 Rajesh Narasimamurthy
Rajesh Narasimamurthy David M. Virshup
David M. Virshup- Program in Cancer and Stem Cell Biology, Duke-NUS Medical School, Singapore, Singapore
An approximately 24-h biological timekeeping mechanism called the circadian clock is present in virtually all light-sensitive organisms from cyanobacteria to humans. The clock system regulates our sleep–wake cycle, feeding–fasting, hormonal secretion, body temperature, and many other physiological functions. Signals from the master circadian oscillator entrain peripheral clocks using a variety of neural and hormonal signals. Even centrally controlled internal temperature fluctuations can entrain the peripheral circadian clocks. But, unlike other chemical reactions, the output of the clock system remains nearly constant with fluctuations in ambient temperature, a phenomenon known as temperature compensation. In this brief review, we focus on recent advances in our understanding of the posttranslational modifications, especially a phosphoswitch mechanism controlling the stability of PER2 and its implications for the regulation of temperature compensation.
The main advantage of having an intact circadian clock system is to anticipate and alert our physiological mechanisms to prepare for daily changes in the environment imposed by light–dark cycle of the earth. At the organism level, the circadian clock is a hierarchical multioscillator network, where in mammals, the suprachiasmatic nuclei (SCN) is the master oscillator. The SCN in the hypothalamus of brain is entrained by the light–dark cycle through the eye and neuronal retinal ganglion cells. Synchronized highly interconnected neurons in the SCN oscillate and transmit their rhythm to peripheral oscillators such as liver, lung, and kidney via systemic cues including neuronal, neuroendocrine, and behavioral pathways. This clock network entrains physiological processes including the sleep–wake cycle, liver metabolism, and body temperature (1–3). At the molecular level, the circadian clock is composed of transcriptional and translational feedback loops that oscillate in cycles of approximately 24-h to create the circadian rhythms we see at the organism level. In the core loop, the positive transcriptional activators Clock and Bmal1 bind to E-box motifs and activate the expression of many targets, including their own negative regulators, Period (Per1, 2, and 3) and Cryptochromes (Cry1 and Cry2). As the negative feedback proteins Per and Cry increase in abundance, they multimerize, enter into the nucleus, and bind to the heterodimeric Clock and Bmal1 complex to inhibit their transcriptional activity. This generates a 24-h cycle that is cell autonomous. This clock machinery is broadly functional in all mammalian tissues (1–3).
The three major hallmarks of circadian clocks are their ~24-h oscillation in the absence of any external stimuli, entrainment by external stimuli, and temperature compensation. Entrainment allows the master clock to synchronize with seasonally and geographically changing light–dark cycles. In mammals, light entrains the central clock via retinal ganglion cells that communicate with the SCN via the retinal–hypothalamic tract. Homeothermic animals such as mammals maintain a nearly constant body temperature with a narrow range of fluctuations in most part of the body, whereas poikilotherms such as frogs have body temperature, which can vary in wide range (4, 5). However, even in mammals, peripheral clocks can be entrained by small daily oscillations in internal body temperature (1, 3, 6).
Although the circadian clock system can be entrained by fluctuations in temperature, it remains fairly resistant to ambient temperature-induced changes in circadian period (5, 7). According to the Arrhenius equation of temperature dependence on reaction rate, in any (bio)chemical reaction, a rise in temperature increases the rate of the reaction (8), which eventually reduces the reaction time. But in the case of the circadian biochemical system, in spite of changes in ambient temperature, the period length remains essentially constant at approximately 24-h. Thus, Pittendrigh demonstrated that the Drosophila rhythm of eclosion (emergence of the adult fly from the pupa) retained a 24-h rhythmicity in total darkness over a temperature range of 16–26°C (5). This phenomenon is referred to as temperature compensation (5, 9). The temperature compensation of circadian period is evolutionarily conserved from light-sensitive cyanobacteria to homeothermic mammals, and surprisingly, even an in vitro circadian clock reconstituted with KaiABC proteins of cyanobacteria shows temperature compensation between 25 and 35°C, suggesting that it is a core design feature of the molecular clock (5, 10–13). More recently, using tissue explants and cell culture, it has been demonstrated that temperature compensation is a tissue and cell autonomous property. For example, the circadian oscillators controlling melatonin synthesis in the retina of golden hamsters are temperature compensated between 27 and 33°C (14), and Per1Luc fibroblasts maintain ~24-h period length despite changes in temperature over the range of 28.5–36.5°C (12). These findings also confirm peripheral cells as bona fide model systems to study the temperature compensation mechanism of the circadian clock (12).
Models of Temperature Compensation
How the active process of temperature compensation is achieved by organisms is an area of intense research interest to both chronobiologists and mathematical modelers. Hastings and Sweeney almost 60 years ago proposed that temperature compensation could be achieved if two temperature-dependent reactions oppose each other, although at the time there was no inkling of what those reactions might be (9). This conceptual model was extended by Ruoff with the notion that positive and negative feedback loops of the oscillators might act as the opposing reactions and lead to temperature compensation in any kinetic oscillator model (15). As specific molecular members of the clock were identified, Hong et al. first proposed that PER protein dimerization might regulate temperature compensation (16). Ten years later, as the complexity of the clock mechanism became clearer, many of the newly described regulatory steps have been tested in mathematical models of the clock to assess their potential contribution to temperature compensation. For example, Hong et al. suggested that switch-like mechanisms acting on sensitive parameters such as phosphorylation, ubiquitination, or complex formation controlling PER protein might regulate temperature compensation (17). Others have suggested using modeling that the concentration of a rate-limiting enzyme involved in processes like phosphorylation can determine temperature compensation (18).
In Neurospora, the core clock gene frequency (frq) undergoes alternative splicing that is temperature sensitive. The resulting two isoforms have opposing effects on clock speed and was once proposed to underlie temperature compensation (19–21). More recently, casein kinase 2 in Neurospora was implicated in temperature compensation. Decreased CK2 activity, or mutation of a specific CK2 phosphorylation site, leads to altered temperature compensation, probably due to an altered balance of phosphorylation at distinct sites. Interestingly, CK2 itself had a normal Q10, i.e., its activity changed twofold with a 10°C increase in temperature (22). In this system, casein kinase 1 (CK1) was important for clock speed but not temperature compensation. Although these studies have provided some insights for understanding the mechanisms of temperature compensation, either they lack good experimental evidence to support their mathematical model or these models are not tested in mammalian system.
Phosphorylation of PER2 Controls Clock Speed
Many of the mathematical models suggested that temperature compensation could be due to two opposing reactions acting on a rate-limiting step of the circadian clock machinery (9, 18). The reversible multisite phosphorylation of PER2 is a potential target in this regard due to its rate-limiting role in regulating clock speed (Figure 1) (23, 24). The importance of phosphorylation in the control of circadian rhythms was demonstrated first by the finding of short- and long-period mutations in Drosophila that both mapped to the Dbt kinase gene, the ortholog of mammalian CK1δ and CK1ε (25, 26). CK1 is a family of serine/threonine kinases with seven different isoforms in mammals that are encoded by distinct genes (α, β, γ1, γ2, γ3, δ, and ε), which are involved in diverse biological functions including circadian rhythms, Wnt signaling, membrane trafficking, cytoskeleton maintenance, DNA replication, DNA damage response, RNA metabolism, and parasitic infections (23, 27–30). The first circadian clock phenotype in mammals was found in tau hamsters with 20-h short period (31). Later, it was identified that a missense mutation in hamster CK1εtau(Arg178Cys) is to underlie the short-period phenotype of the tau hamster (32). Subsequently, point mutation of a CK1δ/ε-regulated motif in human PER2 [S662G, familial advanced sleep phase (FASP) site] (33) and a point mutation of CK1δ were found in families with FASP syndrome (34). A body of evidence suggests that CK1δ is the major driver of clock timing, but that CK1ε plays an important role as well.
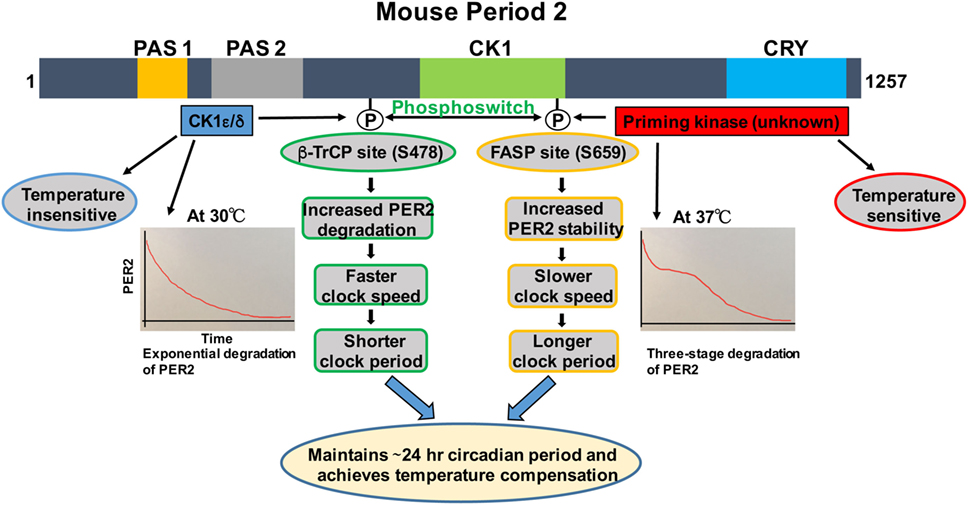
Figure 1. Regulation of PER2 phosphorylation, degradation, and its role in temperature compensation by the phosphoswitch mechanism. Lower temperature increases relative phosphorylation at the β-TrCP site of PER2, leading to faster degradation and shorter period. Higher temperature increases relative familial advanced sleep phase (FASP) site phosphorylation, enhancing PER2 stability and lengthening the period. The degradation pattern of PER2 at 30°C is largely exponential, while at 37°C, three-phase degradation is seen. This has important implications for temperature compensation (see text for details). Domain architectures are shown in colors. PAS1, PAS domain 1 (orange); PAS 2, PAS domain 2 (grey); CK1, Casein kinase 1-binding domain (green); CRY, Cry binding site (blue).
The mechanism by which CK1 regulates phosphorylation of PER2 is complex and is slowly being teased apart. Phosphorylation of PER2 by CK1ε leads to recruitment of the ubiquitin ligase, β-TrCP, and proteasomal-mediated degradation of PER2 (35). But the impact of CK1ε activity on the clock speed has been puzzling, due to opposing observations that reduced CK1 activity shorten (32, 34) and lengthen the circadian period (35). To solve this puzzle, mathematical modeling was applied and then experimentally confirmed the non-intuitive prediction that the short-period tau mutation of CK1ε is in fact functionally a gain of function, not a loss of function mutation. It was further reported that the CK1εtau is a highly specific gain of function for its substrate PER2, which gets phosphorylated and degraded much faster, resulting in a faster clock and shorter circadian period (36). These studies emphasized the value of combining experimental studies with predictive mathematical models to advance our understanding of the clock and how changes in kinase activity can alter the clock.
A Phosphoswitch Regulates PER2 Degradation
We and others have shown that there are two phosphorylation sites, the FASP and the β-TrCP site, regulating stability of mammalian PER2 (Figure 1) (35, 37). The FASP site is a missense mutation at S662G (S659 in mouse) associated with FASPS, which prevents priming phosphorylation by an unknown priming kinase. Priming phosphorylation of S659 (FASP site) is required for the phosphorylation of four immediate downstream serines of PER2 (659-SVVSLTSQCSYSS-671) by CK1ε/δ (33, 37). The second functional phosphorylation site is β-TrCP site that is also a CK1ε-dependent phosphorylation site (S478 in mPER2), but that seems to be independent of priming phosphorylation (35). It has been identified that surprisingly PER2 undergoes three distinct stages of degradation upon addition of the protein synthesis inhibitor cycloheximide during the PER2 accumulation phase (CT 14–26) of the circadian cycle. Mathematical modeling predicts that a phosphoswitch generates the three-stage degradation of PER2 (38). Accordingly, the first rapid decay phase is β-TrCP site phosphorylation dependent, the second slow plateau phase is dependent on FASP site phosphorylation, and in the third and falling phase, PER2 protein is degraded in a CK1δ/ε-independent manner that is not well understood. Importantly, the model was experimentally confirmed (38). Further experiments showed that CK1εtau has decreased activity on the FASP site, leading to an increased activity on the β-TrCP (S478) site. This explains how CK1εtau is a gain of function on phosphorylation at S478 and further supports the phosphoswitch between the two sites (the FASP and the β-TrCP site) (38).
Period2 Phosphoswitch Unravels the Mechanism of Temperature Compensation
Before CK1ε was even identified as a clock component, its role in temperature compensation was suggested by the observation that retinas from tau mutant hamsters have significantly impaired temperature compensation (14). Isojima et al. subsequently reported that unlike virtually all other kinases, CK1ε/δ are temperature insensitive (39). Therefore, they proposed that CK1ε/δ-dependent phosphorylation process might play a central role in temperature compensation of the circadian clock (39). Indeed, in further study, the CK1ε/δ phosphorylation of a β-TrCP peptide was temperature insensitive (39). The mathematical model of Kim and Forger, building on the pioneering work of Forger and Peskin in understanding the mammalian clock system using mathematical tools (40–42), predicted a potential role for the phosphoswitch mechanism in temperature compensation. A key feature of the model requires that there are two sites involved in the phosphoswitch, the FASP and the β-TrCP sites (Figure 1) (38). Since CK1 is relatively temperature insensitive (39), the model assume that priming of the FASP site has normal temperature sensitivity, i.e., its activity increases with increasing temperature, while CK1ε/δ phosphorylation of the β-TrCP site is temperature insensitive, i.e., the rate of phosphorylation is constant regardless of temperature. Incorporating this differential kinase temperature sensitivity into the mathematical model indeed predicted that this could underlie temperature compensation. This model was then experimentally tested in immortalized Per2Luc mouse embryonic fibroblasts (MEFs). It was found that at higher temperatures, increased FASP site phosphorylation by the priming kinase leads to slow second-phase degradation and more accumulation of PER2, eventually lengthening and compensating period length. Similarly, Per2Luc MEFs at 30°C showed a marked decrease in second-phase degradation, whereas first-stage degradation remained intact. These findings underscore the importance of the relative rates of phosphorylation of the two phosphoswitch sites in temperature compensation (38). Additional experiments indirectly tested if an intact phosphoswitch mechanism is necessary for temperature overcompensation. An abnormal temperature compensation was observed in CK1εtau; Per2Luc MEFs, and also in Per2Luc MEFs treated with a CK1ε/δ inhibitor, further supporting a role for CK1ε/δ and an intact phosphoswitch mechanism as a prerequisite for temperature compensation. The studies also support the value of a robust mathematical model that makes testable predictions about complex systems when biological intuition has reached its limits (38).
Recently, it has been reported that cells with knockouts of specific circadian clock components retain temperature compensation (43). The authors concluded that temperature compensation is likely determined by a rate-limiting process(es) that are temperature sensitive, consistent with the phosphoswitch mechanism (43). Another mathematical model for temperature compensation has recently proposed a temperature insulation mechanism where oscillation period is determined by very few temperature-independent or only slightly temperature-dependent parameters, but where other parameters remain strongly temperature dependent (44). This model is analogous to the proposed phosphoswitch mechanism in which the CK1ε/δ is temperature independent or slightly dependent, whereas the priming kinase is temperature dependent (38).
There are a number of unresolved issues. The priming kinase has not been identified yet. It also remains unclear what happens to PER2 phosphorylation over the full 24-h day, in part because the methods to study this in mammalian systems are not suitably sensitive. This is relevant to another unsolved question: how PER2 is degraded in the third phase of three phase decay, when neither CK1 nor proteasome inhibitors impact PER2 loss? Moreover, further study is necessary to understand whether fluctuations in body temperature, which can entrain the clock, do so in part via the phosphoswitch mechanism in addition to the proposed heat shock factor 1 (HSF1) mechanism (7, 45). Finally, it is also important to address whether the mechanisms regulating temperature compensation in peripheral cells and the central pacemaker (SCN) cells are the same and whether temperature-induced changes in peripheral clocks can feed back to the central clock.
The Outlook
It is remarkable that the complex yet robust phenomenon of temperature compensation is regulated by subtle differences in phosphorylation of the same protein at different sites. Notably, this finding is in general agreement with predictions of earlier mathematical models that suggested that opposing outputs with switch-like mechanisms might control temperature compensation (9, 17). In the future, it will be important to identify the priming kinase that plays a central role in the phosphoswitch model. This phosphoswitch mechanism of temperature compensation may be a core feature of clocks in many species, as a similar interaction of phosphorylation sites is operative in Drosophila and Neurospora as well (46–48).
Author Contributions
RN and DV were involved in conceptualizing, researching and writing this review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful for the support by the National Medical Research Council, Singapore.
References
1. Takahashi JS, Hong H-K, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet (2008) 9:764–75. doi: 10.1038/nrg2430
2. Liu AC, Lewis WG, Kay SA. Mammalian circadian signaling networks and therapeutic targets. Nat Chem Biol (2007) 3:630–9. doi:10.1038/nchembio.2007.37
3. Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol (2010) 72:517–49. doi:10.1146/annurev-physiol-021909-135821
4. Refinetti R. The circadian rhythm of body temperature. Front Biosci (Landmark Ed) (2010) 15:564–94. doi:10.2741/3634
5. Pittendrigh CS. On temperature independence in the clock system controlling emergence time in Drosophila. Proc Natl Acad Sci U S A (1954) 40:1018–29. doi:10.1073/pnas.40.10.1018
6. Edgar RS, Green EW, Zhao Y, van Ooijen G, Olmedo M, Qin X, et al. Peroxiredoxins are conserved markers of circadian rhythms. Nature (2012) 485:459–64. doi:10.1038/nature11088
7. Buhr ED, Yoo S-H, Takahashi JS. Temperature as a universal resetting cue for mammalian circadian oscillators. Science (2010) 330:379–85. doi:10.1126/science.1195262
8. Arrhenius S. Ueber die Reaktionsgeschwindigkeit bei der Inversion von Rohrzucker durch Saeuren. Zeitschrift fuer physikalische Chemie (1889) 4:226–48.
9. Hastings JW, Sweeney BM. On the mechanism of temperature independence in a biological clock. Proc Natl Acad Sci U S A (1957) 43:804–11. doi:10.1073/pnas.43.9.804
10. Zimmerman WF, Pittendrigh CS, Pavlidis T. Temperature compensation of the circadian oscillation in Drosophila pseudoobscura and its entrainment by temperature cycles. J Insect Physiol (1968) 14:669–84. doi:10.1016/0022-1910(68)90226-6
11. Ishiura M, Kutsuna S, Aoki S, Iwasaki H, Andersson CR, Tanabe A, et al. Expression of a gene cluster kaiABC as a circadian feedback process in cyanobacteria. Science (1998) 281:1519–23. doi:10.1126/science.281.5382.1519
12. Izumo M, Johnson CH, Yamazaki S. Circadian gene expression in mammalian fibroblasts revealed by real-time luminescence reporting: temperature compensation and damping. Proc Natl Acad Sci U S A (2003) 100:16089–94. doi:10.1073/pnas.2536313100
13. Nakajima M, Imai K, Ito H, Nishiwaki T, Murayama Y, Iwasaki H, et al. Reconstitution of circadian oscillation of cyanobacterial KaiC phosphorylation in vitro. Science (2005) 308:414–5. doi:10.1126/science.1108451
14. Tosini G, Menaker M. The tau mutation affects temperature compensation of hamster retinal circadian oscillators. Neuroreport (1998) 9:1001–5. doi:10.1097/00001756-199804200-00009
15. Ruoff P. Introducing temperature – compensation in any reaction kinetic oscillator model. J Interdiscipl Cycle Res (1992) 23:92–9. doi:10.1080/09291019209360133
16. Hong CI, Tyson JJ. A proposal for temperature compensation of the circadian rhythm in Drosophila based on dimerization of the per protein. Chronobiol Int (1997) 14:521–9. doi:10.3109/07420529709001473
17. Hong CI, Conrad ED, Tyson JJ. A proposal for robust temperature compensation of circadian rhythms. Proc Natl Acad Sci U S A (2007) 104:1195–200. doi:10.1073/pnas.0601378104
18. Hatakeyama TS, Kaneko K. Generic temperature compensation of biological clocks by autonomous regulation of catalyst concentration. Proc Natl Acad Sci U S A (2012) 109:8109–14. doi:10.1073/pnas.1120711109
19. Colot HV, Loros JJ, Dunlap JC. Temperature-modulated alternative splicing and promoter use in the Circadian clock gene frequency. Mol Biol Cell (2005) 16:5563–71. doi:10.1091/mbc.E05-08-0756
20. Akman OE, Locke JC, Tang S, Carré I, Millar AJ, Rand DA. Isoform switching facilitates period control in the Neurospora crassa circadian clock. Mol Syst Biol (2008) 4:164. doi:10.1038/msb.2008.5
21. Diernfellner A, Colot HV, Dintsis O, Loros JJ, Dunlap JC, Brunner M. Long and short isoforms of Neurospora clock protein FRQ support temperature-compensated circadian rhythms. FEBS Lett (2007) 581:5759–64. doi:10.1016/j.febslet.2007.11.043
22. Mehra A, Shi M, Baker CL, Colot HV, Loros JJ, Dunlap JC. A role for casein kinase 2 in the mechanism underlying circadian temperature compensation. Cell (2009) 137:749–60. doi:10.1016/j.cell.2009.03.019
23. Gallego M, Virshup DM. Post-translational modifications regulate the ticking of the circadian clock. Nat Rev Mol Cell Biol (2007) 8:139–48. doi:10.1038/nrm2106
24. Hirano A, Fu Y-H, Ptáček LJ. The intricate dance of post-translational modifications in the rhythm of life. Nat Struct Mol Biol (2016) 23:1053–60. doi:10.1038/nsmb.3326
25. Kloss B, Price JL, Saez L, Blau J, Rothenfluh A, Wesley CS, et al. The Drosophila clock gene double-time encodes a protein closely related to human casein kinase Iepsilon. Cell (1998) 94:97–107. doi:10.1016/S0092-8674(00)81225-8
26. Price JL, Blau J, Rothenfluh A, Abodeely M, Kloss B, Young MW. Double-time is a novel Drosophila clock gene that regulates PERIOD protein accumulation. Cell (1998) 94:83–95. doi:10.1016/S0092-8674(00)81224-6
27. Knippschild U, Gocht A, Wolff S, Huber N, Löhler J, Stöter M. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal (2005) 17:675–89. doi:10.1016/j.cellsig.2004.12.011
28. Liu J, Carvalho LP, Bhattacharya S, Carbone CJ, Kumar KG, Leu NA, et al. Mammalian casein kinase 1alpha and its leishmanial ortholog regulate stability of IFNAR1 and type I interferon signaling. Mol Cell Biol (2009) 29:6401–12. doi:10.1128/MCB.00478-09
29. Price MA. CKI, there’s more than one: casein kinase I family members in Wnt and Hedgehog signaling. Genes Dev (2006) 20:399–410. doi:10.1101/gad.1394306
30. Cheong JK, Virshup DM. Casein kinase 1: complexity in the family. Int J Biochem Cell Biol (2011) 43:465–9. doi:10.1016/j.biocel.2010.12.004
31. Ralph MR, Menaker M. A mutation of the circadian system in golden hamsters. Science (1988) 241:1225–7. doi:10.1126/science.3413487
32. Lowrey PL, Shimomura K, Antoch MP, Yamazaki S, Zemenides PD, Ralph MR, et al. Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science (2000) 288:483–92. doi:10.1126/science.288.5465.483
33. Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, et al. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science (2001) 291:1040–3. doi:10.1126/science.1057499
34. Xu Y, Padiath QS, Shapiro RE, Jones CR, Wu SC, Saigoh N, et al. Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature (2005) 434:640–4. doi:10.1038/nature03453
35. Eide EJ, Woolf MF, Kang H, Woolf P, Hurst W, Camacho F, et al. Control of mammalian circadian rhythm by CKI-regulated proteasome-mediated PER2 degradation. Mol Cell Biol (2005) 25:2795–807. doi:10.1128/MCB.25.7.2795-2807.2005
36. Gallego M, Eide EJ, Woolf MF, Virshup DM, Forger DB. An opposite role for tau in circadian rhythms revealed by mathematical modeling. Proc Natl Acad Sci U S A (2006) 103:10618–23. doi:10.1073/pnas.0604511103
37. Shanware NP, Hutchinson JA, Kim SH, Zhan L, Bowler MJ, Tibbetts RS. Casein Kinase 1-dependent phosphorylation of familial advanced sleep phase syndrome-associated residues controls PERIOD 2 stability. J Biol Chem (2011) 286:12766–74. doi:10.1074/jbc.M111.224014
38. Zhou M, Kim JK, Eng GW, Forger DB, Virshup DM. A period2 phosphoswitch regulates and temperature compensates circadian period. Mol Cell (2015) 60:77–88. doi:10.1016/j.molcel.2015.08.022
39. Isojima Y, Nakajima M, Ukai H, Fujishima H, Yamada RG, Masumoto KH, et al. CKIepsilon/delta-dependent phosphorylation is a temperature-insensitive, period-determining process in the mammalian circadian clock. Proc Natl Acad Sci U S A (2009) 106:15744–9. doi:10.1073/pnas.0908733106
40. Forger DB, Peskin CS. Model based conjectures on mammalian clock controversies. J Theor Biol (2004) 230:533–9. doi:10.1016/j.jtbi.2004.04.041
41. Forger DB, Peskin CS. A detailed predictive model of the mammalian circadian clock. Proc Natl Acad Sci U S A (2003) 100:14806–11. doi:10.1073/pnas.2036281100
42. Forger DB, Peskin CS. Stochastic simulation of the mammalian circadian clock. Proc Natl Acad Sci U S A (2005) 102:321–4. doi:10.1073/pnas.0408465102
43. Tsuchiya Y, Umemura Y, Minami Y, Koike N, Hosokawa T, Hara M, et al. Effect of multiple clock gene ablations on the circadian period length and temperature compensation in mammalian cells. J Biol Rhythms (2016) 31:48–56. doi:10.1177/0748730415613888
44. Wu L, Ouyang Q, Wang H. Robust network topologies for generating oscillations with temperature-independent periods. PLoS One (2017) 12:e0171263. doi:10.1371/journal.pone.0171263
45. Saini C, Morf J, Stratmann M, Gos P, Schibler U. Simulated body temperature rhythms reveal the phase-shifting behavior and plasticity of mammalian circadian oscillators. Genes Dev (2012) 26:567–80. doi:10.1101/gad.183251.111
46. Chiu JC, Ko HW, Edery I. NEMO/NLK phosphorylates PERIOD to initiate a time-delay phosphorylation circuit that sets circadian clock speed. Cell (2011) 145:357–70. doi:10.1016/j.cell.2011.04.002
47. Garbe DS, Fang Y, Zheng X, Sowcik M, Anjum R, Gygi SP, et al. Cooperative interaction between phosphorylation sites on PERIOD maintains circadian period in Drosophila. PLoS Genet (2013) 9:e1003749. doi:10.1371/journal.pgen.1003749
Keywords: circadian clock, temperature compensation, phosphorylation, phosphoswitch, period2
Citation: Narasimamurthy R and Virshup DM (2017) Molecular Mechanisms Regulating Temperature Compensation of the Circadian Clock. Front. Neurol. 8:161. doi: 10.3389/fneur.2017.00161
Received: 03 February 2017; Accepted: 05 April 2017;
Published: 27 April 2017
Edited by:
Timo Partonen, National Institute for Health and Welfare, FinlandReviewed by:
Kristin Eckel-Mahan, University of Texas Health Science Center at Houston, USAAndré Malan, Centre national de la recherche scientifique (CNRS), France
Copyright: © 2017 Narasimamurthy and Virshup. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David M. Virshup, david.virshup@duke-nus.edu.sg