Nipype: a flexible, lightweight and extensible neuroimaging data processing framework in Python
- 1 Neuroinformatics and Computational Neuroscience Doctoral Training Centre, School of Informatics, University of Edinburgh, Edinburgh, UK
- 2 Helen Wills Neuroscience Institute, University of California, Berkeley, CA, USA
- 3 Department of Psychology, University of California, Berkeley, CA, USA
- 4 Department of Psychological and Brain Sciences, Dartmouth College, Hanover, NH, USA
- 5 Department of Brain and Cognitive Sciences, Massachusetts Institute of Technology, Cambridge, MA, USA
- 6 McGovern Institute for Brain Research, Massachusetts Institute of Technology, Cambridge, MA, USA
- 7 Research Laboratory of Electronics, Massachusetts Institute of Technology, Cambridge, MA, USA
Current neuroimaging software offer users an incredible opportunity to analyze their data in different ways, with different underlying assumptions. Several sophisticated software packages (e.g., AFNI, BrainVoyager, FSL, FreeSurfer, Nipy, R, SPM) are used to process and analyze large and often diverse (highly multi-dimensional) data. However, this heterogeneous collection of specialized applications creates several issues that hinder replicable, efficient, and optimal use of neuroimaging analysis approaches: (1) No uniform access to neuroimaging analysis software and usage information; (2) No framework for comparative algorithm development and dissemination; (3) Personnel turnover in laboratories often limits methodological continuity and training new personnel takes time; (4) Neuroimaging software packages do not address computational efficiency; and (5) Methods sections in journal articles are inadequate for reproducing results. To address these issues, we present Nipype (Neuroimaging in Python: Pipelines and Interfaces; http://nipy.org/nipype), an open-source, community-developed, software package, and scriptable library. Nipype solves the issues by providing Interfaces to existing neuroimaging software with uniform usage semantics and by facilitating interaction between these packages using Workflows. Nipype provides an environment that encourages interactive exploration of algorithms, eases the design of Workflows within and between packages, allows rapid comparative development of algorithms and reduces the learning curve necessary to use different packages. Nipype supports both local and remote execution on multi-core machines and clusters, without additional scripting. Nipype is Berkeley Software Distribution licensed, allowing anyone unrestricted usage. An open, community-driven development philosophy allows the software to quickly adapt and address the varied needs of the evolving neuroimaging community, especially in the context of increasing demand for reproducible research.
Introduction
Over the past 20 years, advances in non-invasive in vivo neuroimaging have resulted in an explosion of studies investigating human cognition in health and disease. Current imaging studies acquire multi-modal image data (e.g., structural, diffusion, functional) and combine these with non-imaging behavioral data, patient and/or treatment history, and demographic and genetic information. Several sophisticated software packages (e.g., AFNI, BrainVoyager, FSL, FreeSurfer, Nipy, R, SPM) are used to process and analyze such extensive data. In a typical analysis, algorithms from these packages, each with its own set of parameters, process the raw data. However, data collected for a single study can be diverse (highly multi-dimensional) and large, and algorithms suited for one dataset may not be optimal for another. This complicates analysis methods and makes data exploration and inference challenging, and comparative analysis of new algorithms difficult.
Current Problems
Here we outline issues that hinder replicable, efficient, and optimal use of neuroimaging analysis approaches.
No uniform access to neuroimaging analysis software and usage information
For current multi-modal datasets, researchers typically resort to using different software packages for different components of the analysis. However, these different software packages are accessed, and interfaced with, in different ways, such as: shell scripting (FSL, AFNI, Camino), MATLAB (SPM), and Python (Nipy). This has resulted in a heterogeneous set of software with no uniform way to use these tools or execute them. With the primary focus on algorithmic improvement, academic software development often lacks a rigorous software engineering framework that involves extensive testing and documentation and ensures compatibility with other tools. This often necessitates extensive interactions with the authors of the software to understand their parameters, their quirks, and their usage.
No framework for comparative algorithm development and dissemination
Except for some large software development efforts (e.g., SPM, FSL, FreeSurfer), most algorithm development happens in-house and stays within the walls of a lab, without extensive exposure or testing. Furthermore, testing comparative efficacy of algorithms often requires significant effort (Klein et al., 2010). In general, developers create software for a single package (e.g., VBM8 for SPM), create a standalone cross-platform tool (e.g., Mricron), or simply do not distribute the software or code (e.g., normalization software used for registering architectonic atlases to MNI single subject template – Hömke, 2006).
Personnel turnover in laboratories often limits methodological continuity and training new personnel takes time
Most cognitive neuroscience laboratories aim to understand some aspect of cognition. Although a majority of such laboratories gather and analyze neuroimaging data, very few of them have the personnel with the technical expertise to understand methodological development and modify laboratory procedures to adopt new tools. Lab personnel with no prior imaging experience often learn by following online tutorials, taking organized courses or, as is most often the case, by learning from existing members of the lab. While this provides some amount of continuity, understanding different aspects of neuroimaging has a steep learning curve, and steeper when one takes into account the time and resources needed to learn the different package interfaces and algorithms.
Neuroimaging software packages do not address computational efficiency
The primary focus of neuroimaging analysis algorithms is to solve problems (e.g., registration, statistical estimation, tractography). While some developers focus on algorithmic or numerical efficiency, most developers do not focus on efficiency in the context of running multiple algorithms on multiple subjects, a common scenario in neuroimaging analysis. Creating an analysis workflow for a particular study is an iterative process dependent on the quality of the data and participant population (e.g., neurotypical, presurgical, etc.). Researchers usually experiment with different methods and their parameters to create a workflow suitable for their application, but no suitable framework currently exists to make this process efficient. Furthermore, very few of the available neuroimaging tools take advantage of the growing number of parallel hardware configurations (multi-core, clusters, clouds, and supercomputers).
Method sections of journal articles are often inadequate for reproducing results
Several journals (e.g., PNAS, Science, PLoS) require mandatory submission of data and scripts necessary to reproduce results of a study. However, most current method sections do not have sufficient details to enable a researcher knowledgeable in the domain to reproduce the analysis process. Furthermore, as discussed above, typical neuroimaging analyses integrate several tools and current analysis software do not make it easy to reproduce all the analysis steps in the proper order. This leaves a significant burden on the user to satisfy these journal requirements as well as ensure that analysis details are preserved with the intent to reproduce.
Current Solutions
There have been several attempts to address these issues by creating pipeline systems (for comparison see Table 1). Taverna (Oinn et al., 2006), VisTrails (Callahan et al., 2006) are general pipelining systems with excellent support for web-services, but they do not address problems specific to neuroimaging. BrainVisa (Cointepas et al., 2001), MIPAV (McAuliffe et al., 2001), SPM include their own batch processing tools, but do not allow mixing components from other packages. Fiswidgets (Fissell et al., 2003), a promising initial approach, appears to have not been developed and does not support state of the art methods. A much more extensive and feature rich solution is the LONI Pipeline (Rex et al., 2003; Dinov et al., 2009, 2010). It provides an easy to use graphical interface for choosing processing steps or nodes from a predefined library and defining their dependencies and parameters. Thanks to an advanced client–server architecture, it also has extensive support for parallel execution on an appropriately configured cluster (including data transfer, pausing execution, and combining local and remote software). Additionally, the LONI Pipeline saves information about executed steps (such as software origin, version, and architecture) providing provenance information (Mackenzie-Graham et al., 2008).
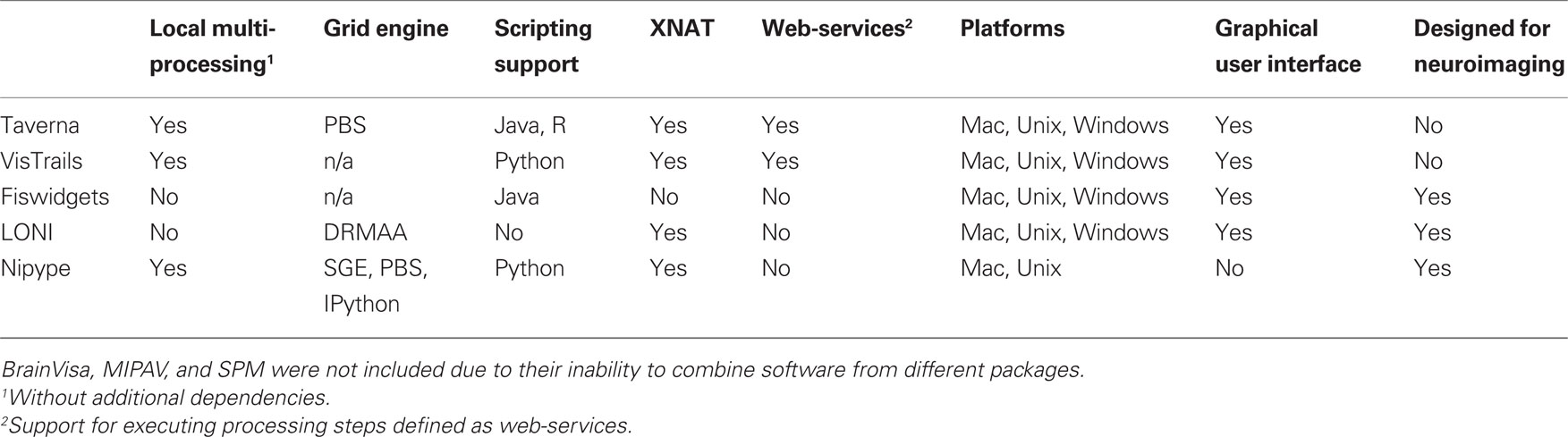
Table 1. Feature comparison of selected pipeline frameworks.
However, the LONI Pipeline does not come without limitations. Processing nodes are defined using eXtensible Markup Language (XML). This “one size fits all” method makes it easy to add new nodes as long as they are well-behaved command lines. However, many software packages do not meet this criterion. For example, SPM, written in MATLAB, does not provide a command line interface. Furthermore, for several command line programs, arguments are not easy to describe in the LONI XML schema (e.g., ANTS – Avants and Gee, 2004). Although it provides a helpful graphical interface, the LONI Pipeline environment does not provide an easy option to script a workflow or for rapidly exploring parametric variations within a workflow (e.g., VisTrails). Finally, due to restrictive licensing, it is not straightforward to modify and redistribute the modifications.
To address issues with existing workflow systems and the ones described earlier, we present Nipype (Neuroimaging in Python: Pipelines and Interfaces), an open-source, community-developed, Python-based software package that easily interfaces with existing software for efficient analysis of neuroimaging data and rapid comparative development of algorithms. Nipype uses a flexible, efficient, and general purpose programming language – Python – as its foundation. Processing modules and their inputs and outputs are described in an object-oriented manner providing the flexibility to interface with any type of software (not just well-behaved command lines). The workflow execution engine has a plug-in architecture and supports both local execution on multi-core machines and remote execution on clusters. Nipype is distributed with a Berkeley Software Distribution (BSD) license allowing anyone to make changes and redistribute it. Development is done openly with collaborators from many different labs, allowing adaptation to the varied needs of the neuroimaging community.
Implementation Details
Nipype consists of three components (see Figure 1): (1) interfaces to external tools that provide a unified way for setting inputs, executing, and retrieving outputs; (2) a workflow engine that allows creating analysis pipelines by connecting inputs and outputs of interfaces as a directed acyclic graph (DAG); and (3) plug-ins that execute workflows either locally or in a distributed processing environment (e.g., Torque1, SGE/OGE). In the following sections, we describe key architectural components and features of this software.
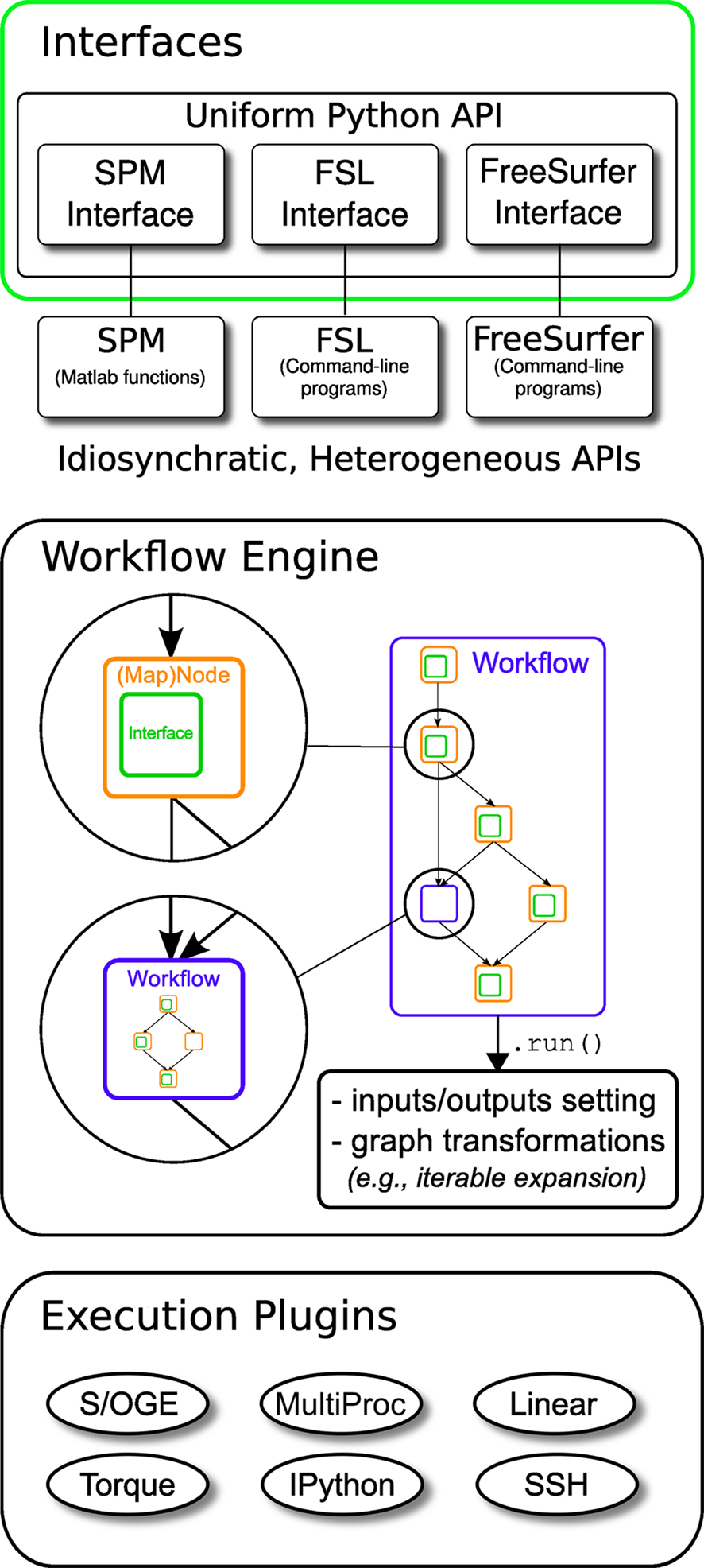
Figure 1. Architecture overview of the Nipype framework. Interfaces are wrapped with Nodes or MapNodes and connected together as a graph within a Workflow. Workflows themselves can act as a Node inside another Workflow, supporting a composite design pattern. The dependency graph is transformed before being executed by the engine component. Execution is performed by one of the plug-ins. Currently Nipype supports serial and parallel (both local multithreading and cluster) execution.
Interfaces
Interfaces form the core of Nipype. The goal of Interfaces2 is to provide a uniform mechanism for accessing analysis tools from neuroimaging software packages (e.g., FreeSurfer, FSL, SPM). Interfaces can be used directly as a Python object, incorporated into custom Python scripts or used interactively in a Python console. For example, there is a Realign Interface that exposes the SPM realignment routine, while the MCFLIRT Interface exposes the FSL realignment routine. In addition, one can also implement an algorithm in Python within Nipype and expose it as an Interface. Interfaces are flexible and can accommodate the heterogeneous software that needs to be supported, while providing unified and uniform access to these tools for the user. Since, there is no need for the underlying software to be changed (recompiled or adjusted to conform to a certain standard), developers can continue to create software using the computer language of their choice.
An Interface definition consists of: (a) input parameters, their types (e.g., file, floating point value, list of integers, etc…) and dependencies (e.g., does input “a” require input “b”); (b) outputs and their types, (c) how to execute the underlying software (e.g., run a MATLAB script, or call a command line program); and (d) a mapping which defines the outputs that are produced given a particular set of inputs. Using an object-oriented approach, we minimize redundancy in interface definition by creating a hierarchy of base Interface classes (see Figure 2) to encapsulate common functionality (e.g., Interfaces that call command line programs are derived from the CommandLine class, which provides methods to translate Interface inputs into command line parameters and for calling the command). Source code of an example Interface is shown in Listing 1.
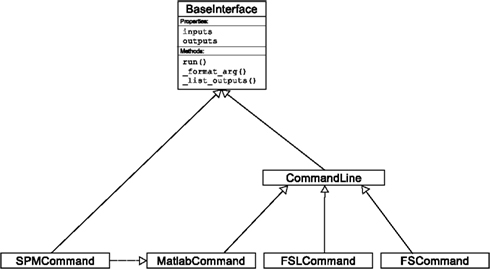
Figure 2. Simplified hierarchy of Interface classes. An object-oriented design is used to reduce code redundancy by defining common functionality in base classes, and makes adding new interfaces easier and quicker. MatlabCommand, FSLCommand, and FSCommand extend the CommandLine class to provide functionality specific to executing MATLAB, FSL, and FreeSurfer programs. The SPMCommand class defines functions that simplify wrapping SPM functionality. The dashed line indicates that the SPMCommand class uses the MatlabCommand class to execute the SPM matlab scripts generated by the SPM interfaces.
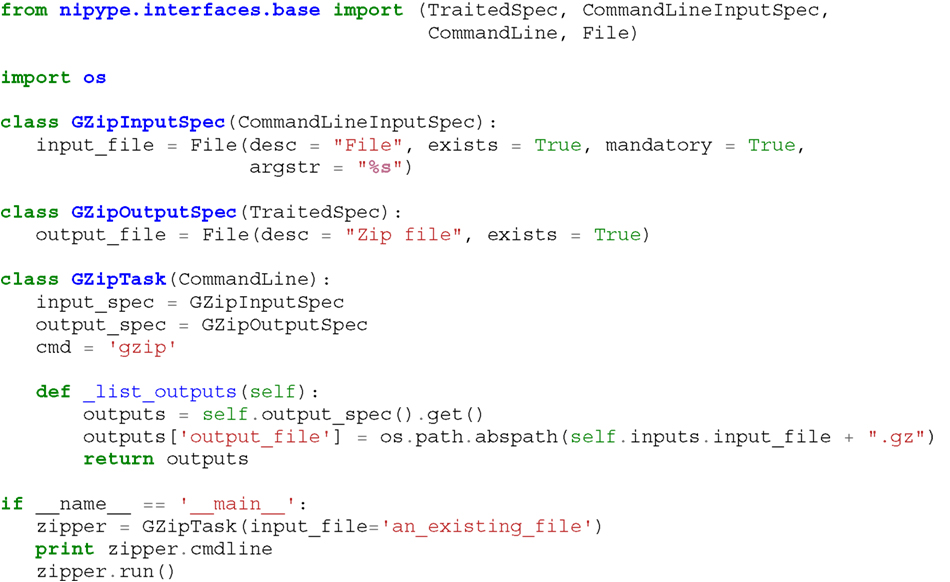
Listing 1. An example interface wrapping the gzip command line tool and a usage example. This Interface takes a file name as an input, calls gzip to compress it and returns a name of the compressed output file.
We use Enthought Traits3 to create a formal definition for Interface inputs and outputs, to define input constraints (e.g., type, dependency, whether mandatory) and to provide validation (e.g., file existence). This allows malformed or underspecified inputs to be detected prior to executing the underlying program. The input definition also allows specifying relations between inputs. Often, some input options should not be set together (mutual exclusion) while other inputs need to be set as a group (mutual inclusion). Part of the input specification for the “bet” (Brain Extraction Tool) program from FSL is shown in Listing 2.

Listing 2. Part of the input specification for the Brain Extraction Tool (BET) Interface. Full specification covers 18 different arguments. Each attribute of this class is a Traits object which defines an input and its data type (i.e., list of integers), constraints (i.e., length of the list), dependencies (when for example setting one option is mutually exclusive with another, see the xor parameter), and additional parameters (such as argstr and position which describe how to convert an input into a command line argument).
Currently, Nipype (version 0.4) is distributed with a wide range of interfaces (see Table 2). Adding new Interfaces is simply a matter of writing a Python class definition as was shown in Listing 1. When a formal specification of inputs and outputs are provided by the underlying software, Nipype can support these programs automatically. For example, the Slicer command line execution modules come with an XML specification that allows Nipype to wrap them without creating individual interfaces.
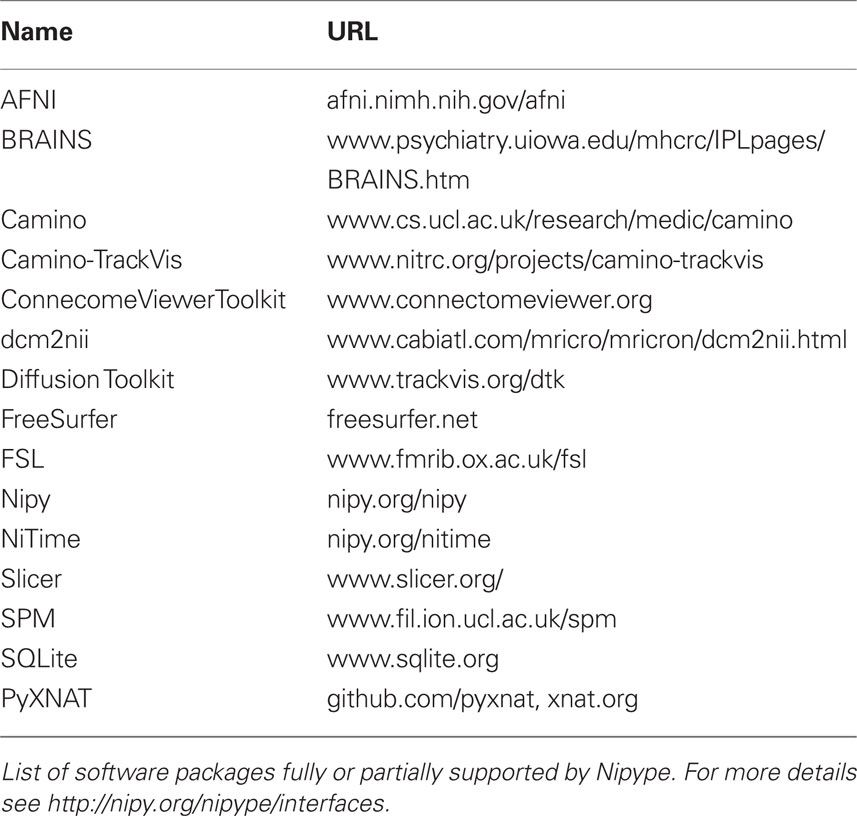
Table 2. Supported software.
Nodes, Mapnodes, and Workflows
Nipype provides a framework for connecting Interfaces to create a data analysis Workflow. In order for Interfaces to be used in a Workflow they need to be encapsulated in either Node or MapNode objects. Node and MapNode objects provide additional functionality to Interfaces. For example, creating a hash of the input state, caching of results, and the ability to iterate over inputs. Additionally, they execute the underlying interfaces in their own uniquely named directories (almost like a sandbox), thus providing a mechanism to isolate and track the outputs resulting from execution of the Interfaces. These mechanisms allow not only for provenance tracking, but aid in efficient pipeline execution.
The MapNode class is a sub-class of Node that implements a MapReduce-like architecture (Dean and Ghemawat, 2008). Encapsulating an Interface within a MapNode allows Interfaces that normally operate on a single input to execute the Interface on multiple inputs. When a MapNode executes, it creates a separate instance of the underlying Interface for every value of an input list and executes these instances independently. When all instances finish running, their results are collected into a list and exposed through the MapNode’s outputs (see Figure 4D). This approach improves granularity of the Workflow and provides easy support for Interfaces that can only process one input at a time. For example, the FSL “bet” program can only run on a single input, but wrapping the BET Interface in a MapNode allows running “bet” on multiple inputs.
A Workflow object captures the processing stages of a pipeline and the dependencies between these processes. Interfaces encapsulated into Node or MapNode objects can be connected together within a Workflow. By connecting outputs of some Nodes to inputs of others, the user implicitly specifies dependencies. These are represented internally as a DAG. The current semantics of Workflow do not allow conditionals and hence the graph needs to be acyclic. Workflows themselves can be a node of the Workflow graph (see Figure 1). This enables a hierarchical architecture and encourages Workflow reuse. The Workflow engine validates that all nodes have unique names, ensures that there are no cycles, and prevents connecting multiple outputs to a given input. For example in an fMRI processing Workflow, preprocessing, model fitting, and visualization of results can be implemented as individual Workflows connected together in a main Workflow. This not only improves clarity of designed Workflows but also enables easy exchange of whole subsets. Common Workflows can be shared across different studies within and across laboratories thus reducing redundancy and increasing consistency.
While a neuroimaging processing pipeline could be implemented as a Bash, MATLAB, or a Python script, Nipype explicitly implements a pipeline as a graph. This makes it easy to follow what steps are being executed and in what order. It also makes it easier to go back and change things by simply reconnecting different outputs and inputs or by inserting new Nodes/MapNodes. This alleviates the tedious component of scripting where one has to manually ensure that the inputs and outputs of different processing calls match and that operations do not overwrite each other’s outputs.
A Workflow provides a detailed description of the processing steps and how data flows between Interfaces. Thus it is also a source of provenance information. We encourage users to provide Workflow definitions (as scripts or graphs) as supplementary material when submitting articles. This ensures that at least the data processing part of the published experiment is fully reproducible. Additionally, exchange of Workflows between researchers stimulates efficient use of methods and experimentation.
Example – Building a Workflow from Scratch
In this section, we describe how to create and extend a typical fMRI processing Workflow. A typical fMRI Workflow can be divided into two sections: (1) preprocessing and (2) modeling. The first one deals with cleaning data from confounds and noise and the second one fits a model to the cleaned data based on the experimental design. The preprocessing stage in this Workflow will consist of only two steps: (1) motion correction (aligns all volumes in a functional run to the mean realigned volume) and (2) smoothing (convolution with a 3D Gaussian kernel). We use SPM Interfaces to define the processing Nodes.
from nipype.pipeline.engine import Node, Workflow
realign = Node(interface=spm.Realign(),
name=“realign”)
realign.inputs.register_to_mean = True
smooth = Node(interface=spm.Smooth(),
name=“smooth”)
smooth.inputs.fwhm = 4
We create a Workflow to include these two Nodes and define the data flow from the output of the realign Node (realigned_files) to the input of the smooth Node (in_files). This creates a simple preprocessing workflow (see Figure 3).
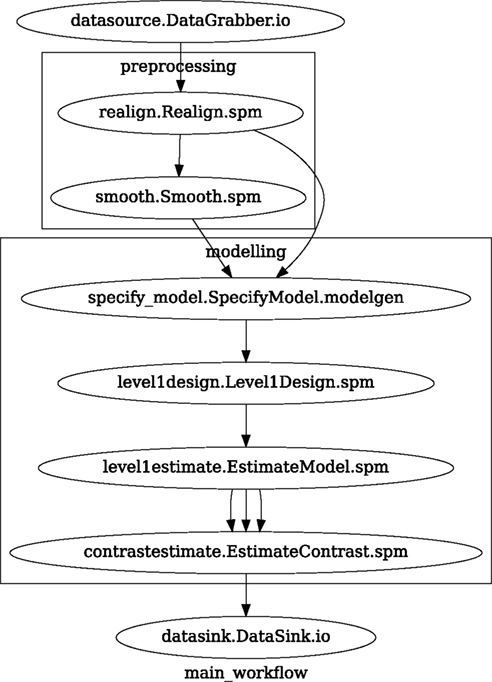
Figure 3. Graph depicting the processing steps and dependencies for a first level functional analysis workflow. Every output–input connection is represented with a separate arrow. Nodes from every subworkflow are grouped in boxes with labels corresponding to the name of the subworkflow. Such graphs can be automatically generated from a Workflow definition and provide a quick overview of the pipeline.
preprocessing = Workflow
(name=“preprocessing”)
preprocessing.connect(realign,
“realigned_files”, smooth, “in_files”)
A modeling Workflow is constructed in an analogous manner, by first defining Nodes for model design, model estimation, and contrast estimation. We again use SPM Interfaces for this purpose. However, Nipype adds an extra abstraction Interface for model specification whose output can be used to create models in different packages (e.g., SPM, FSL, and Nipy). The nodes of this Workflow are: SpecifyModel (Nipype model abstraction Interface), Level1Design (SPM design definition), ModelEstimate, and ContrastEstimate. The connected modeling Workflow can be seen on Figure 3.
We create a master Workflow that connects the preprocessing and modeling Workflows, adds the ability to select data for processing (using DataGrabber Interface) and a DataSink Node to save the outputs of the entire Workflow. Nipype allows connecting Nodes between Workflows. We will use this feature to connect realignment_parameters and smoothed_files to modeling workflow.
The DataGrabber Interface allows the user to define flexible search patterns which can be parameterized by user defined inputs (such as subject ID, session, etc.). This Interface can adapt to a wide range of directory organization and file naming conventions. In our case we will parameterize it with subject ID. In this way we can run the same Workflow for different subjects. We automate this by iterating over a list of subject IDs, by setting the iterables property of the DataGrabber Node for the input subject_id. The DataGrabber Node output is connected to the realign Node from preprocessing Workflow.
DataSink on the other side provides means for storing selected results in a specified location. It supports automatic creation of folders, simple substitutions, and regular expressions to alter target filenames. In this example we store the statistical (T maps) resulting from contrast estimation.
A Workflow defined this way (see Figure 3, for full code see Supplementary Material) is ready to run. This can be done by calling run() method of the master Workflow.
If the run() method is called twice, the Workflow input hashing mechanism ensures that none of the Nodes are executed during the second run if the inputs remain the same. If, however, a highpass filter parameter of specify_model is changed, some of the Nodes (but not all) would have to rerun. Nipype automatically determines which Nodes require rerunning.
Iterables – Parameter Space Exploration
Nipype provides a flexible approach to prototype and experiment with different processing strategies, through the unified and uniform access to a variety of software packages (Interfaces) and creating data flows (Workflows). However, for various neuroimaging tasks, there is often a need to explore the impact of variations in parameter settings (e.g., how do different amounts of smoothing affect group statistics, what is the impact of spline interpolation over trilinear interpolation). To enable such parametric exploration, Nodes have an attribute called iterables.
When an iterable is set on a Node input, the Node, and its subgraph are executed for each value of the iterable input (see Figure 4 iterables_vs_mapnode). Iterables can also be set on multiple inputs of a Node (e.g., somenode.iterables = [(“input1,” [1,2,3]), (“input2,” [“a,” “b”])]). In such cases, every combination of those values is used as a parameter set (the prior example would result in the following parameter sets: (1, “a”), (1, “b”), (2, “a”), etc…). This feature is especially useful to investigate interactions between parameters of intermediate stages with respect to the final results of a workflow. A common use-case of iterables is to execute the same Workflow for many subjects in an fMRI experiment and to simultaneously look at the impact of parameter variations on the results of the Workflow.
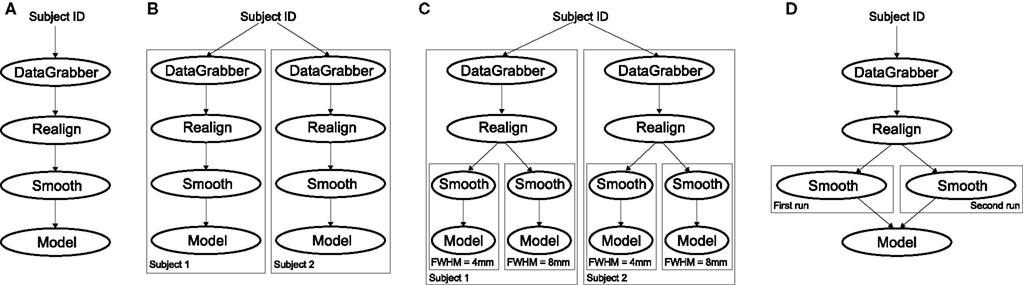
Figure 4. Workflow modification using iterables and MapNodes. If we take the processing pipeline (A) and set iterables parameter of DataGrabber to a list of two subjects, Nipype will effectively execute graph (B). Identical processing will be applied to every subject from the list. Iterables can be used in a graph on many levels. For example, setting iterables on Smooth FWHM to a list of 4 and 8 mm will result in graph (C). In contrast to iterables, MapNode branches within a node of the graph and also merges the results of the branches, effectively performing a MapReduce operation (D).
It is important to note that unlike MapNode, which creates copies of the underlying interface for every element of an input of type list, iterables operate on the subgraph of a node and create copies not only of the node but also of all the nodes dependent on it (see Figure 4).
Parallel Distribution and Execution Plug-Ins
Nipype supports executing Workflows locally (in series or parallel) or on load-balanced grid-computing clusters (e.g., SGE, Torque, or even via SSH) through an extensible plug-in interface. No change is needed to the Workflow to switch between these execution modes. One simply calls the Workflow’s run function with a different plug-in and its arguments. Very often different components of a Workflow can be executed in parallel and even more so when the same Workflow is being repeated on multiple parameters (e.g., subjects). Adding support for additional cluster management systems does not require changes in Nipype, but simply writing a plug-in extension conforming to the plug-in API.
The Workflow engine sends an execution graph to the plug-in. Executing the Workflow in series is then simply a matter of performing a topological sort on the graph and running each node in the sorted order. However, Nipype also provides additional plug-ins that use Python’s multi-processing module, use IPython (includes SSH-based, SGE, LSF, PBS, among others) and provide native interfaces to SGE or PBS/Torque clusters. For all of these, the graph structure defines the dependencies as well as which nodes can be executed in parallel at any given stage of execution.
One of the biggest advantages of Nipype’s execution system is that parallel execution using local multi-processing plug-in does not require any additional software (such as cluster managers like SGE) and therefore makes prototyping on a local multi-core workstations easy. However for bigger studies and complex Workflows, a high-performance computing cluster can provide substantial improvements in execution time. Since there is a clear separation between definition of the Workflow and its execution, Workflows can be executed in parallel (locally or on a cluster) without any modification. Transitioning from developing a processing pipeline on a single subject on a local workstation to executing it on a bigger cohort on a cluster is therefore seamless.
Rerunning workflows has also been optimized. When a Node or MapNode is run, the framework will actually execute the underlying interface only if inputs have changed relative to prior execution. If not, it will simply return cached results.
The Function Interface
One of the Interfaces implemented in Nipype requires special attention: The Function Interface. Its constructor takes as arguments Python function pointer or code, list of inputs, and list of outputs. This allows running any Python code as part of a Workflow. When combined with libraries such as Nibabel (neuroimaging data input and output), Numpy/Scipy (array representation and processing) and scikits-learn or PyMVPA (machine learning and data mining) the Function Interface provides means for rapid prototyping of complex data processing methods. In addition, by using the Function Interface users can avoid writing their own Interfaces which is especially useful for ad hoc solutions (e.g., calling an external program that has not yet been wrapped as an Interface).
Workflow Visualization
To be able to efficiently manage and debug Workflows, one has tohave access to a graphical representation. Using graphviz (Ellson et al., 2002), Nipype generates static graphs representing Nodes and connections between them. In the current version four types of graphs are supported: orig – does not expand inner Workflows, flat – expands inner workflows, exec – expands workflows and iterables, and hierarchical – expands workflows but maintains their hierarchy. Graphs can be saved in a variety of file formats including Scalable Vector Graphics (SVG) and Portable Network Graphics (PNG; see Figures 3 and 6).
Configuration Options
Certain options concerning verbosity of output and execution efficiency can be controlled through configuration files or variables. These include, among others, hash_method and remove_unecessary_outputs. As explained before, rerunning a Workflow only recomputes those Nodes whose inputs have changed since the last run. This is achieved by recording a hash of the inputs. For files there are two ways of calculating the hash (controlled by the hash_method config option): timestamp – based only on the size and modification time and content – based on the content of the file. The first one is faster, but does not deal with the situation when an identical copy overwrites the file. The second one can be slower especially for big files, but can tell that two files are identical even if they have different modification times. To allow efficient recomputation Nipype has to store outputs of all Nodes. This can generate a significant amount of data for typical neuroimaging studies. However, not all outputs of every Node are used as inputs to other Nodes or relevant to the final results. Users can decide to remove those outputs (and save some disk space) by setting the remove_unecessary_outputs to True. These and other configuration options provide a mechanism to streamline the use of Nipype for different applications.
Deployment
Nipype supports GNU/Linux and Mac OS X operating systems (OS). A recent Internet survey based study (Hanke and Halchenko, 2011) showed that GNU/Linux is the most popular platform in the neuroimaging community and together with Mac OS X is used by over 70% of neuroimagers. There are not theoretical reasons why Nipype should not work on Windows (Python is a cross-platform language), but since most of the supported software (for example FSL) requires a Unix based OS, Nipype has not been tested on this platform.
We currently provide three ways of deploying Nipype on a new machine: manual installation from sources4, PyPi repository5, and from package repositories on Debian-based systems. Manual installation involves downloading a source code archive and running a standard Python installation script (distutils). This way the user has to take care of installing all of the dependencies. Installing from PyPI repository lifts this constraint by providing dependency information and automatically installing required packages. Nipype is available from standard repositories on recent Debian and Ubuntu releases. Moreover, NeuroDebian6 (Hanke et al., 2010) repository provides the most recent releases of Nipype for Debian-based systems and a NeuroDebian Virtual Appliance making it easy to deploy Nipype and other imaging tools in a virtual environment on other OS, e.g., Windows. In addition to providing all core dependencies and automatic updates NeuroDebian also provides many of the software packages supported by Nipype (AFNI, FSL, Mricron, etc.), making deployment of heterogeneous Nipype pipelines more straightforward.
Development
Nipype is trying to address the problem of interacting with the ever changing universe of neuroimaging software in a sustainable manner. Therefore the way its development is managed is a part of the solution. Nipype is distributed under BSD license which allows free copying, modification, and distribution and additionally meets all the requirements of open-source definition (see Open-Source Initiative7) and Debian Free Software Guidelines8. Development is carried out openly through distributed version control system (git via GitHub9) in an online community. The current version of the source code together with complete history is accessible to everyone. Discussions between developers and design decisions are done on an open access mailing list. This setup encourages a broader community of developers to join the project and allows sharing of the development resources (effort, money, information, and time).
In these previous paragraphs, we presented key features of Nipype that facilitate rapid development and deployment of analysis procedures in laboratories, and address all of the issues described earlier. In particular, Nipype provides: (1) uniform access to neuroimaging analysis software and usage information; (2) a framework for comparative algorithm development and dissemination; (3) an environment for methodological continuity and paced training of new personnel in laboratories; (4) computationally efficient execution of neuroimaging analysis; and (5) a mechanism to capture the data processing details in compact scripts and graphs. In the following section, we provide examples to demonstrate these solutions.
Usage Examples
Uniform Access to Tools, Their Usage, and Execution
Users access Interfaces by importing them from Nipype modules. Each neuroimaging software distribution such as FSL, SPM, Camino, etc., has a corresponding module in the nipype.interfaces namespace.
from nipype.interfaces.camino import DTIFit
The help() function for each interface prints the inputs and the outputs associated with the interface.
>>> DTIFit.help()
Inputs
------
Mandatory:
in_file: voxel-order data filename
scheme_file: Camino scheme file
(b values / vectors, see camino.fsl2scheme)
Optional:
args: Additional parameters to the command
environ: Environment variables (default={})
ignore_exception: Print an error message
instead of throwing an exception in case
the interface fails to run (default=False)
non_linear: Use non-linear fitting instead
of the default linear regression to the log
measurements.
out_file: None
Outputs
-------
tensor_fitted: path/name of 4D volume in voxel
order
The output of the help() function is standardized across all Interfaces. It is automatically generated based on the traited input and output definitions and includes information about required inputs, types, and default value. Alternatively, extended information is available in the form of auto-generated HTML documentation on the Nipype website (see Figure 5). This extended information includes examples that demonstrate how the interface can be used.

Figure 5. HTML help page for dtfit command from Camino. This was generated based on the Interface code: description and example was taken from the class docstring and inputs/outputs were list was created using traited input/output specification.
For every Interface, input values are set through the inputs field:
fit.inputs.scheme_file = ‘A.scheme’
fit.inputs.in_file = \
‘tensor_fitted_data.Bfloat’
When trying to set an invalid input type (for example a non-existing input file, or a number instead of a string) the Nipype framework will display an error message. Input validity checking before actual Workflow execution saves time. To run an Interface user needs to call run() method:
fit.run()
At this stage the framework checks if all mandatory inputs are set and all input dependencies are satisfied, generating an error if either of these conditions are not met.
Nipype standardizes running and accessing help information irrespective of whether the underlying software is a MATLAB program, a command line tool, or Python module. The framework deals with translating inputs into appropriate form (e.g., command line arguments or MATLAB scripts) for executing the underlying tools in the right way, while presenting the user with a uniform interface.
A Framework for Comparative Algorithm Development and Dissemination
Uniform semantics for interfacing with a wide range of processing methods not only opens the possibility for richer Workflows, but also allows comparing algorithms that are designed to solve the same problem across and within such diverse Workflows. Typically, such an exhaustive comparison can be time-consuming, because of the need to deal with interfacing different software packages. Nipype simplifies this process by standardizing the access to the software. Additionally, the iterables mechanism allows users to easily extend such comparisons by providing a simple mechanism to test different parameter sets.
Accuracy or efficiency of algorithms can be determined in an isolated manner by comparing their outputs or execution time or memory consumption on a given set of data. However, researchers typically want to know how different algorithms used at earlier stages of processing might influence the final output or statistics they are interested in. As an example of such use, we have compared voxelwise isotropic, voxelwise anisotropic, and surface based smoothing all for two levels of FWHM – 4 and 8 mm. First one is the standard convolution with Gaussian kernel as implemented in SPM. Second one involves smoothing only voxels of similar intensity in attempt to retain structure. This was implemented in SUSAN from FSL (Smith, 1992). Third method involves reconstructing surface of the cortex and smoothing along it (Hagler et al., 2006). This avoids bleeding of signal over sulci.
Establishing parameters from data and smoothing using SUSAN is already built into Nipype as a Workflow. It can be created using create_susan_smooth() function. It has similar inputs and outputs as SPM Smooth Interface. Smoothing on a surface involves doing a full cortical reconstruction from T1 volume using FreeSurfer (Fischl et al., 1999) followed by coregistering functional images to the reconstructed surface using BBRegister (Greve and Fischl, 2009). Finally, the surface smoothing algorithm from FreeSurfer is called.
Smoothed EPI volumes (direct/local influence) and statistical maps (indirect/global influence), along with the pipeline used to generate them can be found in Figures 6 and 7. Full code used to generate this data can be found in the Supplementary Material. This comparison serves only to demonstrate Nipype capabilities; a comparison between smoothing methods is outside of the scope of this paper.
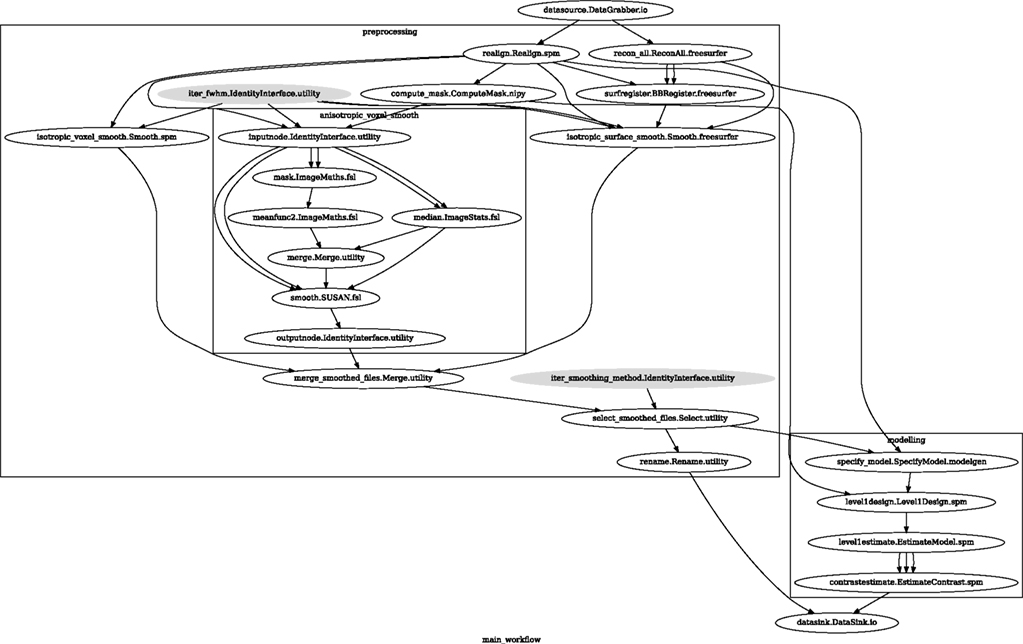
Figure 6. Graph showing the workflow used for the smoothing methods and parameters comparison. The gray shaded nodes have iterables parameter set. This allows easy iteration over all combinations of FWHM and smoothing algorithms used in the comparison.
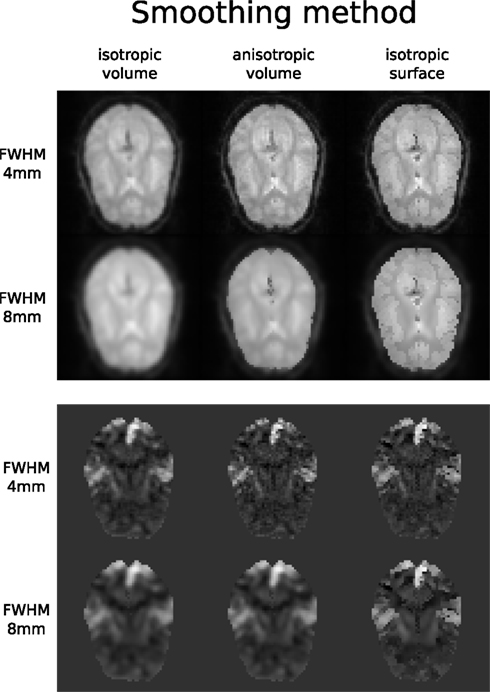
Figure 7. Influence of different smoothing methods and their parameters. Upper half shows direct influence of smoothing on the EPI sequence (slice 16, volume 0, run 2). Lower half shows indirect influence of smoothing on the T maps (same slice) of the main contrast.
Algorithm comparison is not the only way Nipype can be useful for a neuroimaging methods researcher. It is in the interest of every methods developer to make his or hers work most accessible. This usually means providing ready to use implementations. However, because the field is so diverse, software developers have to provide several packages (SPM toolbox, command line tool, C++ library, etc.) to cover the whole user base. With Nipype, a developer can create one Interface and expose a new tool, written in any language, to a greater range of users, knowing it will work with the wide range of software currently supported by Nipype.
A good example of such scenario is ArtifactDetection toolbox10. This piece of software uses EPI timeseries and realignment parameters to find timepoints (volumes) that are most likely artifacts and should be removed (by including them as confound regressors in the design matrix). The tool was initially implemented as a MATLAB script, compatible only with SPM and used locally within the lab. The current Nipype interface can work with SPM or FSL Workflows, thereby not limiting its users to SPM.
An Environment for Methodological Continuity and Paced Training of New Personnel in Laboratories
Neuroimaging studies in any laboratory typically use similar data processing methods with possibly different parameters. Nipype Workflows can be very useful in dividing the data processing into reusable building blocks. This not only improves the speed of building new Workflows but also reduces the number of potential errors, because a well tested piece of code is being reused (instead of being reimplemented every time). Since a Workflow definition is an abstract and simplified representation of the data processing stream, it is much easier to describe and hand over to new project personnel. Furthermore, a data independent Workflow definition (see Figure 8) enables sharing Workflows within and across research laboratories. Nipype provides a high-level abstraction mechanism for exchanging knowledge and expertise between researchers focused on methods in neuroimaging and those interested in applications.
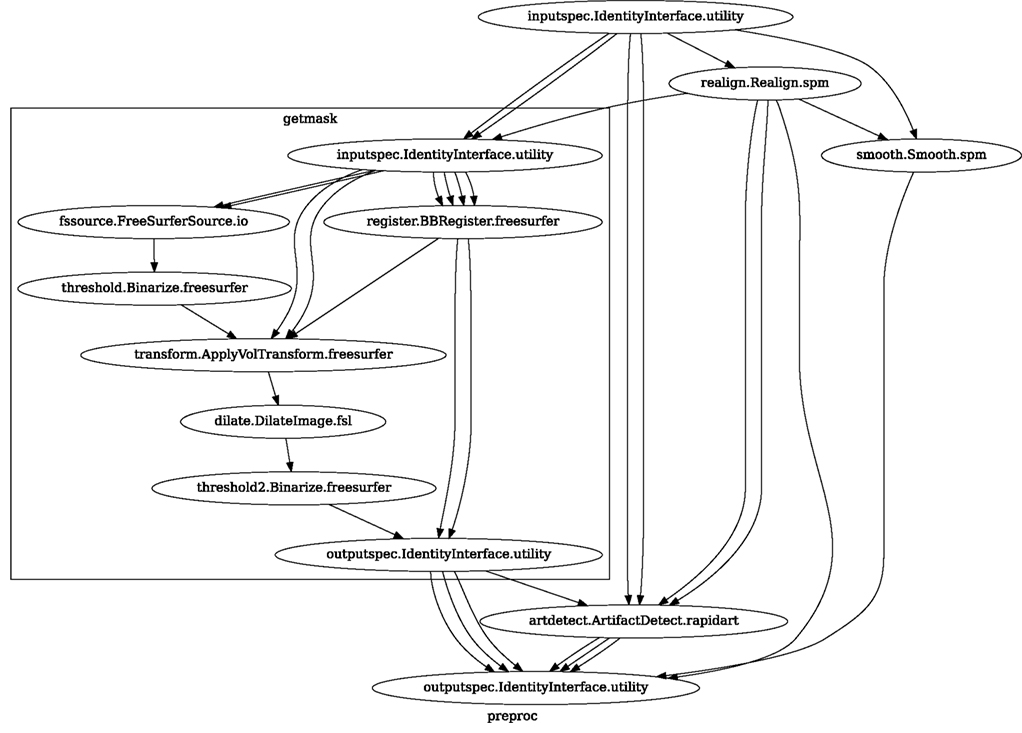
Figure 8. create_spm_preproc() functions returns this reusable, data independent Workflow. It implements typical fMRI preprocessing with smoothing (SPM), motion correction (SPM), artifact detection (Nipype), and coregistration (FreeSurfer). Inputs and outputs are grouped using IdentityInterfaces. Thanks to this, changes in the configuration of the nodes will not break backward compatibility. For full source code see Supplementary Material.
The uniform access to Interfaces and the ease of use of Workflows in Nipype helps with training new staff. Composition provided by Workflows allows users to gradually increase the level of details when learning how to perform neuroimaging analysis. For example user can start with a “black box” Workflow that does analysis from A to Z, and gradually learn what the sub-components (and their sub-components) do. Playing with Interfaces in an interactive console is also a great way to learn how different algorithms work with different parameters without having to understand how to set them up and execute them.
Computationally Efficient Execution of Neuroimaging Analysis
A computationally efficient execution allows for multiple rapid iterations to optimize a Workflow for a given application. Support for optimized local execution (running independent processes in parallel, rerunning only those steps that have been influenced by the changes in parameters or dependencies since the last run) and exploration of parameter space eases Workflow development. The Nipype package provides a seamless and flexible environment for executing Workflows in parallel on a variety of environments from local multi-core workstations to high-performance clusters. In the SPM workflow for single subject functional data analysis (see Figure 9), only a few components can be parallelized. However, running this Workflow across several subjects provides room for embarrassingly parallel execution. Running this Workflow in distributed mode for 69 subjects on a compute cluster (40 cores distributed across 6 machines) took 1 h and 40 min relative to the 32-min required to execute the analysis steps in series for a single subject on the same cluster. The difference from the expected runtime of 64 min (32 min for the first 40 subjects and another 32 min for the remaining 29 subjects) stems from disk I/O and other network and processing resource bottlenecks.
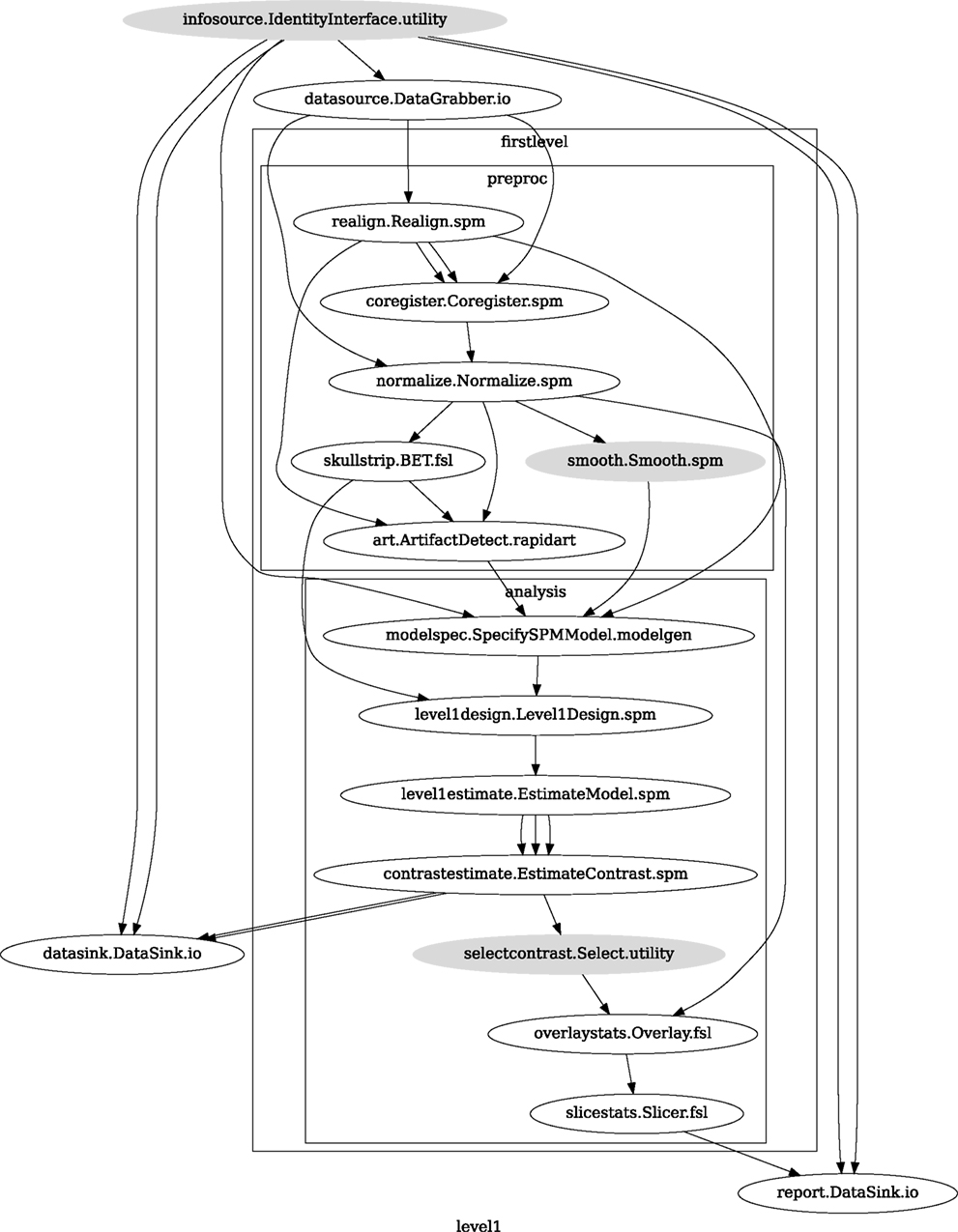
Figure 9. Single subject fMRI Workflow used for benchmarking parallel execution.
Captures Details of Analysis Required to Reproduce Results
The graphs and code presented in the examples above capture all the necessary details to rerun the analysis. Any user, who has the same versions of the tools installed on their machine and access to the data and scripts, will be able to reproduce the results of the study. For example, running Nipype within the NeuroDebian framework can provide access to specific versions of the underlying tools. This provides an easy mechanism to be compliant with the submitting data and scripts/code mandates of journals such as PNAS and Science.
Discussion
Current neuroimaging software offer users an incredible opportunity to analyze their data in different ways, with different underlying assumptions. However, this heterogeneous collection of specialized applications creates several problems: (1) No uniform access to neuroimaging analysis software and usage information; (2) No framework for comparative algorithm development and dissemination; (3) Personnel turnover in laboratories often limit methodological continuity and training new personnel takes time; (4) Neuroimaging software packages do not address computational efficiency; and (5) Method sections of journal articles are often inadequate for reproducing results.
We addressed these issues by creating Nipype, an open-source, community-developed initiative under the umbrella of Nipy. Nipype, solves these issues by providing uniform Interfaces to existing neuroimaging software and by facilitating interaction between these packages within Workflows. Nipype provides an environment that encourages interactive exploration of algorithms from different packages (e.g., SPM, FSL), eases the design of Workflows within and between packages, and reduces the learning curve necessary to use different packages. Nipype is addressing limitations of existing pipeline systems and creating a collaborative platform for neuroimaging software development in Python, a high-level scientific computing language.
We use Python for several reasons. It has extensive scientific computing and visualization support through packages such as SciPy, NumPy, Matplotlib, and Mayavi (Pérez et al., 2010; Millman and Aivazis, 2011). The Nibabel package provides support for reading and writing common neuroimaging file formats (e.g., NIFTI, ANALYZE, and DICOM). Being a high-level language, Python supports rapid prototyping, is easy to learn and adopt and is available across all major OS. At the same time Python allows to seamlessly bind with C code (using Weave package) for improved efficiency of critical subroutines.
Python is also known to be a good choice for the first programming language to learn (Zelle, 1999) and is chosen as the language for introductory programming at many schools and universities11. Being a generic and free language, with various extensions available “out of the box,” it has allowed many researchers to start implementing and sharing their ideas with minimal knowledge of Python, while learning more of the language and programming principles along the way. Many such endeavors later on became popular community-driven FOSS projects, attracting users and contributors, and even outlasting the involvement of the original authors. Python has already been embraced by the neuroscientific community and is rapidly gaining popularity (Bednar, 2009; Goodman and Brette, 2009). The Connectome Viewer Toolkit (Gerhard et al., 2011), Dipy (Garyfallidis et al., 2011), NiBabel12, Nipy13, NiTime (Rokem et al., 2009), PyMVPA (Hanke et al., 2009), PyXNAT (Schwartz et al., 2011), and Scikits-Learn14 are just a few examples of neuroimaging related software written in Python. Nipype, based on Python, thus has immediate access to this extensive community and its software, technological resources and support structure.
Nipype provides a formal and flexible framework to accommodate the diversity of imaging software. Within the neuroimaging community, not all software is limited to well-behaved command line tools. Furthermore, a number of these tools do not have well defined inputs, outputs, or usage help. Although, currently we use Enthought Traits to define inputs and outputs of interfaces, such definitions could be easily translated into instances of XML schemas compatible with other pipeline frameworks. On the other hand, when a tool provides a formal XML description of their inputs and outputs (e.g., Slicer 3D, BRAINS), it is possible to take these definitions and automatically generate Nipype wrappers for those classes.
Nipype development welcomes input and contributions from the community. The source code is freely distributed under a BSD license allowing anyone any use of the software and Nipype conforms to the Open Software Definition of the Open-Source Initiative. Development process is fully transparent and encourages contributions from users from all around the world. The diverse and geographically distributed user and developer base makes Nipype a flexible project that takes into account needs of many scientists.
Improving openness, transparency, and reproducibility of research has been a goal of Nipype since its inception. A Workflow definition is, in principle, sufficient to reproduce the analysis. Since it was used to analyze the data, it is more detailed and accurate than a typical methods description in a paper, but also has the advantage of being reused and shared within and across laboratories. Accompanying a publication with a formal definition of the processing pipeline (such as a Nipype script) increases reproducibility and transparency of research. The Interfaces and Workflows of Nipype capture neuroimaging analysis knowledge and the evolution of methods. Although, at the execution level, Nipype already captures a variety of provenance information, this aspect can be improved by generating provenance reports defined by a standardized XML schema (Mackenzie-Graham et al., 2008).
Increased diversity of neuroimaging data processing software has made systematic comparison of performance and accuracy of underlying algorithms essential (for examples, see Klein et al., 2009, 2010). However, a platform for comparing algorithms, either by themselves or in the context of an analysis workflow, or determining optimal workflows in a given application context (e.g., Churchill et al., 2011), does not exist. Furthermore, in this context of changing hardware and software, traditional analysis approaches may not be suitable in all contexts (e.g., data from 32-channel coils which show a very different sensitivity profile, or data from children). Nipype can make such evaluations, design of optimal workflows, and investigations easier (as demonstrated via the smoothing example above), resulting in more efficient data analysis for the community.
Summary
We presented Nipype, an extensible Python library and framework that provides interactive manipulation of neuroimaging data through uniform Interfaces and enables reproducible, distributed analysis using the Workflow system. Nipype has encouraged the scientific exploration of different algorithms and associated parameters, eased the development of Workflows within and between packages and reduced the learning curve associated with understanding the algorithms, APIs, and user interfaces of disparate packages. An open, community-driven development philosophy provides the flexibility required to address the diverse needs in neuroimaging analysis. Overall, Nipype represents an effort toward collaborative, open-source, reproducible, and efficient neuroimaging software development and analysis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
A list of people who have contributed code to the project is available at http://github.com/nipy/nipype/contributors. We thank Fernando Perez, Matthew Brett, Gael Varoquaux, Jean-Baptiste Poline, Bertrand Thirion, Alexis Roche, and Jarrod Millman for technical and social support and for design discussions. We would like to thank Prof. John Gabrieli’s laboratory at MIT for testing Nipype through its evolutionary stages, in particular, Tyler Perrachione and Gretchen Reynolds. We would also like to thank the developers of FreeSurfer, FSL, and SPM for being supportive of the project and providing valuable feedback on technical issues. We would like to thank James Bednar, Stephan Gerhard, and Helen Ramsden for providing feedback during the preparation of the manuscript. Satrajit Ghosh would like to acknowledge support from NIBIB R03 EB008673 (PI: Ghosh and Whitfield-Gabrieli), the Ellison Medical Foundation, Katrien Vander Straeten, and Amie Ghosh. Krzysztof Gorgolewski would like to thank his supervisors Mark Bastin, Cyril Pernet, and Amos Storkey for their support during this project.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/neuroinformatics/10.3389/fninf.2011.00013/abstract
Footnotes
- ^http://www.clusterresources.com/products/torque-resource-manager.php
- ^Throughout the rest of the paper we are going to use upper case for referring to classes (such as Interfaces, Workflows, etc…) and lower case to refer to general concepts.
- ^http://code.enthought.com/projects/traits/
- ^http://nipy.org/nipype/
- ^http://pypi.python.org/pypi/nipype/
- ^http://neuro.debian.net
- ^http://www.opensource.org/docs/osd
- ^http://www.debian.org/social_contract#guidelines
- ^http://github.com/nipy/nipype
- ^http://www.nitrc.org/projects/artifact_detect/
- ^http://wiki.python.org/moin/SchoolsUsingPython
- ^http://nipy.sourceforge.net/nibabel/
- ^http://nipy.sourceforge.net/nipy/
- ^http://scikit-learn.sourceforge.net
References
Avants, B., and Gee, J. C. (2004). Geodesic estimation for large deformation anatomical shape averaging and interpolation. Neuroimage 23(Suppl. 1), S139–S150.
Bednar, J. A. (2009). Topographica: building and analyzing map-level simulations from Python, C/C++, MATLAB, NEST, or NEURON components. Front. Neuroinform. 3:8.
Callahan, S. P., Freire, J., Santos, E., Scheidegger, C. E., Silva, C. T., and Vo, H. T. (2006). “VisTrails: visualization meets data management,” in Proceedings of the 2006 ACM SIGMOD International Conference on Management of Data, Chicago, IL, 745–747.
Churchill, N. W., Oder, A., Abdi, H., Tam, F., Lee, W., Thomas, C., Ween, J. E., Graham, S. J., and Strother, S. C. (2011). Optimizing preprocessing and analysis pipelines for single-subject fMRI. I. Standard temporal motion and physiological noise correction methods. Hum. Brain Mapp. [Epub ahead of print].
Cointepas, Y., Mangin, J., Garnero, L., and Poline, J. (2001). BrainVISA: software platform for visualization and analysis of multi-modality brain data. Neuroimage 13, 98.
Dean, J., and Ghemawat, S. (2008). MapReduce: simplified data processing on large clusters. Commun. ACM 51, 1–13.
Dinov, I., Lozev, K., Petrosyan, P., Liu, Z., Eggert, P., Pierce, J., Zamanyan, A., Chakrapani, S., Van Horn, J., Parker, D. S., Magsipoc, R., Leung, K., Gutman, B., Woods, R., and Toga, A. (2010). Neuroimaging study designs, computational analyses and data provenance using the LONI pipeline. PLoS ONE 5, e13070.
Dinov, I. D., Van Horn, J. D., Lozev, K. M., Magsipoc, R., Petrosyan, P., Liu, Z., Mackenzie-Graham, A., Eggert, P., Parker, D. S., and Toga, A. W. (2009). Efficient, distributed and interactive neuroimaging data analysis using the LONI pipeline. Front. Neuroinform. 3:22.
Ellson, J., Gansner, E., Koutsofios, L., North, S., and Woodhull, G. (2002). “Graphviz – open source graph drawing tools,” in Proceeding of 9th Internation Symposium on Graph Drawing, Vienna, 594–597.
Fischl, B., Sereno, M. I., and Dale, A. M. (1999). Cortical surface-based analysis. II: inflation, flattening, and a surface-based coordinate system. Neuroimage 9, 195–207.
Fissell, K., Tseytlin, E., Cunningham, D., Iyer, K., Carter, C. S., Schneider, W., and Cohen, J. D. (2003). A graphical computing environment for neuroimaging analysis. Neuroinformatics 1, 111–125.
Garyfallidis, E., Brett, M., Amirbekian, B., Nguyen, C., Yeh, F.-C., Halchenko, Y., and Nimmo-Smith, I. (2011). “Dipy – a novel software library for diffusion MR and tractography,” in 17th Annual Meeting of the Organization for Human Brain Mapping, Quebec City, QC.
Gerhard, S., Daducci, A., Lemkaddem, A., Meuli, R., Thiran, J. P., and Hagmann, P. (2011). The connectome viewer toolkit: an open source framework to manage, analyze, and visualize connectomes. Front. Neuroinform. 5:3.
Greve, D. N., and Fischl, B. (2009). Accurate and robust brain image alignment using boundary-based registration. Neuroimage 48, 63–72.
Hagler, D. J., Saygin, A. P., and Sereno, M. I. (2006). Smoothing and cluster thresholding for cortical surface-based group analysis of fMRI data. Neuroimage 33, 1093–1103.
Hanke, M., Halchenko, Y. O., Haxby, J. V., and Pollmann, S. (2010). “Improving efficiency in cognitive neuroscience research with NeuroDebian,” in Cognitive Neuroscience Society, Montréal.
Hanke, M., Halchenko, Y. O., Sederberg, P. B., Hanson, S. J., Haxby, J. V., and Pollmann, S. (2009). PyMVPA: a Python toolbox for multivariate pattern analysis of fMRI data. Neuroinformatics 7, 37–53.
Hömke, L. (2006). A multigrid method for anisotropic PDEs in elastic image registration. Numer. Linear Algebra Appl. 13, 215–229.
Klein, A., Andersson, J., Ardekani, B. A., Ashburner, J., Avants, B., Chiang, M. C., Christensen, G. E., Collins, D. L., Gee, J., Hellier, P., Song, J. H., Jenkinson, M., Lepage, C., Rueckert, D., Thompson, P., Vercauteren, T., Woods, R. P., Mann, J. J., and Parsey, R. V. (2009). Evaluation of 14 nonlinear deformation algorithms applied to human brain MRI registration. Neuroimage 46, 786–802.
Klein, A., Ghosh, S. S., Avants, B., Yeo, B. T., Fischl, B., Ardekani, B., Gee, J. C., Mann, J. J., and Parsey, R. V. (2010). Evaluation of volume-based and surface-based brain image registration methods. Neuroimage 51, 214–220.
Mackenzie-Graham, A. J., Van Horn, J. D., Woods, R. P., Crawford, K. L., and Toga, A. W. (2008). Provenance in neuroimaging. Neuroimage 42, 178–195.
McAuliffe, M. J., Lalonde, F. M., McGarry, D., Gandler, W., Csaky, K., and Trus, B. L. (2001). “Medical image processing, analysis and visualization in clinical research,” in Proceedings 14th IEEE Symposium on Computer-Based Medical Systems, Bethesda, MD, 381–386.
Millman, K. J., and Aivazis, M. (2011). Python for scientists and engineers. Comput. Sci. Eng. 13, 9–12.
Oinn, T., Greenwood, M., Addis, M. J., Alpdemir, M. N., Ferris, J., Glover, K., Goble, C., Goderis, A., Hull, D., Marvin, D. J., Li, P., Lord, P., Pocock, M. R., Senger, M., Stevens, R., Wipat, A., and Wroe, C. (2006). Taverna: lessons in creating a workflow environment for the life sciences. Concurr. Comput. 18, 1067–1100.
Pérez, F., Granger, B. E., and Hunter, J. D. (2010). Python: an ecosystem for scientific computing. Comput. Sci. Eng. 13, 13–21.
Rex, D. E., Ma, J. Q., and Toga, A. W. (2003). The LONI pipeline processing environment. Neuroimage 19, 1033–1048.
Rokem, A., Trumpis, M., and Perez, F. (2009). “Nitime: time-series analysis for neuroimaging data,” in Proceedings of the 8th Python in Science Conference, Pasadena.
Schwartz, Y., Barbot, A., Vincent, F., Thyreau, B., Varoquaux, G., Thirion, B., and Poline, J. (2011). “PyXNAT: a Python interface for XNAT,” in 17th Annual Meeting of the Organization for Human Brain Mapping, Quebec City, QC.
Keywords: neuroimaging, data processing, workflow, pipeline, reproducible research, Python
Citation: Gorgolewski K, Burns CD, Madison C, Clark D, Halchenko YO, Waskom ML, Ghosh SS (2011) Nipype: a flexible, lightweight and extensible neuroimaging data processing framework in Python. Front. Neuroinform. 5:13. doi: 10.3389/fninf.2011.00013
Received: 23 June 2011; Accepted: 23 July 2011;
Published online: 22 August 2011.
Edited by:
Andrew P. Davison, CNRS, FranceCopyright: © 2011 Gorgolewski, Burns, Madison, Clark, Halchenko, Waskom, Ghosh. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Krzysztof Gorgolewski, School of Informatics, University of Edinburgh, Informatics Forum, 10 Crichton Street, Edinburgh EH8 9AB, UK. e-mail: krzysztof.gorgolewski@gmail.com