 Xiaoyu Liu1
Xiaoyu Liu1 Ning Quan1,2*
Ning Quan1,2*
- 1College of Medicine, Institute for Behavioral Medicine Research, The Ohio State University, Columbus, OH, United States
- 2Division of Biosciences, College of Dentistry, The Ohio State University, Columbus, OH, United States
Activation of microglia and expression of the inflammatory cytokine interleukin-1 (IL-1) in the CNS have become almost synonymous with neuroinflammation. In numerous studies, increased CNS IL-1 expression and altered microglial morphology have been used as hallmarks of CNS inflammation. A central concept of how CNS IL-1 and microglia influence functions of the nervous system was derived from the notion initially generated in the peripheral immune system: IL-1 stimulates monocyte/macrophage (the peripheral counterparts of microglia) to amplify inflammation. It is increasingly clear, however, CNS IL-1 acts on other targets in the CNS and microglia participates in many neural functions that are not related to immunological activities. Further, CNS exhibits immunological privilege (although not as absolute as previously thought), rendering amplification of inflammation within CNS under stringent control. This review will analyze current literature to evaluate the contribution of immunological and non-immunological aspects of microglia/IL-1 interaction in the CNS to gain insights for how these aspects might affect health and disease in the nervous tissue.
Introduction
Changes in microglial morphology are one of the most common findings of neuropathology in almost all CNS diseases. Long regarded as the resident immune cells in the immunologically temperate environment of the CNS, the resting spider-shaped microglia become deramified and amoeboid in activated states (1). This shape shift has been observed in acute brain injury (2), various neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease (3), CNS autoimmune diseases such as multiple sclerosis (MS) (4, 5), convulsive disorders such as epilepsy (6), and even affective disorders including major depression (7, 8), anxiety disorders (9), and autism (10). These evidences led to the hypothesis that microglial activation is a significant common cause of neuropathology in these diseases (11), although microglial morphological changes alone may not always reflect the precise activation status (12, 13) among the variegated states that these cells can adapt.
Another salient-related observation in CNS diseases is the increased expression of the inflammatory cytokine interleukin-1 (IL-1). IL-1 is a master regulator of inflammatory reactions in the immune system, capable of activating innate immunity by inducing the expression of numerous inflammatory cytokines and chemokines, eliciting leukocyte infiltration into the inflammatory loci, increasing phagocytic and bactericidal activity of immune cells, enhancing activity of the complement system, and facilitating the activation of the adaptive immune responses (14). Correlations of plasma or CNS levels of IL-1 and disease severities in the abovementioned CNS diseases have been widely reported (15–20), although there are also many reports that fail to show correlation between plasma IL-1 level and the presence of disease symptoms in these diseases (21, 22).
Combining increased IL-1 expression and microglial activation as a composite indicator of pathogenesis in CNS diseases seems to be an attractive idea because it might overcome the shortcomings of using each of them separately. In peripheral tissues, increased IL-1 expression is tightly linked with macrophage activation during inflammation (23); in the CNS, neuroinflammation may not display the entire panoply of peripheral inflammation, e.g., swelling may not occur during neuroinflammation, but increased expression of IL-1 by brain tissue together with morphological changes in microglia appear to be a frequently observed phenomenon in both human neuropathology and animal models of brain diseases (16, 24–28). Adding increased brain IL-1 expression to supplement microglial morphology changes would further specify that the changes in microglia are part of the inflammatory microgliosis.
The villainization of microglial activation and CNS IL-1 expression, however, has been countered by the teleological argument: is the CNS designed to have microglial activation and IL-1 expression just to cause pathology (29)? Such incredulity has been substantiated by the facts that blocking inflammatory microglial activation can lead to the exacerbation of some symptoms of certain CNS diseases (30–34) and clinical benefits of drug treatments for reducing microglial activation or IL-1 activity have been demonstrated recently in stroke patients (35), but the utility of this strategy for the treatment of the vast majority of the above-mentioned diseases remains to be firmly established after it has been advocated for at least 10 years.
Besides their pathogenic roles, functions of microglial activation and IL-1 expression in CNS development, repair, and physiological activity have been intensely studied recently. This endeavor has yielded tremendous advances, revealing many new areas of understanding on the non-immunological functions of IL-1 and microglia in the CNS (11, 36–39). These new findings in the realm of the positive contributions of microglia and IL-1 in the CNS educe the critical inquiry: how would the immunological and the non-immunological aspects of IL-1 and microglial functions coordinate or disrupt each other to affect health and disease?
The Promises and Limitations of the Inflammatory Paradigm
Although current literature is beginning to shed light on the multifaceted roles played by microglia and CNS IL-1, the simple inflammatory paradigm, viz., increased CNS IL-1 expression together with microglial activation amplifies neuroinflammation and causes neuropathology, has accrued formidable experimental support. The following rationales have propelled the research in this area: (1) inflammatory process is designed to sequester and kill infectious pathogens and contain necrotic tissue damage; this entails the induction of proinflammatory cytokines and chemokines, recruitment of leukocytes, and the production of bactericidal reactive oxygen species (ROS), all potentially neurotoxic, (2) CNS is immunologically privileged site, bystander neuronal casualty from inflammation is likely to cause irreversible damage to this delicate tissue which lacks significant regenerative potential and expandable volume, and (3) neuroinflammation could lead to CNS autoimmunity resulting in attacks by immune cells to CNS antigens which are normally dormant.
The induction of CNS expression of proinflammatory cytokines including IL-1 has been shown in animal models of acute brain injury (40–43), Alzheimer’s disease (44, 45), Parkinson’s disease (25), CNS autoimmunity (46), anxiety disorder (47–50), major depression (51–53), and autism (54). In vitro studies were the first to show that inflammatory cytokines, especially IL-1 and TNF-α, can cause neuronal death by the direct effects of these cytokines on neurons or indirectly by glial production of neurotoxic substances (55–58). Similarly, a few chemokines have also been found to possess neurotoxic activity. CXCL4 (59), was the first to be identified in this regard; more recent studies also found CCL11 (60), CXCL2 (61) can exert neurotoxic effects on cultured neurons.
Neurotoxicity from infiltration of peripheral leukocytes has also been documented. Typically, in experimental conditions that resulted in leukocyte infiltration into the brain, the infiltrated peripheral myeloid cells show higher expression levels of proinflammatory cytokines than resident glial cells (62–64). Thus, entrance of peripheral leukocytes into the CNS may represent a more severe type of CNS inflammation. Reduction of leukocyte infiltration by blocking vascular adhesion molecules or chemokine activity has been shown to improve outcomes in acute brain injury (65, 66) and CNS autoimmune diseases (67, 68). Interestingly, although infiltration of peripheral leukocytes into the CNS is generally not a common observation in human affective disorders, this phenomenon occurs in several animal models of stress- or inflammation-induced depression and/or anxiety (69, 70). Preventing CNS infiltration of IL-1 expressing leukocytes protected animals from displaying depressive and/or anxiety-like behaviors in these models (64, 71).
Other studies demonstrated a pathogenic role of oxidative stress. Blocking inflammation-induced production of ROS or ROS activity alleviates neural damage in cerebral ischemia (72–74) and cerebral hemorrhage (75), reduces depressive and anxiety-like behaviors caused by peripheral inflammatory stimulation (76), lessens certain symptoms induced in an Alzheimer’s mouse model (77). In addition, ROS production and antioxidant defense imbalance has been observed in acute brain injury (78, 79), inflammation-induced depression and anxiety, and neurodegenerative diseases (80, 81). These evidences support the hypothesis that oxidant/antioxidant imbalance downstream of IL-1-stimulated microglial activation is a common feature for both acute and chronic neuropathology and their attendant psychopathology (82, 83).
The possibility of bystander damage of CNS inflammation is best demonstrated in situations of CNS infection. Initially, post-infectious neurological dysfunction was thought as a consequence of permanent damage caused by the invading pathogens and the specific immune responses to the pathogen (84). However, patients who survived CNS infection sometimes show deficits implicating brain regions beyond the foci of the initial infection (85) and animal studies show chronic neuroinflammation may persist after the acute infectious pathogens have been eradicated (86). Thus, off-target inflammatory activity may contribute to post-infectious neuropathology.
Further bolstering the case for malignant inflammatory effects are the findings that endogenous CNS antigens that normally do not induce autoimmune attacks can be turned susceptible when CNS inflammation is present. In experimental autoimmune encephalitis (EAE), the brain endothelial receptor for IL-1 (IL-1R1) and infiltration of myeloid cells expressing IL-1β was found to be required for the induction of illness (63). Because IL-1β-expressing myeloid cells are involved in inflammatory activity, not antigen specific immunity, these results point to the importance of inflammation in facilitating autoimmune activity of the CNS. Dysregulation of microglia may also contribute to the pathogenesis of PANDAS (Pediatric Autoimmune Neuropsychiatric Disorders Associated with Streptococcal Infections) which was thought to be caused by the induction of post-infectious cross-reactive autoantibodies against CNS tissue (87–89). Therefore, neuroinflammation might augment autoimmune activity-related neuropathology.
A major recent advance in the field of inflammation is the discovery of inflammasomes. Inflammasomes are protein complexes that act as intracellular sensors for the disruption of homeostasis (90). They include NOD like receptors and ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain). Inflammasomes regulate IL-1 and IL-18 activity by regulating caspase-1, which cleaves inactive pro-IL-1 and pro-IL-18 to derive the active IL-1 and IL-18. This intermediate step allows preformed pro-IL-1 and pro-IL-18 to be quickly activated, ensures inflammation occurs through the priming stage (the synthesis of pro-IL-1 and molecules of the Inflammasomes) and the activation stage (the generation of mature inflammatory cytokines), thus providing a mechanism that requires “two-hit” to induce inflammation, allowing finer control of the timing and the magnitude of inflammatory cascade. In addition, inflammasomes are sensitive to stimulations by internal disturbance, such as misfolded or aggregated proteins and aberrant products of energy metabolism, broadening the range of inflammation inducers beyond infectious stimuli (90). In ischemic or hemorrhagic stroke models, expression of the NLRP3, a known microglial inflammasome (91, 92) component, was increased, and specific blockade of NLRP3 reduced stroke induced neural damage and functional deficits (93, 94). Several NLRP3 component proteins were also induced in the pathological tissues in Alzheimer’s disease (95). Aggregated or fibrillary α-synuclein, a known pathogenic factor for Parkinson’s disease also stimulates the activation of NLRP3 (96). Activation of NLRP3 has been documented in depression and anxiety and both pharmacological blockade of NLRP3 or gene deletion of NLRP3 reduces depressive behavior and anxiety in animal models of these disorders (97, 98).
The inflammatory paradigm, increased brain IL-1 expression and microglial activation drives the progression of CNS diseases, has gained further momentum from studies that used drugs to inhibit IL-1 activity and/or microglial activation. A naturally occurring antagonist for IL-1 is the IL-1 receptor antagonist (IL-1ra). A recent meta-analysis shows treatment with IL-1ra reduces infarct volume by 36% in animal models of cerebral stroke with more reliable efficacy if the drug is delivered into the cerebral ventricle than into the blood (99). IL-1ra was also effective in blocking stress-induced depression and anxiety (51, 52, 100), and in improving clinical outcomes in experimental epilepsy (101, 102). Minocycline, a tetracycline derived antibiotics, has been found to inhibit inflammatory microglial activation (103). Specifically, activated microglia could differentiate into multiple activated states: the most inflammatory type is designated as M1 and the most anti-inflammatory type is designated as M2. Besides changes in morphology, M1 microglia express inflammatory cytokines including IL-1, TNF-α, and iNOS, whereas the M2 microglia express TGF-β, IL-4 or IL-10, and arginase 1. Treatment with minocycline selectively inhibits M1 microglial activation (104). Pretreatment with minocycline provides neuroprotection against excitotoxicity (105), oxidative stress (106), reduces symptoms in animal models of Parkinson’s disease (107), cerebral stroke (108), epilepsy (109), and stress-induced depression (110). In EAE, a model of MS, minocycline treatment was found to be effective in reducing disease severity and histological outcomes when used in combination with other conventional treatments (111–116) or alone (117, 118). The promise of using drugs against IL-1 and microglial activation to treat CNS diseases is attested by the current clinical trials that use IL-1ra to treat cerebral stroke (119–121), fatigue in Sojegren’s syndrome (122), and minocycline to treat cerebral stroke (123, 124), cerebral hemorrhage (125), Parkinson’s disease (126, 127), epilepsy (128), bipolar and treatment resistant depression (129, 130), and schizophrenia (131). These trials have generated promising results, although large scale clinical tests are still needed. In human MS trials, minocycline treatment reduced MS lesion detected by MRI (132) and reduced the risk of conversion of patients with first demyelinating event from progressing to MS (133).
The notion that all CNS diseases can be effectively treated by inhibition of IL-1 driven microglial activation, of course, is overly simplistic. A dramatic cautionary tale is supplied by a study that investigated the role of IL-1β in Alzheimer’s disease. Beta-amyloid aggregation in this disease causes the formation of senile plaques. Transgenic overexpression of IL-1β unexpectedly reduced plaque formation, despite inducing robust neuroinflammation (134). In another surprising study, chronic unpredictable stress induced depressive-like behavior; stimulating rather than inhibiting microglia provided anti-depressant effects (135). These results highlight the limitation of the inflammatory paradigm and suggest non-immunological functions of IL-1 and microglia should be examined.
IL-1 and Microglia as Neuromodulators
The neurophysiological functions of IL-1 were first investigated in temperature-sensitive neurons because IL-1 was identified as the endogenous pyrogen that mediates fever after bacterial infection. In the temperature control center of the brain, the preoptic area of the hypothalamus, IL-1 decreased the sensitivity of warm-sensitive neurons, but increased the sensitivity of cold-sensitive neurons, thereby modulating the thermoregulatory circuits in a manner consistent with its pyrogenic role (136, 137). This IL-1 activity is not related to neuroinflammation but could be an indirect effect because it can be blocked by inhibitors of cyclooxygenase, which catalyzes prostaglandin production downstream of IL-1 signaling (138). IL-1 may even mediate neurophysiological effects under sterile condition. A good example here is its role in regulating normal sleep. IL-1 is expressed in the brain with a diurnal rhythm, and increased expression of IL-1 is associated with increased spontaneous sleep whereas inhibition of IL-1 activity reduces sleep (139). Interestingly, neuronal IL-1 expression and indirect activation of neurons by CNS IL-1 may underlie the sleep promoting effects of IL-1 as it can promote synchronization of sensory neurons (140). Other indirect electrophysiological effects of IL-1 have been reported in neurons of the supraoptic (138) and paraventricular nucleus of the hypothalamus (141). Direct effects of IL-1 on neuronal excitability have also been reported (138); but the mechanism for this function remains unclear. IL-1 was found to inhibit Ca+ channel currents (142), reduce GABA A receptor-mediated response (143), inhibit NMDA receptor-mediated synaptic transmission (144), activate non-selective cationic conductance (145), potentiate voltage-dependent sodium currents in nociceptive neurons (146, 147), and increase voltage-gated potassium currents (148), depending on the different types of neurons studied. In the dentate gyrus, IL-1 may facilitate or inhibit the generation of long-term potentiation (LTP) (149, 150), a critical neural mechanism for learning and memory. LTP occurs as persistent increases of synaptic strength after high-frequency synaptic stimulation, thus potentially coding for learning or memory processes. Interestingly, the learning process itself causes hippocampal expression of non-inflammatory levels of IL-1, which in term, helps maintain LTP (150, 151). These scattered reports of IL-1-mediated neurophysiological effect appear incongruent at first glance, but an emerging theme is IL-1 can modulate sensory system of the nervous system in order to modulate perception and learning. It should be noted that such modulation may have time-dependent and concentration-dependent variable effects. Acute IL-1 effects may heighten perception and learning whereas chronic IL-1 effects may reduce sensory function, retard learning, and cause fatigue (147, 148, 152–154). Similarly, low levels of IL-1 may facilitate memory whereas high levels of IL-1 or complete blockade of IL-1 signaling may impair memory (155). One difficulty in the past is to identify IL-1 receptor expressing neurons and the observed neuromodulatory effects of IL-1 may be attributed to the indirect action of IL-1 that might elicit neural active substances such as nitric oxide (156), ATP (157), or prostaglandins (145). Recently, we have developed a knockin mouse line that allowed the tracking of IL-1 receptor expression cells in a cell type specific manner. We now have unpublished results that show IL-1 type 1 receptor is preferentially expressed in numerous sensory brain regions.
Another neuromodulatory role of IL-1 is on neurogenesis. Reduced production of new neurons in adult hippocampus has been linked with the pathogenesis of depression (51). This role of IL-1 was initially observed in animal models of interferon-γ (IFN-γ) treatment. IFN-γ is used to treat hepatitis C but has the unfortunate side effect of causing depression. In a rat model of IFN-γ-induced depression, hippocampal IL-1β expression and reduced neurogenesis in dentate gyrus was induced and administration of IL-1ra blocked these effects together with the depressive behavior (158). This mechanism is also operative in chronic stress induced depression: chronic mild stress was found to induce IL-1β expression in the hippocampus, reduce neurogenesis, and cause depressive like behavior in wild-type mice. These changes were absent in IL-1 receptor knockout mice or transgenic mice that express IL-1ra in the brain (159). That IL-1 driven microglial activation may be involved in this phenomenon is further supported by the evidence that inhibition of NFκB activation blocked the antineurogenic and depressive effects of the stress (160). Brain IL-1 is known to induce microglial NFκB activation (161). It should be noted that chronic mild stress dose not induce leukocyte infiltration into the brain; thus this IL-1-mediated microglial activation may not represent an immunological neuroinflammation. In addition, the antineurogenic effect of IL-1 may also be concentration dependent as IL-1 can facilitate neuronal survival by promoting the expression of nerve growth factors (NGFs) (162).
Induction of neurotrophic factors is one of the early observations on IL-1-mediated non-immunological neural effects (163, 164). In traumatic brain injury, increased NGF expression follows the increased expression of IL-1 in the wounded tissue. Injection of IL-1ra blocked NGF and the associate neuroreparative responses (165). IL-1 has also been found to stimulate neurotrophin-3 and brain derived neurotrophic factor, supporting neuronal survival and neurite growth (166, 167). However, interaction between IL-1 and the neurotrophic factors can also be a double-edged sword. Systemic IL-1, not central IL-1, have been reported to reduce hippocampal BDNF expression (168); while acute intracerebral IL-1 caused the expression of neurotrophic factors and neuroprotection, subacute IL-1 (4 days of IL-1 injection) caused the opposite effects (169); IL-1 can also increase neuronal vulnerability by increasing the surface expression of the p75 neurotrophin receptor (170).
Physiological activities of IL-1 in the brain also include neuroendocrine functions. Psychological and metabolic stress induced ACTH and glucocorticoid responses were reduced in IL-1 receptor knockouts or transgenic mice overexpressing brain IL-1ra (171). Intracerebral administration of IL-1 is known to induce CRH release (172) and psychological stress has been shown to induce brain IL-1 expression (173). Therefore, brain IL-1 could mediate physiological response to stress by stimulating the production of the immunosuppressive hormone glucocorticoid. In addition, IL-1 acting in the brain can stimulate brain metabolism despite hypoglycemia. Neuronal IL-1 synthesis was found to be induced by stimulation of AMPA receptors on neurons and the resulting release of IL-1 can stimulate glucose uptake by neurons in an autocrine or paracrine fashion (174). It is interesting to speculate that these physiological activities of IL-1 might coordinate with the immune activities of IL-1 such that hyper-inflammation may be prevented and brain energy usage may be spared even when immune activity might be energetically costly. It is interesting to note that the neuroendocrine function of IL-1 may be evolutionary conserved from invertebrates. In molluscs, CRF causes the production of biogenic amines as a stress response. This response is significantly reduced by IL-1 (175). Thus, this non-immunological IL-1 activity may have an ancient origin.
The non-immunological activities of microglia have been reviewed extensively. The readers are referred to these excellent reviews (11, 36, 176–178). Briefly, emerging evidences show microglia perform surveillance function during “resting state,” prune excessive synapse during development, contribute to adult neurogenesis, support neuronal survival, and modulate neurotransmission. Current research using advanced techniques in molecular biology, imaging and immunology has also identified significant heterogeneity in brain microglia in terms of morphology, gene expression profile, and cellular origin and fate (179). Some characteristics of subsets of microglia appear to be tightly linked with the potential neural function of these cells. For example, microglia from neurogenic regions are capable of substantial proliferation whereas microglia from non-neurogenic regions are not (180). Analysis of microglial expression patterns suggests that microglia from cerebellum and hippocampus appear immunologically more vigilant than microglia from other brain regions. Within Basal ganglia, microglia were found to show regional specific morphology, cell number, expression profile and activity relevant to motor activity and motion control, shaped by local cues (181). These findings demonstrate non-immunological functions of the microglia could be influenced by the specific neural circuitry they modulate. From this perspective, it is interesting to note that chronic unpredictable stress causes depression in association with a reduction of microglia numbers in hippocampus and stimulation of microglial activation by LPS or M-CSF restored microglia numbers and ameliorated stress-induced depression. In another study, chronic unpredictable stress was found to activate microglial cells in association with elevated CSF-1 expression in the prefrontal cortex (PFC), increase microglial phagocytosis of neuronal elements, and reduce dendritic spine density. Viral vector mediated knockdown of CSF-1 in the PFC blocked these effects and stress-induced anxiety- and depressive-like behavior (182). In neurodegenerative disease models, microglial production of proinflammatory cytokines and growth factors has been found to mediate neuroprotection against excitotoxicity (183, 184). In addition, microglia-mediated synaptic stripping was found to be neuroprotective following acute neural injury (185, 186). On the other hand, abnormal synaptic pruning have been observed in obsessive-compulsive disorder (OCD), indicating this mechanism might be pathogenic in OCD (187). These findings show non-immunological activities of microglia can be either neuroprotective or pathogenic depending on the specific circumstances.
The Big Picture
The dizzying progress made in the field of CNS IL-1 and microglia has produced great excitement and confusion. It is clear CNS IL-1 and microglia have both immunological and non-immunological functions. These two types of functions may be separated not only by the physical barrier, such as the blood brain barrier, but also by an invisible barrier: the activation threshold of inflammatory cytokines. For example, IL-1 is able to activate neurons at 1,000-fold lower concentration than that is required for the activation of non-neuronal cells (188). It is possible that low levels of IL-1 acts in the CNS to perform non-immunological functions including non-immunological activation of microglia, which are involved in the remodeling of the CNS tissue. Higher concentration of IL-1 could engage non-neuronal cells of the CNS to produce neuroinflammation. Interestingly, although microglia is the main source of IL-1 production in the brain without infiltrated leukocytes, IL-1 does not directly stimulate microglial cell to produce IL-1 (189). Our unpublished results show IL-1 receptor is not expressed on resting microglia and CNS IL-1 induce microglia to produce IL-1 indirectly via cells of the blood-brain barrier and cells of CSF-brain barrier. The separation of the immunological and non-immunological functions of CNS IL-1 and microglia may be compromised during neural injury or aberrant neural activity. Thus the integrated perspective suggests that the disruption of the proper separation and coordination of the immunological and the non-immunological functions of CNS IL-1 and microglia might be a new way to think about the pathogenic potential of these two critical factors in CNS diseases.
Another important insight is that the detrimental effects of IL-1 and microglial activation does not always stem from immunological functions of these factors. A series studies from Centonze’s group showed that IL-1 and TNFα can cause hyper-excitation in neurons, causing excitotoxicity in MS (190). In addition, they found IL-1 could cause anxiety by blocking neuronal cannabinoid receptor 1-mediated control of GABAergic synapses (49, 100, 191, 192). Thus, aberrant non-immunological function of IL-1 can also contribute to disease progression.
The complex contribution of CNS IL-1 and microglia argues against a one size-fits-all approach to target these factors in treatment without careful considerations for the different phases of pathological processes. For acute brain tissue injury, blocking IL-1 activity and microglial activation at the early phase of the disease could be beneficial as this might dampen excessive neuroinflammation (15, 193); however, blocking later expression of low levels of IL-1 related to its promotion of clearing of debris and wound healing (194) may not be advisable. In chronic degenerative diseases, blockade of CNS IL-1 activity and microglial activation may also need to be titrated, such that the excessive activation of these factors may be attenuated, but the physiological neuroregulatory functions can be preserved. In an extreme case of an animal model of major depression, loss of microglia in hippocampus has been found to be a cause and stimulation of microglia proliferation can effectively alleviate behavioral symptoms of depression (195). Thus, one cannot assume that microglial activation, not deficiency, is always the cause of CNS diseases. The integrated perspective of microglial activation and IL-1 activity in the brain in regards to the pathogenesis of CNS diseases is presented in Figure 1 in which the immunological and non-immunological functions of IL-1 and microglial activation are seen as an integrated whole and dysregulation of either types of functions alone or in combination may contribute to disease progression.
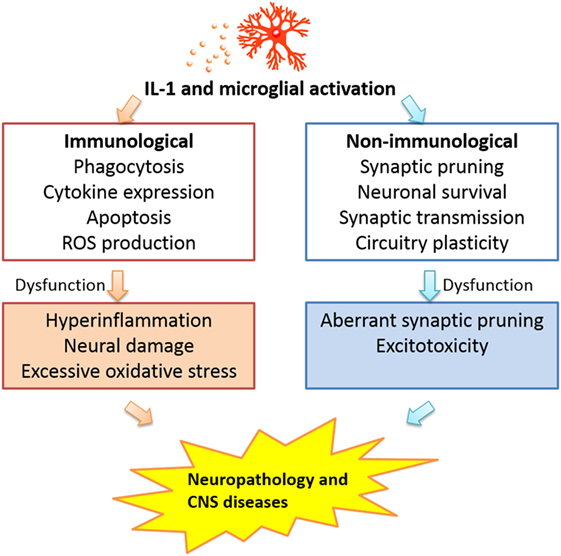
Figure 1. Interleukin-1 activity and microglial activation may influence CNS pathogenesis via dysregulation of either the immunological or the non-immunological functions of these factors.
Author Contributions
NQ and XL cowrote this review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by National Institute of Health (NIH) grants R01-MH-109165 to NQ.
References
1. Graeber MB, Streit WJ. Microglia: immune network in the CNS. Brain Pathol (1990) 1:2. doi:10.1111/j.1750-3639.1990.tb00630.x
2. Kawabori M, Yenari MA. The role of the microglia in acute CNS injury. Metab Brain Dis (2015) 30:381. doi:10.1007/s11011-014-9531-6
3. Thompson KK, Tsirka SE. The diverse roles of microglia in the neurodegenerative aspects of central nervous system (CNS) autoimmunity. Int J Mol Sci (2017) 18:E504. doi:10.3390/ijms18030504
4. Shemer A, Jung S. Differential roles of resident microglia and infiltrating monocytes in murine CNS autoimmunity. Semin Immunopathol (2015) 37:613. doi:10.1007/s00281-015-0519-z
5. Goldmann T, Prinz M. Role of microglia in CNS autoimmunity. Clin Dev Immunol (2013) 2013:208093. doi:10.1155/2013/208093
6. Eyo UB, Murugan M, Wu LJ. Microglia-neuron communication in epilepsy. Glia (2017) 65:5. doi:10.1002/glia.23006
7. Ramirez K, Fornaguera-Trias J, Sheridan JF. Stress-induced microglia activation and monocyte trafficking to the brain underlie the development of anxiety and depression. Curr Top Behav Neurosci (2017) 31:155. doi:10.1007/7854_2016_25
8. Beumer W, Gibney SM, Drexhage RC, Pont-Lezica L, Doorduin J, Klein HC, et al. The immune theory of psychiatric diseases: a key role for activated microglia and circulating monocytes. J Leukoc Biol (2012) 92:959. doi:10.1189/jlb.0212100
9. Shigemori T, Sakai A, Takumi T, Itoh Y, Suzuki H. Altered microglia in the amygdala are involved in anxiety-related behaviors of a copy number variation mouse model of autism. J Nippon Med School (2015) 82:92. doi:10.1272/jnms.82.92
10. Takano T. Role of microglia in autism: recent advances. Dev Neurosci (2015) 37:195. doi:10.1159/000398791
11. Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med (2017) 23:1018. doi:10.1038/nm.4397
12. Catalin B, Stopper L, Balseanu TA, Scheller A. The in situ morphology of microglia is highly sensitive to the mode of tissue fixation. J Chem Neuroanatomy (2017) 86:59. doi:10.1016/j.jchemneu.2017.08.007
13. Morrison HW, Filosa JA. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation (2013) 10:4. doi:10.1186/1742-2094-10-4
14. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol (2009) 27:519. doi:10.1146/annurev.immunol.021908.132612
15. Hewett SJ, Jackman NA, Claycomb RJ. Interleukin-1beta in central nervous system injury and repair. Eur J Neurodegener Dis (2012) 1:195.
16. Gui WS, Wei X, Mai CL, Murugan M, Wu LJ, Xin WJ, et al. Interleukin-1beta overproduction is a common cause for neuropathic pain, memory deficit, and depression following peripheral nerve injury in rodents. Mol Pain (2016) 12:1744806916646784. doi:10.1177/1744806916646784
17. Mrak RE, Griffin WS. Interleukin-1 and the immunogenetics of Alzheimer disease. J Neuropathol Exp Neurol (2000) 59:471. doi:10.1093/jnen/59.6.471
18. Bessler H, Djaldetti R, Salman H, Bergman M, Djaldetti M. IL-1 beta, IL-2, IL-6 and TNF-alpha production by peripheral blood mononuclear cells from patients with Parkinson’s disease. Biomed Pharmacother (1999) 53:141. doi:10.1016/S0753-3322(99)80079-1
19. Qin XY, Zhang SP, Cao C, Loh YP, Cheng Y. Aberrations in peripheral inflammatory cytokine levels in Parkinson disease: a systematic review and meta-analysis. JAMA Neurol (2016) 73:1316. doi:10.1001/jamaneurol.2016.2742
20. Careaga M, Rogers S, Hansen RL, Amaral DG, Van de Water J, Ashwood P. Immune endophenotypes in children with autism spectrum disorder. Biol Psychiatry (2017) 81:434. doi:10.1016/j.biopsych.2015.08.036
21. Pirttila T, Mehta PD, Frey H, Wisniewski HM. Alpha 1-antichymotrypsin and IL-1 beta are not increased in CSF or serum in Alzheimer’s disease. Neurobiol Aging (1994) 15:313. doi:10.1016/0197-4580(94)90026-4
22. Pecorelli A, Cervellati F, Belmonte G, Montagner G, Waldon P, Hayek J, et al. Cytokines profile and peripheral blood mononuclear cells morphology in Rett and autistic patients. Cytokine (2016) 77:180. doi:10.1016/j.cyto.2015.10.002
23. Schultz RM. Interleukin 1 and interferon-gamma: cytokines that provide reciprocal regulation of macrophage and T cell function. Toxicol Pathol (1987) 15:333. doi:10.1177/019262338701500311
24. Zhang QS, Heng Y, Yuan YH, Chen NH. Pathological alpha-synuclein exacerbates the progression of Parkinson’s disease through microglial activation. Toxicol Lett (2017) 265:30. doi:10.1016/j.toxlet.2016.11.002
25. Sznejder-Pacholek A, Joniec-Maciejak I, Wawer A, Ciesielska A, Mirowska-Guzel D. The effect of alpha-synuclein on gliosis and IL-1alpha, TNFalpha, IFNgamma, TGFbeta expression in murine brain. Pharmacol Rep (2017) 69:242. doi:10.1016/j.pharep.2016.11.003
26. Alyu F, Dikmen M. Inflammatory aspects of epileptogenesis: contribution of molecular inflammatory mechanisms. Acta neuropsychiatrica (2017) 29:1. doi:10.1017/neu.2016.47
27. Stojakovic A, Paz-Filho G, Arcos-Burgos M, Licinio J, Wong ML, Mastronardi CA. Role of the IL-1 pathway in dopaminergic neurodegeneration and decreased voluntary movement. Mol Neurobiol (2016) 54(6):4486–95. doi:10.1007/s12035-016-9988-x
28. Hou Y, Xie G, Liu X, Li G, Jia C, Xu J, et al. Minocycline protects against lipopolysaccharide-induced cognitive impairment in mice. Psychopharmacology (2016) 233:905. doi:10.1007/s00213-015-4169-6
29. Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia (2002) 40:133. doi:10.1002/glia.10154
30. Brooke SM, Sapolsky RM. Glucocorticoid exacerbation of gp120 neurotoxicity: role of microglia. Exp Neurol (2002) 177:151. doi:10.1006/exnr.2002.7956
31. Jin WN, Shi SX, Li Z, Li M, Wood K, Gonzales RJ, et al. Depletion of microglia exacerbates postischemic inflammation and brain injury. J Cereb Blood Flow Metab (2017) 37:2224. doi:10.1177/0271678X17694185
32. Zanier ER, Marchesi F, Ortolano F, Perego C, Arabian M, Zoerle T, et al. Fractalkine receptor deficiency is associated with early protection but late worsening of outcome following brain trauma in mice. J Neurotrauma (2016) 33:1060. doi:10.1089/neu.2015.4041
33. Ongerth T, Russmann V, Fischborn S, Boes K, Siegl C, Potschka H. Targeting of microglial KCa3.1 channels by TRAM-34 exacerbates hippocampal neurodegeneration and does not affect ictogenesis and epileptogenesis in chronic temporal lobe epilepsy models. Eur J Pharmacol (2014) 740:72. doi:10.1016/j.ejphar.2014.06.061
34. Ducruet AF, Sosunov SA, Visovatti SH, Petrovic-Djergovic D, Mack WJ, Connolly ES Jr, et al. Paradoxical exacerbation of neuronal injury in reperfused stroke despite improved blood flow and reduced inflammation in early growth response-1 gene-deleted mice. Neurol Res (2011) 33:717. doi:10.1179/1743132810Y.0000000022
35. Veltkamp R, Gill D. Clinical trials of immunomodulation in ischemic stroke. Neurotherapeutics (2016) 13:791. doi:10.1007/s13311-016-0458-y
36. Salter MW, Beggs S. Sublime microglia: expanding roles for the guardians of the CNS. Cell (2014) 158:15. doi:10.1016/j.cell.2014.06.008
37. Singhal G, Baune BT. Microglia: an interface between the loss of neuroplasticity and depression. Front Cell Neurosci (2017) 11:270. doi:10.3389/fncel.2017.00270
38. Vezzani A, Maroso M, Balosso S, Sanchez MA, Bartfai T. IL-1 receptor/toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav Immun (2011) 25:1281. doi:10.1016/j.bbi.2011.03.018
39. Spulber S, Bartfai T, Schultzberg M. IL-1/IL-1ra balance in the brain revisited – evidence from transgenic mouse models. Brain Behav Immun (2009) 23:573. doi:10.1016/j.bbi.2009.02.015
40. Lu KT, Wu CY, Yen HH, Peng JH, Wang CL, Yang YL. Bumetanide administration attenuated traumatic brain injury through IL-1 overexpression. Neurol Res (2007) 29:404. doi:10.1179/016164107X204738
41. Lu KT, Wang YW, Wo YY, Yang YL. Extracellular signal-regulated kinase-mediated IL-1-induced cortical neuron damage during traumatic brain injury. Neurosci Lett (2005) 386:40. doi:10.1016/j.neulet.2005.05.057
42. Saito K, Suyama K, Nishida K, Sei Y, Basile AS. Early increases in TNF-alpha, IL-6 and IL-1 beta levels following transient cerebral ischemia in gerbil brain. Neurosci Lett (1996) 206:149. doi:10.1016/S0304-3940(96)12460-5
43. Wang X, Yue TL, Barone FC, White RF, Gagnon RC, Feuerstein GZ. Concomitant cortical expression of TNF-alpha and IL-1 beta mRNAs follows early response gene expression in transient focal ischemia. Mol Chem Neuropathol (1994) 23:103. doi:10.1007/BF02815404
44. Cacabelos R, Barquero M, Garcia P, Alvarez XA, Varela de Seijas E. Cerebrospinal fluid interleukin-1 beta (IL-1 beta) in Alzheimer’s disease and neurological disorders. Methods Findings Exp Clin Pharmacol (1991) 13:455.
45. Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, et al. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J Immunol (2011) 187:6539. doi:10.4049/jimmunol.1100620
46. Pare A, Mailhot B, Levesque SA, Lacroix S. Involvement of the IL-1 system in experimental autoimmune encephalomyelitis and multiple sclerosis: breaking the vicious cycle between IL-1beta and GM-CSF. Brain Behav Immun (2017) 62:1. doi:10.1016/j.bbi.2016.07.146
47. Cragnolini AB, Schioth HB, Scimonelli TN. Anxiety-like behavior induced by IL-1beta is modulated by alpha-MSH through central melanocortin-4 receptors. Peptides (2006) 27:1451. doi:10.1016/j.peptides.2005.10.020
48. Song C, Li X, Leonard BE, Horrobin DF. Effects of dietary n-3 or n-6 fatty acids on interleukin-1beta-induced anxiety, stress, and inflammatory responses in rats. J Lipid Res (2003) 44:1984. doi:10.1194/jlr.M300217-JLR200
49. Rossi S, Sacchetti L, Napolitano F, De Chiara V, Motta C, Studer V, et al. Interleukin-1beta causes anxiety by interacting with the endocannabinoid system. J Neurosci (2012) 32:13896. doi:10.1523/JNEUROSCI.1515-12.2012
50. Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, et al. beta-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci (2011) 31:6277. doi:10.1523/JNEUROSCI.0450-11.2011
51. Koo JW, Duman RS. Evidence for IL-1 receptor blockade as a therapeutic strategy for the treatment of depression. Curr Opin Invest Drugs (2009) 10:664.
52. Huang QJ, Jiang H, Hao XL, Minor TR. Brain IL-1 beta was involved in reserpine-induced behavioral depression in rats. Acta Pharmacol Sinica (2004) 25:293.
53. Maes M, Song C, Yirmiya R. Targeting IL-1 in depression. Expert Opin Ther Targets (2012) 16:1097. doi:10.1517/14728222.2012.718331
54. Wu H, Wang X, Gao J, Liang S, Hao Y, Sun C, et al. Fingolimod (FTY720) attenuates social deficits, learning and memory impairments, neuronal loss and neuroinflammation in the rat model of autism. Life Sci (2017) 173:43. doi:10.1016/j.lfs.2017.01.012
55. Chao CC, Hu S, Ehrlich L, Peterson PK. Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: involvement of nitric oxide and of N-methyl-D-aspartate receptors. Brain Behav Immun (1995) 9:355. doi:10.1006/brbi.1995.1033
56. Westmoreland SV, Kolson D, Gonzalez-Scarano F. Toxicity of TNF alpha and platelet activating factor for human NT2N neurons: a tissue culture model for human immunodeficiency virus dementia. J Neurovirol (1996) 2:118. doi:10.3109/13550289609146545
57. Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci (2003) 23:8692.
58. Thornton P, Pinteaux E, Gibson RM, Allan SM, Rothwell NJ. Interleukin-1-induced neurotoxicity is mediated by glia and requires caspase activation and free radical release. J Neurochem (2006) 98:258. doi:10.1111/j.1471-4159.2006.03872.x
59. Zheng J, Thylin MR, Ghorpade A, Xiong H, Persidsky Y, Cotter R, et al. Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. J Neuroimmunol (1999) 98:185. doi:10.1016/S0165-5728(99)00049-1
60. Parajuli B, Horiuchi H, Mizuno T, Takeuchi H, Suzumura A. CCL11 enhances excitotoxic neuronal death by producing reactive oxygen species in microglia. Glia (2015) 63:2274. doi:10.1002/glia.22892
61. De Paola M, Buanne P, Biordi L, Bertini R, Ghezzi P, Mennini T. Chemokine MIP-2/CXCL2, acting on CXCR2, induces motor neuron death in primary cultures. Neuroimmunomodulation (2007) 14:310. doi:10.1159/000123834
62. Wohleb ES, Patterson JM, Sharma V, Quan N, Godbout JP, Sheridan JF. Knockdown of interleukin-1 receptor type-1 on endothelial cells attenuated stress-induced neuroinflammation and prevented anxiety-like behavior. J Neurosci (2014) 34:2583. doi:10.1523/JNEUROSCI.3723-13.2014
63. Levesque SA, Paré A, Mailhot B, Bellver-Landete V, Kébir H, Lécuyer MA, et al. Myeloid cell transmigration across the CNS vasculature triggers IL-1beta-driven neuroinflammation during autoimmune encephalomyelitis in mice. J Exp Med (2016) 213:929. doi:10.1084/jem.20151437
64. McKim DB, Weber MD, Niraula A, Sawicki CM, Liu X, Jarrett BL, et al. Microglial recruitment of IL-1beta-producing monocytes to brain endothelium causes stress-induced anxiety. Mol Psychiatry (2017). doi:10.1038/mp.2017.64
65. Allen C, Thornton P, Denes A, McColl BW, Pierozynski A, Monestier M, et al. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J Immunol (2012) 189:381. doi:10.4049/jimmunol.1200409
66. Kong LL, Wang ZY, Han N, Zhuang XM, Wang ZZ, Li H, et al. Neutralization of chemokine-like factor 1, a novel C-C chemokine, protects against focal cerebral ischemia by inhibiting neutrophil infiltration via MAPK pathways in rats. J Neuroinflammation (2014) 11:112. doi:10.1186/1742-2094-11-112
67. Proudfoot AE, de Souza AL, Muzio V. The use of chemokine antagonists in EAE models. J Neuroimmunol (2008) 198:27. doi:10.1016/j.jneuroim.2008.04.007
68. Coisne C, Mao W, Engelhardt B. Cutting edge: natalizumab blocks adhesion but not initial contact of human T cells to the blood-brain barrier in vivo in an animal model of multiple sclerosis. J Immunol (2009) 182:5909. doi:10.4049/jimmunol.0803418
69. Sawicki CM, McKim DB, Wohleb ES, Jarrett BL, Reader BF, Norden DM, et al. Social defeat promotes a reactive endothelium in a brain region-dependent manner with increased expression of key adhesion molecules, selectins and chemokines associated with the recruitment of myeloid cells to the brain. Neuroscience (2015) 302:151. doi:10.1016/j.neuroscience.2014.10.004
70. Aguilar-Valles A, Kim J, Jung S, Woodside B, Luheshi GN. Role of brain transmigrating neutrophils in depression-like behavior during systemic infection. Mol Psychiatry (2014) 19:599. doi:10.1038/mp.2013.137
71. Wohleb ES, Powell ND, Godbout JP, Sheridan JF. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci (2013) 33:13820. doi:10.1523/JNEUROSCI.1671-13.2013
72. Liou KT, Shen YC, Chen CF, Tsao CM, Tsai SK. Honokiol protects rat brain from focal cerebral ischemia-reperfusion injury by inhibiting neutrophil infiltration and reactive oxygen species production. Brain Res (2003) 992:159. doi:10.1016/j.brainres.2003.08.026
73. Malaplate-Armand C, Ferrari L, Masson C, Siest G, Batt AM. Astroglial CYP1B1 up-regulation in inflammatory/oxidative toxic conditions: IL-1beta effect and protection by N-acetylcysteine. Toxicol Lett (2003) 138:243. doi:10.1016/S0378-4274(02)00417-4
74. Wei J, Fang W, Sha L, Han D, Zhang R, Hao X, et al. XQ-1H suppresses neutrophils infiltration and oxidative stress induced by cerebral ischemia injury both in vivo and in vitro. Neurochem Res (2013) 38:2542. doi:10.1007/s11064-013-1176-z
75. Sukumari-Ramesh S, Alleyne CH Jr. Post-injury administration of tert-butylhydroquinone attenuates acute neurological injury after intracerebral hemorrhage in mice. J Mol Neurosci (2016) 58:525. doi:10.1007/s12031-016-0722-y
76. Sulakhiya K, Keshavlal GP, Bezbaruah BB, Dwivedi S, Gurjar SS, Munde N, et al. Lipopolysaccharide induced anxiety- and depressive-like behaviour in mice are prevented by chronic pre-treatment of esculetin. Neurosci Lett (2016) 611:106. doi:10.1016/j.neulet.2015.11.031
77. Melov S, Wolf N, Strozyk D, Doctrow SR, Bush AI. Mice transgenic for Alzheimer disease beta-amyloid develop lens cataracts that are rescued by antioxidant treatment. Free Rad Biol Med (2005) 38:258. doi:10.1016/j.freeradbiomed.2004.10.023
78. Dore S. Decreased activity of the antioxidant heme oxygenase enzyme: implications in ischemia and in Alzheimer’s disease. Free Rad Biol Med (2002) 32:1276. doi:10.1016/S0891-5849(02)00805-5
79. Kelly PJ, Morrow JD, Ning M, Koroshetz W, Lo EH, Terry E, et al. Oxidative stress and matrix metalloproteinase-9 in acute ischemic stroke: the biomarker evaluation for antioxidant therapies in stroke (BEAT-stroke) study. Stroke (2008) 39:100. doi:10.1161/STROKEAHA.107.488189
80. Gibson GE, Zhang H, Sheu KR, Park LC. Differential alterations in antioxidant capacity in cells from Alzheimer patients. Biochim Biophys Acta (2000) 1502:319. doi:10.1016/S0925-4439(00)00057-0
81. Bourdel-Marchasson I, Delmas-Beauvieux MC, Peuchant E, Richard-Harston S, Decamps A, Reignier B, et al. Antioxidant defences and oxidative stress markers in erythrocytes and plasma from normally nourished elderly Alzheimer patients. Age Ageing (2001) 30:235. doi:10.1093/ageing/30.3.235
82. Hall ED, Vaishnav RA, Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurotherapeutics (2010) 7:51. doi:10.1016/j.nurt.2009.10.021
83. Talarowska M, Szemraj J, Berk M, Maes M, Galecki P. Oxidant/antioxidant imbalance is an inherent feature of depression. BMC Psychiatry (2015) 15:71. doi:10.1186/s12888-015-0454-5
84. Clarke P, Leser JS, Quick ED, Dionne KR, Beckham JD, Tyler KL. Death receptor-mediated apoptotic signaling is activated in the brain following infection with West Nile virus in the absence of a peripheral immune response. J Virol (2014) 88:1080. doi:10.1128/JVI.02944-13
85. Gupta K, Banerjee A, Saggar K, Ahluwalia A, Saggar K. A prospective study of magnetic resonance imaging patterns of central nervous system infections in pediatric age group and young adults and their clinico-biochemical correlation. J Pediatric Neurosci (2016) 11:46. doi:10.4103/1817-1745.181244
86. Kashyap RS, Deshpande PS, Ramteke SR, Panchbhai MS, Purohit HJ, Taori GM, et al. Changes in cerebrospinal fluid cytokine expression in tuberculous meningitis patients with treatment. Neuroimmunomodulation (2010) 17:333. doi:10.1159/000292023
87. Nicolini H, López Y, Genis-Mendoza AD, Manrique V, Lopez-Canovas L, Niubo E, et al. Detection of anti-streptococcal, antienolase, and anti-neural antibodies in subjects with early-onset psychiatric disorders. Actas Esp Psiquiatr (2015) 43:35.
88. Yaddanapudi K, Hornig M, Serge R, De Miranda J, Baghban A, Villar G, et al. Passive transfer of streptococcus-induced antibodies reproduces behavioral disturbances in a mouse model of pediatric autoimmune neuropsychiatric disorders associated with streptococcal infection. Mol Psychiatry (2010) 15:712. doi:10.1038/mp.2009.77
89. Frick L, Pittenger C. Microglial dysregulation in OCD, Tourette syndrome, and PANDAS. J Immunol Res (2016) 2016:8606057. doi:10.1155/2016/8606057
90. Patel MN, Carroll RG, Galván-Peña S, Mills EL, Olden R, Triantafilou M, et al. Inflammasome priming in sterile inflammatory disease. Trends Mol Med (2017) 23:165. doi:10.1016/j.molmed.2016.12.007
91. Scheiblich H, Schlütter A, Golenbock DT, Latz E, Martinez-Martinez P, Heneka MT. Activation of the NLRP3 inflammasome in microglia: the role of ceramide. J Neurochem (2017) 143(5):534–50. doi:10.1111/jnc.14225
92. Gustin A, Kirchmeyer M, Koncina E, Felten P, Losciuto S, Heurtaux T, et al. NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One (2015) 10:e0130624. doi:10.1371/journal.pone.0130624
93. Ye X, Shen T, Hu J, Zhang L, Zhang Y, Bao L, et al. Purinergic 2X7 receptor/NLRP3 pathway triggers neuronal apoptosis after ischemic stroke in the mouse. Exp Neurol (2017) 292:46. doi:10.1016/j.expneurol.2017.03.002
94. Yang SJ, Shao GF, Chen JL, Gong J. The NLRP3 inflammasome: an important driver of neuroinflammation in hemorrhagic stroke. Cell Mol Neurobiol (2017). doi:10.1007/s10571-017-0526-9
95. Saresella M, La Rosa F, Piancone F, Zoppis M, Marventano I, Calabrese E, et al. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener (2016) 11:23. doi:10.1186/s13024-016-0088-1
96. Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, et al. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS One (2013) 8:e55375. doi:10.1371/journal.pone.0055375
97. Kaufmann FN, Costa AP, Ghisleni G, Diaz AP, Rodrigues ALS, Peluffo H, et al. NLRP3 inflammasome-driven pathways in depression: clinical and preclinical findings. Brain Behav Immun (2017) 64:367. doi:10.1016/j.bbi.2017.03.002
98. Iwata M, Ota KT, Duman RS. The inflammasome: pathways linking psychological stress, depression, and systemic illnesses. Brain Behav Immun (2013) 31:105. doi:10.1016/j.bbi.2012.12.008
99. McCann SK, Cramond F, Macleod MR, Sena ES. Systematic review and meta-analysis of the efficacy of interleukin-1 receptor antagonist in animal models of stroke: an update. Transl Stroke Res (2016) 7:395. doi:10.1007/s12975-016-0489-z
100. Gentile A, Fresegna D, Musella A, Sepman H, Bullitta S, De Vito F, et al. Interaction between interleukin-1beta and type-1 cannabinoid receptor is involved in anxiety-like behavior in experimental autoimmune encephalomyelitis. J Neuroinflammation (2016) 13:231. doi:10.1186/s12974-016-0682-8
101. Vezzani A, Moneta D, Conti M, Richichi C, Ravizza T, De Luigi A, et al. Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proc Natl Acad Sci U S A (2000) 97:11534. doi:10.1073/pnas.190206797
102. Noe FM, Polascheck N, Frigerio F, Bankstahl M, Ravizza T, Marchini S, et al. Pharmacological blockade of IL-1beta/IL-1 receptor type 1 axis during epileptogenesis provides neuroprotection in two rat models of temporal lobe epilepsy. Neurobiol Dis (2013) 59:183. doi:10.1016/j.nbd.2013.07.015
103. Möller T, Bard F, Bhattacharya A, Biber K, Campbell B, Dale E, et al. Critical data-based re-evaluation of minocycline as a putative specific microglia inhibitor. Glia (2016) 64:1788. doi:10.1002/glia.23007
104. Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K, et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis (2013) 4:e525. doi:10.1038/cddis.2013.54
105. Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci (2001) 21:2580.
106. Kraus RL, Pasieczny R, Lariosa-Willingham K, Turner MS, Jiang A, Trauger JW. Antioxidant properties of minocycline: neuroprotection in an oxidative stress assay and direct radical-scavenging activity. J Neurochem (2005) 94:819. doi:10.1111/j.1471-4159.2005.03219.x
107. Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, et al. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci (2002) 22:1763.
108. Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG. Microglia potentiate damage to blood-brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke (2006) 37:1087. doi:10.1161/01.STR.0000206281.77178.ac
109. Abraham J, Fox PD, Condello C, Bartolini A, Koh S. Minocycline attenuates microglia activation and blocks the long-term epileptogenic effects of early-life seizures. Neurobiol Dis (2012) 46:425. doi:10.1016/j.nbd.2012.02.006
110. Wong ML, Inserra A, Lewis MD, Mastronardi CA, Leong L, Choo J, et al. Inflammasome signaling affects anxiety- and depressive-like behavior and gut microbiome composition. Mol Psychiatry (2016) 21:797. doi:10.1038/mp.2016.46
111. Giuliani F, Metz LM, Wilson T, Fan Y, Bar-Or A, Yong VW. Additive effect of the combination of glatiramer acetate and minocycline in a model of MS. J Neuroimmunol (2005) 158:213. doi:10.1016/j.jneuroim.2004.09.006
112. Giuliani F, Fu SA, Metz LM, Yong VW. Effective combination of minocycline and interferon-beta in a model of multiple sclerosis. J Neuroimmunol (2005) 165:83. doi:10.1016/j.jneuroim.2005.04.020
113. Luccarini I, Ballerini C, Biagioli T, Biamonte F, Bellucci A, Rosi MC, et al. Combined treatment with atorvastatin and minocycline suppresses severity of EAE. Exp Neurol (2008) 211:214. doi:10.1016/j.expneurol.2008.01.022
114. Chen X, Hu X, Zou Y, Pi R, Liu M, Wang T, et al. Combined treatment with minocycline and prednisone attenuates experimental autoimmune encephalomyelitis in C57 BL/6 mice. J Neuroimmunol (2009) 210:22. doi:10.1016/j.jneuroim.2009.02.016
115. Chen X, Pi R, Liu M, Ma X, Jiang Y, Liu Y, et al. Combination of methylprednisolone and minocycline synergistically improves experimental autoimmune encephalomyelitis in C57 BL/6 mice. J Neuroimmunol (2010) 226:104. doi:10.1016/j.jneuroim.2010.05.039
116. Faissner S, Mahjoub Y, Mishra M, Haupeltshofer S, Hahn JN, Gold R, et al. Unexpected additive effects of minocycline and hydroxychloroquine in models of multiple sclerosis: prospective combination treatment for progressive disease? Mult Scler (2017) 1:1352458517728811. doi:10.1177/1352458517728811
117. Herrmann I, Kellert M, Spreer A, Gerber J, Eiffert H, Prinz M, et al. Minocycline delays but does not attenuate the course of experimental autoimmune encephalomyelitis in Streptococcus pneumoniae-infected mice. J Antimicrob Chemother (2007) 59:74. doi:10.1093/jac/dkl446
118. Nikodemova M, Lee J, Fabry Z, Duncan ID. Minocycline attenuates experimental autoimmune encephalomyelitis in rats by reducing T cell infiltration into the spinal cord. J Neuroimmunol (2010) 219:33. doi:10.1016/j.jneuroim.2009.11.009
119. Ogungbenro K, Hulme S, Rothwell N, Hopkins S, Tyrrell P, Galea J. Study design and population pharmacokinetic analysis of a phase II dose-ranging study of interleukin-1 receptor antagonist. J Pharmacokinet Pharmacodyn (2016) 43:1. doi:10.1007/s10928-015-9450-0
120. Funk DJ, HayGlass KT, Koulack J, Harding G, Boyd A, Brinkman R. A randomized controlled trial on the effects of goal-directed therapy on the inflammatory response open abdominal aortic aneurysm repair. Crit Care (2015) 19:247. doi:10.1186/s13054-015-0974-x
121. Helmy A, Guilfoyle MR, Carpenter KL, Pickard JD, Menon DK, Hutchinson PJ. Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: a phase II randomized control trial. J Cereb Blood Flow Metab (2014) 34:845. doi:10.1038/jcbfm.2014.23
122. Norheim KB, Harboe E, Goransson LG, Omdal R. Interleukin-1 inhibition and fatigue in primary Sjogren’s syndrome – a double blind, randomised clinical trial. PLoS One (2012) 7:e30123. doi:10.1371/journal.pone.0030123
123. Kohler E, Prentice DA, Bates TR, Hankey GJ, Claxton A, van Heerden J, et al. Intravenous minocycline in acute stroke: a randomized, controlled pilot study and meta-analysis. Stroke (2013) 44:2493. doi:10.1161/STROKEAHA.113.000780
124. Lampl Y, Boaz M, Gilad R, Lorberboym M, Dabby R, Rapoport A, et al. Minocycline treatment in acute stroke: an open-label, evaluator-blinded study. Neurology (2007) 69:1404. doi:10.1212/01.wnl.0000277487.04281.db
125. Fouda AY, Newsome AS, Spellicy S, Waller JL, Zhi W, Hess DC, et al. Minocycline in acute cerebral hemorrhage: an early phase randomized trial. Stroke (2017) 48:2885. doi:10.1161/STROKEAHA.117.018658
126. LeWitt PA, Taylor DC. Protection against Parkinson’s disease progression: clinical experience. Neurotherapeutics (2008) 5:210. doi:10.1016/j.nurt.2008.01.007
127. Investigators NN-P. A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology (2006) 66:664. doi:10.1212/01.wnl.0000201252.57661.e1
128. Lang N, Rothkegel H, Terney D, Antal A, Paulus W. Minocycline exerts acute inhibitory effects on cerebral cortex excitability in humans. Epilepsy Res (2013) 107:302. doi:10.1016/j.eplepsyres.2013.09.006
129. Soczynska JK, Kennedy SH, Alsuwaidan M, Mansur RB, Li M, McAndrews MP, et al. A pilot, open-label, 8-week study evaluating the efficacy, safety and tolerability of adjunctive minocycline for the treatment of bipolar I/II depression. Bipolar Disord (2017) 19:198. doi:10.1111/bdi.12496
130. Husain MI, Chaudhry IB, Husain N, Khoso AB, Rahman RR, Hamirani MM, et al. Minocycline as an adjunct for treatment-resistant depressive symptoms: a pilot randomised placebo-controlled trial. J Psychopharmacol (2017) 31:1166. doi:10.1177/0269881117724352
131. Ghanizadeh A, Dehbozorgi S, OmraniSigaroodi M, Rezaei Z. Minocycline as add-on treatment decreases the negative symptoms of schizophrenia; a randomized placebo-controlled clinical trial. Recent Pat Inflamm Allergy Drug Discov (2014) 8:211. doi:10.2174/1872213X08666141029123524
132. Zhang Y, Metz LM, Yong VW, Bell RB, Yeung M, Patry DG, et al. Pilot study of minocycline in relapsing-remitting multiple sclerosis. Can J Neurol Sci (2008) 35:185. doi:10.1017/S0317167100008611
133. Metz LM, Li DKB, Traboulsee AL, Duquette P, Eliasziw M, Cerchiaro G, et al. Trial of minocycline in a clinically isolated syndrome of multiple sclerosis. N Engl J Med (2017) 376:2122. doi:10.1056/NEJMoa1608889
134. Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O’Banion MK. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest (2007) 117:1595. doi:10.1172/JCI31450
135. Kreisel T, Frank MG, Licht T, Reshef R, Ben-Menachem-Zidon O, Baratta MV, et al. Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol Psychiatry (2014) 19:699. doi:10.1038/mp.2013.155
136. Nakashima T, Hori T, Mori T, Kuriyama K, Mizuno K. Recombinant human interleukin-1 beta alters the activity of preoptic thermosensitive neurons in vitro. Brain Res Bull (1989) 23:209. doi:10.1016/0361-9230(89)90149-4
137. Hori T, Shibata M, Nakashima T, Yamasaki M, Asami A, Asami T, et al. Effects of interleukin-1 and arachidonate on the preoptic and anterior hypothalamic neurons. Brain Res Bull (1988) 20:75. doi:10.1016/0361-9230(88)90010-X
138. Li Z, Inenage K, Kawano S, Kannan H, Yamashita H. Interleukin-1 beta directly excites hypothalamic supraoptic neurons in rats in vitro. Neuroreport (1992) 3:91. doi:10.1097/00001756-199201000-00024
139. Krueger JM, Fang J, Taishi P, Chen Z, Kushikata T, Gardi J. Sleep. A physiologic role for IL-1 beta and TNF-alpha. Ann N Y Acad Sci (1998) 856:148. doi:10.1111/j.1749-6632.1998.tb08323.x
140. Hallett H, Churchill L, Taishi P, De A, Krueger JM. Whisker stimulation increases expression of nerve growth factor- and interleukin-1beta-immunoreactivity in the rat somatosensory cortex. Brain Res (2010) 1333:48. doi:10.1016/j.brainres.2010.03.048
141. Saphier D, Ovadia H. Selective facilitation of putative corticotropin-releasing factor-secreting neurones by interleukin-1. Neurosci Lett (1990) 114:283. doi:10.1016/0304-3940(90)90577-V
142. Plata-Salaman CR, French-Mullen JM. Interleukin-1 beta inhibits Ca2+ channel currents in hippocampal neurons through protein kinase C. Eur J Pharmacol (1994) 266:1. doi:10.1016/0922-4106(94)90202-X
143. Pringle AK, Gardner CR, Walker RJ. Reduction of cerebellar GABAA responses by interleukin-1 (IL-1) through an indomethacin insensitive mechanism. Neuropharmacology (1996) 35:147. doi:10.1016/0028-3908(95)00161-1
144. Coogan A, O’Connor JJ. Inhibition of NMDA receptor-mediated synaptic transmission in the rat dentate gyrus in vitro by IL-1 beta. Neuroreport (1997) 8:2107. doi:10.1097/00001756-199707070-00004
145. Ferri CC, Yuill EA, Ferguson AV. Interleukin-1beta depolarizes magnocellular neurons in the paraventricular nucleus of the hypothalamus through prostaglandin-mediated activation of a non selective cationic conductance. Regul Pept (2005) 129:63. doi:10.1016/j.regpep.2005.01.004
146. Liu L, Yang TM, Liedtke W, Simon SA. Chronic IL-1beta signaling potentiates voltage-dependent sodium currents in trigeminal nociceptive neurons. J Neurophysiol (2006) 95:1478. doi:10.1152/jn.00509.2005
147. Zhou C, Qi C, Zhao J, Wang F, Zhang W, Li C, et al. Interleukin-1beta inhibits voltage-gated sodium currents in a time- and dose-dependent manner in cortical neurons. Neurochem Res (2011) 36:1116. doi:10.1007/s11064-011-0456-8
148. Tabarean IV, Korn H, Bartfai T. Interleukin-1beta induces hyperpolarization and modulates synaptic inhibition in preoptic and anterior hypothalamic neurons. Neuroscience (2006) 141:1685. doi:10.1016/j.neuroscience.2006.05.007
149. Ikegaya Y, Delcroix I, Iwakura Y, Matsuki N, Nishiyama N. Interleukin-1beta abrogates long-term depression of hippocampal CA1 synaptic transmission. Synapse (2003) 47:54. doi:10.1002/syn.10154
150. del Rey A, Balschun D, Wetzel W, Randolf A, Besedovsky HO. A cytokine network involving brain-borne IL-1beta, IL-1ra, IL-18, IL-6, and TNFalpha operates during long-term potentiation and learning. Brain Behav Immunity (2013) 33:15. doi:10.1016/j.bbi.2013.05.011
151. Depino AM, Alonso M, Ferrari C, del Rey A, Anthony D, Besedovsky H, et al. Learning modulation by endogenous hippocampal IL-1: blockade of endogenous IL-1 facilitates memory formation. Hippocampus (2004) 14:526. doi:10.1002/hipo.10164
152. Stemkowski PL, Noh MC, Chen Y, Smith PA. Increased excitability of medium-sized dorsal root ganglion neurons by prolonged interleukin-1beta exposure is K(+) channel dependent and reversible. J Physiol (2015) 593:3739. doi:10.1113/JP270905
153. Wu MD, Montgomery SL, Rivera-Escalera F, Olschowka JA, O’Banion MK. Sustained IL-1beta expression impairs adult hippocampal neurogenesis independent of IL-1 signaling in nestin+ neural precursor cells. Brain Behav Immun (2013) 32:9. doi:10.1016/j.bbi.2013.03.003
154. Yadlapati S, Efthimiou P. Impact of IL-1 inhibition on fatigue associated with autoinflammatory syndromes. Mod Rheumatol (2016) 26:3. doi:10.3109/14397595.2015.1069459
155. Goshen I, Kreisel T, Ounallah-Saad H, Renbaum P, Zalzstein Y, Ben-Hur T, et al. A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology (2007) 32:1106. doi:10.1016/j.psyneuen.2007.09.004
156. Akama KT, Van Eldik LJ. Beta-amyloid stimulation of inducible nitric-oxide synthase in astrocytes is interleukin-1beta- and tumor necrosis factor-alpha (TNFalpha)-dependent, and involves a TNFalpha receptor-associated factor- and NFkappaB-inducing kinase-dependent signaling mechanism. J Biol Chem (2000) 275:7918.
157. Sperlagh B, Baranyi M, Hasko G, Vizi ES. Potent effect of interleukin-1 beta to evoke ATP and adenosine release from rat hippocampal slices. J Neuroimmunol (2004) 151:33. doi:10.1016/j.jneuroim.2004.02.004
158. Kaneko N, Kudo K, Mabuchi T, Takemoto K, Fujimaki K, Wati H, et al. Suppression of cell proliferation by interferon-alpha through interleukin-1 production in adult rat dentate gyrus. Neuropsychopharmacology (2006) 31:2619. doi:10.1038/sj.npp.1301137
159. Goshen I, Kreisel T, Ben-Menachem-Zidon O, Licht T, Weidenfeld J, Ben-Hur T, et al. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry (2008) 13:717. doi:10.1038/sj.mp.4002055
160. Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci U S A (2010) 107:2669. doi:10.1073/pnas.0910658107
161. Ferreira R, Xapelli S, Santos T, Silva AP, Cristóvão A, Cortes L, et al. Neuropeptide Y modulation of interleukin-1{beta} (IL-1{beta})-induced nitric oxide production in microglia. J Biol Chem (2010) 285:41921. doi:10.1074/jbc.M110.164020
162. Carlson NG, Wieggel WA, Chen J, Bacchi A, Rogers SW, Gahring LC. Inflammatory cytokines IL-1 alpha, IL-1 beta, IL-6, and TNF-alpha impart neuroprotection to an excitotoxin through distinct pathways. J Immunol (1999) 163:3963.
163. Friedman WJ. Interactions of interleukin-1 with neurotrophic factors in the central nervous system: beneficial or detrimental? Mol Neurobiol (2005) 32:133. doi:10.1385/MN:32:2:133
164. Heese K, Hock C, Otten U. Inflammatory signals induce neurotrophin expression in human microglial cells. J Neurochem (1998) 70:699. doi:10.1046/j.1471-4159.1998.70020699.x
165. DeKosky ST, Styren SD, O’Malley ME, Goss JR, Kochanek P, Marion D, et al. Interleukin-1 receptor antagonist suppresses neurotrophin response in injured rat brain. Ann Neurol (1996) 39:123. doi:10.1002/ana.410390118
166. Boato F, Hechler D, Rosenberger K, Lüdecke D, Peters EM, Nitsch R, et al. Interleukin-1 beta and neurotrophin-3 synergistically promote neurite growth in vitro. J Neuroinflammation (2011) 8:183. doi:10.1186/1742-2094-8-183
167. Correale J, Villa A. The neuroprotective role of inflammation in nervous system injuries. J Neurol (2004) 251:1304. doi:10.1007/s00415-004-0649-z
168. Lapchak PA, Araujo DM, Hefti F. Systemic interleukin-1 beta decreases brain-derived neurotrophic factor messenger RNA expression in the rat hippocampal formation. Neuroscience (1993) 53:297. doi:10.1016/0306-4522(93)90196-M
169. Song C, Zhang Y, Dong Y. Acute and subacute IL-1beta administrations differentially modulate neuroimmune and neurotrophic systems: possible implications for neuroprotection and neurodegeneration. J Neuroinflammation (2013) 10:59. doi:10.1186/1742-2094-10-59
170. Choi S, Friedman WJ. Interleukin-1beta enhances neuronal vulnerability to proNGF-mediated apoptosis by increasing surface expression of p75(NTR) and sortillin. Neuroscience (2014) 257:11. doi:10.1016/j.neuroscience.2013.10.058
171. Goshen I, Yirmiya R, Iverfeldt K, Weidenfeld J. The role of endogenous interleukin-1 in stress-induced adrenal activation and adrenalectomy-induced adrenocorticotropic hormone hypersecretion. Endocrinology (2003) 144:4453. doi:10.1210/en.2003-0338
172. Barbanel G, Ixart G, Szafarczyk A, Malaval F, Assenmacher I. Intrahypothalamic infusion of interleukin-1 beta increases the release of corticotropin-releasing hormone (CRH 41) and adrenocorticotropic hormone (ACTH) in free-moving rats bearing a push-pull cannula in the median eminence. Brain Res (1990) 516:31–6. doi:10.1016/0006-8993(90)90893-G
173. Gadek-Michalska A, Bugajski J. Interleukin-1 (IL-1) in stress-induced activation of limbic-hypothalamic-pituitary adrenal axis. Pharmacol Rep (2010) PR 62:969. doi:10.1016/S1734-1140(10)70359-5
174. Del Rey A, Verdenhalven M, Lörwald AC, Meyer C, Hernangómez M, Randolf A, et al. Brain-borne IL-1 adjusts glucoregulation and provides fuel support to astrocytes and neurons in an autocrine/paracrine manner. Mol Psychiatry (2016) 21:1309. doi:10.1038/mp.2015.174
175. Ottaviani E, Caselgrandi E, Franceschi C. Cytokines and evolution: in vitro effects of IL-1 alpha, IL-1 beta, TNF-alpha and TNF-beta on an ancestral type of stress response. Biochem Biophys Res Commun (1995) 207:288. doi:10.1006/bbrc.1995.1185
176. Mosser CA, Baptista S, Arnoux I, Audinat E. Microglia in CNS development: shaping the brain for the future. Prog Neurobiol (2017) 1:149–50. doi:10.1016/j.pneurobio.2017.01.002
177. Ziebell JM, Adelson PD, Lifshitz J. Microglia: dismantling and rebuilding circuits after acute neurological injury. Metab Brain Dis (2015) 30:393. doi:10.1007/s11011-014-9539-y
178. Chen Z, Trapp BD. Microglia and neuroprotection. J Neurochem (2016) 136(Suppl 1):10. doi:10.1111/jnc.13062
179. Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci (2011) 14:1227. doi:10.1038/nn.2923
180. Marshall GP II, Deleyrolle LP, Reynolds BA, Steindler DA, Laywell ED. Microglia from neurogenic and non-neurogenic regions display differential proliferative potential and neuroblast support. Front Cell Neurosci (2014) 8:180. doi:10.3389/fncel.2014.00180
181. De Biase LM, Schuebel KE, Fusfeld ZH, Jair K, Hawes IA, Cimbro R, et al. Local cues establish and maintain region-specific phenotypes of basal ganglia microglia. Neuron (2017) 95:341. doi:10.1016/j.neuron.2017.06.020
182. Wohleb ES, Terwilliger R, Duman CH, Duman RS. Stress-induced neuronal colony stimulating factor 1 provokes microglia-mediated neuronal remodeling and depressive-like behavior. Biol Psychiatry (2018) 83(1):38–49. doi:10.1016/j.biopsych.2017.05.026
183. Masuch A, Shieh CH, van Rooijen N, van Calker D, Biber K. Mechanism of microglia neuroprotection: Involvement of P2X7, TNFalpha, and valproic acid. Glia (2016) 64:76. doi:10.1002/glia.22904
184. Arroba AI, Alvarez-Lindo N, van Rooijen N, de la Rosa EJ. Microglia-mediated IGF-I neuroprotection in the rd10 mouse model of retinitis pigmentosa. Invest Ophthalmol Vis Sci (2011) 52:9124. doi:10.1167/iovs.11-7736
185. Chen Z, Jalabi W, Hu W, Park HJ, Gale JT, Kidd GJ, et al. Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nat Commun (2014) 5:4486. doi:10.1038/ncomms5486
186. Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X, et al. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci (2012) 32:11706. doi:10.1523/JNEUROSCI.0730-12.2012
187. Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci (2014) 17:400. doi:10.1038/nn.3641
188. Huang Y, Smith DE, Ibanez-Sandoval O, Sims JE, Friedman WJ. Neuron-specific effects of interleukin-1beta are mediated by a novel isoform of the IL-1 receptor accessory protein. J Neurosci (2011) 31:18048–59. doi:10.1523/JNEUROSCI.4067-11.2011
189. An Y, Chen Q, Quan N. Interleukin-1 exerts distinct actions on different cell types of the brain in vitro. J Inflammation Res (2011) 2011:11.
190. Rossi S, Furlan R, De Chiara V, Motta C, Studer V, Mori F, et al. Interleukin-1beta causes synaptic hyperexcitability in multiple sclerosis. Ann Neurol (2012) 71:76–83. doi:10.1002/ana.22512
191. De Chiara V, Motta C, Rossi S, Studer V, Barbieri F, Lauro D, et al. Interleukin-1beta alters the sensitivity of cannabinoid CB1 receptors controlling glutamate transmission in the striatum. Neuroscience (2013) 250:232. doi:10.1016/j.neuroscience.2013.06.069
192. Rossi S, Studer V, Motta C, De Chiara V, Barbieri F, Bernardi G, et al. Inflammation inhibits GABA transmission in multiple sclerosis. Mult Scler (2012) 18:1633. doi:10.1177/1352458512440207
193. Yang Y, Salayandia VM, Thompson JF, Yang LY, Estrada EY, Yang Y. Attenuation of acute stroke injury in rat brain by minocycline promotes blood-brain barrier remodeling and alternative microglia/macrophage activation during recovery. J Neuroinflammation (2015) 12:26. doi:10.1186/s12974-015-0245-4
194. Giulian D, Chen J, Ingeman JE, George JK, Noponen M. The role of mononuclear phagocytes in wound healing after traumatic injury to adult mammalian brain. J Neurosci (1989) 9:4416.
Keywords: cytokine, neuroinflammation, brain, neuromodulation, IL-1R1
Citation: Liu X and Quan N (2018) Microglia and CNS Interleukin-1: Beyond Immunological Concepts. Front. Neurol. 9:8. doi: 10.3389/fneur.2018.00008
Received: 31 October 2017; Accepted: 05 January 2018;
Published: 23 January 2018
Edited by:
Bert A. ’T Hart, Biomedical Primate Research Centre, NetherlandsReviewed by:
Robert Adam Harris, Karolinska Institute (KI), SwedenFred Lühder, University of Göttingen, Germany
Copyright: © 2018 Liu and Quan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ning Quan, quan.14@osu.edu