 Rodi Zutt1
Rodi Zutt1 Joke M. Dijk2
Joke M. Dijk2 Kathryn J. Peall1,3Hans Speelman2
Kathryn J. Peall1,3Hans Speelman2 Yasmine E. M. Dreissen2
Yasmine E. M. Dreissen2 Maria Fiorella Contarino2,4†
Maria Fiorella Contarino2,4† Marina A. J. Tijssen1*†
Marina A. J. Tijssen1*†
- 1Department of Neurology, University Medical Center Groningen, Groningen, Netherlands
- 2Department of Neurology, Academic Medical Center, University of Amsterdam, Amsterdam, Netherlands
- 3MRC Centre for Neuropsychiatric Genetics and Genomics, Institute of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, UK
- 4Department of Neurology, Haga Teaching Hospital, Den Haag, Netherlands
Introduction: Myoclonus–dystonia (M–D) is a young onset movement disorder typically involving myoclonus and dystonia of the upper body. A proportion of the cases are caused by mutations to the autosomal dominantly inherited, maternally imprinted, epsilon-sarcoglycan gene (SGCE). Despite several sets of diagnostic criteria, identification of patients most likely to have an SGCE mutation remains difficult.
Methods: Forty consecutive patients meeting pre-existing diagnostic clinical criteria for M–D underwent a standardized clinical examination (20 SGCE mutation positive and 20 negative). Each video was reviewed and systematically scored by two assessors blinded to mutation status. In addition, the presence and coexistence of myoclonus and dystonia was recorded in four body regions (neck, arms, legs, and trunk) at rest and with action.
Results: Thirty-nine patients were included in the study (one case was excluded owing to insufficient video footage). Based on previously proposed diagnostic criteria, patients were subdivided into 24 “definite,” 5 “probable,” and 10 “possible” M–D. Motor symptom severity was higher in the SGCE mutation-negative group. Myoclonus and dystonia were most commonly observed in the neck and upper limbs of both groups. Truncal dystonia with action was significantly seen more in the mutation-negative group (p < 0.05). Coexistence of myoclonus and dystonia in the same body part with action was more commonly seen in the mutation-negative cohort (p < 0.05).
Conclusion: Truncal action dystonia and coexistence of myoclonus and dystonia in the same body part with action might suggest the presence of an alternative mutation in patients with M–D.
Introduction
Myoclonus–dystonia (M–D) is a rare movement disorder, characteristically with onset in the first two decades of life (1). Motor features are typically of myoclonic jerks, predominantly involving the upper body, although also involving the lower limbs, face, and larynx in up to a quarter of cases (2–4). The dystonic component most frequently involves the neck and upper limbs (writer’s cramp) (5, 6). Both the myoclonus and dystonia may be exacerbated by posture, action, or stress. Presentation and progression of motor symptoms may vary, ranging from an early childhood-onset form starting with upper body or lower limb involvement and progressing to upper limbs involvement to a later-onset form, with predominant upper body symptoms and frequent cervical involvement. The clinical course can be stable or show a progressive course, with increasing severity and/or spreading of symptoms (7). Alcohol consumption is widely reported to improve motor symptoms, particularly the myoclonus, resulting in excess alcohol consumption in some cases (8, 9). Several studies have also identified psychiatric symptoms in M–D cohorts, including anxiety, panic attacks, and obsessive–compulsive disorder (10–12).
Myoclonus–dystonia is inherited in an autosomal dominant fashion with mutations in the maternally imprinted SGCE gene (DYT11) observed in a proportion of cases (13–15). At present, clinical discrimination of SGCE mutation-positive from mutation-negative M–D cases remains difficult. Previous studies have shown the frequency of SGCE mutations in M–D cohorts to vary between 21 and 85% dependent on the inclusion criteria employed (3, 4, 6, 16–19). Several factors have been proposed as predictors of an SGCE mutation, including motor symptom onset <20 years, a positive family history of a similar movement disorder, and comorbid psychiatric symptoms (19–21).
A classification system has been developed to determine the likelihood of an SGCE mutation in individual cases, with subgroups “definite,” “probable,” or “possible,” based on the distribution of motor symptoms, age at onset, and presence or absence of a family history (Table S1 in Supplementary Material) (18). Refinement of these diagnostic criteria has been proposed to include a positive family history with specific paternal transmission and normal brain imaging (5), or the combination of young onset motor symptoms with psychiatric features (20). However, it remains difficult to identify those patients most likely to have an SGCE mutation.
The aim of this study is to determine whether the characteristics of motor signs observed during clinical examination can be of help in identifying carriers of a SGCE mutation. Particular emphasis was placed on whether coexistence of myoclonus and dystonia in the same body part was helpful in distinguishing those with and without an SGCE mutation. We hypothesized that SGCE mutation-negative patients with “jerky dystonia” would more often present with jerky movements superimposed on the dystonic posture in the same body part, whereas for those with an SGCE mutation the myoclonus and dystonia would be evident independently and in different body regions.
Materials and Methods
Following informed consent, 40 consecutive M–D patients from the movement disorders service at the Academic Medical Center, Amsterdam, The Netherlands were recruited for the study (20 SGCE positive and 20 SGCE negative). Participants were divided into one of the three diagnostic categories, “definite,” “probable,” and “possible,” according to previously proposed diagnostic criteria (Table S1 in Supplementary Material). All participants underwent a videotaped clinical examination, which in the majority of cases followed a standardized protocol (n = 31), and the remaining cases were examined as part of routine clinical practice (n = 9). Each videotaped examination was subsequently assessed by two of the three independent movement disorder experts (Maria Fiorella Contarino, Hans Speelman, and Joke M. Dijk), blinded to the genetic status of the participant.
The motor section of the Burke–Fahn–Marsden Dystonia Rating Scale (BFMDRS) and sections 2 (myoclonus at rest) and 4 (action myoclonus) of the Unified Myoclonus Rating Scale (UMRS) were used to assess motor symptom severity (22). In addition, the presence and coexistence of myoclonus and dystonia were recorded in four body regions (neck, arms, legs, and trunk), at rest and with action. In the absence of adequate video footage to allow evaluation of a specific body region in individual patients, the score for this region was omitted.
The Ethical Board of the Academic Medical Center of Amsterdam approved the study.
Statistical Analysis
Clinical features were analyzed using the Fisher’s exact test or Student’s t-test where appropriate. Inter-rater reliability was assessed using intraclass correlation coefficients (ICCs) (two way mixed, consistency, and average measures). ICC results were further classified as; 0.91–1: excellent, 0.71 and 0.9: good, 0.51 and 0.70: moderate, <0.5: poor, and <0.3: very poor (23). The inter-rater reliability of the new evaluation tool was reported both in absolute agreement and percentage of agreement between raters.
Results
Demographic Characteristics
A full summary of the demographic characteristics of this cohort is reported in Table 1. Due to the consecutive nature of recruitment, mutation-positive and -negative groups were not matched for gender and age at the onset of motor symptoms. The SGCE mutation-positive cohort included a greater number of cases with motor symptom onset <20 years and a positive family history. There were no significant differences in demographic characteristics between the groups.
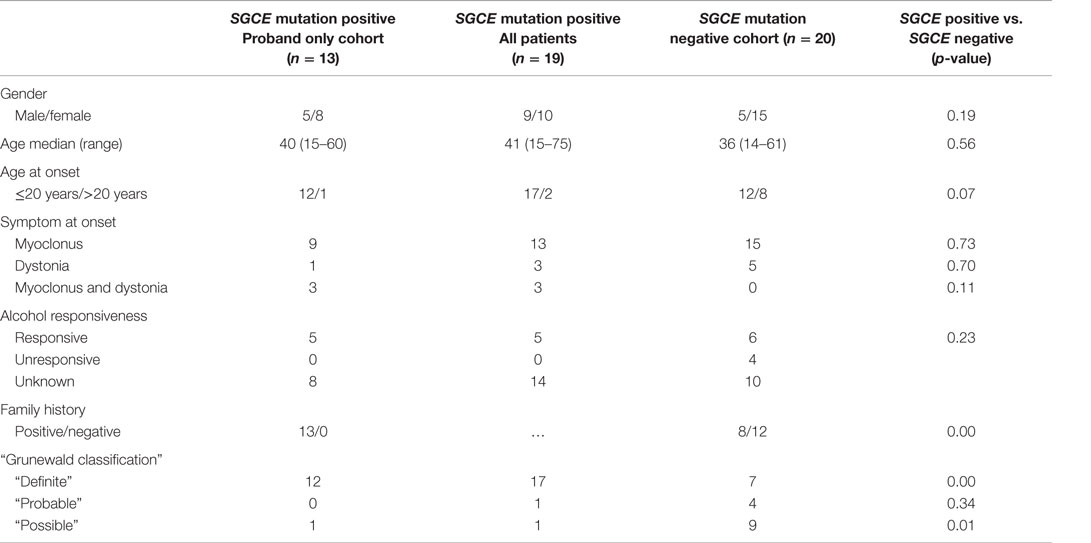
Table 1. Demographic characteristics.
Overall, 39 patients (25F: 14M) with a clinical M–D phenotype were included in the study. Nineteen had an SGCE mutation (one mutation-positive case was excluded owing to insufficient video footage) and 20 patients were mutation negative. Median age at examination was 39 years (range: 14–75). Myoclonus was the presenting feature in 28 cases, dystonia in 8 and both myoclonus and dystonia were observed at symptom onset in 3. Details of the cognitive and psychological characteristics of this cohort have been published elsewhere (12).
The SGCE mutation-positive cohort (n = 19, 10F: 9M) included 13 probands and had a median age at examination of 41 years (range: 15–75). Seventeen cases had onset of symptoms <20 years of age with single cases developing motor symptoms in each of the 30- to 40-year and 40- to 50-year age brackets. Applying Grunewald diagnostic criteria, this group was further subdivided into 17 “definite,” 1 “probable,” and 1 “possible” cases.
The SGCE mutation-negative cohort (n = 20, 15F: 5M) had a median age at examination of 36 years (range: 14–61). Motor symptom onset was <20 years in 12 cases, 30–40 years in 4 cases, 40–50 years in 3 cases, and >50 years in a single participant. With application of the same diagnostic criteria, this group was sub-divided into 7 “definite,” 4 “probable,” and 9 “possible.”
Due to a sub-optimal video footage, 11/76 (at rest) and 11/76 (with action) dystonia and 2/76 (at rest) and 3/76 (with action) myoclonus video assessment sections were scored as missing in the SGCE mutation-positive cohort. In the mutation-negative group, 2/80 (at rest) and 3/80 (with action) dystonia while none of the myoclonus assessment sections incomplete.
Distribution of Symptoms
Myoclonus and dystonia were most commonly observed in the neck and arms in both mutation positive and negative groups. Comparison of the SGCE mutation-positive probands and the mutation-negative group identified significant difference with truncal dystonia during action [8/19 (SGCE mutation-negative) 0/13 (SGCE mutation-positive probands only); p = 0.01, OR 0.01, 95% CI (0.00, 0.74)]. This difference was preserved when extended to include the entire mutation-positive group: truncal dystonia [8/19 (SGCE mutation-negative) 1/17 (SGCE mutation-positive); p = 0.02, OR 0.09, 95% CI (0.00, 0.88)] (Tables 2 and 3; Figure S1 in Supplementary Material).
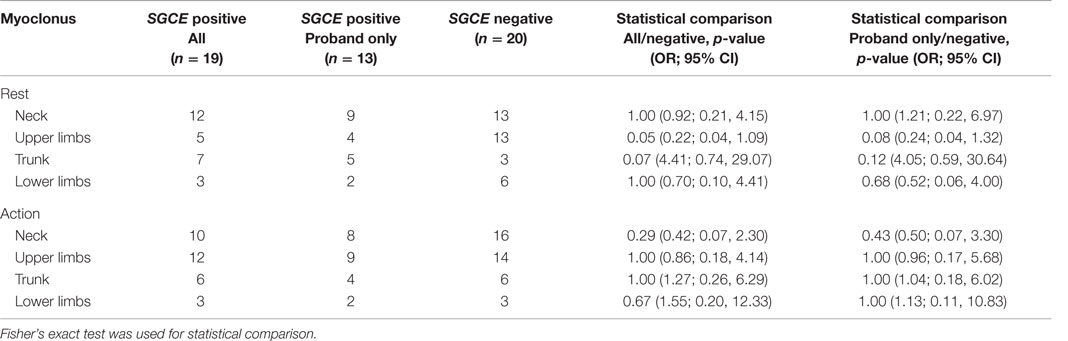
Table 2. Distribution of myoclonus at rest and with action.
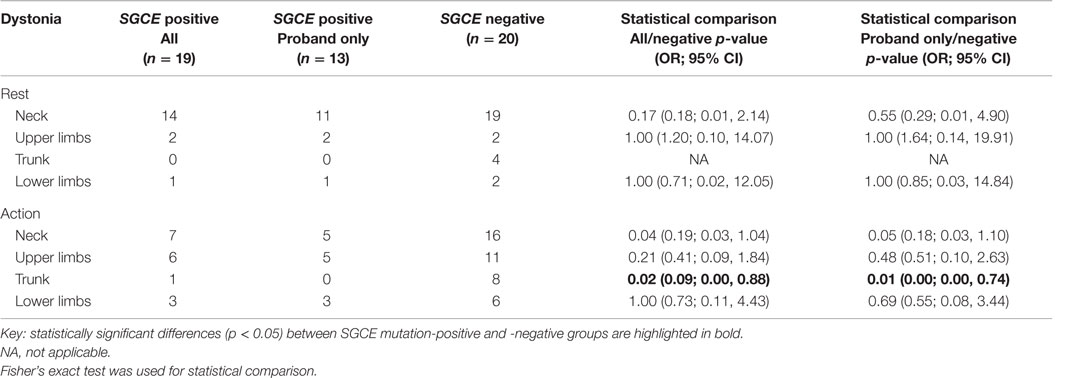
Table 3. Distribution of dystonia at rest and with action.
Coexistence of Myoclonus and Dystonia
At rest, there was no significant difference between the proband-only mutation-positive group and those without an SGCE mutation in the coexistence of myoclonus and dystonia in the same body part, either overall or by individual body part. Overall assessment with action showed a significant difference between the two groups [p = 0.01, OR = 0.11, 95% CI (0.01, 0.73)], being the coexistence more common in the mutation-negative group, although no difference was observed between individual body parts. Inclusion of the entire mutation-positive cohort showed similar results overall [p = 0.01, OR 0.13, 95% CI (0.02, 0.71)] and a trend toward significance when examining the cervical region [p = 0.09, OR = 0.26, 95% CI (0.05, 1.26)]. A full summary of the rates and distribution of coexistent myoclonus and dystonia can be seen in Table 4.
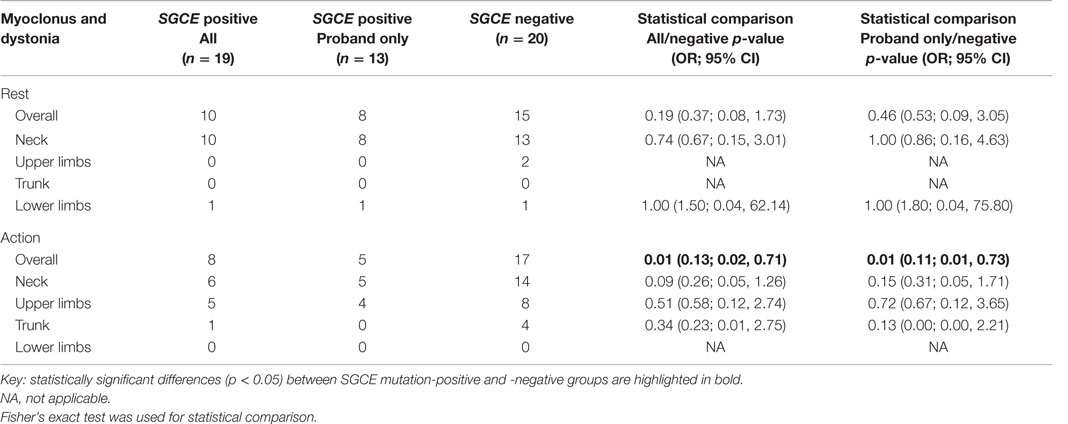
Table 4. Comparison of co-existent myoclonus and dystonia in the same body region in SGCE mutation-positive and -negative cohorts.
Severity of Myoclonus and Dystonia with Use of BFMDRS and UMRS Rating Scales
The median total BFMDRS score was significantly higher in the SGCE mutation negative group [6/120 (range: 4–47)] vs. 3.5/120 (range: 0–11) than in the mutation-positive group (p < 0.05). A higher median UMRS total score was also observed in the SGCE-negative patients [25/240 (range: 0–92)] compared to 14.5/240 (range: 0–80) in the mutation-positive group (p > 0.05), although this difference was not statistically significant.
Inter-Rater Agreement
Two assessors evaluated the SGCE mutation-negative group using both BFMDRS and UMRS rating scales, achieving an inter-rater concordance of “good” [ICC BFMDRS = 0.91 (95% CI: 0.74–0.97)/ICC UMRS = 0.87 (95% CI: 0.60–0.96)]. Each rating scale was scored by a single assessor during evaluation of the mutation-positive patients (Table S2 in Supplementary Material). In evaluating co-occurrence of myoclonus and dystonia, overall agreement between the two assessors was 88% at rest and 84% with action. Evaluation of individual body parts showed the strongest concordance when assessing the truncal region (94%, 34/36) and the lowest rate of agreement when evaluating movements of the neck with action (64%, 23/36). A summary of the positive agreement between assessors can be seen in Table S3 in Supplementary Material.
Discussion
This study examined the distribution and coexistence of myoclonus and dystonia, at rest and with action, as a predictive factor in determining the presence of an SGCE mutation in patients with an M–D phenotype. We have demonstrated that truncal dystonia and coexistence of myoclonus and dystonia in the same body region with action are more frequently observed in those without an SGCE mutation.
Application of the Grunewald diagnostic criteria to this study cohort did not clearly distinguish between those with and without an SGCE mutation. Seven on those without a mutation were deemed to be in the “definite” diagnostic category, while a two individuals with mutations were placed, one each, in the “probable” and “possible” groups. In keeping with the current diagnostic criteria, myoclonus and dystonia were most frequently observed in the neck and arms of both mutation positive and negative cohorts, with onset of symptoms <20 years being more frequent in those with an SGCE mutation (17/19 vs. 12/20) (5, 18). A positive family history was more frequently observed among those with an SGCE mutation and therefore increased the number of cases in the “definite” diagnostic category. The mutation-positive cohort consisted of 13 probands with an additional 6 affected cases recruited from two families, reflecting an inherent recruitment bias from a specialist tertiary movement disorder service. It could also be argued that by recruiting multiple members of the same kindred, additional genetic and environmental factors may also be contributing to their motor phenotype. However, little difference in results was observed when comparing both the proband only and complete SGCE mutation-positive cohort to the mutation-negative group, suggesting that any potential additional factors had little effect in the outcome of this study. In addition, multiple case reports and case series have demonstrated significant intra-familial motor variability among those with SGCE mutations (4).
It is worth mentioning that mutations in other genes, including KCTD17, THD, and RELN, have been recently associated with M–D, although confirmation in a larger number of families is still needed (24–26). Available data suggest that the phenotype associated with these mutations might slightly differ from that associated with SGCE mutation. For example the KCTD17 gene mutation is characterized by a milder myoclonus affecting the upper limbs and progressive dystonia spreading from the craniocervical region to other sites. The patients in our cohort were not screened for these mutations, which could potentially account for some of the SCGE-negative cases.
Although multiple previous reports have commented on worsening of motor features with action in M–D cohorts, none of the previous studies have directly compared the nature and frequency of the movement disorder between an SGCE mutation-positive cohort and a suitable mutation-negative control group, both at rest and with action. Overall, no difference between the two groups was observed at rest; however, coexistence of myoclonus and dystonia in the same body area was significantly more frequent with action in the mutation-negative cohort (p < 0.05) with a trend toward significance observed in the cervical region with action (p = 0.09). These observations of coexistent myoclonus and dystonia in the same body region in those without an SGCE mutation may reflect a “jerky” dystonia rather than myoclonus. These two forms of hyperkinesias are known to be difficult to differentiate, both in clinical practice and assessment of videotaped examination. It may be contributory to include neurophysiological testing in future studies to aid in differentiating between these two forms of movement disorder (27).
Multi-rater comparison of clinical cases can potentially result in significant variability of clinical opinion. Overall inter-rater agreement between the movement disorder specialists involved in this study was good. Disparities in scoring were predominantly observed in the cervical region where dystonic “overflow” or movement of other body parts can cause diagnostic difficulty. These results highlight the notoriously difficult task of hyperkinetic movement disorder phenomenology, particularly when two or more movement disorder subtypes may coexist in the same body region. This study can be regarded as a pilot study due to the relatively small number of patients in each study group ultimately preventing further and more elaborate statistic analysis of the available results. Future studies will require large, multicenter collaboration in order to enable recruitment of sufficiently large and diverse cohorts to allow definitive conclusions to be drawn.
Conclusion
The results of this pilot study suggest that the presence of truncal dystonia and the coexistence of myoclonus and dystonia in the same body region with action reduce the likelihood of SGCE mutation in patients with an M–D phenotype. Larger series are needed to confirm our preliminary findings before they can be translated into clinical practice. These results highlight the importance of examining movement disorders both at rest and with action during clinical assessment, particularly, when selecting those patients to undergo specific genetic testing.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Dr. E. Foncke and Dr. S. van der Salm for performing some of the videos.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fneur.2016.00072
References
1. Foncke EM, Gerrits MC, van Ruissen F, Baas F, Hedrich K, Tijssen CC, et al. Distal myoclonus and late onset in a large dutch family with myoclonus-dystonia. Neurology (2006) 67(9):1677–80. doi: 10.1212/01.wnl.0000242880.49051.1f
3. Roze E, Apartis E, Clot F, Dorison N, Thobois S, Guyant-Marechal L, et al. Myoclonus-dystonia: clinical and electrophysiologic pattern related to SGCE mutations. Neurology (2008) 70(13):1010–6. doi:10.1212/01.wnl.0000297516.98574.c0
4. Nardocci N, Zorzi G, Barzaghi C, Zibordi F, Ciano C, Ghezzi D, et al. Myoclonus-dystonia syndrome: clinical presentation, disease course, and genetic features in 11 families. Mov Disord (2008) 23(1):28–34. doi:10.1002/mds.21715
5. Kinugawa K, Vidailhet M, Clot F, Apartis E, Grabli D, Roze E. Myoclonus-dystonia: an update. Mov Disord (2009) 24(4):479–89. doi:10.1002/mds.22425
6. Asmus F, Zimprich A, Tezenas Du Montcel S, Kabus C, Deuschl G, Kupsch A, et al. Myoclonus-dystonia syndrome: epsilon-sarcoglycan mutations and phenotype. Ann Neurol (2002) 52(4):489–92. doi:10.1002/ana.10325
7. Peall KJ, Kurian MA, Wardle M, Waite AJ, Hedderly T, Lin JP, et al. SGCE and myoclonus dystonia: motor characteristics, diagnostic criteria and clinical predictors of genotype. J Neurol (2014) 261(12):2296–304. doi:10.1007/s00415-014-7488-3
8. Klein C, Liu L, Doheny D, Kock N, Müller B, de Carvalho Aguiar P, et al. Epsilon-sarcoglycan mutations found in combination with other dystonia gene mutations. Ann Neurol (2002) 52(5):675–9. doi:10.1002/ana.10358
10. Foncke EM, Cath D, Zwinderman K, Smit J, Schmand B, Tijssen M. Is psychopathology part of the phenotypic spectrum of myoclonus-dystonia?: a study of a large dutch M-D family. Cogn Behav Neurol (2009) 22(2):127–33. doi:10.1097/WNN.0b013e3181a7228f
11. Peall KJ, Smith DJ, Kurian MA, Wardle M, Waite AJ, Hedderly T, et al. SGCE mutations cause psychiatric disorders: clinical and genetic characterization. Brain (2013) 136(Pt 1):294–303. doi:10.1093/brain/aws308
12. van Tricht MJ, Dreissen YE, Cath D, Dijk JM, Contarino MF, van der Salm SM, et al. Cognition and psychopathology in myoclonus-dystonia. J Neurol Neurosurg Psychiatry (2012) 83(8):814–20. doi:10.1136/jnnp-2011-301386
13. Müller B, Hedrich K, Kock N, Dragasevic N, Svetel M, Garrels J, et al. Evidence that paternal expression of the epsilon-sarcoglycan gene accounts for reduced penetrance in myoclonus-dystonia. Am J Hum Genet (2002) 71(6):1303–11. doi:10.1086/344531
14. Grabowski M, Zimprich A, Lorenz-Depiereux B, Kalscheuer V, Asmus F, Gasser T, et al. The epsilon-sarcoglycan gene (SGCE), mutated in myoclonus-dystonia syndrome, is maternally imprinted. Eur J Hum Genet (2003) 11(2):138–44. doi:10.1038/sj.ejhg.5200938
15. Zimprich A, Grabowski M, Asmus F, Naumann M, Berg D, Bertram M, et al. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat Genet (2001) 29(1):66–9. doi:10.1038/ng709
16. Valente EM, Edwards MJ, Mir P, DiGiorgio A, Salvi S, Davis M, et al. The epsilon-sarcoglycan gene in myoclonic syndromes. Neurology (2005) 64(4):737–9. doi:10.1212/01.WNL.0000151979.68010.9B
17. Raymond D, Saunders-Pullman R, de Carvalho Aguiar P, Schule B, Kock N, Friedman J, et al. Phenotypic spectrum and sex effects in eleven myoclonus-dystonia families with epsilon-sarcoglycan mutations. Mov Disord (2008) 23(4):588–92. doi:10.1002/mds.21785
18. Grünewald A, Djarmati A, Lohmann-Hedrich K, Farrell K, Zeller JA, Allert N, et al. Myoclonus-dystonia: significance of large SGCE deletions. Hum Mutat (2008) 29(2):331–2. doi:10.1002/humu.9521
19. Tezenas du Montcel S, Clot F, Vidailhet M, Roze E, Damier P, Jedynak CP, et al. Epsilon sarcoglycan mutations and phenotype in french patients with myoclonic syndromes. J Med Genet (2006) 43(5):394–400. doi:10.1136/jmg.2005.036780
20. Carecchio M, Magliozzi M, Copetti M, Ferraris A, Bernardini L, Bonetti M, et al. Defining the epsilon-sarcoglycan (SGCE) gene phenotypic signature in myoclonus-dystonia: a reappraisal of genetic testing criteria. Mov Disord (2013) 28:787–94. doi:10.1002/mds.25506
21. Gerrits MC, Foncke EM, de Haan R, Hedrich K, van de Leemput YL, Baas F, et al. Phenotype-genotype correlation in dutch patients with myoclonus-dystonia. Neurology (2006) 66(5):759–61. doi:10.1212/01.wnl.0000201192.66467.a3
22. Burke RE, Fahn S, Marsden CD, Bressman SB, Moskowitz C, Friedman J. Validity and reliability of a rating scale for the primary torsion dystonias. Neurology (1985) 35(1):73–7. doi:10.1212/WNL.35.1.73
23. Fermanian J. Measuring agreement between 2 observers: a quantitative case. Rev Epidemiol Sante Publique (1984) 32(6):408–13.
24. Mencacci NE, Rubio-Agusti I, Zdebik A, Asmus F, Ludtmann MH, Ryten M, et al. A missense mutation in KCTD17 causes autosomal dominant myoclonus-dystonia. Am J Hum Genet (2015) 96(6):938–47. doi:10.1016/j.ajhg.2015.04.008
25. Stamelou M, Mencacci NE, Cordivari C, Batla A, Wood NW, Houlden H, et al. Myoclonus-dystonia syndrome due to tyrosine hydroxylase deficiency. Neurology (2012) 79(5):435–41. doi:10.1212/WNL.0b013e318261714a
26. Groen JL, Ritz K, Jalalzadeh H, van der Salm SM, Jongejan A, Mook OR, et al. RELN rare variants in myoclonus-dystonia. Mov Disord (2015) 30(3):415–9. doi:10.1002/mds.26070
Keywords: myoclonus–dystonia, myoclonus, dystonia, SGCE mutations, motor characteristics
Citation: Zutt R, Dijk JM, Peall KJ, Speelman H, Dreissen YEM, Contarino MF and Tijssen MAJ (2016) Distribution and Coexistence of Myoclonus and Dystonia as Clinical Predictors of SGCE Mutation Status: A Pilot Study. Front. Neurol. 7:72. doi: 10.3389/fneur.2016.00072
Received: 17 June 2015; Accepted: 27 April 2016;
Published: 13 May 2016
Edited by:
Pille Taba, University of Tartu, EstoniaReviewed by:
Francesca Morgante, University of Messina, ItalyAngelo Quartarone, University of Messina, Italy
Copyright: © 2016 Zutt, Dijk, Peall, Speelman, Dreissen, Contarino and Tijssen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marina A. J. Tijssen, m.a.j.de.koning-tijssen@umcg.nl
†Maria Fiorella Contarino and Marina A. J. Tijssen contributed equally to the work.