 Joshua M. Garcia1†
Joshua M. Garcia1† Stephanie A. Stillings1†
Stephanie A. Stillings1† Jenna L. Leclerc2,3†
Jenna L. Leclerc2,3† Harrison Phillips4
Harrison Phillips4 Nancy J. Edwards5,6
Nancy J. Edwards5,6 Steven A. Robicsek4,7,8
Steven A. Robicsek4,7,8 Brian L. Hoh7Spiros Blackburn9
Brian L. Hoh7Spiros Blackburn9 Sylvain Doré2,3,10,11,12,13*
Sylvain Doré2,3,10,11,12,13*
- 1College of Medicine, University of Florida, Gainesville, FL, United States
- 2Department of Anesthesiology, College of Medicine, Center for Translational Research in Neurodegenerative Disease, University of Florida, Gainesville, FL, United States
- 3Department of Neuroscience, College of Medicine, University of Florida, Gainesville, FL, United States
- 4Department of Anesthesiology, University of Florida, Gainesville, FL, United States
- 5Department of Neurology, University of California, San Francisco, CA, United States
- 6Department of Neurosurgery, University of California, San Francisco, CA, United States
- 7Department of Neurosurgery, University of Florida, Gainesville, FL, United States
- 8Department of Neuroscience, University of Florida, Gainesville, FL, United States
- 9Department of Neurosurgery, University of Texas, Houston, TX, United States
- 10Department of Neurology, Center for Translational Research in Neurodegenerative Disease, McKnight Brain Institute, University of Florida, Gainesville, FL, United States
- 11Department of Psychology, Center for Translational Research in Neurodegenerative Disease, McKnight Brain Institute, University of Florida, Gainesville, FL, United States
- 12Department of Psychiatry, Center for Translational Research in Neurodegenerative Disease, McKnight Brain Institute, University of Florida, Gainesville, FL, United States
- 13Department of Pharmaceutics, Center for Translational Research in Neurodegenerative Disease, McKnight Brain Institute, University of Florida, Gainesville, FL, United States
Interleukin-10 (IL-10) is an important anti-inflammatory cytokine expressed in response to brain injury, where it facilitates the resolution of inflammatory cascades, which if prolonged causes secondary brain damage. Here, we comprehensively review the current knowledge regarding the role of IL-10 in modulating outcomes following acute brain injury, including traumatic brain injury (TBI) and the various stroke subtypes. The vascular endothelium is closely tied to the pathophysiology of these neurological disorders and research has demonstrated clear vascular endothelial protective properties for IL-10. In vitro and in vivo models of ischemic stroke have convincingly directly and indirectly shown IL-10-mediated neuroprotection; although clinically, the role of IL-10 in predicting risk and outcomes is less clear. Comparatively, conclusive studies investigating the contribution of IL-10 in subarachnoid hemorrhage are lacking. Weak indirect evidence supporting the protective role of IL-10 in preclinical models of intracerebral hemorrhage exists; however, in the limited number of clinical studies, higher IL-10 levels seen post-ictus have been associated with worse outcomes. Similarly, preclinical TBI models have suggested a neuroprotective role for IL-10; although, controversy exists among the several clinical studies. In summary, while IL-10 is consistently elevated following acute brain injury, the effect of IL-10 appears to be pathology dependent, and preclinical and clinical studies often paradoxically yield opposite results. The pronounced and potent effects of IL-10 in the resolution of inflammation and inconsistency in the literature regarding the contribution of IL-10 in the setting of acute brain injury warrant further rigorously controlled and targeted investigation.
Introduction
Stroke and traumatic brain injury (TBI) are devastating acute neurological disorders that can result in high mortality rates or long-lasting disability. Approximately 87% of strokes are ischemic and 13% are hemorrhagic, with 10 and 3% of the latter representing intracerebral hemorrhage (ICH) and subarachnoid hemorrhage (SAH), respectively (1). Stroke is the fourth most common cause of death in the United States, and ischemic stroke (IS) in particular is the seventh most frequent emergency department presentation (2, 3). TBI and concussions have over twice the incidence of all strokes combined (4), with more than three million people in the United States alone living with long-term disability as a result of TBI (5). Collectively, stroke and TBI have very few treatments, and despite advances in clinical management of these disorders, they are still associated with significant disability and mortality (6, 7).
Inflammation plays a central role in the pathophysiology of stroke and TBI and can have both protective and harmful effects on brain tissue (8–15). Although there are some distinct differences in the inflammatory cascades following the various types of acute brain injury, there are also numerous commonalities. Acute neuroinflammation is characterized by the activation of resident central nervous system (CNS) immune surveillance glial cells that release cytokines, chemokines, and other immunologic mediators, which facilitate the recruitment of peripheral cells such as monocytes, neutrophils, and lymphocytes (8, 9, 12, 15). Collectively, this initial response is helpful in the clearance of toxic entities and the restoration and repair of damaged tissue. However, during the resolution phase, with an uncontrolled and prolonged inflammatory response, secondary damage results from overactivation of this inflammatory surge and release of additional factors that led to breakdown of the blood–brain barrier (BBB), cerebral edema, cerebral hypertension, and ischemia.
Interleukin-10 is generally known as an anti-inflammatory cytokine that exerts a plethora of immunomodulatory functions during an inflammatory response and is particularly important during the resolution phase. Expression of IL-10 in the brain increases with CNS pathology, promoting neuronal and glial cell survival, and dampening of inflammatory responses via a number of signaling pathways (16). IL-10 was originally described as cytokine synthesis inhibitory factor and in addition to attenuating the synthesis of proinflammatory cytokines, IL-10 also limits inflammation by reducing cytokine receptor expression and inhibiting receptor activation (16). Furthermore, IL-10 has potent and diverse effects on essentially all hematopoetic cells that infiltrate the brain following injury. For example, IL-10 reduces the activation and effector functions of T cells, monocytes, and macrophages, ultimately ending the inflammatory response to injury (17). The structure, function, and regulation of IL-10 have been extensively reviewed elsewhere, including a review of IL-10 in the brain (16–20), although not in the context of the various forms of acute brain injury. Please refer to the aforementioned reviews for additional details, including the potential cellular sources, target cells, signal transduction, and mode of action of IL-10.
Given the intriguing multifactorial role of IL-10 in the resolution of inflammatory cascades that are important for promoting neurologic recovery from acute brain injury, here we present a comprehensive literature review of preclinical and clinical studies in this area. We focus on the contribution of IL-10 in modulating various important parameters and pathophysiologic processes important for IS, SAH, ICH, and TBI outcomes, and whether IL-10 has therapeutic or biomarker potential. A better understanding of the many functions of IL-10 in the brain after injury, particularly in the resolution phase of inflammatory processes, will promote our knowledge of the pathophysiology of these debilitating disorders and guide future development of novel therapeutic approaches.
Vascular Endothelium, Remodeling, and Dysfunction
The neurovascular unit, including the vascular endothelium, has become a therapeutic target of interest in the various types of acute brain injury (14, 21–24). IL-10 has an assortment of functions acting at the vascular and endothelial level, such as modulating vascular remodeling, reducing leukocyte adhesion and extravasation, mitigating leukocyte–endothelial interactions that facilitate coagulation, promoting vasodilatation via increased production of nitric oxide, and direct protection of the endothelium from oxidative stress via the downregulation of harmful reactive oxygen species (ROS)-producing enzymes, and/or the upregulation of antioxidant pathways.
IL-10 in Preclinical Studies
Interleukin-10 deficiency results in a spectrum of problems with the vasculature, including everything from vascular and endothelial damage from increased oxidative stress and inflammation to deleterious remodeling and an inability of the vasculature to respond to physiological demands such as the requirement for vasodilation. IL-10 inhibits Nox1, a subunit of NADPH oxidase, which plays a role in degenerative vascular remodeling by generating ROS, causing oxidative stress (25). IL-10 knockout (IL-10−/−) mice have higher levels of Nox1, Nox2, and p22phox (two additional NADPH oxidase components) and superoxide production and display decreased aortic medial thickness, a loss of smooth muscle cells, and increased vascular collagen deposition, indicating harmful vascular remodeling with IL-10 deficiency (25, 26). Administration of a superoxide scavenger in IL-10−/− mice prevented vascular remodeling, suggesting the oxidative stress-dependent mechanism (ROS formation by NADPH oxidase) of injurious vascular remodeling with IL-10 deficiency (26). Additionally, IL-10 deficiency led to increased levels of matrix metalloproteinase-9 (MMP-9) in aortic smooth muscle cells, IL-6 in aortas, and the vasoconstrictor endothelin-1 in plasma (25).
Interleukin-10 attenuates endothelial dysfunction and vasoconstriction mediated by ROS, endothelin-1 (27), angiotensin II (28), and by ischemia-reperfusion injury (29), among other mediators, and these protective mechanisms may become more important with age. Old, but not young, IL-10−/− mice have diminished vasodilatory responses to acetylcholine, while the nitroprusside response is intact, suggesting endothelial rather than vascular smooth muscle dysfunction (26). Additionally, viral transduction of canine basilar arteries with IL-10 increases vasodilatory responses and reduces levels of ICAM-1 and VCAM-1, further supporting the protective role of IL-10 on the endothelium (29). IL-10 has also been shown to act via the AKT pathway to decrease TNFα-directed ceramide synthesis, resulting in lower levels of ROS and ICAM-1 (30). In aortic rings from IL-10−/− mice, TNFα reduces endothelial nitric oxide synthase expression and vasodilatory ability, and IL-10 administration restores function, providing a protective effect (31). A similar experiment showed that murine aortic rings treated with angiotensin II showed impaired relaxation that was reversible with IL-10 administration (32). High cyclooxygenase-2 activity also plays a role in these stiffer vessels seen in IL-10−/− mice, ultimately resulting in decreased vascular relaxation, impaired cardiac function, and a larger heart size (33).
The source of endothelial protective IL-10 may come from CD4+CD25+ regulatory T cells (Tregs) (34), as well as the various other types of IL-10-producing cells, including B cells (see the “IS” section below). For example, hypertensive IL-10−/− mice transduced with hypertensive wildtype (WT) Tregs have a better vasodilatory response to acetylcholine and lower levels of NADPH oxidase, whereas hypertensive IL-10−/− Tregs does not confer protection on hypertensive WT mice (34). In middle-aged spontaneously hypertensive rats that display features similar to early-stage human cerebral small vessel disease, there is a decreased level of IL-10 in the cerebrospinal fluid and other immune changes (35).
Of interest, inflammation mediated by toxic bacterial agents, such as lipopolysaccharide (LPS), is more severe in IL-10−/− mice (36). Surprisingly, cerebral bacteremia may result in so much endothelial damage in the absence of IL-10 as to cause ICH and death shortly following peak bacterial loads (37). These findings are associated with increases in the FAS/FAS-ligand apoptotic pathway, which IL-10 reverses (37). However, the effects of IL-10 seem to be specific to the noxious stimuli generating its production. For example, IL-10 produced in response to both LPS and Borrelia burgdorferi reduces lymphocyte endothelial migration and blunts endothelial production of chemokines; however, IL-10 produced in response to IL-1β and TNFα does not show such effects (38).
Summary of Evidence Describing the Role of IL-10 in Protecting the Vasculature
Given the intimate tie between the vascular endothelium and pathophysiology of all forms of acute brain injury, it is important to consider the effects of IL-10 on the vasculature. The above studies have shown that following injury, the inflammatory system works on many different levels to cause endothelial cell damage and vascular dysfunction. IL-10 appears to play a central and multifaceted role in attenuating these effects and facilitating the resolution phase of the inflammatory system. In contrast, low or absent IL-10 leads to several changes in gene expression that ultimately results in deleterious vascular remodeling and impaired vascular relaxation in response to physiologic mediators (Figure 1), outcomes that would exacerbate secondary brain damage following acute injury. In summary, IL-10 plays a crucial role in restoring vascular function following injury to the vasculature, similar to that which occurs after IS, SAH, ICH, and TBI.
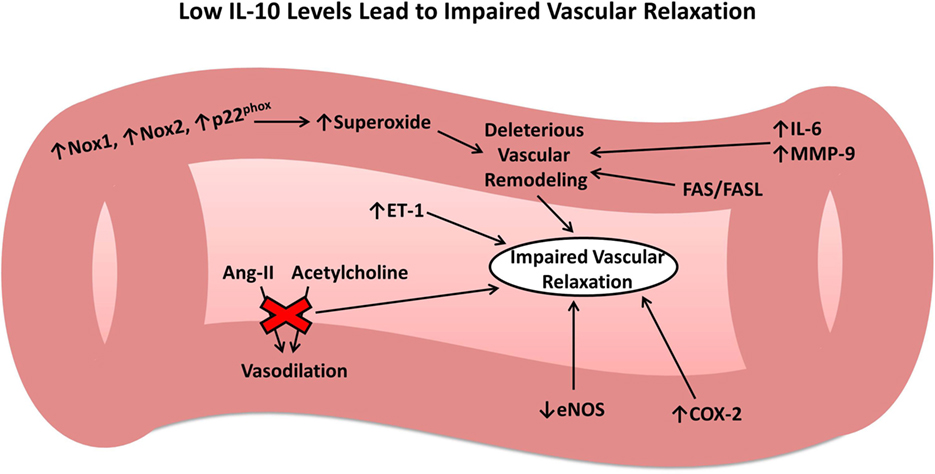
Figure 1. Role of interleukin-10 (IL-10) in vascular remodeling and dysfunction. Low or absent IL-10 results in numerous changes to the vasculature leading to harmful vascular remodeling and impaired vascular relaxation in response to important physiologic mediators.
Ischemic Stroke
Ischemic stroke occurs when a local thrombus or embolus occludes in a cerebral vessel and obstructs blood flow to the brain. Approximately 800,000 people per year in the United States experience a new or recurrent stroke (1) and approximately 33% of these patients will die within 1 year post-stroke (39). Consequently, IS is a significant cause of morbidity, particularly in the elderly, where approximately half of these patients will have enduring moderate-to-severe neurologic deficits (40). Tables 1–3 provide a summary of the IL-10 preclinical and clinical IS studies described below.
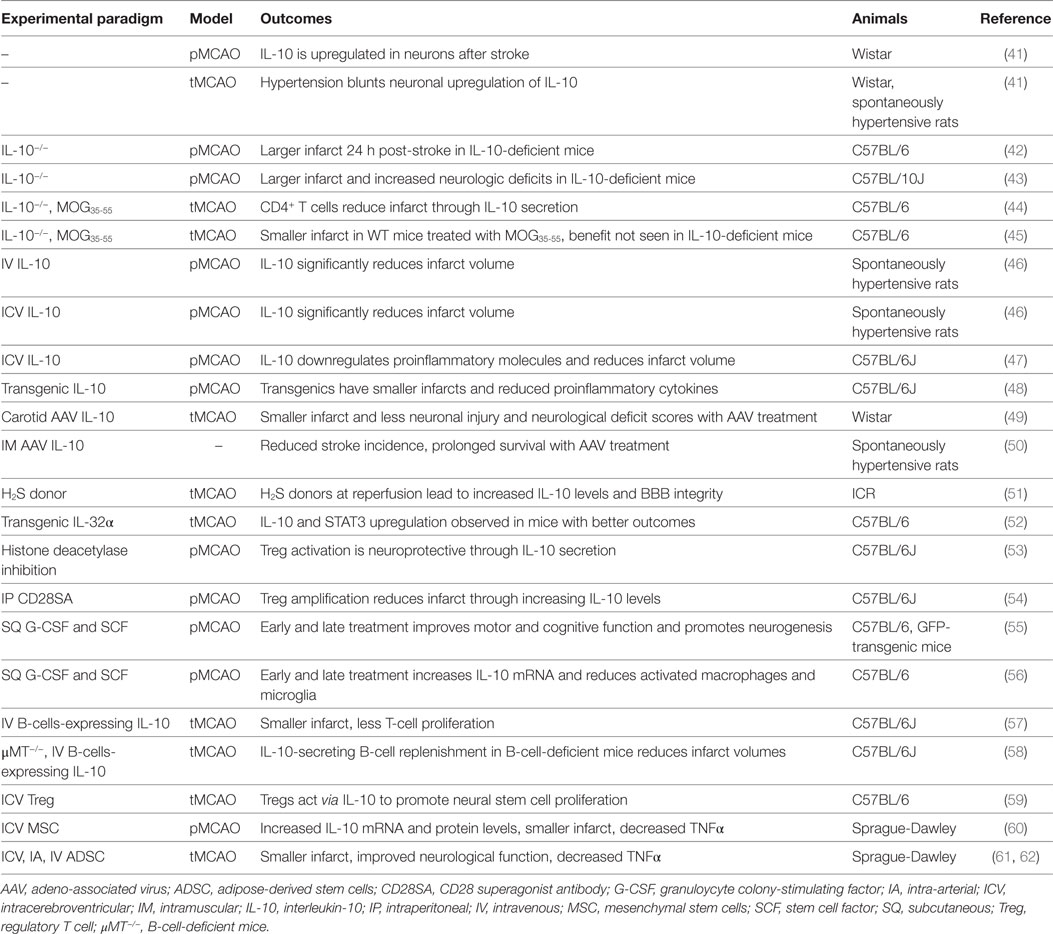
Table 1. Summary of IL-10 ischemic stroke preclinical studies.
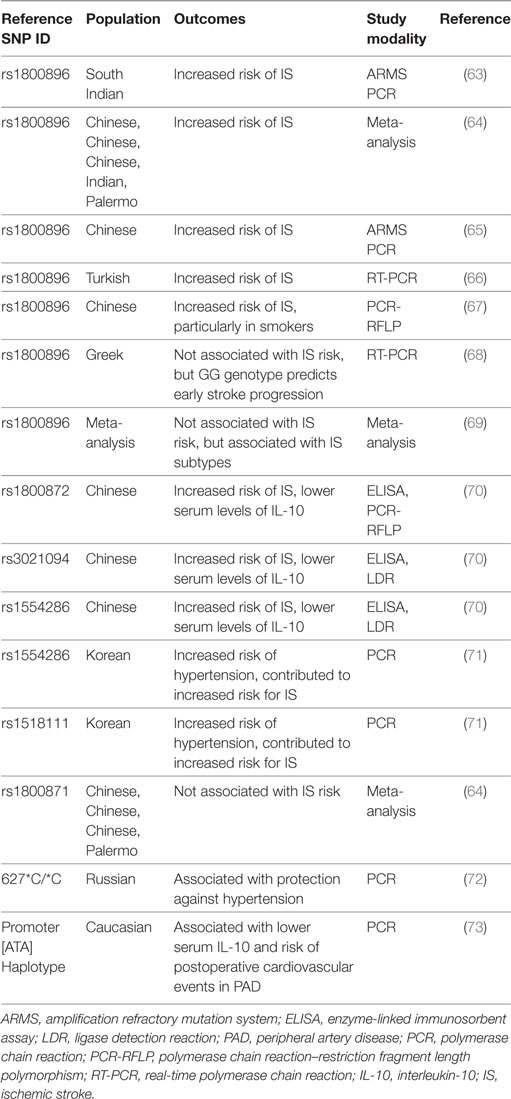
Table 2. Summary of clinical ischemic stroke studies investigating IL-10 genetic polymorphisms.
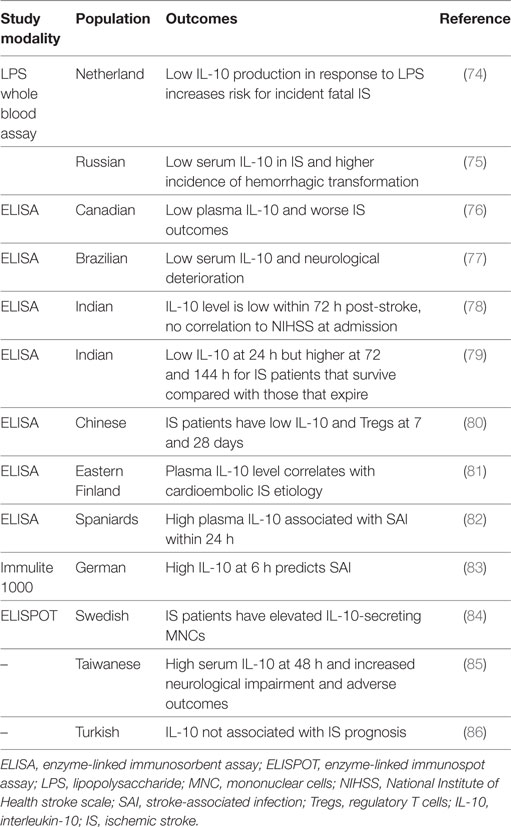
Table 3. Summary of non-genetic IL-10 ischemic stroke clinical studies.
IL-10 in In Vitro Models
Cortical neuron cultures from IL-10−/− mice are more susceptible to neurotoxicity following excitotoxicity and combined oxygen–glucose deprivation, and administration of exogenous IL-10 provides neuroprotection to cultures from both WT and knockout strains (42). This IL-10-mediated protection was later separately shown to be via the IL-10 receptor on cortical neurons and PI3K/AKT and STAT3 signal transduction pathways (87). The transcription factor Nrf2 is expressed widely throughout the body and upregulates the expression of numerous antioxidant genes in response to oxidative stress (88, 89). In astroglia preconditioned to oxidative stress, neuroprotection from oxidative stress–glucose deprivation was reported to be via an Nrf2/IL-10-dependent mechanism (90). Lipocalin-2 is significantly increased in neurons following stroke and is proposed to signal stress from injured neurons to supporting glia (91). Lipocalin-2 treatment of microglia results in glial activation, IL-10 release, and enhanced phagocytosis, and conditioned media from these microglia protect neurons from oxygen–glucose deprivation (91).
IL-10 in Preclinical Models
In a permanent middle cerebral artery occlusion (MCAO) model, IL-10−/− C57BL/6 mice have 30% larger infarct volumes compared with WT mice 24 h post-stroke (42). In this model, WT and IL-10−/− mice display similar degrees of proinflammatory molecules within the first 6 h. However, after 4 days, the IL-10−/− mice have higher levels of proinflammatory molecules that persist through the end of the first week after stroke (43).
Both systemic intravenous (IV) and central intracerebroventricular (ICV) exogenous administration of IL-10 reduces infarct volumes following permanent MCAO (46). Similarly, in a transgenic C57BL/6J mouse model overexpressing IL-10 (56% increase in the brain and 200% increase in the plasma), infarct volumes were reduced by 40% 4 days following permanent MCAO (48). In the same study, these findings were associated with upregulation of free radical scavengers such as glutathione and manganese superoxide dismutase, reduced activity of the proapoptotic protein caspase 3, and downregulation of IL-1β, IFN-γ, and TNFα 1 day post-stroke (48). Injection of a recombinant adeno-associated viral vector serotype 1 expressing IL-10 into the cerebral artery of rats 3 weeks prior to MCAO results in elevated IL-10 serum levels 3 weeks after injection and decreases neurologic deficit scores, infarct volume, and neuronal injury (49). Systemic IL-10 overexpression in stroke-prone spontaneously hypertensive rats reduces the incidence of stroke, decreases stroke-associated symptoms, and improves survival (50). In Wistar rats, IL-10 is upregulated in viable neurons in the ischemic brain following permanent and transient MCAO, and hypertension blunts this response, potentially contributing to the worse outcomes in the hypertensive setting (41).
Interleukin-10 appears to exert its anti-inflammatory effects in part by downregulation of NFκB (92). Administration of hydrogen sulfide donors at the time of reperfusion protects BBB integrity after ischemia/reperfusion and is accompanied by enhanced IL-10 expression, reduced NFκB nuclear translocation, and MMP-9 and NOX4 activity (51). Moreover, upregulation of IL-10 and the STAT3 pathway and downregulation of NFκB have been proposed as the mechanisms for improved outcomes following cerebral ischemia in mice that overexpress IL-32α (52). A recent whole genome array in C57BL/6 mice showed that stroke upregulated 347 genes 24 h after reperfusion. In this study, ICV injection of IL-10 5 min after occlusion downregulated 341 of these genes (47).
Immune cells, including T and B cells, are important in attenuating neuroinflammation via the modulation of various cytokines and chemokines, with IL-10 playing a central immunomodulatory role (53, 58). Tolerizing mice to oligodendrocyte glycoprotein prior to MCAO reduces infarct size by 70% at 24 h and by 50% at 72 h, but this effect was not seen in IL-10−/− mice (45). This protection is specifically mediated by IL-10 secreted from CD4+ T cells, which were able to protect mice from stroke only when originating from WT mice (44). Elevated levels of IL-10 also reduce the number of CD11b+ cells that may contribute to secondary infarct expansion via nitric oxide pathways (45). Histone deacetylase plays a role in activating the expression of Foxp3 on Tregs, which secrete IL-10, and IL-10 was suggested as the main mediator of attenuated infarct volume and behavioral deficits, reduced levels of proinflammatory cytokines, and increased number of Tregs in the brain of MCAO mice with histone deacetylase inhibition (53). Similarly, expansion of the Treg cell population in the CNS via administration of a CD28 superagonist monoclonal antibody at onset of reperfusion reduced infarct size 7 days following MCAO, and its effect was attributed to an increased amount of IL-10 (54). Transferring IL-10-producing B cells into B cell-deficient mice 24 h after MCAO reduced infarct size, the amount of T cells and monocytes in the brain parenchyma, and the peripheral proinflammatory milieu (58). A follow-up to this study showed similar results in B cell-sufficient mice (57). Notably, IL-10-producing B cells also upregulated the number of Tregs (58), perhaps indicating a positive feedback loop between B cells and Tregs, both of which are neuroprotective via IL-10 production.
Neurogenesis is a highly debated topic for potentially enhancing neural recovery following IS. Injection of activated Tregs into the lateral ventricle of C57BL/6 mice after 60 min of transient MCAO promotes neural stem cell proliferation in the subventricular zone in ischemic and normal mouse brains. However, this effect was eliminated by blocking IL-10 with a neutralizing antibody, indicating that activated Tregs act via IL-10 to promote neural stem cell proliferation (59). Hematopoietic cytokines such as GCSF and stem cell factor have been shown to promote neurogenesis (55) and also may be responsible for providing the initial signals to produce IL-10 in the setting of IS (56). Treatment with these cytokines early (1–10 days) and later (11–20 days) after MCAO markedly elevates IL-10 mRNA levels, reduces the levels of activated microglia/macrophages, and does not change proinflammatory cytokine expression in C57BL/6J mice (56). Another study where bone marrow-derived mesenchymal stem cells were transplanted into the lateral ventricle of Sprague-Dawley rats before permanent MCAO afforded similar results, where IL-10 mRNA and protein levels are increased for up to 4 days post-stroke, TNFα is decreased, infarct volumes are smaller, and neurologic function is preserved (60). Adipose tissue-derived stem cells trigger similar effects as mesenchymal stem cells (61) and are most effective when delivered intravenously at 24 h after MCAO (62). Thus, either administration of stem cells themselves or hematopoietic cytokines seem to improve outcomes following IS in part through increasing IL-10 levels.
IL-10 in Clinical Studies
Several epidemiologic studies have analyzed the genetic contribution of IL-10 to IS risk and outcome. A study in a Chinese population showed that lower basal serum levels of IL-10, an IL-10 promoter single nucleotide polymorphism (SNP) rs1800872 (AA vs. AC + CC), and IL-10 intron SNPs rs1554286 (TT vs. CT + CC) and rs3021094 (CC/CA vs. AA) are all associated with higher risk for IS (70). Similarly, subjects with low IL-10 production in response to LPS stimulation have a higher risk for fatal stroke (74). A study in a South Indian population showed an increased risk of IS with the A allele of the IL-10 promotor SNP rs1800896 (A vs. G), which is associated with low IL-10 production (63). In agreement, a recent meta-analysis showed that the IL-10 1082G/A SNP (rs1800896) is associated with increased risk of IS, but not the 819 C/T SNP (rs1800871) (64, 65). Several other studies further revealed that the IL-10 1082G/A SNP is associated with susceptibility for an IS (66, 67, 93, 94). However, in a different study, the IL-10 1082G/A SNP was not associated with the occurrence of an IS, but the GG genotype predicted early stroke progression and functional dependency independent of other standard risk factors (68). Similarly, another study showed that the IL-10 1082G/A SNP was significantly associated with the risk for specific subtypes of IS, but no significant association was found with the overall risk for an IS (69). A study in a Korean population showed that rs1518111 and rs1554286 are not associated with IS per se, but are associated with hypertension in the risk of IS (71). Similar results were seen in a Russian population, where the IL-10 627*C/*C genotype is protective against hypertension in male patients with stroke (72). One other study in an Eastern Finland population showed that IL-10 plasma concentration independently correlates with cardioembolic high-risk sources, suggesting its usefulness in improving diagnosis of stroke etiology (81). Lastly, in Caucasian patients with peripheral artery disease receiving elective revascularization, the IL-10ATA haplotypes that are associated with lower serum IL-10 levels correlate with a high risk for postoperative cardiovascular events (73).
The relative levels of IL-10 at various times following IS differ from controls and appear to have important implications on IS patient outcomes. For example, at 48 h after IS, mean serum IL-10 is significantly higher than in healthy or at-risk controls, and high IL-10 sera levels independently correlate with severe neurologic deficits (NIHSS ≥12) at 48 h post-stroke and predict major adverse clinical outcomes (recurrent IS, any cause of death, NIHSS ≥12) at 90 days (85). However, in another study, IL-10 levels within the first 72 h after IS were lower than in controls and no significant correlation with NIHSS at admission was observed (78). As compared to those who die, IS patients who improve have been shown to have lower IL-10 at 24 h, but increased levels at 72 h and 144 h (79). In other studies, low IL-10 levels are associated with acute neurologic decline post-stroke (76, 77). Yet another small study found no correlation with IL-10 and other cytokines and IS patient prognosis (86). IS patients with low levels of IL-10 had a higher incidence of hemorrhagic transformation (75), perhaps providing a link between IL-10 and endothelial integrity. At 7 and 28 days, IS patients appear to have lower number of Tregs and IL-1 and higher relative levels of proinflammatory cytokines such as IL-6 and IL-1β (80). In the Leiden 85-plus study, patients with a history of stroke displayed lower IL-10 production in response to LPS stimulation than subjects without stroke (74). No correlation between IL-10 levels and infarct volume has been found (81, 86). Considerable variation exists among the aforementioned studies and is likely due to the different study populations, relatively small cohort sizes, differences in study methodology, and the consistency or lack thereof for identifying and correcting for covariates. Additional large and controlled prospective studies are needed to establish the link between IL-10 and IS severity, recovery, and outcomes.
Ischemic stroke may induce a functional immunosuppressive state that may persist for several weeks, wherein patients may be more susceptible to infection, and infection is a leading cause of the high mortality rates seen long after an IS (82, 95). The temporal IL-10 profile appears to be different between those that develop stroke-associated infection (82, 83). Two studies have shown that high IL-10 levels at admission or 6 h following ischemia is an independent predictor of infection (82, 83). In one of those studies, TNFα and the TNFα/IL-10 ratio was decreased (82). Another study found that the levels of IL-10-secreting mononuclear cells (MNCs) were highly elevated in IS patients compared with healthy individuals (84). Thus, the immunomodulatory role of IL-10 may be an important consideration in understanding infectious processes and mortality after IS.
Summary and Comparison of the Role of IL-10 in Preclinical and Clinical IS Studies
Ischemic stroke invokes a robust local inflammatory response in the brain that significantly contributes to secondary brain damage and poor outcomes. With IL-10 representing a canonical anti-inflammatory cytokine that intriguingly has a special role in the resolution of inflammation, it is not surprising that many studies investigating the role of IL-10 in IS have been performed. In vitro models have provided evidence for a neuroprotective function of IL-10 under hypoxic and excitotoxic conditions. In IS preclinical models, IL-10 reduces infarct volume via its protective effects on the vascular endothelium and attenuation of inflammatory cascades. Multiple epidemiologic genetic studies have in general shown that polymorphisms in the IL-10 gene that lower IL-10 levels increase the risk of IS. However, clinical studies measuring IL-10 levels after IS are more varied with regard to the role of IL-10 in predicting neurologic outcomes and complications. Even with all of the studies described here, it is clear that additional work is needed to clarify the role of IL-10 in predicting the risk for IS, modulating IS outcomes, and whether IL-10 has biomarker and/or therapeutic potential after IS.
Subarachnoid Hemorrhage
Subarachnoid hemorrhage affects an estimated 7.2–9.0 per 100,000 people/year in the United States (96), and most cases are due to the rupture of a cerebral aneurysm resulting in bleeding within the subarachnoid space (97). Although approximately 15% of people die within a few hours from the bleed and a proportion will never make it to a hospital (98), even those who initially survive the bleed can have significant morbidity and mortality. In addition to early brain injury that occurs at the time of SAH, patients can also develop neurological deficits or die from delayed cerebral ischemia (DCI), which is often secondary to cerebral vasospasm (CV) (99, 100). Among the possible mechanisms for CV and DCI (13, 100–110), inflammation is a common element. IL-10 has not been extensively studied in the context of SAH, despite its proposed pleiotropic immunomodulatory effects relevant to SAH pathophysiology. Tables 4 and 5 provide a summary of the IL-10 preclinical and clinical SAH studies described below.
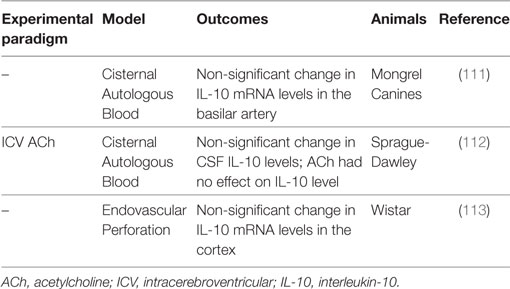
Table 4. Summary of IL-10 subarachnoid hemorrhage preclinical studies.
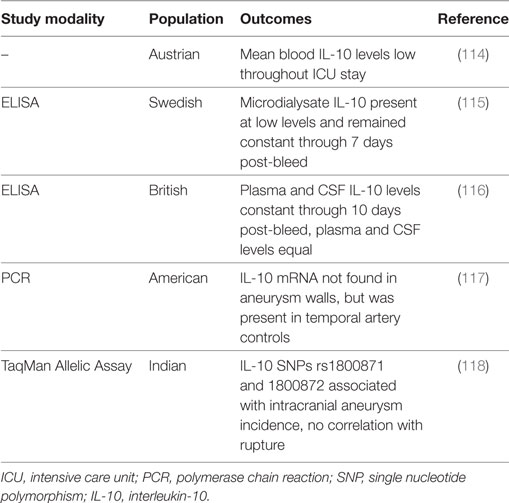
Table 5. Summary of IL-10 subarachnoid hemorrhage clinical studies.
IL-10 in Preclinical Models
Temporal changes in IL-10 are seen following experimental SAH, although these are not statistically significant (111–113). After autologous blood infusion into the cisterna magna of canines, IL-10 mRNA expression in the basilar artery tends to be increased for up to 14 days post-ictus (111). Similarly, IL-10 tends to be increased in the CSF at 6 h and then declines, but remains elevated through 48 h after autologous blood injection into the cisterna magna of male adult Sprague-Dawley rats (112). Interestingly, although acetylcholine was able to attenuate CV in this model, it had no effect on IL-10 levels (112). Last in an endovascular perforation model of SAH in Wistar rats, trends toward higher IL-10 mRNA in the cortex are seen at 48 h (113). In contrast to the less impressive changes in IL-10, these studies have shown highly increased levels of proinflammatory cytokines such as IL-1α (111), IL-1β (113), IL-6 (111, 112), IL-8 (111), and TNFα (111) at 48 h following experimental SAH. Indeed, Aihara and colleagues postulated that the anti-inflammatory effects of IL-10 were overwhelmed by these proinflammatory cytokines such that IL-10 was unable to counteract the deleterious inflammatory processes involved in CV pathophysiology and thereby improve SAH outcomes (111).
IL-10 in Clinical Studies
Studies in humans have also not shown significant changes in IL-10 after SAH (114–117). One study reported that IL-10 mRNA expression was virtually non-existent in aneurysm walls, although it was present in the walls of temporal artery controls (117). In microdialysate, when detectable, IL-10 was present in extremely small concentrations and remained at relatively constant levels through 7 days post-bleed (115). Similarly, IL-10 levels were constant through at least 10 days post-bleed in paired plasma and CSF samples, and the levels were not significantly different between the CSF and plasma (means were below 10 pg/ml) (116). In another study, mean IL-10 blood levels throughout the intensive care unit stay were also quite low (mean ≈11 pg/ml) (114). However, a study in an Indian population showed that IL-10 SNPs rs1800871 and rs1800872 are significantly associated with the incidence of intracranial aneurysms, independent of epidemiological factors, although it is unclear whether this increases the risk for aneurysm rupture and thus the incidence of SAH (118). Additional larger controlled studies are necessary to clarify the association of IL-10 with aneurysm formation and rupture and the role of IL-10 after SAH, including its prognostic and diagnostic predictive potential for SAH outcomes.
Summary and Comparison of the Role of IL-10 in Preclinical and Clinical SAH Studies
In comparison with the other forms of acute brain injury described herein, substantially fewer studies have explored the contribution of IL-10 in the setting of SAH. Preclinical studies have shown non-significant increases in IL-10 after SAH, accompanied by robust changes in proinflammatory cytokines, suggesting an imbalance in the inflammatory milieu that could possibly contribute to prolonged disease course and worse SAH outcomes. In addition, clinical studies have not shown significant changes in IL-10 after SAH, but no studies have investigated whether IL-10 affects clinical outcomes or complications such as CV and/or DCI, or whether IL-10 has therapeutic or biomarker potential. It is clear that additional work is needed to understand how IL-10 changes in the various biocompartments (serum, CSF, brain) after SAH and the resulting implications on SAH pathophysiology.
Intracerebral Hemorrhage
Intracerebral hemorrhage is characterized by bleeding within the brain parenchyma and can result from various etiologies such as trauma, vascular malformations, medical therapies such as anticoagulants, amyloid angiopathy, and most commonly, hypertension. ICH has a poor prognosis, and there are currently no therapies to decrease the 30-day mortality rate of 35–50% (119). At present, the only available interventions include supportive care, and in some select cases, invasive surgery to evacuate hematomas (120), yet, it is well established that inflammation is a key player in the overall brain damage and edema after ICH that results in enduring neurological deficits. Tables 6 and 7 provide a summary of the IL-10 preclinical and clinical ICH studies described below.
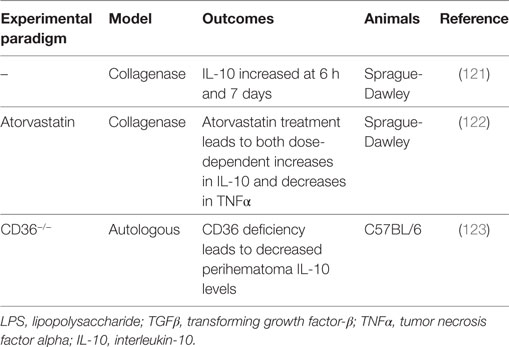
Table 6. Summary of IL-10 intracerebral hemorrhage preclinical studies.
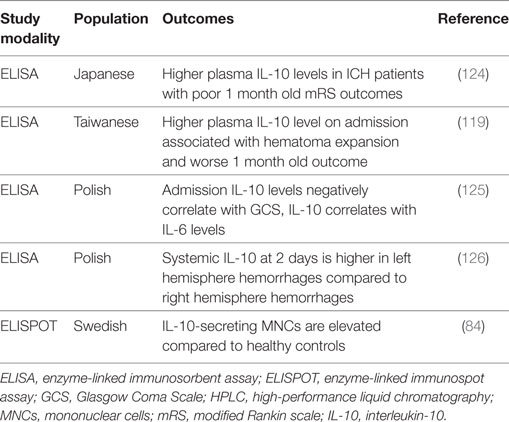
Table 7. Summary of IL-10 intracerebral hemorrhage clinical studies.
IL-10 in Preclinical Models
Microglia, a primary player in immune surveillance and the initiation of inflammation in the CNS (127), become activated shortly after ICH (12) and secrete proinflammatory cytokines such as TNFα, IL-6, and IL-1β (127). These microglia also secrete IL-10, which acts on astrocytes in the brain to redirect their focus from the production of a proinflammatory cytokine profile toward the production of TGFβ (127). TGFβ then feeds back to act on the microglia and attenuate their proinflammatory response. IL-10 can also increase CD36 expression and thereby enhance the erythrophagocytic ability of microglia, and with CD36 deficiency, mRNA levels of TNFα and IL-1β are higher and IL-10 levels are significantly lower in the perihematomal tissues (123). Additionally, IL-10 levels are significantly increased at 6 h and 7 days after ICH with respect to sham, suggesting that IL-10 may have early and late influences on ICH outcomes (121).
Interestingly, a study in IL-10−/− mice suggests that the presence of IL-10 is protective against the development of cerebral hemorrhage (128). Cerebral hemorrhage and edema are attenuated by anti-TNFα therapy (128), indicating that the main protective effect of IL-10 is derived from its inhibition on the production of proinflammatory cytokines, namely, TNFα (129–131). This same effect is found in adult male Sprague-Dawley rats, where statins (2, 5, 10 mg/kg) produce dose-dependent increases in IL-10 levels and are associated with dose-dependent decreases in TNFα, as well as fewer activated microglia, sensorimotor deficits, and inflammation and less edema 3 days after ICH (122).
IL-10 in Clinical Studies
Only a couple of studies have looked at IL-10 genetic polymorphisms and the risk for developing ICH. The IL-10 1082G/A SNP has been identified as a risk factor for ICH in a North Indian population (132). This SNP was not associated with the risk for hemorrhagic presentation of brain arteriovenous malformations (133).
Interleukin-10 and the amount of IL-10-secreting MNCs are elevated in peripheral blood 2 days following ICH (84, 125). Another study found that systemic IL-10 release was significantly higher in ICH patients with left hemisphere hemorrhage compared with those with right hemisphere hemorrhage at 2 days, whereas no such correlation was found for IL-6 (126). Because dysfunction of the autonomic nervous system is a common ICH complication (134), the autonomic nervous system can be regulated asymmetrically, and the sympathetic nervous system can induce a notable increase in systemic IL-10 release, this study posits autonomic dysfunction as a potential mechanism for asymmetrical regulation of IL-10 after ICH (126).
At admission, IL-10 levels are negatively correlated with the Glasgow Coma Scale (GCS), although a stronger negative correlation was found for IL-6 and GCS (125). At 2 days post-bleed, IL-10 levels correlate with IL-6 levels, and IL-6 levels correlate with hematoma volume and mass effect, but IL-10 does not (125). Hematoma expansion is a major cause of morbidity and mortality after ICH, and inflammation may be associated with its pathogenesis (119). In spontaneous ICH, higher plasma IL-10 levels on admission are associated with hematoma expansion and worse 30-day outcomes (119). Another study also found that plasma IL-10 levels were higher in ICH patients with poor outcome (modified Rankin Scores of 3–6) at 1 month (124), although this study did not utilize a multivariate model.
Summary and Comparison of the Role of IL-10 in Preclinical and Clinical ICH Studies
Similar to IL-10 and SAH, few studies have explored the contribution of IL-10 in the setting of ICH, although IL-10 appears to be elevated following experimental and clinical ICH. In preclinical models, IL-10 exerts a protective effect against spontaneous cerebral hemorrhage by downregulating TNFα, a key proinflammatory cytokine, and some weak evidence exists suggesting that IL-10 is neuroprotective after ICH. Clinically, very little is evident regarding the contribution of IL-10 polymorphisms. However, other studies generally have found that IL-10 levels correlate with the extent of initial brain injury and have prognostic value, suggesting both diagnostic and prognostic biomarker potential for IL-10 after ICH.
Traumatic Brain Injury
Traumatic brain injury can result from a direct force like a concussion, an indirect force from a blast injury, or a penetrating injury. TBI is a prevalent clinical problem (9), with an estimated two million people in the United States sustaining a TBI each year (135, 136), leading to an astounding estimated 30.5% of injury-related deaths (137). TBI has substantial long-term effects on patients (137, 138) and imposes a significant financial burden (9, 135, 136, 138). The outcome for a TBI patient is determined by the severity of the initial injury, which is related to the mechanism of primary injury and degree of physical force, and secondary injury, linked to neuroinflammation (8, 9, 139). It is proposed that the majority of damage from TBI is the product of secondary damage, which is closely associated with the production and recruitment of proinflammatory cytokines and other inflammatory mediators (8, 140). Tables 8 and 9 provide a summary of the IL-10 preclinical and clinical TBI studies described below.
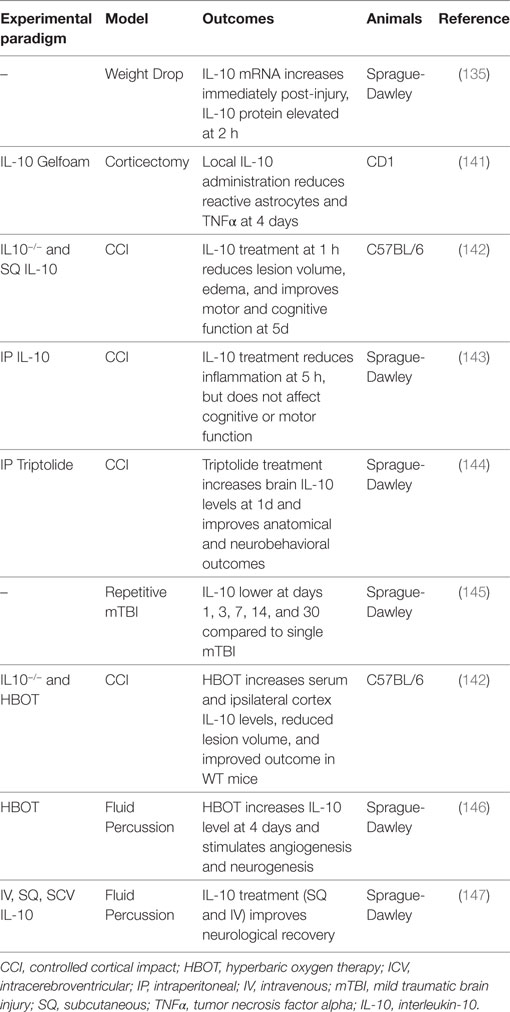
Table 8. Summary of IL-10 traumatic brain injury preclinical studies.
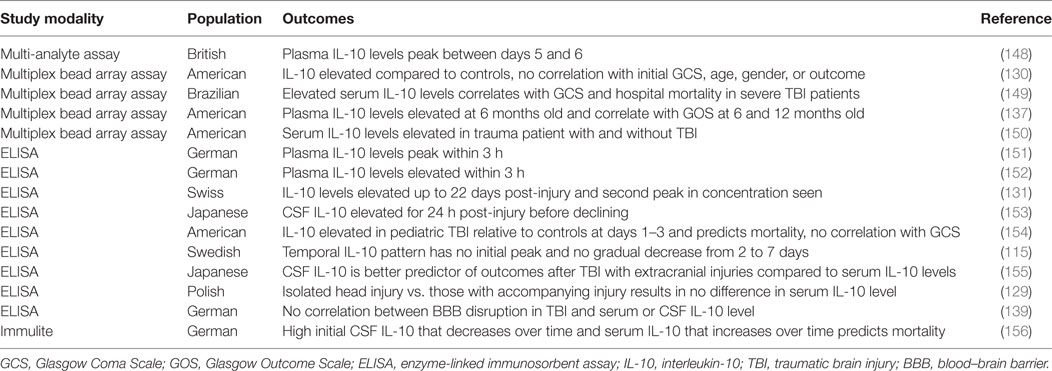
Table 9. Summary of IL-10 traumatic brain injury clinical studies.
IL-10 in Preclinical Models
Understanding the temporal relationship between the injury and elaboration of IL-10 provides insight into its immunomodulatory role after TBI. A study in adult male Sprague-Dawley rats found that brain IL-10 mRNA expression increases immediately following TBI, whereas IL-10 protein levels are stable initially, and only begin to rise rapidly after 2 h (135). These findings suggest that the surge in IL-10 levels is due to an increase in local IL-10 synthesis rather than systemic IL-10 entering through a leaky BBB. IL-10 protein levels continue to rise through 24 h, although at a slower rate (135). No change in plasma IL-10 levels is seen throughout this timeframe (135). Following repetitive mild TBI, TNFα and IL-6 are higher, and the expression of IL-10 is lower when compared to a single TBI group on days 1, 3, 7, 14, and 30 post-injury (145).
Studies with IL-10−/− mice have suggested that IL-10 is beneficial after TBI, although IL-10 administered after TBI as a therapeutic agent has provided mixed results depending on the experimental paradigm and administration route. At 4 weeks after TBI, 8-week-old IL-10−/− C57BL/6 female mice have larger lesion volumes, increased edema and inflammation, worse motor and cognitive function, and higher levels of BBB dysfunction and apoptosis after controlled cortical impact-induced TBI (142). Additionally, using the lateral fluid percussion TBI model, SQ IL-10 (100 µg) administered 1 h after TBI results in a significant reduction in lesion volume and edema at 5 days, as well as improved recovery of motor and cognitive function (142). In another study using the lateral fluid percussion model and adult male Sprague-Dawley rats, both SQ and IV IL-10 (100 µg) improved neurological recovery, but ICV dosing (1 or 6 µg) did not (147). Although not statistically significant, survival rates in the SQ and IV groups were higher than in the controls, and the ICV group had dose-dependent lower survival rates (147). IV administration also reduced TNFα and IL-1β expression in the injured cortex and hippocampus (147). Although these studies have shown that systemic IL-10 treatment reduces neuroinflammation and improves neurological recovery (142, 147), a different study found that while systemically administered IL-10 (5 µg) does reduce inflammation (75% reduction in neutrophil accumulation) at 5 h, there is no improvement in motor or cognitive recovery (143). Last, central IL-10 administration following corticectomy in adult female CD1 mice results in a reduction in the number of reactive astrocytes and TNFα levels at 4 days (141), corroborating previous studies showing a reduction in neuroinflammation with IL-10 treatment.
A couple of studies with drugs or therapeutic regimens have demonstrated improved TBI outcomes through increased IL-10 protein levels and IL-10-dependent mechanisms. TBI has a significant ischemic injury component associated with poor cerebral blood flow (157). Hyperbaric oxygen (HBO) therapy has been suggested to provide some therapeutic benefit in conditions where poor blood flow and hypoxia lead to secondary tissue injury, and HBO therapy following TBI improves outcomes (142, 158). HBO therapy increases IL-10 at 4 days in adult Sprague-Dawley rats and mice above the levels induced by TBI alone in both the lateral fluid percussion (146) and the controlled cortical impact (142) models. The reported beneficial effects of HBO therapy after TBI are thought to be through an IL-10-dependent mechanism, as the smaller lesion volumes, less edema, improved motor and cognitive recovery, decreased inflammation, reduced apoptosis and BBB dysfunction 4 weeks after TBI with HBO therapy are only seen in WT mice and not in IL-10−/− mice. Triptolide, an anti-inflammatory molecule, given immediately after TBI significantly increases IL-10 levels in the brain after 1 day and attenuates increases in proinflammatory cytokines; improves neurobehavioral outcomes; and reduces edema, contusion volume, and apoptosis (144).
IL-10 in Clinical Studies
The temporal profile of IL-10 in clinical TBI studies is not conclusive. Early reports found that plasma IL-10 levels peak within the first 3 h (151, 152), while a later study showed the peak is between 5 and 6 days post-injury (148). IL-10 levels may remain elevated for up to 22 days (131, 139) or even up to 6 months (137), and in some cases, there is a second peak in concentration (131, 152). In contrast to this general pattern of a rise in IL-10 levels followed by a gradual decline in the CSF and plasma (153), others have been unable to identify any pattern of IL-10 levels in microdialysate (115). Another study in patients with a severely disrupted BBB found high initial CSF IL-10 levels that decreased over time and serum IL-10 levels that increased over time (156), supporting a heightened intrathecal IL-10 synthesis after TBI with overflow of the cytokine into the systemic circulation (137), augmenting baseline systemic IL-10 levels. However, there is inconsistency in the literature as to whether IL-10 levels are more dramatically increased in the CSF or serum following TBI (131, 139), rendering it difficult to determine the source of increased IL-10 levels. This discrepancy likely relates to isolated head injury versus multiple injuries, as one study found that serum levels were higher than CSF levels in patients with additional injuries; however, in patients with isolated TBI, CSF concentrations were greater than or equal to the corresponding serum levels (155). Isolated head injuries have also been shown to result in either no difference in serum IL-10 levels (129) or lower IL-10 levels than that seen with multiple injuries (151, 152). Moreover, serum levels of IL-10 are elevated in trauma patients with and without brain injury (150, 159). Thus, the heterogeneous nature of clinical TBI patients may be in part responsible for the inconclusive patterns and biocompartmental distribution of IL-10 following TBI.
Similarly, it seems that clinical severity measures and TBI complications such as GCS, various Injury Scores/Scales, and BBB dysfunction/disruption have not been definitively correlated with IL-10 levels. Although most studies found no correlation with GCS (129, 130, 153–155) or Injury Severity Score (129, 155) and IL-10 levels, a minority endorse a significant association (149, 151). Blood–brain barrier BBB dysfunction/disruption is one component of secondary brain damage after TBI (8, 135, 139, 147, 160), and although more severe BBB dysfunction is evident in non-survivors of TBI (156), no significant correlation can be made between IL-10 levels in either the serum or CSF and the degree of BBB disruption (131, 139, 156). Disruption to BBB integrity often contributes to elevated intracranial pressure (ICP), another common complication of TBI (140, 161) that is associated with poor outcomes (136, 140). Although high ICP coupled with sympathetic activation has been shown to increase systemic IL-10 levels (162), and some studies have demonstrated higher IL-10 levels in patients with both high ICP and unfavorable outcomes (155, 156), not all studies have found such an association between ICP and IL-10 levels (131, 150). Although some interleukins exhibit a stronger and more prolonged response in females (115), no studies have found a clear association between IL-10 levels and gender (115, 130). One study has shown that increased IL-10 levels after TBI are associated with age, such that increased levels after TBI are found with ages less than 4 years (154).
Interleukin-10 is elevated after TBI and exhibits a more prolonged response when compared to other cytokines (115), and although many have attempted to correlate IL-10 levels with outcomes, this effort has not been reliably successful (129, 130, 136, 139, 150, 153, 163–166). However, a few studies did find that elevated IL-10 concentrations are associated with unfavorable outcomes (137, 149, 154–156). Higher CSF IL-10 in pediatric patients (154) and elevated serum and CSF IL-10 levels in adult patients were significantly associated with mortality (156). Non-survivors demonstrate higher serum IL-10 levels at admission that continued to rise over the next 24 h, whereas survivors have lower IL-10 serum levels at admission that subsequently decrease (156). Additionally, non-survivors have higher CSF IL-10 levels at admission; however, both the non-survivors and survivors demonstrate decreasing CSF IL-10 levels over time (156). In another study, elevated IL-10 levels at 10 or 30 h after TBI were 6 and 5 times more frequently associated with hospital mortality, independent of GCS, age, and systemic trauma (149). It appears that early IL-10 correlates best with outcome, as both admission CSF and serum IL-10 levels within the first 30 h correlate with mortality, whereas later time points do not (149, 156). Increased CSF and serum levels correlate with a GCS score less than 4 (137, 155). Additionally, in a multivariate analysis, one study found that a higher IL-6/IL-10 ratio from 2 weeks to 3 months was significantly associated with a GCS score less than 4 at 6 months (78). Hypothermia is known to attenuate the proinflammatory response following TBI147; however, monitoring of IL-10 levels in severe TBI patients divided into hypothermia and normothermia treatment groups revealed no significant differences in CSF or serum IL-10 levels, nor outcomes in pediatric or adult patients (130, 153, 167).
It is well documented that IL-10 levels in humans and experimental models increase in both the serum and CSF shortly after TBI (8, 129, 135, 151, 152, 168) and remain elevated for many days (131, 152) followed by a slow decline. However, due to methodological variations in studies, the prognostic and diagnostic value of this cytokine remains unclear.
Summary and Comparison of the Role of IL-10 in Preclinical and Clinical TBI Studies
Traumatic brain injury is a heterogenous type of acute brain injury that involves a complex interplay of both direct primary injury and secondary injury, the latter of which is closely linked to neuroinflammatory processes. Preclinical studies have shown that IL-10 reduces neuroinflammation following brain trauma and, in general, IL-10 treatment improves neurological outcomes after TBI. Clinically, several studies have shown that IL-10 levels increase after TBI, although the temporal profile of IL-10 levels and whether IL-10 correlates with initial injury severity is less clear. Regarding the prognostic potential, it appears that IL-10 has the most utility in predicting mortality after TBI, although given the controversy in the literature, future work is necessary to further define the role of IL-10 in predicting other outcomes.
Conclusion
Interleukin-10 is significantly elevated following brain injury and appears to play a variety of roles depending on the type of acute neurologic insult, where it interacts with each condition’s overlapping, yet distinct, pathophysiology and secondary complications. High IL-10 levels tend to predict worse outcomes after hemorrhagic brain injury, whereas the converse is true for brain ischemia, low IL-10 levels resulting from SNPs increase the risk for IS and low levels after IS predict worse outcome. While IL-10 appears to have prognostic value, comparatively far fewer studies report on diagnostic potential. In the limited work in the literature, significant controversy exists where some report no correlation with measures of initial brain injury severity and others report significant correlations. From a therapeutic perspective, preclinical models have shown that IL-10 administration after IS and TBI lend better outcomes, although no work has been done in this area for SAH or ICH. Intertwined with all these acute pathologic processes are the effects of IL-10 on the vasculature, where it is crucial for protection. Finally, the varied effects and roles of IL-10 after IS, SAH, ICH, and TBI likely stems from pathology-dependent differences in the temporal balance of pro- and anti-inflammatory mediators. The latter point is particularly important in the context of IL-10, given its pleiotropic immunomodulatory functions that polarize the inflammatory system to an anti-inflammatory phenotype, aiding in the resolution of the neuroinflammation. It is likely that the ability of IL-10 to overcome the proinflammatory milieu is temporally different between the various forms of acute brain injury, with delays resulting in prolonged inflammation that exacerbates secondary brain damage, leading to worse outcomes. Novel therapies targeted to control inflammation will hinge on an understanding of the complex balance of the pro- and anti-inflammatory mediators, of which IL-10 plays a central overarching role. This represents an exciting avenue of research that will hopefully usher unique immunomodulatory therapies, changing the management of patients with acute neurologic injury from supportive measures to active therapeutic care to improve patient outcomes.
Author Contributions
JG and SS drafted the manuscript. JL and SD made substantial contributions to the conception and design of the work and wrote the manuscript. JL, HP, NE, SR, BH, SB, and SD revised the manuscript. All authors have approved the manuscript for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank members of the Dorelab, the University of Florida Center for Translational Research in Neurodegenerative Disease, and the McKnight Brain Institute. We also thank Corey Astrom for her expert editorial assistance.
Funding
This work was supported by grants from the McKnight Brain Research Foundation, Brain and Spinal Cord Injury Research Trust Fund (SD), and National Institutes of Health T35HL007489 (JG and SS), F31NS086441 (JL), AHA33450010 (SD), R01NS046400 (SD), R01AT007429 (SD), and R21NS095166 (BH, SB, and SD).
References
1. Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics – 2014 update: a report from the American Heart Association. Circulation (2014) 129(3):e28–292. doi: 10.1161/01.cir.0000441139.02102.80
2. Sauser K, Burke JF, Reeves MJ, Barsan WG, Levine DA. A systematic review and critical appraisal of quality measures for the emergency care of acute ischemic stroke. Ann Emerg Med (2014) 64(3):235–44.e5. doi:10.1016/j.annemergmed.2014.01.034
3. Centers for Disease C, Prevention. Prevalence of stroke – United States, 2006–2010. MMWR Morb Mortal Wkly Rep (2012) 61(20):379–82.
4. Ma VY, Chan L, Carruthers KJ. Incidence, prevalence, costs, and impact on disability of common conditions requiring rehabilitation in the United States: stroke, spinal cord injury, traumatic brain injury, multiple sclerosis, osteoarthritis, rheumatoid arthritis, limb loss, and back pain. Arch Phys Med Rehabil (2014) 95(5):986–95.e1. doi:10.1016/j.apmr.2013.10.032
5. Zaloshnja E, Miller T, Langlois JA, Selassie AW. Prevalence of long-term disability from traumatic brain injury in the civilian population of the United States, 2005. J Head Trauma Rehabil (2008) 23(6):394–400. doi:10.1097/01.HTR.0000341435.52004.ac
6. Namjoshi DR, Good C, Cheng WH, Panenka W, Richards D, Cripton PA, et al. Towards clinical management of traumatic brain injury: a review of models and mechanisms from a biomechanical perspective. Dis Model Mech (2013) 6(6):1325–38. doi:10.1242/dmm.011320
7. Lee WI, Mitchell P, Dowling R, Yan B. Clinical factors are significant predictors of outcome post intra-arterial therapy for acute ischaemic stroke: a review. J Neuroradiol (2013) 40(5):315–25. doi:10.1016/j.neurad.2013.09.002
8. Cederberg D, Siesjo P. What has inflammation to do with traumatic brain injury? Childs Nerv Syst (2010) 26(2):221–6. doi:10.1007/s00381-009-1029-x
9. Patterson ZR, Holahan MR. Understanding the neuroinflammatory response following concussion to develop treatment strategies. Front Cell Neurosci (2012) 6:58. doi:10.3389/fncel.2012.00058
10. Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics (2010) 7(1):22–30. doi:10.1016/j.nurt.2009.10.016
11. Giunta B, Obregon D, Velisetty R, Sanberg PR, Borlongan CV, Tan J. The immunology of traumatic brain injury: a prime target for Alzheimer’s disease prevention. J Neuroinflammation (2012) 9:185. doi:10.1186/1742-2094-9-185
12. Wang J, Doré S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab (2007) 27(5):894–908. doi:10.1038/sj.jcbfm.9600403
13. Dumont AS, Dumont RJ, Chow MM, Lin CL, Calisaneller T, Ley KF, et al. Cerebral vasospasm after subarachnoid hemorrhage: putative role of inflammation. Neurosurgery (2003) 53(1):123–33; discussion 33–5. doi:10.1227/01.NEU.0000068863.37133.9E
14. Miller BA, Turan N, Chau M, Pradilla G. Inflammation, vasospasm, and brain injury after subarachnoid hemorrhage. Biomed Res Int (2014) 2014:384342. doi:10.1155/2014/384342
15. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med (2011) 17(7):796–808. doi:10.1038/nm.2399
16. Strle K, Zhou JH, Shen WH, Broussard SR, Johnson RW, Freund GG, et al. Interleukin-10 in the brain. Crit Rev Immunol (2001) 21(5):427–49. doi:10.1615/CritRevImmunol.v21.i5.20
17. Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol (2001) 19:683–765. doi:10.1146/annurev.immunol.19.1.683
18. MacKenzie KF, Pattison MJ, Arthur JS. Transcriptional regulation of IL-10 and its cell-specific role in vivo. Crit Rev Immunol (2014) 34(4):315–45. doi:10.1615/CritRevImmunol.2014010694
19. Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol (2010) 10(3):170–81. doi:10.1038/nri2711
20. Mosser DM, Zhang X. Interleukin-10: new perspectives on an old cytokine. Immunol Rev (2008) 226:205–18. doi:10.1111/j.1600-065X.2008.00706.x
21. del Zoppo GJ, Hallenbeck JM. Advances in the vascular pathophysiology of ischemic stroke. Thromb Res (2000) 98(3):73–81. doi:10.1016/S0049-3848(00)00218-8
22. Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis (2008) 32(2):200–19. doi:10.1016/j.nbd.2008.08.005
23. Alves JL. Blood-brain barrier and traumatic brain injury. J Neurosci Res (2014) 92(2):141–7. doi:10.1002/jnr.23300
24. Keep RF, Zhou N, Xiang J, Andjelkovic AV, Hua Y, Xi G. Vascular disruption and blood-brain barrier dysfunction in intracerebral hemorrhage. Fluids Barriers CNS (2014) 11:18. doi:10.1186/2045-8118-11-18
25. Dammanahalli JK, Wang X, Sun Z. Genetic interleukin-10 deficiency causes vascular remodeling via the upregulation of Nox1. J Hypertens (2011) 29(11):2116–25. doi:10.1097/HJH.0b013e32834b22a0
26. Kinzenbaw DA, Chu Y, Pena Silva RA, Didion SP, Faraci FM. Interleukin-10 protects against aging-induced endothelial dysfunction. Physiol Rep (2013) 1(6):e00149. doi:10.1002/phy2.149
27. Zemse SM, Hilgers RH, Simkins GB, Rudic RD, Webb RC. Restoration of endothelin-1-induced impairment in endothelium-dependent relaxation by interleukin-10 in murine aortic rings. Can J Physiol Pharmacol (2008) 86(8):557–65. doi:10.1139/Y08-049
28. Didion SP, Kinzenbaw DA, Schrader LI, Chu Y, Faraci FM. Endogenous interleukin-10 inhibits angiotensin II-induced vascular dysfunction. Hypertension (2009) 54(3):619–24. doi:10.1161/HYPERTENSIONAHA.109.137158
29. Kang H, Yang PY, Rui YC. Adenovirus viral interleukin-10 inhibits adhesion molecule expressions induced by hypoxia/reoxygenation in cerebrovascular endothelial cells. Acta Pharmacol Sin (2008) 29(1):50–6. doi:10.1111/j.1745-7254.2008.00718.x
30. Huet O, Laemmel E, Fu Y, Dupic L, Aprico A, Andrews KL, et al. Interleukin 10 antioxidant effect decreases leukocytes/endothelial interaction induced by tumor necrosis factor alpha. Shock (2013) 39(1):83–8. doi:10.1097/SHK.0b013e318278ae36
31. Zemse SM, Chiao CW, Hilgers RH, Webb RC. Interleukin-10 inhibits the in vivo and in vitro adverse effects of TNF-alpha on the endothelium of murine aorta. Am J Physiol Heart Circ Physiol (2010) 299(4):H1160–7. doi:10.1152/ajpheart.00763.2009
32. Zemse SM, Hilgers RH, Webb RC. Interleukin-10 counteracts impaired endothelium-dependent relaxation induced by ANG II in murine aortic rings. Am J Physiol Heart Circ Physiol (2007) 292(6):H3103–8. doi:10.1152/ajpheart.00456.2006
33. Sikka G, Miller KL, Steppan J, Pandey D, Jung SM, Fraser CD III, et al. Interleukin 10 knockout frail mice develop cardiac and vascular dysfunction with increased age. Exp Gerontol (2013) 48(2):128–35. doi:10.1016/j.exger.2012.11.001
34. Kassan M, Galan M, Partyka M, Trebak M, Matrougui K. Interleukin-10 released by CD4(+)CD25(+) natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arterioscler Thromb Vasc Biol (2011) 31(11):2534–42. doi:10.1161/ATVBAHA.111.233262
35. Kaiser D, Weise G, Moller K, Scheibe J, Posel C, Baasch S, et al. Spontaneous white matter damage, cognitive decline and neuroinflammation in middle-aged hypertensive rats: an animal model of early-stage cerebral small vessel disease. Acta Neuropathol Commun (2014) 2:169. doi:10.1186/s40478-014-0169-8
36. Gunnett CA, Heistad DD, Berg DJ, Faraci FM. IL-10 deficiency increases superoxide and endothelial dysfunction during inflammation. Am J Physiol Heart Circ Physiol (2000) 279(4):H1555–62.
37. Londono D, Carvajal J, Strle K, Kim KS, Cadavid D. IL-10 Prevents apoptosis of brain endothelium during bacteremia. J Immunol (2011) 186(12):7176–86. doi:10.4049/jimmunol.1100060
38. Lisinski TJ, Furie MB. Interleukin-10 inhibits proinflammatory activation of endothelium in response to Borrelia burgdorferi or lipopolysaccharide but not interleukin-1beta or tumor necrosis factor alpha. J Leukoc Biol (2002) 72(3):503–11.
39. Appelros P, Nydevik I, Viitanen M. Poor outcome after first-ever stroke: predictors for death, dependency, and recurrent stroke within the first year. Stroke (2003) 34(1):122–6. doi:10.1161/01.STR.0000047852.05842.3C
40. Kelly-Hayes M, Beiser A, Kase CS, Scaramucci A, D’Agostino RB, Wolf PA. The influence of gender and age on disability following ischemic stroke: the Framingham study. J Stroke Cerebrovasc Dis (2003) 12(3):119–26. doi:10.1016/S1052-3057(03)00042-9
41. Fouda AY, Kozak A, Alhusban A, Switzer JA, Fagan SC. Anti-inflammatory IL-10 is upregulated in both hemispheres after experimental ischemic stroke: hypertension blunts the response. Exp Transl Stroke Med (2013) 5(1):12. doi:10.1186/2040-7378-5-12
42. Grilli M, Barbieri I, Basudev H, Brusa R, Casati C, Lozza G, et al. Interleukin-10 modulates neuronal threshold of vulnerability to ischaemic damage. Eur J Neurosci (2000) 12(7):2265–72. doi:10.1046/j.1460-9568.2000.00090.x
43. Perez-de Puig I, Miro F, Salas-Perdomo A, Bonfill-Teixidor E, Ferrer-Ferrer M, Marquez-Kisinousky L, et al. IL-10 deficiency exacerbates the brain inflammatory response to permanent ischemia without preventing resolution of the lesion. J Cereb Blood Flow Metab (2013) 33(12):1955–66. doi:10.1038/jcbfm.2013.155
44. Frenkel D, Huang Z, Maron R, Koldzic DN, Moskowitz MA, Weiner HL. Neuroprotection by IL-10-producing MOG CD4+ T cells following ischemic stroke. J Neurol Sci (2005) 233(1–2):125–32. doi:10.1016/j.jns.2005.03.022
45. Frenkel D, Huang Z, Maron R, Koldzic DN, Hancock WW, Moskowitz MA, et al. Nasal vaccination with myelin oligodendrocyte glycoprotein reduces stroke size by inducing IL-10-producing CD4+ T cells. J Immunol (2003) 171(12):6549–55. doi:10.4049/jimmunol.171.12.6549
46. Spera PA, Ellison JA, Feuerstein GZ, Barone FC. IL-10 reduces rat brain injury following focal stroke. Neurosci Lett (1998) 251(3):189–92. doi:10.1016/S0304-3940(98)00537-0
47. Liesz A, Bauer A, Hoheisel JD, Veltkamp R. Intracerebral interleukin-10 injection modulates post-ischemic neuroinflammation: an experimental microarray study. Neurosci Lett (2014) 579:18–23. doi:10.1016/j.neulet.2014.07.003
48. de Bilbao F, Arsenijevic D, Moll T, Garcia-Gabay I, Vallet P, Langhans W, et al. In vivo over-expression of interleukin-10 increases resistance to focal brain ischemia in mice. J Neurochem (2009) 110(1):12–22. doi:10.1111/j.1471-4159.2009.06098.x
49. Liang QJ, Jiang M, Wang XH, Le LL, Xiang M, Sun N, et al. Pre-existing interleukin 10 in cerebral arteries attenuates subsequent brain injury caused by ischemia/reperfusion. IUBMB Life (2015) 67(9):710–9. doi:10.1002/iub.1429
50. Nomoto T, Okada T, Shimazaki K, Yoshioka T, Nonaka-Sarukawa M, Ito T, et al. Systemic delivery of IL-10 by an AAV vector prevents vascular remodeling and end-organ damage in stroke-prone spontaneously hypertensive rat. Gene Ther (2009) 16(3):383–91. doi:10.1038/gt.2008.151
51. Wang Y, Jia J, Ao G, Hu L, Liu H, Xiao Y, et al. Hydrogen sulfide protects blood-brain barrier integrity following cerebral ischemia. J Neurochem (2014) 129(5):827–38. doi:10.1111/jnc.12695
52. Hwang CJ, Yun HM, Jung YY, Lee DH, Yoon NY, Seo HO, et al. Reducing effect of IL-32alpha in the development of stroke through blocking of NF-kappaB, but enhancement of STAT3 pathways. Mol Neurobiol (2015) 51(2):648–60. doi:10.1007/s12035-014-8739-0
53. Liesz A, Zhou W, Na SY, Hammerling GJ, Garbi N, Karcher S, et al. Boosting regulatory T cells limits neuroinflammation in permanent cortical stroke. J Neurosci (2013) 33(44):17350–62. doi:10.1523/JNEUROSCI.4901-12.2013
54. Na SY, Mracsko E, Liesz A, Hunig T, Veltkamp R. Amplification of regulatory T cells using a CD28 superagonist reduces brain damage after ischemic stroke in mice. Stroke (2015) 46(1):212–20. doi:10.1161/STROKEAHA.114.007756
55. Kawada H, Takizawa S, Takanashi T, Morita Y, Fujita J, Fukuda K, et al. Administration of hematopoietic cytokines in the subacute phase after cerebral infarction is effective for functional recovery facilitating proliferation of intrinsic neural stem/progenitor cells and transition of bone marrow-derived neuronal cells. Circulation (2006) 113(5):701–10. doi:10.1161/CIRCULATIONAHA.105.563668
56. Morita Y, Takizawa S, Kamiguchi H, Uesugi T, Kawada H, Takagi S. Administration of hematopoietic cytokines increases the expression of anti-inflammatory cytokine (IL-10) mRNA in the subacute phase after stroke. Neurosci Res (2007) 58(4):356–60. doi:10.1016/j.neures.2007.04.006
57. Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. Treatment of experimental stroke with IL-10-producing B-cells reduces infarct size and peripheral and CNS inflammation in wild-type B-cell-sufficient mice. Metab Brain Dis (2014) 29(1):59–73. doi:10.1007/s11011-013-9474-3
58. Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. IL-10-producing B-cells limit CNS inflammation and infarct volume in experimental stroke. Metab Brain Dis (2013) 28(3):375–86. doi:10.1007/s11011-013-9413-3
59. Wang J, Xie L, Yang C, Ren C, Zhou K, Wang B, et al. Activated regulatory T cell regulates neural stem cell proliferation in the subventricular zone of normal and ischemic mouse brain through interleukin 10. Front Cell Neurosci (2015) 9:361. doi:10.3389/fncel.2015.00361
60. Liu N, Chen R, Du H, Wang J, Zhang Y, Wen J. Expression of IL-10 and TNF-alpha in rats with cerebral infarction after transplantation with mesenchymal stem cells. Cell Mol Immunol (2009) 6(3):207–13. doi:10.1038/cmi.2009.28
61. Du HW, Liu N, Wang JH, Zhang YX, Chen RH, Xiao YC. [The effects of adipose-derived stem cell transplantation on the expression of IL-10 and TNF-alpha after cerebral ischaemia in rats]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi (2009) 25(11):998–1001.
62. Du G, Liu Y, Dang M, Zhu G, Su R, Fan Y, et al. Comparison of administration routes for adipose-derived stem cells in the treatment of middle cerebral artery occlusion in rats. Acta Histochem (2014) 116(6):1075–84. doi:10.1016/j.acthis.2014.05.002
63. Munshi A, Rajeshwar K, Kaul S, Al-Hazzani A, Alshatwi AA, Sai Babu M, et al. Interleukin-10-1082 promoter polymorphism and ischemic stroke risk in a South Indian population. Cytokine (2010) 52(3):221–4. doi:10.1016/j.cyto.2010.09.013
64. Yin GT, Ma YT, Zheng YY, Yang YN, Li XM, Fu ZY, et al. Polymorphisms of interleukin-10 genes on the risk of ischemic stroke in a meta-analysis. Int J Clin Exp Med (2015) 8(2):1888–95.
65. Jiang XH, Lin KX, Zhang YX, Chen RH, Liu N. Correlating interleukin-10 promoter gene polymorphisms with human cerebral infarction onset. Neural Regen Res (2015) 10(11):1809–13. doi:10.4103/1673-5374.170308
66. Ozkan A, Silan F, Uludag A, Degirmenci Y, Ozisik Karaman HI. Tumour necrosis factor alpha, interleukin 10 and interleukin 6 gene polymorphisms of ischemic stroke patients in south Marmara region of Turkey. Int J Clin Exp Pathol (2015) 8(10):13500–4.
67. He W, Song H, Ding L, Li C, Dai L, Gao S. Association between IL-10 gene polymorphisms and the risk of ischemic stroke in a Chinese population. Int J Clin Exp Pathol (2015) 8(10):13489–94.
68. Marousi S, Ellul J, Antonacopoulou A, Gogos C, Papathanasopoulos P, Karakantza M. Functional polymorphisms of interleukin 4 and interleukin 10 may predict evolution and functional outcome of an ischaemic stroke. Eur J Neurol (2011) 18(4):637–43. doi:10.1111/j.1468-1331.2010.03228.x
69. Kumar P, Yadav AK, Misra S, Kumar A, Chakravarty K, Prasad K. Role of interleukin-10 (1082A/G) gene polymorphism with the risk of ischemic stroke: a meta-analysis. Neurol Res (2016) 38(9):823–30. doi:10.1080/01616412.2016.1202395
70. Xie G, Myint PK, Zaman MJ, Li Y, Zhao L, Shi P, et al. Relationship of serum interleukin-10 and its genetic variations with ischemic stroke in a Chinese general population. PLoS One (2013) 8(9):e74126. doi:10.1371/journal.pone.0074126
71. Park HK, Kim DH, Yun DH, Ban JY. Association between IL10, IL10RA, and IL10RB SNPs and ischemic stroke with hypertension in Korean population. Mol Biol Rep (2013) 40(2):1785–90. doi:10.1007/s11033-012-2232-5
72. Timasheva YR, Nasibullin TR, Zakirova AN, Mustafina OE. Association of interleukin-6, interleukin-12, and interleukin-10 gene polymorphisms with essential hypertension in Tatars from Russia. Biochem Genet (2008) 46(1–2):64–74. doi:10.1007/s10528-007-9130-x
73. Stoica AL, Stoica E, Constantinescu I, Uscatescu V, Ginghina C. Interleukin-6 and interleukin-10 gene polymorphism, endothelial dysfunction, and postoperative prognosis in patients with peripheral arterial disease. J Vasc Surg (2010) 52(1):103–9. doi:10.1016/j.jvs.2010.01.088
74. van Exel E, Gussekloo J, de Craen AJ, Bootsma-van der Wiel A, Frolich M, Westendorp RG. Inflammation and stroke: the Leiden 85-Plus study. Stroke (2002) 33(4):1135–8. doi:10.1161/01.STR.0000014206.05597.9E
75. Nikiforova Postnikova TA, Doronin BM, Peskov SA. [Blood serum cytokines as predictors of hemorrhagic transformation of ischemic stroke]. Zh Nevrol Psikhiatr Im S S Korsakova (2014) 114(3 vypusk 2 Insul’t):20–6.
76. Vila N, Castillo J, Davalos A, Esteve A, Planas AM, Chamorro A. Levels of anti-inflammatory cytokines and neurological worsening in acute ischemic stroke. Stroke (2003) 34(3):671–5. doi:10.1161/01.STR.0000057976.53301.69
77. Protti GG, Gagliardi RJ, Forte WC, Sprovieri SR. Interleukin-10 may protect against progressing injury during the acute phase of ischemic stroke. Arq Neuropsiquiatr (2013) 71(11):846–51. doi:10.1590/0004-282X20130168
78. Singh HV, Pandey A, Shrivastava AK, Raizada A, Singh SK, Singh N. Prognostic value of neuron specific enolase and IL-10 in ischemic stroke and its correlation with degree of neurological deficit. Clin Chim Acta (2013) 419:136–8. doi:10.1016/j.cca.2013.02.014
79. Nayak AR, Kashyap RS, Purohit HJ, Kabra D, Taori GM, Daginawala HF. Evaluation of the inflammatory response in sera from acute ischemic stroke patients by measurement of IL-2 and IL-10. Inflamm Res (2009) 58(10):687–91. doi:10.1007/s00011-009-0036-4
80. Hu Y, Zheng Y, Wu Y, Ni B, Shi S. Imbalance between IL-17A-producing cells and regulatory T cells during ischemic stroke. Mediators Inflamm (2014) 2014:813045. doi:10.1155/2014/813045
81. Arponen O, Muuronen A, Taina M, Sipola P, Hedman M, Jakala P, et al. Acute phase IL-10 plasma concentration associates with the high risk sources of cardiogenic stroke. PLoS One (2015) 10(4):e0120910. doi:10.1371/journal.pone.0120910
82. Chamorro A, Amaro S, Vargas M, Obach V, Cervera A, Torres F, et al. Interleukin 10, monocytes and increased risk of early infection in ischaemic stroke. J Neurol Neurosurg Psychiatry (2006) 77(11):1279–81. doi:10.1136/jnnp.2006.100800
83. Worthmann H, Tryc AB, Dirks M, Schuppner R, Brand K, Klawonn F, et al. Lipopolysaccharide binding protein, interleukin-10, interleukin-6 and C-reactive protein blood levels in acute ischemic stroke patients with post-stroke infection. J Neuroinflammation (2015) 12:13. doi:10.1186/s12974-014-0231-2
84. Pelidou SH, Kostulas N, Matusevicius D, Kivisakk P, Kostulas V, Link H. High levels of IL-10 secreting cells are present in blood in cerebrovascular diseases. Eur J Neurol (1999) 6(4):437–42. doi:10.1046/j.1468-1331.1999.640437.x
85. Chang LT, Yuen CM, Liou CW, Lu CH, Chang WN, Youssef AA, et al. Link between interleukin-10 level and outcome after ischemic stroke. Neuroimmunomodulation (2010) 17(4):223–8. doi:10.1159/000290038
86. Sahan M, Sebe A, Acikalin A, Akpinar O, Koc F, Ay MO, et al. Acute-phase reactants and cytokines in ischemic stroke: do they have any relationship with short-term mortality? Eur Rev Med Pharmacol Sci (2013) 17(20):2773–7.
87. Sharma S, Yang B, Xi X, Grotta JC, Aronowski J, Savitz SI. IL-10 directly protects cortical neurons by activating PI-3 kinase and STAT-3 pathways. Brain Res (2011) 1373:189–94. doi:10.1016/j.brainres.2010.11.096
88. Zhu J, Wang H, Fan Y, Lin Y, Zhang L, Ji X, et al. Targeting the NF-E2-related factor 2 pathway: a novel strategy for glioblastoma (review). Oncol Rep (2014) 32(2):443–50. doi:10.3892/or.2014.3259
89. Leonardo CC, Doré S. Dietary flavonoids are neuroprotective through Nrf2-coordinated induction of endogenous cytoprotective proteins. Nutr Neurosci (2011) 14(5):226–36. doi:10.1179/1476830511Y.0000000013
90. Segev-Amzaleg N, Trudler D, Frenkel D. Preconditioning to mild oxidative stress mediates astroglial neuroprotection in an IL-10-dependent manner. Brain Behav Immun (2013) 30:176–85. doi:10.1016/j.bbi.2012.12.016
91. Xing C, Wang X, Cheng C, Montaner J, Mandeville E, Leung W, et al. Neuronal production of lipocalin-2 as a help me signal for glial activation. Stroke (2014) 45(7):2085–92. doi:10.1161/STROKEAHA.114.005733
92. Driessler F, Venstrom K, Sabat R, Asadullah K, Schottelius AJ. Molecular mechanisms of interleukin-10-mediated inhibition of NF-κB activity: a role for p50. Clin Exp Immunol (2004) 135(1):64–73. doi:10.1111/j.1365-2249.2004.02342.x
93. Jin J, Li W, Peng L, Chen J, Li R, Wu P, et al. Relationship between interleukin-10 1082A/G polymorphism and risk of ischemic stroke: a meta-analysis. PLoS One (2014) 9(4):e94631. doi:10.1371/journal.pone.0094631
94. Li WZ, Gao CY, He WL, Zhang HM. Association of the interleukin-10 gene 1082A/G genetic polymorphism with risk of ischemic stroke in a Chinese population. Genet Mol Res (2016) 15(1). doi:10.4238/gmr.15017541
95. Zhang S, He WB, Chen NH. Causes of death among persons who survive an acute ischemic stroke. Curr Neurol Neurosci Rep (2014) 14(8):467. doi:10.1007/s11910-014-0467-3
96. Rincon F, Rossenwasser RH, Dumont A. The epidemiology of admissions of nontraumatic subarachnoid hemorrhage in the United States. Neurosurgery (2013) 73(2):217–22; discussion 2–3. doi:10.1227/01.neu.0000430290.93304.33
97. Marder CP, Narla V, Fink JR, Tozer Fink KR. Subarachnoid hemorrhage: beyond aneurysms. AJR Am J Roentgenol (2014) 202(1):25–37. doi:10.2214/AJR.12.9749
98. Connolly ES Jr, Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke (2012) 43(6):1711–37. doi:10.1161/STR.0b013e3182587839
99. Velat GJ, Kimball MM, Mocco JD, Hoh BL. Vasospasm after aneurysmal subarachnoid hemorrhage: review of randomized controlled trials and meta-analyses in the literature. World Neurosurg (2011) 76(5):446–54. doi:10.1016/j.wneu.2011.02.030
100. Sabri M, Lass E, Macdonald RL. Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res Treat (2013) 2013:394036. doi:10.1155/2013/394036
101. Caner B, Hou J, Altay O, Fujii M, Zhang JH. Transition of research focus from vasospasm to early brain injury after subarachnoid hemorrhage. J Neurochem (2012) 123(Suppl 2):12–21. doi:10.1111/j.1471-4159.2012.07939.x
102. Sozen T, Tsuchiyama R, Hasegawa Y, Suzuki H, Jadhav V, Nishizawa S, et al. Role of interleukin-1beta in early brain injury after subarachnoid hemorrhage in mice. Stroke (2009) 40(7):2519–25. doi:10.1161/STROKEAHA.109.549592
103. Macdonald RL, Weir BK. Cerebral vasospasm and free radicals. Free Radic Biol Med (1994) 16(5):633–43. doi:10.1016/0891-5849(94)90064-7
104. Imperatore C, Germano A, D’Avella D, Tomasello F, Costa G. Effects of the radical scavenger AVS on behavioral and BBB changes after experimental subarachnoid hemorrhage. Life Sci (2000) 66(9):779–90. doi:10.1016/S0024-3205(99)00651-7
105. Schwartz AY, Sehba FA, Bederson JB. Decreased nitric oxide availability contributes to acute cerebral ischemia after subarachnoid hemorrhage. Neurosurgery (2000) 47(1):208–14; discussion 14–5. doi:10.1097/00006123-200007000-00042
106. Afshar JK, Pluta RM, Boock RJ, Thompson BG, Oldfield EH. Effect of intracarotid nitric oxide on primate cerebral vasospasm after subarachnoid hemorrhage. J Neurosurg (1995) 83(1):118–22. doi:10.3171/jns.1995.83.1.0118
107. Vergouwen MD, Algra A, Rinkel GJ. Endothelin receptor antagonists for aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis update. Stroke (2012) 43(10):2671–6. doi:10.1161/STROKEAHA.112.666693
108. Hishikawa T, Ono S, Ogawa T, Tokunaga K, Sugiu K, Date I. Effects of deferoxamine-activated hypoxia-inducible factor-1 on the brainstem after subarachnoid hemorrhage in rats. Neurosurgery (2008) 62(1):232–40; discussion 40–1. doi:10.1227/01.NEU.0000311082.88766.33
109. Macdonald RL, Weir BK. A review of hemoglobin and the pathogenesis of cerebral vasospasm. Stroke (1991) 22(8):971–82. doi:10.1161/01.STR.22.8.971
110. Ohnishi H, Iihara K, Kaku Y, Yamauchi K, Fukuda K, Nishimura K, et al. Haptoglobin phenotype predicts cerebral vasospasm and clinical deterioration after aneurysmal subarachnoid hemorrhage. J Stroke Cerebrovasc Dis (2013) 22(4):520–6. doi:10.1016/j.jstrokecerebrovasdis.2013.02.005
111. Aihara Y, Kasuya H, Onda H, Hori T, Takeda J. Quantitative analysis of gene expressions related to inflammation in canine spastic artery after subarachnoid hemorrhage. Stroke (2001) 32(1):212–7. doi:10.1161/01.STR.32.1.212
112. Song JN, Zhang M, Li DD, Li M, An JY, Cheng MF, et al. Dynamic expression of the suppressor of cytokine signaling-3 and cytokines in the cerebral basilar artery of rats with subarachnoid hemorrhage, and the effect of acetylcholine. Acta Neurochir (Wien) (2014) 156(5):941–9; discussion 9. doi:10.1007/s00701-014-1998-8
113. Kooijman E, Nijboer CH, van Velthoven CT, Mol W, Dijkhuizen RM, Kesecioglu J, et al. Long-term functional consequences and ongoing cerebral inflammation after subarachnoid hemorrhage in the rat. PLoS One (2014) 9(3):e90584. doi:10.1371/journal.pone.0090584
114. Broessner G, Lackner P, Fischer M, Beer R, Helbok R, Pfausler B, et al. Influence of prophylactic, endovascularly based normothermia on inflammation in patients with severe cerebrovascular disease: a prospective, randomized trial. Stroke (2010) 41(12):2969–72. doi:10.1161/STROKEAHA.110.591933
115. Mellergard P, Aneman O, Sjogren F, Saberg C, Hillman J. Differences in cerebral extracellular response of interleukin-1beta, interleukin-6, and interleukin-10 after subarachnoid hemorrhage or severe head trauma in humans. Neurosurgery (2011) 68(1):12–9; discussion 9. doi:10.1227/NEU.0b013e3181ef2a40
116. Hopkins SJ, McMahon CJ, Singh N, Galea J, Hoadley M, Scarth S, et al. Cerebrospinal fluid and plasma cytokines after subarachnoid haemorrhage: CSF interleukin-6 may be an early marker of infection. J Neuroinflammation (2012) 9:255. doi:10.1186/1742-2094-9-255
117. Jayaraman T, Berenstein V, Li X, Mayer J, Silane M, Shin YS, et al. Tumor necrosis factor alpha is a key modulator of inflammation in cerebral aneurysms. Neurosurgery (2005) 57(3):558–64; discussion 64. doi:10.1227/01.NEU.0000170439.89041.D6
118. Sathyan S, Koshy LV, Srinivas L, Easwer HV, Premkumar S, Nair S, et al. Pathogenesis of intracranial aneurysm is mediated by proinflammatory cytokine TNFA and IFNG and through stochastic regulation of IL10 and TGFB1 by comorbid factors. J Neuroinflammation (2015) 12:135. doi:10.1186/s12974-015-0354-0
119. Wang KW, Cho CL, Chen HJ, Liang CL, Liliang PC, Tsai YD, et al. Molecular biomarker of inflammatory response is associated with rebleeding in spontaneous intracerebral hemorrhage. Eur Neurol (2011) 66(6):322–7. doi:10.1159/000332027
120. Broderick J, Connolly S, Feldmann E, Hanley D, Kase C, Krieger D, et al. Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: a guideline from the American Heart Association/American Stroke Association stroke council, high blood pressure research council, and the quality of care and outcomes in research interdisciplinary working group. Stroke (2007) 38(6):2001–23.
121. Wasserman JK, Zhu X, Schlichter LC. Evolution of the inflammatory response in the brain following intracerebral hemorrhage and effects of delayed minocycline treatment. Brain Res (2007) 1180:140–54. doi:10.1016/j.brainres.2007.08.058
122. Ewen T, Qiuting L, Chaogang T, Tao T, Jun W, Liming T, et al. Neuroprotective effect of atorvastatin involves suppression of TNF-alpha and upregulation of IL-10 in a rat model of intracerebral hemorrhage. Cell Biochem Biophys (2013) 66(2):337–46. doi:10.1007/s12013-012-9453-z
123. Fang H, Chen J, Lin S, Wang P, Wang Y, Xiong X, et al. CD36-mediated hematoma absorption following intracerebral hemorrhage: negative regulation by TLR4 signaling. J Immunol (2014) 192(12):5984–92. doi:10.4049/jimmunol.1400054
124. Oto J, Suzue A, Inui D, Fukuta Y, Hosotsubo K, Torii M, et al. Plasma proinflammatory and anti-inflammatory cytokine and catecholamine concentrations as predictors of neurological outcome in acute stroke patients. J Anesth (2008) 22(3):207–12. doi:10.1007/s00540-008-0639-x
125. Dziedzic T, Bartus S, Klimkowicz A, Motyl M, Slowik A, Szczudlik A. Intracerebral hemorrhage triggers interleukin-6 and interleukin-10 release in blood. Stroke (2002) 33(9):2334–5. doi:10.1161/01.STR.0000027211.73567.FA
126. Dziedzic T, Slowik A, Klimkowicz A, Szczudlik A. Asymmetrical modulation of interleukin-10 release in patients with intracerebral hemorrhage. Brain Behav Immun (2003) 17(6):438–41. doi:10.1016/S0889-1591(03)00064-3
127. Norden DM, Fenn AM, Dugan A, Godbout JP. TGFbeta produced by IL-10 redirected astrocytes attenuates microglial activation. Glia (2014) 62(6):881–95. doi:10.1002/glia.22647
128. Sanni LA, Jarra W, Li C, Langhorne J. Cerebral edema and cerebral hemorrhages in interleukin-10-deficient mice infected with Plasmodium chabaudi. Infect Immun (2004) 72(5):3054–8. doi:10.1128/IAI.72.5.3054-3058.2004
129. Dziurdzik P, Krawczyk L, Jalowiecki P, Kondera-Anasz Z, Menon L. Serum interleukin-10 in ICU patients with severe acute central nervous system injuries. Inflamm Res (2004) 53(8):338–43. doi:10.1007/s00011-004-1265-1
130. Buttram SD, Wisniewski SR, Jackson EK, Adelson PD, Feldman K, Bayir H, et al. Multiplex assessment of cytokine and chemokine levels in cerebrospinal fluid following severe pediatric traumatic brain injury: effects of moderate hypothermia. J Neurotrauma (2007) 24(11):1707–17. doi:10.1089/neu.2007.0349
131. Csuka E, Morganti-Kossmann MC, Lenzlinger PM, Joller H, Trentz O, Kossmann T. IL-10 levels in cerebrospinal fluid and serum of patients with severe traumatic brain injury: relationship to IL-6, TNF-alpha, TGF-beta1 and blood-brain barrier function. J Neuroimmunol (1999) 101(2):211–21. doi:10.1016/S0165-5728(99)00148-4
132. Kumar P, Kumar A, Sagar R, Misra S, Faruq M, Suroliya V, et al. Association between interleukin-10 1082G/A gene polymorphism and risk of stroke in the North Indian population: a case-control study. J Stroke Cerebrovasc Dis (2016) 25(2):461–8. doi:10.1016/j.jstrokecerebrovasdis.2015.10.020
133. Pawlikowska L, Tran MN, Achrol AS, McCulloch CE, Ha C, Lind DL, et al. Polymorphisms in genes involved in inflammatory and angiogenic pathways and the risk of hemorrhagic presentation of brain arteriovenous malformations. Stroke (2004) 35(10):2294–300. doi:10.1161/01.STR.0000141932.44613.b1
134. Hamann GF, Strittmatter M, Hoffmann KH, Holzer G, Stoll M, Keshevar T, et al. Pattern of elevation of urine catecholamines in intracerebral haemorrhage. Acta Neurochir (Wien) (1995) 132(1–3):42–7. doi:10.1007/BF01404846
135. Kamm K, Vanderkolk W, Lawrence C, Jonker M, Davis AT. The effect of traumatic brain injury upon the concentration and expression of interleukin-1beta and interleukin-10 in the rat. J Trauma (2006) 60(1):152–7. doi:10.1097/01.ta.0000196345.81169.a1
136. Lenzlinger PM, Morganti-Kossmann MC, Laurer HL, McIntosh TK. The duality of the inflammatory response to traumatic brain injury. Mol Neurobiol (2001) 24(1–3):169–81. doi:10.1385/MN:24:1-3:169
137. Kumar RG, Boles JA, Wagner AK. Chronic inflammation after severe traumatic brain injury: characterization and associations with outcome at 6 and 12 months postinjury. J Head Trauma Rehabil (2015) 30(6):369–81. doi:10.1097/HTR.0000000000000067
138. Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Front Neurol (2013) 4:18. doi:10.3389/fneur.2013.00018
139. Maier B, Schwerdtfeger K, Mautes A, Holanda M, Muller M, Steudel WI, et al. Differential release of interleukines 6, 8, and 10 in cerebrospinal fluid and plasma after traumatic brain injury. Shock (2001) 15(6):421–6. doi:10.1097/00024382-200115060-00002
140. Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol (2008) 7(8):728–41. doi:10.1016/S1474-4422(08)70164-9
141. Balasingam V, Yong VW. Attenuation of astroglial reactivity by interleukin-10. J Neurosci (1996) 16(9):2945–55.
142. Chen X, Duan XS, Xu LJ, Zhao JJ, She ZF, Chen WW, et al. Interleukin-10 mediates the neuroprotection of hyperbaric oxygen therapy against traumatic brain injury in mice. Neuroscience (2014) 266:235–43. doi:10.1016/j.neuroscience.2013.11.036
143. Kline AE, Bolinger BD, Kochanek PM, Carlos TM, Yan HQ, Jenkins LW, et al. Acute systemic administration of interleukin-10 suppresses the beneficial effects of moderate hypothermia following traumatic brain injury in rats. Brain Res (2002) 937(1–2):22–31. doi:10.1016/S0006-8993(02)02458-7
144. Lee HF, Lee TS, Kou YR. Anti-inflammatory and neuroprotective effects of triptolide on traumatic brain injury in rats. Respir Physiol Neurobiol (2012) 182(1):1–8. doi:10.1016/j.resp.2012.01.016
145. Gao H, Han Z, Bai R, Huang S, Ge X, Chen F, et al. The accumulation of brain injury leads to severe neuropathological and neurobehavioral changes after repetitive mild traumatic brain injury. Brain Res (2017) 1657:1–8. doi:10.1016/j.brainres.2016.11.028
146. Lin KC, Niu KC, Tsai KJ, Kuo JR, Wang LC, Chio CC, et al. Attenuating inflammation but stimulating both angiogenesis and neurogenesis using hyperbaric oxygen in rats with traumatic brain injury. J Trauma Acute Care Surg (2012) 72(3):650–9. doi:10.1097/TA.0b013e31823c575f
147. Knoblach SM, Faden AI. Interleukin-10 improves outcome and alters proinflammatory cytokine expression after experimental traumatic brain injury. Exp Neurol (1998) 153(1):143–51. doi:10.1006/exnr.1998.6877
148. Helmy A, Antoniades CA, Guilfoyle MR, Carpenter KL, Hutchinson PJ. Principal component analysis of the cytokine and chemokine response to human traumatic brain injury. PLoS One (2012) 7(6):e39677. doi:10.1371/journal.pone.0039677
149. Schneider Soares FM, Menezes de Souza N, Liborio Schwarzbold M, Paim Diaz A, Costa Nunes J, Hohl A, et al. Interleukin-10 is an independent biomarker of severe traumatic brain injury prognosis. Neuroimmunomodulation (2012) 19(6):377–85. doi:10.1159/000342141
150. Stein DM, Lindell A, Murdock KR, Kufera JA, Menaker J, Keledjian K, et al. Relationship of serum and cerebrospinal fluid biomarkers with intracranial hypertension and cerebral hypoperfusion after severe traumatic brain injury. J Trauma (2011) 70(5):1096–103. doi:10.1097/TA.0b013e318216930d
151. Hensler T, Sauerland S, Bouillon B, Raum M, Rixen D, Helling HJ, et al. Association between injury pattern of patients with multiple injuries and circulating levels of soluble tumor necrosis factor receptors, interleukin-6 and interleukin-10, and polymorphonuclear neutrophil elastase. J Trauma (2002) 52(5):962–70.
152. Hensler T, Sauerland S, Riess P, Hess S, Helling HJ, Andermahr J, et al. The effect of additional brain injury on systemic interleukin (IL)-10 and IL-13 levels in trauma patients. Inflamm Res (2000) 49(10):524–8. doi:10.1007/s000110050626
153. Hayakata T, Shiozaki T, Tasaki O, Ikegawa H, Inoue Y, Toshiyuki F, et al. Changes in CSF S100B and cytokine concentrations in early-phase severe traumatic brain injury. Shock (2004) 22(2):102–7. doi:10.1097/01.shk.0000131193.80038.f1
154. Bell MJ, Kochanek PM, Doughty LA, Carcillo JA, Adelson PD, Clark RS, et al. Interleukin-6 and interleukin-10 in cerebrospinal fluid after severe traumatic brain injury in children. J Neurotrauma (1997) 14(7):451–7. doi:10.1089/neu.1997.14.451
155. Shiozaki T, Hayakata T, Tasaki O, Hosotubo H, Fuijita K, Mouri T, et al. Cerebrospinal fluid concentrations of anti-inflammatory mediators in early-phase severe traumatic brain injury. Shock (2005) 23(5):406–10. doi:10.1097/01.shk.0000161385.62758.24
156. Kirchhoff C, Buhmann S, Bogner V, Stegmaier J, Leidel BA, Braunstein V, et al. Cerebrospinal IL-10 concentration is elevated in non-survivors as compared to survivors after severe traumatic brain injury. Eur J Med Res (2008) 13(10):464–8.
157. Mustafa AG, Alshboul OA. Pathophysiology of traumatic brain injury. Neurosciences (Riyadh) (2013) 18(3):222–34.
158. Prakash A, Parelkar SV, Oak SN, Gupta RK, Sanghvi BV, Bachani M, et al. Role of hyperbaric oxygen therapy in severe head injury in children. J Pediatr Neurosci (2012) 7(1):4–8. doi:10.4103/1817-1745.97610
159. Neidhardt R, Keel M, Steckholzer U, Safret A, Ungethuem U, Trentz O, et al. Relationship of interleukin-10 plasma levels to severity of injury and clinical outcome in injured patients. J Trauma (1997) 42(5):863–70; discussion 70–1. doi:10.1097/00005373-199705000-00017
160. Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response by head injury. Injury (2007) 38(12):1392–400. doi:10.1016/j.injury.2007.10.005
161. Sellmann T, Miersch D, Kienbaum P, Flohe S, Schneppendahl J, Lefering R, et al. The impact of arterial hypertension on polytrauma and traumatic brain injury. Dtsch Arztebl Int (2012) 109(49):849–56. doi:10.3238/arztebl.2012.0849
162. Woiciechowsky C, Volk HD. Increased intracranial pressure induces a rapid systemic interleukin-10 release through activation of the sympathetic nervous system. Acta Neurochir Suppl (2005) 95:373–6. doi:10.1007/3-211-32318-X_76
163. Woiciechowsky C, Asadullah K, Nestler D, Eberhardt B, Platzer C, Schoning B, et al. Sympathetic activation triggers systemic interleukin-10 release in immunodepression induced by brain injury. Nat Med (1998) 4(7):808–13. doi:10.1038/nm0798-808
164. Woiciechowsky C, Schoning B, Cobanov J, Lanksch WR, Volk HD, Docke WD. Early IL-6 plasma concentrations correlate with severity of brain injury and pneumonia in brain-injured patients. J Trauma (2002) 52(2):339–45.
165. Lo TY, Jones PA, Minns RA. Pediatric brain trauma outcome prediction using paired serum levels of inflammatory mediators and brain-specific proteins. J Neurotrauma (2009) 26(9):1479–87. doi:10.1089/neu.2008-0753
166. Lo TY, Jones PA, Minns RA. Combining coma score and serum biomarker levels to predict unfavorable outcome following childhood brain trauma. J Neurotrauma (2010) 27(12):2139–45. doi:10.1089/neu.2010.1387
167. Shiozaki T, Kato A, Taneda M, Hayakata T, Hashiguchi N, Tanaka H, et al. Little benefit from mild hypothermia therapy for severely head injured patients with low intracranial pressure. J Neurosurg (1999) 91(2):185–91. doi:10.3171/jns.1999.91.2.0185
Keywords: concussion, endothelium, intracerebral hemorrhage, ischemia, stroke, subarachnoid hemorrhage, traumatic brain injury, vasculature
Citation: Garcia JM, Stillings SA, Leclerc JL, Phillips H, Edwards NJ, Robicsek SA, Hoh BL, Blackburn S and Doré S (2017) Role of Interleukin-10 in Acute Brain Injuries. Front. Neurol. 8:244. doi: 10.3389/fneur.2017.00244
Received: 09 January 2017; Accepted: 17 May 2017;
Published: 12 June 2017
Edited by:
Jean-Claude Baron, Université Paris Descartes, FranceReviewed by:
Arthur Liesz, Ludwig-Maximilians-Universität München, GermanyEmmanuel Pinteaux, University of Manchester, United Kingdom
Maria-Grazia De Simoni, Istituto Di Ricerche Farmacologiche Mario Negri, Italy
Copyright: © 2017 Garcia, Stillings, Leclerc, Phillips, Edwards, Robicsek, Hoh, Blackburn and Doré. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylvain Doré, sdore@ufl.edu
†These authors have contributed equally to this work.