 Mohammed Aftab Alam
Mohammed Aftab Alam
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 28 June 2016
Sec. Stroke
Volume 7 - 2016 | https://doi.org/10.3389/fneur.2016.00094
This article is part of the Research Topic Neuroimaging and Computational Model Guided Post-Stroke Non-Invasive Brain Stimulation Therapy View all 5 articles
Aims: Recent studies indicate that anti-inflammatory drugs, act as a double-edged sword, not only exacerbating secondary brain injury but also contributing to neurological recovery after stroke. Our aim is to explore whether there is a beneficial role for neuroprotection and functional recovery using anti-inflammatory drug along with neurorehabilitation therapy using transcranial direct current stimulation (tDCS) and repetitive transcranial magnetic stimulation (rTMS), so as to improve functional recovery after ischemic stroke.
Methods: We develop a computational systems biology approach from preclinical data, using ordinary differential equations, to study the behavior of both phenotypes of microglia, such as M1 type (pro-inflammatory) vis-à-vis M2 type (anti-inflammatory) under anti-inflammatory drug action (minocycline). We explore whether pharmacological treatment along with cerebral stimulation using tDCS and rTMS is beneficial or not. We utilize the systems pathway analysis of minocycline in nuclear factor kappa beta (NF-κB) signaling and neurorehabilitation therapy using tDCS and rTMS that act through brain-derived neurotrophic factor (BDNF) and tropomyosin-related kinase B (TrkB) signaling pathways.
Results: We demarcate the role of neuroinflammation and immunomodulation in post-stroke recovery, under minocycline activated-microglia and neuroprotection together with improved neurogenesis, synaptogenesis, and functional recovery under the action of rTMS or tDCS. We elucidate the feasibility of utilizing rTMS/tDCS to increase neuroprotection across the reperfusion stage during minocycline administration. We delineate that the signaling pathways of minocycline by modulation of inflammatory genes in NF-κB and proteins activated by tDCS and rTMS through BDNF, TrkB, and calmodulin kinase (CaMK) signaling. Utilizing systems biology approach, we show that the activation pathways for pharmacotherapy (minocycline) and neurorehabilitation (rTMS applied to ipsilesional cortex and tDCS) results into increased neuronal and synaptic activity that commonly occur through activation of N-methyl-d-aspartate receptors. We construe that considerable additive neuroprotection effect would be obtained and delayed reperfusion injury can be remedied, if one uses multimodal intervention of minocycline together with tDCS and rTMS.
Conclusion: Additive beneficial effect is, thus, noticed for pharmacotherapy along with neurorehabilitation therapy, by maneuvering the dynamics of immunomodulation using anti-inflammatory drug and cerebral stimulation for augmenting the functional recovery after stroke, which may engender clinical applicability for enhancing plasticity, rehabilitation, and neurorestoration.
Recent investigations have reported that immune responses to inflammation are non-specific systemic infections associated with progression of neurodegenerative diseases via activation of macrophages (1). Minocycline is a tetracycline antibiotic having several properties, such as anti-inflammatory, anti-apoptosis, free radical scavenger, and protein misfolding (2). The therapeutic effects of minocycline in preclinical models of neurodegenerative diseases showed direct neuroprotection and reduction of microglial inflammatory responses (3). It has been reported in in vivo studies that minocycline blocks the adhesion of leukocytes to cerebrovascular endothelial cells induced by lipopolysaccharides, as well as tumor necrosis factor-α (TNF-α) production in the brain (4). In vitro studies have reported the anti-inflammatory effects of minocycline for neuroprotection (5) and in macrophages (6). Neuroprotective effects of minocycline include reduction of macrophage activation, prevention of the potentiation of ischemia-like injury to astrocytes and endothelial cells consolidating the brain tissue parenchyma (7). Although, the anti-inflammatory effects of minocycline are known to some extent, the direct effects of neuroprotection have not been well investigated in neurodegenerative diseases.
Several studies have shown that the physiological neuroprotection mechanisms that occur after stroke are targeted through various signaling pathways. Several studies suggest that the mechanisms associated with either reducing the size of infarct or enabling neurorestoration, involve the following entities: (i) anti-high mobility group box-1 activity (8); (ii) NF-κB (9); (iii) mammalian target of rapamycin (mTOR) inhibitor (10, 11); (iv) stimulation of toll-like receptors (TLR2 and TLR4) prior to brain ischemia (12, 13), (v) c-Jun N-terminal kinase (JNK) inhibitor (14); (vi) p38 mitogen-activated protein kinase (p38 MAPK) inhibitor (15); (vii) MEK1 pathway (16); (viii) MAPP/MEK/ERK inhibitor (17); and (ix) Minocycline-induced reduction of LPS-stimulated p38 MAPK activation, and stimulation of the phosphoinositide 3-kinase (PI3K)/Akt pathway (18).
Currently, little is known about endogenous counter regulatory immune mechanisms that can induce neurorestoration. The glycogen synthase kinase-3β (AKT/GSK-3β) pathway has been recognized as a protective pathway against cerebral ischemic injury. In cerebral ischemia models, it has been shown that remote limb conditioning does indeed activate and upregulate the pro-survival AKT pathway (19) and long-term protection against cerebral ischemia is afforded by limb post-conditioning that is associated with AKT, MAPK, phosphatidylinositol 3-kinase (PI3K), and protein kinase C (PKC) signaling pathways (20). NF-κB transcription factor family members, such as p50, p65/RelA in the hippocampus, are regulated by metabotropic glutamate receptor signaling and c-Rel transcription factor is responsible for the formation and maintenance of long-term memory (21). Minocycline directly inhibits matrix metalloproteinase (MMP)-9 activation through NF-κB pathway (22). In silico modeling of anti-inflammatory response has been reported for endotoxins (LPS) and corticosteroids by activating TLRs in NF-κB (23).
Taken together, the modulation of cell survival and death signaling by hypoxic/ischemic preconditioning appears to be capable of targeting multiple levels of signaling cascades. Several inhibitors targeted the point of convergence through distinct and interacting signaling pathways (crosstalk mechanism) for inflammation by activating macrophages that lead to neuroprotection. Also, cerebral stimulation-based transcranial magnetic stimulation and direct current stimulation enhances brain-derived neurotrophic factor (BDNF) and tropomyosin-related kinase B (TrkB) signaling (24, 25). In this study, we harness the convergent signaling pathways of pharmacotherapy (anti-inflammatory, immunomodulatory) and neurorehabilitation therapy (functional recovery) for efficient post-stroke neurorestoration by experimental and systems-level approach. We modeled using the systems biology approach of minocycline modulation of MMPs through NF-κB signaling pathway, a master regulator of inflammatory responses along with neurorehabilitation-based activation in BDNF and TrkB signaling.
There are numerous cellular responses, stress events, neuronal death, and inflammatory mechanisms, including over-activation of microglia as shown in Figure 1. We utilized and targeted anti-inflammatory drug in our approach. We formulated various cell kinetics interactions utilizing the systems biology platform for pharmacotherapy (Module 1) and neurorehabilitation (Module 2)-based activation through different signaling pathways, such as NF-κB, BDNF, and TrkB. To formulate and model the effect of therapeutic interventions, we activated the corresponding signal transduction factor accordingly. For minocycline, activation input was in the form of a constant single impulse or step function, while for the transcranial direct current stimulation (tDCS) and repetitive transcranial magnetic stimulation (rTMS) inputs, we used repeated series of electrical or magnetic pulses involved in cerebral stimulation. The chemical reactions of the modules were constructed using the framework of ordinary differential equations (ODEs), which were then solved by numerical computation using the Runge–Kutta method.
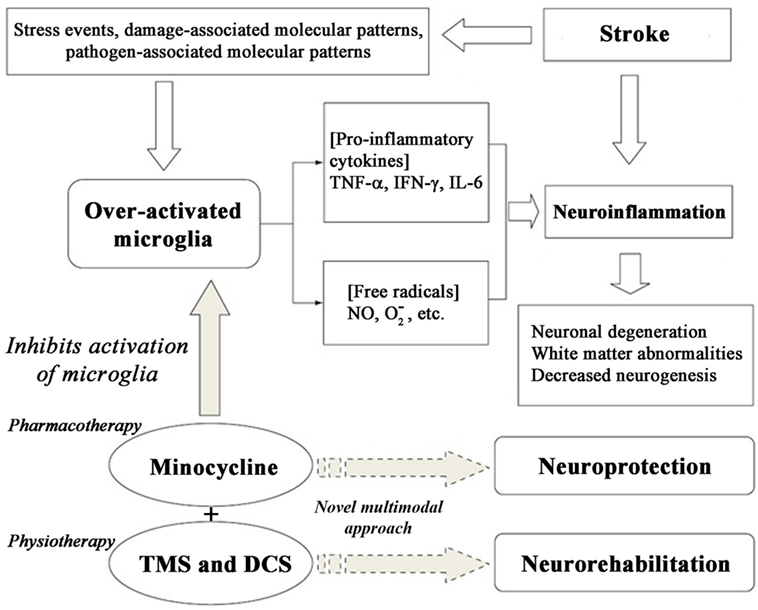
Figure 1. Cascade of cell signaling events post-stroke and enabling neuroprotection using minocycline.
We formulated and elucidated the cascade of cell signaling events using ODEs that were solved using ODE23 solver in MatlabR2013a. We modeled the inflammatory mechanism of minocycline through regulation of genes and proteins that significantly corresponded to NF-κB transcriptome as shown in Figure 2. Studies have suggested that minocycline-induced suppression of NF-κB activity is mediated by the inhibition of M1 microglia and activation of M2 microglia. All the sequential chemical processes are explained in Module 1. The in vitro and in vivo studies have shown that both the acute and chronic doses of minocycline lead to suppression of p65 phosphorylation and nuclear translocation accompanied by downregulation of NF-κB activity and endogenous MMP9 protein levels (22).
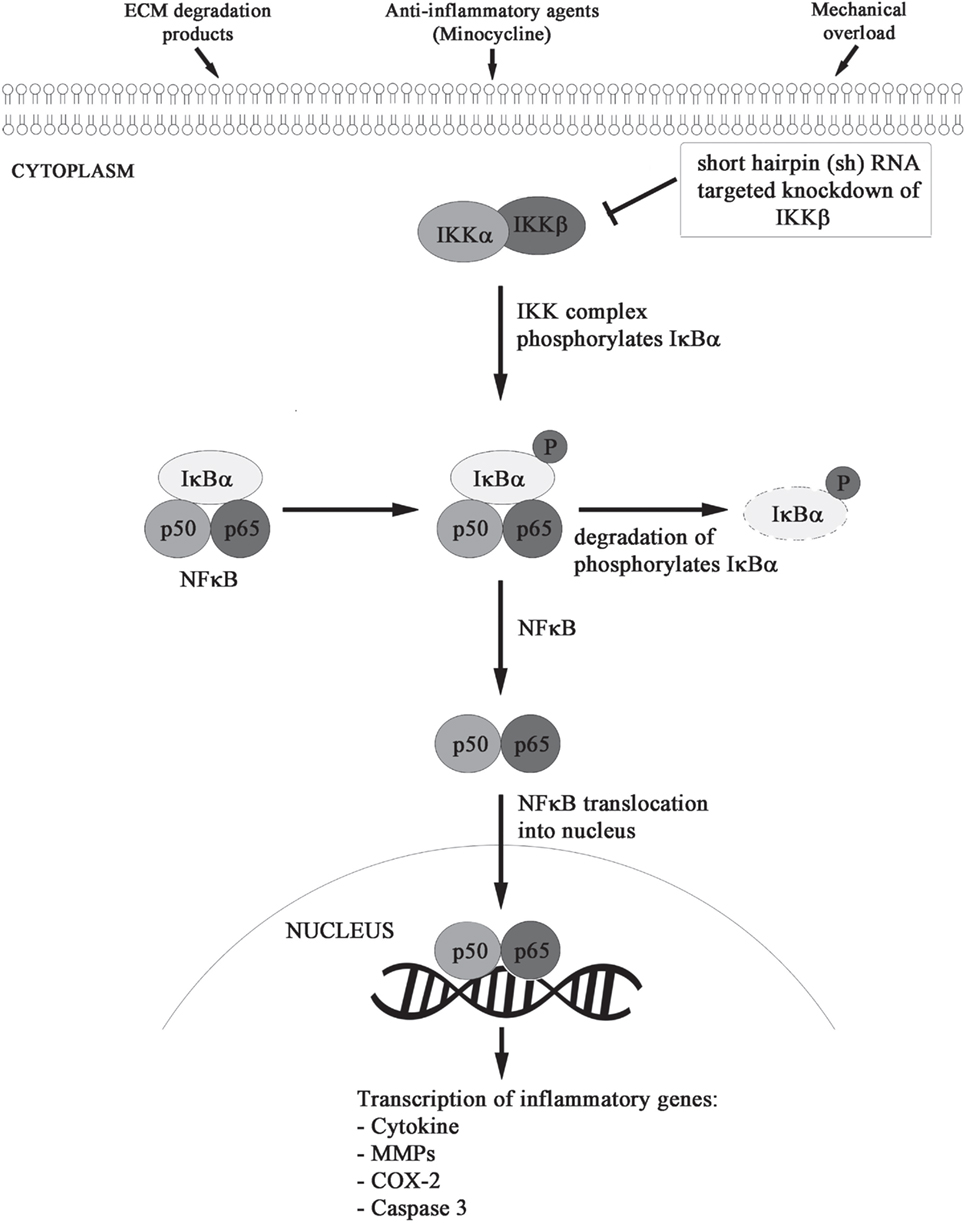
Figure 2. Minocycline-activated anti-inflammatory response through NF-κB signaling pathway.
Using the dynamics of known NF-κB stages (26, 27), the action of minocycline via the signaling pathway was represented as shown in Module 1. The sequence depicts a chronological order of the formation of different biochemical species (mentioned on the right side after % symbol) with their corresponding reaction kinetic equation (on the left side).
The non-zero initial concentration of the chemical species and the association and dissociation rate constants are taken from previous experiments (23, 26) given in APPENDIX (Table A1). The ODE equations shown in Module 1 were solved using ODE23 solver in MatlabR2013a.
To elucidate the process of neurorehabilitation therapy using tDCS and rTMS via BDNF and TrkB pathway. We constructed the upstream and downstream dynamics of the pathway using currently available data (28). The corresponding chemical reactions are formulated as ODE and shown in Module 2.
Brain-derived neurotrophic factor activates TrkB through several downstream signaling pathways, such as AKT, CaMK, Ras/Raf/MEK/ERK leading to cell survival, growth, and neuroplasticity as shown in Figure 3. BDNF activates TrkB stimulation via phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) and also activates proteins like Shc, Grb-2, and Gab-1. The PI3K is also activated by binding to Ras homolog enriched by brain glutamine triphosphate (Ras-GTP). TMS was delivered to male Sprague Dawley rats using 1600 stimuli at 5 Hz in four blocks of each 400 stimuli in 2.5 min with 1 min inter-block interval (24). Anodal tDCS was applied with current density of 0.04 mA/cm2, total charge of 0.048 C/cm2 for 20 min (25). Activated PI3K phosphorylates PtdIns[3,4]P2 (PIP2) and PtdIns[3,4,5] P3 (PIP3), and then PIP3 is dephosphorylated by phosphotase and tensin homolog (PTEN). PIP3 activates AKT (also known as protein kinase B) leading to the formation of Rheb-GTP, a regulator of rapamycin (TOR) (29, 30). AKT needs phosphorylation twice to become active. PIP3 binds to AKT and recruits it in the membrane and does the same for Pyruvate dehydrogenase lipoamide kinase isozyme 1 (PDK1).
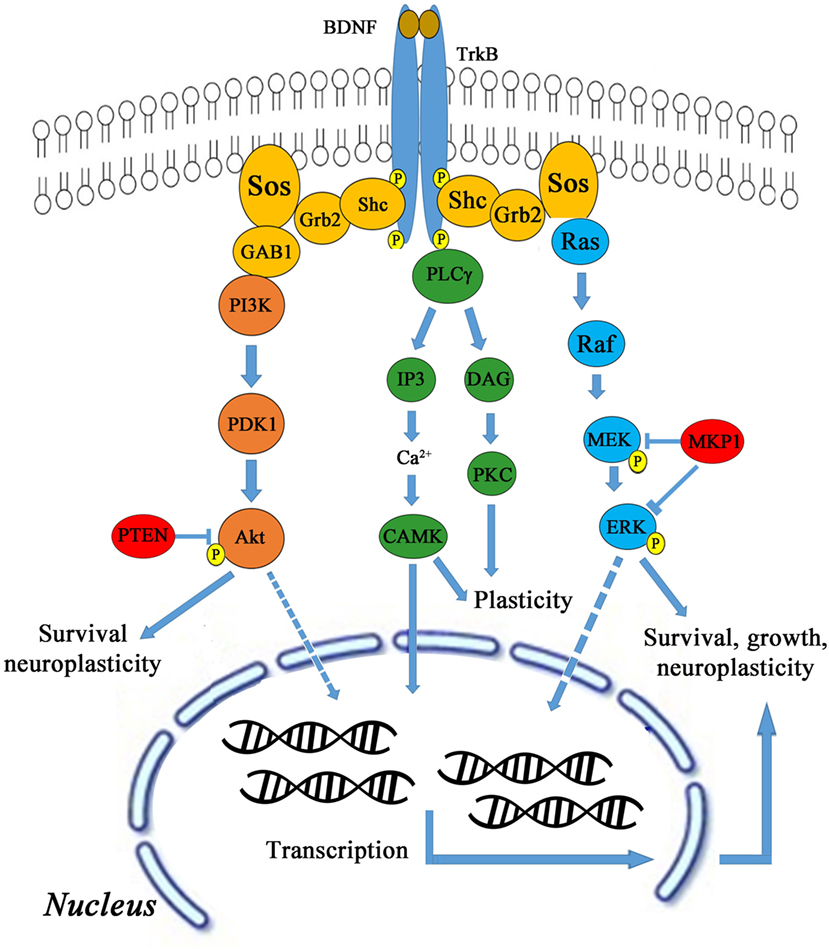
Figure 3. BDNF and TrkB signaling pathway.
In the membrane, PDK1–PIP3 complex phosphorylates AKT on Thr308 and further phosphorylates on Ser-473 by PIP3-PDK2. Both these phosphorylates are reversed by protein phosphatase 2A (PP2A). The doubly phosphorylated AKT becomes active and phosphorylates tuberous sclerosis 1,2 (TSC1–TSC2), which in turn regulates Rheb-GTP levels. The unphosphorylated complex has GTPase-activating protein (GAP)-type properties toward Rheb, while TSC1–TSC2 hydrolyzes Rheb-GTP to Rheb-GDP (31). The active AKT inactivates TSC1–TSC2 and only Rheb-GTP remains. Therefore, the net effect is, PIP3 activates AKT leading to activation of downstream target TOR. Furthermore, Rheb-GTP activates TOR and the converged input from MAPK leads to S6K activation, forming active 40S for translation (32, 33). The Rheb-GTP binds to the TOR_complex to stimulate kinase activity through phosphorylation in each kinase, viz. MAPK, TOR_complex, and PDK1. Next, dephosphorylation occurs at all the sites by PP2A. The double- and triple-phosphorylated S6K becomes active and phosphorylates S6, which is a subunit of 40S ribosomal protein.
Another possible signaling mechanism is phospholipase C gamma (PLC-γ) that in turn cleaves PIP2, diacylglycerol (DAG), and inositol trisphosphate (IP3). DAG remains bound to membrane, whereas IP3 is released as a soluble structure into the cytosol. Then IP3 diffuses through cytosol and binds to IP3 receptors, particularly calcium channels in a smooth endoplasmic reticulum, and then activates Ca2+ in calmodulin kinase (CaMK) activity. Correspondingly, DAG activates PKC for plasticity (34). CaMKIII is activated by Ca2+ and inactivated by S6K. CaMKIII inhibits eukaryotic elongation factor 2 (eEF2), the net effect of Ca2+ on elongation factor is inhibitory and of S6K is excitatory. eEF2 is the major substrate for CaMKIII. CaMKIII catalyzes eEF2 phosphorylation at thr-56 and thr-58, strictly in the presence of Ca2+ and CaM (35). CaMKIII phosphorylates eEF2 to render it inactive. Dephosphorylation of eEF2 by PP2A restores its activity.
The S6K phosphorylates CaMKIII at Ser-366 and decreases its activity and thereby increases the level of eEF2 (36). Calmodulin dependence of CaMKIII activity was measured in terms of eEF2 phosphorylation (37). Next, the active S6K is added to CaMKIII and time course of CaMKIII is measured. Then, active eEF2-thr-36 is measured with respect to time. The dose–response for 40S complex is measured for different 40S concentrations (38), the dephosphorylation reaction and the formation of phosphorylated eEF2 (28). The 40S and eEF2 bind to form the translation complex, leading to protein synthesis. MAPK is downstream of PI3K as well as CaM-Ca4. The MAPK activity is closely related to synaptic activity leading to Ca2+ influx. The alternate mechanism is through downstream signaling of Ras/Raf/MEK/ERK pathway for cell survival, growth, and neuroplasticity.
The non-zero initial concentration of chemical species is given in APPENDIX (Table A2). All these parameters are taken from the experimental studies related to memory, learning, and synaptic plasticity through BDNF and TrkB signaling. The implications of tDCS and rTMS in the present study are to enhance the post-stroke motor-learning along with functional recovery in ischemic stroke patients. The association and dissociation rate constants are taken from experimental studies as given in APPENDIX (Table A3). The aforesaid equations are given in Module 2, which were solved by ODE23 solver in MatlabR2013a.
We simulated the biochemical processes of both Modules 1 and 2. Our observations and inferences are explained in details below.
The results of the signaling activation by the pharmacological agent minocycline are shown in Figure 4. A sub-optimal dose of 4.5 mg kg−1 was chosen from a dose escalation study of minocycline in stroke patients (81). Our findings delineate the inflammatory mechanism of minocycline through regulation of genes and proteins that correspond to the NF-κB transcriptome. Our simulation has shown the molecular mechanism of minocycline that could be attributed to modulation of NF-κB signaling. Minocycline suppressed NF-κB activation in neurons and glial cells and was correlated with attenuation of IκBα kinase (IKK) activation, IκBα phosphorylation and degradation, and p65 phosphorylation and nuclear translocation. The inhibition of IKK was found to be associated with suppression of activated-microglia (neurotoxic M1 microglia) and correspondingly enhanced the restorative microglia (neuroprotective M2 microglia). Furthermore, the simulation demonstrated that minocycline upregulated TNF-α expression. Enforced TNF-α expression induced NF-κB activity and minocycline rescued through inhibition of iNOS and NO production in cells. Our simulated results are consistent with the experimental findings that suppressed NF-κB activation and abrogated the inhibitory effect of minocycline on the transcription factor, TNF-α. These results suggest that minocycline led to suppression of p65 phosphorylation and nuclear translocation accompanied by downregulation of NF-κB activity and endogenous MMP9 protein levels and its target, PTGS2 gene.
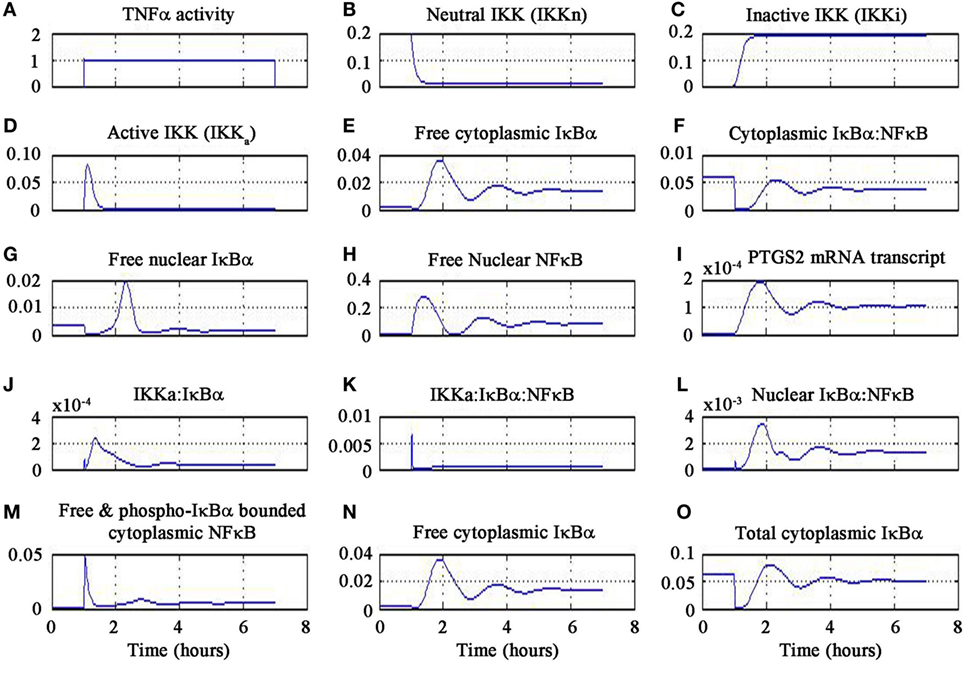
Figure 4. Minocycline-induced TNF-α activity in NF-κB pathway.
In the simulation, a minocycline dose of 4.5 mg kg−1 induced TNF-α activity for 6 h as shown in Figure 4A. The neutral IKK activity gradually reduced from 60 to 90 min and then asymptotically saturated downwards to baseline as shown in Figure 4B. Similarly, the inactive form of IKK increased from 60 to 90 min and then saturated to baseline as shown in Figure 4C. The active IKK expression increased from 60 to 90 min, peaked at 90 min and then fell gradually till 120 min and saturated downwards to basal level as shown in Figure 4D. Free cytoplasmic IκBα increased from 60 to 90 min, peaked at 120 min and then gradually fell till 180 min and oscillated as shown in Figure 4E. The cytoplasmic IκBα|NF-κB activity initially fell till 60 min, then peaked at 120 min, fell down till 180 min, and then showed oscillatory behavior as shown in Figure 4F. The free nuclear IκBα increased from 60 to 140 min, fell down till 180 min and finally damped as in Figure 4G. The free nuclear NF-κB activity increased from 60 to 90 min, fell down from 90 to 120 min, and then showed oscillatory behavior as in Figure 4H.
The PTGS2 gene transcription increased from 60 to 120 min, fell down till 180 min, and then oscillated as shown in Figure 4I. The expression of IKKa|IκBα complex increased from 60 to 90 min, gradually fell till 180 min, and then oscillated as in Figure 4J. The expression of IKKa|IκBα|NF-κB complex peaked at 60 min and then fell afterwards as shown in Figure 4K. The nuclear IκBα|NF-κB expression increased from 60 to 120 min and then gradually fell till 180 min and oscillated as in Figure 4L. Free and phosphorylated IκBα bounded to cytoplasmic NF-κB peaked at 60 min, fell gradually till 90 min and damped as in Figure 4M. Free cytoplasmic IκBα expression increased from 60 to 120 min then gradually fell till 180 min, and finally oscillated as in Figure 4N. The total cytoplasmic IκBα activity initially fell down till 60 min and then onward gradually increased till 120 min, and finally showed oscillatory behavior as in Figure 4O. It is important to note that the temporal profile for the chemical species will not change for different concentrations of minocycline.
The simulations showed increased activity of TrkB*2 from 0 to 500 s, and then fell till 1000 s, and then asymptotically saturated downwards to a definitive level as shown in Figure 5A. The Shc* activity increased linearly when plotted against BDNF till the BDNF concentration reached 2 nM as shown in Figure 5B. The PLC-γ activity increased linearly when plotted against BDNF till the concentration of 3.7 nM as in Figure 5C. Furthermore, the PIP3 activity linearly increased as shown in Figure 5D, while the cell elongation factor (eEF2) eEF2thr-56 showed sigmoid relationship when plotted against low concentrations of the calcium-binding protein Calmodulin, CaM (measured in terms of negative log) as in Figure 5E. eEF2 activity was observed to linearly increase with time as in Figure 5F. We observed that 40S complex activity displayed a declining sigmoidal function with an increased activity till 300 nM when plotted against 40S concentration, followed by constant activity (very slowly declining) from 300 to 600 nM, and thereafter finally the activity rises beyond 600 nM as in Figure 5G.
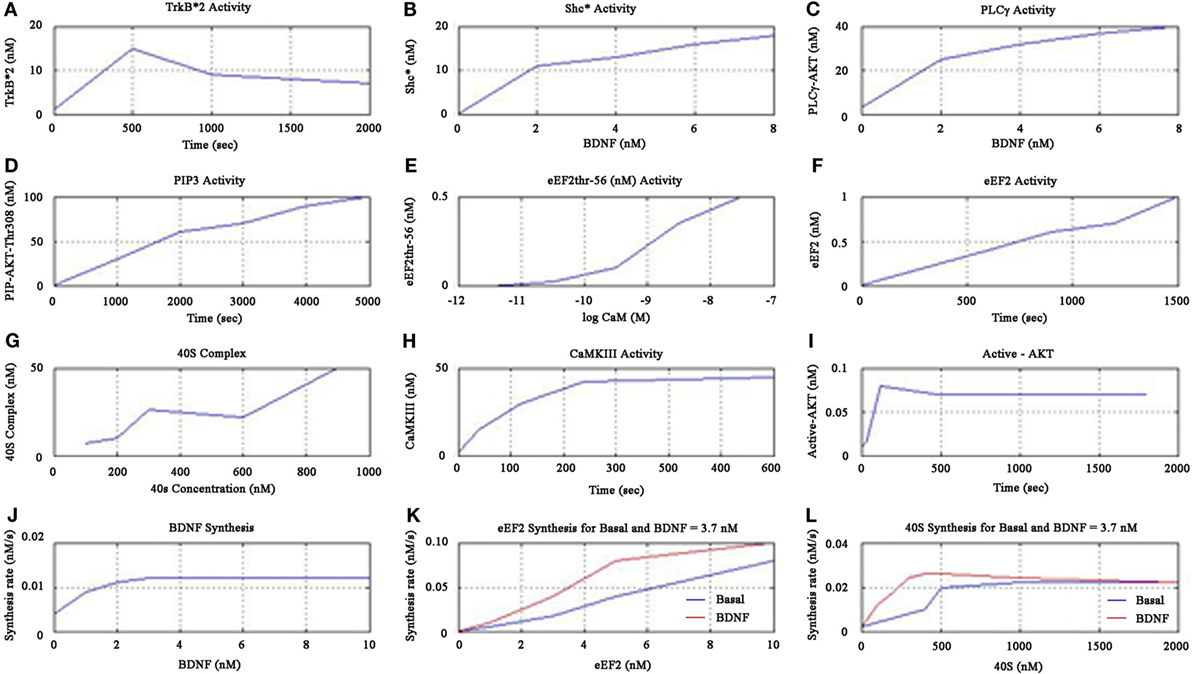
Figure 5. Cerebral stimulation induced BDNF and TrkB signaling.
Continuing the formulation further, we note that the kinase CaMKIII activity increased till 240 s and then saturated upwards as shown in Figure 5H. We found active AKT increase till 100 s, decreased after 480 s and then downward saturation to baseline as in Figure 5I. We inferred that the BDNF synthesis rate was maximum for the concentration of 3.7 nM as shown in Figure 5J. Moreover, we noticed eEF2 synthesis rate linearly increased when plotted against eEF2 concentration, for both basal kinase activity and for BDNF concentration of 3.7 nM (Figure 5K). We found that the synthesis rate of eEF2 for BDNF concentration of 3.7 nM was double the basal kinase level, when the eEF2 concentration is 5 nM as shown in Figure 5K. We also showed that the 40S synthesis rate peaked at 500 nM of 40S concentration for both basal kinase level and for BDNF level of 3.7 nM, and 40S synthesis rate was higher for BDNF level of 3.7 nM than that for basal kinase level, as shown in Figure 5L.
We also found that protein synthesis rate is constant for all BDNF concentrations for Ca2+ = 0.5 nM and notice that increase of synthesis rate occurs beyond 0.1 nM concentration for Ca2+ = 0.08 nM. This suggests that BDNF = 3.7 nM and lower Ca2+ concentration leads to higher protein synthesis rate as shown in Figure 6A. We plotted the percentage of kinase enzymes CaMKIII and MAPK activity against Ca2+ concentration and found MAPK activity peaked for Ca2+ = 0.5 nM, while CaMK activity peaked for Ca2+ = 1.0 nM as shown in Figure 6B. The protein synthesis rate for constant CamKIII = 0.6 μM gradually decreased for both basal kinase activity and for BDNF level of 3.7 nM, till Ca2+ = 0.1 μM. Thereafter, the protein synthesis activity was constant for higher Ca2+ concentrations. However, the synthesis rate was initially higher for BDNF level of 3.7 nM than for basal kinase activity, for lower values of Ca2+ concentration (<0.1 μM), as in Figure 6C.
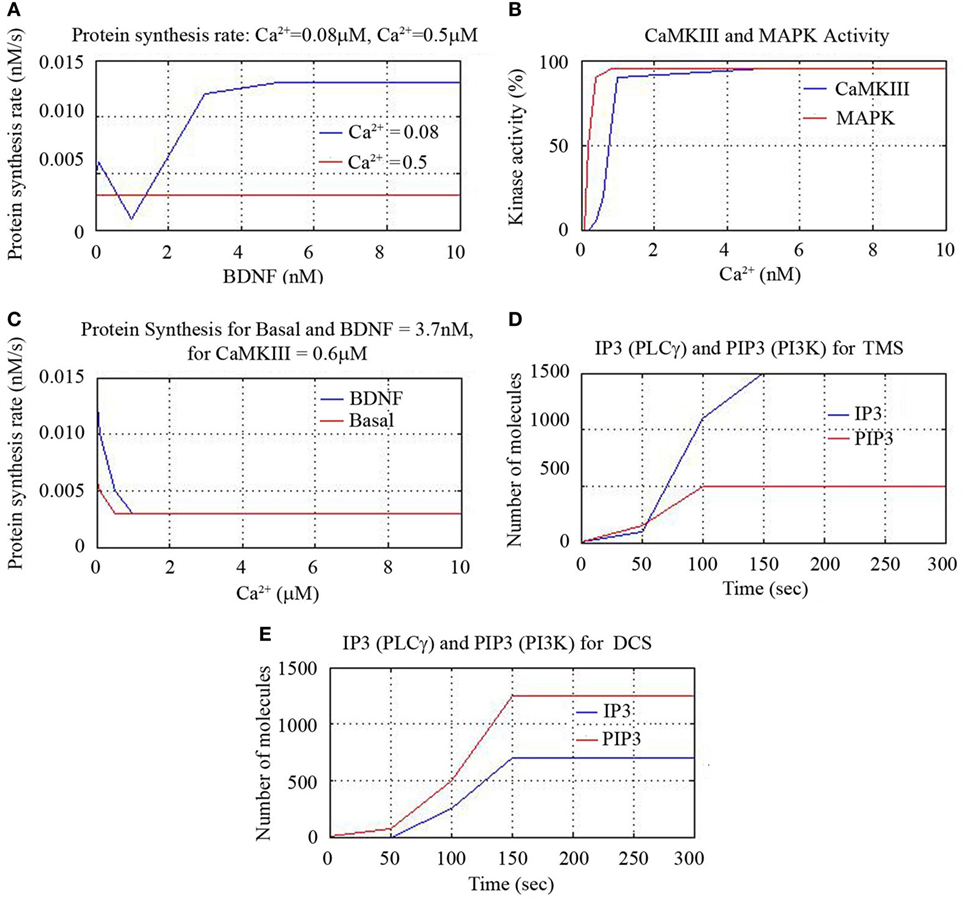
Figure 6. Cerebral stimulation induced IP3, PIP3, and CaMKIII activity.
As mentioned earlier, we activated the corresponding signal transduction factor accordingly (inositol triphosphate IP3, and the related compound, phosphatidyl-IP3, viz. PIP3). First, the initial activation of IP3 through phospholipase C (PLC-γ) and the activation of PIP3 through PI3K were plotted for TMS and DCS. TMS showed higher level of IP3 than PIP3 with higher number of reactant molecules generated till 150 s, and then the number of molecules saturated to a definitive level as in Figure 6D. DCS showed higher PIP3 than IP3 with an increased number of molecules till 150 s as in Figure 6E.
Thus, we have shown that both TMS and DCS activated CaMKIII that shows the cell survival and synaptic plasticity. To draw a perspective, we elucidated that all the modalities (minocycline, tDCS, rTMS) have different pathways with common activation of N-methyl-d-aspartate (NMDA) receptors. Therefore, pharmacotherapy and neurorehabilitation can be incisively combined for additive beneficial effect.
Oxidative stress and inflammation are the two major pathophysiological mechanisms involved during early and late phases of ischemic stroke. Brain tissue is not armed with antioxidant defenses, so reactive oxygen species and other free radicals/oxidants released by inflammatory cells, threaten tissue viability in the core ischemic region. This study targets the molecular aspects of inflammation in ischemic stroke and offers potential therapeutic strategies that target neuroinflammation and modulates the innate immune system. In addition to antibiotic activity, minocycline exhibits anti-inflammatory responses in both monocytes and macrophages. Earlier studies have reported that minocycline causes significant reduction in the inflammatory response of LPS-challenged monocytes, reducing LPS-induced transcription of pro-inflammatory TNF-α, interleukin-1 beta (IL-1β) (82), interleukin-6 (IL-6), and cyclooxygenase-2 (COX-2) (83), the LPS stimulated TNF-α, IL-6, and PGE2 release. Minocycline inhibited LPS-induced activation of the lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1), NF-κB, LPS-induced TNF-α factor (LITAF) and the Nur77 nuclear receptor (20). It has been reported that the neuroprotective effects of minocycline are associated with inhibition of iNOS induction and NO production in glial cells, which are mediated by the LPS-induced production of TNF-α (84).
The regulation of inflammatory genes and proteins in NF-κB signaling pathway are MMPs and Cox-2. MMPs are a family of zinc-dependent proteases responsible for the degradation of extracellular matrix proteins and capable of causing bioactive molecules. Cox-2 inhibitors are non-steroidal anti-inflammatory drugs (NSAIDs). The in vitro models showed direct neuroprotection by minocycline as well as inhibition of other inflammatory mediators, such as iNOS, MMPs, TNF-α, and tissue plasminogen activator (tPA). However, some earlier studies have reported expression of inflammatory cytokines, such as TNF-α in ischemic neurons (85) and tPA (86), and generation of neuronal tPA has been thought to mediate microglial activation. Thus, these studies suggested that neurons may be capable of secreting inflammatory mediators themselves from classical immune cells, including microglia and other glial cells, such as astrocytes. Also the neuronal survival depends on the endogenous or exogenous tPA, which can be either neuroprotective or neurotoxic. Similar conclusions could be made of neuronal generation of iNOS and MMPs. MMPs 2 and 9 levels are elevated a few hours after ischemia (87) and maintain increased activity for days after the onset of stroke (88). The specific class of agents inhibits these enzymes, reverts the breakdown of laminin (89), and prevents increased barrier permeability, edema, and hemorrhage after ischemic stroke (90).
Ischemic stroke activates a complex cascade of events and tetracycline antibiotics may exert anti-inflammatory effects by multiple mechanisms. The models of brain injury, which involve matrix degradation and vascular instability, targets inhibition of proteolytic cascade. However, there are limitations for the usage of MMP inhibitors as therapeutic agents because of their poor solubility. Minocycline has been shown to be a neuroprotective agent by inhibiting MMP2 and MMP9 activation by ischemia (91). The dysregulation of the proteolytic cascade at the level of the endothelial and microglial cells is one of the common pathological mechanisms of ischemic brain damage. Minocycline targets through the interference of this cascade and might be the key pathway for neuroprotection and salvage of injured tissue. Animal models of permanent cerebral ischemia showed that minocycline treatment was protective and beneficial in cerebral ischemia by mechanisms that are not linked to the production of free radicals during reperfusion. Understanding inflammatory interactions of all brain cell types is needed to identify how they may be manipulated to provide neuroprotection. Studies of minocycline effects on brain cells in addition to microglia are timely, as minocycline has shown efficacy in clinical studies for various neurodegenerative disorders (92, 93) and ischemic stroke (81, 94, 95). Thus, further investigations are needed for a better understanding of the complexities of immune responses of various types of cell bodies following brain injury.
The primary mechanism of tDCS for inducing cortical excitability shifts is a sub-threshold modulation of the neuronal resting membrane potential. The anodal stimulation outcome is a sub-threshold depolarization, while cathodal stimulation hyperpolarizes neuronal membranes. During a short tDCS, which elicits no after-effects (stimulation duration of about 4 s), synaptic mechanisms are not involved, as shown by the absence of effect of NMDA receptor antagonists, the GABA agonist lorazepam and the monoamine reuptake blocker amphetamine on the tDCS-induced excitability changes under these conditions. Furthermore, the after-effects are not due to reverberating electrical circuits or other purely electrical phenomena, as shown in early animal experiments. They depend on modifications of NMDA receptor efficacy, since these are blocked by the NMDA receptor antagonist dextromethorphan, but prolonged by the partial NMDA receptor agonist d-cycloserine (96). The tDCS polarity-dependent shift of NMDA receptor function seems to be initiated by the respective membrane potential shift and probably by the accompanying cortical activity modification, because it is prevented by the sodium channel blocker (carbamazepine). Likewise, tDCS requires the neurophysiological experimental studies for the selection of electrode montage in stroke (97). Furthermore, in silico modeling studies are required to understand the neuromodulatory effects of tDCS and helps in determining the optimal levels of anodal and cathodal stimulation that is beneficial for stroke patients.
Earlier studies also reported a decrease in IP 3 receptor mRNA level in the cortex at 4 h after a 45 min ischemic insult; however, lesser change was noticed for IP3 3-kinase mRNA levels during the initial 8 h after reperfusion (98). Also, IP3 receptor mRNA level in the peri-infarct areas was also reduced, specifically at a delayed time of reperfusion. The staining of cortical neurons in the infarct area showed morphological changes between 4 and 8 h after reperfusion (99). Thus, the decrease in IP3 receptor mRNA level probably occurred at a time prior to alteration of neuron morphology. A recent study showed that DCS stimulation-induced vasodilation occurs without IP3 receptor activation (100).
Brain-derived neurotrophic factor induces the transformation of early- to late-phase long-term potentiation (LTP) in the presence of protein synthesis inhibitors and BDNF–TrkB signaling is involved in synaptic tagging (101). BDNF activates the RAS/RAF signaling pathway and produces ERK, promoting gene transcription through cAMP-response element binding protein (CREB) in the cell nucleus. NMDA activation on the cell membrane produces a calcium influx that activates calcium CaMK IV, phosphorylating glycogen synthase kinase 3B (GSK3B) and allowing B-catenin to activate CREB. Recent studies have provided different mechanistic explanation for the early and late phases of LTP. The mechanism for later phases of LTP required for memory storage is the complex of CaMK signaling with the NMDA receptor (102). Furthermore, neurophysiological and in silico modeling studies are required to understand the neuromodulating effects of tDCS, and help in determining the optimal levels of anodal and cathodal stimulation that is beneficial for stroke patients.
Earlier study have reported the effects of rTMS and illustrated the mechanisms of rTMS in regulating cognitive capacity. rTMS enhances spatial memory behavior, neuron and synapse morphology in the hippocampus, and synaptic protein markers and BDNF/TrkB in normal aging mice (103). Aging mice inhibited the activation of BDNF–TrkB signaling pathway and showed hippocampal-dependent cognitive impairment in relative to adult animals, with the reduced transcription and expression of synaptic protein markers, such as growth-associated protein 43 (GAP43), synaptophysin (SYN), post-synaptic density protein 95 (PSD95), including decreased synapse density, and PSD thickness. Surprisingly, low-intensity rTMS (110% average resting motor threshold intensity, 1 Hz) triggered the activation of BDNF and TrkB, upregulated the level of synaptic protein markers, increased synapse density and PSD thickness, and reversed the spatial cognition dysfunction in aging mice. On the other hand, high-intensity rTMS (150% average resting motor threshold intensity, 1 Hz) was harmful, inducing reduction of PSDs thickness, disordered synaptic structure, as well as reduction in the number of synapses, and downregulation of BDNF–TrkB and synaptic proteins. The aging-induced cognitive deficits depends on the intensity level of TMS, which are closely associated with hippocampal structural synaptic plasticity that plays an important role in regulating cognitive behavior via changing structural synaptic plasticity through BDNF signaling. The effect of rTMS on functional recovery and its underlying molecular mechanism has been studied by assessing proteins associated with neural plasticity in a sub-acute ischemic rat model (104). A total of 3,500 impulses with 10 Hz frequency were applied to the ipsilesional cortex from post-operative day 4 over a 2-week period. The study reported that rTMS group showed more functional improvement on the beam balance test and the immunohistochemistry analysis showed stronger Bcl-2 and weaker Bax expression when compared with the sham group and noticed no significant difference in the expressions of NMDA and MAP-2 (104). However, rTMS may increase or decrease motor cortical excitability mostly depending on the characteristics of the stimulation protocol (105). Non-invasive cortical stimulation upregulates excitability in M1 lesioned hemisphere and downregulates in M1 intact hemisphere that could contribute to correcting abnormalities in inter-hemispheric inhibition identified after stroke (106). Thus, both the pharmacotherapy and neurorehabilitation therapy converge through the NMDA receptor activation.
One of the challenges for antibiotic therapy is the need to identify the optimum dose level. Recently, a reliable and feasible approach of dose estimation using chemical binding kinetics was proposed by Abel et al. (107). Another challenge is determining the impact of combinational multiple drug therapies on several proteins. A recent study suggested an approach using linear superposition of their responses to individual therapy (108). Utilizing these approaches, we can develop in silico models that could provide insight into the design of clinical trials of immunomodulatory therapies, ranging from optimal patient-specific dose selection, and duration of proposed therapeutic interventions (109).
In this paper, we utilize a systems biology approach to maneuver the dynamics of immunomodulation using linear combination of doses of pharmacotherapy (minocycline) in NF-κB signaling pathway along with adjuvant neurorehabilitation therapy (rTMS or tDCS) through BDNF and TrkB signaling pathway stimulation for enhancing synaptic plasticity, rehabilitation, and neurorestoration. Thus, a thorough understanding and modeling of pathways, along with the optimal therapeutic doses could lead us to prevent cell death at an earlier stage and with a higher chance of maintaining the long-term viability of the cell protection after stroke. To conclude, further studies are required to dissect, report, and analyze these pathways toward the goal of improving neurorestoration by multimodal therapy for stroke and related neurovascular disorders.
The research was conceived and planned by PR, jointly with inputs from MA and VR. The numerical experimentation was performed by VR and the biological studies investigated by MA. All authors examined and evaluated the data. VR, MA, and PR wrote the manuscript. All authors read and approved the final manuscript and have no conflict of interests.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Rajiv Ramaswamy (National Brain Research Centre, India) for proof reading and necessary language editing.
Glue – Grant Scheme and Tata Innovation Program, Department of Biotechnology, Government of India; Department of Information Technology, Government of India; and National Brain Research Centre for supporting this work.
1. Perry VH, Newman TA, Cunningham C. The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci (2003) 4:103–12. doi: 10.1038/nrn1032
2. Noble W, Garwood CJ, Hanger DP. Minocycline as a potential therapeutic agent in neurodegenerative disorders characterised by protein misfolding. Prion (2009) 3:78–83. doi:10.4161/pri.3.2.8820
3. Stirling DP, Koochesfahani KM, Steeves JD, Tetzlaff W. Minocycline as a neuroprotective agent. Neuroscientist (2005) 11:308–22. doi:10.1177/1073858405275175
4. Zhou H, Lapointe BM, Clark SR, Zbytnuik L, Kubes P. A requirement for microglial TLR4 in leukocyte recruitment into brain in response to lipopolysaccharide. J Immunol (2006) 177:8103–10. doi:10.4049/jimmunol.177.11.8103
5. Huang WC, Qiao Y, Xu L, Kacimi R, Sun X, Giffard RG, et al. Direct protection of cultured neurons from ischemia-like injury by minocycline. Anat Cell Biol (2010) 43:325–31. doi:10.5115/acb.2010.43.4.325
6. Dunston CR, Griffiths HR, Lambert PA, Staddon S, Vernallis AB. Proteomic analysis of the anti-inflammatory action of minocycline. Proteomics (2011) 11:42–51. doi:10.1002/pmic.201000273
7. Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG. Microglia potentiate damage to blood-brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke (2006) 37:1087–93. doi:10.1161/01.STR.0000206281.77178.ac
8. Qiu J, Nishimura M, Wang Y, Sims JR, Qiu S, Savitz SI, et al. Early release of HMGB-1 from neurons after the onset of brain ischemia. J Cereb Blood Flow Metab (2008) 28:927–38. doi:10.1038/sj.jcbfm.9600582
9. Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab (2011) 13:11–22. doi:10.1016/j.cmet.2010.12.008
10. Chauhan A, Sharma U, Jagannathan NR, Reeta KH, Gupta YK. Rapamycin protects against middle cerebral artery occlusion induced focal cerebral ischemia in rats. Behav Brain Res (2011) 225:603–9. doi:10.1016/j.bbr.2011.08.035
11. Carloni S, Girelli S, Scopa C, Buonocore G, Longini M, Balduini W. Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy (2010) 6:366–77. doi:10.4161/auto.6.3.11261
12. Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation (2007) 115:1599–608. doi:10.1161/CIRCULATIONAHA.106.603431
13. Ziegler G, Harhausen D, Schepers C, Hoffmann O, Röhr C, Prinz V, et al. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun (2007) 359:574–9. doi:10.1016/j.bbrc.2007.05.157
14. Wang LW, Tu YF, Huang CC, Ho CJ. JNK signaling is the shared pathway linking neuroinflammation, blood-brain barrier disruption, and oligodendroglial apoptosis in the white matter injury of the immature brain. J Neuroinflammation (2012) 9:175. doi:10.1186/1742-2094-9-175
15. Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, et al. Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med Res Rev (2001) 21:129–45. doi:10.1002/1098-1128(200103)21:2<129::AID-MED1003>3.0.CO;2-H
16. Wang X, Wang H, Xu L, Rozanski DJ, Sugawara T, Chan PH, et al. Significant neuroprotection against ischemic brain injury by inhibition of the MEK1 protein kinase in mice: exploration of potential mechanism associated with apoptosis. J Pharmacol Exp Ther (2003) 304:172–8. doi:10.1124/jpet.102.040246
17. Maddahi A, Edvinsson L. Enhanced expressions of microvascular smooth muscle receptors after focal cerebral ischemia occur via the MAPK MEK/ERK pathway. BMC Neurosci (2008) 9:85. doi:10.1186/1471-2202-9-85
18. Pang T, Wang J, Benicky J, Saavedra JM. Minocycline ameliorates LPS-induced inflammation in human monocytes by novel mechanisms including LOX-1, Nur77 and LITAF inhibition. Biochim Biophys Acta (2012) 1820:503–10. doi:10.1016/j.bbagen.2012.01.011
19. Qi ZF, Luo YM, Liu XR, Wang RL, Zhao HP, Yan F, et al. AKT/GSK3β-dependent autophagy contributes to the neuroprotection of limb remote ischemic postconditioning in the transient cerebral ischemic rat model. CNS Neurosci Ther (2012) 18:965–73. doi:10.1111/cns.12016
20. Gao X, Zhang H, Takahashi T, Hsieh J, Liao J, Steinberg GK, et al. The Akt signaling pathway contributes to postconditioning’s protection against stroke; the protection is associated with the MAPK and PKC pathways. J Neurochem (2008) 105:943–55. doi:10.1111/j.1471-4159.2008.05218.x
21. O’Riordan KJ, Huang IC, Pizzi M, Spano P, Boroni F, Egli R, et al. Regulation of nuclear factor κB in the hippocampus by group I metabotropic glutamate receptors. J Neurosci (2006) 2006(26):4870–9. doi:10.1523/JNEUROSCI.4527-05.2006
22. Switzer JA, Hess DC, Ergul A, Waller JL, Machado LS, Portik-Dobos V, et al. Matrix metalloproteinase-9 in an exploratory trial of intravenous minocycline for acute ischemic stroke. Stroke (2011) 42:2633–5. doi:10.1161/STROKEAHA.111.618215
23. Foteinou PT, Calvano SE, Lowry SF, Androulakis IP. In silico simulation of corticosteroids effect on an NFkB-dependent physicochemical model of systemic inflammation. PLoS One (2009) 4:e4706. doi:10.1371/journal.pone.0004706
24. Wang HY, Crupi D, Liu J, Stucky A, Cruciata G, Di Rocco A, et al. Repetitive transcranial magnetic stimulation enhances BDNF-TrkB signaling in both brain and lymphocyte. J Neurosci (2011) 31:11044–54. doi:10.1523/JNEUROSCI.2125-11.2011
25. Fritsch B, Reis J, Martinowich K, Schambra HM, Ji Y, Cohen LG, et al. Direct current stimulation promotes BDNF-dependent synaptic plasticity: potential implications for motor learning. Neuron (2010) 66:198–204. doi:10.1016/j.neuron.2010.03.035
26. Lipniacki T, Paszek P, Brasier AR, Luxon B, Kimmel M. Mathematical model of NF-kappaB regulatory module. J Theor Biol (2004) 228:195–215. doi:10.1016/j.jtbi.2004.01.001
27. Ulgen BO, Field MG, Qureshi W, Knight RA, Stephanou A, Latchman DS, et al. The role of minocycline in ischemia-reperfusion injury: a comprehensive review of an old drug with new implications. Recent Pat Cardiovasc Drug Discov (2011) 6:123–32. doi:10.2174/157489011795933783
28. Jain P, Bhalla US. Signaling logic of activity-triggered dendritic protein synthesis: an mTOR gate but not a feedback switch. PLoS Comput Biol (2009) 5:e1000287. doi:10.1371/journal.pcbi.1000287
29. Yuen EC, Mobley WC. Early BDNF, NT-3, and NT-4 signaling events. Exp Neurol (1999) 159:297–308. doi:10.1006/exnr.1999.7148
30. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol (1997) 7:261–9. doi:10.1016/S0960-9822(06)00122-9
31. Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev (2003) 17:1829–34. doi:10.1101/gad.1110003
32. Isotani S, Hara K, Tokunaga C, Inoue H, Avruch J, Yonezawa K. Immunopurified mammalian target of rapamycin phosphorylates and activates p70 S6 kinase alpha in vitro. J Biol Chem (1999) 274:34493–8. doi:10.1074/jbc.274.48.34493
33. Alessi DR, Kozlowski MT, Weng QP, Morrice N, Avruch J. 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr Biol (1998) 8:69–81. doi:10.1016/S0960-9822(98)70037-5
34. Worley PF, Baraban JM, Snyder SH. Inositol 1,4,5-trisphosphate receptor binding: autoradiographic localization in rat brain. J Neurosci (1989) 9:339–46.
35. Redpath NT, Price NT, Severinov KV, Proud CG. Regulation of elongation factor-2 by multisite phosphorylation. Eur J Biochem (1993) 213:689–99. doi:10.1111/j.1432-1033.1993.tb17809.x
36. Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J (2001) 20:4370–9. doi:10.1093/emboj/20.16.4370
37. Mitsui K, Brady M, Palfrey HC, Nairn AC. Purification and characterization of calmodulin-dependent protein kinase III from rabbit reticulocytes and rat pancreas. J Biol Chem (1993) 268:13422–33.
38. Parkhurst KM, Hileman RE, Saha D, Gupta NK, Parkhurst LJ. Thermodynamic characterization of the cooperativity of 40S complex formation during the initiation of eukaryotic protein synthesis. Biochemistry (1994) 33:15168–77. doi:10.1021/bi00254a028
39. Kafitz KW, Rose CR, Thoenen H, Konnerth A. Neurotrophin-evoked rapid excitation through TrkB receptors. Nature (1999) 401:918–21. doi:10.1038/44847
40. Atwal JK, Massie B, Miller FD, Kaplan DR. The TrkB-Shc site signals neuronal survival and local axon growth via MEK and P13-kinase. Neuron (2000) 27:265–77. doi:10.1016/S0896-6273(00)00035-0
41. Aksan I, Kurnaz ML. A computer-based model for the regulation of mitogen activated protein kinase (MAPK) activation. J Recept Signal Transduct Res (2003) 23:197–209. doi:10.1081/RRS-120025203
42. Hayakawa-Yano Y, Nishida K, Fukami S, Gotoh Y, Hirano T, Nakagawa T, et al. Epidermal growth factor signaling mediated by grb2 associated binder1 is required for the spatiotemporally regulated proliferation of olig2-expressing progenitors in the embryonic spinal cord. Stem Cells (2007) 25:1410–22. doi:10.1634/stemcells.2006-0584
43. Ma L, Janetopoulos C, Yang L, Devreotes PN, Iglesias PA. Two complementary, local excitation, global inhibition mechanisms acting in parallel can explain the chemoattractant-induced regulation of PI(3,4,5)P3 response in dictyostelium cells. Biophys J (2004) 87:3764–74. doi:10.1529/biophysj.104.045484
44. Chodniewicz D, Alteraifi AM, Zhelev DV. Experimental evidence for the limiting role of enzymatic reactions in chemoattractant-induced pseudopod extension in human neutrophils. J Biol Chem (2004) 279:24460–6. doi:10.1074/jbc.M312764200
45. Yamada KM, Araki M. Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis. J Cell Sci (2001) 114:2375–82.
46. Lee FS, Kim AH, Khursigara G, Chao MV. The uniqueness of being a neurotrophin receptor. Curr Opin Neurobiol (2001) 11:281–6. doi:10.1016/S0959-4388(00)00209-9
47. Barde YA. Neurobiology: neurotrophin channels excitement. Nature (2002) 419:683–4. doi:10.1038/419683a
48. Barnea A, Roberts J, Croll SD. Continuous exposure to brain-derived neurotrophic factor is required for persistent activation of TrkB receptor, the ERK signaling pathway, and the induction of neuropeptide Y production in cortical cultures. Brain Res (2004) 1020:106–17. doi:10.1016/j.brainres.2004.06.018
49. Obermeier A, Lammers R, Wiesmuller KH, Jung G, Schlessinger J, Ullrich A. Identification of Trk binding sites for SHC and phosphatidylinositol 3’-kinase and formation of a multimeric signaling complex. J Biol Chem (1993) 268:22963–6.
50. Laminet AA, Apell G, Conroy L, Kavanaugh WM. Affinity, specificity, and kinetics of the interaction of the SHC phosphotyrosine binding domain with asparagine-X-X-phosphotyrosine motifs of growth factor receptors. J Biol Chem (1996) 271:264–9. doi:10.1074/jbc.271.1.264
51. Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, et al. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell (2000) 103:931–43. doi:10.1016/S0092-8674(00)00196-3
52. Carpenter CL, Duckworth BC, Auger KR, Cohen B, Schaffhausen BS, Cantley LC. Purification and characterization of phosphoinositide 3-kinase from rat liver. J Biol Chem (1990) 265:19704–11.
53. Koh G, Teong HF, Clement MV, Hsu D, Thiagarajan PS. A decompositional approach to parameter estimation in pathway modeling: a case study of the Akt and MAPK pathways and their crosstalk. Bioinformatics (2006) 22:e271–80. doi:10.1093/bioinformatics/btl264
54. Alessi DR, Cohen P. Mechanism of activation and function of protein kinase B. Curr Opin Genet Dev (1998) 8:55–62. doi:10.1016/S0959-437X(98)80062-2
55. Barnett SF, Defeo-Jones D, Fu S, Hancock PJ, Haskell KM, Jones RE, et al. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem J (2005) 385:399–408. doi:10.1042/BJ20041140
56. Kikuchi S, Fujimoto K, Kitagawa N, Fuchikawa T, Abe M, Oka K, et al. Kinetic simulation of signal transduction system in hippocampal long-term potentiation with dynamic modeling of protein phosphatase 2A. Neural Netw (2003) 16:1389–98. doi:10.1016/j.neunet.2003.09.002
57. Geist RT, Gutmann DH. The tuberous sclerosis 2 gene is expressed at high levels in the cerebellum and developing spinal cord. Cell Growth Differ (1995) 6:1477–83.
58. James SR, Downes CP, Gigg R, Grove SJ, Holmes AB, Alessi DR. Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem J (1996) 315:709–13. doi:10.1042/bj3150709
59. Biondi RM, Cheung PC, Casamayor A, Deak M, Currie RA, Alessi DR. Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J (2000) 19:979–88. doi:10.1093/emboj/19.5.979
60. Gottschalk WA, Jiang H, Tartaglia N, Feng L, Figurov A, Lu B. Signaling mechanisms mediating BDNF modulation of synaptic plasticity in the hippocampus. Learn Mem (1999) 6:243–56.
61. Mai L, Jope RS, Li X. BDNF-mediated signal transduction is modulated by GSK3beta and mood stabilizing agents. J Neurochem (2002) 82:75–83. doi:10.1046/j.1471-4159.2002.00939.x
62. Dijkhuizen PA, Ghosh A. BDNF regulates primary dendrite formation in cortical neurons via the PI3-kinase and MAP kinase signaling pathways. J Neurobiol (2005) 62:278–88. doi:10.1002/neu.20100
63. Zu YL, Wu F, Gilchrist A, Ai Y, Labadia ME, Huang CK. The primary structure of a human MAP kinase activated protein kinase 2. Biochem Biophys Res Commun (1994) 200:1118–24. doi:10.1006/bbrc.1994.1566
64. Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, et al. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem (1995) 270:815–22. doi:10.1074/jbc.270.2.815
65. Jakubowicz T, Leader DP. Activation of a ribosomal protein S6 kinase in mouse fibroblasts during infection with herpesvirus. Eur J Biochem (1987) 168:371–6. doi:10.1111/j.1432-1033.1987.tb13429.x
66. Haystead TA, Haystead CM, Hu C, Lin TA, Lawrence JC Jr. Phosphorylation of PHAS-I by mitogen-activated protein (MAP) kinase. Identification of a site phosphorylated by MAP kinase in vitro and in response to insulin in rat adipocytes. J Biol Chem (1994) 269:23185–91.
67. Toral-Barza L, Zhang WG, Lamison C, Larocque J, Gibbons J, Yu K. Characterization of the cloned full-length and a truncated human target of rapamycin: activity, specificity, and enzyme inhibition as studied by a high capacity assay. Biochem Biophys Res Commun (2005) 332:304–10. doi:10.1016/j.bbrc.2005.04.117
68. Flotow H, Thomas G. Substrate recognition determinants of the mitogen-activated 70K S6 kinase from rat liver. J Biol Chem (1992) 267:3074–8.
69. Zhang WG, Shor B, Yu K. Identification and characterization of a constitutively T-loop phosphorylated and active recombinant S6K1: expression, purification, and enzymatic studies in a high capacity non-radioactive TR-FRET Lance assay. Protein Expr Purif (2006) 46:414–20. doi:10.1016/j.pep.2005.08.012
70. McKendrick L, Morley SJ, Pain VM, Jagus R, Joshi B. Phosphorylation of eukaryotic initiation factor 4E (eIF4E) at Ser209 is not required for protein synthesis in vitro and in vivo. Eur J Biochem (2001) 268:5375–85. doi:10.1046/j.0014-2956.2001.02478.x
71. Duncan R, Milburn SC, Hershey JW. Regulated phosphorylation and low abundance of HeLa cell initiation factor eIF-4F suggest a role in translational control. Heat shock effects on eIF-4F. J Biol Chem (1987) 262:380–8.
72. Eberwine J, Miyashiro K, Kacharmina JE, Job C. Local translation of classes of mRNAs that are targeted to neuronal dendrites. Proc Natl Acad Sci U S A (2001) 98:7080–5. doi:10.1073/pnas.121146698
73. Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci U S A (1998) 95:1432–7. doi:10.1073/pnas.95.4.1432
74. Karim MM, Hughes JM, Warwicker J, Scheper GC, Proud CG, McCarthy JE. A quantitative molecular model for modulation of mammalian translation by the eIF4E-binding protein 1. J Biol Chem (2001) 276:20750–7. doi:10.1074/jbc.M011068200
75. Ptushkina M, von der Haar T, Karim MM, Hughes JM, McCarthy JE. Repressor binding to a dorsal regulatory site traps human eIF4E in a high cap-affinity state. EMBO J (1999) 18:4068–75. doi:10.1093/emboj/18.14.4068
76. Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem (1999) 68:913–63. doi:10.1146/annurev.biochem.68.1.913
77. von der Haar T, Hughes JM, Manjarul Karim M, Ptushkina M, McCarthy JE. Translation initiation and surface plasmon resonance: new technology applied to old questions. Biochem Soc Trans (2002) 30:155–62. doi:10.1042/bst0300155
78. Scheper GC, van Kollenburg B, Hu J, Luo Y, Goss DJ, Proud CG. Phosphorylation of eukaryotic initiation factor 4E markedly reduces its affinity for capped mRNA. J Biol Chem (2002) 277:3303–9. doi:10.1074/jbc.M103607200
79. Marin P, Nastiuk KL, Daniel N, Girault JA, Czernik AJ, Glowinski J, et al. Glutamate-dependent phosphorylation of elongation factor-2 and inhibition of protein synthesis in neurons. J Neurosci (1997) 17:3445–54.
80. Ryazanov AG. Ca2+/calmodulin-dependent phosphorylation of elongation factor 2. FEBS Lett (1987) 214:331–4. doi:10.1016/0014-5793(87)80081-9
81. Fagan SC, Waller JL, Nichols FT, Edwards DJ, Pettigrew LC, Clark WM, et al. Minocycline to improve neurologic outcome in stroke (MINOS): a dose-finding study. Stroke (2010) 41:2283–7. doi:10.1161/STROKEAHA.110.582601
82. Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J (2003) 17:2115–7. doi:10.1096/fj.03-0329fje
83. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol (2011) 31:986–1000. doi:10.1161/ATVBAHA.110.207449
84. Lee SM, Yune TY, Kim SJ, Kim YC, Oh YJ, Markelonis GJ, et al. Minocycline inhibits apoptotic cell death via attenuation of TNF-alpha expression following iNOS/NO induction by lipopolysaccharide in neuron/glia co-cultures. J Neurochem (2004) 91:568–78. doi:10.1111/j.1471-4159.2004.02780.x
85. Liu T, Clark RK, McDonnell PC, Young PR, White RF, Barone FC, et al. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke (1994) 25:1481–8. doi:10.1161/01.STR.25.7.1481
86. Siao CJ, Fernandez SR, Tsirka SE. Cell type-specific roles for tissue plasminogen activator released by neurons or microglia after excitotoxic injury. J Neurosci (2003) 23:3234–42. doi: 0270-6474/03/233234-09$15.00/0
87. Gasche Y, Fujimura M, Morita-Fujimura Y, Copin JC, Kawase M, Massengale J, et al. Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: a possible role in blood-brain barrier dysfunction. J Cereb Blood Flow Metab (1999) 19:1020–8. doi:10.1097/00004647-199909000-00010
88. Clark AW, Krekoski CA, Bou SS, Chapman KR, Edwards DR. Increased gelatinase A (MMP-2) and gelatinase B (MMP-9) activities in human brain after focal ischemia. Neurosci Lett (1997) 238:53–6. doi:10.1016/S0304-3940(97)00859-8
89. Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY, et al. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci (2005) 25:6401–8. doi:10.1523/JNEUROSCI.1563-05.2005
90. Lapchak PA, Chapman DF, Zivin JA. Metalloproteinase inhibition reduces thrombolytic (tissue plasminogen activator)-induced hemorrhage after thromboembolic stroke. Stroke (2000) 31:3034–40. doi:10.1161/01.STR.31.12.3034
91. Machado LS, Kozak A, Ergul A, Hess DC, Borlongan CV, Fagan SC. Delayed minocycline inhibits ischemia-activated matrix metalloproteinases 2 and 9 after experimental stroke. BMC Neurosci (2006) 7:56. doi:10.1186/1471-2202-7-56
92. Blum D, Chtarto A, Tenenbaum L, Brotchi J, Levivier M. Clinical potential of minocycline for neurodegenerative disorders. Neurobiol Dis (2004) 17:359–66. doi:10.1016/j.nbd.2004.07.012
93. Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C, et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol (2007) 6:1045–53. doi:10.1016/S1474-4422(07)70270-3
94. Lampl Y, Boaz M, Gilad R, Lorberboym M, Dabby R, Rapoport A, et al. Minocycline treatment in acute stroke: an open-label, evaluator-blinded study. Neurology (2007) 69:1404–10. doi:10.1212/01.wnl.0000277487.04281.db
95. Srivastava MVP, Bhasin A, Bhatia R, Garg A, Gaikwad S, Prasad K, et al. Efficacy of minocycline in acute ischemic stroke: a single-blinded, placebo-controlled trial. Neurol India (2012) 60:23–8. doi:10.4103/0028-3886.93584
96. Nitsche M, Antal A, Paulus W. Induction and modulation of neuroplasticity by transcranial direct current stimulation, transcranial brain stimulation for treatment of psychiatric disorders. In: Marcolin MA, Padberg F, editors. Advances in Biological Psychiatry Basel. (Vol. 23), Munich: Karger (2007). p. 172–86.
97. Mahmoudi H, Borhani Haghighi A, Petramfar P, Jahanshahi S, Salehi Z, Fregni F. Transcranial direct current stimulation: electrode montage in stroke. Disabil Rehabil (2011) 33:1383–8. doi:10.3109/09638288.2010.532283
98. Zhang SX, Zhang JP, Fletcher DL, Zoeller RT, Sun GY. In situ hybridization of mRNA expression for IP3 receptor and IP3-3-kinase in rat brain after transient focal cerebral ischemia. Brain Res Mol Brain Res (1995) 32:252–60. doi:10.1016/0169-328X(95)00085-7
99. Sun GY, Zhang JP, Lin TA, Lin TN, He YY, Hsu CY. Inositol trisphosphate, polyphosphoinositide turnover, and high-energy metabolites in focal cerebral ischemia and reperfusion. Stroke (1995) 26:1893–900. doi:10.1161/01.STR.26.10.1893
100. Nizar K, Uhlirova H, Tian P, Saisan PA, Cheng Q, Reznichenko L, et al. In vivo stimulus-induced vasodilation occurs without IP3 receptor activation and may precede astrocytic calcium increase. J Neurosci (2013) 33:8411–22. doi:10.1523/JNEUROSCI.3285-12.2013
101. Lu Y, Christian K, Lu B. BDNF: a key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol Learn Mem (2008) 89:312–23. doi:10.1016/j.nlm.2007.08.018
102. Sanhueza M, Lisman J. The CaMKII/NMDAR complex as a molecular memory. Mol Brain (2013) 6:10. doi:10.1186/1756-6606-6-10
103. Ma J, Zhang Z, Kang L, Geng D, Wang Y, Wang M, et al. Repetitive transcranial magnetic stimulation (rTMS) influences spatial cognition and modulates hippocampal structural synaptic plasticity in aging mice. Exp Gerontol (2014) 58:256–68. doi:10.1016/j.exger.2014.08.011
104. Yoon KJ, Lee YT, Han TR. Mechanism of functional recovery after repetitive transcranial magnetic stimulation (rTMS) in the subacute cerebral ischemic rat model: neural plasticity or anti-apoptosis? Exp Brain Res (2011) 214:549–56. doi:10.1007/s00221-011-2853-2
105. Di Lazzaro V, Profice P, Pilato F, Dileone M, Oliviero A, Ziemann U. The effects of motor cortex rTMS on corticospinal descending activity. Clin Neurophysiol (2010) 121:464–73. doi:10.1016/j.clinph.2009.11.007
106. Hummel FC, Cohen LG. Non-invasive brain stimulation: a new strategy to improve neurorehabilitation after stroke? Lancet Neurol (2006) 5:708–12. doi:10.1016/S1474-4422(06)70525-7
107. Abel Zur, Wiesch P, Abel S, Gkotzis S, Ocampo P, Engelstädter J, et al. Classic reaction kinetics can explain complex patterns of antibiotic action. Sci Transl Med (2015) 7:287ra273. doi:10.1126/scitranslmed.aaa8760
108. Geva-Zatorsky N, Dekel E, Cohen AA, Danon T, Cohen L, Alon U. Protein dynamics in drug combinations: a linear superposition of individual-drug responses. Cell (2010) 140:643–51. doi:10.1016/j.cell.2010.02.011
109. Clermont G, Bartels J, Kumar R, Constantine G, Vodovotz Y, Chow C. In silico design of clinical trials: a method coming of age. Crit Care Med (2004) 32:2061–70. doi:10.1097/01.CCM.0000142394.28791.C3

Table A1. Chemical species for Module 1.

Table A2. The following association and dissociation rate constants for module I are taken from Ref. (23, 26).

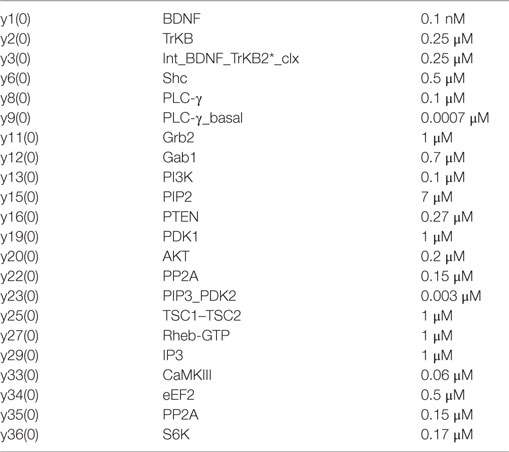
Table A3. Chemical species and initial concentration for Module 2.

Table A4. The following association and dissociation rate constants for Module 2 are taken from Ref. (29–33, 37, 39–80).

Module 1. Pharmacotherapy (minocycline) activation through TNF-α in NF-κB signaling pathway.

Module 2. We now formulate the ordinary differential equations for the above chemical reactions (Eqs 1–37).
Keywords: stroke, neuroprotection, rehabilitation, minocycline, direct current stimulation, transcranial magnetic stimulation
Citation: Alam MA, Subramanyam Rallabandi VP and Roy PK (2016) Systems Biology of Immunomodulation for Post-Stroke Neuroplasticity: Multimodal Implications of Pharmacotherapy and Neurorehabilitation. Front. Neurol. 7:94. doi: 10.3389/fneur.2016.00094
Received: 22 January 2016; Accepted: 07 June 2016;
Published: 28 June 2016
Edited by:
Anirban Dutta, Leibniz-Institut für Arbeitsforschung an der TU Dortmund, GermanyReviewed by:
Raju Surampudi Bapi, University of Hyderabad, IndiaCopyright: © 2016 Alam, Subramanyam Rallabandi and Roy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prasun K. Roy, pkroy@nbrc.ac.in
†Equal first authors.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.