
- Laboratoire de Physiopathologie des Maladies du Système Nerveux Central, UMR CNRS-7224 CNRS et UMRS-INSERM 952, Université Pierre et Marie Curie-Paris 6, Paris, France
The ternary complex factor (TCF) Elk-1 is a transcription factor that regulates immediate early gene (IEG) expression via the serum response element (SRE) DNA consensus site. Elk-1 is associated with a dimer of serum response factor (SRF) at the SRE site, and its phosphorylation occurs at specific residues in response to mitogen-activated protein kinases (MAPKs), including c-Jun-N terminal kinase (JNK), p38/MAPK, and extracellular-signal regulated kinase (ERK). This phosphorylation event is critical for triggering SRE-dependent transcription. Although MAPKs are fundamental actors for the instatement and maintenance of memory, and much investigation of their downstream signaling partners have been conducted, no data yet clearly implicate Elk-1 in these processes. This is partly due to the complexity of Elk-1 sub-cellular localization, and hence functions, within neurons. Elk-1 is present in its resting state in the cytoplasm, where it colocalizes with mitochondrial proteins or microtubules. In this particular sub-cellular compartment, overexpression of Elk-1 is toxic for neuronal cells. When phosphorylated by the MAPK/ERK, Elk-1 translocates to the nucleus where it is implicated in regulating chromatin remodeling, SRE-dependent transcription, and neuronal differentiation. Another post-translational modification is the conjugation to SUMO (Small Ubiquitin-like MOdifier), which relocalizes Elk-1 in the cytoplasm. Thus, Elk-1 plays a dual role in neuronal functions: pro-apoptotic within the cytoplasm, and pro-differentiation within the nucleus. To address the role of Elk-1 in the brain, one must be aware of its multiple facets, and design molecular tools that will shut down Elk-1 expression, trafficking, or activation, in specific neuronal compartments. We summarize in this review the known molecular functions of Elk-1, its regulation in neuronal cells, and present evidence of its possible implication in model systems of synaptic plasticity, learning, but also in neurodegenerative diseases.
Elk-1 Modulates Gene Transcription
Elk-1 Belongs to the Ets Family of Ternary Complex Factors
Elk-1 is a member of the Ets (E twenty-six) oncogene family of transcription factors that includes nuclear phosphoproteins involved in many biological processes, such as cell growth, differentiation and survival, hematopoiesis, angiogenesis, wound healing, cancer, and inflammation (Sharrocks, 2001).
The Ets transcription factor family is defined by the presence of a highly conserved DNA-binding domain (DBD, the Ets domain) comprised of 85 amino acids. This family can be divided into subfamilies according to the homology of their Ets domain (Laudet et al., 1999), and comprises at least 27 different transcription factors in mammals. Two-thirds are expressed in most adult tissues, albeit with different levels of expression (Hollenhorst et al., 2004). A chromatin immunoprecipitation coupled with genome wide promoter microarrays was used to uncover the mechanisms regulating specificity within the Ets family (Hollenhorst et al., 2007). Frequent redundant occupancy was detected that correlated with housekeeping classes of genes, whereas specific binding examples represented more specialized genes.
Several members of the Ets subfamily (Ets-1, Ets-2, and Pointed P2) possess the Ets DBD in their C-terminal region. In the case of the TCF subfamily this motif is located in the N-terminus end of the protein.
Originally described in HeLa cell nuclear extracts, TCFs form a ternary complex with two molecules of SRF on the consensus DNA binding region called SRE at the c-fos promoter (Herrera et al., 1989; Shaw et al., 1989). First called p62 due to its 62000 Da molecular weight (Shaw et al., 1989), it was then shown to be homologous to Elk-1 (Ets-like transcription factor) that had been cloned previously (Rao et al., 1989; Hipskind et al., 1991). Two other related cDNAs were also identified, Sap-1 (SRF accessory protein 1)/Elk-4 (Dalton and Treisman, 1992) and Net/Erp/Sap-2/Elk-3 (Giovane et al., 1994; Lopez et al., 1994; Nozaki et al., 1996). The three TCFs, Net, Elk-1, and Sap-1 are distinct gene products with different chromosomal localizations in both the mouse and human genomes (Giovane et al., 1995).
Each of the TCFs contains four conserved domains with high sequence and functional similarities, named A-D (Figure 1). The A domain, localized in the N-terminal region, corresponds to the Ets DBD. This motif contains a nuclear localization signal (NLS) as well as a nuclear export signal (NES) that is deleted in a smaller, neuronal, isoform of Elk-1 called short-Elk-1 (sElk-1; Vanhoutte et al., 2001). The B box domain, which enables TCFs to interact co-operatively with SRF and promotes ternary complex formation, is located downstream of the Ets domain (Treisman et al., 1992; Hill et al., 1993). The D domain contains the DEJL (docking site for ERK and JNK LXL) consensus sequence that permits the binding to MAP kinases of ERK, JNK, and p38MAPK families. The C-terminal end of the protein corresponds to the transcriptional activation domain, or C domain, containing the multiple S/TP motifs targeted by activated MAP kinases (Marais et al., 1993; Janknecht et al., 1994). Within this region lies a docking domain, the FXFP or DEF (docking site for ERK FXFP) motif, which is specifically involved in docking to ERK (Sharrocks et al., 2000; Fantz et al., 2001). In Elk-1, the DEF domain is critically required for its selective phosphorylation by the MAPK kinase/ERK (Jacobs et al., 1999).
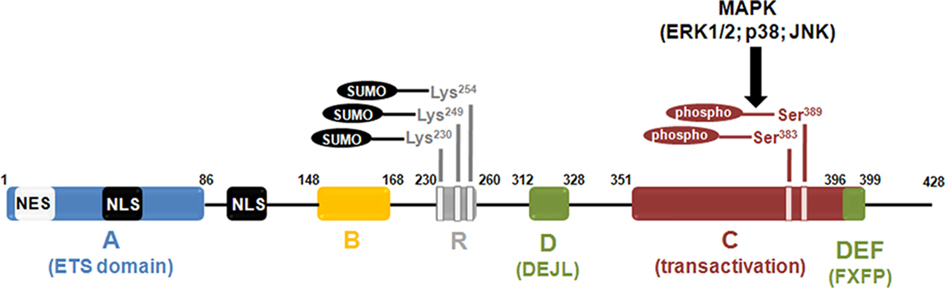
Figure 1. Diagram illustrating functional domains and major post-translational modifications of the Elk-1 protein. The ETS domain (or A) domain lies within the n-terminus end of the protein and is responsible for Elk-1 binding to DNA. The B domain is involved in the binding of Elk-1 to a dimer of its cofactor, the SRF. The R domain is crucial for the repression of Elk-1 transcriptional activity. It contains the lysine residues that are susceptible to be SUMOylated, a post-translational event that reinforce the repression exerted by the R domain. The C (or transactivation) domain contains the amino acids that are phosphorylated by MAP kinases. Among these residues, phosphorylation of Serine 383 and Serine 389 is a crucial event to activate Elk-1-mediated transcription. The two domains depicted in green are involved in the binding of Elk-1 to activated MAP kinases. The D (or DEJL) domain is responsible for the binding of Elk-1 to activated MAP kinases of the ERK, JNK, and p38 subtypes. The DEF (or FXFP) domain is more specific since it is only required for the binding of Elk-1 to activated ERK. The NES and NLS motifs are involved in nuclear export and import of Elk-1, respectively.
Two variants of Elk-1 have been described so far, ΔElk-1 and sElk-1. ΔElk-1 is an alternative splicing variant lacking the SRF interaction domain and a part of the Elk-1 DBD (Rao and Reddy, 1993). sElk-1 is a neuronal specific isoform of Elk-1 that arises from an internal translation start site in its sequence, and corresponds to a protein lacking the first 54 amino acids of the DBD (Vanhoutte et al., 2001). Thus, both truncated proteins present different DNA binding properties than full-length protein.
Classes of Genes Targeted by Elk-1
The best-characterized Elk-1 target is the proto-oncogene c-fos (Hipskind et al., 1991; Gille et al., 1992; Hill et al., 1993). Its promoter is regulated rapidly and transiently by mitogens via the SRE (Treisman et al., 1992), which binds the ternary complex formed by two molecules of SRF and one of the TCFs (Hill et al., 1993). Inhibiting formation or activity of this ternary complex leads to down-regulation of c-fos expression induced by serum. Conversely, increasing the activity of the ternary complex, through phosphorylation of TCFs, increases transcription. The recruitment of Elk-1 to SRE is made by a combination of protein–protein and protein–DNA interactions, via its Ets domain and B box, respectively. The determinant residues in SRF–Elk1 interaction are Tyrosine 153, Tyrosine 159, and Phenylalanine 162 (Ling et al., 1998). These residues represent a large fraction of Elk-1’s surface interaction with SRF and increase the intrinsic binding activity of Elk-1 toward SRE sites.
Serum response elements are also located in the promoters of other IEGs, such as junB, egr-1 (early growth response gene-1, also called zif268 or NGF-IA), and Nur77, the murine homolog of the human NR4A1 gene (Treisman et al., 1992). The recent identification of the repertoire of Elk-1 target genes reveals that a significant number of them encode transcription factors but also components of the basal transcriptional machinery, such as the TATA binding protein (TBP; Boros et al., 2009a). Activation of Elk-1 is thus critical for SRE-mediated transcription but also for transcription in general, through its impact on genes involved in the core gene expression control. Interestingly, the srf gene itself has been identified as an Elk-1 target (Kasza et al., 2005).
Transcriptional Activity Induced by Elk-1 Phosphorylation
The transcriptional activity of Elk-1 depends on its phosphorylation, which is regulated by the MAP kinase pathway. Elk-1 is activated through phosphorylation by the three classes of MAP kinases, ERK, JNK, and p38 (Cavigelli et al., 1995; Gille et al., 1995; Whitmarsh et al., 1995; Enslen et al., 1998; Cruzalegui et al., 1999). While the ERK cascade is activated by growth factors and mitogens, the JNK and p38 cascades respond to cytokines and stress. Phosphorylation by MAP kinases can occur on at least nine residues in the C domain of Elk-1 (Cruzalegui et al., 1999), two of them, Serine 383 and Serine 389, being crucial for the transcriptional activity of Elk-1. Once mutated in non-phosphorylable alanines, SRE-dependent gene induction by Elk-1 is totally blocked (Janknecht et al., 1993). Moreover, the Serine 383 is particularly important for the transactivating properties of Elk-1, because a single mutation of this residue evokes a total loss of these properties (Gille et al., 1995). In Elk-1, the DEF motif is responsible for direct ERK-induced phosphorylation of this residue. At the molecular level, the C-terminal phosphorylation of Elk-1 leads to a conformational change in the overall tertiary structure of Elk-1 (Yang et al., 1999). This, in turn, results in an increased binding to SRF via the B box, and overall increased DNA binding of Elk-1 to the 5′CAGGA motif of the SRE.
Once activated by phosphorylation, Elk-1 promotes, in addition to enhanced DNA binding to the SRE, a recruitment of coactivators, including CREB binding protein (CBP), p300, or the Srb mediator. CBP directly interacts with Elk-1 in vitro and in vivo. This interaction occurs between the Elk-1 C domain and the N-terminus of CBP. However, functional interaction occurs only after signaling-driven phosphorylation of Elk-1 (Janknecht and Nordheim, 1996). The N-terminal domain of Elk-1 can also interact with CBP via its bromodomain (Nissen et al., 2001). Like CBP, p300 (a CBP-related protein) acts like a coactivator in complexes that contain Elk-1 (Li et al., 2003). Both CBP and p300 possess an intrinsic histone acetyltransferase (HAT) activity or associate with HATs that acetylate core histones and relieve repression of transcription through chromatin decompaction. Stimulation of HAT activity mediated by Elk-1 recruitment is particularly relevant for the c-fos promoter since analysis of its chromatin structure revealed a nucleosome positioned nearby the SRE site (Herrera et al., 1997). The role of Elk-1 in histone acetylation at the c-fos promoter was also studied in the context of cortical neurons treated with nerve growth factor. The increase of Elk-1 phosphorylation and c-fos induction is induced by poly-ADP-ribosylated PARP-1, which mediates acetylation of core histone H4 (Cohen-Armon et al., 2007). A molecular bridge between Elk-1 and RNA polymerase II is the Srb mediator complex, which plays a role in Elk-1-dependent gene transactivation (Boyer et al., 1999). The local nucleosomal structure also facilitates the recruitment of a second transcription factor, the nuclear factor I (also known as NF1, NF-1, or CTF, for CAAT box transcription factor), which induces the recruitment of the basal transcriptional machinery components TFIIB and RNA polymerase II to the c-fos promoter after MAP kinase activation induced by phorbol 12-myristate 13-acetate (PMA; O’Donnell et al., 2008; White and Sharrocks, 2010). Recently, it was shown that ERK and mitogen- and stress-activated kinase (MSK), a kinase for histone H3 downstream ERK, is recruited to the SRE promoter complex in vitro and in vivo (Zhang et al., 2008). Their recruitment in vitro correlates with Elk-1 binding to activated ERK via its DEJL domain. Of interest, the recruitment of both ERK and MSK to the DNA promoter region is stimulated in vivo by mitogens, and correlates with histone H3 phosphorylation. These events are impaired by Elk-1 knockdown, thus clearly implicating Elk-1 as a molecular partner for the recruitment of MSK at vicinity of nucleosomes in DNA promoter regions.
In conclusion, Elk-1 phosphorylation plays a dual role in gene regulations, by promoting chromatin remodeling through histone acetylation and phosphorylation, and via the recruitment of coactivators of the basal transcriptional machinery.
Transcriptional Repression by Elk-1 Dephosphorylation and Sumoylation
Elk-1 activity depends on the equilibrium between kinases and phosphatases. Calcineurin (PP2B) is the major phosphatase of Elk-1 identified suggesting an important role of nuclear calcium in Elk-1 dephosphorylation (Sugimoto et al., 1997; Tian and Karin, 1999). Consequently, Cyclosporin A, a PP2B inhibitor, blocks Elk-1 dephosphorylation in vitro and enhances Serine 383 phosphorylation (Tian and Karin, 1999).The temporal window in which PP2B is active for Elk-1 dephosphorylation has not been investigated so far. However, given the transient phosphorylation of Elk-1 in response to mitogenic stimuli, one can speculate that kinase activity precedes phosphatase-mediated effects on Serine 383 phosphorylation state.
Another repression mechanism involves the recruitment of co-repressor complexes, many of them containing histone deacetylase activities (HDACs), which leads to compaction of the chromatin at the level of promoters. One example is the mSin3A–HDAC complex, which binds to the N-terminal transcriptional repression domain of Elk-1 (Yang et al., 2001). This recruitment is delayed following EGF stimulation and corresponds kinetically to the shut-off of target promoters. Activation of the ERK pathway, and specifically phosphorylation of Elk-1 at Serine 383, in vivo or in vitro, evokes an enhanced binding of the mSin3A–HDAC complex, and hence histone deacetylation and chromatin compaction. Thus, Elk-1 represents a key element that can act sequentially as an activator and then a repressor of c-fos transcription.
The functionally active Elk-1 is also regulated by SUMOylation. Yang et al. (2002)described the R motif, a novel class of repression domain of Elk-1 that is located upstream the transactivation domain (Figure 1). This domain maintains Elk-1 in an inactive state prior to its activation by ERKs. Elk-1 contains three potential SUMOylation sites, the Lysines 230, 249, and 254 lying within the R motif. In basal conditions, Elk-1 is SUMOylated and inactive (Yang et al., 2003). SUMOylated Elk-1 recruits HDAC to the c-fos and egr-1 promoters and evokes their repression (Yang and Sharrocks, 2004). The repression induced by SUMOylation of Elk-1, is reversed by activation of ERK, which causes SUMO and HDAC release. In the derepression process, the SUMO E3 ligase PIASxα, plays a key role in promoting SUMO loss of Elk-1. This PIASxα-mediated coactivation of Elk-1 depends on the binding of PIASxα to Elk-1, independently of the E3 activity of PIASxα (Yang and Sharrocks, 2005). PIASxα also reduces the recruitment of HDAC-2 to the c-fos promoter, leading to increased histone H4 acetylation and consequent transactivation of this IEG. The activity of PIASxα toward HDAC-2 removal is abolished upon activation of the stress-activated MAPK pathways, despite Elk-1 phosphorylation (Yang and Sharrocks, 2006). Finally, Elk-1 can be ubiquitylated in vitro, and hence degraded by the proteasomal machinery (Fuchs et al., 1997).
Sub-Cellular Distribution and Regulation of Elk-1 Functions in Neuronal Cells
Expression of Elk-1 in the CNS
A strong expression of Elk-1 mRNA in the brain of humans and rodents was first described by global analyses (Price et al., 1995). A more detailed analysis in the rat brain revealed a widespread expression in most brain regions, within neuronal and not glial cell types (Sgambato et al., 1998a). Elk-1 is restricted to the nucleus in non-neuronal cells (Janknecht et al., 1994; Pingoud et al., 1994), a result expected for a transcription factor, but expressed in both cytoplasmic and neuritic compartments in addition to the nucleus in mature neurons (Sgambato et al., 1998a; Vanhoutte et al., 2001). This work also led us to characterize of a neuron-specific isoform of Elk-1, named sElk-1, the expression of which is restricted to the nucleus. sElk-1 arises from an internal translation start site in the Elk-1 sequence, which generates a protein lacking the first 54 amino acids of the Elk-1 DBD. This truncated isoform of Elk-1 antagonizes Elk-1-mediated transactivation of SRE and promotes neuronal differentiation. The strictly nuclear expression of sElk-1 is due to deletion of a NES that was identified within the Ets domain (Vanhoutte et al., 2001).
Since then, the presence of Elk-1 in the cytoplasm of neurons or neuroblastomas was confirmed, but Elk-1 functions in this particular compartment remain elusive. Studies form Eberwine and colleagues demonstrated that Elk-1 binds to the mitochondrial permeability transition pore (PTP) complex, an interaction being reinforced in experimental conditions triggering apoptosis of cortical neurons (Barrett et al., 2006a). They showed that overexpression of Elk-1 in primary neurons decreases cell viability, whereas Elk-1 siRNA-mediated knockdown increases cell survival. This decrease in viability induced by Elk-1 overexpression is blocked with application of a PTP inhibitor. These results suggested that one extranuclear function for Elk-1 was pro-apoptotic. By means of a region-directed phototransfection of Elk-1 mRNA, the same group made the interesting observation that overexpression of Elk-1 in distal dendrites was toxic whereas its presence, when restricted to the cell body, did not trigger neuronal death (Barrett et al., 2006b). Recently, a direct interaction of Elk-1 with tubulin has been demonstrated (Demir et al., 2009), however the physiological role of this binding to neuronal microtubules is not yet defined.
Regulation of Elk-1 Cyto-Nuclear Shuttling by Translational Modifications
Although mature neurons are post-mitotic cells, ERKs are strongly expressed and activated by multiple extracellular stimuli, including neurotransmitters, neuromodulators, and neurotrophic factors (for review Valjent et al., 2001a; Thomas and Huganir, 2004). A large body of evidence implicates this signaling pathway, along with downstream substrates, in learning and memory. Yet, there is no direct evidence implicating Elk-1 in these processes. An ERK-dependent Serine 383 phosphorylation of Elk-1 has been found in nuclear, somatic, and dendritic compartments upon electrical stimulation of the cortico-striatal pathway in vivo (Sgambato et al., 1998a,b). In this model system, ERKs activation is involved in the induction of zif268, c-fos, and mkp-1 (MAP kinase phosphatase 1). ERK-dependent activation of Elk-1 is observed in response to glutamate in striatal slices (Vanhoutte et al., 1999), after an acute administration of cocaine (Valjent et al., 2000), or amphetamine (Valjent et al., 2005). ERK-induced Elk-1 phosphorylation is also detectable in hippocampal neurons after induction of long-term potentiation (LTP; Davis et al., 2000) or during contextual fear conditioning (Sananbenesi et al., 2002). The finding that Elk-1 is activated in the cytoplasm suggested that it may have a local role and/or translocate to the nucleus upon phosphorylation by ERKs. Using total Elk-1 immunolabeling Caboche and colleagues found that indeed Elk-1 translocates to the nucleus upon activation by cocaine in vivo, or glutamate in vitro (Lavaur et al., 2007). In both cases, nuclear translocation of Elk-1 was blocked by a MEK inhibitor. Furthermore, using a cell-penetrating peptide that specifically interferes with the DEF docking domain of Elk-1 (TAT–DEF–Elk-1), and as such blocks Elk-1 phosphorylation induced by ERKs, we observed retention of Elk-1 within the cytoplasm. The TAT–DEF–Elk-1 peptide does not interfere with ERK activity toward other substrates, such as MSK-1. This approach, combined with site-directed mutagenesis in the Elk-1 Serine 383 and Serine 389 phosphorylation sites, demonstrated that Elk-1 translocation from the cytoplasm to the nucleus is tightly linked to its activation by ERKs (Lavaur et al., 2007). The nuclear translocation of activated Elk-1 was further confirmed in neuroblastoma cells treated with high concentration of serum that activates the ERK/Elk-1 module (Demir et al., 2009). In a follow up study, we clarified some of the molecular mechanisms involved in the trafficking of activated ERK and Elk-1 proteins from distal neurites toward the nucleus in vitro. This retrograde trafficking induced by glutamate involves a recruitment of activated ERK and Elk-1 to macromolecular complexes comprising markers of clathrin-dependent endocytosis, originating, at least in part, from the endocytosis of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (Trifilieff et al., 2009).
Thus, by triggering its nuclear translocation, phosphorylation of Elk-1 on Serine 383 and Serine 389 is likely to disrupt Elk-1 binding to microtubules and mitochondria and orients Elk-1 toward transcriptional properties at the SRE promoter regions.
Conversely, SUMOylation of lysine residues lying within the R domain of Elk-1 also prompts the export of Elk-1 from the nucleus toward the cytoplasm (Salinas et al., 2004). The physiological and/or pathological stimuli that regulate the SUMO pathway in neurons are not yet defined. Nevertheless, by homology with their contrasting roles in transactivation or repression of transcription in non-neuronal cells (Yang et al., 2003), phosphorylation and SUMOylation seems to exert opposing roles on Elk-1 trafficking between the cytoplasm and the nucleus in neuronal cell types. Another residue, Threonine 417 is phosphorylated in neuronal cells by a still unknown kinase. When mutated on this residue, Elk-1 overexpression in the cytoplasm is not toxic anymore in cultured hippocampal neurons. Interestingly, increased cytoplasmic phosphorylation of Threonine 417 is observed in various brain tissues from patients with neurodegenerative diseases (Sharma et al., 2010).
Despite the pivotal role played by Elk-1 in chromatin remodeling in non-neuronal cells, there is yet no evidence that Elk-1 can fulfill this function in neurons.
Parsing Nuclear Functions of Elk-1 in Neuronal Cells
The complexity of Elk-1 localization and functions within neuronal cells prompted us to design new molecular approaches for specific impairment of ERK-dependent phosphorylation and nuclear translocation. We used the TAT–DEF–Elk-1 peptide that selectively impairs Elk-1 phosphorylation by ERK on Serine 383 and Serine 389 residues. Importantly, the specific inhibition of Elk-1 phosphorylation, and subsequent nuclear translocation, was associated with a defect in glutamate-induced expression of IEGs bearing SRE site(s) on their promoter, such as c-fos, junB, and zif 268 (Lavaur et al., 2007).
Consistent with the known role of Elk-1 as a transcriptional activation of the srf gene (Kasza et al., 2005), chronic blockade of Elk-1 phosphorylation was associated with a decrease of SRF expression in neurons (Lavaur et al., 2007). This was particularly interesting given that SRF was shown to behave as a sensor of cytoskeleton actin dynamics (Knoll and Nordheim, 2009). Indeed, conditional srf knock-out mice in the forebrain present a reduced expression level of actin, an impairment of axonal guidance and defects in growth cones morphology (Knoll et al., 2006). Interestingly, when Elk-1 phosphorylation is inhibited, SRF and actin levels are down regulated and growth cone collapse occurs (Lavaur et al., 2007). Thus, inhibition of Elk-1 phosphorylation by ERK or knock out of the srf gene leads to somewhat overlapping phenotypes. This is consistent with SRF being a critical cofactor for Elk-1 transcriptional activity at SRE sites (Shaw et al., 1989). However, Elk-1 is also able to regulate transcription of defined set of genes in a SRF-independent fashion (Boros et al., 2009a,b). This raises the questions as to whether phenotypes induced by Elk-1 inhibition are due to the associated down-regulation of SRF and/or to mechanisms involving Elk-1 activation independently of SRF.
Altogether these data clearly indicates that post-translational modifications of Elk-1 play a crucial role in the control of both Elk-1 cytoplasm-to-nucleus shuttling and Elk-1 functions (Figure 2). In PC12 cells, SUMO conjugation to Elk-1 increases its shuttling to cytoplasmic compartments, and inhibits spontaneous neuronal differentiation induced by wild-type Elk-1 (Salinas et al., 2004). Within the cytoplasm, Elk-1 tends to be toxic, at least when it is associated with the mitochondrial PTP or expressed in the distal dendrites (Barrett et al., 2006a,b). As opposed to these deleterious effects of Elk-1 in the cytoplasm, ERK-induced phosphorylation of Elk-1 on Serine 383 and Serine 389 forces Elk-1 translocation to the nucleus, where it plays an important role in SRE-dependent transcription, promotes neuronal differentiation, and regulates the cytoskeleton dynamics (Lavaur et al., 2007). The pivotal role played by Elk-1 in IEGs induction, downstream of ERK activation raises the question as to whether Elk-1 phosphorylation is involved in synaptic plasticity, learning, and long-term behavioral adaptations.
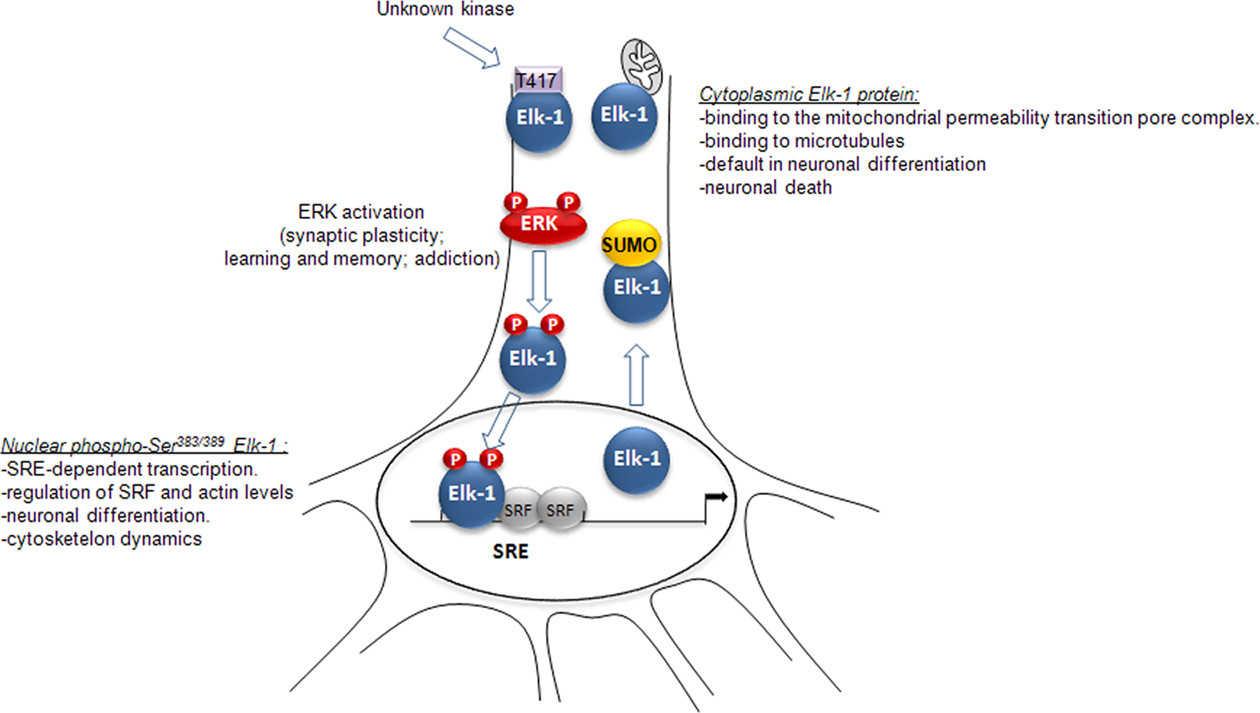
Figure 2 Synthetic view of the impact of Elk-1 post-translational modifications on its cytoplasm-to-nucleus trafficking and functions in neurons (see main text for details).
Elk-1, a Molecular Partner of ERK in Synaptic Plasticity and Complex Behaviors
Elk-1 Phosphorylation and Neuronal Plasticity
Elk-1 is a target of ERK but also of JNK and p38 MAPK, which are all involved in diverse physiopathological forms of neuronal plasticity. Mice deficient for the elk-1 gene present a mild alteration in their phenotype when compared to wild-type littermate (Cesari et al., 2004). Within the brain, no alteration in the proteomic display was found, but a modest decrease in c-Fos and Zif268 expression was observed in the hippocampus and cortex after kainate-induced seizure. No deficits in the immunological response were found in these mice. Our studies, using elk-1 knock-out mice, showed no gross alteration of brain histology and no modification of cocaine-induced gene regulation in the striatum (personal observation). As suggested by Nordheim and colleagues (Cesari et al., 2004), this mild phenotype could be attributed to a functional redundancy between Elk-1 and other TCF members.
An ERK-dependent Serine 383 and Serine 389 phosphorylation of Elk-1 has been observed in various models of synaptic plasticity. LTP is a possible cellular mechanism for information storage. The ERK signaling pathway is critical for this synaptic plasticity (Sweatt, 2004), probably because it controls the induction of some IEGs, including zif268 (Davis et al., 2000). In vivo, zif268 mRNAs are induced within the dentate gyrus (DG) immediately after a high frequency tetanus of the perforant path. In this model, a concomitant hyperphosphorylation of ERK, along with Elk-1 and the transcription factor CREB, can be found in the same region. Pharmacological blockade of MEK inhibits Elk-1 and CREB phosphorylation as well as zif268 mRNA induction and LTP (Davis et al., 2000). This study provided the first in vivo evidence that ERK activation is critical for LTP. Afterward, the essential role of Zif268 in LTP was established. Mice deficient for Zif268 show a deficit in late, but not early, LTP and long-term memory (Jones et al., 2001). Similarly, Thiels et al. (2002) showed a robust increase in nuclear ERK and Elk-1 phosphorylation after electrical NMDA-dependent long-term depression (LTD) induction in the hippocampal CA1 area. The MEK inhibitor SL327 totally abolished Elk-1 hyperphosphorylation and NMDA-dependent LTD, thus confirming that Elk-1 activation depends on ERK and is associated with LTD in vivo. In accordance with these results, Lindecke et al. (2006) reported a strong hyperphosphorylation of Elk-1 after LTD induced by a stimulation of metabotropic glutamate receptors (mGluR) on mouse organotypic hippocampal slices. Interestingly, mGluR-induced LTD at CA3–CA1 synapses was previously described as dependent on p38 activity in mouse hippocampal slices (Bolshakov et al., 2000). Future studies are needed to further delineate a possible involvement of p38/Elk-1 module in mGluR-induced hippocampal LTD.
Elk-1 Phosphorylation in Models of Learning
In light of the growing evidence that MAPKs are involved in the formation of long-term memory, Berman et al. (1998) showed that aversive taste-learning experience-induced transient phosphorylation of ERK, JNK, and Elk-1 (but not p38 nor AKT) in the insular cortex (IC). Microinfusion of the MEK inhibitor in the IC totally abrogates Elk-1 phosphorylation and partially impairs the acquisition of the conditioned-taste aversion. In a further study, local microinjection of pharmacological antagonists revealed that taste-induced Elk-1 activation in the IC depends on glutamatergic, dopaminergic, and acetylcholinergic receptors stimulation (Berman, 2003). Altogether, these results indicates that taste-dependent Elk-1 phosphorylation in the IC depends on ERK, which contributes to the acquisition of the long-term taste memory. Similar results were obtained in the visual cortex, where Elk-1 is transiently phosphorylated by a visual stimulation applied after a period of dark rearing (Kaminska et al., 1999). ERK, but not JNK activation, was temporally matched to the onset of Elk-1 phosphorylation, whereas p38 phosphorylation was unaltered in this paradigm. One-trial avoidance learning is also associated with a specific increase in the phosphorylation of ERK, Elk-1, CREB, and c-Fos expression in the hippocampus (Cammarota et al., 2000). Microinfusion of a NMDA receptor antagonist in the hippocampal CA1 area impairs the activation of the ERK signaling cascade as well as the acquisition of one-trial avoidance learning. A coincident hyperphosphorylation of ERK, Elk-1, and RSK-1 is also observed in the CA3 hippocampal subfield and the DG 30 min after context-dependent fear conditioning (Sananbenesi et al., 2002).
Elk-1 has been associated with food aversion conditioned reflex, which depends on serotonin in edible snail (Grinkevich et al., 2004). The authors used two different models of serotonin-depleted animals displaying altered abilities to develop the defense reflex. In both models, experience-induced ERK and Elk-1 phosphorylation are significantly altered as compared to controls. Altogether, these results suggest that an alteration of serotonin metabolism leads to the disruption of ERK and Elk-1 activity possibly accounting for the edible snail inability to develop defense reflex.
In order to elucidate the role of Elk-1 interaction with DNA, decoy oligonucleotides were used to block either Elk-1 or SRF binding to DNA. These oligonucleotides were microinjected in the hippocampus of rats before being trained to the Morris water maze (Dash et al., 2005). Interestingly, sequestration of SRF, but not Elk-1, is sufficient to disrupt long-term spatial memory. These results suggest that Elk-1 interaction with DNA per se is not necessary for spatial memory formation. However, a specific role of Elk-1 in such memory process cannot be totally ruled out. In fact, phosphorylation of Elk-1 by ERK can trigger multiple molecular events that do not specifically require binding of Elk-1 to DNA, including recruitment of MSK-1 and subsequent phosphorylation of histones (Zhang et al., 2008), increased binding to SRF, or functional interactions with CBP, and recruitment of the basal transcriptional machinery. Whether these molecular events are disrupted using the decoy oligonucleotides directed against binding of Elk-1 to DNA remains to be established. In conclusion, in the absence of specific pharmacological inhibitors, or genetic models aimed at specifically blunting Elk-1 Serine 383 phosphorylation, we cannot conclude about the causative link between this phosphorylation event, long-term neuronal plasticity and learning.
A cross-talk between ERK and PI3K/SGK1 (serum- and glucocorticoid-inducible kinase 1) signaling pathways for the regulation of Elk-1 transcriptional activity arises from a recent work. Using the Morris water maze and microinjecting the negative mutant of SGK1 in the hippocampal CA1 of rats before the training sessions, the authors found an impairment of mRNAs encoding CREB and phosphorylation of SRF, along with an impaired induction of Zif268 and memory formation (Tyan et al., 2008). Interestingly, in vitro results revealed that active SGK1 may negatively regulate Elk-1 transcriptional activity through phosphorylation of Elk-1 at Serine 159 and Threonine 160, which interrupts Elk-1 binding to SRF. Thus, future work will be required to shed light on the relevance of these findings in the context of long-term memory formation.
Elk-1 Activation and Drugs of Abuse
The ERK pathway is a central actor in addiction (Lu et al., 2006). Our group pioneered the observation that ERK is activated upon acute cocaine administration in mice (Valjent et al., 2000). We found a strong and transient hyperphosphorylation of ERK and Elk-1 in the striatum of mice after an acute cocaine administration (Valjent et al., 2000). Pretreatment with SL327, a MEK inhibitor that can cross the blood–brain barrier, abolished Elk-1 phosphorylation, the induction of Zif268 and c-Fos, and the establishment of conditioned place preference (CPP) induced by cocaine. These results were recently confirmed in rats (Jenab et al., 2005; Sun et al., 2007).
Then, a common feature of drugs of abuse was shown, i.e., a transient activation of ERK in the striatum and extended amygdala (Valjent et al., 2004). Thus, nicotine, Δ9-THC (the psychoactive molecule of cannabis), morphine, and amphetamine were shown to activate ERK in these brain areas, in a dopamine-dependent manner (Valjent et al., 2004). Increased phosphorylation of Elk-1 was further demonstrated in the striatum of mice upon Δ9-THC (Valjent et al., 2001a,b) and amphetamine (Choe et al., 2002) treatments, and in the hippocampal CA1 area after acute nicotine administration (Nuutinen et al., 2007). Acute ethanol exposure triggered Elk-1 hyperphosphorylation in subregions of the amygdala (Pandey et al., 2008). To date, in vivo activation of Elk-1 by morphine has not been investigated. However, a recent study used small interfering RNAs for Elk-1 and CREB in HEK cells expressing the rat μ-opioid receptor (Ligeza et al., 2008). Silencing of Elk-1 and CREB activity diminished the cellular responses to withdrawal signals and chronic opioid treatment in this in vitro system.
Besides Elk-1, ERK controls the phosphorylation levels of the transcription factor CREB, which plays an important role in the expression of genes involved in synaptic plasticity and drug addiction (Carlezon Jr. et al., 1998; for review, see Nestler, 2004). CREB phosphorylation at Serine 133 is not catalyzed directly by ERK but by protein kinases activated by ERK, including two isoforms of mitogen-activated protein kinase-activated protein kinase 1 (MAPK-APK1; also called ribosome S6 kinases, RSKs; De Cesare et al., 1998; Xing et al., 1998) and mitogen- and stress-activated protein kinases (MSKs; Deak et al., 1998; Arthur and Cohen, 2000). Activation of MSKs is also implicated in histone H3 phosphorylation (Soloaga et al., 2003) and thus in chromatin reorganization (Nowak and Corces, 2004).
Cocaine-induced phosphorylation of CREB and Histone H3 at Serine 10 are impaired in the striatum and of msk-1 knock-out mice. In these mice, the induction by cocaine of c-Fos and Dyn but not Zif268 expression is altered (Brami-Cherrier et al., 2005). These mice display a partial decrease of locomotor sensitization, with a slight increase of CPP. This study demonstrates that alterations of the ERK/MSK-1/CREB signaling module only partially recapitulate the molecular and behavioral alterations observed with SL327, which abrogates both locomotor sensitization and CPP induced by cocaine (Valjent et al., 2000, 2006). Because MSK-1 cannot target some important SRE-driven IEGs (including Zif268) induced by cocaine, we suspect that an ERK/Elk-1 module may be involved in some important molecular and behavioral adaptations to cocaine, and by extension most drugs of abuse.
The ERK pathway is also critical for the retrieval (or expression) of a previously established CPP, in the absence of drug administration. Hyperphosphorylation of ERK and Elk-1 in the nucleus accumbens (NAcc) is associated with the expression of methamphetamine- and cocaine-induced CPP (Mizoguchi et al., 2004; Miller and Marshall, 2005). Local inhibition of ERK blocks the expression these behaviors. MEK inhibition during memory retrieval resulted in a long lasting amnesic effect (at least 2 weeks post-infusion) indicating that ERK is required for both the establishment and retrieval of memory. Analyzing the specific role of Elk-1 phosphorylation on Serine 383 in these two paradigms might be critical.
Phosphorylation of Elk-1 in Pathophysiological Conditions
As illustrated in this section, Elk-1 phosphorylation has been shown to be modulated in various CNS diseases.
Alzheimer’s disease
Accumulation of amyloid-β (Aβ) peptides initiates and/or contributes to Alzheimer’s disease (AD). Tong et al. (2004) showed that sublethal concentrations of Aβ interfere with BDNF-induced activation of Elk-1 in cultured cortical neurons and results in an altered SRE-driven gene regulation, which is likely to account for increased neuronal vulnerability. Pastorcic and Das (2003) defined Elk-1 as a potent repressor of transcription of the presenilin 1 gene (PS1) encoding for a protein required for the final step of the sequential proteolytic processing of amyloid precursor protein (APP), which produces highly amyloidogenic variants of Aβ (Aβ42/43 polypeptide). PS1 is genetically linked to the majority of cases of early-onset of familial Alzheimer’s disease (FAD). Collectively, these data highlight the intriguing link connecting Aβ, Elk-1, and PS1. Future investigations should aim at deciphering if Aβ derepresses PS1 expression by inhibiting Elk-1 activity in AD and/or FAD pathogenesis.
Huntington’s disease
Huntington’s disease (HD) is a neurodegenerative disorder due to abnormal polyglutamine expansion in the N-terminal region of huntingtin (Htt) protein. This expansion causes protein aggregation, transcriptional dysregulations, neuronal dysfunction, and finally death. ERK, Elk-1, and CREB were reported to be hyperphosphorylated in the striatum of R6/2 transgenic, a mouse model of HD (Roze et al., 2008). However, transcriptional activity at the c-fos promoter was impaired, probably because of MSK-1 expression deficiency in this mouse model. Interestingly, deficiency of MSK-1 expression was also found in the striatum, but not cortex, of HD post-mortem brain patients. Histone H3 and MSK-1 phosphorylation, along with transcriptional dysregulations, may account for striatal degeneration in R6/2 mice. Restoring MSK-1 expression reversed neuronal dysfunctions induced by expanded Htt, in vitro (Roze et al., 2008). Given the key role of Elk-1 in the recruitment of MSK-1 at the vicinity of histone H3 (Zhang et al., 2008), it would be interesting to analyze a possible synergy between Elk-1 and MSK-1 in neuronal protection in HD.
Synucleinopathies
α-Synuclein forms aggregate in neurons in Parkinson’s disease (PD) and mainly in oligodendrocytes in multiple system atrophy (MSA). α-Synuclein interacts indirectly with Elk-1 via ERK, and prominently attenuates Elk-1 phosphorylation on Serine 383 along with c-Fos induction in response to EGF in Neuro2A cells (Iwata et al., 2001). Nevertheless, no causative link between this phosphorylation event and the degenerative process has been described so far in PD or MSA.
The accumulation of a cytoplasmic and toxic form of Elk-1 has been observed in post-mortem brain from patients affected with each of the three neurodegenerative diseases mentioned above. An increased cytoplasmic phosphorylation of Elk-1 on Threonine 417 was found to colocalize with inclusions in PD (Lewy body), AD, and HD (Sharma et al., 2010).
Down syndrome
Age-dependent alterations of ERK activity were observed in brains from Ts65Dn mice, a transgenic mice model of Down syndrome (DS; Gardiner, 2003). A decrease of calcineurin activity, the major phosphatase for Elk-1, was also observed in the fetal DS brain and aged Ts65Dn mice. Expression of SUMO3 that inhibits Elk-1 activity by triggering its sumoylation in vitro, was increased in the hippocampus of the adult DS patient (Gardiner, 2006). DS is thus associated with alterations of ERK, calcineurin, and SUMO pathways, which all converge and act antagonistically on Elk-1 activity. This raises questions as to whether Elk-1 functions might be altered in rodent models of DS and, if so, what are the potential behavioral consequences of such deregulation.
Depression
Extracellular-signal regulated kinase mRNA and protein levels were found to be decreased in the prefrontal cortex and hippocampus, but not the cerebellum, in post-mortem brains of depressed suicide subjects (Dwivedi et al., 2001). As a consequence, a decrease in ERK-mediated Elk-1 phosphorylation was observed in these brain areas. These results indicate that depression and suicidal behavior are associated with altered ERK signaling. Future work will be needed to determine a potential involvement of Elk-1 in the pathophysiology of depression.
Concluding Remarks and Future Directions
If the aforementioned studies pointed out the tight correlation existing between Elk-1 phosphorylation at Serine 383 with several models of synaptic plasticity, experience-learning, drug addiction, and some pathological processes, a precise role for Elk-1 in such in vivo models remains elusive. For this purpose, Elk-1 deficient mice were generated by homologous recombination in embryonic stem cells (Cesari et al., 2004). Unfortunately, these mice appeared to be phenotypically indistinguishable from their wild-type littermates. These results suggested compensatory activities by other TCFs such as SAP1 and/or NET. Given the large range of ERK substrates, one cannot use global inhibitors of this pathway to address specifically the role of Elk-1 phosphorylation in neuronal plasticity or mnesic processes.
Recently, we designed a synthetic cell-penetrating peptide which specifically interferes with ERK-dependent activation of Elk-1 in vitro. This peptide efficiently impedes Elk-1 phosphorylation, without interfering with ERK, MSK-1, or CREB activation, in response to glutamate in cultured striatal neurons (Lavaur et al., 2007). Importantly, since TAT penetrating sequence has previously been reported to efficiently cross the blood–brain barrier (Schwarze et al., 1999), this bioengineered peptide represents a promising alternative to investigate in vivo Elk-1 mediated biological functions. Another challenging issue regarding Elk-1 functions within the brain is to further investigate the other post-translational modifications described so far in non-neuronal cells, including SUMOylation and ubiquitination. In other words, it will important to know whether these modifications occur in response to neurotransmitter stimulation, in physiological or pathophysiological conditions, and what precise role they play in the trafficking and functions of Elk-1. Finally, in light of recent data in the literature (Zhang et al., 2008), it is of major importance to determine a possible role of Elk-1 in epigenetic responses and chromatin remodeling. Unraveling the spatio-temporal regulation of Elk-1 downstream of ERK may give further insights into the role of this signaling module in neuronal plasticity, memory formation, and drug addiction, but also in neuronal survival in neurodegenerative processes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Our laboratory is supported by Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale, Agence Nationale pour la Recherche (ANR-08-BLAN). Antoine Besnard and Beatriz Galan-Rodriguez are recipients of fellowships from Fondation pour la Recherche Médicale and Ecole des Neurosciences de Paris, respectively. Authors are grateful to members of the laboratory for helpful discussions and critical reading of the manuscript.
References
Arthur, J. S., and Cohen, P. (2000). MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells. FEBS Lett. 482, 44–48.
Barrett, L. E., Sul, J. Y., Takano, H., Van Bockstaele, E. J., Haydon, P. G., and Eberwine, J. H. (2006a). Region-directed phototransfection reveals the functional significance of a dendritically synthesized transcription factor. Nat. Methods 3, 455–460.
Barrett, L. E., Van Bockstaele, E. J., Sul, J. Y., Takano, H., Haydon, P. G., and Eberwine, J. H. (2006b). Elk-1 associates with the mitochondrial permeability transition pore complex in neurons. Proc. Natl. Acad. Sci. U.S.A. 103, 5155–5160.
Berman, D. E. (2003). Modulation of taste-induced Elk-1 activation by identified neurotransmitter systems in the insular cortex of the behaving rat. Neurobiol. Learn. Mem. 79, 122–126.
Berman, D. E., Hazvi, S., Rosenblum, K., Seger, R., and Dudai, Y. (1998). Specific and differential activation of mitogen-activated protein kinase cascades by unfamiliar taste in the insular cortex of the behaving rat. J. Neurosci. 18, 10037–10044.
Bolshakov, V. Y., Carboni, L., Cobb, M. H., Siegelbaum, S. A., and Belardetti, F. (2000). Dual MAP kinase pathways mediate opposing forms of long-term plasticity at CA3–CA1 synapses. Nat. Neurosci. 3, 1107–1112.
Boros, J., O’Donnell, A., Donaldson, I. J., Kasza, A., Zeef, L., and Sharrocks, A. D. (2009a). Overlapping promoter targeting by Elk-1 and other divergent ETS-domain transcription factor family members. Nucleic Acids Res. 37, 7368–7380.
Boros, J., Donaldson, I. J., O’Donnell, A., Odrowaz, Z. A., Zeef, L., Lupien, M., Meyer, C. A., Liu, X. S., Brown, M., and Sharrocks, A. D. (2009b). Elucidation of the ELK1 target gene network reveals a role in the coordinate regulation of core components of the gene regulation machinery. Genome Res. 19, 1963–1973.
Boyer, T. G., Martin, M. E., Lees, E., Ricciardi, R. P., and Berk, A. J. (1999). Mammalian Srb/mediator complex is targeted by adenovirus E1A protein. Nature 399, 276–279.
Brami-Cherrier, K., Valjent, E., Herve, D., Darragh, J., Corvol, J. C., Pages, C., Arthur, S. J., Girault, J. A., and Caboche, J. (2005). Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J. Neurosci. 25, 11444–11454.
Cammarota, M., Bevilaqua, L. R., Ardenghi, P., Paratcha, G., Levi de Stein, M., Izquierdo, I., and Medina, J. H. (2000). Learning-associated activation of nuclear MAPK, CREB and Elk-1, along with Fos production, in the rat hippocampus after a one-trial avoidance learning: abolition by NMDA receptor blockade. Brain Res. Mol. Brain Res. 76, 36–46.
Carlezon, W. A. Jr., Thome, J., Olson, V. G., Lane-Ladd, S. B., Brodkin, E. S., Hiroi, N., Duman, R. S., Neve, R. L., and Nestler, E. J. (1998). Regulation of cocaine reward by CREB. Science 282, 2272–2275.
Cavigelli, M., Dolfi, F., Claret, F. X., and Karin, M. (1995). Induction of c-fos expression through JNK-mediated TCF/Elk-1 phosphorylation. EMBO J. 14, 5957–5964.
Cesari, F., Brecht, S., Vintersten, K., Vuong, L. G., Hofmann, M., Klingel, K., Schnorr, J. J., Arsenian, S., Schild, H., Herdegen, T., Wiebel, F. F., and Nordheim, A. (2004). Mice deficient for the ets transcription factor elk-1 show normal immune responses and mildly impaired neuronal gene activation. Mol. Cell. Biol. 24, 294–305.
Choe, E. S., Chung, K. T., Mao, L., and Wang, J. Q. (2002). Amphetamine increases phosphorylation of extracellular signal-regulated kinase and transcription factors in the rat striatum via group I metabotropic glutamate receptors. Neuropsychopharmacology 27, 565–575.
Cohen-Armon, M., Visochek, L., Rozensal, D., Kalal, A., Geistrikh, I., Klein, R., Bendetz-Nezer, S., Yao, Z., and Seger, R. (2007). DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Mol. Cell 25, 297–308.
Cruzalegui, F. H., Cano, E., and Treisman, R. (1999). ERK activation induces phosphorylation of Elk-1 at multiple S/T-P motifs to high stoichiometry. Oncogene 18, 7948–7957.
Dalton, S., and Treisman, R. (1992). Characterization of SAP-1, a protein recruited by serum response factor to the c-fos serum response element. Cell 68, 597–612.
Dash, P. K., Orsi, S. A., and Moore, A. N. (2005). Sequestration of serum response factor in the hippocampus impairs long-term spatial memory. J. Neurochem. 93, 269–278.
Davis, S., Vanhoutte, P., Pages, C., Caboche, J., and Laroche, S. (2000). The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J. Neurosci. 20, 4563–4572.
Deak, M., Clifton, A. D., Lucocq, L. M., and Alessi, D. R. (1998). Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 17, 4426–4441.
De Cesare, D., Jacquot, S., Hanauer, A., and Sassone-Corsi, P. (1998). Rsk-2 activity is necessary for epidermal growth factor-induced phosphorylation of CREB protein and transcription of c-fos gene. Proc. Natl. Acad. Sci. U.S.A. 95, 12202–12207.
Demir, O., Korulu, S., Yildiz, A., Karabay, A., and Kurnaz, I. A. (2009). Elk-1 interacts with neuronal microtubules and relocalizes to the nucleus upon phosphorylation. Mol. Cell. Neurosci. 40, 111–119.
Dwivedi, Y., Rizavi, H. S., Roberts, R. C., Conley, R. C., Tamminga, C. A., and Pandey, G. N. (2001). Reduced activation and expression of ERK1/2 MAP kinase in the post-mortem brain of depressed suicide subjects. J. Neurochem. 77, 916–928.
Enslen, H., Raingeaud, J., and Davis, R. J. (1998). Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J. Biol. Chem. 273, 1741–1748.
Fantz, D. A., Jacobs, D., Glossip, D., and Kornfeld, K. (2001). Docking sites on substrate proteins direct extracellular signal-regulated kinase to phosphorylate specific residues. J. Biol. Chem. 276, 27256–27265.
Fuchs, S. Y., Xie, B., Adler, V., Fried, V. A., Davis, R. J., and Ronai, Z. (1997). c-Jun NH2-terminal kinases target the ubiquitination of their associated transcription factors. J. Biol. Chem. 272, 32163–32168.
Gardiner, K. (2003). Predicting pathway perturbations in Down syndrome. J. Neural Transm. Suppl. 67, 21–37.
Gardiner, K. (2006). Transcriptional dysregulation in Down syndrome: predictions for altered protein complex stoichiometries and post-translational modifications, and consequences for learning/behavior genes ELK, CREB, and the estrogen and glucocorticoid receptors. Behav. Genet. 36, 439–453.
Gille, H., Sharrocks, A. D., and Shaw, P. E. (1992). Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at c-fos promoter. Nature 358, 414–417.
Gille, H., Strahl, T., and Shaw, P. E. (1995). Activation of ternary complex factor Elk-1 by stress-activated protein kinases. Curr. Biol. 5, 1191–1200.
Giovane, A., Pintzas, A., Maira, S. M., Sobieszczuk, P., and Wasylyk, B. (1994). Net, a new ets transcription factor that is activated by Ras. Genes Dev. 8, 1502–1513.
Giovane, A., Sobieszczuk, P., Mignon, C., Mattei, M. G., and Wasylyk, B. (1995). Locations of the ets subfamily members net, elk1, and sap1 (ELK3, ELK1, and ELK4) on three homologous regions of the mouse and human genomes. Genomics 29, 769–772.
Grinkevich, L. N., Lisachev, P. D., Gudzik, K. A., Grinkevich, V. V., and Kharchenko, O. A. (2004). Comparative analysis of the activation of the Elk-1 transcription factor in the central nervous system of animals with different learning capacities. Dokl. Biol. Sci. 397, 269–271.
Herrera, R. E., Nordheim, A., and Stewart, A. F. (1997). Chromatin structure analysis of the human c-fos promoter reveals a centrally positioned nucleosome. Chromosoma 106, 284–292.
Herrera, R. E., Shaw, P. E., and Nordheim, A. (1989). Occupation of the c-fos serum response element in vivo by a multi-protein complex is unaltered by growth factor induction. Nature 340, 68–70.
Hill, C. S., Marais, R., John, S., Wynne, J., Dalton, S., and Treisman, R. (1993). Functional analysis of a growth factor-responsive transcription factor complex. Cell 73, 395–406.
Hipskind, R. A., Rao, V. N., Mueller, C. G., Reddy, E. S., and Nordheim, A. (1991). Ets-related protein Elk-1 is homologous to the c-fos regulatory factor p62TCF. Nature 354, 531–534.
Hollenhorst, P. C., Jones, D. A., and Graves, B. J. (2004). Expression profiles frame the promoter specificity dilemma of the ETS family of transcription factors. Nucleic Acids Res. 32, 5693–5702.
Hollenhorst, P. C., Shah, A. A., Hopkins, C., and Graves, B. J. (2007). Genome-wide analyses reveal properties of redundant and specific promoter occupancy within the ETS gene family. Genes Dev. 21, 1882–1894.
Iwata, A., Miura, S., Kanazawa, I., Sawada, M., and Nukina, N. (2001). alpha-Synuclein forms a complex with transcription factor Elk-1. J. Neurochem. 77, 239–252.
Jacobs, D., Glossip, D., Xing, H., Muslin, A. J., and Kornfeld, K. (1999). Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 13, 163–175.
Janknecht, R., Ernst, W. H., Pingoud, V., and Nordheim, A. (1993). Activation of ternary complex factor Elk-1 by MAP kinases. EMBO J. 12, 5097–5104.
Janknecht, R., and Nordheim, A. (1996). MAP kinase-dependent transcriptional coactivation by Elk-1 and its cofactor CBP. Biochem. Biophys. Res. Commun. 228, 831–837.
Janknecht, R., Zinck, R., Ernst, W. H., and Nordheim, A. (1994). Functional dissection of the transcription factor Elk-1. Oncogene 9, 1273–1278.
Jenab, S., Festa, E. D., Nazarian, A., Wu, H. B., Sun, W. L., Hazim, R., Russo, S. J., and Quinones-Jenab, V. (2005). Cocaine induction of ERK proteins in dorsal striatum of Fischer rats. Brain Res. Mol. Brain Res. 142, 134–138.
Jones, M. W., Errington, M. L., French, P. J., Fine, A., Bliss, T. V., Garel, S., Charnay, P., Bozon, B., Laroche, S., and Davis, S. (2001). A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat. Neurosci. 4, 289–296.
Kaminska, B., Kaczmarek, L., Zangenehpour, S., and Chaudhuri, A. (1999). Rapid phosphorylation of Elk-1 transcription factor and activation of MAP kinase signal transduction pathways in response to visual stimulation. Mol. Cell. Neurosci. 13, 405–414.
Kasza, A., O’Donnell, A., Gascoigne, K., Zeef, L. A., Hayes, A., and Sharrocks, A. D. (2005). The ETS domain transcription factor Elk-1 regulates the expression of its partner protein, SRF. J. Biol. Chem. 280, 1149–1155.
Knoll, B., Kretz, O., Fiedler, C., Alberti, S., Schutz, G., Frotscher, M., and Nordheim, A. (2006). Serum response factor controls neuronal circuit assembly in the hippocampus. Nat. Neurosci. 9, 195–204.
Knoll, B., and Nordheim, A. (2009). Functional versatility of transcription factors in the nervous system: the SRF paradigm. Trends Neurosci. 32, 432–442.
Laudet, V., Hanni, C., Stehelin, D., and Duterque-Coquillaud, M. (1999). Molecular phylogeny of the ETS gene family. Oncogene 18, 1351–1359.
Lavaur, J., Bernard, F., Trifilieff, P., Pascoli, V., Kappes, V., Pages, C., Vanhoutte, P., and Caboche, J. (2007). A TAT–DEF–Elk-1 peptide regulates the cytonuclear trafficking of Elk-1 and controls cytoskeleton dynamics. J. Neurosci. 27, 14448–14458.
Li, Q. J., Yang, S. H., Maeda, Y., Sladek, F. M., Sharrocks, A. D., and Martins-Green, M. (2003). MAP kinase phosphorylation-dependent activation of Elk-1 leads to activation of the co-activator p300. EMBO J. 22, 281–291.
Ligeza, A., Wawrzczak-Bargiela, A., Kaminska, D., Korostynski, M., and Przewlocki, R. (2008). Regulation of ERK1/2 phosphorylation by acute and chronic morphine – implications for the role of cAMP-responsive element binding factor (CREB)-dependent and Ets-like protein-1 (Elk-1)-dependent transcription; small interfering RNA-based strategy. FEBS J. 275, 3836–3849.
Lindecke, A., Korte, M., Zagrebelsky, M., Horejschi, V., Elvers, M., Widera, D., Prullage, M., Pfeiffer, J., Kaltschmidt, B., and Kaltschmidt, C. (2006). Long-term depression activates transcription of immediate early transcription factor genes: involvement of serum response factor/Elk-1. Eur. J. Neurosci. 24, 555–563.
Ling, Y., West, A. G., Roberts, E. C., Lakey, J. H., and Sharrocks, A. D. (1998). Interaction of transcription factors with serum response factor. Identification of the Elk-1 binding surface. J. Biol. Chem. 273, 10506–10514.
Lopez, M., Oettgen, P., Akbarali, Y., Dendorfer, U., and Libermann, T. A. (1994). ERP, a new member of the ets transcription factor/oncoprotein family: cloning, characterization, and differential expression during B-lymphocyte development. Mol. Cell. Biol. 14, 3292–3309.
Lu, L., Koya, E., Zhai, H., Hope, B. T., and Shaham, Y. (2006). Role of ERK in cocaine addiction. Trends Neurosci. 29, 695–703.
Marais, R., Wynne, J., and Treisman, R. (1993). The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell 73, 381–393.
Miller, C. A., and Marshall, J. F. (2005). Molecular substrates for retrieval and reconsolidation of cocaine-associated contextual memory. Neuron 47, 873–884.
Mizoguchi, H., Yamada, K., Mizuno, M., Mizuno, T., Nitta, A., Noda, Y., and Nabeshima, T. (2004). Regulations of methamphetamine reward by extracellular signal-regulated kinase 1/2/ets-like gene-1 signaling pathway via the activation of dopamine receptors. Mol. Pharmacol. 65, 1293–1301.
Nestler, E. J. (2004). Molecular mechanisms of drug addiction. Neuropharmacology 47(Suppl. 1), 24–32.
Nissen, L. J., Gelly, J. C., and Hipskind, R. A. (2001). Induction-independent recruitment of CREB-binding protein to the c-fos serum response element through interactions between the bromodomain and Elk-1. J. Biol. Chem. 276, 5213–5221.
Nowak, S. J., and Corces, V. G. (2004). Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet. 20, 214–220.
Nozaki, M., Onishi, Y., Kanno, N., Ono, Y., and Fujimura, Y. (1996). Molecular cloning of Elk-3, a new member of the Ets family expressed during mouse embryogenesis and analysis of its transcriptional repression activity. DNA Cell Biol. 15, 855–862.
Nuutinen, S., Barik, J., Jones, I. W., and Wonnacott, S. (2007). Differential effects of acute and chronic nicotine on Elk-1 in rat hippocampus. Neuroreport 18, 121–126.
O’Donnell, A., Yang, S. H., and Sharrocks, A. D. (2008). MAP kinase-mediated c-fos regulation relies on a histone acetylation relay switch. Mol. Cell 29, 780–785.
Pandey, S. C., Zhang, H., Ugale, R., Prakash, A., Xu, T., and Misra, K. (2008). Effector immediate-early gene arc in the amygdala plays a critical role in alcoholism. J. Neurosci. 28, 2589–2600.
Pastorcic, M., and Das, H. K. (2003). Ets transcription factors ER81 and Elk1 regulate the transcription of the human presenilin 1 gene promoter. Brain Res. Mol. Brain Res. 113, 57–66.
Pingoud, V., Zinck, R., Hipskind, R. A., Janknecht, R., and Nordheim, A. (1994). Heterogeneity of ternary complex factors in HeLa cell nuclear extracts. J. Biol. Chem. 269, 23310–23317.
Price, M. A., Rogers, A. E., and Treisman, R. (1995). Comparative analysis of the ternary complex factors Elk-1, SAP-1a and SAP-2 (ERP/NET). EMBO J. 14, 2589–2601.
Rao, V. N., Huebner, K., Isobe, M., ar-Rushdi, A., Croce, C. M., and Reddy, E. S. (1989). elk, tissue-specific ets-related genes on chromosomes X and 14 near translocation breakpoints. Science 244, 66–70.
Rao, V. N., and Reddy, E. S. (1993). Delta elk-1, a variant of elk-1, fails to interact with the serum response factor and binds to DNA with modulated specificity. Cancer Res. 53, 215–220.
Roze, E., Betuing, S., Deyts, C., Marcon, E., Brami-Cherrier, K., Pages, C., Humbert, S., Merienne, K., and Caboche, J. (2008). Mitogen- and stress-activated protein kinase-1 deficiency is involved in expanded-huntingtin-induced transcriptional dysregulation and striatal death. FASEB J. 22, 1083–1093.
Salinas, S., Briancon-Marjollet, A., Bossis, G., Lopez, M. A., Piechaczyk, M., Jariel-Encontre, I., Debant, A., and Hipskind, R. A. (2004). SUMOylation regulates nucleo-cytoplasmic shuttling of Elk-1. J. Cell Biol. 165, 767–773.
Sananbenesi, F., Fischer, A., Schrick, C., Spiess, J., and Radulovic, J. (2002). Phosphorylation of hippocampal Erk-1/2, Elk-1, and p90-Rsk-1 during contextual fear conditioning: interactions between Erk-1/2 and Elk-1. Mol. Cell. Neurosci. 21, 463–476.
Schwarze, S. R., Ho, A., Vocero-Akbani, A., and Dowdy, S. F. (1999). In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285, 1569–1572.
Sgambato, V., Pages, C., Rogard, M., Besson, M. J., and Caboche, J. (1998a). Extracellular signal-regulated kinase (ERK) controls immediate early gene induction on corticostriatal stimulation. J. Neurosci. 18, 8814–8825.
Sgambato, V., Vanhoutte, P., Pages, C., Rogard, M., Hipskind, R., Besson, M. J., and Caboche, J. (1998b). In vivo expression and regulation of Elk-1, a target of the extracellular-regulated kinase signaling pathway, in the adult rat brain. J. Neurosci. 18, 214–226.
Sharma, A., Callahan, L. M., Sul, J. Y., Kim, T. K., Barrett, L., Kim, M., Powers, J. M., Federoff, H., and Eberwine, J. A. (2010). Neurotoxic phosphoform of Elk-1 associates with inclusions from multiple neurodegenerative diseases. PLoS ONE 5, e9002. doi: 10.1371/journal.pone.0009002
Sharrocks, A. D. (2001). The ETS-domain transcription factor family. Nat. Rev. Mol. Cell Biol. 2, 827–837.
Sharrocks, A. D., Yang, S. H., and Galanis, A. (2000). Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem. Sci. 25, 448–453.
Shaw, P. E., Schroter, H., and Nordheim, A. (1989). The ability of a ternary complex to form over the serum response element correlates with serum inducibility of the human c-fos promoter. Cell 56, 563–572.
Soloaga, A., Thomson, S., Wiggin, G. R., Rampersaud, N., Dyson, M. H., Hazzalin, C. A., Mahadevan, L. C., and Arthur, J. S. (2003). MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 22, 2788–2797.
Sugimoto, T., Stewart, S., and Guan, K. L. (1997). The calcium/calmodulin-dependent protein phosphatase calcineurin is the major Elk-1 phosphatase. J. Biol. Chem. 272, 29415–29418.
Sun, W. L., Zhou, L., Hazim, R., Quinones-Jenab, V., and Jenab, S. (2007). Effects of acute cocaine on ERK and DARPP-32 phosphorylation pathways in the caudate-putamen of Fischer rats. Brain Res. 1178, 12–19.
Sweatt, J. D. (2004). Mitogen-activated protein kinases in synaptic plasticity and memory. Curr. Opin. Neurobiol. 14, 311–317.
Thiels, E., Kanterewicz, B. I., Norman, E. D., Trzaskos, J. M., and Klann, E. (2002). Long-term depression in the adult hippocampus in vivo involves activation of extracellular signal-regulated kinase and phosphorylation of Elk-1. J. Neurosci. 22, 2054–2062.
Thomas, G. M., and Huganir, R. L. (2004). MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183.
Tian, J., and Karin, M. (1999). Stimulation of Elk1 transcriptional activity by mitogen-activated protein kinases is negatively regulated by protein phosphatase 2B (calcineurin). J. Biol. Chem. 274, 15173–15180.
Tong, L., Balazs, R., Thornton, P. L., and Cotman, C. W. (2004). Beta-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J. Neurosci. 24, 6799–6809.
Treisman, R., Marais, R., and Wynne, J. (1992). Spatial flexibility in ternary complexes between SRF and its accessory proteins. EMBO J. 11, 4631–4640.
Trifilieff, P., Lavaur, J., Pascoli, V., Kappes, V., Brami-Cherrier, K., Pages, C., Micheau, J., Caboche, J., and Vanhoutte, P. (2009). Endocytosis controls glutamate-induced nuclear accumulation of ERK. Mol. Cell. Neurosci. 41, 325–336.
Tyan, S. W., Tsai, M. C., Lin, C. L., Ma, Y. L., and Lee, E. H. (2008). Serum- and glucocorticoid-inducible kinase 1 enhances zif268 expression through the mediation of SRF and CREB1 associated with spatial memory formation. J. Neurochem. 105, 820–832.
Valjent, E., Caboche, J., and Vanhoutte, P. (2001a). Mitogen-activated protein kinase/extracellular signal-regulated kinase induced gene regulation in brain: a molecular substrate for learning and memory? Mol. Neurobiol. 23, 83–99.
Valjent, E., Pages, C., Rogard, M., Besson, M. J., Maldonado, R., and Caboche, J. (2001b). Delta 9-tetrahydrocannabinol-induced MAPK/ERK and Elk-1 activation in vivo depends on dopaminergic transmission. Eur. J. Neurosci. 14, 342–352.
Valjent, E., Corvol, J. C., Pages, C., Besson, M. J., Maldonado, R., and Caboche, J. (2000). Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J. Neurosci. 20, 8701–8709.
Valjent, E., Corvol, J. C., Trzaskos, J. M., Girault, J. A., and Herve, D. (2006). Role of the ERK pathway in psychostimulant-induced locomotor sensitization. BMC Neurosci. 7, 20. doi: 10.1186/1471-2202-7-20
Valjent, E., Pages, C., Herve, D., Girault, J. A., and Caboche, J. (2004). Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur. J. Neurosci. 19, 1826–1836.
Valjent, E., Pascoli, V., Svenningsson, P., Paul, S., Enslen, H., Corvol, J. C., Stipanovich, A., Caboche, J., Lombroso, P. J., Nairn, A. C., Greengard, P., Herve, D., and Girault, J. A. (2005). Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc. Natl. Acad. Sci. U.S.A. 102, 491–496.
Vanhoutte, P., Barnier, J. V., Guibert, B., Pages, C., Besson, M. J., Hipskind, R. A., and Caboche, J. (1999). Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol. Cell. Biol. 19, 136–146.
Vanhoutte, P., Nissen, J. L., Brugg, B., Gaspera, B. D., Besson, M. J., Hipskind, R. A., and Caboche, J. (2001). Opposing roles of Elk-1 and its brain-specific isoform, short Elk-1, in nerve growth factor-induced PC12 differentiation. J. Biol. Chem. 276, 5189–5196.
White, R. J., and Sharrocks, A. D. (2010). Coordinated control of the gene expression machinery. Trends Genet. 26, 214–220.
Whitmarsh, A. J., Shore, P., Sharrocks, A. D., and Davis, R. J. (1995). Integration of MAP kinase signal transduction pathways at the serum response element. Science 269, 403–407.
Xing, J., Kornhauser, J. M., Xia, Z., Thiele, E. A., and Greenberg, M. E. (1998). Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol. Cell. Biol. 18, 1946–1955.
Yang, S. H., Bumpass, D. C., Perkins, N. D., and Sharrocks, A. D. (2002). The ETS domain transcription factor Elk-1 contains a novel class of repression domain. Mol. Cell. Biol. 22, 5036–5046.
Yang, S. H., Jaffray, E., Hay, R. T., and Sharrocks, A. D. (2003). Dynamic interplay of the SUMO and ERK pathways in regulating Elk-1 transcriptional activity. Mol. Cell 12, 63–74.
Yang, S. H., and Sharrocks, A. D. (2004). SUMO promotes HDAC-mediated transcriptional repression. Mol. Cell 13, 611–617.
Yang, S. H., and Sharrocks, A. D. (2005). PIASx acts as an Elk-1 coactivator by facilitating derepression. EMBO J. 24, 2161–2171.
Yang, S. H., and Sharrocks, A. D. (2006). PIASxalpha differentially regulates the amplitudes of transcriptional responses following activation of the ERK and p38 MAPK pathways. Mol. Cell 22, 477–487.
Yang, S. H., Shore, P., Willingham, N., Lakey, J. H., and Sharrocks, A. D. (1999). The mechanism of phosphorylation-inducible activation of the ETS-domain transcription factor Elk-1. EMBO J. 18, 5666–5674.
Yang, S. H., Vickers, E., Brehm, A., Kouzarides, T., and Sharrocks, A. D. (2001). Temporal recruitment of mSin3A-histone deacetylase corepressor complex to the ETS domain transcription factor Elk-1. Mol. Cell. Biol. 21, 2802–2814.
Keywords: ERK signaling, gene regulation, chromatin remodeling, brain plasticity, memory formation, long-term neuronal adaptation
Citation: Besnard A, Galan-Rodriguez B, Vanhoutte P and Caboche J (2011) Elk-1 a transcription factor with multiple facets in the brain. Front. Neurosci. 5:35. doi: 10.3389/fnins.2011.00035
Received: 30 December 2010;
Paper pending published: 22 January 2011;
Accepted: 04 March 2011;
Published online: 16 March 2011.
Edited by:
Shannon K. Mcweeney, Oregon Clinical and Translational Research Institute, USAReviewed by:
Claudio V. Mello, Oregon Health and Science University, USAHans P. Herzel, Humboldt University, Germany
Copyright: © 2011 Besnard, Galan-Rodriguez, Vanhoutte and Caboche. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Jocelyne Caboche and Peter Vanhoutte, Laboratoire de Physiopathologie des Maladies du Système Nerveux Central, UMR CNRS-7224 CNRS et UMRS-INSERM 952, Université Pierre et Marie Curie-Paris 6, Paris, France. e-mail: jocelyne.caboche@snv.jussieu.fr; peter.vanhoutte@snv.jussieu.fr