 Sigrun Lange1,2*
Sigrun Lange1,2*
- 1Department of Pharmacology, UCL School of Pharmacy, London, UK
- 2Department of Biomedical Sciences, University of Westminster, London, UK
Oxygen deprivation and infection are major causes of perinatal brain injury leading to cerebral palsy and other neurological disabilities. The identification of novel key factors mediating white and gray matter damage are crucial to allow better understanding of the specific contribution of different cell types to the injury processes and pathways for clinical intervention. Recent studies in the Rice–Vannucci mouse model of neonatal hypoxic ischemia (HI) have highlighted novel roles for calcium-regulated peptidylarginine deiminases (PADs) and demonstrated neuroprotective effects of pharmacological PAD inhibition following HI and synergistic infection mimicked by lipopolysaccharide stimulation.
Introduction
Hypoxia and synergistic infection are major causes of perinatal brain injury (1–3), contributing to neurological disabilities affecting 1–8 cases per 1000 live births in the Western world and up to 26 per 1000 live births in underdeveloped countries (4, 5). In the acute phase of hypoxic–ischemic encephalopathy (HIE), cerebral blood flow is decreased, resulting in reduced oxygen and glucose delivery to the brain and anaerobic metabolism. Subsequent adenosine triphosphate (ATP) depletion leads to intracellular accumulation of sodium, water, and calcium due to reduced transcellular transport (6, 7). The cell releases excitatory glutamate upon membrane depolarization, allowing calcium to flow into the cell via N-methyl-d-aspartate (NMDA)-gated channels. The resulting peroxidation of free fatty acids by oxygen free radicals leads to more cellular damage. The culmination of energy failure, acidosis, glutamate release, lipid peroxidation, and toxic effects of nitric oxide (NO) leads to cell death via necrosis and apoptosis (7–9). The re-oxygenation stage following a neonatal hypoxic ischemia (HI) insult is followed by an intermediate “grace period” with little overt metabolic, nuclear magnetic resonance (NMR), or histological abnormalities, and only then by secondary energy failure, inflammation, apoptotic, necrotic and/or autophagic cell death, and axonal degeneration (10–13). During this intermediate phase, changes in cellular transcription, de novo protein synthesis, and posttranslational modifications all play pivotal roles (14–16). The tertiary phase of HIE during the months after the acute insult involves late cell death, remodeling of the injured brain, and astrogliosis (Figure 1).
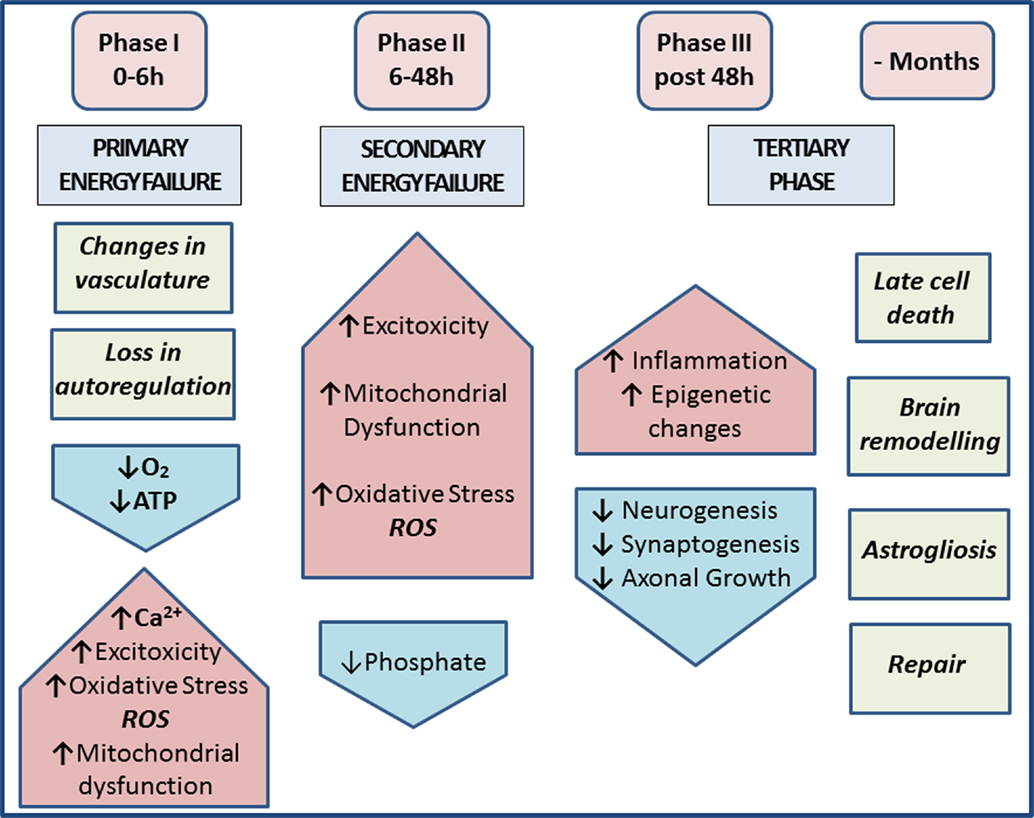
Figure 1. Phases of injury post-HIE. Phase I at 0–6 h post-insult includes vasculature changes and primary energy failure. This results in loss of autoregulation and severe lowering of the systemic arterial blood pressure. This causes decrease in oxygen, depletion of ATP, increased excitotoxicity, raised intracellular calcium, oxidative stress (ROS), and mitochondrial dysfunction. Phase II at 6–48 h post-insult and secondary energy failure lead to continued excitotoxicity, mitochondrial dysfunction, and oxidative stress; reduced phosphate levels. Phase III post 48 h shows injury to microglia, neurons, and astrocytes and also leads to continuous release of cytokines and other detrimental factors causing chronic inflammation, which in turn lead to epigenetic changes, as well as impairment of synaptogenesis, axonal growth, and neurogenesis. The tertiary phase continues months after injury involving late cell death, brain remodeling, and astrogliosis [based on Ref. (7, 17)].
Experimental studies in HIE animal models have shown roles for epigenetic mechanisms (18, 19), pH changes (20–22), and the tumor necrosis factor (TNF) gene cluster of cytokines in a combined inflammatory and HI brain insult (23). Clinical studies have shown protective therapeutic effects of hypothermia (24), which remains the standard care for HIE. Recent translational studies demonstrated significantly enhanced neuroprotection following co-therapy of hypothermia with xenon (25) or melatonin (26). Therefore, novel targets for synergistic therapies to enhance post-insult neuroprotection are of considerable interest.
Peptidylarginine Deiminases
The peptidylarginine deiminase (PAD) enzymes are a family of five calcium-dependent mammalian isozymes (PAD1, 2, 3, 4/5, and 6), located within a 355-kb gene cluster on human chromosome 1p36.13 (27–29) and mouse chromosome 4E1 with intron and exon boundaries well conserved throughout the evolution (27, 30). Each PAD isozyme has a unique tissue distribution pattern and substrate preferences (28, 31). PADs cause posttranslational protein deimination by modifying positively charged protein arginine residues into hydrophilic but uncharged citrullines, producing a loss of one positive charge per conversion and release of ammonia (30). The incidental disruption of ionic and hydrogen bonds within the substrate proteins affects protein structure, function, and protein–protein interactions (Figure 2). Main target proteins identified include nuclear histones (32), cytoskeletal, and structural proteins, such as components of the myelin sheath, intermediate filaments and associated adaptor proteins, extracellular components including fibrin and fibronectin (19, 33), and the chemokines (34–36). Intrinsically disordered proteins and β-turns are particularly prone to deimination (33). Deimination also has autoregulatory activity on PAD enzymes themselves and also upstream cytokines and chemokines, such as TNFα, C–X–C motif chemokine (CXCL) 8, and CXCL10 (37, 38). PADs are involved in physiological processes during development – including nerve growth and differentiation, gene regulation, regulation of pluripotency, and cell death (39–43). PAD dysregulation is associated with various inflammatory and degenerative diseases (30, 33, 34). PADs are also implicated in cancer (44–46), including through regulation of microvesicular (MV) release (47), which also is related to cerebral hypoxia induced by acute ischemic stroke (48, 49). PAD2 is considered the main central nervous system (CNS) isozyme (50), but PAD3 and PAD4 are also detected in neuronal tissue (19, 51, 52). PAD4 is the only isozyme with a designated nuclear translocation site, but the nuclear translocation of PAD2 and PAD3 has also been reported (19, 43, 44, 47). A novel role for PADs in acute CNS damage, and significant neuroprotective measures using pharmacological pan-PAD inhibition (Cl-amidine), was first described in a chick model of spinal cord injury (19). This neuroprotective effect was shown to be translatable to the Rice–Vannucci neonatal HI mouse model, resulting in significant protection of neuronal tissue and reduced microglial activation and cell death (53).
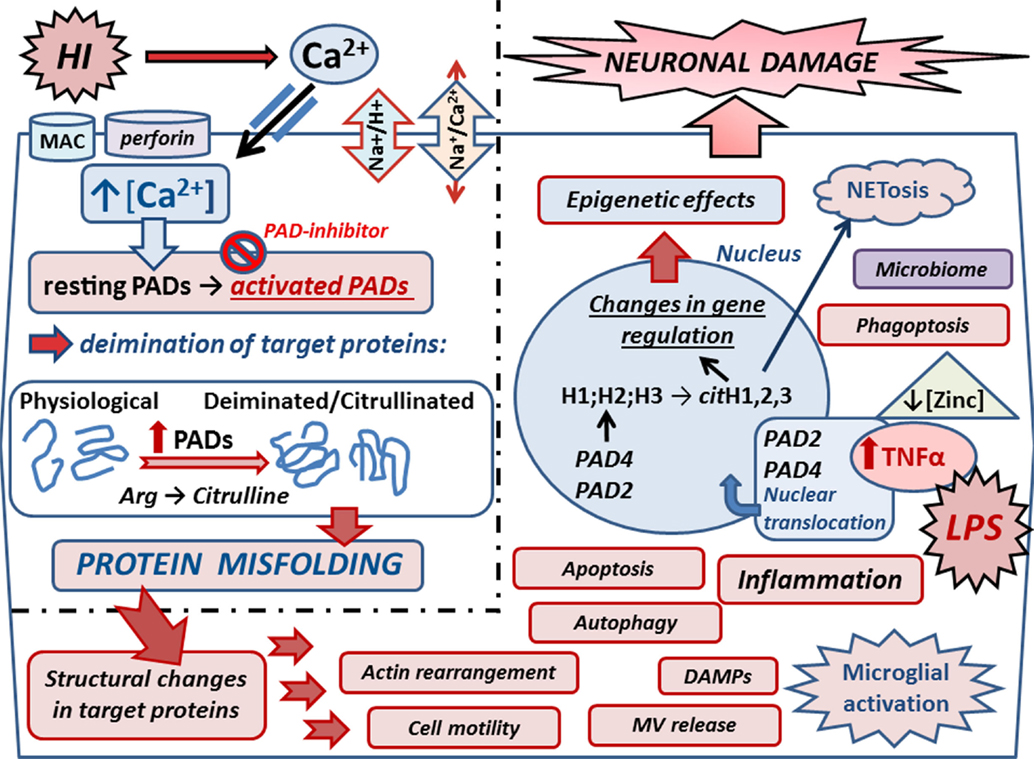
Figure 2. Mechanisms of peptidylarginine deiminases (PADs) in HI and synergistic infection. Upon HIE, Ca2+ entry is facilitated via reversal of the Na+/Ca2+ exchanger due to over activation of the Na+/H+ exchanger (NHE). Ca2+ entry can also be facilitated due to membranolytic pathways including the complement membrane attack complex (MAC) and perforin. Increased cytosolic Ca2+ triggers the neurotoxic cascade, which includes activation of the Ca2+-dependent PAD enzymes. Upon increased intracellular Ca2+, resting PADs are activated and catalyze the conversion of arginines into citrullines in target proteins. Protein deimination causes conformational changes and protein misfolding, affecting protein function and protein–protein interaction. Actin and cytoskeletal rearrangement is affected causing changes in cell motility, microvesicular (MV) release, autophagy, and phagoptosis. Deiminated neo-epitopes result in damage-associated molecular patterns (DAMPs) and contribute to inflammatory processes, such as microglial activation and downstream cascades. Inflammation causes upregulation of TNFα, which also is induced upon infection as mimicked by LPS stimulation in the HI/LPS synergy model. Raised TNFα levels cause translocation of PADs into the nucleus where they deiminate histones and affect gene regulation and epigenetics. Histone deimination also leads to extracellular trap formation (NETosis), which forms part of the microbial defense but can also cause the generation of neo-epitopes, leading to auto-inflammatory responses. Zinc is hypothesized to be a regulator of PADs, and as zinc levels are reduced in sites of infection, this regulatory role is lost, leading to increased protein deimination during infection. Individual variations of the neonatal microbiome may add to the complexity of the interplay of microbial defenses via NETosis and impact microglial activation and other mechanisms that remain to be elucidated. PAD inhibition with the pan-PAD inhibitior Cl-amidine has been successful in neuroprotection in the Rice–Vannucci mouse model of HI and HI/LPS synergy model, significantly reducing microglial activation, neuronal loss, and cell death. Refined inhibitors of PAD activation thus pose promising combinatory treatment options in synergy with current hypothermia measures.
PADs in Epigenetics
Epigenetic roles for PADs in gene regulation are supported in both HIE and HIE/lipopolysaccharide (LPS) synergy models where histone deimination, caused by PADs translocated to the nucleus, was abolished upon PAD inhibition. This paralleled the significant reduction observed in brain tissue loss, particularly in the cortex and hippocampus, which were the main areas affected (53). The LPS/HI model of infectious/ischemic form of brain damage is known to be strongly dependent on TNFα and related family of cytokines as global gene deletion of the whole TNF cluster of cytokines completely abolishes the synergistic, damage-enhancing effect of LPS on HI insult (23). In a transgenic multiple sclerosis murine model, exposure to elevated levels of TNFα-elicited nuclear translocation of PAD4 (54). In the same vein, PAD activity induced by HI insult and LPS stimulation increased neuronal damage following HI insult with beneficial effect of PAD inhibition. This indicates that LPS-induced upregulation of macrophage TNFα and related cytokines may act as upstream signals to PADs and histone deimination in HIE as well as in other forms of brain damage [(52); Figure 2].
PADs in Phagoptosis
Microglia, the professional brain phagocytes, are strongly activated in HIE and can partly reduce inflammation by phagocytosis of dead/dying neurons and neuronal debris (9, 55–57). Microglia have also been shown to phagocytose live neurons, neutrophils, neural progenitors, and glioma cells causing death of the engulfed cell (58–62). Some phosphatidylserine (PS)-exposing neurons may be able to reverse their “eat-me” signals but still be phagocytosed via phagoptosis (62), which may dominate in conditions in which toll-like receptors (TLRs) are activated, as through LPS stimulation (58, 59). The roles of TLRs differ in the immature and adult brain following brain damage (63). Microglial activation can cause neurotoxicity by various other mechanisms, including NO generation by inducible NO synthase (iNOS), which inhibits neuronal mitochondria (64), or oxidant formation by phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, causing direct neurotoxicity (62, 65). In areas of ischemia and hypoxia, phagocytosis of dead or dying neurons may decrease inflammation by clearing harmful cellular components. Following mild ischemia, blocking phagocytosis could be beneficial by preventing the phagocytosis of stressed, but viable, neurons (59). Viable neurons in the post-ischemic brain are labeled by annexin V, indicating PS exposure that peaks at 3 days after transient ischemia and reverses thereafter (66), making these PS-exposed neurons targets for phagocytosis (59). The presence of phospholipids has been shown to reduce the calcium dependence of PAD2 by almost twofold in vitro (67), indicating that PAD activation may be facilitated in the presence of PS in vivo. In HIE, reduced microglial activation was observed 48 h post-insult in PAD inhibitor-treated animals in all brain regions and could be crucial for the protection of PS-exposed neurons against phagoptosis. This may be reflected in the significant reduction observed in neuronal loss in the PAD inhibitor-treated animals (53). Actin polymerization, facilitated by deimination and required for this mechanism, may be inhibited with PAD blockers as recently described in the regulation of cellular MV release (47). Actin, vimentin, and other cytoskeletal proteins were identified to be deiminated in acute CNS damage (19), while PAD3 inhibition has been shown to cause cytoskeleton disassembly in human neural stem cells as well as apoptosis-inducing factor (AIF) translocation to the nucleus (43). AIF has been shown to be a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia (68).
PADs in Autophagy – Deiminated Neo-Epitopes
Autophagy is a cellular death mechanism recently implicated in HIE as a potential therapeutic target (69). This lysosomal pathway for intracellular degradation of macromolecules and organelles is important in the maintenance of cellular survival and homeostasis (70). Results are conflicting as pharmacological inhibition of autophagy was reported as neuroprotective (71), while studies inducing autophagy immediately after injury indicated it to be an endogenous neuroprotective mechanism (72). Recently, PAD4 was implicated in the induction of autophagy in hepatocellular carcinoma (73). Deiminated peptides were also shown to be processed in autophagy vesicles of antigen-presenting cells, indicating a linkage between autophagy and autoreactivity through the generation of deiminated neo-epitopes (74, 75), which in HIE could contribute to neuroinflammatory responses.
PAD Activation via Membranolytic Pathways
Perforin and complement mediate membranolytic pathways of the cellular and humoral immune response and use a common “membrane attack complex component/perforin” (MACPF) domain to form pores in membranes to exert their potent anti-pathogen activity (76). Roles for both pathways have been described in the generation of hypercitrullinated proteins in rheumatoid arthritis (RA) (77). In a rodent study of HIE, the administration of complement C9 appeared detrimental (78) – which makes it tempting to speculate that hypercitrullinated proteins may be generated in HI in a similar fashion and contribute to microglial activation and neuroinflammatory damage.
Perforin oligomerizes in a Ca2+-dependent manner to form pores on target cells, triggering influx of calcium (79). In RA, this mechanism induced hypercitrullination in neutrophils, which express the PAD2, PAD3, and PAD4 isozymes, with each isozyme generating a distinct pattern of protein deimination (77). Again, it is tempting to speculate whether perforin contributes to neuroinflammatory responses in HIE.
Zinc – A Regulator of PAD Activation?
Zinc is hypothesized to be an upregulatory modulator of calcium-dependent activation of posttranslationally acting PADs in autoimmune diseases (80). Zinc has previously been shown to inhibit the calcium-induced activation of PAD4 (81), and one can speculate that it may have a regulatory role for PADs in HIE. Putative links between zinc deficiency and HIE has been touched upon (82) in the context of effects of oxidative stress in childhood and neonatal disorders. The role of zinc may be particularly interesting with respect to HIE and synergistic infections as the zinc concentration is thought to be downregulated upon infection (80). This may cause downstream activation of the PAD cascade and further contributes to neuroinflammatory responses in HIE (Figure 2). Whether supplementing neonates with zinc as a preventive measure is open to debate.
PADs in Neutrophil Extracellular Trap Formation (NETosis) – Interplay with the Microbiome
Recent studies have highlighted increasingly diverse roles for histone deimination including in NETosis, which forms part of the innate immune response in microbial defense and is implicated in autoimmunity (83). NETosis has recently received attention in neonatal infections (84) as neonatal neutrophils from umbilical cord were shown to differ significantly from adult controls in proteomic analysis and function including delayed NETosis (85). NETosis is dependent on the PAD4 isozyme (86, 87), which upon neutrophil activation, catalyzes arginine citrullination in three of the four core histones (88, 89). PAD4-null mice have been shown to fail to produce NETs, which coincides with lack of histone3 hypercitrullination (90). NETs are composed of DNA strands associated with antibacterial granular proteins, including deiminated histones, neutrophil elastase, myeloperoxidase, lactoferrin, and defensins (91). During inflammation, NETs can alert the immune system to danger by activating DNA receptors, such as TLR-9, helping in the recruitment of immune cells. TLR-9 activation via NETs in dendritic cells produces interferons, which can in turn initiate autoimmune responses (92, 93). Also, proteolytic and oxidative processing of proteins during NETosis can generate neoantigens (91), and NETosis has indeed been connected to several autoimmune diseases (92–97).
Neonatal neutrophils can form robust cellular aggregates and are able to release NETs in response to fungal hyphae when presented with extracellular matrix (84). In preeclampsia placentas, NETs have been shown to be in close contact with the syncytiotrophoblast, putatively obstructing the intervillous space, reducing blood flow, and leading to hypoxia in the fetus (98). NETosis can also be induced by activated endothelial cells and cause damage of the endothelium leading to more severe preeclampsia (99).
In the murine HIE/infection synergy model, LPS stimulation prior to neonatal HI insult may activate NETosis, which can result in further damage of surrounding tissue – emphasizing that this innate defense mechanism may be a double-edged sword when toxicity is directed against own tissues (53). It is interesting to note that PAD inhibitor effectively reduced histone deimination, which coincided with significantly reduced neuronal damage and cell death (53). As NET formation has been shown to require functional tubulin and actin filaments (100) and PAD blocker inhibits actin and tubulin deimination (19, 47), this may affect the ability of NET formation.
The capability of premature and healthy term infant cord neutrophils to form NETs is based on material from cords donated after cesarean section (84, 86). Labor deliveries are reported to increase the surface expression of TLRs on monocytes, and interleukin 8-induced neutrophil chemotaxis has been shown to be enhanced due to the stress of birth (101). As the effect of the microbiome in health and disease is gaining ever increasing attention, differences in the maternal and neonatal microbiome are emerging as crucial in response to infection, development, and neurological developmental disorders (102–104). It is tempting to speculate how individual differences in the neonatal microbiome may affect the capability of dealing with infections via mechanisms, such as NETosis – also in the wider context of cesarean sections versus vaginal delivery and breastfeeding – both of which influence the microbial colonization of the gut and the composition of the microflora (105, 106). This is particularly interesting with respect to emerging evidence of the importance of the neonatal gut–brain axis (107), a link between microglial maturation and the microbiome (108), sexual dimorphism in microglial numbers and response to HIE (109), and the connection between the maternal microbiome and pregnancy outcomes, including preterm births (110). Future studies comparing the effect of cesarean section versus labor delivery on the neonatal response to fungal infections and other pathogens will provide better understanding of the role of NETosis and PADs in neonatal HIE and synergistic infections.
pH Changes and PAD Activity
Brain intracellular pH (pHi) is implicated in HIE (20–22). The Na+/H+ exchanger, which regulates pHi and cell volume, is over-activated during HIE, resulting in Ca2+ entry via reversal of the Na+/Ca2+ exchanger. Increased cytosolic Ca2+ triggers the neurotoxic cascade, which includes activation of the Ca2+-dependent PAD enzymes and downstream protein deimination (Figure 2). Activation of the Na+/H+ exchanger also leads to rapid normalization of pHi and to an alkaline shift. Brain pHi changes are closely involved in the control of cell death after injury as alkalosis enhances excitability, while a mild acidosis has the opposite effect (21). The degree of brain alkalosis in 78 babies with neonatal encephalopathy was related to the severity of brain injury (21). The effect of pH on PAD-catalyzed reaction and its kinetics has been studied to some extent (81). PAD4 has pKa values of ~6.2 and ~7.9, the pH optimum being pH 7.6, while calcium dependence of PAD4 is essentially pH-independent over the range of pH 6.0–8.5 (81). The PAD activation pathway should thus be stable over a relatively wide pH range.
Hypothermia and PAD Inhibition – A Synergistic Treatment Option
Cooling brain or whole body to 33°C (moderate hypothermia) is currently the only strategy with a clearly demonstrated clinical benefit in babies with HIE. Therapeutic hypothermia promotes neuroprotection by several mechanisms including a decrease in intracellular calcium influx, which may improve serum Ca2+ levels and homeostasis (111–114). Excessive Ca2+ influx during and after a significant HI insult promotes depolarization of the mitochondria and abnormal excitatory receptor activity promoting further Ca2+ entry into the cells (113). Post-insult hypothermia suppresses this intrinsic pathway (114) and should also affect the Ca2+-regulated PAD activation pathway as calcium binding to PAD4 promotes the bioactive conformation, increasing PAD4 activity by 10,000-fold (115). The effects of cooling on calcium levels was investigated in a piglet model where plasma calcium increased to levels compared with normothermic animals within 3 h of hypothermia and could thus aid Ca2+ homeostasis in HIE (116). As several injury-associated signals, e.g., phosphorylated extracellular signal-regulated kinases (ERK), are increased following hypothermic intervention (117), combined therapies are desirable for increased neuroprotective effects. Hypothermia has been combined with adjunct therapy including xenon, erythropoietin, melatonin, and stem cells for substantial neuroprotection (118–125).
The success of reducing brain injury responses with pharmacological PAD inhibition in murine mouse models of HIE is promising as the treatment proved translatable between both animal and neuronal injury models. The effects observed on changes in histone deimination suggest a role for epigenetic regulation in response to CNS injury, while changes in deimination of cytoskeletal components could affect apoptosis, cell motility, and the ability of injured neurons to regrow axons (19, 43, 53). While the pan-PAD inhibitor Cl-amidine (126) still remains the most used experimental inhibitor to date, the therapeutic potential and generation of selective PAD inhibitors is receiving ever increasing attention (52, 127–131).
Conclusion
A novel role for calcium-dependent PAD is highlighted in neural impairment in neonatal HIE, emphasizing their role as promising new candidates for drug-directed intervention in neurotrauma. During hypoxic ischemic insult, PADs are activated due to calcium dysregulation and putative upregulatory changes in zinc balance, which is decreased upon synergistic infection. Deiminated proteins are irreversibly modified due to changes of protein-bound arginines into citrullines, leading to protein misfolding and changes in protein function. In the case of synergistic infection, TNFα is upregulated and causes nuclear translocation of PAD enzymes, resulting in histone deimination and downstream changes in gene regulation. Mechanisms of PADs in HIE also touch upon the interplay of the microbiome, cesarean versus vaginal deliveries, and how neutrophil and microglial responses to pathogens could be affected. Isozyme-specific PAD inhibition through refined pharmacological intervention poses a promising combinatory treatment option in synergy with current head-cooling approaches.
Author Contributions
The author confirms being the sole contributor to this manuscript and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This publication was supported by the European Union: a component of the Human Brain Project (HBP-604102).
References
1. Wu YW, Colford JM Jr. Chorioamnionitis as a risk factor for cerebral palsy: a meta analysis. JAMA (2000) 284(11):1417–24. doi: 10.1001/jama.284.11.1417
2. Hagberg H, Peebles D, Mallard C. Models of white matter injury: comparison of infectious, hypoxic-ischaemic, and excitotoxic insults. Ment Retard Dev Disabil Res Rev (2002) 8(1):30–8. doi:10.1002/mrdd.10007
3. Mallard C, Welin AK, Peebles D, Hagberg H, Kjellmer I. White matter injury following systemic endotoxemia or asphyxia in the fetal sheep. Neurochem Res (2003) 28:215–23. doi:10.1023/A:1022368915400
4. Perlman JM. Intervention strategies for neonatal hypoxic-ischemic cerebral injury. Clin Ther (2006) 28:1353–65. doi:10.1016/j.clinthera.2006.09.005
5. Kurinczuk JJ, White-Koning M, Badawi N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum Dev (2010) 86(6):329–38. doi:10.1016/j.earlhumdev.2010.05.010
6. Wassink G, Gunn ER, Drury PP, Bennet L, Gunn AJ. The mechanisms and treatment of asphyxia encephalopathy. Front Neurosci (2014) 8:40. doi:10.3389/fnins.2014.00040
7. Douglas-Escobar M, Weiss MD. Hypoxic-ischemic encephalopathy: a review for the clinician. JAMA Pediatr (2015) 169(4):397–403. doi:10.1001/jamapediatrics.2014.3269
8. Ferriero DM. Neonatal brain injury. N Engl J Med (2004) 351(19):1985–95. doi:10.1056/NEJMra041996
9. Rocha-Ferreira E, Hristova M. Antimicrobial peptides and complement in neonatal hypoxia-ischemia induced brain damage. Front Immunol (2015) 6:56. doi:10.3389/fimmu.2015.00056
10. Wyatt JS, Edwards AD, Azzopardi D, Reynolds EO. Magnetic resonance and near infrared spectroscopy for investigation of perinatal hypoxic-ischaemic brain injury. Arch Dis Child (1989) 64:953–63. doi:10.1136/adc.64.7_Spec_No.953
11. Stys PK. Anoxic and ischemic injury of myelinated axons in CNS white matter: from mechanistic concepts to therapeutics. J Cereb Blood Flow Metab (1998) 18:2–25. doi:10.1097/00004647-199801000-00002
12. Dragunow M, Young D, Hughes P, MacGibbon G, Lawlor P, Singleton K, et al. Is c-Jun involved in nerve cell death following status epilepticus and hypoxic-ischaemic brain injury? Mol Brain Res (1993) 18:347–52. doi:10.1016/0169-328X(93)90101-T
13. Adhami F, Schloemer A, Kuan CY. The roles of autophagy in cerebral ischemia. Autophagy (2007) 3:42–4. doi:10.4161/auto.3412
14. Culman J, Zhao Y, Gohlke P, Herdegen T. PPAR-gamma: therapeutic target for ischemic stroke. Trends Pharmacol Sci (2007) 28:244–9. doi:10.1016/j.tips.2007.03.004
15. Pirianov G, Brywe KG, Mallard C, Edwards AD, Flavell RA, Hagberg H, et al. Deletion of the c-Jun N-terminal kinase 3 gene protects neonatal mice against cerebral hypoxic-ischaemic injury. J Cereb Blood Flow Metab (2007) 27(5):1022–32. doi:10.1038/sj.jcbfm.9600413
16. Yi JH, Park SW, Kapadia R, Vemuganti R. Role of transcription factors in mediating post-ischemic cerebral inflammation and brain damage. Neurochem Int (2007) 50:1014–27. doi:10.1016/j.neuint.2007.04.019
17. Dixon BJ, Reis C, Ho WM, Tang J, Zhang JH. Neuroprotective strategies after neonatal hypoxic ischemic encephalopathy. Int J Mol Sci (2015) 16:22368–401. doi:10.3390/ijms160922368
18. Kumral A, Tuzun F, Tugyan K, Ozbal S, Yılmaz O, Yesilirmak CD, et al. Role of epigenetic regulatory mechanisms in neonatal hypoxic-ischemic brain injury. Early Hum Dev (2012) 89:165–73. doi:10.1016/j.earlhumdev.2012.09.016
19. Lange S, Gögel S, Leung KY, Vernay B, Nicholas AP, Causey CP, et al. Protein deiminases: new players in the developmentally regulated loss of neural regenerative ability. Dev Biol (2011) 355(2):205–14. doi:10.1016/j.ydbio.2011.04.015
20. Robertson NJ, Stafler P, Battini R, Cheong J, Tosetti M, Bianchi MC, et al. Brain lactic alkalosis in Aicardi-Goutières syndrome. Neuropediatrics (2004) 35:20–6. doi:10.1055/s-2004-815787
21. Uria-Avellanal C, Robertson NJ. Na+/H+ exchangers and intracellular pH in perinatal brain injury. Transl Stroke Res (2014) 5(1):79–98. doi:10.1007/s12975-013-0322-x
22. Kendall GS, Hristova M, Zbarsky V, Clements A, Peebles DM, Robertson NJ, et al. Distribution of pH changes in mouse neonatal hypoxic-ischaemic insult. Dev Neurosci (2011) 33:505–18. doi:10.1159/000333850
23. Kendall GS, Hristova M, Horn S, Dafou D, Acosta-Saltos A, Almolda B, et al. TNF gene cluster deletion abolishes lipopolysaccharide-mediated sensitization of the neonatal brain to hypoxic ischemic insult. Lab Invest (2011) 91:328–41. doi:10.1038/labinvest.2010.192
24. Wyatt JS, Gluckman PD, Liu PY, Azzopardi D, Ballard R, Edwards AD, et al. Determinants of outcomes after head cooling for neonatal encephalopathy. Pediatrics (2007) 119:912–21. doi:10.1542/peds.2006-2839
25. Faulkner S, Bainbridge A, Kato T, Chandrasekaran M, Kapetanakis AB, Hristova M, et al. Xenon augmented hypothermia reduces early lactate/N-acetylaspartate and cell death in perinatal asphyxia. Ann Neurol (2011) 70:133–50. doi:10.1002/ana.22387
26. Robertson NJ, Faulkner S, Fleiss B, Bainbridge A, Andorka C, Price D, et al. Melatonin augments hypothermic neuroprotection in a perinatal asphyxia model. Brain (2013) 136:90–105. doi:10.1093/brain/aws285
27. Guerrin M, Ishigami A, Méchin MC, Nachat R, Valmary S, Sebbag M, et al. cDNA cloning, gene organization and expression analysis of human peptidylarginine deiminase type I. Biochem J (2003) 370(Pt 1):167–74. doi:10.1042/bj20020870
28. Chavanas S, Méchin MC, Takahara H, Kawada A, Nachat R, Serre G, et al. Comparative analysis of the mouse and human peptidylarginine deiminase gene clusters reveals highly conserved non-coding segments and a new human gene, PADI6. Gene (2004) 330:19–27. doi:10.1016/j.gene.2003.12.038
29. Chavanas S, Méchin MC, Nachat R, Adoue V, Coudane F, Serre G, et al. Peptidylarginine deiminases and deimination in biology and pathology: relevance to skin homeostasis. J Dermatol Sci (2006) 44(2):63–72. doi:10.1016/j.jdermsci.2006.07.004
30. Vossenaar ER, Zendman AJ, van Venrooij WJ, Pruijn GJ. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays (2003) 25(11):1106–18. doi:10.1002/bies.10357
31. Balandraud N, Gouret P, Danchin EG, Blanc M, Zinn D, Roudier J, et al. A rigorous method for multigenic families’ functional annotation: the peptidyl arginine deiminase (PADs) proteins family example. BMC Genomics (2005) 6:153. doi:10.1186/1471-2164-6-153
32. Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science (2004) 306:279–83. doi:10.1126/science.1101400
33. György B, Tóth E, Tarcsa E, Falus A, Buzás EI. Citrullination: a posttranslational modification in health and disease. Int J Biochem Cell Biol (2006) 38:1662–77. doi:10.1016/j.biocel.2006.03.008
34. Loos T, Mortier A, Gouwy M, Ronsse I, Put W, Lenaerts JP, et al. Citrullination of CXCL10 and CXCL11 by peptidylarginine deiminase: a naturally occurring posttranslational modification of chemokines and new dimension of immunoregulation. Blood (2008) 112(7):2648–56. doi:10.1182/blood-2008-04-149039
35. Proost P, Loos T, Mortier A, Schutyser E, Gouwy M, Noppen S, et al. Citrullination of CXCL8 by peptidylarginine deiminase alters receptor usage, prevents proteolysis, and dampens tissue inflammation. J Exp Med (2008) 205(9):2085–97. doi:10.1084/jem.20080305
36. Loos T, Opdenakker G, Van Damme J, Proost P. Citrullination of CXCL8 increases this chemokine’s ability to mobilize neutrophils into the blood circulation. Haematologica (2009) 94(10):1346–53. doi:10.3324/haematol.2009.006973
37. Moelants EA, Van Damme J, Proost P. Detection and quantification of citrullinated chemokines. PLoS One (2011) 6(12):e28976. doi:10.1371/journal.pone.0028976
38. Moelants EA, Mortier A, Grauwen K, Ronsse I, Van Damme J, Proost P. Citrullination of TNF-α by peptidylarginine deiminases reduces its capacity to stimulate the production of inflammatory chemokines. Cytokine (2013) 61(1):161–7. doi:10.1016/j.cyto.2012.09.011
39. Hagiwara T, Nakashima K, Hirano H, Senshu T, Yamada M. Deimination of arginine residues in nucleophosmin/B23 and histones in HL-60 granulocytes. Biochem Biophys Res Commun (2002) 290(3):979–83. doi:10.1006/bbrc.2001.6303
40. Horibata S, Coonrod SA, Cherrington BD. Role for peptidylarginine deiminase enzymes in disease and female reproduction. J Reprod Dev (2012) 58(3):274–82. doi:10.1262/jrd.2011-040
41. Li P, Yao H, Zhang Z, Li M, Luo Y, Thompson PR, et al. Regulation of p53 target gene expression by peptidylarginine deiminase 4. Mol Cell Biol (2008) 28(15):4745–58. doi:10.1128/MCB.01747-07
42. Christophorou MA, Castelo-Branco G, Halley-Stott RP, Oliveira CS, Loos R, Radzisheuskaya A, et al. Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature (2014) 507(7490):104–8. doi:10.1038/nature12942
43. U KP, Subramanian V, Nicholas AP, Thompson PR, Ferretti P. Modulation of calcium-induced cell death in human neural stem cells by the novel peptidylarginine deiminase-AIF pathway. Biochim Biophys Acta (2014) 1843:1162–71. doi:10.1016/j.bbamcr.2014.02.018
44. Cherrington BD, Zhang X, McElwee JL, Morency E, Anguish LJ, Coonrod SA. Potential role for PAD2 in gene regulation in breast cancer cells. PLoS One (2012) 7:e41242. doi:10.1371/journal.pone.0041242
45. Witalison EE, Cui X, Causey CP, Thompson PR, Hofseth LJ. Molecular targeting of protein arginine deiminases to suppress colitis and prevent colon cancer. Oncotarget (2015) 6(34):36053–62. doi:10.18632/oncotarget.5937
46. Cui YY, Yan L, Zhou J, Zhao S, Zheng YB, Sun BH, et al. The role of peptidylarginine deiminase 4 in ovarian cancer cell tumorigenesis and invasion. Tumour Biol (2015). doi:10.1007/s13277-015-4363-5
47. Kholia S, Jorfi S, Thompson PR, Nicholas AP, Inal JM, Lange S. A novel role for peptidylarginine deiminases in microvesicle release reveals therapeutic potential of PAD inhibition in sensitizing prostate cancer cells to chemotherapy. J Extracell Vesicles (2015) 4:26192. doi:10.3402/jev.v4.26192
48. Cherian P, Hankey GJ, Eikelboom JW, Thom J, Baker RI, McQuillan A, et al. Endothelial and platelet activation in acute ischemic stroke and its etiological subtypes. Stroke (2003) 34(9):2132–7. doi:10.1161/01.STR.0000086466.32421.F4
49. Simak J, Gelderman MP, Yu H, Wright V, Baird AE. Circulating endothelial microparticles in acute ischemic stroke: a link to severity, lesion volume and outcome. J Thromb Haemost (2006) 4(6):1296–302. doi:10.1111/j.1538-7836.2006.01911.x
50. Wood DD, Ackerley CA, Brand BV, Zhang L, Raijmakers R, Mastronardi FG, et al. Myelin localization of peptidylarginine deiminases 2 and 4: comparison of PAD2 and PAD4 activities. Lab Invest (2008) 88(4):354–64. doi:10.1038/labinvest.3700748
51. Keilhoff G, Prell T, Langnaese K, Mawrin C, Simon M, Fansa H, et al. Expression pattern of peptidylarginine deiminase in rat and human Schwann cells. Dev Neurobiol (2008) 68(1):101–14. doi:10.1002/dneu.20578
52. Ferretti P, U KP, Vagaska B, Merchant R, Matthews CJ, Marson CM. Discovery of a structurally novel, drug-like and potent inhibitor of peptidylarginine deiminase. Med Chem Commun (2013) 4:1109–13. doi:10.1039/c3md00091e
53. Lange S, Rocha-Ferreira E, Thei L, Mawjee P, Bennett K, Thompson PR, et al. Peptidylarginine deiminases: novel drug targets for prevention of neuronal damage following hypoxic ischemic insult (HI) in neonates. J Neurochem (2014) 130(4):555–62. doi:10.1111/jnc.12744
54. Mastronardi FG, Wood DD, Mei J, Raijmakers R, Tseveleki V, Dosch HM, et al. Increased citrullination of histone H3 in multiple sclerosis brain and animal models of demyelination: a role for tumor necrosis factor-induced peptidylarginine deiminase 4 translocation. J Neurosci (2006) 26(44):11387–96. doi:10.1523/JNEUROSCI.3349-06.2006
55. Neumann J, Sauerzweig S, Rönicke R, Gunzer F, Dinkel K, Ullrich O, et al. Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J Neurosci (2008) 28(23):5965–75. doi:10.1523/JNEUROSCI.0060-08.2008
56. Baburamani AA, Supramaniam VG, Hagberg H, Mallard C. Microglia toxicity in preterm brain injury. Reprod Toxicol (2014) 48:106–12. doi:10.1016/j.reprotox.2014.04.002
57. Hagberg H, Gressens P, Mallard C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann Neurol (2012) 71(4):444–57. doi:10.1002/ana.22620
58. Neher JJ, Neniskyte U, Zhao JW, Bal-Price A, Tolkovsky AM, Brown GC. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J Immunol (2011) 186(8):4973–83. doi:10.4049/jimmunol.1003600
59. Neher JJ, Emmrich JV, Fricker M, Mander PK, Théry C, Brown GC. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci U S A (2013) 110:E4098–107. doi:10.1073/pnas.1308679110
60. Fricker M, Oliva-Martín MJ, Brown GC. Primary phagocytosis of viable neurons by microglia activated with LPS or Aβ is dependent on calreticulin/LRP phagocytic signalling. J Neuroinflammation (2012) 9:196. doi:10.1186/1742-2094-9-196
61. Kopatz J, Beutner C, Welle K, Bodea LG, Reinhardt J, Claude J, et al. Siglec-h on activated microglia for recognition and engulfment of glioma cells. Glia (2013) 61(7):1122–33. doi:10.1002/glia.22501
62. Brown GC, Neher JJ. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol (2010) 41(2–3):242–7. doi:10.1007/s12035-010-8105-9
63. Mallard C, Wang X, Hagberg H. The role of toll-like receptors in perinatal brain injury. Clin Perinatol (2009) 36(4):763–72. doi:10.1016/j.clp.2009.07.009
64. Neniskyte U, Brown GC. Analysis of microglial production of reactive oxygen and nitrogen species. Methods Mol Biol (2013) 1041:103–11. doi:10.1007/978-1-62703-520-0_12
65. Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci (2007) 8(1):57–69. doi:10.1038/nrn2038
66. Mari C, Karabiyikoglu M, Goris ML, Tait JF, Yenari MA, Blankenberg FG. Detection of focal hypoxic-ischemic injury and neuronal stress in a rodent model of unilateral MCA occlusion/reperfusion using radiolabeled annexin V. Eur J Nucl Med Mol Imaging (2004) 31(5):733–9. doi:10.1007/s00259-004-1473-5
67. Musse AA, Polverini E, Raijmakers R, Harauz G. Kinetics of human peptidylarginine deiminase 2 (hPAD2) – reduction of Ca2+ dependence by phospholipids and assessment of proposed inhibition by paclitaxel side chains. Biochem Cell Biol (2008) 86(5):437–47. doi:10.1139/o08-124
68. Zhu C, Wang X, Huang Z, Qiu L, Xu F, Vahsen N, et al. Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ (2007) 14(4):775–84. doi:10.1038/sj.cdd.4402053
69. Ginet V, Puyal J, Clarke PG, Truttmann AC. Enhancement of autophagic flux after neonatal cerebral hypoxia-ischemia and its region-specific relationship to apoptotic mechanisms. Am J Pathol (2009) 175:1962–74. doi:10.2353/ajpath.2009.090463
70. Northington FJ, Chavez-Valdez R, Martin LJ. Neuronal cell death in neonatal hypoxia-ischemia. Ann Neurol (2011) 36(5):743–58. doi:10.1002/ana.22419
71. Li Q, Li H, Roughton K, Wang X, Kroemer G, Blomgren K, et al. Lithium reduces apoptosis and autophagy after neonatal hypoxia-ischemia. Cell Death Dis (2010) 1:e56. doi:10.1038/cddis.2010.33
72. Balduini W, Carloni S, Buonocore G. Autophagy in hypoxia-ischemia induced brain injury. Mater Fetal Neonatal Med (2012) 25:30–4. doi:10.3109/14767058.2012.663176
73. Fan T, Zhang C, Zong M, Zhao Q, Yang X, Hao C, et al. Peptidylarginine deiminase IV promotes the development of chemoresistance through inducing autophagy in hepatocellular carcinoma. Cell Biosci (2014) 4:49. doi:10.1186/2045-3701-4-49
74. Ireland JM, Unanue ER. Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4 T cells. J Exp Med (2011) 208(13):2625–32. doi:10.1084/jem.20110640
75. Ireland JM, Unanue ER. Processing of proteins in autophagy vesicles of antigen-presenting cells. Autophagy (2012) 8(3):429–30. doi:10.4161/auto.19261
76. McCormack R, de Armas L, Shiratsuchi M, Podack ER. Killing machines: three pore-forming proteins of the immune system. Immunol Res (2013) 57(1–3):268–78. doi:10.1007/s12026-013-8469-9
77. Romero V, Fert-Bober J, Nigrovic PA, Darrah E, Haque UJ, Lee DM, et al. Immune-mediated pore-forming pathways induce cellular hypercitrullination and generate citrullinated autoantigens in rheumatoid arthritis. Sci Transl Med (2013) 5(209):209ra150. doi:10.1126/scitranslmed.3006869
78. Imm MD, Feldhoff PW, Feldhoff RC, Lassiter HA. The administration of complement component C9 augments post-ischemic cerebral infarction volume in neonatal rats. Neurosci Lett (2002) 325(3):175–8. doi:10.1016/S0304-3940(02)00271-9
79. Law RH, Lukoyanova N, Voskoboinik I, Caradoc-Davies TT, Baran K, Dunstone MA, et al. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature (2010) 468(7322):447–51. doi:10.1038/nature09518
80. Stenberg P, Roth B. Zinc is the modulator of the calcium-dependent activation of post-translationally acting thiol-enzymes in autoimmune diseases. Med Hypotheses (2015) 84(4):331–5. doi:10.1016/j.mehy.2015.01.022
81. Kearney P, Bhatia M, Jones NG, Yuan L, Glascock MC, Cathings KL, et al. Kinetic characterization of protein arginine deiminase 4: a transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry (2005) 44(31):10570–82. doi:10.1021/bi050292m
82. Sharda B. Free radicals: emerging challenge in environmental health research in childhood and neonatal disorders. Int J Environ Res Public Health (2006) 3(3):286–91. doi:10.3390/ijerph2006030035
83. Dwivedi N, Radic M. Citrullination of autoantigens implicates NETosis in the induction of autoimmunity. Ann Rheum Dis (2014) 73(3):483–91. doi:10.1136/annrheumdis-2013-203844
84. Byrd AS, O’Brien XM, Laforce-Nesbitt SS, Parisi VE, Hirakawa MP, Bliss JM, et al. NETosis in neonates: evidence of a reactive oxygen species-independent pathway in response to fungal challenge. J Infect Dis (2016) 213(4):634–9. doi:10.1093/infdis/jiv435
85. Zhu J, Zhang H, Guo T, Li W, Li H, Zhu Y, et al. Quantitative proteomics reveals differential biological processes in healthy neonatal cord neutrophils and adult neutrophils. Proteomics (2014) 14(13–14):1688–97. doi:10.1002/pmic.201400009
86. Yost CC, Cody MJ, Harris ES, Thornton NL, McInturff AM, Martinez ML, et al. Impaired neutrophil extracellular trap (NET) formation: a novel innate immune deficiency of human neonates. Blood (2009) 113:6419–27. doi:10.1182/blood-2008-07-171629
87. Marcos V, Nussbaum C, Vitkov L, Hector A, Wiedenbauer EM, Roos D, et al. Delayed but functional neutrophil extracellular trap formation in neonates. Blood (2009) 114:4908–11. doi:10.1182/blood-2009-09-242388
88. Neeli I, Khan SN, Radic M. Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol (2008) 180(3):1895–902. doi:10.4049/jimmunol.180.3.1895
89. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol (2009) 184(2):205–13. doi:10.1083/jcb.200806072
90. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med (2010) 207(9):1853–62. doi:10.1084/jem.20100239
91. Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol (2012) 198(5):773–83. doi:10.1083/jcb.201203170
92. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med (2011) 3(73):73ra20. doi:10.1126/scitranslmed.3001201
93. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med (2011) 3(73):73ra19. doi:10.1126/scitranslmed.3001180
94. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol (2011) 187(1):538–52. doi:10.4049/jimmunol.1100450
95. Wright HL, Moots RJ, Edwards SW. The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol (2014) 10(10):593–601. doi:10.1038/nrrheum.2014.80
96. Spengler J, Lugonja B, Jimmy Ytterberg A, Zubarev RA, Creese AJ, Pearson MJ, et al. Release of active peptidyl arginine deiminases by neutrophils can explain production of extracellular citrullinated autoantigens in rheumatoid arthritis synovial fluid. Arthritis Rheumatol (2015) 67(12):3135–45. doi:10.1002/art.39313
97. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med (2009) 15(6):623–5. doi:10.1038/nm.1959
98. Gupta AK, Hasler P, Holzgreve W, Hahn S. Neutrophil NETs: a novel contributor to preeclampsia-associated placental hypoxia? Semin Immunopathol (2007) 29(2):163–7. doi:10.1007/s00281-007-0073-4
99. Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett (2010) 584(14):3193–7. doi:10.1016/j.febslet.2010.06.006
100. Neeli I, Dwivedi N, Khan S, Radic M. Regulation of extracellular chromatin release from neutrophils. J Innate Immun (2009) 1(3):194–201. doi:10.1159/000206974
101. Yektaei-Karin E, Moshfegh A, Lundahl J, Berggren V, Hansson LO, Marchini G. The stress of birth enhances in vitro spontaneous and IL-8-induced neutrophil chemotaxis in the human newborn. Pediatr Allergy Immunol (2007) 18(8):643–51. doi:10.1111/j.1399-3038.2007.00578.x
102. Romano-Keeler J, Weitkamp JH. Maternal influences on fetal microbial colonization and immune development. Pediatr Res (2015) 77(1–2):189–95. doi:10.1038/pr.2014.163
103. Goyal DK, Miyan JA. Neuro-immune abnormalities in autism and their relationship with the environment: a variable insult model for autism. Front Endocrinol (2015) 5:29. doi:10.3389/fendo.2014.00029
104. Keunen K, van Elburg RM, van Bel F, Benders MJ. Impact of nutrition on brain development and its neuroprotective implications following preterm birth. Pediatr Res (2015) 77(1–2):148–55. doi:10.1038/pr.2014.171
105. Azad MB, Konya T, Persaud RR, Guttman DS, Chari RS, Field CJ, et al. Impact of maternal intrapartum antibiotics, method of birth and breastfeeding on gut microbiota during the first year of life: a prospective cohort study. BJOG (2015). doi:10.1111/1471-0528.13601
106. Latuga MS, Stuebe A, Seed PC. A review of the source and function of microbiota in breast milk. Semin Reprod Med (2014) 32(1):68–73. doi:10.1055/s-0033-1361824
107. Sherman MP, Zaghouani H, Niklas V. Gut microbiota, the immune system, and diet influence the neonatal gut–brain axis. Pediatr Res (2015) 77(1–2):127–35. doi:10.1038/pr.2014.161
108. Erny D, Hrabě de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci (2015) 18(7):965–77. doi:10.1038/nn.4030
109. Mirza MA, Ritzel R, Xu Y, McCullough LD, Liu F. Sexually dimorphic outcomes and inflammatory responses in hypoxic-ischemic encephalopathy. J Neuroinflammation (2015) 12:32. doi:10.1186/s12974-015-0251-6
110. Fox C, Eichelberger K. Maternal microbiome and pregnancy outcomes. Fertil Steril (2015) 104(6):1358–63. doi:10.1016/j.fertnstert.2015.09.037
111. Alonso-Alconada D, Broad KD, Bainbridge A, Chandrasekaran M, Faulkner SD, Kerenyi Á, et al. Brain cell death is reduced with cooling by 3.5°C to 5°C but increased with cooling by 8.5°C in a piglet asphyxia model. Stroke (2015) 46(1):275–8. doi:10.1161/STROKEAHA.114.007330
112. Prempunpong C, Efanov I, Sant’Anna G. Serum calcium concentrations and incidence of hypocalcemia in infants with moderate or severe hypoxic-ischemic encephalopathy: effect of therapeutic hypothermia. Early Hum Dev (2015) 91(9):535–40. doi:10.1016/j.earlhumdev.2015.06.008
113. Zipfel GJ, Babcock DJ, Lee JM, Choi DW. Neuronal apoptosis after CNS injury: the roles of glutamate and calcium. J Neurotrauma (2000) 17(10):857–69. doi:10.1089/neu.2000.17.857
114. Zhu C, Wang X, Cheng X, Qiu L, Xu F, Simbruner G, et al. Post-ischemic hypothermia-induced tissue protection and diminished apoptosis after neonatal cerebral hypoxia-ischemia. Brain Res (2004) 996(1):67–75. doi:10.1016/j.brainres.2003.10.013
115. Knuckley B, Causey CP, Jones JE, Bhatia M, Dreyton CJ, Osborne TC, et al. Substrate specificity and kinetic studies of PADs 1, 3, and 4 identify potent and selective inhibitors of protein arginine deiminase 3. Biochemistry (2010) 49:4852–63. doi:10.1021/bi100363t
116. Karlsson M, Tooley JR, Satas S, Hobbs CE, Chakkarapani E, Stone J, et al. Delayed hypothermia as selective head cooling or whole body cooling does not protect brain or body in newborn pig subjected to hypoxia-ischemia. Pediatr Res (2008) 64(1):74–8. doi:10.1203/PDR.0b013e318174efdd
117. Choi JS, Park J, Suk K, Moon C, Park YK, Han HS. Mild hypothermia attenuates intercellular adhesion molecule-1 induction via activation of extracellular signal-regulated kinase-1/2 in a focal cerebral ischemia model. Stroke Res Treat (2011) 2011:846716. doi:10.4061/2011/846716
118. Ma D, Hossain M, Chow A, Arshad M, Battson RM, Sanders RD, et al. Xenon and hypothermia combine to provide neuroprotection from neonatal asphyxia. Ann Neurol (2005) 58(2):182–93. doi:10.1002/ana.20547
119. Martin JL, Ma D, Hossain M, Xu J, Sanders RD, Franks NP, et al. Asynchronous administration of xenon and hypothermia significantly reduces brain infarction in the neonatal rat. Br J Anaesth (2007) 98(2):236–40. doi:10.1093/bja/ael340
120. David HN, Haelewyn B, Rouillon C, Lecoq M, Chazalviel L, Apiou G, et al. Neuroprotective effects of xenon: a therapeutic window of opportunity in rats subjected to transient cerebral ischemia. FASEB J (2008) 22(4):1275–86. doi:10.1096/fj.07-9420com
121. Dingley J, Tooley J, Liu X, Scull-Brown E, Elstad M, Chakkarapani E, et al. Xenon ventilation during therapeutic hypothermia in neonatal encephalopathy: a feasibility study. Pediatrics (2014) 133(5):809–18. doi:10.1542/peds.2013-0787
122. Zhu C, Kang W, Xu F, Cheng X, Zhang Z, Jia L, et al. Erythropoietin improved neurologic outcomes in newborns with hypoxic-ischemic encephalopathy. Pediatrics (2009) 124(2):e218–26. doi:10.1542/peds.2008-3553
123. Wu YW, Bauer LA, Ballard RA, Ferriero DM, Glidden DV, Mayock DE, et al. Erythropoietin for neuroprotection in neonatal encephalopathy: safety and pharmacokinetics. Pediatrics (2012) 130(4):683–91. doi:10.1542/peds.2012-0498
124. Lee MY, Kuan YH, Chen HY, Chen TY, Chen ST, Huang CC, et al. Intravenous administration of melatonin reduces the intracerebral cellular inflammatory response following transient focal cerebral ischemia in rats. J Pineal Res (2007) 42(3):297–309. doi:10.1111/j.1600-079X.2007.00420.x
125. Cotten CM, Murtha AP, Goldberg RN, Grotegut CA, Smith PB, Goldstein RF, et al. Feasibility of autologous cord blood cells for infants with hypoxic-ischemic encephalopathy. J Pediatr (2014) 164(5):973.e–9.e. doi:10.1016/j.jpeds.2013.11.036
126. Luo Y, Knuckley B, Bhatia M, Pellechia PJ, Thompson PR. Activity-based protein profiling reagents for protein arginine deiminase 4 (PAD4): synthesis and in vitro evaluation of a fluorescently labeled probe. J Am Chem Soc (2006) 128(45):14468–9. doi:10.1021/ja0576233
127. Slack JL, Causey CP, Thompson PR. Protein arginine deiminase 4: a target for an epigenetic cancer therapy. Cell Mol Life Sci (2011) 68(4):709–20. doi:10.1007/s00018-010-0480-x
128. Bozdag M, Dreker T, Henry C, Tosco P, Vallaro M, Fruttero R, et al. Novel small molecule protein arginine deiminase 4 (PAD4) inhibitors. Bioorg Med Chem Lett (2013) 23(3):715–9. doi:10.1016/j.bmcl.2012.11.102
129. Wei L, Wasilewski E, Chakka SK, Bello AM, Moscarello MA, Kotra LP. Novel inhibitors of protein arginine deiminase with potential activity in multiple sclerosis animal model. J Med Chem (2013) 56(4):1715–22. doi:10.1021/jm301755q
130. Subramanian V, Knight JS, Parelkar S, Anguish L, Coonrod SA, Kaplan MJ, et al. Design, synthesis, and biological evaluation of tetrazole analogs of Cl-amidine as protein arginine deiminase inhibitors. J Med Chem (2015) 58(3):1337–44. doi:10.1021/jm501636x
Keywords: hypoxic–ischemic encephalopathy, peptidylarginine deiminases, deimination/citrullination, NETosis, microglia, microvesicles, epigenetics, microbiome
Citation: Lange S (2016) Peptidylarginine Deiminases as Drug Targets in Neonatal Hypoxic–Ischemic Encephalopathy. Front. Neurol. 7:22. doi: 10.3389/fneur.2016.00022
Received: 01 December 2015; Accepted: 09 February 2016;
Published: 22 February 2016
Edited by:
Pedro M. Pimentel-Coelho, Universidade Federal do Rio de Janeiro, BrazilReviewed by:
Susan Cohen, Medical College of Wisconsin, USAHong-Shuo Sun, University of Toronto, Canada
Copyright: © 2016 Lange. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sigrun Lange, s.lange@westminster.ac.uk, sigrun.lange@ucl.ac.uk