 Andrea Kistner
Andrea Kistner Eugénie Lhommée
Eugénie Lhommée Paul Krack
Paul Krack- 1Movement Disorder Unit, Department of Psychiatry and Neurology, University Hospital Grenoble, Grenoble, France
- 2Unité 836, Équipe 11, INSERM, Grenoble Institut des Neurosciences, Grenoble, France
- 3Joseph Fourier University, Grenoble, France
Typical body weight changes are known to occur in Parkinson’s disease (PD). Weight loss has been reported in early stages as well as in advanced disease and malnutrition may worsen the clinical state of the patient. On the other hand, an increasing number of patients show weight gain under dopamine replacement therapy or after surgery. These weight changes are multifactorial and involve changes in energy expenditure, perturbation of homeostatic control, and eating behavior modulated by dopaminergic treatment. Comprehension of the different mechanisms contributing to body weight is a prerequisite for the management of body weight and nutritional state of an individual PD patient. This review summarizes the present knowledge and highlights the necessity of evaluation of body weight and related factors, as eating behavior, energy intake, and expenditure in PD.
Introduction
Parkinson’s disease (PD) is a chronic neurodegenerative disease with diffuse α-synuclein deposits in the neural system (1). The most prevalent symptoms in early disease are mainly due to progressive degeneration of the dopaminergic nigrostriatal and mesocorticolimbic pathways with motor (akinesia, rigidity, tremor) and non-motor (apathy, anxiety, depression) symptoms (2). The disease is also characterized by the presence of non-motor vegetative symptoms explained by a synucleinopathy of central and peripheral vegetative system (3) and in its advanced stages by dementia, which correlates with cortical deposits of alpha-synuclein (intracellular Lewy bodies and Lewy neurites) (4). On top of the dopaminergic system, serotonergic, noradrenergic, and cholinergic nuclei in the brainstem projecting to the cortex are also impaired by the diffuse synucleinopathy which starts in the lower brainstem. Typically, the first alpha-synuclein deposits are found in the vagal nerve with a gastroparesis and constipation starting before the first motor symptoms and leading the patients to consult before a diagnosis of PD can be made (5). According to the clinical stage, body weight of a given patient may considerably change during the course of the disease raising the risk for both disease-related under-nutrition and treatment-related overweight.
Body weight is determined by many factors including genetic, epigenetic, metabolic, and environmental components and under physiological conditions homeostatic behavioral adaptations protect against weight gain as well as weight loss (6).
However, regulation of body weight seems to be more effective in response to weight loss than to weight gain (7). Weight gain is the result of a positive energy balance, which means that energy intake exceeds expenditure, resulting in accumulation of fat. Although this equation seems rather simple maintaining a constant body weight is a complex physiological process comprising internal and external, homeostatic and hedonic, and neurological and metabolic factors. The fine regulation of these systems is hindered by the «obesogenic» environment characterized by increased availability of large amounts of palatable and energy-dense foods and presence of powerful food cues, together with minimal physical activity. The result is the increasing prevalence of obesity in western communities (8).
The situation of PD patients should be seen against this background with additional factors in relation to the severity of the disease and dopaminergic treatment: these factors include (1) perturbation of hypothalamic metabolic regulation, (2) alteration of energy expenditure (EE) (through tremor, rigidity, dyskinesia, physical activity including hyperactivity, sleep disorders), and (3) alteration of intake (i.e., perturbation of swallowing, gastrointestinal disorders, alteration of eating behavior).
The aim of this review is:
• To highlight pathophysiological mechanisms implicated in nutrition and leading to body weight fluctuations in PD.
• To summarize available data about body weight fluctuations in PD (literature until January 2014).
• To link observed body weight fluctuations to possible mechanisms in order to improve future patient care of PD patients.
Physiological Mechanisms
Homeostatic Control of Food Intake
Homeostatic control of food intake and energy metabolism is assured by a network of several hypothalamic nuclei (Figure 1) [for review see Ref. (9–11)].
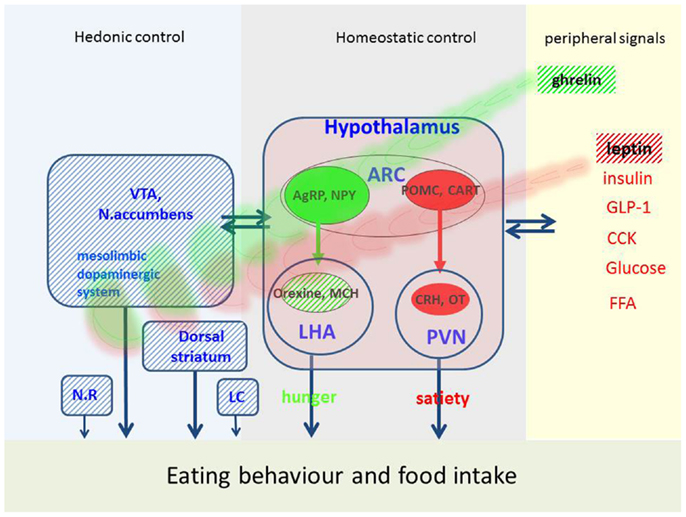
Figure 1. Possible factors implicated in eating behavior and food intake in PD. Eating behavior is regulated by hedonic, homeostatic, and peripheral signals. This figure represents the main mechanisms, which are disturbed in PD (hatched areas) and may affect eating behavior. VTA, ventral-tegmental area; ARC, arcuate nucleus; NR, nucleus raphe; LC, locus coeruleus; PVN, paraventricular nucleus; LHA, lateral hypothalamus; AgRP, agouti-gene related peptide; NPY, neuropeptide Y; MCH, melanin-concentration hormone; POMC, proopiomelanocortin; CART, cocaine-and-amphetamine-regulated transcript; CRH, corticotropin-releasing hormone; OT, oxytocin; GLP-1, glucagon-like peptide; CCK, cholecystokinin; FFA, free fatty acids.
Central pathway of hunger
The neuronal «pathway of hunger» includes neurons in the arcuate nucleus containing orexigenic peptides (i.e., agouti-gene-related peptide, neuropeptide Y). These neurons project to the lateral hypothalamus called «hunger center» which contains neuronal populations producing orexigenic peptides like melanin-concentration hormone and orexins (also called hypocretin). The name orexin refers to the Greek term for appetite (orexis). Orexin increases appetite, arousal, and spontaneous physical activity and therefore plays an important role in energy balance. Loss of orexin leads to loss of appetite and a reduced metabolism. Orexins are thought to provide a link between energy balance, behavioral arousal, and reward (10, 12) and play a role in thermogenesis (13).
Both orexin and melanin-concentration hormone neurons are selectively lost in PD which is correlated with the clinical stage and severity of disease (14–16). Thus, deficiency of orexigenic peptides could contribute to the weight changes in PD. However, the exact mechanisms in PD are complex and far from being understood.
Central pathway of satiety
Another neuronal population in the arcuate nucleus is encoding satiety. These neurons produce numerous anorexigenic peptides, such as α-melanocyte-stimulating hormone, derived from a common precursor, called pro-opiomelanocortin (POMC). Together with their receptor, these peptides constitute the melanocortin system which plays an important role in energy homeostasis. Interactions of this system with dopamine neurons in the nucleus accumbens are described (11). Another central satiety signal is cocaine-and-amphetamine-regulated transcript (CART) which was also shown to interact with hedonic circuits and dopamine (11). These peptides project to the «satiety center» located in the paraventricular nucleus which contains other anorexigenic peptides such as corticotropin-releasing hormone (CRH) and stimulation of these neurons reduces appetite. Today, a large number of central signals are identified and the list is still growing [for review see Ref. (9–11)].
Peripheral signals
This network assures a bidirectional homeostasis in response to peripheral signals reflecting the actual nutritional stage. The «satiety pathway» is activated by several factors, i.e., gastric dilatation, intestinal peptides liberated in postprandial state (cholecystokinin, glucagon-like peptide, peptide YY), metabolites as fatty acids and glucose and hormones as insulin and leptin. The latter is a hormone synthesized and liberated by adipose tissue. Serum levels of leptin reflect the degree of adipose tissue and high levels reduce food intake. Accordingly, in PD patients who lost body weight leptin tends to be low (16–18) and increases when body weight increases, e.g., after DBS surgery (19). In mice lacking D2 receptors, an enhanced hypothalamic leptin signaling has been shown (20) arguing for an alteration of this mechanism in PD which could explain weight loss in spite of low leptin levels.
Hunger and appetite may be induced by ghrelin, which is the only peripheral hunger signal thus far identified. Ghrelin is a peptide synthesized and liberated from the gastric mucosa in fasting state (10). In addition to actions in the hypothalamus, ghrelin stimulates appetite via receptors located in mesolimbic circuits (9). In PD, plasma ghrelin levels are low and paradoxically correlated with BMI (17). A reduced postprandial ghrelin response was shown in early stages of PD (21) which is not modified by dopamine treatment or acute STN-stimulation (22, 23). Thus, low levels of ghrelin may contribute to weight loss in PD (24). Six months after STN-stimulation, ghrelin was shown to increase (25) which is coherent with the concomitant weight gain (see later).
Dopaminergic Control of Eating Behavior
The hypothalamic control of food intake is modulated by the dopaminergic system and both systems are modulated by homeostatic orexigenic and anorexigenic signals such as ghrelin and leptin (26, 27). Dopamine and the dopamine D2 receptor play a central role in motivated behavior including feeding behavior (28, 29). However, the role of the dopaminergic system in feeding behavior is very complex and not completely understood. It seems to exert different actions in separate circuits and in the pattern of release (phasic versus chronic release) (26, 30).
Exposure to food and food-related cues results in an activation of the mesolimbic dopamine system and especially the projection from the ventral tegmental area to the nucleus accumbens [for review see Ref. (10, 30)]. This led to the hypothesis that the mesolimbic dopamine system mediated pleasure associated with eating [for review see Ref. (26, 27, 30)]. This idea is strongly challenged since it was shown that dopaminergic depletion of nucleus accumbens does not blunt the hedonic response to pleasant food and dopamine is not required for “liking” of food (28, 31). In line with these results, dopamine-deficient mice still demonstrate a marked preference for sucrose over water (31).
Instead, dopamine is necessary for motivational processes, referred to as “incentive salience” or “wanting” [for review see Ref. (28)]. Accordingly, increasing the synaptic dopamine by inhibition of the DA transporter or administration of amphetamine increase the motivation for high effort activities [for review see Ref. (30)]. This motivation can be measured in laboratory animals with running activity or lever pressing in order to acquire foodstuff. Lack of mesolimbic dopamine reduces the activity of rodents to acquire foodstuff, for example running activity or working to get access to food (26, 28, 32). On the other hand, when palatable food is abundant and may be acquired without effort, food intake of dopamine-deficient rodents remains stable or even increases (32).
Other dopaminergic pathways are implicated in eating-related behaviors: in dopamine-deficient mice, restoration of dopamine in dorsal striatum rescues feeding in these animals that otherwise would die of starvation (26). It seems that a regulated dopamine release in dorsal striatum is essential for normal feeding in mice (33) and humans (34). The dopamine release in the dorsal striatum following feeding correlated with the experienced pleasure in healthy humans (35). Obese people have low striatal dopamine D2 receptor availability (36), and low dorsal striatal presynaptic dopamine synthesis capacity was correlated with overeating behavior in a PET study with healthy volunteers (37).
These observations led to the “reward deficiency hypothesis for overeating.” According to this hypothesis, overeating may be considered as a “therapy” of low dopaminergic state leading to weight gain and obesity [for review see Ref. (29)]. However, as stated by Berridge et al. (28), the decrease of D2 receptors in obesity could also be a downregulation following overeating.
On the other hand, too much dopamine signaling was shown to inhibit feeding in mice, demonstrated with non-specific dopamine receptor agonists, DAT inhibitors, or amphetamines which increase the synaptic dopamine via an inhibition of presynaptic dopamine reuptake (26). In healthy adults, the amphetamine and DAT inhibitor methylphenidate reduces eating and food intake by one-third (38, 39) and weight loss was shown in PD patients treated with methylphenidate (40). In hypothalamic pathways, dopamine inhibits feeding (28), and a tonic inhibition of orexin-producing neurons in the lateral hypothalamus by dopaminergic hotspots in the nucleus accumbens has been described [for review see Ref. (29)]. However, in PD treatment with dopamine agonists may induce in some cases compulsive eating behavior leading to weight gain (41). This eating behavior is often referred to as “binge-eating disorder (BED)” and is considered to be an impulse control disorder (ICD). The underlying mechanism is thought to be an activation of mesolimbic dopaminergic pathways, especially in ventral striatum and nucleus accumbens, mediated by D3 receptors (42).
Taken together the present knowledge, we may conclude:
(1) Dopamine is necessary for motivational salience and efforts linked to alimentation, like shopping and preparing meals.
(2) In physiological conditions, dopamine over-signaling inhibits feeding.
(3) In some cases, a dopamine overstimulation may increase motivation for food leading to a drive to eat, foraging behavior, and overeating (craving or binge-eating), known to occur in PD patients under treatment with dopamine agonists (see later).
(4) In hypo-dopaminergic state, taste perception and appreciation of the foodstuff (liking) is maintained. Thus, as feeding does not require any effort and highly palatable food is easily available, snacking can be maintained even in apathetic patients who lost motivation for any other physical activity.
(5) Thus, both hypo- and hyperdopaminergic traits may lead to overeating with subsequent weight gain. Eating behavior in both cases may be different.
Serotonergic and Noradrenergic Modulation of Energy Metabolism and Appetite
Other aminergic systems such as serotonergic or noradrenergic systems are mutually connected with the hypothalamus and may influence homeostatic metabolic regulation. Both systems are affected by alpha-synucleinopathy (43). The noradrenergic locus coeruleus (LC) as well as serotonergic neurons express high amounts of orexin receptor and dense orexin fiber projections (44). Loss of LC neurons had been described in PD (45). In the 6-OHDA rat model, degeneration of LC produces a transient drop in body weight which could be reversed by DBS-STN (46). This had led to the hypothesis that weight variations in PD could be modulated by noradrenergic interaction between LC, STN, and hypothalamus (47).
Serotonin plays a role in eating behavior and high cerebral levels may improve mood, depression, and sleep (48). Cerebral serotonin biosynthesis is favored by its precursor, the essential amino acid tryptophan in the presence of dietary carbohydrates. This mechanism is triggered via the insulin response which enhances cerebral uptake of tryptophan (49). Tryptophan is a constituent of many protein-containing foods. The positive effect of carbohydrates, especially those with high glycemic index such as sucrose, on mood could be the reason why efforts to lose weight are doomed to failure in obesity (49) which is often associated with depression (50). Neurodegeneration of the serotonergic system with low levels of serotonin in PD (48) may explain the pronounced preference for all kind of sweets and increased intake of chocolate in PD patients (51).
Enteric Nervous System and Gastrointestinal Disorders
Gastrointestinal functions are regulated by the enteric nervous system, a neuronal network organized in two plexuses, myenteric and submucosal, which control gut motility and secretion (5).
In PD, the enteral nervous system is affected by alpha-synucleinopathy which may explain the high incidence of gastrointestinal disorders, beginning in pre-motor stages of the disease. The most frequent symptom is chronic constipation affecting up to 89% of PD patients [for review see Ref. (52)]. The primary cause for constipation is impaired peristalsis with slow colonic transit due to neurodegeneration of myenteric neurons, which may be modulated by dopamine (52). In some patients, constipation is secondary to abnormal coordination of the rectoanal reflex with paradoxical contraction of the puborectalis muscle, which leads to defecatory dysfunction (5, 52). Loss of serotonergic neurons in raphe nucleus is thought to be involved in this clinical feature (52). Gastroparesis is characterized by slowed emptying of food into the small bowel leading to postprandial fullness, early satiety, nausea, vomiting, and bloating (53). Gastrointestinal disorders affect the quality of life and may limit food intake thus contributing to mal- and under-nutrition in PD (24).
Parkinson’s Disease
Weight Changes in Pre-Motor PD
In prospective American cohort or case–control and Chinese epidemiological studies, a decrease of body weight was reported several years prior to diagnosis (5 pounds in the 4 year preceding diagnosis) (54–57) (Table 1). On the other hand, large Finnish and Japanese cohort studies reported a weight gain in pre-diagnostic PD (58, 59). The same result was found in the Honolulu Heart Program which included Americans of Japanese origin (60). No association between PD and BMI before or at disease onset was reported for the Greece EPIC population (61), the UK-based general Practice Research Database (62), and in Italian case–control studies (63, 64). As degeneration of the dopaminergic system begins years before diagnosis (65), BMI variation may reflect a dysregulation of dopaminergic control of eating behavior rather than modification of energy metabolism in pre-motor stages of the disease. Apathy, depression, and anxiety are frequent in de novo PD (66, Bichon et al., personal communication) and eating disorders may also appear in response to these negative emotional state. In the general population, a strong association between depression and overweight has been described (50, 67) which may be due to sub-threshold eating disorders described as “emotional eating” (68), increased “snacking” (69), or increased sweet preference (70). Alterations of eating behavior have been described in de novo PD, prior to treatment (Bichon et al., personal communication).
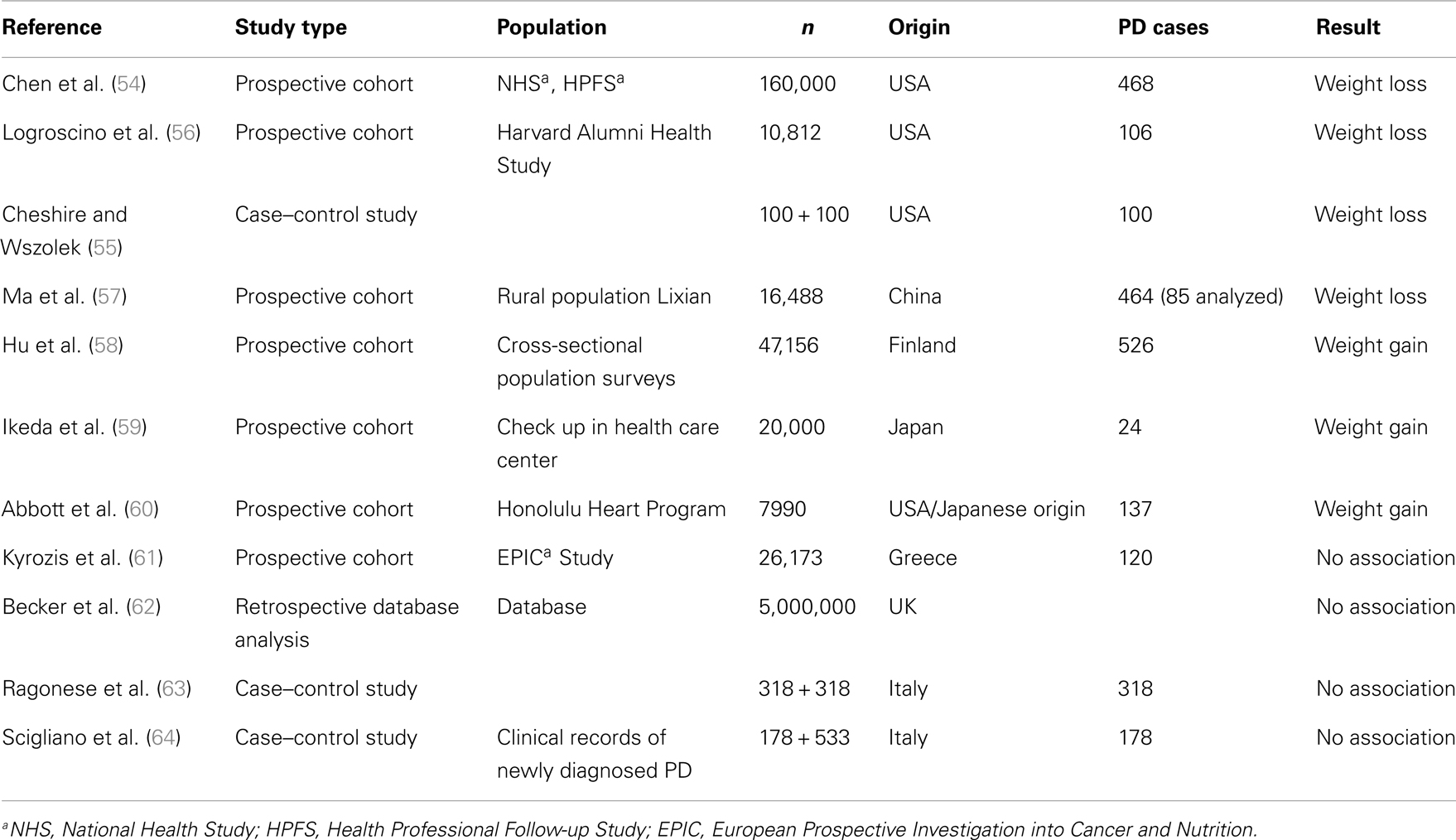
Table 1. Body weight modification in pre-motor PD.
Furthermore, disturbance of smell and taste may alter preference for foodstuff. In fact, nutritional factors have been correlated with “PD-risk”: in a Chinese study, meat consumption was inversely associated with PD (57) and in a Japanese study, dietary glycemic index was inversely correlated with PD-risk (71). An association of dietary milk protein with PD-risk is established for large prospective US cohorts (72), but a case–control study in Japan could not confirm this association (73). As dietary patterns are very different between Asian and western populations, these findings might reflect cultural variations of eating behavior in pre-motor PD. A recent meta-analysis found significant negative associations with PD for smoking, coffee drinking, and alcohol consumption which may represent a modification of preferences in early PD (74).
Prevalence of Malnutrition in PD
Weight loss in PD has been reported since the first publication of James Parkinson in 1817. A recent meta-analysis on BMI in PD reported a lower BMI of PD patients than controls (pooled difference: −1.73 kg/m2), which is related to disease severity (75). Average weight loss is about 3.6 kg 8 years after diagnosis (54) or 6 kg in one decade (76). Both fat mass and lean body mass were reported to be reduced in PD patients who lost weight (18, 77). It should be outlined that a lower average BMI does not mean that many PD patients are at risk for malnutrition. In spite of a decline of body weight, during disease progression patients may be overweight (see later). Prevalence of underweight depends on the used assessment tool and ranges from 0 to 24% (4–5% in the control group), while 3–60% of PD patients were reported to be at risk of malnutrition (77). However, the use of the mini nutritional assessment, a valid nutrition assessment tool, resulted in malnutrition rates of only 0–2% while 20–34% were at risk of malnutrition (77). Malnutrition is associated with disease severity (78).
Predictors of Weight Loss in PD
Increased energy expenditure
Despite eventual weight loss, PD patients increase their energy intake by about 350 kcal/day, mainly due to increased carbohydrate intake (54, 79) (Figure 2). This suggests that weight loss is induced by increased energy expenditure (EE). Indeed, metabolic studies had shown that resting EE is increased in PD (about 20–51% of control subjects) in ON and OFF-medication state. The main factors for this increase seem to be dyskinesia and rigidity (80–87). Consequently, when patients with severe dyskinesia were excluded, resting EE was not increased (87–89). Dyskinesia and rigidity as well as tremor may be considered as spontaneous physical activity, like standing or fidgeting. In healthy volunteers, it was shown that spontaneous physical activity may account for EE up to 700 kcal/day (90). If this is not compensated by energy intake, weight loss is inevitable.
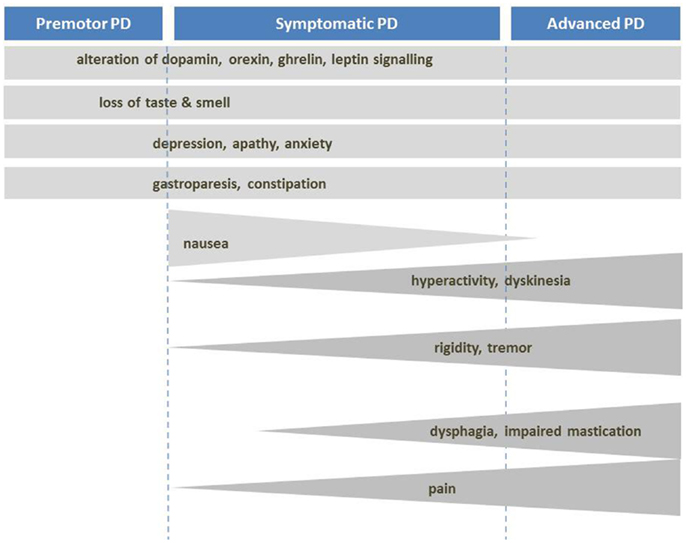
Figure 2. Mechanisms of body weight loss in PD. Mechanisms of body weight loss in Parkinson’s disease according to the stage of the disease. Factors with important contribution to weight loss are dark grey.
Impaired homeostatic regulation of energy metabolism
Taken into account that weight loss may take place before the onset of motor symptoms, other disease-related factors should be considered. As described above, several disorders of the hypothalamic regulation are known in PD, namely a massive loss of hypothalamic orexin-producing neurons, occurring prior to the onset of drug treatment (15, 16, 91). As orexin is involved both in appetite and spontaneous physical activity (92), its decrease may contribute to a decrease in food intake and physical activity. Weight loss could be promoted by impaired bioenergetics due to mitochondrial dysfunction, as shown in a mouse model of PD. In this model, which has a loss-of-function mutation of the mitochondrial protein kinase (PINK1) causing a genetic form of PD, significant weight reduction occurred in pre-motor stage (93).
Ghrelin, the gastric «hunger hormone» is reduced in PD and has even been considered as a potential biomarker of the disease (21). This could be due to impaired gastric mobility and contribute to weight loss in all stages of the disease [for review see Ref. (24)]. Furthermore, evidence from studies with rodents indicate that hypothalamic leptin signaling (which acts as a satiety signal) might be enhanced in PD (20).
Impact of dopaminergic treatment
As described above, dopamine has anorectic effects in the hypothalamic arcuate nucleus leading to a suppression of appetite and food intake (28). Likewise, substances which increase the synaptic dopamine by inhibition of the presynaptic dopamine transporter like amphetamine have anorectic effects (38).
In addition, dopaminergic treatment, especially apomorphine, may induce nausea and vomiting thus limiting energy intake, mainly on introduction of treatment (94). However, in the long-term, the treatment is mostly tolerated and may induce dyskinesia and behavioral side effects including physical hyperactivity both leading to increased EE.
Other factors
Other factors include impairment of gastrointestinal function (dysphagia, delayed gastric emptying, constipation, mal-absorption), disturbed hand–mouth-coordination, and other motor symptoms limiting activities of daily living or a decline in cognitive functions (95–99). Medical conditions such as infection, bone fractures, and malignant diseases may be other factors (99). Levodopa is an amino acid and its intestinal absorption and cerebral uptake competes with dietary amino acids thus may be impaired by dietary protein. A low-protein diet may increase its bioavailability. Patients with severe postprandial OFF-periods are often advised for a “protein-redistribution diet” which is based on the protein restriction throughout the day whereas the daily protein ration is consumed at dinner. These diets may further worsen the nutritional state of the patient (100). Some authors reported a link of PD with impaired glucose homeostasis (101, 102) but presently there is no clear evidence in this context [for review see Ref. (103)] and a recent meta-analysis confirmed a lack of relationship between PD and diabetes (104).
Weight Gain and Overweight in PD
In the pre-levodopa era, PD was a disease of malnutrition and even 20 years ago, it was rare to encounter obese PD patients (98) (Figure 3). Today, in spite of a decreased average BMI, individual PD patients may be overweight (75). In fact, prevalence of overweight in PD was reported to be about 60% in Italy (105) and 50% in Germany (80). In France and USA, more than 50% of the patients selected for DBS, i.e., patients with advanced disease suffering from motor complications presented an overweight with a BMI >25 kg/m2 (106–108). These data are close to the prevalence of overweight and obesity in the general population in western countries which is between 50 and 70% (109). We can state that modern pharmacotherapy together with overall increase in overweight in modern society seems to have changed the phenotype of the PD patients. Modern treatment for PD as agonists and DBS may have an additional impact.
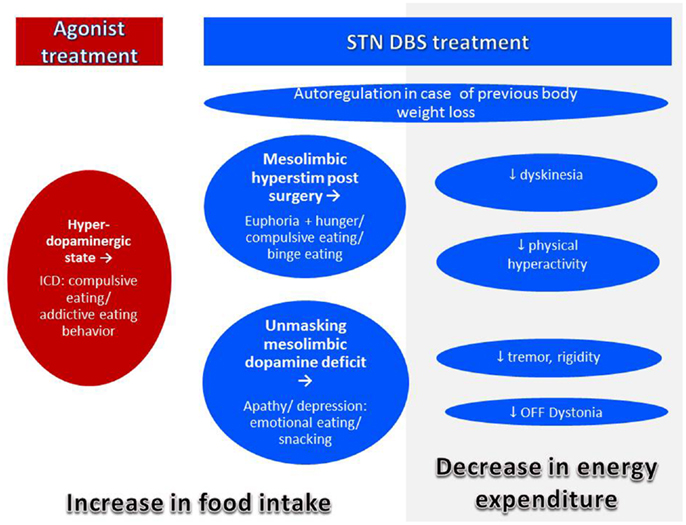
Figure 3. Mechanisms of body weight gain in PD. Body weight gain may occur with treatment by agonist or DBS. Both agonist and DBS treatment may lead to modifications of eating behavior leading to increased food intake. In addition, with successful DBS treatment, energy expenditure decreases by improvement of symptoms leading to body weight gain even in the absence of eating disorders.
Weight gain with dopamine replacement therapy
As stated above, levodopa may be considered as an anorexigenic agent. In contrast, dopamine replacement therapy (DRT) and in particular D2/D3-selective dopamine agonists may lead to behavioral changes, ICDs, and among them changes in eating behavior, referred to as BED (110) and explained by an activation of the “reward system” mediated by D3 receptors (42). Eating disorders were described in up to 15% of patients treated with dopamine agonist leading to weight gain and sometimes to obesity (110–120). Nocturnal eating is frequent and related to sleep disorders in patients treated with agonists (121). Other alterations of eating behavior are described as “insatiable craving,” “compulsive overeating,” “binge-eating” with uncontrollable and compulsive traits as common feature (41, 122–126). In patients with mood fluctuations, eating of snacks occurred more likely in “ON” state while they experienced euphoria (127). Lowering or discontinuation of dopamine agonist may reverse the symptoms (41).
Weight gain after surgery for PD
Weight gain after pallidotomy for PD was first published in 1953 (128–130) (Table 2). Since then many authors reported increased body weight after surgery for PD, mostly STN-DBS (131, 132), but also stimulation of GPi (107, 133–135). In case of STN-DBS, weight gain is present in the majority of patients, is rapid, that is occurs mainly within the first 3 months (131, 136, 137). In the long-term, body weight tends to stabilize in many patients (138, 139) and some patients may lose weight again, but mean weight remains higher than before surgery (136, 140). In individual patients, excessive weight gain leading to obesity was described (106). Weight gain seems to be independent of target (GPi or STN) and procedure (lesion, stimulation, uni-or bilateral) (107, 134, 141). However, in a recent study, STN was more associated with weight gain as GPi (142). In any case, weight gain is achieved by a positive energy balance for which several mechanisms may play a role.
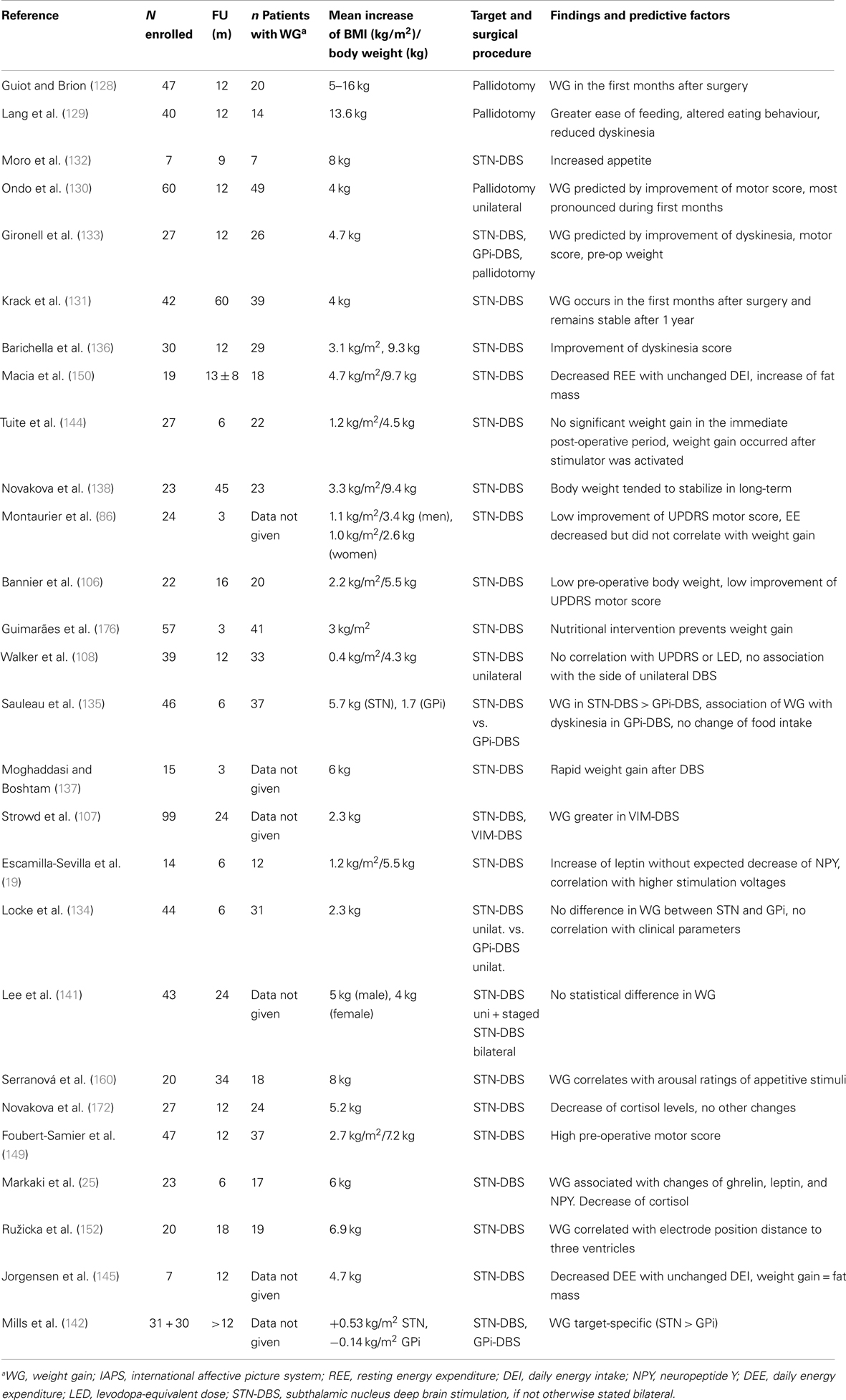
Table 2. Weight gain in PD after surgical treatment: possible mechanisms and predictive factors.
Normalization of body weight after previous weight loss. A compensation of previous weight loss in underweight patients is normal and desirable. In humans as in animals, a period of starvation results in hyperphagia related to the extent to which body fat was previously depleted. Therefore, a drive to overeat seems to be part of a regulatory system to restore fat mass (6). The phenomenon of “rebound adiposity” in PD after STN-DBS was described by Dulloo and Montani (143). Accordingly, weight gain after DBS in PD may be correlated with the pre-operative weight, and initially underweight patients take more weight (106, 133). However, weight gain often exceeds previous weight loss by far (144). Thus, additional mechanisms have to be considered.
Reduction of energy expenditure with unchanged intake. After STN-DBS, a significant decrease (7–13%) of daily EE was reported (86, 145). A decrease of EE of 13% without adaptation of intake would lead to a weight gain of 20 kg after 1 year (145). This decrease of EE after successful STN-DBS may be explained by:
• a reduced resting EE (87) following improvement of rigidity and tremor,
• a reduction of levodopa-induced dyskinesia (146),
• reduction of OFF-period dystonia (147),
• a reduction of levodopa-induced behavioral hyperactivity (121),
• an improvement of sleep pattern and nocturnal hyperactivity (121, 148).
Accordingly, correlations of weight gain with reduction of motor symptoms, reduction and severity of OFF-periods, LEDD reduction, and reduction of levodopa-induced dyskinesia (133, 136, 149, 150) had been described. Despite improved motor response and decreased EE after DBS, 80% of the patients do not increase their physical activity nor change their eating habits (136). This imbalance between intake and expenditure leads inevitably to weight gain, mostly due to an increase in fat mass (86, 106, 150).
Direct impact of STN-stimulation on adjacent brain regions. A direct effect of STN-stimulation on the hypothalamus by current diffusion has been hypothesized as indeed the lateral hypothalamus is very close to the medial limbic tip of the STN (151). In a recent study, weight gain after chronic DBS of STN was inversely related to the distance of the contacts from the wall of the third ventricle (152). However, this observation could not distinguish between current diffusion to the hypothalamus or to the mesolimbic part of the STN. In the rat, a lesion of the STN without lesioning the hypothalamus leads to impulsive feeding behavior (153), strongly arguing for a lesioning effect of the mesolimbic STN. The most impressive clinical behavioral changes may be observed immediately after stimulation has been switched on, while patients are still in the hospital. Patients in the immediate post-operative state may experience a euphoric state induced by STN-DBS, characterized by disinhibition, hyperactivity, and increased appetite. This condition spontaneously recovers within few weeks or months and is thought to be linked to both the lesional effect of surgery with an edema of the STN and to the long-term response of mesolimbic effects of dopaminergic medication (154). The most important gain weight occurs in the first weeks and months until stabilizing. The inverse is the case for stimulation settings, which start very low, and then gradually are increased over time in order to avoid behavioral changes (154). The volume of current diffusion in neural tissue does not increase in a linear way with increase of current setting. On the contrary, there is sharp exponential decrease of efficiency with the distance to the electrode (155). If weight gain were to be explained by current diffusion to the hypothalamus, the weight gain should increase over time with increasing stimulation parameters. Furthermore, high-frequency stimulation or lesion of the lateral hypothalamus has anorectic effects, as shown in rats (156) and obese humans, respectively (157). Altogether, these are strong argument against current diffusion to the hypothalamus as the explanation of weight increase after surgery for STN-DBS.
Weight gain in PD patients treated with STN-DBS is accompanied by increasing levels of leptin reflecting the increasing degree of adipose tissue (19, 25). In normal conditions, high leptin levels have an anorectic function on the hypothalamus by downregulating the expression of orexigenic neuropeptide Y in the arcuate nucleus, a mechanism which ensures body weight stability (158). It was shown that in PD patients treated with STN-DBS, neuropeptide Y levels increase despite high leptin levels and it was argued that DBS interferes with the inhibitory action of leptin in the hypothalamus (19, 25). However, 3 or 6 months after STN surgery, basal levels of hormones of the hypothalamic–adrenal, –gonadal, and –somatotropic axis were normal and hypothalamic function in STN-DBS was considered to be normal (159). These observations argue for a direct effect of STN-stimulation to the mesolimbic STN which has an influence on motivation for food intake in rats (153). Accordingly in PD patients with STN-DBS, an increased sensitivity to food reward cues was reported which correlated with post-operative weight gain (160). However, as is the case for other behavioral effects of STN-DBS, some tolerance effect developing over the first months after surgery is likely (154). It is possible that STN-DBS interferes with homeostatic hypothalamic regulation, but not related to current diffusion toward the hypothalamus.
Eating disorders. As described above, treatment with D2/D3-specific dopamine agonists may lead to compulsive eating behavior, which disappears after discontinuation of treatment (Table 3). Alteration of eating behavior has also been described in PD patients treated with STN-DBS (113, 121, 161). Due to missing classification and nomenclature, hyperphagia is often classified as BED, the only eating behavior disorder (beside bulimia and anemia nervosa) which is described in DSM-IV. BED includes recurrent and frequent bulimic episodes with lack of control and marked distress. There is no compensatory behavior as vomiting. BED is considered as an ICD (110) and as such is part of scales assessing ICD such as the Questionnaire for Impulsive–Compulsive Disorders in PD (162). However, these scales do not inquire about the frequency and amount eaten and therefore do not allow a BED diagnosis. Consequently, they report high false-positive rates (120). In general, assessment of eating behavior in PD in the literature is not systematic and may range from simple telephone interviews, patients self-reports, retrospective database research of key words to different psychological scales.
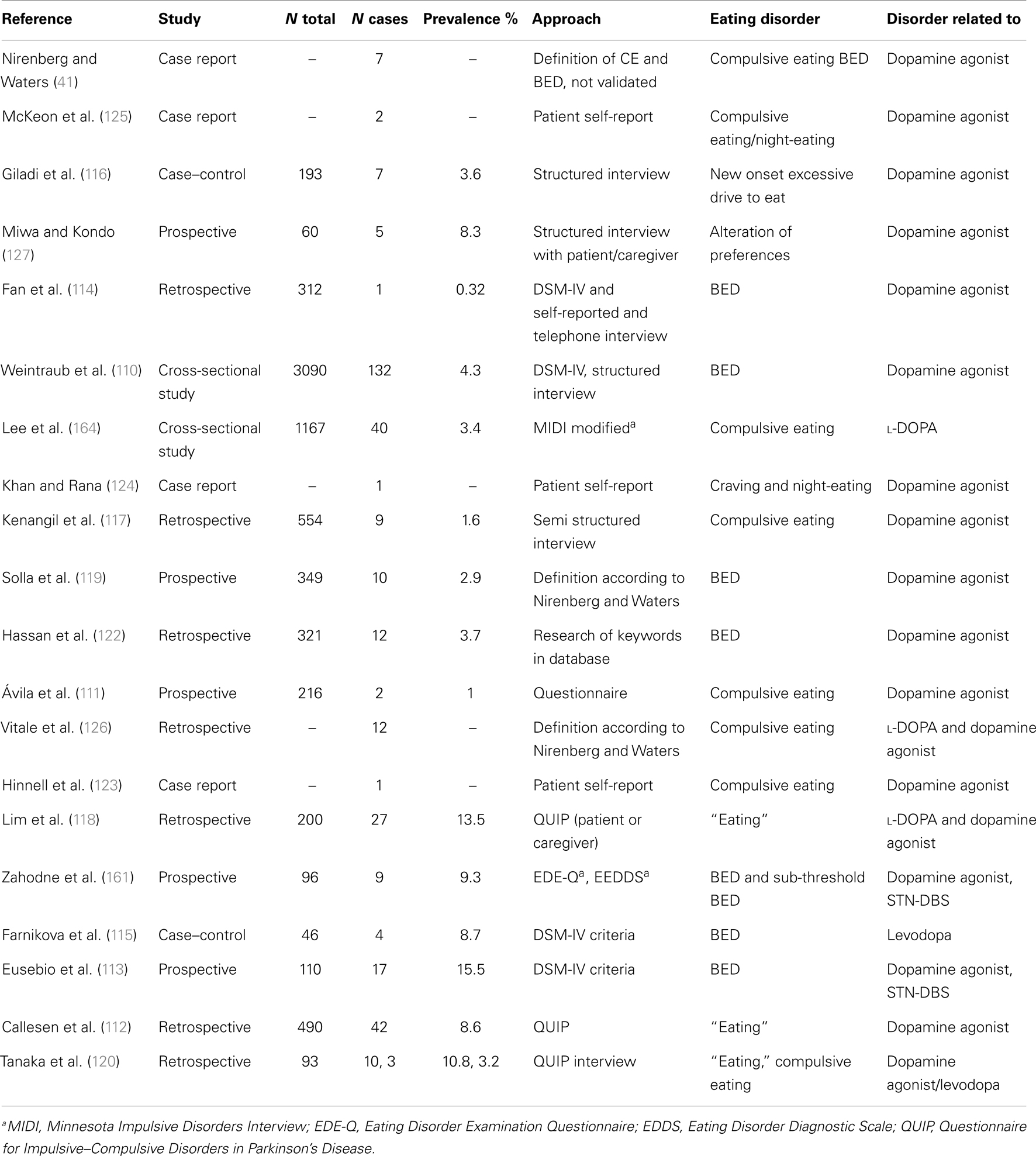
Table 3. Eating disorders in PD.
When employing scales which assess DSM-IV criteria for BED such as the Eating Disorder Examination Questionnaire (EDE-Q), BED is reported to be rather rare in PD: prevalence was about 1% in a small sample of patients (161), comparable with its occurrence in the general population of 1.4% (163). In contrast, prevalence of sub-threshold pathologic eating behavior (“compulsive eating”) was reported to be between 3.4 and 4.5% in PD (110, 164) and seems to increase after STN-DBS (161). We argue that alterations of eating behavior disorders in PD are mostly not BED but comprise a large spectrum of sub threshold pathologic variants of normal eating behavior, described as «snacking», «night-eating», «sweet preferences», «craving», «compulsive eating» which may not all be impulsive. This may explain why in STN-DBS-treated patients, a marked decrease of ICD was described whereas the prevalence of eating disorders decreases less (121) or even increases significantly (113). In fact, DBS-STN was an independent predictor of sub-threshold eating disorders in a small sample of patients (161).
Impulse control disorders are psychiatric conditions characterized by motivation-based behaviors that involve repetitive reward-based activities and loss of control (165). In PD, ICDs are linked to dopamine dysregulation (165) and hyperdopaminergic conditions (121). Successful surgery allows for a marked decrease of dopaminergic treatment (166), leading to disappearance of hyperdopaminergic behavior (gambling, hypersexuality, buying) with exception of eating disorders (113, 121). This condition is often associated with apathy and hypoactivity (121). In these patients, eating may be the only pleasure-generating activity which does not require any effort and is therefore compatible with apathy, which is defined as a decrease in motivation (167) in opposition with ICD which reflects excess motivation oriented toward pleasurable activities (165). In the absence of dopamine, the hedonic response (“liking”) and the perception of taste is conserved (28). Moreover, feeding of palatable food increases dopamine levels in dorsal (35) and ventral striatum including nucleus accumbens (168). Hyperphagia leading to obesity in hypo-dopaminergic conditions such as ADHD had been interpreted as unconscious “self-therapy” in order to normalize mesolimbic dopamine concentrations (169). Thus, hyperphagia could be related to a relative hypo-dopaminergic state which is the case for many PD patients in the post-operative period on chronic DBS when successful stimulation allows for marked decrease in dopaminergic treatment (121, 166). This hypothesis is compatible with a laboratory study which could show that rats previously treated for 5 days with l-DOPA gain 15% more weight than control rats during 12 weeks ad libitum feeding. The authors argue that overeating after dopamine withdrawal might be a side effect of dopaminergic stimulation, (170) and this side effect can easily be explained by a downregulation of the dopaminergic system during dopaminergic treatment followed by a withdrawal syndrome. Of note, a withdrawal state in addiction to cocaine, a direct dopamine increasing drug via inhibition of the dopamine transporter, is characterized by progressive apathy over a period of several weeks and its severity correlates with a progressive striatal dopamine depletion (171).
Given the frequency of apathy after STN-DBS, we propose that the vast majority of eating disorders that appear following DBS in PD should therefore not be considered as ICD but interpreted as sub-threshold pathologic behavior in order to compensate for low dopaminergic signaling. In order to differentiate this particular eating behavior from the compulsive eating observed in patients treated with high dose D2/D3 dopamine agonists, we propose to call this behavior “hypo-dopaminergic snacking.”
Hypo-dopaminergic states more rarely occur after GPi-DBS which does not allow for reduction of levodopa. This may explain why weight gain after GPi-DBS on average is less important than after STN-DBS, and mostly due to the reduction of dyskinesia directly related to GPi-DBS (135).
Other factors.
Improvement of gastric function by STN-DBS and the role of ghrelin. STN-DBS can improve the gastric function in PD and thus improve upper gastrointestinal symptoms such as heavy feeling in the stomach, bloating, nausea or feeling sick, and belching (22). This finding may explain the increased levels of the gastric orexigenic peptide ghrelin in PD patients treated by STN-DBS (25) leading to increased hunger. However, the role of ghrelin in STN-DBS remains unproven since three other authors could not confirm increased levels (22, 23, 172).
Alteration of the serotonergic system. STN-DBS reduces the firing rate of serotonergic neurons in raphe nucleus (173). As serotonin is involved in control of appetite, this may have an impact on eating behavior and increased snacking of sweet foods may be due to a lack of serotonin.
Altered thermogenesis. Centrally-regulated thermogenesis is an important factor in maintaining stable body weight and obesity resistance. Adaptive thermogenesis takes place in brown adipose tissue and the neuropeptide orexin is a key driver (13, 92, 174). Low levels of orexin are common in PD (see above). Although this peptide is investigated in PD mainly with regard to sleep–wake rhythm, orexin deficiency may have an impact on EE and obesity resistance. In fact, PD patients are intolerant to high temperature and drenching sweats is a non-motor symptom which disappears after surgery (175).
Strategies for Maintaining a Stable Body Weight in PD
As weight gain may be desirable or deleterious, the patient’s individual situation should be thoroughly evaluated. Before intervention the following factors should be assessed:
• Actual BMI and previous weight loss, normal weight, previous fluctuations of body weight, and eating disorders.
• Estimation of pre- and post-surgery EE: motor symptoms, dyskinesia, physical activity.
• Actual alimentation, eating habits, and eating disorders.
• Psychological assessment: apathy, depression, hyperactivity.
• Quality of sleep (night-eating disorder).
In DBS patients, nutritional intervention has been shown to be effective (176) and should be performed routinely (95). As weight gain occurs essentially in the first months after surgery, information and dietetic guidance of the patient should start before surgery. As energy requirement is often diminished after successful surgery, an energy-reduced diet should take place and be maintained lifelong. Patients should be encouraged to control their body weight regularly, to supervise their alimentation, and to practice regular physical exercise. These measures should be considered as an adaptation of lifestyle rather than short-time diet.
Recent changes of eating behavior should be taken seriously. Severe hyperphagia with compulsive (craving, binge-eating) or night-eating may improve by discontinuation of agonist treatment. On the other hand, disorders including emotional eating may occur in depressive or hypo-dopaminergic patients treated with STN-DBS. In these patients, a deficit of motivation renders dietary approaches difficult and intervention should first be focused on pharmacological treatment of apathy. Indeed, in selected obese subjects with apathy, it has been shown that treatment with methylphenidate in combination with a weight-loss program was more effective than the weight loss program alone (177). In healthy and obese adults, methylphenidate reduces dietary energy intake by about 20% (38, 39). Thus, alterations of dopaminergic signaling may be an important factor in obesity management of PD.
Concluding Remarks
In general, body weight gain results from dysregulation of the balance between energy requirement and energy input, the latter is reflected in eating behavior. In PD, dysregulation may be due to alterations of (i) hypothalamic regulation, (ii) energy expenditure, or (iii) dopaminergic signaling. Consequently, different pathomechanisms may account for alteration of eating behavior in PD. Disruption of homeostatic control results in increased appetite and hunger and may be accompanied by compulsive eating behavior. Weight gain despite unaltered eating may argue for reduced energy expenditure. Hyperdopaminergic eating behavior is merely characterized by compulsive and nocturnal eating whereas hyperphagia in hypo-dopaminergic state is part of the hypo-dopaminergic behavior accompanied by apathy and characterized by random snacking and emotional eating.
Understanding the eating behavior may therefore be a window on the underlying factors for weight gain. Any intervention, if pharmacological, behavioral, or nutritional should focus on analysis of the patient’s energy expenditure and a detailed analysis of eating behavior.
Conflict of Interest Statement
Paul Krack received research grant from Lundbeck, Orkyn, Novartis, Medtronic, LVL, St. Jude; travel costs/honoraria from Euthérapie, Lundbeck, Boehringer Ingelheim, Novartis, UCB, Medtronic, Orkyn, Abbott, Orion, TEVA, Boston Scientific. The other authors declare no conflicts of interest.
References
1. Braak H, Braak E. Pathoanatomy of Parkinson’s disease. J Neurol (2000) 247(Suppl 2):II3–10. doi: 10.1007/PL00007758
2. Agid Y, Arnulf I, Bejjani P, Bloch F, Bonnet AM, Damier P, et al. Parkinson’s disease is a neuropsychiatric disorder. Adv Neurol (2003) 91:365–70.
3. Chaudhuri KR, Healy DG, Schapira AH. Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol (2006) 5:235–45. doi:10.1016/S1474-4422(06)70373-8
4. Aarsland D, Kurz MW. The epidemiology of dementia associated with Parkinson’s disease. J Neurol Sci (2010) 289:18–22. doi:10.1016/j.jns.2009.08.034
5. Lebouvier T, Chaumette T, Paillusson S, Duyckaerts C, Bruley des Varannes S, et al. The second brain and Parkinson’s disease. Eur J Neurosci (2009) 30:735–41. doi:10.1111/j.1460-9568.2009.06873.x
6. Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature (2006) 443:289–95. doi:10.1038/nature05026
7. Muller MJ, Bosy-Westphal A, Heymsfield SB. Is there evidence for a set point that regulates human body weight? F1000 Med Rep (2010) 2:59. doi:10.3410/M2-59
8. Hill JO, Peters JC. Environmental contributions to the obesity epidemic. Science (1998) 280:1371–4. doi:10.1126/science.280.5368.1371
9. Williams KW, Elmquist JK. From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nat Neurosci (2012) 15:1350–5. doi:10.1038/nn.3217
10. Benarroch EE. Neural control of feeding behavior: overview and clinical correlations. Neurology (2010) 74:1643–50. doi:10.1212/WNL.0b013e3181df0a3f
11. Harrold JA, Dovey TM, Blundell JE, Halford JC. CNS regulation of appetite. Neuropharmacology (2012) 63:3–17. doi:10.1016/j.neuropharm.2012.01.007
12. Kotz CM. Integration of feeding and spontaneous physical activity: role for orexin. Physiol Behav (2006) 88:294–301. doi:10.1016/j.physbeh.2006.05.031
13. Butterick TA, Billington CJ, Kotz CM, Nixon JP. Orexin: pathways to obesity resistance? Rev Endocr Metab Disord (2013) 14:357–64. doi:10.1007/s11154-013-9259-3
14. Drouot X, Moutereau S, Lefaucheur JP, Palfi S, Covali-Noroc A, Margarit L, et al. Low level of ventricular CSF orexin-A is not associated with objective sleepiness in PD. Sleep Med (2011) 12:936–7. doi:10.1016/j.sleep.2011.08.002
15. Fronczek R, Overeem S, Lee SY, Hegeman IM, van Pelt J, van Duinen SG, et al. Hypocretin (orexin) loss in Parkinson’s disease. Brain (2007) 130:1577–85. doi:10.1093/brain/awm090
16. Thannickal TC, Lai YY, Siegel JM. Hypocretin (orexin) and melanin concentrating hormone loss and the symptoms of Parkinson’s disease. Brain (2008) 131:e87. doi:10.1093/brain/awm221
17. Fiszer U, Michałowska M, Baranowska B, Wolinska-Witort E, Jeske W, Jethon M, et al. Leptin and ghrelin concentrations and weight loss in Parkinson’s disease. Acta Neurol Scand (2010) 121:230–6. doi:10.1111/j.1600-0404.2009.01185.x
18. Lorefalt B, Toss G, Granerus AK. Weight loss, body fat mass, and leptin in Parkinson’s disease. Mov Disord (2009) 24:885–90. doi:10.1002/mds.22466
19. Escamilla-Sevilla F, Pérez-Navarro MJ, Muñoz-Pasadas M, Sáez-Zea C, Jouma-Katati M, Piédrola-Maroto G, et al. Change of the melanocortin system caused by bilateral subthalamic nucleus stimulation in Parkinson’s disease. Acta Neurol Scand (2011) 124:275–81. doi:10.1111/j.1600-0404.2010.01469.x
20. Kim KS, Yoon YR, Lee HJ, Yoon S, Kim SY, Shin SW, et al. Enhanced hypothalamic leptin signaling in mice lacking dopamine D2 receptors. J Biol Chem (2010) 285:8905–17. doi:10.1074/jbc.M109.079590
21. Unger MM, Möller JC, Mankel K, Eggert KM, Bohne K, Bodden M, et al. Postprandial ghrelin response is reduced in patients with Parkinson’s disease and idiopathic REM sleep behaviour disorder: a peripheral biomarker for early Parkinson’s disease? J Neurol (2011) 258:982–90. doi:10.1007/s00415-010-5864-1
22. Arai E, Arai M, Uchiyama T, Higuchi Y, Aoyagi K, Yamanaka Y, et al. Subthalamic deep brain stimulation can improve gastric emptying in Parkinson’s disease. Brain (2012) 135:1478–85. doi:10.1093/brain/aws086
23. Corcuff JB, Perlemoine C, Macia F, Tison F, Coman I, Guehl D, et al. Subthalamic nucleus stimulation in patients with Parkinson’s disease does not increase serum ghrelin levels. Br J Nutr (2006) 95:1028–9. doi:10.1079/BJN20051678
24. Marrinan S, Emmanuel AV, Burn DJ. Delayed gastric emptying in Parkinson’s disease. Mov Disord (2014) 29:23–32. doi:10.1002/mds.25708
25. Markaki E, Ellul J, Kefalopoulou Z, Trachani E, Theodoropoulou A, Kyriazopoulou V, et al. The role of ghrelin, neuropeptide Y and leptin peptides in weight gain after deep brain stimulation for Parkinson’s disease. Stereotact Funct Neurosurg (2012) 90:104–12. doi:10.1159/000335045
26. Palmiter RD. Is dopamine a physiologically relevant mediator of feeding behavior? Trends Neurosci (2007) 30:375–81. doi:10.1016/j.tins.2007.06.004
27. Volkow ND, Wang GJ, Tomasi D, Baler RD. The addictive dimensionality of obesity. Biol Psychiatry (2013) 73:811–8. doi:10.1016/j.biopsych.2012.12.020
28. Berridge KC, Ho CY, Richard JM, Di Felice Antonio AG. The tempted brain eats: pleasure and desire circuits in obesity and eating disorders. Brain Res (2010) 1350:43–64. doi:10.1016/j.brainres.2010.04.003
29. Kenny PJ. Reward mechanisms in obesity: new insights and future directions. Neuron (2011) 69:664–79. doi:10.1016/j.neuron.2011.02.016
30. Salamone JD, Correa M. Dopamine and food addiction: lexicon badly needed. Biol Psychiatry (2013) 73:e15–24. doi:10.1016/j.biopsych.2012.09.027
32. McClure SM, Daw ND, Montague PR. A computational substrate for incentive salience. Trends Neurosci (2003) 26:423–8. doi:10.1016/S0166-2236(03)00177-2
33. Sotak BN, Hnasko TS, Robinson S, Kremer EJ, Palmiter RD. Dysregulation of dopamine signaling in the dorsal striatum inhibits feeding. Brain Res (2005) 1061:88–96. doi:10.1016/j.brainres.2005.08.053
34. Volkow ND, Wang GJ, Fowler JS, Logan J, Jayne M, Franceschi D, et al. “Non-hedonic” food motivation in humans involves dopamine in the dorsal striatum and methylphenidate amplifies this effect. Synapse (2002) 44:175–80. doi:10.1002/syn.10075
35. Small DM, Jones-Gotman M, Dagher A. Feeding-induced dopamine release in dorsal striatum correlates with meal pleasantness ratings in healthy human volunteers. Neuroimage (2003) 19:1709–15. doi:10.1016/S1053-8119(03)00253-2
36. Wang GJ, Volkow ND, Logan J, Pappas NR, Wong CT, Zhu W, et al. Brain dopamine and obesity. Lancet (2001) 357:354–7. doi:10.1016/S0140-6736(00)03643-6
37. Wilcox CE, Braskie MN, Kluth JT, Jagust WJ. Overeating behavior and striatal dopamine with 6-[F]-fluoro-L-m-tyrosine PET. J Obes (2010) 2010:909348. doi:10.1155/2010/909348
38. Goldfield GS, Lorello C, Doucet E. Methylphenidate reduces energy intake and dietary fat intake in adults: a mechanism of reduced reinforcing value of food? Am J Clin Nutr (2007) 86:308–15.
39. Leddy JJ, Epstein LH, Jaroni JL, Roemmich JN, Paluch RA, Goldfield GS, et al. Influence of methylphenidate on eating in obese men. Obes Res (2004) 12:224–32. doi:10.1038/oby.2004.29
40. Moreau C, Delval A, Defebvre L, Dujardin K, Duhamel A, Petyt G, et al. Methylphenidate for gait hypokinesia and freezing in patients with Parkinson’s disease undergoing subthalamic stimulation: a multicentre, parallel, randomised, placebo-controlled trial. Lancet Neurol (2012) 11:589–96. doi:10.1016/S1474-4422(12)70106-0
41. Nirenberg MJ, Waters C. Compulsive eating and weight gain related to dopamine agonist use. Mov Disord (2006) 21:524–9. doi:10.1002/mds.20757
42. Vilas D, Pont-Sunyer C, Tolosa E. Impulse control disorders in Parkinson’s disease. Parkinsonism Relat Disord (2012) 18(Suppl 1):S80–4. doi:10.1016/S1353-8020(11)70026-8
43. Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol (2013) 9:13–24. doi:10.1038/nrneurol.2012.242
44. Eriksson KS, Sergeeva OA, Haas HL, Selbach O. Orexins/hypocretins and aminergic systems. Acta Physiol (Oxf) (2010) 198:263–75. doi:10.1111/j.1748-1716.2009.02015.x
45. Marien MR, Colpaert FC, Rosenquist AC. Noradrenergic mechanisms in neurodegenerative diseases: a theory. Brain Res Brain Res Rev (2004) 45:38–78. doi:10.1016/j.brainresrev.2004.02.002
46. Guimarães J, Moura E, Silva E, Aguiar P, Garrett C, Vieira-Coelho MA. Locus coeruleus is involved in weight loss in a rat model of Parkinson’s disease: an effect reversed by deep brain stimulation. Brain Stimul (2013) 6:845–55. doi:10.1016/j.brs.2013.06.002
47. Guimarães J, Moura E, Vieira-Coelho MA, Garrett C. Weight variation before and after surgery in Parkinson’s disease: a noradrenergic modulation? Mov Disord (2012) 27:1078–82. doi:10.1002/mds.25063
48. Fox SH, Chuang R, Brotchie JM. Serotonin and Parkinson’s disease: on movement, mood, and madness. Mov Disord (2009) 24:1255–66. doi:10.1002/mds.22473
49. Wurtman RJ, Wurtman JJ. Brain serotonin, carbohydrate-craving, obesity and depression. Obes Res (1995) 3(Suppl 4):477S–80S. doi:10.1002/j.1550-8528.1995.tb00215.x
50. Luppino FS, de Wit LM, Bouvy PF, Stijnen T, Cuijpers P, Penninx BW, et al. Overweight, obesity, and depression: a systematic review and meta-analysis of longitudinal studies. Arch Gen Psychiatry (2010) 67:220–9. doi:10.1001/archgenpsychiatry.2010.2
51. Wolz M, Kaminsky A, Löhle M, Koch R, Storch A, Reichmann H. Chocolate consumption is increased in Parkinson’s disease. Results from a self-questionnaire study. J Neurol (2009) 256:488–92. doi:10.1007/s00415-009-0118-9
52. Cersosimo MG, Benarroch EE. Pathological correlates of gastrointestinal dysfunction in Parkinson’s disease. Neurobiol Dis (2012) 46:559–64. doi:10.1016/j.nbd.2011.10.014
53. Enweluzo C, Aziz F. Gastroparesis: a review of current and emerging treatment options. Clin Exp Gastroenterol (2013) 6:161–5. doi:10.2147/CEG.S50236
54. Chen H, Zhang SM, Hernan MA, Willett WC, Ascherio A. Weight loss in Parkinson’s disease. Ann Neurol (2003) 53:676–9. doi:10.1002/ana.10577
55. Cheshire WP Jr, Wszolek ZK. Body mass index is reduced early in Parkinson’s disease. Parkinsonism Relat Disord (2005) 11:35–8. doi:10.1016/j.parkreldis.2004.07.001
56. Logroscino G, Sesso HD, Paffenbarger RS Jr, Lee IM. Body mass index and risk of Parkinson’s disease: a prospective cohort study. Am J Epidemiol (2007) 166:1186–90. doi:10.1093/aje/kwm211
57. Ma L, Zhang L, Gao XH, Chen W, Wu YP, Wang Y, et al. Dietary factors and smoking as risk factors for PD in a rural population in China: a nested case-control study. Acta Neurol Scand (2006) 113:278–81. doi:10.1111/j.1600-0404.2005.00571.x
58. Hu G, Jousilahti P, Nissinen A, Antikainen R, Kivipelto M, Tuomilehto J. Body mass index and the risk of Parkinson disease. Neurology (2006) 67:1955–9. doi:10.1212/01.wnl.0000247052.18422.e5
59. Ikeda K, Kashihara H, Tamura M, Kano O, Iwamoto K, Iwasaki Y. Body mass index and the risk of Parkinson disease. Neurology (2007) 68:2156. doi:10.1212/01.wnl.0000269477.49238.ec
60. Abbott RD, Ross GW, White LR, Nelson JS, Masaki KH, Tanner CM, et al. Midlife adiposity and the future risk of Parkinson’s disease. Neurology (2002) 59:1051–7. doi:10.1212/WNL.59.7.1051
61. Kyrozis A, Ghika A, Stathopoulos P, Vassilopoulos D, Trichopoulos D, Trichopoulou A. Dietary and lifestyle variables in relation to incidence of Parkinson’s disease in Greece. Eur J Epidemiol (2013) 28:67–77. doi:10.1007/s10654-012-9760-0
62. Becker C, Brobert GP, Johansson S, Jick SS, Meier CR. Diabetes in patients with idiopathic Parkinson’s disease. Diabetes Care (2008) 31:1808–12. doi:10.2337/dc08-0479
63. Ragonese P, D’Amelio M, Callari G, Di Benedetto N, Palmeri B, Mazzola MA, et al. Body mass index does not change before Parkinson’s disease onset. Eur J Neurol (2008) 15:965–8. doi:10.1111/j.1468-1331.2008.02236.x
64. Scigliano G, Musicco M, Soliveri P, Piccolo I, Ronchetti G, Girotti F. Reduced risk factors for vascular disorders in Parkinson disease patients: a case-control study. Stroke (2006) 37:1184–8. doi:10.1161/01.STR.0000217384.03237.9c
65. Gaig C, Tolosa E. When does Parkinson’s disease begin? Mov Disord (2009) 24(Suppl 2):S656–64. doi:10.1002/mds.22672
66. Aarsland D, Brønnick K, Alves G, Tysnes OB, Pedersen KF, Ehrt U, et al. The spectrum of neuropsychiatric symptoms in patients with early untreated Parkinson’s disease. J Neurol Neurosurg Psychiatry (2009) 80:928–30. doi:10.1136/jnnp.2008.166959
67. Blaine B. Does depression cause obesity? A meta-analysis of longitudinal studies of depression and weight control. J Health Psychol (2008) 13:1190–7. doi:10.1177/1359105308095977
68. Konttinen H, Männistö S, Sarlio-Lähteenkorva S, Silventoinen K, Haukkala A. Emotional eating, depressive symptoms and self-reported food consumption. A population-based study. Appetite (2010) 54:473–9. doi:10.1016/j.appet.2010.01.014
69. Kawada T, Suzuki S. Depressive state, aging, and prevalence of snacking: a preliminary study. Psychogeriatrics (2011) 11:247–8. doi:10.1111/j.1479-8301.2011.00365.x
70. Rose N, Koperski S, Golomb BA. Mood food: chocolate and depressive symptoms in a cross-sectional analysis. Arch Intern Med (2010) 170:699–703. doi:10.1001/archinternmed.2010.78
71. Murakami K, Miyake Y, Sasaki S, Tanaka K, Fukushima W, Kiyohara C, et al. Dietary glycemic index is inversely associated with the risk of Parkinson’s disease: a case-control study in Japan. Nutrition (2010) 26:515–21. doi:10.1016/j.nut.2009.05.021
72. Chen H, Zhang SM, Hernán MA, Willett WC, Ascherio A. Diet and Parkinson’s disease: a potential role of dairy products in men. Ann Neurol (2002) 52:793–801. doi:10.1002/ana.10381
73. Miyake Y, Tanaka K, Fukushima W, Sasaki S, Kiyohara C, Tsuboi Y, et al. Lack of association of dairy food, calcium, and vitamin D intake with the risk of Parkinson’s disease: a case-control study in Japan. Parkinsonism Relat Disord (2011) 17:112–6. doi:10.1016/j.parkreldis.2010.11.018
74. Noyce AJ, Bestwick JP, Silveira-Moriyama L, Hawkes CH, Giovannoni G, Lees AJ, et al. Meta-analysis of early non-motor features and risk factors for Parkinson disease. Ann Neurol (2012) 72:893–901. doi:10.1002/ana.23687
75. van der Marck MA, Dicke HC, Uc EY, Kentin ZH, Borm GF, Bloem BR, et al. Body mass index in Parkinson’s disease: a meta-analysis. Parkinsonism Relat Disord (2012) 18:263–7. doi:10.1016/j.parkreldis.2011.10.016
76. Uc EY, Struck LK, Rodnitzky RL, Zimmerman B, Dobson J, Evans WJ. Predictors of weight loss in Parkinson’s disease. Mov Disord (2006) 21:930–6. doi:10.1002/mds.20837
77. Sheard JM, Ash S, Silburn PA, Kerr GK. Prevalence of malnutrition in Parkinson’s disease: a systematic review. Nutr Rev (2011) 69:520–32. doi:10.1111/j.1753-4887.2011.00413.x
78. Sheard JM, Ash S, Mellick GD, Silburn PA, Kerr GK. Markers of disease severity are associated with malnutrition in Parkinson’s disease. PLoS One (2013) 8:e57986. doi:10.1371/journal.pone.0057986
79. Davies KN, King D, Davies H. A study of the nutritional status of elderly patients with Parkinson’s disease. Age Ageing (1994) 23:142–5. doi:10.1093/ageing/23.suppl_2.P11-b
80. Bachmann CG, Zapf A, Brunner E, Trenkwalder C. Dopaminergic treatment is associated with decreased body weight in patients with Parkinson’s disease and dyskinesias. Eur J Neurol (2009) 16:895–901. doi:10.1111/j.1468-1331.2009.02617.x
81. Broussolle E, Borson F, Gonzalez de Suso JM, Chayvialle JA, Beylot M, Chazot G. Increase of energy expenditure in Parkinson’s disease. Rev Neurol (Paris) (1991) 147:46–51.
82. Capecci M, Petrelli M, Emanuelli B, Millevolte M, Nicolai A, Provinciali L, et al. Rest energy expenditure in Parkinson’s disease: role of disease progression and dopaminergic therapy. Parkinsonism Relat Disord (2013) 19:238–41. doi:10.1016/j.parkreldis.2012.10.016
83. Levi S, Cox M, Lugon M, Hodkinson M, Tomkins A. Increased energy expenditure in Parkinson’s disease. BMJ (1990) 301:1256–7. doi:10.1136/bmj.301.6763.1256
84. Markus HS, Cox M, Tomkins AM. Raised resting energy expenditure in Parkinson’s disease and its relationship to muscle rigidity. Clin Sci (Lond) (1992) 83:199–204.
85. Markus HS, Tomkins AM, Stern GM. Increased prevalence of undernutrition in Parkinson’s disease and its relationship to clinical disease parameters. J Neural Transm Park Dis Dement Sect (1993) 5:117–25. doi:10.1007/BF02251202
86. Montaurier C, Morio B, Bannier S, Derost P, Arnaud P, Brandolini-Bunlon M, et al. Mechanisms of body weight gain in patients with Parkinson’s disease after subthalamic stimulation. Brain (2007) 130:1808–18. doi:10.1093/brain/awm113
87. Perlemoine C, Macia F, Tison F, Coman I, Guehl D, Burbaud P, et al. Effects of subthalamic nucleus deep brain stimulation and levodopa on energy production rate and substrate oxidation in Parkinson’s disease. Br J Nutr (2005) 93:191–8. doi:10.1079/BJN20041297
88. Delikanaki-Skaribas E, Trail M, Wong WW, Lai EC. Daily energy expenditure, physical activity, and weight loss in Parkinson’s disease patients. Mov Disord (2009) 24:667–71. doi:10.1002/mds.22372
89. Toth MJ, Fishman PS, Poehlman ET. Free-living daily energy expenditure in patients with Parkinson’s disease. Neurology (1997) 48:88–91. doi:10.1212/WNL.48.1.88
90. Galgani J, Ravussin E. Energy metabolism, fuel selection and body weight regulation. Int J Obes (Lond) (2008) 32(Suppl 7):S109–19. doi:10.1038/ijo.2008.246
91. Drouot X, Moutereau S, Nguyen JP, Lefaucheur JP, Créange A, Remy P, et al. Low levels of ventricular CSF orexin/hypocretin in advanced PD. Neurology (2003) 61:540–3. doi:10.1212/01.WNL.0000078194.53210.48
92. Kotz C, Nixon J, Butterick T, Perez-Leighton C, Teske J, Billington C. Brain orexin promotes obesity resistance. Ann N Y Acad Sci (2012) 1264:72–86. doi:10.1111/j.1749-6632.2012.06585.x
93. Gispert S, Ricciardi F, Kurz A, Azizov M, Hoepken HH, Becker D, et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS One (2009) 4:e5777. doi:10.1371/journal.pone.0005777
94. Koller WC. Levodopa in the treatment of Parkinson’s disease. Neurology (2000) 55(11 Suppl 4):S2–7; discussion S8–12.
95. Barichella M, Cereda E, Pezzoli G. Major nutritional issues in the management of Parkinson’s disease. Mov Disord (2009) 24:1881–92. doi:10.1002/mds.22705
96. Bachmann CG, Trenkwalder C. Body weight in patients with Parkinson’s disease. Mov Disord (2006) 21:1824–30. doi:10.1002/mds.21068
97. Heetun ZS, Quigley EM. Gastroparesis and Parkinson’s disease: a systematic review. Parkinsonism Relat Disord (2012) 18:433–40. doi:10.1016/j.parkreldis.2011.12.004
98. Jankovic J, Wooten M, Van der Linden C, Jansson B. Low body weight in Parkinson’s disease. South Med J (1992) 85:351–4. doi:10.1097/00007611-199204000-00005
99. Kashihara K. Weight loss in Parkinson’s disease. J Neurol (2006) 253(Suppl 7):VII38–41. doi:10.1007/s00415-006-7009-0
100. Cereda E, Barichella M, Pedrolli C, Pezzoli G. Low-protein and protein-redistribution diets for Parkinson’s disease patients with motor fluctuations: a systematic review. Mov Disord (2010) 25:2021–34. doi:10.1002/mds.23226
101. Lipman IJ, Boykin ME, Flora RE. Glucose intolerance in Parkinson’s disease. J Chronic Dis (1974) 27:573–9. doi:10.1016/0021-9681(74)90031-9
102. Sandyk R. The relationship between diabetes mellitus and Parkinson’s disease. Int J Neurosci (1993) 69:125–30. doi:10.3109/00207459309003322
103. Cai H, Cong WN, Ji S, Rothman S, Maudsley S, Martin B. Metabolic dysfunction in Alzheimer’s disease and related neurodegenerative disorders. Curr Alzheimer Res (2012) 9:5–17. doi:10.2174/156720512799015064
104. Cereda E, Barichella M, Pedrolli C, Klersy C, Cassani E, Caccialanza R, et al. Diabetes and risk of Parkinson’s disease: a systematic review and meta-analysis. Diabetes Care (2011) 34:2614–23. doi:10.2337/dc11-1584
105. Barichella M, Marczewska A, Vairo A, Canesi M, Pezzoli G. Is underweightness still a major problem in Parkinson’s disease patients? Eur J Clin Nutr (2003) 57:543–7. doi:10.1038/sj.ejcn.1601581
106. Bannier S, Montaurier C, Derost PP, Ulla M, Lemaire JJ, Boirie Y, et al. Overweight after deep brain stimulation of the subthalamic nucleus in Parkinson disease: long term follow-up. J Neurol Neurosurg Psychiatry (2009) 80:484–8. doi:10.1136/jnnp.2008.158576
107. Strowd RE, Cartwright MS, Passmore LV, Ellis TL, Tatter SB, Siddiqui MS. Weight change following deep brain stimulation for movement disorders. J Neurol (2010) 257:1293–7. doi:10.1007/s00415-010-5509-4
108. Walker HC, Lyerly M, Cutter G, Hagood J, Stover NP, Guthrie SL, et al. Weight changes associated with unilateral STN DBS and advanced PD. Parkinsonism Relat Disord (2009) 15:709–11. doi:10.1016/j.parkreldis.2009.01.009
109. WHO. Non-Communicable Diseases Country Profiles 2011. Geneva: World Health Organization (2011).
110. Weintraub D, Koester J, Potenza MN, Siderowf AD, Stacy M, Voon V, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol (2010) 67:589–95. doi:10.1001/archneurol.2010.65
111. Ávila A, Cardona X, Bello J, Maho P, Sastre F, Martín-Baranera M. Impulse control disorders and punding in Parkinson’s disease: the need for a structured interview. Neurologia (2011) 26:166–72. doi:10.1016/j.nrl.2010.09.007
112. Callesen MB, Weintraub D, Damholdt MF, Møller A. Impulsive and compulsive behaviors among Danish patients with Parkinson’s disease: prevalence, depression, and personality. Parkinsonism Relat Disord (2014) 20:22–6. doi:10.1016/j.parkreldis.2013.09.006
113. Eusebio A, Witjas T, Cohen J, Fluchère F, Jouve E, Régis J, et al. Subthalamic nucleus stimulation and compulsive use of dopaminergic medication in Parkinson’s disease. J Neurol Neurosurg Psychiatry (2013) 84:868–74. doi:10.1136/jnnp-2012-302387
114. Fan W, Ding H, Ma J, Chan P. Impulse control disorders in Parkinson’s disease in a Chinese population. Neurosci Lett (2009) 465:6–9. doi:10.1016/j.neulet.2009.06.074
115. Farnikova K, Obereigneru R, Kanovsky P, Prasko J. Comparison of personality characteristics in Parkinson disease patients with and without impulse control disorders and in healthy volunteers. Cogn Behav Neurol (2012) 25:25–33. doi:10.1097/WNN.0b013e31824b4103
116. Giladi N, Weitzman N, Schreiber S, Shabtai H, Peretz C. New onset heightened interest or drive for gambling, shopping, eating or sexual activity in patients with Parkinson’s disease: the role of dopamine agonist treatment and age at motor symptoms onset. J Psychopharmacol (2007) 21:501–6. doi:10.1177/0269881106073109
117. Kenangil G, Ozekmekçi S, Sohtaoglu M, Erginöz E. Compulsive behaviors in patients with Parkinson’s disease. Neurologist (2010) 16:192–5. doi:10.1097/NRL.0b013e31819f952b
118. Lim SY, Tan ZK, Ngam PI, Lor TL, Mohamed H, Schee JP, et al. Impulsive-compulsive behaviors are common in Asian Parkinson’s disease patients: assessment using the QUIP. Parkinsonism Relat Disord (2011) 17:761–4. doi:10.1016/j.parkreldis.2011.07.009
119. Solla P, Cannas A, Floris GL, Orofino G, Costantino E, Boi A, et al. Behavioral, neuropsychiatric and cognitive disorders in Parkinson’s disease patients with and without motor complications. Prog Neuropsychopharmacol Biol Psychiatry (2011) 35:1009–13. doi:10.1016/j.pnpbp.2011.02.002
120. Tanaka K, Wada-Isoe K, Nakashita S, Yamamoto M, Nakashima K. Impulsive compulsive behaviors in Japanese Parkinson’s disease patients and utility of the Japanese version of the Questionnaire for impulsive-compulsive disorders in Parkinson’s disease. J Neurol Sci (2013) 331:76–80. doi:10.1016/j.jns.2013.05.013
121. Lhommée E, Klinger H, Thobois S, Schmitt E, Ardouin C, Bichon A, et al. Subthalamic stimulation in Parkinson’s disease: restoring the balance of motivated behaviours. Brain (2012) 135:1463–77. doi:10.1093/brain/aws078
122. Hassan A, Bower JH, Kumar N, Matsumoto JY, Fealey RD, Josephs KA, et al. Dopamine agonist-triggered pathological behaviors: surveillance in the PD clinic reveals high frequencies. Parkinsonism Relat Disord (2011) 17:260–4. doi:10.1016/j.parkreldis.2011.01.009
123. Hinnell C, Hulse N, Martin A, Samuel M. Hypersexuality and compulsive over-eating associated with transdermal dopamine agonist therapy. Parkinsonism Relat Disord (2011) 17:295–6. doi:10.1016/j.parkreldis.2011.01.010
124. Khan W, Rana AQ. Dopamine agonist induced compulsive eating behaviour in a Parkinson’s disease patient. Pharm World Sci (2010) 32:114–6. doi:10.1007/s11096-009-9358-0
125. McKeon A, Josephs KA, Klos KJ, Hecksel K, Bower JH, Michael Bostwick J, et al. Unusual compulsive behaviors primarily related to dopamine agonist therapy in Parkinson’s disease and multiple system atrophy. Parkinsonism Relat Disord (2007) 13:516–9. doi:10.1016/j.parkreldis.2007.04.004
126. Vitale C, Santangelo G, Trojano L, Verde F, Rocco M, Grossi D, et al. Comparative neuropsychological profile of pathological gambling, hypersexuality, and compulsive eating in Parkinson’s disease. Mov Disord (2011) 26:830–6. doi:10.1002/mds.23567
127. Miwa H, Kondo T. Alteration of eating behaviors in patients with Parkinson’s disease: possibly overlooked? Neurocase (2008) 14:480–4. doi:10.1080/13554790802495324
128. Guiot G, Brion S. Traitement des mouvements anormaux par la coagulation pallidale. Technique et résultats. Rev Neurol (Paris) (1953) 89:578–580.
129. Lang AE, Lozano A, Tasker R, Duff J, Saint-Cyr J, Trépanier L. Neuropsychological and behavioral changes and weight gain after medial pallidotomy. Ann Neurol (1997) 41:834–6. doi:10.1002/ana.410410624
130. Ondo WG, Ben-Aire L, Jankovic J, Lai E, Contant C, Grossman R. Weight gain following unilateral pallidotomy in Parkinson’s disease. Acta Neurol Scand (2000) 101:79–84. doi:10.1034/j.1600-0404.2000.101002079.x
131. Krack P, Batir A, Van Blercom N, Chabardes S, Fraix V, Ardouin C, et al. Five-year follow-up of bilateral stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med (2003) 349:1925–34. doi:10.1056/NEJMoa035275
132. Moro E, Scerrati M, Romito LM, Roselli R, Tonali P, Albanese A. Chronic subthalamic nucleus stimulation reduces medication requirements in Parkinson’s disease. Neurology (1999) 53:85–90. doi:10.1212/WNL.53.1.85
133. Gironell A, Pascual-Sedano B, Otermin P, Kulisevsky J. Weight gain after functional surgery for Parkinson’s disease. Neurologia (2002) 17:310–6.
134. Locke MC, Wu SS, Foote KD, Sassi M, Jacobson CE, Rodriguez RL, et al. Weight changes in subthalamic nucleus vs globus pallidus internus deep brain stimulation: results from the COMPARE Parkinson disease deep brain stimulation cohort. Neurosurgery (2011) 68:1233–7. doi:10.1227/NEU.0b013e31820b52c5 discussion 1237-8.
135. Sauleau P, Leray E, Rouaud T, Drapier S, Drapier D, Blanchard S, et al. Comparison of weight gain and energy intake after subthalamic versus pallidal stimulation in Parkinson’s disease. Mov Disord (2009) 24:2149–55. doi:10.1002/mds.22765
136. Barichella M, Marczewska AM, Mariani C, Landi A, Vairo A, Pezzoli G. Body weight gain rate in patients with Parkinson’s disease and deep brain stimulation. Mov Disord (2003) 18:1337–40. doi:10.1002/mds.10543
137. Moghaddasi M, Boshtam M. Weight changes in Parkinson’s disease patients after subthalamic nucleus deep brain stimulation surgery. Acta Neurol Belg (2010) 110:311–3.
138. Novakova L, Ruzicka E, Jech R, Serranova T, Dusek P, Urgosik D. Increase in body weight is a non-motor side effect of deep brain stimulation of the subthalamic nucleus in Parkinson’s disease. Neuro Endocrinol Lett (2007) 28:21–5.
139. Rodriguez-Oroz MC, Moro E, Krack P. Long-term outcomes of surgical therapies for Parkinson’s disease. Mov Disord (2012) 27:1718–28. doi:10.1002/mds.25214
140. Castrioto A, Lozano AM, Poon YY, Lang AE, Fallis M, Moro E. Ten-year outcome of subthalamic stimulation in Parkinson disease: a blinded evaluation. Arch Neurol (2011) 68:1550–6. doi:10.1001/archneurol.2011.182
141. Lee EM, Kurundkar A, Cutter GR, Huang H, Guthrie BL, Watts RL, et al. Comparison of weight changes following unilateral and staged bilateral STN DBS for advanced PD. Brain Behav (2011) 1:12–8. doi:10.1002/brb3.9
142. Mills KA, Scherzer R, Starr PA, Ostrem JL. Weight change after globus pallidus internus or subthalamic nucleus deep brain stimulation in Parkinson’s disease and dystonia. Stereotact Funct Neurosurg (2012) 90:386–93. doi:10.1159/000340071
143. Dulloo AG, Montani JP. Obesity in Parkinson’s disease patients on electrotherapy: collateral damage, adiposity rebound or secular trends? Br J Nutr (2005) 93:417–9. doi:10.1079/BJN20041337
144. Tuite PJ, Maxwell RE, Ikramuddin S, Kotz CM, Billington CJ, Laseski MA, et al. Weight and body mass index in Parkinson’s disease patients after deep brain stimulation surgery. Parkinsonism Relat Disord (2005) 11:247–52. doi:10.1016/j.parkreldis.2005.01.006
145. Jorgensen HU, Werdelin L, Lokkegaard A, Westerterp KR, Simonsen L. Free-living energy expenditure reduced after deep brain stimulation surgery for Parkinson’s disease. Clin Physiol Funct Imaging (2012) 32:214–20. doi:10.1111/j.1475-097X.2011.01079.x
146. Krack P, Limousin P, Benabid AL, Pollak P. Chronic stimulation of subthalamic nucleus improves levodopa-induced dyskinesias in Parkinson’s disease. Lancet (1997) 350:1676–1676. doi:10.1016/S0140-6736(05)64273-0
147. Krack P, Pollak P, Limousin P, Benazzouz A, Deuschl G, Benabid AL. From off-period dystonia to peak-dose chorea. The clinical spectrum of varying subthalamic nucleus activity. Brain (1999) 122(Pt 6):1133–46. doi:10.1093/brain/122.6.1133
148. Arnulf I, Leu-Semenescu S. Sleepiness in Parkinson’s disease. Parkinsonism Relat Disord (2009) 15(Suppl 3):S101–4. doi:10.1016/S1353-8020(09)70792-8
149. Foubert-Samier A, Maurice S, Hivert S, Guelh D, Rigalleau V, Burbaud P, et al. A long-term follow-up of weight changes in subthalamic nucleus stimulated Parkinson’s disease patients. Rev Neurol (Paris) (2012) 168:173–6. doi:10.1016/j.neurol.2011.04.006
150. Macia F, Perlemoine C, Coman I, Guehl D, Burbaud P, Cuny E, et al. Parkinson’s disease patients with bilateral subthalamic deep brain stimulation gain weight. Mov Disord (2004) 19:206–12. doi:10.1002/mds.10630
151. Haynes WI, Haber SN. The organization of prefrontal-subthalamic inputs in primates provides an anatomical substrate for both functional specificity and integration: implications for basal ganglia models and deep brain stimulation. J Neurosci (2013) 33:4804–14. doi:10.1523/JNEUROSCI.4674-12.2013
152. Ružicka F, Jech R, Nováková L, Urgošík D, Vymazal J, Ružicka E. Weight gain is associated with medial contact site of subthalamic stimulation in Parkinson’s disease. PLoS One (2012) 7:e38020. doi:10.1371/journal.pone.0038020
153. Baunez C, Amalric M, Robbins TW. Enhanced food-related motivation after bilateral lesions of the subthalamic nucleus. J Neurosci (2002) 22:562–8.
154. Castrioto A, Lhommée E, Moro E, Krack P. Mood and behavioural effects of subthalamic stimulation in Parkinson’s disease. Lancet Neurol (2014) 13(3):287–305. doi:10.1016/S1474-4422(13)70294-1
155. Butson CR, Maks CB, McIntyre CC. Sources and effects of electrode impedance during deep brain stimulation. Clin Neurophysiol (2006) 117:447–54. doi:10.1016/j.clinph.2005.10.007
156. Sani S, Jobe K, Smith A, Kordower JH, Bakay RA. Deep brain stimulation for treatment of obesity in rats. J Neurosurg (2007) 107:809–13. doi:10.3171/JNS-07/10/0809
157. Quaade F, Vaernet K, Larsson S. Stereotaxic stimulation and electrocoagulation of the lateral hypothalamus in obese humans. Acta Neurochir (Wien) (1974) 30:111–7. doi:10.1007/BF01405759
158. Pandit R, de Jong JW, Vanderschuren LJ, Adan RA. Neurobiology of overeating and obesity: the role of melanocortins and beyond. Eur J Pharmacol (2011) 660:28–42. doi:10.1016/j.ejphar.2011.01.034
159. Seifried C, Boehncke S, Heinzmann J, Baudrexel S, Weise L, Gasser T, et al. Diurnal variation of hypothalamic function and chronic subthalamic nucleus stimulation in Parkinson’s disease. Neuroendocrinology (2013) 97:283–90. doi:10.1159/000343808
160. Serranová T, Jech R, Dušek P, Sieger T, Ružicka F, Urgošík D, et al. Subthalamic nucleus stimulation affects incentive salience attribution in Parkinson’s disease. Mov Disord (2011) 26:2260–6. doi:10.1002/mds.23880
161. Zahodne LB, Susatia F, Bowers D, Ong TL, Jacobson CE IV, Okun MS, et al. Binge eating in Parkinson’s disease: prevalence, correlates and the contribution of deep brain stimulation. J Neuropsychiatry Clin Neurosci (2011) 23:56–62. doi:10.1176/appi.neuropsych.23.1.56
162. Weintraub D, Hoops S, Shea JA, Lyons KE, Pahwa R, Driver-Dunckley ED, et al. Validation of the questionnaire for impulsive-compulsive disorders in Parkinson’s disease. Mov Disord (2009) 24:1461–7. doi:10.1002/mds.22571
163. Kessler RC, Berglund PA, Chiu WT, Deitz AC, Hudson JI, Shahly V, et al. The prevalence and correlates of binge eating disorder in the World Health Organization World Mental Health Surveys. Biol Psychiatry (2013) 73:904–14. doi:10.1016/j.biopsych.2012.11.020
164. Lee JY, Kim JM, Kim JW, Cho J, Lee WY, Kim HJ, et al. Association between the dose of dopaminergic medication and the behavioral disturbances in Parkinson disease. Parkinsonism Relat Disord (2010) 16:202–7. doi:10.1016/j.parkreldis.2009.12.002
165. Okai D, Samuel M, Askey-Jones S, David AS, Brown RG. Impulse control disorders and dopamine dysregulation in Parkinson’s disease: a broader conceptual framework. Eur J Neurol (2011) 18:1379–83. doi:10.1111/j.1468-1331.2011.03432.x
166. Thobois S, Ardouin C, Lhommée E, Klinger H, Lagrange C, Xie J, et al. Non-motor dopamine withdrawal syndrome after surgery for Parkinson’s disease: predictors and underlying mesolimbic denervation. Brain (2010) 133:1111–27. doi:10.1093/brain/awq032
167. Marin RS, Fogel BS, Hawkins J, Duffy J, Krupp B. Apathy: a treatable syndrome. J Neuropsychiatry Clin Neurosci (1995) 7:23–30.
168. Taber MT, Fibiger HC. Feeding-evoked dopamine release in the nucleus accumbens: regulation by glutamatergic mechanisms. Neuroscience (1997) 76:1105–12. doi:10.1016/S0306-4522(96)00450-2
169. Liu LL, Li BM, Yang J, Wang YW. Does dopaminergic reward system contribute to explaining comorbidity obesity and ADHD? Med Hypotheses (2008) 70:1118–20. doi:10.1016/j.mehy.2007.10.012
170. Reinholz J, Skopp O, Breitenstein C, Bohr I, Winterhoff H, Knecht S. Compensatory weight gain due to dopaminergic hypofunction: new evidence and own incidental observations. Nutr Metab (2008) 5: doi:10.1186/1743-7075-5-35
171. Wu JC, Bell K, Najafi A, Widmark C, Keator D, Tang C, et al. Decreasing striatal 6-FDOPA uptake with increasing duration of cocaine withdrawal. Neuropsychopharmacology (1997) 17:402–9. doi:10.1016/S0893-133X(97)00089-4
172. Novakova L, Haluzik M, Jech R, Urgosik D, Ruzicka F, Ruzicka E. Hormonal regulators of food intake and weight gain in Parkinson’s disease after subthalamic nucleus stimulation. Neuro Endocrinol Lett (2011) 32:437–41.
173. Tan SK, Hartung H, Sharp T, Temel Y. Serotonin-dependent depression in Parkinson’s disease: a role for the subthalamic nucleus? Neuropharmacology (2011) 61:387–99. doi:10.1016/j.neuropharm.2011.01.006
174. Seale P. Orexin turns up the heat on obesity. Cell Metab (2011) 14:441–2. doi:10.1016/j.cmet.2011.09.007
175. Krack P, Fraix V, Mendes A, Benabid AL, Pollak P. Postoperative management of subthalamic nucleus stimulation for Parkinson’s disease. Mov Disord (2002) 17:S188–97. doi:10.1002/mds.10163
176. Guimarães J, Matos E, Rosas MJ, Vieira-Coelho A, Borges N, Correia F, et al. Modulation of nutritional state in Parkinsonian patients with bilateral subthalamic nucleus stimulation. J Neurol (2009) 256:2072–8. doi:10.1007/s00415-009-5252-x
Keywords: Parkinson’s disease, body weight, eating behavior, DBS, dopamine, binge-eating disorder
Citation: Kistner A, Lhommée E and Krack P (2014) Mechanisms of body weight fluctuations in Parkinson’s disease. Front. Neurol. 5:84. doi: 10.3389/fneur.2014.00084
Received: 03 January 2014; Paper pending published: 10 April 2014;
Accepted: 16 May 2014; Published online: 02 June 2014.
Edited by:
Ryuji Kaji, Tokushima University Hospital, JapanReviewed by:
Maria Fiorella Contarino, Academic Medical Center, NetherlandsSatoshi Goto, Tokushima University Hospital, Japan
Copyright: © 2014 Kistner, Lhommée and Krack. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Kistner, Department of Neurology, University Hospital Grenoble, BP 217, 38043 Grenoble Cedex 09, France e-mail: akistner@chu-grenoble.fr