively used in adult patients affected by pharmacoresistant epilepsy as well (16). The clinical efficacy of KDs has been suggested by various investigations that were limited by the lack of randomization or a small patient sample (17). One study was based on the prospective analysis of a large (n = 150) sample of children, aged 1–16 years, with more than two refractory seizures/week, followed for 1 year (18). Interestingly, after 6 months of the KD, 71% remained on the diet, and 51% had a more than 50% decrease in seizure frequency (32% had more than 90% seizure reduction). Similar results were reported by a study that involved 20 children (3–16 years), who were treated for 30 months (19). Positive results were also reported in a controlled randomized study (20). However, limitations of the nutritional approach include adverse effects such as nausea, constipation, abdominal pain, or other less frequent complications (20–22). Low compliance with the dietetic regimen is also frequently present (22). Thus, in the long term effective control of seizures was achieved, at best, in approximately 25% of patients under KDs (21).
Dietetic treatments represent a less invasive approach for managing refractory epilepsy. Moreover, the possibility of understanding how KDs work could be of great help in discovering new treatments with possibly minimal side effects and the ability to control seizures unresponsive to currently available drug treatments. Further achievements may result from the discovery of new mechanisms involved in controlling ictogenesis, when studying the effects of KDs. Until now, the reason why ketogenesis is effective in restraining ictogenesis is undetermined. However, several possible mechanisms have been proposed to contribute to KD effects. This review is aimed at defining the current knowledge on the mechanisms putatively involved in the therapeutic effects of KDs. We also analyze the changes in endocrine axes related to fasting and ketogenesis, to tentatively identify the possible contribution of hormone peptides and neuropeptides to the antiepileptic effects of KDs.
KDs: Common Characteristics and Several Differences
The classic KD is a low-carbohydrate/high-fat and normal protein diet that has been used as a treatment for pharmacoresistant epilepsy for a long time (22). The classic KD was initially proposed in 1921 (23) and thereafter was used extensively, although it was partially abandoned when AEDs became fully available. In spite of the development of new effective AEDs, the therapeutic application of the classic KD has survived over the decades as an alternative to drug therapy failure (24). During the course of the last 43 years, other modified KDs have been proposed, and the dietetic approach to epilepsy is being reevaluated as an effective alternative to control seizures refractory to at least two different AEDs. The most recently introduced KDs are all aimed at reducing the lipid content of the diet, in order to increase patient compliance and to limit the side effects (22, 25). Interestingly, a common characteristic of all KDs is maintaining a low-carbohydrate content whereas the fat content is presently less stringent (26).
Among the various types of KDs proposed to ameliorate the balance in diet macronutrients, the use of medium chain triglycerides (MCTs) as an alternative fat source was established at the beginning of the 1970s (27). The main constituents of the MCT diet are medium chain octanoic and decanoic fatty acids, which are absorbed more efficiently than long chain fatty acids and are readily transported to the liver by albumin. Conversely, long chain triglycerides (LCTs) are incorporated into chylomicrons and transported via the thoracic duct into the blood circulation. After hepatic uptake, MCTs are rapidly metabolized by liver mitochondria and, following oxidation, converted to ketone bodies. In contrast, LCTs require carnitine as a carrier to enter the mitochondrial biochemical machinery. These differences in metabolism facilitate more rapid and efficient oxidation of MCTs, supposed to result in a higher ketone yield per kilocalorie of dietary energy than that obtained from LCTs. Therefore, less total fat should be required to achieve the desired level of ketosis. With this method, more proteins and higher carbohydrate content may improve palatability and patient acceptance. However, a recent trial that compared the classic KD with the MCT diet demonstrated significantly higher induction of ketone bodies with the classic KD, but the clinical effectiveness of the two alternative diets was the same (21).
In the last few decades, two other types of KDs have been proposed with success. The modified Atkins diet (MAD) was introduced at the John Hopkins Hospital to restrict the fat content of the classic KD by allowing more carbohydrates and, especially, proteins, without limiting the total caloric intake (25). The MAD is based on the popular weight loss diet first described in 1972 (28), which was adapted to allow carbohydrate intake up to a maximum of 10–20 g/day, and by approximating a fat-to-carbohydrate/protein ratio of 1:1, thus less restrictive compared with the 3:1 or 4:1 ratios of the classic KD. Alternatively, the diet known as “low glycemic index treatment” (LGIT) was first described by Pfeifer and Thiele (29). This diet allows carbohydrate intake up to 40–60 g/day using a selection of carbohydrates with a low (<50) glycemic index. Consumption of carbohydrates with a low glycemic index minimizes the increase in glucose blood levels, keeping glycemia at a lower level after the meal. Food is not weighed but given in portion sizes. Protein, fat, and calorie intake under LGIT is considerably less strictly monitored compared with the classic KD. Unlike the MAD, a high-fat intake is not actively encouraged in LGIT protocols (Table 1).
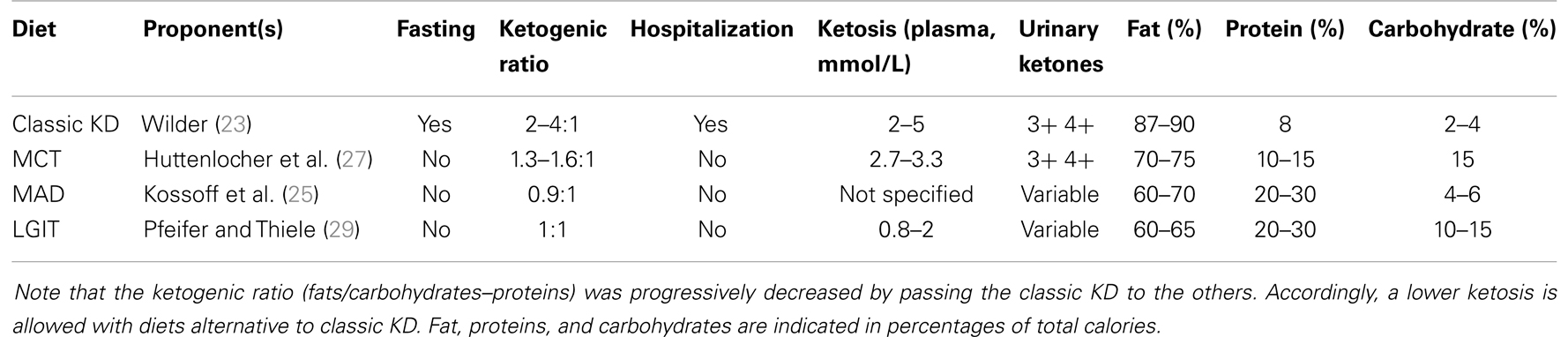
Table 1. Ketogenic diets (KDs) used to treat pharmacoresistant epilepsy are listed.
It is clear that varying the amount of carbohydrates in different diets induces different degrees of ketosis. This variability argues against a critical role of ketogenesis in the efficacy of KDs. A recent study by Neal and collaborators (21) compared the therapeutic effects of different levels of ketosis on seizures in a group of 125 children affected by intractable epilepsy. They were randomized to receive the classic KD or the MCT diet. Consistent with previous reports (30), this study showed that the mean acetoacetate level was significantly higher in the group maintained on the classic KD in the long term, even after 1 year of treatment, with no differences in the antiepileptic efficacy between the KD and MCT treatments. These data do not support the view of a causal relationship between ketosis and seizure control. Indeed, whereas some authors suggest that ketone bodies must be at a sufficient level to guarantee effective control of seizures (31), others disagree and support the view that a strict link between ketosis and seizure control may be questioned (32, 33). Interestingly, other findings support the hypothesis that a decrease in seizure frequency could depend on a fine balance between glucose reduction and ketone body production (34). Thus, ketosis could be just something more than an epiphenomenon of reduced glucose availability, and this latter could be the major player in controlling seizure activity, common to the different dietetic approaches.
Mechanisms Thought to be Involved in KD Effects
During the past few decades, interest in understanding the key mechanism of KDs has steadily been grown. Apparently, the mechanisms of KDs could be easily delineated. This alternative therapeutic approach was designed to mimic the biochemical changes associated with fasting, since fasting was proven to be effective in reducing seizure frequency (22, 23, 25). Thus, the reproduction by dietary manipulation of the ketotic state associated with fasting appears to be the way by which pharmacoresistant epilepsy can be controlled. Ketosis is characterized by increased levels of ketone bodies (β-hydroxybutyric acid, acetoacetic acid, and acetone) that can be achieved by reducing blood glucose levels (35–37). Ketone bodies are the obvious candidates to investigate the mechanism of action by which KDs may control seizure activity. According to Gilbert et al. (31), serum β-hydroxybutyrate levels may correlate with seizure reduction. Although this observation was confirmed by others (38), various studies addressing the anticonvulsant activity of β-hydroxybutyrate failed to demonstrate any relevant effect of this ketone body in different animal models (37, 39–41) or even in vitro (42). However, some investigators demonstrated an anticonvulsant effect of acetoacetate and of its metabolite acetone in various seizure paradigms, including the maximal electroshock test, the subcutaneous pentylenetetrazole test, the amygdala kindling test, and atypical absence seizures, which characterize the Lennox–Gastaut syndrome (43, 44). Interestingly, acetone may act by opening the two-pore-domain potassium channels (K2p), whose characteristic is to hyperpolarize neurons thus limiting neuronal excitability (45). In spite of these encouraging results, in vivo experimental (46) and clinical studies (47) showed that acetone levels did not rise sufficiently to suggest the involvement of ketone bodies in the therapeutic effects of KDs. Specifically, according to proton magnetic resonance spectroscopy studies, acetone levels were around 0.7 mM in the brain of patients maintained on a KD (47) or, based on measurements in the cerebrospinal fluid (48), even lower. In addition, the acetone plasma levels effective in rats must be higher than 2 mM; generally, such levels were never obtained under different KD treatments (46). These findings raise several questions: although investigations of animal models remain the required approach for studying the antiepileptic effects of KDs (49, 50), in every tested animal model the efficacy of KDs has strongly depended on the specific seizure model. Moreover, according to some authors, KDs are not effective in seizure models as they are in patients, with the exception of the 6-Hz corneal stimulation paradigm (51, 52) (Table 2).
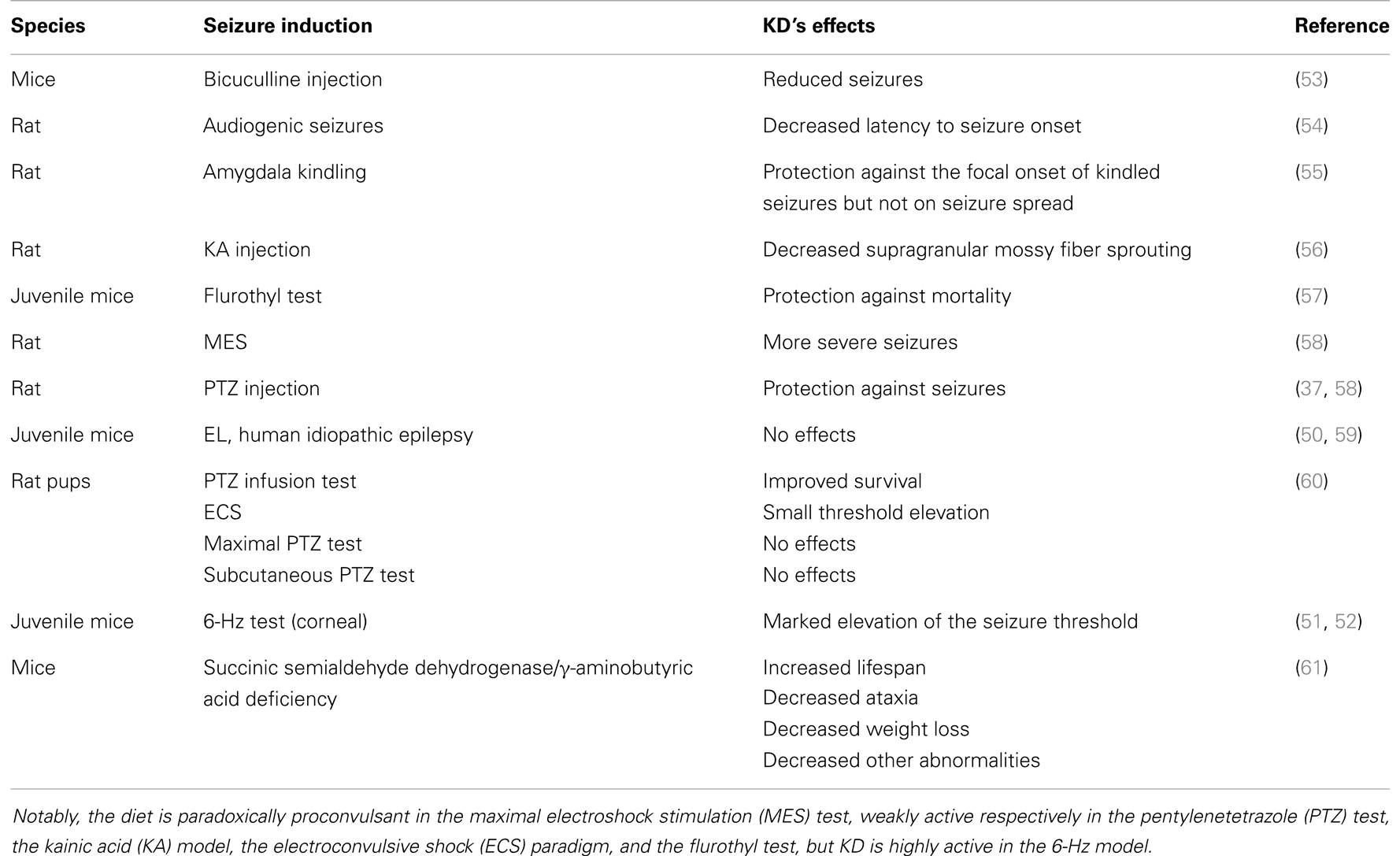
Table 2. Effects of the ketogenic diet (KD) in seizure and epilepsy models.
Inconsistencies between the therapeutic levels of ketone bodies and doses required to produce anticonvulsant activity in in vivo and in vitro models are at the basis of the absence of clear explanations of KD effects. Although evidence of the direct involvement of ketone bodies in KD effects is lacking, changes in the functioning of different neurotransmitters have been described in the presence of ketosis and were extensively analyzed in a recent review (37). Relevant changes in γ-aminobutyric acid (GABA) and glycine levels were demonstrated in the cerebrospinal fluid of patients maintained on KDs, suggesting the enhanced activity of inhibitory neurons (62, 63). Conversely, animal studies failed to demonstrate any change in GABA levels measured in the brain homogenates of rodents maintained on a KD, disconfirming a role for GABA in mediating KD effects (64). Although the overall evidence is against upregulation of GABA by KDs in animal models, electrophysiological recordings demonstrated that network excitability is diminished in animals fed with KDs (65). Another hypothesis is based on the depression of glutamatergic transmission (66), since acetoacetate and, less efficiently, β-hydroxybutyrate can compete with chloride, which is an allosteric activator of vesicular glutamate transport, and this competition could lead to decreased glutamate release. However, the required concentrations of ketone bodies to reduce glutamate release appear to be far from those found in patients (37).
A more recent attempt to identify a mechanism for KDs suggested the involvement of KATP channels, hyperpolarizing ion channels that are opened by low energy levels, i.e., decreased ATP. It has been argued that ketone bodies, which directly enter the Krebs cycle, could reduce glycolytic ATP production thus activating KATP channels (67). Data obtained in in vitro (68, 69) and in vivo (70) experiments supported the involvement of KATP channels. However, the evidence that ATP levels are decreased by KDs is lacking (71–73).
Several other mechanisms have been taken into account, including: (i) cerebral acidosis by itself, tentatively reproduced by the administration of inorganic acids (74). However, animal studies have shown that initial acidosis is then compensated and brain pH results are actually normal (75). Another mechanism is (ii) changes in the accumulation of lipids, such as polyunsaturated fatty acids (PUFAs) (76). PUFAs activate the peroxisome proliferator-activated receptor-α and regulate the transcription of genes that enhance energy metabolism (77). The third mechanism is (iii) increased ATP synthesis leading to the accumulation of adenosine (78). Adenosine stimulates adenosine A1 receptors and may reduce spontaneous seizures, as shown in mice with elevated levels of adenosine (79). The fourth mechanism is (iv) the noradrenergic tone: noradrenaline may modulate the propensity to develop seizures (80, 81), and KDs were shown to enhance noradrenaline tissue levels. Mice lacking the gene for dopamine β-hydroxylase failed to exhibit an elevated flurothyl threshold when fed a KD (82). An intriguing consequence of KD effects on noradrenergic signaling could be the stimulation of the release of food intake modulators, which are known to work in conjunction with hypothalamic neurotransmitters.
New Hypothesis: Peptide Hormones as Possible Mediators of KD Effects
Several peripheral peptides produced in the gut and associated tissues have been suggested to link changes in body metabolism with central nervous system functions and, thus, may be critical regulators in various pathophysiological conditions, including control of neuronal excitability in epilepsy (83–86). Peptide hormones are short molecules composed of approximately 3–100 amino acid residues, characterized by a structure simpler that than for proteins. Some are also synthesized by neurons and termed neuropeptides. Peptides and neuropeptides act through G-protein coupled receptors diffusely expressed in the nervous system. Notably, neuropeptides can act at a distance by diffusing from the releasing site in the extracellular space to interact with extrasynaptic receptors and produce long-lasting effects (87–89). At least one receptor for each peptide hormone has been identified, which means that presumably several hundred receptors can affect a multitude of intracellular transduction pathways, complicating the interpretation of their functions. Many peptides are expressed in neurons that co-express at least one classic transmitter and often more than one neuropeptide (90, 91). Physiologically, peptides mainly behave as neuromodulators with prolonged action on multiple physiological and behavioral actions. The long term effects of peptides depend on the modulation of gene expression via activation of a particular cascade of intracellular signaling molecules (92, 93). However, rapid effects of peptides on neuronal activity by interactions among peptide receptors and ion channels or exchangers have also been described (94, 95). The versatility of peptide hormones and neuropeptide actions explains their multiple physiological effects. In line with the localization of neuropeptides in the hippocampus, amygdala, hypothalamus, striatum, and spinal cord, physiological functions include emotional reactivity, learning and memory, neuroendocrine response to stress, hippocampal synaptic plasticity, and hypothalamic neurotransmission (90, 96–98).
Peptide Hormones Involved in the Control of Metabolism
Metabolism is regulated by various neural and hormonal systems. The latter include hormone peptides that display anorexigenic or orexigenic properties. Peripheral anorexigenic hormones are produced in different tissues, spanning the gut to the pancreas and, finally, including the adipose tissue. Appetite-reducing peptides released by the gut include cholecystokinin (CCK) (99, 100), glucagon-like peptide-1 (GLP-1) (101, 102), peptide YY (PYY) (103, 104), and oxyntomodulin (105, 106). Anorexigenic peptides from the pancreas are the pancreatic polypeptide (PP) (107, 108), glucagon (109), insulin (110, 111), and amylin (112). Finally, hormones that suppress appetite synthesized in the adipose tissue are leptin (113, 114), adiponectin (115), and resistin (116). In contrast to this variety of appetite-suppressing hormones, ghrelin is the only peripheral peptide with orexigenic properties and is mainly produced in the stomach (117, 118). Interestingly, some of the hormones displaying food intake regulatory properties also possess antiseizure effects (86, 119) (Table 3).
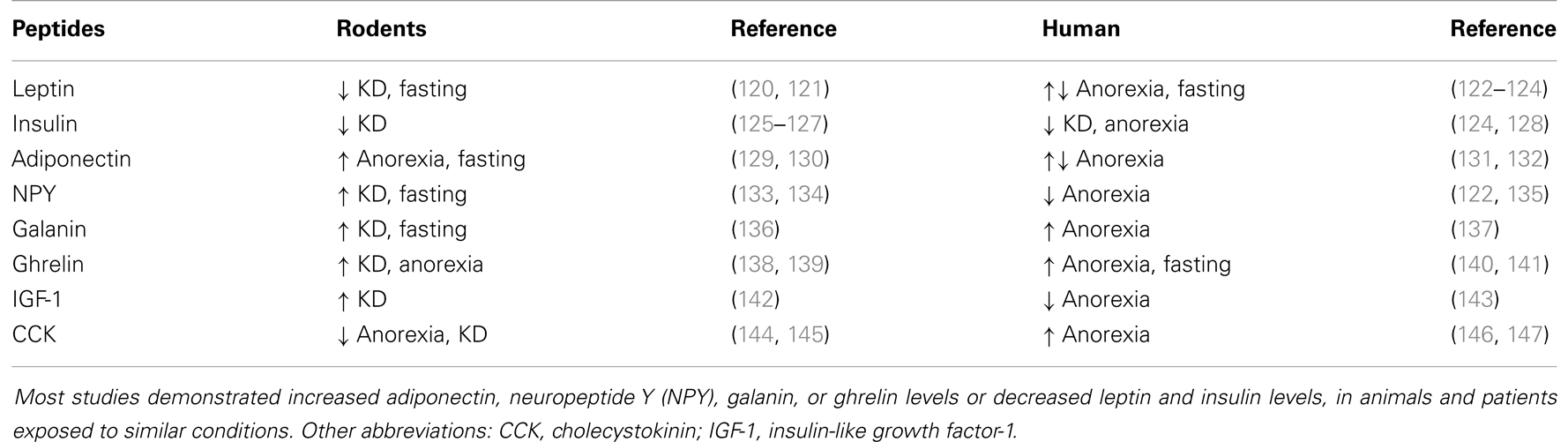
Table 3. Changes in different neuroactive peptide levels in animals and humans during various metabolic states, including anorexia, prolonged fasting, or a ketogenic diet (KD).
The regulation of energy balance depends on the coordination of multiple peripheral and central systems and is tightly modulated by neuropeptides (148, 149). The critical regulation of food intake is thought to occur in the hypothalamus. Neurons in the parvocellular part of the paraventricular hypothalamic nucleus produce the anorectic neuropeptide corticotropin releasing factor (CRF) (150), while those in the lateral hypothalamus produce orexigenic neuropeptides such as orexin and melanin-concentrating hormone (151). The arcuate nucleus of the hypothalamus contains two distinct neuronal populations that produce orexigenic and anorexigenic peptides, which seem to play antagonistic roles in energy balance control and that are regulated by leptin and insulin (152–154). The “anorexigenic” population is located in the lateral subdivision of the arcuate nucleus and produces the anorectic neuropeptides proopiomelanocortin and cocaine- and amphetamine-regulated transcript (155, 156). The “orexigenic” population of neurons is located in the medial subregion of the arcuate nucleus and produces orexigenic neuropeptides such as agouti-related peptide and neuropeptide Y (NPY) (30, 157). The hypothalamic cell groups are specially tuned to sense metabolic changes, to regulate hormonal secretion, energy homeostasis, and metabolism. The mechanisms for controlling food intake during fasting or a KD involve a complex interplay between the peripheral systems controlling gastrointestinal peptide secretion and the central nervous system. These neuronal systems include neuropeptides and peripheral neuroactive peptides such as CRF, opioids, NPY, CCK, orexin, galanin, and leptin, as well as monoamines (serotonin, dopamine, and noradrenaline).
The complex machinery involved in regulating food intake in relation to body metabolism is also strictly linked to the characteristics of the meal. For instance, ghrelin stimulates sugar intake (158), whereas the neuropeptide oxytocin inhibits carbohydrate but not fat intake (159). Galanin, enkephalins (160), and β-endorphin (161) promote the preference for fat meals. Conversely, macronutrients can influence peptide hormone release. This is the case for lipids, which exert a satiating effect that is probably related to the increased secretion of GLP-1, PYY, and CCK (162). Interestingly, in the presence of increased ghrelin levels a high-fat diet can prevent any change in food intake due to hormonal regulation (163). Thus, the classic KD could deeply alter peptide hormone levels and their function by virtue of the prevalent lipid content of the diet. This effect could be further influenced by the presence of ketone bodies or, in some cases, by weight loss. For instance, in the case of weight loss caused by a KD, insulin and leptin are both reduced when compared to pre-diet levels (164). Ghrelin is not affected by weight loss induced by a KD (164), but is increased in the case of anorexia (165, 166) or cachexia (167, 168). Upregulation of ghrelin levels during the course of starvation is expected, since ghrelin production is stimulated by a negative energy balance as a compensatory response.
Peptide Hormones that Modulate Seizures
Peptide hormones and neuropeptides have been extensively investigated in the context of animal models of epilepsy and with clinical studies (Table 4). Lines of evidence point to an important role for peptide hormones and neuropeptides in regulating neuronal activity during seizures. In particular, peptide hormones such as adiponectin, ghrelin, and leptin and neuropeptides such as CCK, galanin, NPY, and somatostatin appear to be promising candidates for future research on new treatments for epilepsy.
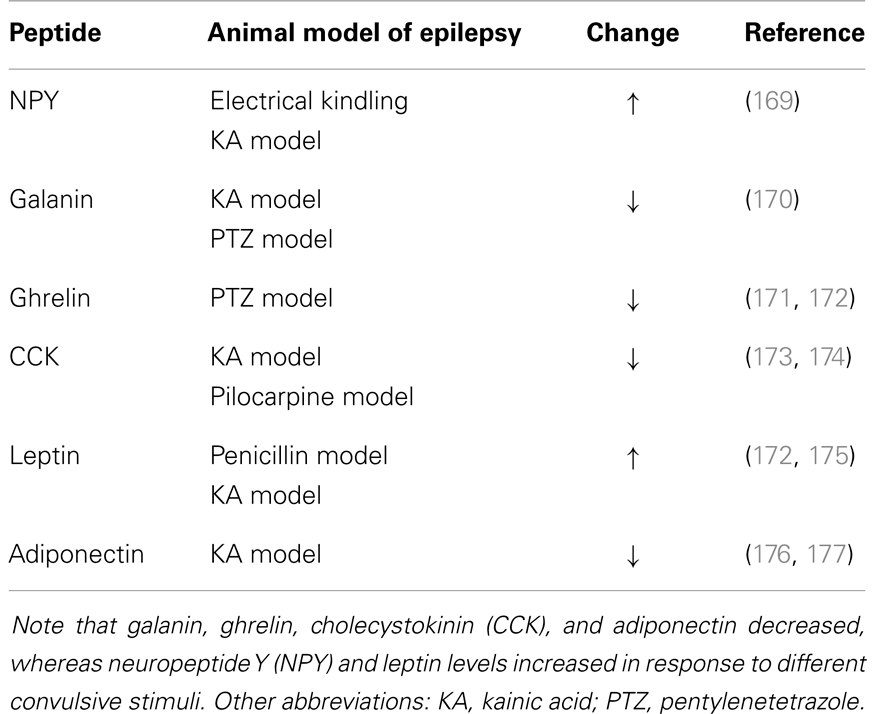
Table 4. Changes in the levels of neuroactive peptides following seizures.
Adiponectin is produced by adipocytes to regulate fat oxidation and sensitivity to insulin. Adiponectin plasma levels decreased in metabolic syndrome, suggesting the involvement of this hormone in the insulin resistance and hyperlipidemia characterizing the syndrome (178). Adiponectin knockout (KO) mice developed vascular injury and inflammation when exposed to a high-fat diet. Conversely, adiponectin protected from cerebral ischemia due to modulatory properties on inflammation (179, 180). Beneficial effects were also demonstrated in the kainate model of temporal lobe epilepsy, in which adiponectin reduced neuronal cell loss (176). Lee et al. (177) investigated the effects of kainate administration in adiponectin KO mice fed a high-fat diet, compared with the wild-type (WT) controls, by monitoring the blood parameters related to altered glucose and lipid metabolism, the epileptic response to kainate and acute tissue damage. Although the authors were not able to document the effects of kainate with electroencephalography, the seizure score was always lower in the WT mice compared with the KO mice, which also were characterized by metabolic changes such as glucose intolerance. In another experiment, kainate was directly injected in the hippocampus, and the damage was examined after 2 weeks. Glial reaction was more intense in the adiponectin KO mice compared with the WT mice, but the neuronal cell loss and granule cell dispersion were also significantly more pronounced in the investigated model of metabolic syndrome. These last findings suggest that a high-fat diet causes hippocampal damage in the long term. To exclude the role of adiponectin deletion in the observed proconvulsive effects of the diet, the authors replicated the same experiments with kainate injection in KO mice fed a standard diet, and observed a response similar to that found in the WT mice. In addition, the authors demonstrated a strong correlation between glucose intolerance, cholesterol, fat mass, free fatty acids, and seizure severity. In contrast, hypertriglyceridemia and body weight were not correlated with epileptic activity (177).
Ghrelin has been suggested to act as an anticonvulsant in various seizure models. In particular, ghrelin was found to delay the onset of myoclonic jerks and tonic generalized seizures induced by pentylenetetrazole (181). However, in the same experiment ghrelin increased the duration of generalized clonic seizures. In the penicillin-induced neocortical seizure model, ghrelin decreased the frequency of interictal spiking activity (182). Furthermore, in the kainate model of status epilepticus (SE), this hormone significantly attenuated seizures and SE duration, resulting in decreased cell loss (183). Additional experiments based on the pilocarpine model confirmed the anticonvulsant effects of ghrelin (119), but this activity was not strong enough to prevent the development of SE (184, 185). Intriguingly, better findings were instead obtained by using ghrelin analogs such as des-acyl ghrelin and EP-80317 (184). Conversely, the ghrelin full agonist JMV-1843 failed to prevent pilocarpine-induced seizures (184, 185). This picture was further complicated by the discovery that KO mice for the ghrelin receptor were resistant to pilocarpine-induced seizures (119). These findings led to the proposal that the growth hormone secretagogue receptor 1a (GHS-R1a), which is endowed with high intrinsic activity, was reduced on the cell surface as a consequence of interaction with ghrelin, and that this phenomenon was probably the explanation for the anticonvulsant effects of ghrelin (119). However, ghrelin is rapidly converted to des-acyl ghrelin, and this metabolite could be responsible for the effects attributed to ghrelin administration (186). Interestingly, des-acyl ghrelin could also modulate other targets through binding to the CD36 scavenger receptor, which is involved in regulating food intake and in signaling cascades that lead to neuronal dysfunction and initiation of cell death (187).
Leptin’s anticonvulsant activity was originally shown in two different models: Xu et al. (188) showed that leptin administered by the intranasal route delayed the effects of the convulsant pentylenetetrazole, as well as reduced seizures evoked by 4-aminopyridine. In addition, the same authors confirmed the anticonvulsant effects of leptin in an in vitro model and suggested that this effect was related to the depression of glutamatergic neurotransmission (188). In agreement with these data, pentylenetetrazole-induced seizures were more severe in leptin KO (ob/ob) mice (189). However, at odds with previous findings, other experiments showed that leptin potentiated neocortical electrographic seizures elicited by penicillin, probably by interfering with the production of nitric oxide (190). Leptin’s proconvulsant activity was also suggested by another study (191), in which leptin pretreatment increased the number of animals exhibiting convulsions in response to N-methyl-d-aspartic acid or kainate administration. Recently, the possible involvement of the cannabinoid CB1 receptor in leptin proconvulsant activity in the penicillin model has also been suggested (175). These discrepancies are still unexplained and could depend on the seizure model. In particular, leptin appears to play a modulatory role rather than to be an anticonvulsant.
The expression of neuropeptides changes dramatically in animal models of epilepsy, especially in the hippocampal formation (Table 4). The implication of these peptides in modulating seizures is likely due to their ability to decrease excitability in the brain. Moreover, their brain concentration changes after spontaneous seizures, and this may contribute to enhanced susceptibility to seizures (169). Importantly, recurrent spontaneous seizures were suppressed by overexpression of NPY in the hippocampus using adeno-associated virus vectors (192). Several studies have investigated the therapeutic potential of these neuropeptides to suppress seizure activity in animals, in various epilepsy models (193). These studies were in agreement with data on NPY or galanin KO mice, which presented an increased propensity to develop seizures (170, 194). Another interesting aspect of neuropeptides is their neuroprotective role after brain injury, a condition that can lead to the development of chronic epilepsy. In particular, NPY promotes neurogenesis in the subventricular zone and the subgranular zone of the adult mouse brain (195, 196). These aspects and others were the focus of recent, extensive reviews about the role of neuropeptides and their receptors (86, 197).
Could Peptide Hormones be Mediators of KD Antiepileptic Effects?
Patients affected by epilepsy and treated with AEDs present with various changes in the plasma levels of hormones related to metabolism, by which patients could also develop metabolic disorders. In a pioneering study of 40 patients treated with valproate, obesity was found in 15 epileptic patients who also displayed increased leptin and insulin levels and decreased ghrelin and adiponectin levels, when compared with subjects with normal body weight (198). In contrast, another investigation suggested ghrelin production increased (+70%) in patients affected by epilepsy (199). These observations were confirmed by examining the effects of valproic acid treatment on ghrelin in a sample of children characterized by increased body weight, body mass index, and height, in which serum ghrelin levels were higher than in controls and negatively correlated with insulin-like growth factor-1 (IGF-1) and insulin-like growth factor binding protein-3 (200). Further studies on ghrelin in patients suffering from epilepsy suggested that ghrelin levels are lower in adults treated with various AEDs (201), as well as in children treated with carbamazepine or valproic acid (202). Finally, a relationship between peptide hormones and seizures has been suggested by a study in which prolactin, nesfatin-1, and ghrelin were evaluated within 5 min after a convulsion: the serum levels of prolactin and nesfatin-1 increased, and interestingly, the ghrelin levels were abated for at least 24 h (172). Since ghrelin is a promising candidate in view of its modulatory properties in seizures (119, 203), the changes in this hormone regulation found in patients with epilepsy are of major interest, whereas other changes in peptide hormone regulation, involving leptin, galanin, and NPY (204, 205), must be clearly defined.
The metabolism of peptide hormones such as ghrelin and leptin is modified by seizures and AEDs, suggesting that they could also be affected by KDs. In spite of the important role peptide hormones have in regulating body metabolism (206–208), only a few studies have examined the changes in peptide hormone levels in humans treated with KDs (164, 209, 210). Fraser et al. (209) analyzed the acute anti-inflammatory effects of a KD in patients affected by rheumatoid arthritis and found significant decreases in serum leptin and IGF-1, similar to those observed during acute starvation. These results were probably explained by the weight loss induced by the diet. Significant changes in various peptide hormones associated with weight loss in obese subjects maintained on a KD were also described (164). More interesting for epilepsy and KD treatment, decreased fasting insulin and leptin levels were reported in children affected by Glut-1 deficiency syndrome and treated with a 3:1 KD, whereas adiponectin levels were unmodified (210). In this last study, the observed changes in insulin and leptin levels were not dependent on weight loss. More data have been obtained by studying animal models: Kinzig and collaborators (138) found that rats maintained on a low-carbohydrate/high-fat diet had increased adiposity when compared with controls, accompanied by increased leptin and ghrelin levels and decreased insulin plasma levels. Thus, insulin, but not leptin, was downregulated as in children affected by Glut-1 deficiency syndrome and maintained on KD therapy (210). The high-fat content of the diet seems to be an important requisite to stimulate leptin production, since the leptin levels of rats maintained on a low-carbohydrate/high-protein diet did not vary (211). More interestingly, ghrelin induces ketone bodies production when administered to humans (212), and ghrelin upregulation by a low-carbohydrate/high-fat diet, as demonstrated in rats (138), could be a critical phenomenon in mediating KD effects.
Ghrelin is an important regulator of energy balance that is released from the stomach in response to fasting (180). In addition to regulating appetite, ghrelin stimulates the release of growth hormone (GH) through the GHS-R1a. Other different ghrelin-related peptides are produced in the stomach, including des-acyl ghrelin, which is also obtained by ghrelin desacylation in the periphery. According to some authors, des-acyl ghrelin represents approximately 90% of total circulating ghrelin-related peptides (213). It regulates the energy balance independently of ghrelin. Indeed des-acyl ghrelin is unable to activate GHS-R1a; thus, this peptide is devoid of GH-releasing properties. Interestingly, fasting alters the secretion of ghrelin and des-acyl ghrelin, but des-acyl ghrelin appears to be a long-lasting signal associated with a negative energy balance. Indeed, when fasting is prolonged and implies a negative energy balance, such as in patients affected by anorexia or cachexia, des-acyl ghrelin is more stably elevated than ghrelin, and the des-acyl ghrelin/ghrelin ratio is also significantly increased (214). Prolonged fasting is mimicked by KD in patients affected by epilepsy. These data suggest that ghrelin-related peptides, especially des-acyl ghrelin, could be stably increased in patients affected by pharmacoresistant epilepsy and receiving a KD treatment. Ghrelin, but not des-acyl ghrelin, modulated the release of GABA via the GHS-R1a receptor (215) and increased the GABAergic tone. This phenomenon probably explains the counteraction of pentylenetetrazole and penicillin proconvulsive activities observed after ghrelin administration (181, 182). However, des-acyl ghrelin, which does not activate GHS-R1a, also displays anticonvulsive properties by unknown mechanisms, as shown in models of pharmacologically induced acute seizure (184). Experiments based on administration of EP-80317, one of the various molecules developed by modifying ghrelin, confirmed the existence of anticonvulsive effects independent of GHS-R1a receptor activation (203). Des-acyl ghrelin was previously considered an inactive ghrelin metabolite. Although des-acyl ghrelin lacks most of the biological properties of ghrelin, des-acyl ghrelin is now recognized as a hormone because it can modulate food intake by a mechanism independent of GHS-R1a stimulation, depending on circadian rhythmicity and on fasting pre-exposure (216, 217). In addition, des-acyl ghrelin depresses gastrointestinal motility independently of ghrelin, by a central mechanism involving the CRF type 2 receptor (218). At the cellular level, des-acyl ghrelin increased medium chain fatty acid uptake in cardiomiocytes, whereas ghrelin was ineffective; conversely, ghrelin inhibited the increase in glucose uptake normally induced by insulin, but des-acyl ghrelin did not. Thus, des-acyl ghrelin appears to regulate the body metabolism in a way different from ghrelin, without causing dramatic changes in food intake or body development. This hypothesis is confirmed by studies on des-acyl ghrelin transgenic mice that showed a limited decrease in linear growth and slightly reduced fat mass, phenomena related to the negative energy balance produced by des-acyl ghrelin (219). These properties make des-acyl ghrelin interesting from a pharmacological point of view, also for the nervous system. Pharmacokinetic experiments showed that des-acyl ghrelin enters the brain by non-saturable transmembrane diffusion and is sequestered once within the central nervous system to exert its activity there (218). Thus, the demonstration that des-acyl ghrelin could mediate the anticonvulsive effects of KD could pose the basis for a pharmacological exploitation of this hormone and its possible analogs in epileptic disorders.
Different ghrelin analogs have been developed by simply modifying the peptidergic structure of this hormone, obtaining GH secretagogues with diverse pharmacological profiles (203, 220, 221). Some molecules have been developed to be orally administered and, in general, present an advantageous pharmacokinetic profile when compared with ghrelin. In particular, JMV-1843 is a potent GHS-R1a agonist that stimulates GH release and food intake, whereas JMV-2959 appears to be an antagonist (221, 222). Hexarelin presents less marked GH stimulating properties, which are completely lost with molecules functionally closer to des-acyl ghrelin, such as EP-80317 (187). Interestingly, EP-80317 interacts with CD36 (223), a type B scavenger receptor involved in internalizing oxidized low-density lipoprotein, which initiates a signaling cascade that regulates microglial recruitment and activation, and subsequently, secretion of inflammatory mediators such as interleukin (IL)-1β and IL-6. These mediators, particularly IL-1β, were shown to promote neuronal overexcitation and seizures (224). Indeed, Bulgarelli et al. (187) demonstrated that EP-80317, hexarelin, and des-acyl ghrelin in the nanomolar range effectively counteract the stimulation of IL-1β and IL-6 synthesis in microglial cells lacking the specific ghrelin receptor GHS-R1a, suggesting the involvement of the CD36 receptor in these effects. However, the expression of this receptor in the brain is limited to a few regions (225), and thus, des-acyl ghrelin anticonvulsive effects, whether mediated through CD36 activation, have to be restricted mainly to microglial cells. Thus, new molecular pathways, possibly important for pharmacoresistant epilepsy, could be implicated in des-acyl ghrelin anticonvulsive effects, but these relationships remain to be investigated in appropriate animal models to elucidate their possible involvement in KD beneficial effects.
Conclusion
Neuroactive peptides involved in the control of metabolism are putative candidates as mediators of KD effects in pharmacoresistant epilepsy (119, 184). Fasting is mimicked by KDs through inducing ketone bodies and by lowering glucose plasma levels, effects that depend on the activation of some endocrine axis and on the suppression of others. The release of peptides such as insulin, leptin, ghrelin, des-acyl ghrelin, and adiponectin is closely related to food intake or fasting. These hormones have also been related to seizure induction (insulin) or suppression (leptin, ghrelin, des-acyl ghrelin, and adiponectin). Thus, several peripherally originated neuroactive peptides could play a role in modulating epileptic activity in the brain, and their changes in response to fasting or KDs are of primary interest in the attempt to clarify which mechanism could be responsible for the efficacy of dietetic approaches to pharmacoresistant epilepsy. A promising hormonal product is represented by des-acyl ghrelin. This neuroactive peptide is increased in catabolic states and was shown to exert anticonvulsant activity in models of SE (184). A des-acyl ghrelin analog, EP-80317, was also effective in preventing seizure induction by pilocarpine when preventively administered in rats (203). In future studies, we suggest that des-acyl ghrelin may deserve thorough investigation as a possible mediator of KD effects.
Identifying KD mediators could represent a significant advancement in treating pharmacoresistant epilepsy. Although patients suffering from pharmacoresistance could take advantage of therapeutic alternatives such as surgical resection of epileptic foci, intracranial or extracranial neural stimulation, or KDs, compliance with these approaches is not optimal. Many reasons could discourage a patient from adopting any of these therapeutic alternatives to drug treatment: (i) the need for invasive surgery; (ii) possible impairment of cognitive and memory functions caused by intracranial or extracranial stimulation; and (iii) gastrointestinal complications related to KDs such as nausea, constipation, and abdominal pain. Clarification of the neuronal and molecular mechanisms of antiepileptic effects of KDs will propose a specific therapeutic solution for pharmacoresistant epilepsy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the Italian Ministry of Health (grant RF-2010-2309921 to Giuseppe Biagini).
References
1. Berg AT, Baca CB, Loddenkemper T, Vickrey BG, Dlugos D. Priorities in pediatric epilepsy research: improving children’s futures today. Neurology (2013) 81:1166–75. doi: 10.1212/WNL.0b013e3182a55fb9
2. Perucca E, French J, Bialer M. Development of new antiepileptic drugs: challenges, incentives, and recent advances. Lancet Neurol (2007) 6:793–804. doi:10.1016/S1474-4422(07)70215-6
3. Wiebe S, Blume WT, Girvin JP, Eliasziw M. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N Engl J Med (2001) 345:311–8. doi:10.1056/NEJM200108023450501
4. Lim ME, Bowen JM, Snead OC, Elliott I, Donner E, Weiss SK, et al. Access to surgery for pediatric patients with medically refractory epilepsy: a systems analysis. Epilepsy Res (2013) 107:286–96. doi:10.1016/j.eplepsyres.2013.08.010
5. Engel J Jr, McDermott MP, Wiebe S, Langfitt JT, Stern JM, Dewar S, et al. Early surgical therapy for drug-resistant temporal lobe epilepsy: a randomized trial. JAMA (2012) 307:922–30. doi:10.1001/jama.2012.220
6. Schachter SC, Saper CB. Vagus nerve stimulation. Epilepsia (1998) 39:677–86. doi:10.1111/j.1528-1157.1998.tb01151
7. Parker AP, Polkey CE, Binnie CD, Madigan C, Ferrie CD, Robinson RO. Vagal nerve stimulation in epileptic encephalopathies. Pediatrics (1999) 103:778–82. doi:10.1542/peds.103.4.778
8. Carius A, Wintermantel A. Vagus nerve stimulation therapy in epilepsy patients: long-term outcome and adverse effects: a retrospective analysis. Nervenarzt (2013) 84:1473–85. doi:10.1007/s00115-013-3923-4
9. Klinkenberg S, van den Bosch CN, Majoie HJ, Aalbers MW, Leenen L, Hendriksen J, et al. Behavioural and cognitive effects during vagus nerve stimulation in children with intractable epilepsy – a randomized controlled trial. Eur J Paediatr Neurol (2013) 17:82–90. doi:10.1016/j.ejpn.2012.07.003
10. Morris IIIGL, Gloss D, Buchhalter J, Mack JK, Nickels K, Harden C. Evidence-based guideline update: vagus nerve stimulation for the treatment of epilepsy. Neurology (2013) 81:1453–9. doi:10.1212/WNL.0b013e3182a393d1
11. Skarpaas TL, Morrell MJ. Intracranial stimulation therapy for epilepsy. Neurotherapeutics (2009) 6:238–43. doi:10.1016/j.nurt.2009.01.022
12. Kerrigan JF, Litt B, Fisher RS, Cranstoun S, French JA, Blum DE, et al. Electrical stimulation of the anterior nucleus of the thalamus for the treatment of intractable epilepsy. Epilepsia (2004) 45:346–54. doi:10.1111/j.0013-9580.2004.01304.x
13. Oommen J, Morrell M, Fisher RS. Experimental electrical stimulation therapy for epilepsy. Curr Treat Options Neurol (2005) 7:261–71. doi:10.1007/s11940-005-0036-9
14. Cordella R, Acerbi F, Marras CE, Carozzi C, Vailati D, Saini M, et al. Risk of seizures during intraoperative electrocortical stimulation of brain motor areas: a retrospective study on 50 patients. Neurol Sci (2013) 34:63–70. doi:10.1007/s10072-012-0968-2
15. Lefevre F, Aronson N. Ketogenic diet for the treatment of refractory epilepsy in children: a systematic review of efficacy. Pediatrics (2000) 105:E46. doi:10.1542/peds.105.4.e46
16. Coppola G, Veggiotti P, Cusmai R, Bertoli S, Cardinali S, Dionisi-Vici C, et al. The ketogenic diet in children, adolescents and young adults with refractory epilepsy: an Italian multicentric experience. Epilepsy Res (2002) 48:221–7. doi:10.1016/S0920-1211(01)00315-1
17. Levy RG, Cooper PN, Giri P. Ketogenic diet and other dietary treatments for epilepsy. Cochrane Database Syst Rev (2012) 3:CD001903. doi:10.1002/14651858.CD001903
18. Freeman JM, Vining EP, Pillas DJ, Pyzik PL, Casey JC, Kelly LM. The efficacy of the ketogenic diet-1998: a prospective evaluation of intervention in 150 children. Pediatrics (1998) 102:1358–63. doi:10.1542/peds.102.6.1358
19. Hartman AL, Vining EP. Clinical aspects of the ketogenic diet. Epilepsia (2007) 48:31–42. doi:10.1111/j.1528-1167.2007.00914.x
20. Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, et al. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol (2008) 7:500–6. doi:10.1016/S1474-4422(08)70092-9
21. Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, et al. A randomized trial of classical and medium-chain triglyceride ketogenic diets in the treatment of childhood epilepsy. Epilepsia (2009) 50:1109–17. doi:10.1111/j.1528-1167.2008.01870
22. Kossoff EH, Zupec-Kania BA, Amark PE. Optimal clinical management of children receiving the ketogenic diet: recommendations of the international ketogenic diet study group. Epilepsia (2009) 50:304–17. doi:10.1111/j.1528-1167.2008.01765.x
24. Stafstrom CE. Dietary approaches to epilepsy treatment: old and new options on the menu. Epilepsy Curr (2004) 4:215–22. doi:10.1111/j.1535-7597.2004.46001.x
25. Kossoff EH, McGrogan JR, Bluml RM, Pillas DJ, Rubenstein JE, Vining EP. A modified Atkins diet is effective for the treatment of intractable pediatric epilepsy. Epilepsia (2006) 47:421–4. doi:10.1111/j.1528-1167.2006.00438.x
26. Chapman KE, Kim DY, Rho JM, Ng YT, Kerrigan JF. Ketogenic diet in the treatment of seizures associated with hypothalamic hamartomas. Epilepsy Res (2011) 94:218–21. doi:10.1016/j.eplepsyres.2011.02.004
27. Huttenlocher PR, Wilbourn AJ, Signore JM. Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology (1971) 21:1097–103. doi:10.1212/WNL.21.11.1097
29. Pfeifer HH, Thiele EA. Low-glycemic-index treatment: a liberalized ketogenic diet for treatment of intractable epilepsy. Neurology (2005) 65:1810–2. doi:10.1212/01.wnl.0000187071.24292.9e
30. Schwartz MW, Woods SC, Porte DJ, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature (2000) 404:661–71. doi:10.1038/35007534
31. Gilbert DL, Pyzik PL, Freeman JM. The ketogenic diet: seizure control correlates better with serum β-hydroxybutyrate than with urine ketones. J Child Neurol (2000) 15:787–90. doi:10.1177/088307380001501203
32. Bough KJ, Matthews PJ, Eagles DA. A ketogenic diet has different effects upon seizures induced by maximal electroshock and by pentylenetetrazole infusion. Epilepsy Res (2000) 38:105–14. doi:10.1016/S0920-1211(99)00079-0
33. Seo JH, Lee YM, Lee JS, Kang HC, Kim HD. Efficacy and tolerability of the ketogenic diet according to lipid:nonlipid ratios-comparison of 3:1 with 4:1 diet. Epilepsia (2007) 48:801–5. doi:10.1111/j.1528-1167.2007.01025.x
34. Seyfried BT, Mantis JG, Todorova MT, Greene AE. Dietary management of epilepsy: role of glucose and ketone bodies. In: Schwartzkroin PA, editor. The Encyclopedia of Basic Epilepsy Research (Vol. 2). San Diego, CA: Elsevier Press/Academic Press (2009). p. 687–93.
35. Appleton DB, DeVivo DC. An animal model for the ketogenic diet. Epilepsia (1974) 15:211–27. doi:10.1111/j.1528-1157.1974.tb04943.x
36. Pan JW, de Graaf RA, Petersen KF, Shulman GI, Hetherington HP, Rothman DL. [2,4-13C2]-β-hydroxybutyrate metabolism in human brain. J Cereb Blood Flow Metab (2002) 22:890–8. doi:10.1097/00004647-200207000-00014
37. McNally MA, Hartman AL. Ketone bodies in epilepsy. J Neurochem (2012) 121:28–35. doi:10.1111/j.1471-4159.2012.07670
38. van Delft R, Lambrechts D, Verschuure P, Hulsman J, Majoie M. Blood beta-hydroxybutyrate correlates better with seizure reduction due to ketogenic diet than do ketones in the urine. Seizure (2010) 19:36–9. doi:10.1016/j.seizure.2009.10.009
39. Likhodii SS, Musa K, Mendonca A, Dell C, Burnham WM, Cunnane SC. Dietary fat, ketosis, and seizure resistance in rats on the ketogenic diet. Epilepsia (2000) 41:1400–10. doi:10.1111/j.1528-1157.2000.tb00115.x
40. Harney JP, Madara J, I’Anson H. Effects of acute inhibition of fatty acid oxidation on latency to seizure and concentrations of β hydroxybutyrate in plasma of rats maintained on calorie restriction and/or the ketogenic diet. Epilepsy Res (2002) 49:239–46. doi:10.1016/S0920-1211(02)00046-3
41. Rho JM, White HS, Anderson G, Donevan S. Acetoacetate, acetone, and dibenzylamine (a contaminant in L-(+)-β-hydroxybutyrate) exhibit direct anticonvulsant actions in vivo. Epilepsia (2002) 43:358–61. doi:10.1046/j.1528-1157.2002.47901.x
42. Samoilova M, Weisspapir M, Abdelmalik P, Velumian AA, Carlen PL. Chronic in vitro ketosis is neuroprotective but not anti-convulsant. J Neurochem (2010) 113:826–35. doi:10.1111/j.1471-4159.2010
43. Likhodii SS, Serbanescu I, Cortez MA, Murphy P, Snead OC, Burnham WM. Anticonvulsant properties of acetone, a brain ketone elevated by the ketogenic diet. Ann Neurol (2003) 54:219–26. doi:10.1002/ana.10634
44. Gasior M, French A, Joy MT, Tang RS, Hartman AL, Rogawski MA. The anticonvulsant activity of acetone, the major ketone body in the ketogenic diet, is not dependent on its metabolites acetol, 1,2-propanediol, methylglyoxal, or pyruvic acid. Epilepsia (2007) 48:793–800. doi:10.1111/j.1528-1167.2007.01026.x
45. Bough KJ, Rho JM. Anticonvulsant mechanisms of the ketogenic diet. Epilepsia (2007) 48:43–58. doi:10.1111/j.1528-1167.2007.00915.x
46. Nylen K, Likhodii SS, Hum KM, Burnham WM. A ketogenic diet and diallyl sulfide do not elevate afterdischarge thresholds in adult kindled rats. Epilepsy Res (2006) 71:23–31. doi:10.1016/j.eplepsyres.2006.05.008
47. Seymour KJ, Bluml S, Sutherling J, Sutherling W, Ross BD. Identification of cerebral acetone by 1H-MRS in patients with epilepsy controlled by ketogenic diet. MAGMA (1999) 8:33–42. doi:10.1007/BF02590633
48. Nordli DR Jr, De Vivo DC. The ketogenic diet revisited: back to the future. Epilepsia (1997) 38:743–9. doi:10.1111/j.1528-1157.1997.tb01460.x
49. Wada K, Kiryu K, Kawata Y, Chiba T, Mizuno K, Okada M, et al. Prognosis and clinical features of intractable epilepsy: a prospective study. Psychiatry Clin Neurosci (1997) 51:233–5. doi:10.1111/j.1440-1819.1997.tb02589.x
50. Todorova MT, Tandon P, Madore RA, Stafstrom CE, Seyfried TN. The ketogenic diet inhibits epileptogenesis in EL mice: a genetic model for idiopathic epilepsy. Epilepsia (2000) 41:933–40. doi:10.1111/j.1528-1157.2000.tb00275.x
51. Hartman AL, Gasior M, Vining EPG, Rogawski MA. The neuropharmacology of the ketogenic diet. Pediatr Neurol (2007) 36:281–92. doi:10.1016/j.pediatrneurol.2007.02.008
52. Hartman AL, Lyle M, Rogawski MA, Gasior M. Efficacy of the ketogenic diet in the 6-Hz seizure test. Epilepsia (2008) 49:334–9. doi:10.1111/j.1528-1167.2007.01430.x
53. Uhlemann ER, Neims AH. Anticonvulsant properties of the ketogenic diet in mice. J Pharmacol Exp Ther (1972) 180:231–8.
54. Mahoney AW, Hendricks DG, Bernhard N, Sisson DV. Fasting and ketogenic diet effects on audiogenic seizures susceptibility of magnesium deficient rats. Pharmacol Biochem Behav (1983) 18:683–7. doi:10.1016/0091-3057(83)90006-0
55. Hori A, Tandon P, Holmes GL, Stafstrom CE. Ketogenic diet: effects on expression of kindled seizures and behavior in adult rats. Epilepsia (1997) 38:750–8. doi:10.1111/j.1528-1157.1997.tb01461.x
56. Muller-Schwarze AB, Tandon P, Liu Z, Yang Y, Holmes GL, Stafstrom CE. Ketogenic diet reduces spontaneous seizures and mossy fiber sprouting in the kainic acid model. Neuroreport (1999) 10:1517–22. doi:10.1097/00001756-199905140-00023
57. Rho JM, Kim DW, Robbins CA, Anderson GD, Schwartzkroin PA. Age-dependent differences in flurothyl seizure sensitivity in mice treated with a ketogenic diet. Epilepsy Res (1999) 37:233–40. doi:10.1016/S0920-1211(99)00068-6
58. Bough KJ, Eagles DA. A ketogenic diet increases the resistance to pentylenetetrazole-induced seizures in the rat. Epilepsia (1999) 40:138–43. doi:10.1111/j.1528-1157.1999.tb02066.x
59. Mantis JG, Centeno NA, Todorova MT, McGowan R, Seyfried TN. Management of multifactorial idiopathic epilepsy in EL mice with caloric restriction and the ketogenic diet: role of glucose and ketone bodies. Nutr Metab (2004) 1:11. doi:10.1186/1743-7075-1-11
60. Thavendiranathan P, Chow C, Cunnane S, McIntyre Burnham W. The effect of the “classic” ketogenic diet on animal seizure models. Brain Res (2003) 959:206–13. doi:10.1016/S0006-8993(02)03744-7
61. Nylen K, Velazquez JL, Likhodii SS, Cortez MA, Shen L, Leshchenko Y, et al. A ketogenic diet rescues the murine succinic semialdehyde dehydrogenase deficient phenotype. Exp Neurol (2008) 210:449–57. doi:10.1016/j.expneurol.2007.11.015
62. Dahlin M, Elfving A, Ungerstedt U, Amark P. The ketogenic diet influences the levels of excitatory and inhibitory amino acids in the CSF in children with refractory epilepsy. Epilepsy Res (2005) 64:115–25. doi:10.1016/j.eplepsyres.2005.03.008
63. Yudkoff M, Daikhin Y, Nissim I, Lazarow A, Nissim I. Ketogenic diet, brain glutamate metabolism and seizure control. Prostaglandins Leukot Essent Fatty Acids (2004) 70:277–85. doi:10.1016/j.plefa.2003.07.005
64. Melo TM, Nehlig A, Sonnewald U. Neuronal-glial interactions in rats fed a ketogenic diet. Neurochem Int (2006) 48:498–507. doi:10.1016/j.neuint.2005.12.037
65. Bough KJ, Schwartzkroin PA, Rho JM. Calorie restriction and ketogenic diet diminish neuronal excitability in rat dentate gyrus in vivo. Epilepsia (2003) 44:752–60. doi:10.1046/j.1528-1157.2003.55502.x
66. Juge N, Gray JA, Omote H, Miyaji T, Inoue T, Hara C, et al. Metabolic control of vesicular glutamate transport and release. Neuron (2010) 68:99–112. doi:10.1016/j.neuron.2010.09.002
67. Ma W, Berg J, Yellen G. Ketogenic diet metabolites reduce firing in central neurons by opening KATP channels. J Neurosci (2007) 27:3618–25. doi:10.1523/JNEUROSCI.0132-07.2007
68. Kawamura M Jr, Ruskin DN, Masino SA. Metabolic autocrine reguation of neurons involves cooperation among pannexin hemichannels, adenosine receptors, and KATP channels. J Neurosci (2010) 30:3886–95. doi:10.1523/JNEUROSCI.0055-10.2010
69. Tanner GR, Lutas A, Martínez-François JR, Yellen G. Single KATP channel opening in response to action potential firing in mouse dentate granule neurons. J Neurosci (2011) 31:8689–96. doi:10.1523/JNEUROSCI.5951-10.2011
70. Koranda JL, Ruskin DN, Masino SA, Blaise JH. A ketogenic diet reduces long-term potentiation in the dentate gyrus of freely behaving rats. J Neurophysiol (2011) 106:662–6. doi:10.1152/jn.00001.2011
71. DeVivo DC, Leckie MP, Ferrendelli JS, McDougal DB. Chronic ketosis and cerebral metabolism. Ann Neurol (1978) 3:331–7. doi:10.1002/ana.410030410
72. Bough KJ, Wetherington J, Hassel B, Pare JF, Gawryluk JW, Greene JG. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol (2006) 60:223–35. doi:10.1002/ana.20899
73. Nylen K, Likhodii S, Burnham WM. The ketogenic diet: proposed mechanisms of action. Neurotherapeutics (2009) 6:402–5. doi:10.1016/j.nurt.2009.01.021
74. Lennox WG, Cobb S. Studies in epilepsy VIII. The clinical effect of fasting. Arch Neurol Psychiatry (1928) 20:771–9. doi:10.1001/archneurpsyc.1928.02210160112009
75. Al-Mudallal AS, LaManna JC, Lust WD, Harik SI. Diet-induced ketosis does not cause cerebral acidosis. Epilepsia (1996) 37:258–61. doi:10.1111/j.1528-1157.1996.tb00022.x
76. Vamecq J, Vallée L, Lesage F, Gressens P, Stables JP. Antiepileptic popular ketogenic diet: emerging twists in an ancient story. Prog Neurobiol (2005) 75:1–28. doi:10.1016/j.pneurobio.2004.11.003
77. Cullingford TE. The ketogenic diet; fatty acids, fatty acid-activated receptors and neurological disorders. Prostaglandins Leukot Essent Fatty Acids (2004) 70:253–64. doi:10.1016/j.plefa.2003.09.008
78. Masino SA, Geiger JD. Are purines mediators of the anticonvulsant/neuroprotective effects of ketogenic diets? Trends Neurosci (2008) 31:273–8. doi:10.1016/j.tins.2008.02.009
79. Masino SA, Theofilas P, Li T, Ruskin DN, Fredholm BB, Geiger JD, et al. A ketogenic diet suppresses seizures in mice through adenosine A1 receptors. J Clin Invest (2011) 121:2679–83. doi:10.1172/JCI57813
80. Giorgi FS, Pizzanelli C, Biagioni F, Murri L, Fornai F. The role of norepinephrine in epilepsy: from the bench to the bedside. Neurosci Biobehav Rev (2004) 28:507–24. doi:10.1016/j.neubiorev.2004.06.008
81. Kaminski RM, Shippenberg TS, Jeffrey MW, Rocha BA. Genetic deletion of the norepinephrine transporter decreases vulnerability to seizures. Neurosci Lett (2005) 382:51–5. doi:10.1016/j.neulet.2005.02.056
82. Szot P, Weinshenker D, Rho JM, Storey TW, Schwartzkroin PA. Norepinephrine is required for the anticonvulsant effect of the ketogenic diet. Brain Res (2001) 129:211–4. doi:10.1016/S0165-3806(01)00213-9
83. Hökfelt T, Mutt V. Neuropeptides. In: Adelman G, Smith BH, editors. Encyclopedia of Neuroscience (Vol. 2). Amsterdam: Elsevier (1999). p. 1423–9.
84. De Wied D, Sigling HO. Neuropeptides involved in the pathophysiology of schizophrenia and major depression. Neurotox Res (2002) 4:453–68. doi:10.1080/10298420290031432
86. Dobolyi A, Kekesi KA, Juhasz G, Szekely AD, Lovas G, Kovacs Z. Receptors of peptides as therapeutic targets in epilepsy research. Curr Med Chem (2014) 21:764–87. doi:10.2174/0929867320666131119154018
87. Agnati LF, Cortelli P, Biagini G, Bjelke B, Fuxe K. Different classes of volume transmission signals exist in the central nervous system and are affected by metabolic signals, temperature gradients and pressure waves. Neuroreport (1994) 6:9–12. doi:10.1097/00001756-199412300-00004
88. Ludwig M, Leng G. Dendritic peptide release and peptide-dependent behaviours. Nat Rev Neurosci (2006) 7:126–36. doi:10.1038/nrn1845
89. Tallent MK. Presynaptic inhibition of glutamate release by neuropeptides: use-dependent synaptic modification. Results Probl Cell Differ (2008) 44:177–200. doi:10.1007/400_2007_037
90. Fuxe K, Li XM, Bjelke B, Hedlund PB, Biagini G, Agnati LF. Possible mechanisms for the powerful actions of neuropeptides. Ann N Y Acad Sci (1994) 739:42–59. doi:10.1111/j.1749-6632.1994.tb19806.x
91. Hökfelt T, Broberger C, Xu ZQ, Sergeyev V, Ubink R, Diez M. Neuropeptides – an overview. Neuropharmacology (2000) 39:1337–56. doi:10.1016/S0028-3908(00)00010-1
92. Gil-Campos M, Aguilera CM, Canete R, Gil A. Ghrelin: a hormone regulating food intake and energy homeostasis. Br J Nutr (2006) 96:201–26. doi:10.1079/BJN20061787
93. Bjorbaek C, Kahn BB. Leptin signalling in the central nervous system and the periphery. Recent Prog Horm Res (2004) 59:305–31. doi:10.1210/rp.59.1.305
94. Acuna-Goycolea C, van den Pol AN. Neuroendocrine proopiomelanocortin neurons are excited by hypocretin/orexin. J Neurosci (2009) 29:1503–13. doi:10.1523/JNEUROSCI.5147-08.2009
95. Hamilton TJ, Xapelli S, Michaelson SD, Larkum ME, Colmers WF. Modulation of distal calcium electrogenesis by neuropeptide Y1 receptors inhibits neorortical long-term depression. J Neurosci (2013) 33:11184–93. doi:10.1523/JNEUROSCI.5595-12.2013
96. Heilig M, Koob GF, Ekman R, Britton KT. Corticotropin-releasing factor and neuropeptide Y: role in emotional integration. Trends Neurosci (1994) 17:80–5. doi:10.1016/0166-2236(94)90079-5
97. Ögren SO, Kuteeva E, Hökfelt T, Kehr J. Galanin receptor antagonists: a potential novel pharmacological treatment for mood disorders. CNS Drugs (2006) 20:633–54. doi:10.2165/00023210-200620080-00003
98. Schwarzer C. 30 Years of dynorphins – new insights on their functions in neuropsychiatric diseases. Pharmacol Ther (2009) 123:353–70. doi:10.1016/j.pharmthera.2009.05.006
99. Muurahainen N, Kissileff HR, Derogatis AJ, Pi-Sunyer FX. Effects of cholecystokinin-octapeptide (CCK-8) on food intake and gastric emptying in man. Physiol Behav (1988) 44:645–9. doi:10.1016/0031-9384(88)90330-7
100. Little TJ, Horowitz M, Feinle-Bisset C. Role of cholecystokinin in appetite control and body weight regulation. Obes Rev (2005) 6:297–306. doi:10.1111/j.1467-789X.2005.00212.x
101. Flint A, Raben A, Astrup A. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest (1998) 101:515–20. doi:10.1172/JCI990
102. Gallwitz B. Anorexigenic effects of GLP-1 and its analogues. Handb Exp Pharmacol (2012) 209:185–207. doi:10.1007/978-3-642-24716-3_8
103. Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, et al. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature (2002) 418:650–4. doi:10.1038/nature00887
104. Karra E, Chandarana K, Batterham RL. The role of peptide YY in appetite regulation and obesity. J Physiol (2009) 587:19–25. doi:10.1113/jphysiol.2008.164269
105. Cohen MA, Ellis SM, Le Roux CW, Batterham RL, Park A, Patterson M, et al. Oxyntomodulin suppresses appetite and reduces food intake in humans. J Clin Endocrinol Metab (2003) 88:4696–701. doi:10.1210/jc.2003-030421
106. Dakin CL, Small CJ, Batterham RL, Neary NM, Cohen MA, Patterson M, et al. Peripheral oxyntomodulin reduces food intake and body weight gain in rats. Endocrinology (2004) 145:2687–95. doi:10.1210/en.2003-1338
107. Batterham RL, Le Roux CW, Cohen MA, Park AJ, Ellis SM, Patterson M. Pancreatic polypeptide reduces appetite and food intake in humans. J Clin Endocrinol Metab (2003) 88:3989–92. doi:10.1210/jc.2003-030630
108. Hankir MK, Parkinson JR, Minnion JS, Addison ML, Bloom SR, Bell JD. Peptide YY 3-36 and pancreatic polypeptide differentially regulate hypothalamic neuronal activity in mice in vivo as measured by manganese-enhanced magnetic resonance imaging. J Neuroendocrinol (2011) 23:371–80. doi:10.1111/j.1365-2826.2011.02111
109. Geary N. Pancreatic glucagon signals postprandial satiety. Neurosci Biobehav Rev (1990) 14:323–38. doi:10.1016/S0149-7634(05)80042-9
110. Porte D Jr, Woods SC. Regulation of food intake and body weight in insulin. Diabetologia (1981) 20:274–80. doi:10.1007/BF00254493
111. Werther GA, Hogg A, Oldfield BJ, McKinley MJ, Figdor R, Allen AM, et al. Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology (1987) 121:1562–70. doi:10.1210/endo-121-4-1562
112. Cline MA, Nandar W, Smith ML, Pittman BH, Kelly M, Rogers JO. Amylin causes anorexigenic effects via the hypothalamus and brain stem in chicks. Regul Pept (2008) 146:140–6. doi:10.1016/j.regpep.2007.09.003
113. Fulton S, Woodside B, Shizgal P. Modulation of brain reward circuitry by leptin. Science (2000) 287:125–8. doi:10.1126/science.287.5450.125
114. Schwartz MW. Central nervous system regulation of food intake. Obesity (2006) 14(Suppl 2):1s–8s. doi:10.1038/oby.2006.275
115. Qi Y, Takahashi N, Hileman SM, Patel HR, Berg AH, Pajvani UB, et al. Adiponectin acts in the brain to decrease body weight. Nat Med (2014) 10:524–9. doi:10.1038/nm1029
116. Tovar S, Nogueiras R, Tung LY, Castañeda TR, Vázquez MJ, Morris A, et al. Central administration of resistin promotes short-term satiety in rats. Eur J Endocrinol (2005) 153:R1–R5. doi:10.1530/eje.1.01999
117. Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab (2001) 86:5992–5. doi:10.1210/jc.86.12.5992
118. Naleid AM, Grace MK, Cummings DE, Levine AS. Ghrelin induces feeding in the mesolimbic reward pathway between the ventral tegmental area and the nucleus accumbens. Peptides (2005) 26:2274–9. doi:10.1016/j.peptides.2005.04.025
119. Portelli J, Michotte Y, Smolders I. Ghrelin: an emerging new anticonvulsant neuropeptide. Epilepsia (2012) 53:585–95. doi:10.1111/j.1528-1167.2012.03423
120. Kinzig KP, Taylor RJ. Maintenance on a ketogenic diet: voluntary exercise, adiposity and neuroendocrine effects. Int J Obes (2009) 33:824–30. doi:10.1038/ijo.2009.109
121. Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, et al. Role of leptin in the neuroendocrine response to fasting. Nature (1996) 382:250–2. doi:10.1038/382250a0
122. Oswiecimska J, Ziora K, Geisler G, Broll-Waska K. Prospective evaluation of leptin and neuropeptide Y (NPY) serum levels in girls with anorexia nervosa. Neuro Endocrinol Lett (2005) 26:301–4.
123. Monteleone P, Castaldo E, Maj M. Neuroendocrine dysregulation of food intake in eating disorders. Regul Pept (2008) 149:39–50. doi:10.1016/j.regpep.2007.10.007
124. Hayes MR, Miller CK, Ulbrecht JS, Mauger JL, Parker-Klees L, Gutschall MD, et al. A carbohydrate-restricted diet alters gut peptides and adiposity signals in men and women with metabolic syndrome. J Nutr (2007) 137:1944–50.
125. Thio LL, Erbayat-Altay E, Rensing N, Yamada KA. Leptin contributes to slower weight gain in juvenile rodents on a ketogenic diet. Pediatr Res (2006) 60:413–7. doi:10.1203/01.pdr.0000238244.54610.27
126. Honors MA, Davenport BM, Kinzig KP. Effects of consuming a high carbohydrate diet after eight weeks of exposure to a ketogenic diet. Nutr Metab (2009) 6:46. doi:10.1186/1743-7075-6-46
127. Badman MK, Kennedy AR, Adams AC, Pissios P, Maratos-Flier E. A very low carbohydrate ketogenic diet improves glucose tolerance in ob/ob mice independently of weight loss. Am J Physiol Endocrinol Metab (2009) 297:E1197–204. doi:10.1152/ajpendo.00357
128. Fery F, Bourdoux P, Christophe J, Balasse EO. Hormonal and metabolic changes induced by an isocaloric isoproteinic ketogenic diet in healthy subjects. Diabete Metab (1982) 8:299–305.
129. Brichard SM, Delporte ML, Lambert M. Adipocytokines in anorexia nervosa: a review focusing on leptin and adiponectin. Horm Metab Res (2003) 35:337–42. doi:10.1055/s-2003-41353
130. Chaolu H, Asakawa A, Ushikai M, Li YX, Cheng KC, Li JB. Effect of exercise and high-fat diet on plasma adiponectin and nesfatin levels in mice. Exp Ther Med (2011) 2:2369–73. doi:10.3892/etm.2011.199
131. Bosy-Westphal A, Brabant G, Haas V, Onur S, Paul T, Nutzinger D. Determinants of plasma adiponectin levels in patients with anorexia nervosa examined before and after weight gain. Eur J Nutr (2005) 44:355–9. doi:10.1007/s00394-005-0533-3
132. Tagami T, Satoh N, Usui T, Yamada K, Shimatsu A, Kuzuya H. Adiponectin in anorexia nervosa and bulimia nervosa. J Clin Endocrinol Metab (2004) 89:1833–7. doi:10.1210/jc.2003-031260
133. Tabb K, Szot P, White SS, Liles LC, Weinshenker D. The ketogenic diet does not alter brain expression of orexigenic neuropeptides. Epilepsy Res (2004) 62:35–9. doi:10.1016/j.eplepsyres.2004.08.002
134. Shiraev T, Chen H, Morris MJ. Differential effects of restricted versus unlimited high-fat feeding in rats on fat mass, plasma hormones and brain appetite regulators. J Neuroendocrinol (2009) 21:602–9. doi:10.1111/j.1365-2826.2009.01877
135. Baranowska B, Wolinska-Witort E, Wasilewska-Dziubinska E, Roguski K, Chmielowska M. Plasma leptin, neuropeptide Y (NPY) and galanin concentrations in bulimia nervosa and in anorexia nervosa. Neuro Endocrinol Lett (2001) 22:356–8.
136. Gundlach AL. Galanin/GALP and galanin receptors: role in central control of feeding, body weight/obesity and reproduction? Eur J Pharmacol (2002) 440:255–68. doi:10.1016/S0014-2999(02)01433-4
137. De Marinis L, Mancini A, Valle D, Bianchi A, Gentilella R, Milardi D, et al. Effects of galanin on growth hormone and prolactin secretion in anorexia nervosa. Metabolism (2000) 49:155–9. doi:10.1016/S0026-0495(00)91091-6
138. Kinzig KP, Scott KA, Hyun J, Bi S, Moran TH. Altered hypothalamic signaling and responses to food deprivation in rats fed a low-carbohydrate diet. Obes Res (2005) 13:1672–82. doi:10.1038/oby.2005.205
139. Nakahara T, Kojima S, Tanaka M, Yasuhara D, Harada T, Sagiyama K, et al. Incomplete restoration of the secretion of ghrelin and PYY compared to insulin after food ingestion following weight gain in anorexia nervosa. J Psychiatr Res (2007) 41:814–20. doi:10.1016/j.jpsychires.2006.07.021
140. Tanaka M, Naruo T, Yasuhara D, Tatebe Y, Nagai N, Shiiya T, et al. Fasting plasma ghrelin levels in subtypes of anorexia nervosa. Psychoneuroendocrinology (2003) 28:829–35. doi:10.1016/S0306-4530(02)00066-5
141. Otto B, Tschop M, Fruhauf E, Heldwein W, Fichter M, Otto C, et al. Postprandial ghrelin release in anorectic patients before and after weight gain. Psychoneuroendocrinology (2005) 30:577–81. doi:10.1016/j.psyneuen.2005.01.009
142. Cheng CM, Kelley B, Wang J, Strauss D, Eagles DA, Bondy CA. A ketogenic diet increases brain insulin-like growth factor receptor and glucose transporter gene expression. Endocrinology (2003) 144:2676–82. doi:10.1210/en.2002-0057
143. Misra M, Klibanski A. Neuroendocrine consequences of anorexia nervosa in adolescents. Endocr Dev (2010) 17:197–214. doi:10.1159/000262540
144. Ueno N, Asakawa A, Satoh Y, Inui A. Increased circulating cholecystokinin contributes to anorexia and anxiety behavior in mice overexpressing pancreatic polypeptide. Regul Pept (2007) 141:8–11. doi:10.1016/j.regpep.2006.12.019
145. Duca FA, Zhong L, Covasa M. Reduced CCK signaling in obese-prone rats fed a high fat diet. Horm Behav (2013) 64:812–7. doi:10.1016/j.yhbeh.2013.09.004
146. Fujimoto S, Inui A, Kiyota N, Seki W, Koide K, Takamiya S, et al. Increased cholecystokinin and pancreatic polypeptide responses to a fat-rich meal in patients with restrictive but not bulimic anorexia nervosa. Biol Psychiatry (1997) 41:1068–70. doi:10.1016/S0006-3223(97)00044-9
147. Keel PK, Wolfe BE, Liddle RA, De Young KP, Jimerson DC. Clinical features and physiological response to a test meal in purging disorder and bulimia nervosa. Arch Gen Psychiatry (2007) 64:1058–66. doi:10.1001/archpsyc.64.9.1058
148. Richard D, Timofeeva E. Energy balance regulation: complex interplay between the autonomic and cognitive/limbic brains to control food intake and thermogenesis. In: Johnson C, editor. Obesity Prevention: The Role of Brain and Society on Individual Behavior. Oxford: Elsevier (2010). p. 299–316.
149. Parker JA, Bloom SR. Hypothalamic neuropeptides and the regulation of appetite. Neuropharmacology (2012) 63:18–30. doi:10.1016/j.neuropharm.2012.02.004
150. Richard D, Timofeeva E. Energy homeostasis: paraventricular nucleus (PVN) system. In: Squire LR, editor. Encyclopedia of Neuroscience. Berlin: Elsevier (2009). p. 1035–41.
151. Barson JR, Morganstern I, Leibowitz SF. Complementary roles of orexin and melanin-concentrating hormone in feeding behaviour. Int J Endocrinol (2013) 2013:983964. doi:10.1155/2013/983964
152. Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron (1999) 22:221–32. doi:10.1016/S0896-6273(00)81084-3
153. Baskin DG, Figlewicz Lattemann D, Seeley RJ, Woods SC, Porte DJ, Schwartz MW. Insulin and leptin: dual adiposity signals to the brain for the regulation of food intake and body weight. Brain Res (1999) 848:114–23. doi:10.1016/S0006-8993(99)01974-5
154. Varela L, Horvath TL. Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep (2012) 13:1079–86. doi:10.1038/embor.2012.174
155. Kristensen P, Judge ME, Thim L, Ribel U, Christjansen KN, Wulff BS, et al. Hypothalamic CART is a new anorectic peptide regulated by leptin. Nature (1998) 393:72–6. doi:10.1038/29993
156. Williams KW, Margatho LO, Lee CE, Choi M, Lee S, Scott MM, et al. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci (2010) 30:2472–9. doi:10.1523/JNEUROSCI.3118-09.2010
157. Wang Q, Liu C, Uchida A, Chuang JC, Walker A, Liu T, et al. Arcuate AgRP neurons mediate orexigenic and glucoregulatory action of ghrelin. Mol Metab (2014) 3:64–72. doi:10.1016/j.molmet.2013.10.001
158. Disse E, Bussier AL, Veyrat-Durebex C, Deblon N, Pfluger PT, Tschöp MH, et al. Peripheral ghrelin enhances sweet taste food consumption and preference, regardless of its caloric content. Physiol Behav (2010) 101:277–81. doi:10.1016/j.physbeh.2010.05.017
159. Olszewski PK, Klockars A, Olszewska AM, Fredriksson R, Schiöth HB, Levine AS. Molecular, immunohistochemical, and pharmacological evidence of oxytocin’s role as inhibitor of carbohydrate but not fat intake. Endocrinology (2010) 151:4736–44. doi:10.1210/en.2010-0151
160. Leibowitz SF, Akabayashi A, Alexander J, Karatayev O, Chang GQ. Puberty onset in female rats: relationship with fat intake, ovarian steroids and the peptides, galanin and enkephalin, in the paraventricular and medial preoptic nuclei. J Neuroendocrinol (2009) 21:538–49. doi:10.1111/j.1365-2826.2009.01870.x
161. Mizushige T, Saitoh K, Manabe Y, Nishizuka T, Taka Y, Eguchi A, et al. Preference for dietary fat induced by release of beta-endorphin in rats. Life Sci (2009) 84:760–5. doi:10.1016/j.lfs.2009.03.003
162. Ogawa N, Ito M, Yamaguchi H, Shiuchi T, Okamoto S, Wakitani K, et al. Intestinal fatty acid infusion modulates food preference as well as calorie intake via the vagal nerve and midbrain-hypothalamic neural pathways in rats. Metabolism (2012) 61:1312–20. doi:10.1016/j.metabol.2012.02.011
163. Gardiner JV, Campbell D, Patterson M, Kent A, Ghatei MA, Bloom SR, et al. The hyperphagic effect of ghrelin is inhibited in mice by a diet high in fat. Gastroenterology (2010) 138:2468–76. doi:10.1053/j.gastro.2010.02.012
164. Sumithran P, Prendergast LA, Delbridge E, Purcell K, Shulkes A, Kriketos A, et al. Ketosis and appetite-mediating nutrients and hormones after weight loss. Eur J Clin Nutr (2013) 67:759–64. doi:10.1038/ejcn.2013.90
165. Shen C, Yu T, Tang ZH, Wu KM. Changes in ghrelin and obestatin levels before and after a meal in children with simple obesity and anorexia. Horm Res Paediatr (2013) 79:341–6. doi:10.1159/000351464
166. Singhal V, Misra M, Klibanski A. Endocrinology of anorexia nervosa in young people: recent insights. Curr Opin Endocrinol Diabetes Obes (2014) 21(1):64–70. doi:10.1097/MED.0000000000000026
167. Guillory B, Splenser A, Garcia J. The role of ghrelin in anorexia-cachexia syndromes. Vitam Horm (2013) 92:61–106. doi:10.1016/B978-0-12-410473-0.00003-9
168. Invernizzi M, Carda S, Cisari C. On behalf of Società Italiana per lo Studio della Sarcopenia e della Disabilità Muscolo-Scheletrica (SISDIM). Possible synergism of physical exercise and ghrelin-agonists in patients with cachexia associated with chronic heart failure. Aging Clin Exp Res (2014). doi:10.1007/s40520-013-0186-7
169. Vezzani A, Sperk G. Overexpression of NPY and Y2 receptors in epileptic brain tissue: an endogenous neuroprotective mechanism in temporal lobe epilepsy? Neuropeptides (2004) 38:245–52. doi:10.1016/j.npep.2004.05.004
170. Mazarati AM, Hohmann JG, Bacon A, Liu H, Sankar R, Steiner RA, et al. Modulation of hippocampal excitability and seizures by galanin. J Neurosci (2000) 20:6276–81.
171. Ataie Z, Golzar MG, Babri SH, Ebrahimi H, Mohaddes G. Does ghrelin level change after epileptic seizure in rats? Seizure (2011) 20:347–9. doi:10.1016/j.seizure.2011.01.001
172. Aydin S, Dag E, Ozkan Y, Arslan O, Koc G, Bek S, et al. Time-dependent changes in the serum levels of prolactin, nesfatin-1 and ghrelin as a marker of epileptic attacks young male patients. Peptides (2011) 32:1276–80. doi:10.1016/j.peptides.2011.04.021
173. Gall C. Seizures induce dramatic and distinctly different changes in enkephalin, dynorphin, and CCK immunoreactivities in mouse hippocampal mossy fibers. J Neurosci (1988) 8:1852–62.
174. Wyeth MS, Zhang N, Mody I, Houser CR. Selective reduction of cholecystokinin-positive basket cell innervation in a model of temporal lobe epilepsy. J Neurosci (2010) 30:8993–9006. doi:10.1523/JNEUROSCI.1183-10.2010
175. Arslan G, Alici SK, Ayyildiz M, Agar E. The role of CB1-receptors in the proconvulsant effect of leptin on penicillin-induced epileptiform activity in rats. CNS Neurosci Ther (2013) 19:222–8. doi:10.1111/cns.12075
176. Jeon BT, Shin HJ, Kim JB, Kim YK, Lee DH, Kim KH, et al. Adiponectin protects hippocampal neurons against kainic acid-induced excitotoxicity. Brain Res Rev (2009) 61:81–8. doi:10.1016/j.brainresrev.2009.05.002
177. Lee EB, Warmann G, Dhir R, Ahima RS. Metabolic dysfunction associated with adiponectin deficiency enhances kainic acid-induced seizure severity. J Neurosci (2011) 31:14361–6. doi:10.1523/JNEUROSCI.3171-11.2011
178. Ahima RS. Metabolic actions of adipocyte hormones: focus on adiponectin. Obesity (2006) 14(Suppl 1):9S–15S. doi:10.1038/oby.2006.276
179. Nishimura M, Izumiya Y, Higuchi A, Shibata R, Qiu J, Kudo C, et al. Adiponectin prevents cerebral ischemic injury through endothelial nitric oxide synthase dependent mechanisms. Circulation (2008) 117:216–23. doi:10.1161/CIRCULATIONAHA.107.725044
180. Chen B, Liao WQ, Xu N, Xu H, Wen JY, Yu CA, et al. Adiponectin protects against cerebral ischemia-reperfusion injury through anti-inflammatory action. Brain Res (2009) 1273:129–37. doi:10.1016/j.brainres.2009.04.002
181. Obay BD, Tasdemir E, Tümer C, Bilgin HM, Sermet A. Antiepileptic effects of ghrelin on pentylenetetrazole-induced seizures in rats. Peptides (2007) 28:1214–9. doi:10.1016/j.peptides.2007.04.003
182. Aslan A, Yildirim M, Ayyildiz M, Güven A, Agar E. The role of nitric oxide in the inhibitory effect of ghrelin against penicillin-induced epileptiform activity in rat. Neuropeptides (2009) 43:295–302. doi:10.1016/j.npep.2009.05.005
183. Lee J, Lim E, Kim Y, Li E, Park S. Ghrelin attenuates kainic acid-induced neuronal cell death in the mouse hippocampus. J Endocrinol (2010) 205:263–70. doi:10.1677/JOE-10-0040
184. Biagini G, Torsello A, Marinelli C, Gualtieri F, Vezzali R, Coco S, et al. Beneficial effects of desacyl-ghrelin, hexarelin and EP-80317 in models of status epilepticus. Eur J Pharmacol (2011) 670:130–6. doi:10.1016/j.ejphar.2011.08.020
185. Lucchi C, Curia G, Vinet J, Gualtieri F, Bresciani E, Locatelli V, et al. Protective but not anticonvulsant effects of ghrelin and JMV-1843 in the pilocarpine model of status epilepticus. PLoS One (2013) 8(8):e72716. doi:10.1371/journal.pone.0072716
186. Biagini G, Locatelli V, Torsello A. Ghrelin anticonvulsive properties: is it a matter of desacylation? Epilepsia (2012) 53:1277–8. doi:10.1111/j.1528-1167.2012.03546.x
187. Bulgarelli I, Tamiazzo L, Bresciani E, Rapetti D, Caporali S, Lattuada D, et al. Desacyl-ghrelin and synthetic GH-secretagogues modulate the production of inflammatory cytokines in mouse microglia cells stimulated by beta-amyloid fibrils. J Neurosci Res (2009) 87:2718–27. doi:10.1002/jnr.22088
188. Xu L, Rensing N, Yang XF, Zhang HX, Thio LL, Rothman SM, et al. Leptin inhibits 4-aminopyridine- and pentylenetetrazole-induced seizures and AMPAR-mediated synaptic transmission in rodents. J Clin Invest (2008) 118:272–80. doi:10.1172/JCI33009
189. Erbayat-Altay E, Yamada KA, Wong M, Thio LL. Increased severity of pentylenetetrazol induced seizures in leptin deficient ob/ob mice. Neurosci Lett (2008) 433:82–6. doi:10.1016/j.neulet.2007.12.051
190. Aslan A, Yildirim M, Ayyildiz M, Güven A, Agar E. Interaction of leptin and nitric oxide pathway on penicillin-induced epileptiform activity in rats. Brain Res (2010) 1321:117–24. doi:10.1016/j.brainres.2010.01.054
191. Lynch JJ III, Shek EW, Castagné V, Mittelstadt SW. The proconvulsant effects of leptin on glutamate receptor-mediated seizures in mice. Brain Res Bull (2010) 82:99–103. doi:10.1016/j.brainresbull.2010.02.003
192. Noè F, Pool AH, Nissinen J, Gobbi M, Bland R, Rizzi M, et al. Neuropeptide Y gene therapy decreases chronic spontaneous seizures in a rat model of temporal lobe epilepsy. Brain (2008) 131:1506–15. doi:10.1093/brain/awn079
193. Mazarati A, Wasterlain CG. Anticonvulsant effects of four neuropeptides in the rat hippocampus during self-sustaining status epilepticus. Neurosci Lett (2002) 331:123–7. doi:10.1016/S0304-3940(02)00847-9
194. Baraban SC. Antiepileptic actions of neuropeptide Y in the mouse hippocampus require Y5 receptors. Epilepsia (2002) 43(Suppl 5):9–13. doi:10.1046/j.1528-1157.43.s.5.13.x
195. Howell OW, Silva S, Scharfman HE, Sosunov AA, Zaben M, Shatya A, et al. Neuropeptide Y is important for basal and seizure-induced precursor cell proliferation in the hippocampus. Neurobiol Dis (2007) 26:174–88. doi:10.1016/j.nbd.2006.12.014
196. Agasse F, Bernardino L, Kristiansen H, Christiansen SH, Ferreira R, Silva B, et al. Neuropeptide Y promotes neurogenesis in murine subventricular zone. Stem Cells (2008) 26:1636–45. doi:10.1634/stemcells.2008-0056
197. Kovac S, Walker MC. Neuropeptides in epilepsy. Neuropeptides (2013) 47:467–75. doi:10.1016/j.npep.2013.10.015
198. Greco R, Latini G, Chiarelli F, Iannetti P, Verrotti A. Leptin, ghrelin, and adiponectin in epileptic patients treated with valproic acid. Neurology (2005) 65:1808–9. doi:10.1212/01.wnl.0000187074.27586.d1
199. Berilgen MS, Mungen B, Ustundag B, Demir C. Serum ghrelin levels are enhanced in patients with epilepsy. Seizure (2006) 15:106–11. doi:10.1016/j.seizure.2005.11.008
200. Gungor S, Yücel G, Akinci A, Tabel Y, Ozerol IH, Yologlu S. The role of ghrelin in weight gain and growth in epileptic children using valproate. J Child Neurol (2007) 22:1384–8. doi:10.1177/0883073807307096
201. Aydin S, Dag E, Ozkan Y, Erman F, Dagli AF, Kilic N, et al. Nesfatin-1 and ghrelin levels in serum and saliva of epileptic patients: hormonal changes can have a major effect on seizure disorders. Mol Cell Biochem (2009) 328:49–56. doi:10.1007/s11010-009-0073-x
202. Prodam F, Bellone S, Casara G, De Rienzo F, Grassino EC, Bonsignori I, et al. Ghrelin levels are reduced in prepubertal epileptic children under treatment with carbamazepine or valproic acid. Epilepsia (2010) 51:312–5. doi:10.1111/j.1528-1167.2009.02307.x
203. Curia G, Lucchi C, Vinet J, Gualtieri F, Marinelli C, Torsello A, et al. Pathophysiogenesis of mesial temporal lobe epilepsy: is prevention of damage antiepileptogenic? Curr Med Chem (2014) 21:663–88. doi:10.2174/0929867320666131119152201
204. Cansu A, Serdaroglu A, Cinaz P. Serum insulin, cortisol, leptin, neuropeptide Y, galanin and ghrelin levels in epileptic children receiving oxcarbazepine. Eur J Paediatr Neurol (2011) 15:527–31. doi:10.1016/j.ejpn.2011.05.004
205. Tokgoz H, Aydin K, Oran B, Kiyici A. Plasma leptin, neuropeptide Y, ghrelin, and adiponectin levels and carotid artery intima media thickness in epileptic children treated with valproate. Childs Nerv Syst (2012) 228:1049–53. doi:10.1007/s00381-012-1788-7
206. Lancha A, Frühbeck G, Gómez-Ambrosi J. Peripheral signalling involved in energy homeostasis control. Nutr Res Rev (2012) 25:223–48. doi:10.1017/S0954422412000145
207. Riediger T. The receptive function of hypothalamic and brainstem centres to hormonal and nutrient signals affecting energy balance. Proc Nutr Soc (2012) 71:463–77. doi:10.1017/S0029665112000778
208. Williams KW, Elmquist JK. From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nat Neurosci (2012) 15:1350–5. doi:10.1038/nn.3217
209. Fraser DA, Thoen J, Bondhus S, Haugen M, Reseland JE, Djøseland O, et al. Reduction in serum leptin and IGF-1 but preserved T-lymphocyte numbers and activation after a ketogenic diet in rheumatoid arthritis patients. Clin Exp Rheumatol (2000) 18:209–14.
210. Rauchenzauner M, Klepper J, Leiendecker B, Luef G, Rostasy K, Ebenbichler C. The ketogenic diet in children with Glut1 deficiency syndrome and epilepsy. J Pediatr (2008) 153:716–8. doi:10.1016/j.jpeds.2008.05.012
211. Kinzig KP, Hargrave SL, Hyun J, Moran TH. Energy balance and hypothalamic effects of a high-protein/low-carbohydrate diet. Physiol Behav (2007) 92:454–60. doi:10.1016/j.physbeh.2007.04.019
212. Huda MS, Dovey TM, Wong SP, English PJ, Halford JC, McCulloch P, et al. Ghrelin does not orchestrate the metabolic changes seen in fasting but has significant effects on lipid mobilisation and substrate utilisation. Eur J Endocrinol (2011) 165:45–55. doi:10.1530/EJE-10-1122
213. Veldhuis JD, Bowers CY. Integrating GHS into the ghrelin system. Int J Pept (2010) 2010: doi:10.1155/2010/879503
214. Ogiso K, Asakawa A, Amitani H, Inui A. Ghrelin and anorexia nervosa: a psychosomatic perspective. Nutrition (2011) 27:988–93. doi:10.1016/j.nut.2011.05.005
215. Vergnano AM, Ferrini F, Salio C, Lossi L, Baratta M, Merighi A. The gastrointestinal hormone ghrelin modulates inhibitory neurotransmission in deep laminae of mouse spinal cord dorsal horn. Endocrinology (2008) 149:2306–12. doi:10.1210/en.2007-1164
216. Toshinai K, Yamaguchi H, Sun Y, Smith RG, Yamanaka A, Sakurai T, et al. Des-acyl ghrelin induces food intake by a mechanism independent of the growth hormone secretagogue receptor. Endocrinology (2006) 147:2306–14. doi:10.1210/en.2005-1357
217. Inhoff T, Wiedenmann B, Klapp BF, Mönnikes H, Kobelt P. Is desacyl ghrelin a modulator of food intake? Peptides (2009) 30:991–4. doi:10.1016/j.peptides.2009.01.019
218. Chen CY, Inui A, Asakawa A, Fujino K, Kato I, Chen CC, et al. Des-acyl ghrelin acts by CRF type 2 receptors to disrupt fasted stomach motility in conscious rats. Gastroenterology (2005) 129:8–25. doi:10.1053/j.gastro.2005.04.015
219. Iwakura H, Hosoda K, Son C, Fujikura J, Tomita T, Noguchi M, et al. Analysis of rat insulin II promoter-ghrelin transgenic mice and rat glucagon promoter-ghrelin transgenic mice. J Biol Chem (2005) 280:15247–56. doi:10.1074/jbc.M411358200
220. Guerlavais V, Boeglin D, Mousseaux D, Oiry C, Heitz A, Deghenghi R, et al. New active series of growth hormone secretagogues. J Med Chem (2003) 46:1191–203. doi:10.1021/jm020985q
221. Moulin A, Demange L, Ryan J, Mousseaux D, Sanchez P, Bergé G, et al. New trisubstituted 1,2,4-triazole derivatives as potent ghrelin receptor antagonists. 3. Synthesis and pharmacological in vitro and in vivo evaluations. J Med Chem (2008) 51:689–93. doi:10.1021/jm701292s
222. Demange L, Boeglin D, Moulin A, Mousseaux D, Ryan J, Bergé G, et al. Synthesis and pharmacological in vitro and in vivo evaluations of novel triazole derivatives as ligands of the ghrelin receptor. 1. J Med Chem (2007) 50:1939–57. doi:10.1021/jm070024h
223. Demers A, McNicoll N, Febbraio M, Servant M, Marleau S, Silverstein R, et al. Identification of the growth hormone-releasing peptide binding site in CD36: a photoaffinity cross-linking study. Biochem J (2004) 382:417–24. doi:10.1042/BJ20040036
224. Balosso S, Maroso M, Sanchez-Alavez M, Ravizza T, Frasca A, Bartfai T, et al. A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1β. Brain (2008) 131:3256–65. doi:10.1093/brain/awn271
Keywords: adiponectin, fasting, ghrelin, ketogenic diet, neuropeptide Y, epilepsy
Citation: Giordano C, Marchiò M, Timofeeva E and Biagini G (2014) Neuroactive peptides as putative mediators of antiepileptic ketogenic diets. Front. Neurol. 5:63. doi: 10.3389/fneur.2014.00063
Received: 19 March 2014; Paper pending published: 09 April 2014;
Accepted: 14 April 2014; Published online: 29 April 2014.
Edited by:
Cara Jean Westmark, University of Wisconsin, USAReviewed by:
Richard Eugene Frye, Children’s Hospital Boston, USA; Harvard University, USADavid Ruskin, Trinity College, USA
Copyright: © 2014 Giordano, Marchiò, Timofeeva and Biagini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Biagini, Laboratorio di Epilettologia Sperimentale, Sezione di Fisiologia e Neuroscienze, Dipartimento di Scienze Biomediche, Metaboliche e Neuroscienze, Università di Modena e Reggio Emilia, Via Campi 287, Modena 41125, Italy e-mail: gbiagini@unimore.it