
- 1 Department of Biological Sciences, Carnegie Mellon University, Pittsburgh, PA, USA
- 2 Center for Neural Basis of Cognition, Pittsburgh, PA, USA
- 3 Department of Neuroscience, University of Pittsburgh, Pittsburgh, PA, USA
Adult-born neurons (ABNs) are added to the olfactory bulb (OB) throughout life in rodents. While many factors have been identified as regulating the survival and integration of ABNs into existing circuitry, the understanding of how these factors affect ABN morphology and connectivity is limited. Here we compare how cell intrinsic [small interfering RNA (siRNA) knock-down of voltage gated sodium channels NaV1.1–1.3] and circuit level (naris occlusion) reductions in activity affect ABN morphology during integration into the OB. We found that both manipulations reduce the number of dendritic spines (and thus likely the number of reciprocal synaptic connections) formed with the surrounding circuitry and inhibited dendritic ramification of ABNs. Further, we identified regions of ABN apical dendrites where the largest and most significant decreases occur following siRNA knock-down or naris occlusion. In siRNA knock-down cells, reduction of spines is observed in proximal regions of the apical dendrite. This suggests that distal regions of the dendrite may remain active independent of NaV1.1–1.3 channel expression, perhaps facilitated by activation of T-type calcium channels and NMDA receptors. By contrast, circuit level reduction of activity by naris occlusion resulted in a global depression of spine number. Together, these results indicate that ABNs retain the ability to develop their typical overall morphological features regardless of experienced activity, and activity modulates the number and location of formed connections.
Introduction
Adult neurogenesis contributes adult-born neurons (ABNs) to the mouse olfactory bulb (OB) throughout life (Altman, 1969). These neurons, generated in the subventricular zone (SVZ), migrate to the OB, and upon arrival become mature neurons of several different types (Bagley et al., 2007). A small fraction of these ABNs mature to become periglomerular neurons, but the majority of ABNs become local circuit interneurons morphologically similar to neonatal granule cells (Carleton et al., 2003; Saghatelyan et al., 2005; Ninkovic et al., 2007). Following their migration, many factors influence the survival of ABNs in the olfactory system (Alonso et al., 2006; Lazarini and Lledo, 2010). Manipulations that reduce activity – naris occlusion (Corotto et al., 1994; Cummings and Brunjes, 1997), benzodiazepine treatment (Yamaguchi and Mori, 2005), over-expression of potassium channels (Kelsch et al., 2009) or axonomy of olfactory receptor neurons (Mandairon et al., 2003) – decrease ABN survival and integration. Conversely, manipulations thought to increase olfactory activity – enrichment of the olfactory environment (Rochefort et al., 2002), odor discrimination training (Alonso et al., 2006; Moreno et al., 2009), or increased excitability (by viral expression of a bacterial sodium channel; Kelsch et al., 2009) – have proven to increase rates of ABN survival and thus integration. While our understanding of the regulation of ABN survival by activity has advanced substantially, how these manipulations affect ABN morphology and connectivity is poorly understood (Saghatelyan et al., 2005; Kelsch et al., 2009). Examination of ABN morphology will provide insight into the number and location of connections established by ABNs during their integration into mature OB circuitry. Information about these connections will, in turn, indicate how much input ABNs are receiving from the existing network, while also revealing the influence these new cells may have on network activity.
Here we compare how two kinds of activity reduction affect the ability of ABNs to integrate into existing circuitry. Specifically, we asked how reducing either cell intrinsic or network-level activity influenced the morphology and connectivity of ABNs, as assessed by measures of EGFP-expressing adult-born granule cell morphology. We observed that both modes of activity reduction (intrinsic and network-level) caused reductions in the number of spines formed by ABNs. These changes were widely distributed across regions of the apical dendritic tree, unlike what was observed in a previous report in which activity was reduced by naris occlusion (Kelsch et al., 2009). Additionally we observed subtle changes in the morphologies of the apical dendrites of activity-reduced ABNs, evidenced by reductions in branching and apical dendrite complexity. These results suggest that activity-reduced ABNs may be less competent to receive synaptic input and thus integrate poorly when activity is low.
Materials and Methods
Stereotaxic Lentivirus Injections
All animal protocols were carried out in accordance with the Carnegie Mellon University Institutional Animal Care and Use Committee guidelines. One of two lentiviral constructs, developed and characterized previously (Komai et al., 2006), were injected into 2-month-old C57B/L6 mice (Taconic Farms). The first viral vector contained a ubiquitin promoter followed by EGFP the second viral vector contained a tandem of U6 promoters, each expressing one shRNA, followed by the ubiquitin promoter expressing EGFP (see Figure 1A). Lentiviral injections were be performed as previously described (Dittgen et al., 2004; Cetin et al., 2006). Briefly, mice were anesthetized by IP injection of a mixture of ketamine (100 mg/kg) and xylazine (10 mg/kg) in water. Animals were kept deeply anesthetized as assessed by monitoring eyelid reflex, vibrissae movements, and pinch withdrawal. Body temperature was maintained at 37°C with a heating pad with digitally monitored feedback control. Two craniotomies 300–500 μm in diameter were drilled above the sub ventricular zone (final coordinates relative to Bregma: anterior −1 mm, lateral ±1 mm, depth 2.2 mm beneath the pia mater). Pulled glass quartz pipettes were broken to an outer diameter of ≈30 μm and inserted at a 27° angle caudal–rostral into the pia. 500 nL of lentivirus was injected slowly over ≈5 min.
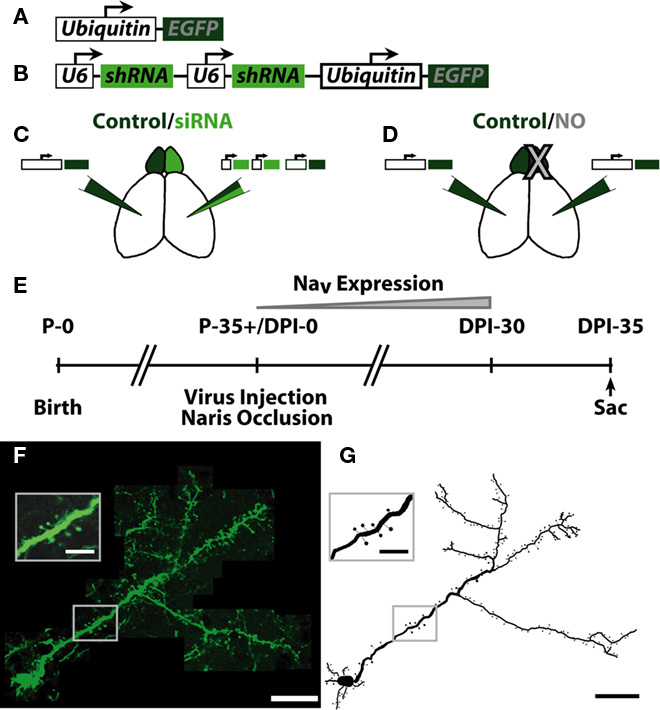
Figure 1. Summary of experimental conditions and injection paradigm. (A) Schematic of EGFP coding lentivirus. EGFP was expressed downstream of the ubiquitin promoter. (B) Schematic of siRNA + EGFP coding lentivirus. Two U6 promoters, each expressing one shRNA against voltage gated sodium channel 1.1–1.3 (NaV1.1–1.3) expression were followed by the ubiquitin promoter expressing EGFP. Each group received the following manipulations: (C) – Control EGFP lentivirus [Control, see (A)] in one hemisphere, siRNA sodium channel knock-down lentivirus [siRNA, see (B)] opposite hemisphere. (D) – Control EGFP lentivirus in both hemispheres, naris occlusion (NO) in one hemisphere. (E) Experimental timeframe. Mice were allowed to survive for 35 days post-injection (DPI)/naris occlusion before sacrifice, allowing for expression of NaV in progenitor cells born the day of injection (DPI-0). (F) Example neuron from individual slices of an image stack. Scale bar, 100 μm. Inset shows resolution of apical dendrite spines. Scale bar, 25 μm. (G) Example ABN reconstruction from images in (F). Scale bar, 100 μm. Inset shows reconstruction of inset in (F). Scale bar, 25 μm.
Unilateral Olfactory Deprivation
Mice were anesthetized by IP injection of a mixture of ketamine (100 mg/kg) and xylazine (10 mg/kg) in water. After animals became non-responsive as monitored by tail pinch their external naris was closed by cauterization. A cautery tool (Fine Science Tools), consisting of a fine loop of platinum wire that is rapidly heated by passage of electrical current, was placed against the nostril until the skin flap that covers the naris was “melted” onto the inner surface. Immediately following the cautery, the area was swabbed with antibacterial/anesthetic gel and the animal was returned to a recovery cage where it was monitored until recovery over the following 48 h. Extent of naris closure was assessed when animals were sacrificed. Only tissue from animals in which the occluded naris remained completely closed was used in immunohistochemistry and analysis.
Immunohistochemistry
Mice were anesthetized as above and perfused transcardially with cold 1% NaCl in 0.1 M phosphate buffer (PB) followed by cold 4% paraformaldehyde in 0.1 M PB. The brains were dissected and fixed in 4% paraformaldehyde in PB overnight at 4°C and then sunk in 30% sucrose at 4°C for 24 h. Each hemisphere of the brain was sliced coronally at 300 μm using an SM2000R cryostat (Leica Microsystems). Incubations were done at room temperature with oscillation. Sections were first incubated in 2% Triton X-100 (Sigma) and 2% normal donkey serum (NDS; Jackson Immuno Research Laboratories) in PB for 1 h. Rabbit anti-GFP antibody (1:1000, Invitrogen) was added in the presence of 2% NDS and 0.1% Triton X-100 in PB and incubated for 1 h. The secondary antibody added was a conjugated Alexa Fluor 488 (1:600, Invitrogen), incubated for 1 h in 2% NDS and 0.1% Triton X-100 in PB. After the addition of the secondary antibody all incubations were performed in the dark. Sections were washed three times between each incubation step in PB for 5 min.
Imaging Procedures
Sections were imaged in their entirety using a Carl Zeiss LSM 510 Meta NLO multi-photon microscope (Carl Zeiss). Excitation of EGFP was provided by a Chameleon Ultra-II femtosecond laser (Coherent) operating at 890 nm. Non-descanned detection was provided by two silicon photomultiplier tubes situated on the back of the fluorescence light train of the Axio Observer Z1 inverted microscope stand. A long-pass 680 nm blocking filter was placed in front of the detection assembly to prevent any stray two-photon excitation. An individual band pass barrier filter (505–550 nm) was placed in front of the detector for fluorescence emission selection. All images were produced using a 40× 1.3 NA Neofluar air objective (Carl Zeiss). Three hundred micrometer thick sections from the first coronal slice directly in front of the accessory OB were imaged in their entirety through the Z-plane (see gray bar in Figure 1A). Excitation and gain were set to maximize the resolution of dendritic spines in the center of the Z direction. Z-steps for each section were set at 0.8 μm.
Granule Cell Reconstructions
Granule cells to be traced were chosen at random from the center 30% of the Z-plane of each slice. The volume used for this randomization was a series of image stacks from the lateral to medial edge of the OB. Image stacks were stitched together with the Neurolucida software package (MBF Bioscience). Cell bodies within the center 30% of the Z direction were then numbered and a random number list was generated based on the total number of cell bodies in the volume. The first five granule cells, corresponding to the order generated by the random number list, were then traced that met the following criteria were traced: (1) Cell body located in the granule cell layer, between the internal plexiform layer and the subependymal zone; (2) Dendritic tree completely contained within the slice; (3) Cells sufficiently bright under fluorescence to allow for tracing of all dendritic spines and fine dendritic processes. Cells not meeting all criteria were skipped and replaced by the following randomly numbered cell in the sequence. On average, the first 21.36 (±11.07) neurons from the random number list provided five cells fulfilling all criteria for tracing.
Statistical Analysis of Traced Cells
All slides and images were coded and the analysis was performed with the experimenter blinded to the conditions of the animal and individual brain hemispheres during analysis. Cells were analyzed using the Neurolucida Explorer software package (MBF Bioscience) quantifying aspects of the apical dendrite, such as the location, number, and branching order of spines and dendritic segments, as well as the location of the various lamina of the bulb. Further analysis was done using both Neurolucida Explorer and custom functions written in Igor Pro (Wavemetrics). A Wilcoxon Rank-Sum test followed by a Bonferroni correction for multiple measures was used for all statistical tests.
Results
EGFP-Encoding Lentivirus Labels Adult-Born Granule Cells
To visualize ABNs in the OB, we injected lentiviruses encoding EGFP into the SVZ of adult mice. This virus infected and labeled neural progenitors, which migrate along the rostral migratory stream (RMS) to the OB and integrate as inhibitory granule cells. To measure the effects of manipulating activity on ABN morphology, we considered two groups of animals (Figures 1C,D). The first group [control/small interfering RNA (siRNA), 4 animals, 20 cells; Figure 1C] received an EGFP-encoding lentivirus in one hemisphere (Figure 1A), and in the opposite hemisphere a lentivirus encoding for EGFP and a siRNA (Figure 1B) that knocked down sodium channel expression (Komai et al., 2006). These animals were otherwise unmanipulated (no naris occlusion). The second group (control/NO, 3 animals, 21 cells; Figure 1D) received injections of a lentivirus encoding EGFP only in both hemispheres followed by occlusion of one external naris on the same day (NO). Animals were allowed to survive for 5 weeks post-virus injection – during which expression of sodium channels increases substantially in control cells (Carleton et al., 2003) – before sacrifice and perfusion (Figure 1E). Following perfusion, thick sections (300 μm) were cut and completely labeled ABNs were imaged in their entirety throughout the depth of the section (Figure 1F). Manual 3-D cellular reconstructions were performed from the acquired image stacks (Figures 1G and 2).
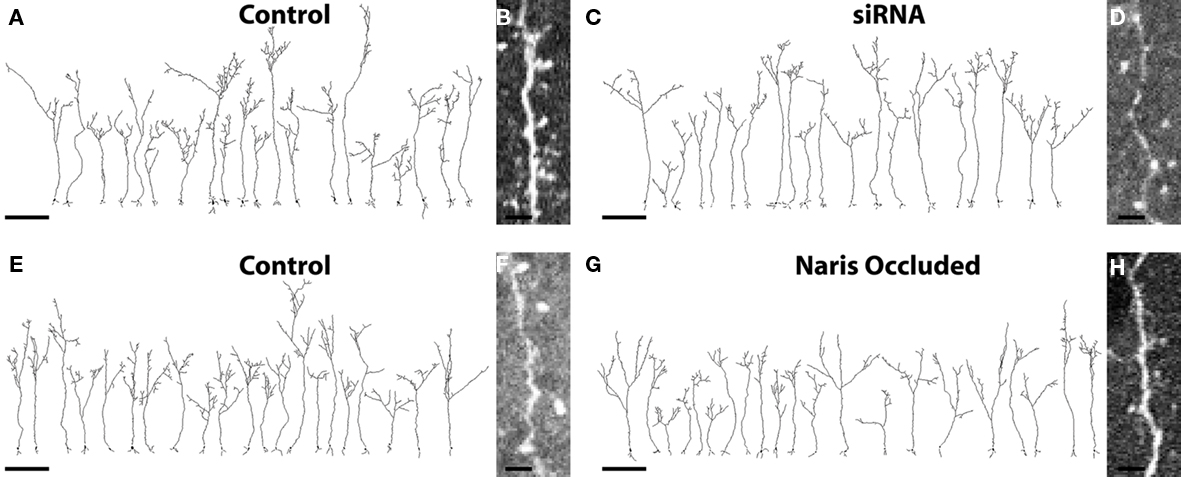
Figure 2. Complete cellular reconstructions of randomly selected EGFP+ ABNs. (A,C) Group 1 cells; four animals, five cells per hemisphere, n = 20 per condition. Scale bar, 100 μm. (B,D) Raw image of dendrite and spines taken from image stacks of control and siRNA cells respectively. Scale bar, 5 μm. (E,G) Group 2 cells; three animals, seven cells per hemisphere, n = 21 per condition. Scale bar, 100 μm. (F,H) Raw image of dendrite and spines taken from image stacks of control and naris-occluded cells respectively. Scale bar, 5 μm.
Reductions in Spine Number
Upon arrival in the OB, ABNs mature into olfactory granule cells (Saghatelyan et al., 2005). Olfactory granule cells are inhibitory interneurons which form dendrodendritic connections with olfactory mitral and tufted cells (Rall et al., 1966) through the formation of dendritic spines (Price and Powell, 1970). From our cellular reconstructions we quantified the number of spines on apical dendrites of mature ABNs to measure the degree to which these cells were able to integrate into mature OB circuitry. Genetic reduction of cell intrinsic activity (siRNA knock-down of sodium channels) led to significant reductions in the total number of apical dendritic spines formed by integrating ABNs (control: 139.8 ± 11.7; siRNA: 106.5 ± 8.4; p = 0.03; n = 20; Figure 3A). Network-level reduction in activity (NO) resulted in a similar decrement in total spine number (control: 122.9 ± 8.0; NO: 92.9 ± 5.9; p = 0.003; n = 21; Figure 3A). The two control groups were not significantly different (p > 0.31).
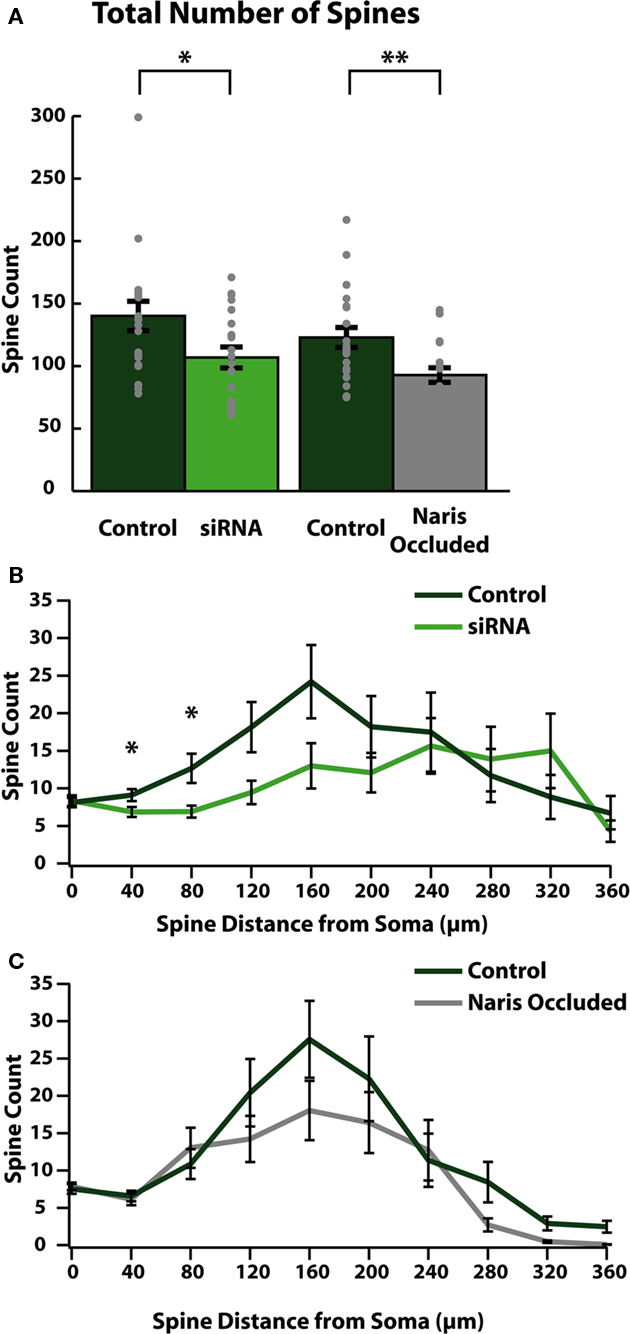
Figure 3. Reducing activity reduces the number of functional synapses formed in ABNs. (A) Total spines per cell (gray dots) and group means (bars with SE) for reconstructed ABNs. Both siRNA knock-down and naris occlusion showed significant reductions in total numbers of spines compared to controls (control/siRNA: p = 0.031, n = 20; control/NO: p = 0.003, Wilcoxon Rank-Sum test, n = 21) in the number of formed synapses. (B,C) Distribution of spines plotted as total number of spines per 40 μm bin vs. distance along apical dendrite path with SE. Significant differences occurred in early segments (40–80 μm) of apical dendrites in siRNA knock-down cells (B); (p < 0.05 and p < 0.05 respectively, n = 20).
We also determined the location along the dendrite where these reductions occurred by plotting the number of spines (spine count) vs. the distance from the soma along the dendritic tree (Figures 3B,C). In siRNA knock-down animals (Figure 3B) we saw significant differences in spine count between control and siRNA knock-down cells beginning as little as 40 μm from the soma (at 40 μm: control: 9.1 ± 0.8 spines/bin, siRNA: 6.9 ± 0.06 spines/bin; p < 0.05; at 80 μm: control: 12.7 ± 1.9 spines/bin, siRNA: 6.9 ± 0.8 spines/bin; p < 0.05; n = 20 cells). Proceeding distally, in siRNA knock-down cells this significant reduction was followed by continued sparseness, although spine counts were similar to control cells in distal segments. Conversely in NO cells, reductions in spine count did not localize to any specific region of the dendrite.
We next evaluated whether changes in dendrite morphology could explain the observed decreases in spine count by comparing differences in average spine densities per unit of dendritic length between control and activity-reduced cells (Figure 4A). We also plotted the distribution of spine density in spines/μm of dendrite along the dendritic tree for both control and activity-reduced cells (Figures 4B,C). This normalization controls for changes in spine density due to changes in dendritic arborization. Comparison of spine density again revealed a small but significant reduction by siRNA knock-down (control: 0.25 ± 0.01 spines/μm; siRNA: 0.21 ± 0.01 spines/μm; p < 0.001; n = 20) and NO (control: 0.23 ± 0.01 spines/μm; NO: 0.21 ± 0.01 spines/μm respectively; p = 0.039; n = 21; Figure 4A). In cells from control/siRNA animals, the spine density distribution shows that this reduction is observed throughout the first 200 μm of the somato-dendritic axis (Figure 4B), with large differences in spine density in proximal (at 40 μm from soma: control: 0.23 ± 0.02 spines/μm; siRNA: 0.16 ± 0.01 spines/μm, p < 0.001; at 80 μm from soma: control: 0.28 ± 0.04 spines/μm, siRNA: 0.16 ± 0.02 spines/μm, p < 0.01; n = 20) regions of the dendrite. There was no significant difference in spine density in distal dendritic regions of siRNA cells (Figure 4B). In control/NO animals, differences in spine density distribution (Figure 4C) were small and not significant.
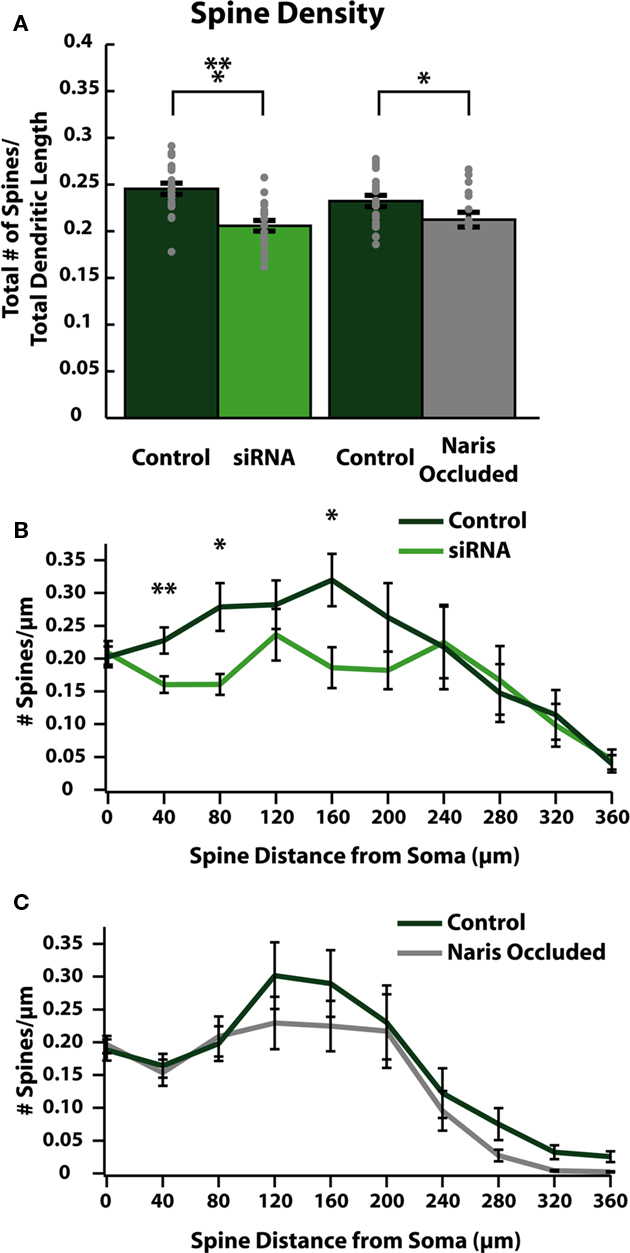
Figure 4. Reducing activity decreases spine densities in specific regions of ABNs. (A) Spine density per cell (gray dots) and group means (bars with SE). Spine density calculated as total spines per cell divided by total apical dendrite length. Both siRNA knock-down and naris-occluded cells show significant overall reductions in spine (control/siRNA: p < 0.001, n = 20; control/NO: p = 0.039, n = 21), without significant difference in total apical dendritic length (see Tables 1 and 2). (B,C) Spine density plotted as total number of spines divided by total apical dendrite length in 40 μm bins vs. distance along apical dendrite path with SE. siRNA knock-down cells (B) show significant decreases in spine density in proximal and intermediate regions of the dendrite (at 40 μm: p < 0.01; at 80 μm: p < 0.05; at 160 μm: p < 0.05; n = 20). No significant differences were observed in naris-occluded cells (C).
Activity Deprivation Causes Reduced Dendritic Branching
To understand how ABNs integrating into the mature circuit might sample input from potential presynaptic partners, we compared how each of the two types of activity reduction affect the morphology and complexity of the apical dendritic trees of integrating ABNs independent of spine distribution (Figure 5). Extensive and elaborate ramification of dendritic trees into zones of potential synaptic input would suggest successful integration of ABNs. Initial comparisons of total dendritic length, total number of branches, average terminal branch distance, and average terminal branch order showed no significant differences in either group (Tables 1 and 2), though in both experimental groups across these measures there was a trend toward reduced dendritic complexity in the activity-reduced groups. While these measures of overall dendritic complexity did not vary significantly, we wanted to carefully investigate the possibility of morphological changes specific to those regions where we saw changes in spine count and spine density.
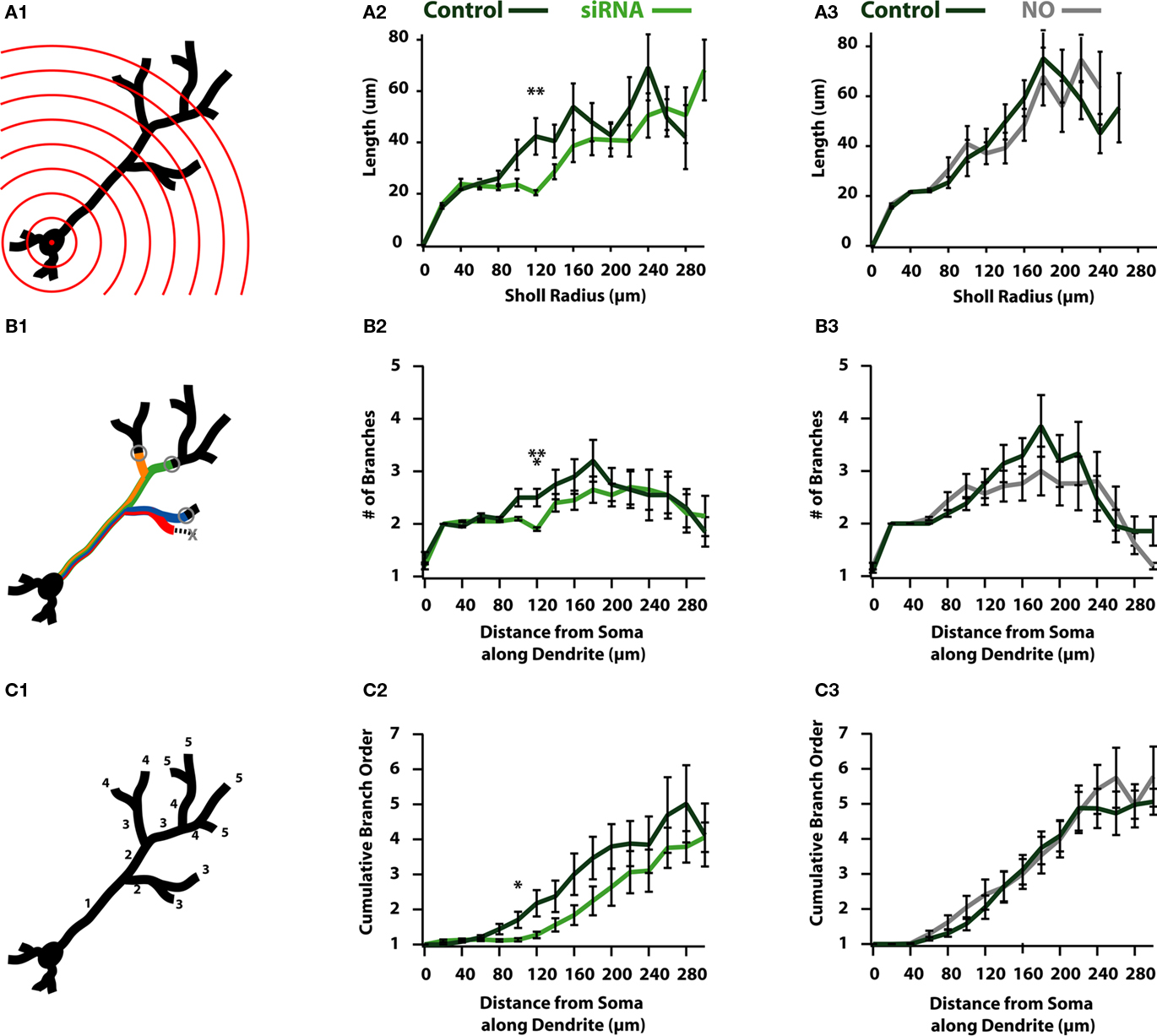
Figure 5. Reductions in activity reduce the overall complexity of newborn ABNs. (A1) Classical Sholl analysis with radii increasing in 20 μm increments. Sholl analysis measures apical dendrite length within each sphere plotted against radius from soma. (A2) siRNA knock-down cells show a significant reduction in length in proximal-intermediate distances from soma (at 120 μm: p < 0.01, n = 20). (A3) NO cells show no significant differences in Sholl analysis. (B1) Modified Sholl analysis of branch intersections. Modified Sholl analysis represents the number of branches at a fixed distance along the apical dendrite (not counting terminated branches). (B2) siRNA knock-down cells show a significant reduction in the number of branches at proximal-intermediate distances from soma (at 120 μm: p < 0.001, n = 20). (B3) NO cells show no significant differences in modified Sholl analysis. (C1) Cumulative branch order along length of apical dendrite. Plotted as the cumulative branch order (where branch order increments by one following bifurcation) vs. distance from soma along apical dendrite length. (C2) siRNA knock-down cells show a significant reduction in cumulative branch order at proximal-intermediate distance from soma (at 100 μm: p < 0.05, n = 20). (C3) NO cells show no differences in cumulative branch order.
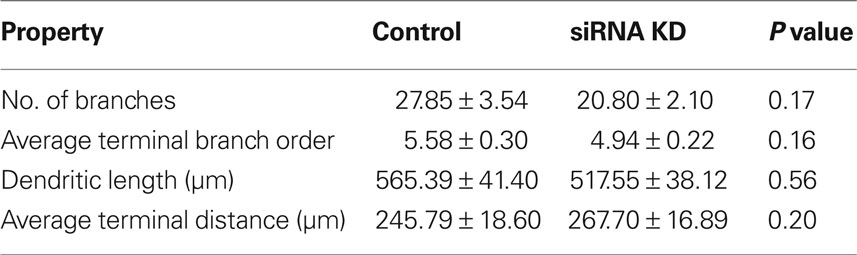
Table 1. Apical dendrite properties – control vs. siRNA knock-down.
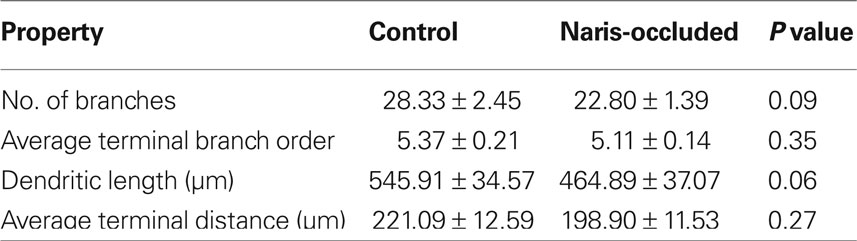
Table 2. Apical dendrite properties – control vs. naris-occluded.
To this end we performed a 3-D Sholl analysis (Figure 5A) to measure the total dendritic length or number of intersections in spheres of increasing radii (Figure 5A1). These measures are reported as a function of sphere radius and are indicative of the degree of branching in neuronal processes (Sholl, 1953), where more complex processes result in more intersections with the “Sholl” sphere as distance increases. Sholl analysis of total dendritic length indicates siRNA knock-down cells have reduced dendritic length beginning in medial regions of the dendrite, showing a significant difference at a radius of 120 μm (control: 42.3 ± 7.1 μm; siRNA: 20.4 ± 1.0 μm; p < 0.01; n = 20; Figure 5A2), indicating that control cells contain more or longer branches than siRNA knock-down cells. Unlike siRNA knock-down cells, NO animals failed to exhibit significant differences in any region of the dendrite (Figure 5A3).
Reductions in the length of dendrites as assayed by the 3-D Sholl analysis may be explained by two scenarios: (1) dendrites in activity-reduced groups are less tortuous than those in the control group, or (2) there is a reduction in the number of branches formed by activity-reduced cells. Qualitatively, there did not appear to be significant differences in the relative tortuosity between control and activity-reduced cells of either group (Figure 2). Therefore we suspected that the differences observed in the distribution of length could be accounted for by differences in branch pattern, i.e., the reduction in activity resulted in a reduction of high-order branching. To test this, we created modified Sholl plots that showed the number of intersections as a function of the distance from the soma along the dendritic tree (Figure 5B1) to form a more accurate picture of the magnitude of branching of the dendritic tree. We observed that siRNA reduced branching at medial distances (at 120 μm: control: 2.5 ± 0.17 branches; siRNA: 1.9 ± 0.03 branches; p < 0.01; n = 20; Figure 5B2). Thus, an exaggerated dendritic tree of an siRNA cell would resemble a control cell stripped of branches at all but the most distal locations. Global activity reduction in NO cells showed no significant difference in branching in our modified Sholl plot (Figure 5B3).
To examine global differences in dendritic complexity between control and activity-reduced cells, we plotted the average branch order against the distance from the soma along the dendritic tree (Figure 5C1). Though differences in total branch number and average terminal branch order were insignificant for both activity reduced groups (Table 1), observing these subtle differences together with the distributions of both the standard (Figure 5A) and modified (Figure 5B) Sholl plots suggest differences in cumulative branch number. Consistent with this prediction, there appeared to be an overall reduction of branching in siRNA knock-down cells over much of the dendrite (Figure 5C2). Branch order was significantly reduced at 100 μm (control: 0.71 ± 0.23; siRNA: 0.14 ± 0.05; p < 0.05; n = 20) although branching recovered at the furthest distal segments (Figure 5C2). However, global activity reduction appeared to have no effect on the distribution of branch order (Figure 5C3). Coupled with the modified Sholl plot (Figure 5B), these data suggest that while global activity reduction does not compromise branch elaboration, it does cause reductions in branch length.
Discussion
Alterations of activity can result either promote or inhibit the survival of ABNs in the OB (Cummings and Brunjes, 1997; Mandairon et al., 2003; Yamaguchi and Mori, 2005; Kelsch et al., 2009; Moreno et al., 2009; Mouret et al., 2009). Here we used a viral vector encoding an siRNA to knock-down sodium channel expression and naris occlusion to reduce the activity of ABNs that migrate to the OB (which we note to reduce survival of ABNs by 44 and 24% respectively, data not shown). Both methods used to reduce neuronal activity resulted in significant decreases in the total number of spines, overall spine density, and several measures of branching complexity. In siRNA knock-down cells we observed that reductions in spine number, spine density, and branching complexity incurred primarily in proximal and intermediate regions of the dendritic tree. However naris-occluded cells did not exhibit significant changes in any specific region along the dendrite. Rather changes in dendritic length and branching were distributed along the length of the dendrite.
Previously published results by Kelsch et al. (2009) reported that naris occlusion resulted in highly localized changes in the number of formed connections of ABNs labeled by a lentiviral vector expressing GFP tagged PSD-95. Specifically, changes seen in ABNs experiencing sensory deprivation by naris occlusion resulted in significant decreases in the number of PSD-95 labeled synapses at distal regions of the dendrite, and significant increases in the number of synapses at proximal regions of the dendrite (Kelsch et al., 2009). This effect does not appear in our results. However, it is important to note that PSD-95-GFP likely labels glutamatergic inputs onto ABNs rather than GABAergic outputs (Sheng, 2001). While Kelsch et al. (2009) did observe output synapses of ABNs via labeling of synaptophysin (SypG), which localizes to presynaptic neurotransmitter vesicles (Sudhof and Jahn, 1991), they reported significant changes in the number of SypG+ clusters at distal regions of the dendrite only. Because our experimental paradigm aimed to look at the effect of naris occlusion on the functional integration of ABNs into the OB, we chose looked at changes in spines, which serve as the primary sites of GABAergic lateral inhibition onto mitral/tufted cells of the OB (Panzanelli et al., 2009). While our dataset appeared to exhibit trends consistent with analysis of SypG clustering by Kelsch et al. (2009) it is unclear why our results did not reach significance.
Conversely, Lin et al. (2010) observed the effects of genetically manipulating excitability on both survival and morphology of ABNs. They noted that neither genetically decreasing excitability (via expression of ESKir2.1) nor genetically increasing excitability (via expression of NaChBac) had any effect on morphology of ABNs (Lin et al., 2010). While these results appear to contrast our own, it is important to note that Lin et al. (2010) altered the excitability of ABNs by inserting ion channels rather than altering expression of endogenous channels. Thus, the degree to which activity was changed may be different in our studies vs. the previous work. Alternatively, homeostatic mechanisms may more effectively compensate for reductions in activity not caused by knock-down of endogenous channels. These subtle and specific manipulations may provide insight into how differences in activity across unmanipulated ABNs can influence their connectivity.
Our results suggest that reduced levels of activity in ABNs adversely affect their ability to integrate into existing OB circuitry. Specifically, fewer spines formed by ABNs experiencing reduced activity indicates fewer connections made with surrounding circuitry, and thus reduced participation in olfactory processing. Further it appears that these two different manipulations of activity differentially affect spine development of ABN dendrites. While it is believed that naris occlusion results in sensory deprivation (and thus reduction of activity) in the OB (Frazier-Cierpial and Brunjes, 1989), the effect of knocking down expression of voltage gated sodium channels (NaV1.1–1.3) in ABNs is less clear.
In mature olfactory granule cells, lateral inhibition onto mitral cells can be evoked by global action potentials (Petreanu and Avarez-Buylla, 2002; Carleton et al., 2003) and low-threshold spiking (Egger, 2008). Global action potentials result in long-lasting depolarizations, mediated by calcium activated non-specific cation currents (Egger, 2008), and NMDAR-dependent calcium influx. This results in prolonged release of GABA at dendrodendritic synapses, and thus more efficient lateral inhibition onto mitral and tufted cell dendrites when compared to inhibition due to low-threshold spiking (Hall and Delaney, 2002). By knocking down NaV expression in ABNs, we hoped to reduce the number of global action potentials and their resulting activity.
Given the role of NaV channels in the physiology of olfactory granule cells, we hypothesize that global spiking of ABNs serves as the primary form of activity in proximal regions of the dendrite (Egger et al., 2003). However at distal regions of the dendrite, highly localized calcium transients due to dense populations of T-type calcium channels in spines facilitate NMDA receptor activation (Egger et al., 2005) and allow for dendrodendritic inhibition independent of sodium channel activation (Isaacson and Strowbridge, 1998; Schoppa et al., 1998). That is, activity in more proximal regions of the dendrite may be more vulnerable to fluctuations of NaV channel expression, where activity in distal synapses may be more electrically isolated from the soma and thus depend only weakly on voltage gated sodium channels. This selective change in activity may be relevant to the differences between our results and those of Kelsch et al. (2009).
One question is whether siRNA knock-down changes overall activity of affected OBs, including uninfected cells. Since our lentiviral injections selectively labeled neural progenitors, only a small fraction of cells in a given OB are infected by the virus; consequently the activity reduction in the infected granule cells is likely to have little effect on overall OB activity. Thus the overall level of input received by new granule cells in the hemisphere injected with siRNA virus is likely to be comparable to cells in the control hemisphere. By contrast, cells in naris-occluded animals experience a reduction of activity via a reduction of synaptic input from sensory neurons to mitral cells. This will directly reduce input to distal dendrites of granule cells and also reduce somatic spiking. Thus changes in the spatial profile of formed spines may reflect differences in the profile of activity in cells under these two different conditions.
We have focused on differences observed in mature ABNs. Therefore we chose a single time point (35 days post-injection), which allowed ample time for ABN maturation following birth. As reported previously, ABNs reach “class 5” morphology (full maturation) 15–30 days after birth (Petreanu and Avarez-Buylla, 2002). Cells in this morphological class have been shown to have adult-like membrane properties and sodium channel expression (Carleton et al., 2003). Given the typical maturation time of ABNs, we do not expect any time points beyond 35 days post-injection to show differences from those reported here, but examining earlier time points could identify when activity reduction begins to affect synapse formation.
We have shown that ABNs can develop nearly normal morphological features, despite manipulations in their activity levels. That is, these neurons adapt to their altered states or environments by altering specific aspects of their morphologies, while maintaining their gross structural features. Thus, major morphological features of these neurons develop in a manner that is largely insensitive to changes in activity, and observed changes are linked to more subtle and possibly highly specific changes in input number and location.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Jennifer Dry-Henich and Greg LaRocca for animal care and laboratory support, and James Fitzpatrick for technical imaging assistance. This work was supported by R01DC0005798 (to Nathan N. Urban), F31DC009968 (to Daniel A. Jimenez), and F32DC010535 (to Richard C. Gerkin).
References
Alonso, M., Viollet, C., Gabellec, M. M., Meas-Yedid, V., Olivo-Marin, J. C., and Lledo, P. M. (2006). Olfactory discrimination learning increases the survival of adult-born neurons in the olfactory bulb. J. Neurosci. 26, 10508–10513.
Altman, J. (1969). Autoradiographic and histological studies of postnatal neurogenesis. IV. Cell proliferation and migration in the anterior forebrain, with special reference to persisting neurogenesis in the olfactory bulb. J. Comp. Neurol. 137, 433–457.
Bagley, J., LaRocca, G., Jimenez, D. A., and Urban, N. N. (2007). Adult neurogenesis and specific replacement of interneuron subtypes in the mouse main olfactory bulb. BMC Neurosci. 8, 92. doi: 10.1186/1471-2202-8-92
Carleton, A., Petreanu, L. T., Lansford, R., varez-Buylla, A., and Lledo, P. M. (2003). Becoming a new neuron in the adult olfactory bulb. Nat. Neurosci. 6, 507–518.
Cetin, A., Komai, S., Eliava, M., Seeburg, P. H., and Osten, P. (2006). Stereotaxic gene delivery in the rodent brain. Nat. Protoc. 1, 3166–3173.
Corotto, F. S., Henegar, J. R., and Maruniak, J. A. (1994). Odor deprivation leads to reduced neurogenesis and reduced neuronal survival in the olfactory bulb of the adult mouse. Neuroscience 61, 739–744.
Cummings, D. M., and Brunjes, P. C. (1997). The effects of variable periods of functional deprivation on olfactory bulb development in rats. Exp. Neurol. 148, 360–366.
Dittgen, T., Nimmerjahn, A., Komai, S., Licznerski, P., Waters, J., Margrie, T. W., Helmchen, F., Denk, W., Brecht, M., and Osten, P. (2004). Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc. Natl. Acad. Sci. U.S.A. 101, 18206–18211.
Egger, V. (2008). Synaptic sodium spikes trigger long-lasting depolarizations and slow calcium entry in rat olfactory bulb granule cells. Eur. J. Neurosci. 27, 2066–2075.
Egger, V., Svoboda, K., and Mainen, Z. F. (2003). Mechanisms of lateral inhibition in the olfactory bulb: efficiency and modulation of spike-evoked calcium influx into granule cells. J. Neurosci. 23, 7551–7558.
Egger, V., Svoboda, K., and Mainen, Z. F. (2005). Dendrodendritic synaptic signals in olfactory bulb granule cells: local spine boost and global low-threshold spike. J. Neurosci. 25, 3521–3530.
Frazier-Cierpial, L., and Brunjes, P. C. (1989). Early postnatal cellular proliferation and survival in the olfactory bulb and rostral migratory stream of normal and unilaterally odor-deprived rats. J. Comp. Neurol. 289, 481–492.
Hall, B. J., and Delaney, K. R. (2002). Contribution of a calcium-activated non-specific conductance to NMDA receptor-mediated synaptic potentials in granule cells of the frog olfactory bulb. J. Physiol. 543, 819–834.
Isaacson, J. S., and Strowbridge, B. W. (1998). Olfactory reciprocal synapses: dendritic signaling in the CNS. Neuron 20, 749–761.
Kelsch, W., Lin, C. W., Mosley, C. P., and Lois, C. (2009). A critical period for activity-dependent synaptic development during olfactory bulb adult neurogenesis. J. Neurosci. 29, 11852–11858.
Komai, S., Licznerski, P., Cetin, A., Waters, J., Denk, W., Brecht, M., and Osten, P. (2006). Postsynaptic excitability is necessary for strengthening of cortical sensory responses during experience-dependent development. Nat. Neurosci. 9, 1125–1133.
Lazarini, F., and Lledo, P. M. (2010). Is adult neurogenesis essential for olfaction? Trends Neurosci. 34, 20–30.
Lin, C. W., Sim, S., Ainsworth, A., Okada, M., Kelsch, W., and Lois, C. (2010). Genetically increased cell-intrinsic excitability enhances neuronal integration into adult brain circuits. Neuron 65, 32–39.
Mandairon, N., Jourdan, F., and Didier, A. (2003). Deprivation of sensory inputs to the olfactory bulb up-regulates cell death and proliferation in the subventricular zone of adult mice. Neuroscience 119, 507–516.
Moreno, M. M., Linster, C., Escanilla, O., Sacquet, J., Didier, A., and Mandairon, N. (2009). Olfactory perceptual learning requires adult neurogenesis. Proc. Natl. Acad. Sci. U.S.A. 106, 17980–17985.
Mouret, A., Lepousez, G., Gras, J., Gabellec, M. M., and Lledo, P. M. (2009). Turnover of newborn olfactory bulb neurons optimizes olfaction. J. Neurosci. 29, 12302–12314.
Ninkovic, J., Mori, T., and Gotz, M. (2007). Distinct modes of neuron addition in adult mouse neurogenesis. J. Neurosci. 27, 10906–10911.
Panzanelli, P., Bardy, C., Nissant, A., Pallotto, M., Sassoe-Pognetto, M., Lledo, P. M., and Fritschy, J. M. (2009). Early synapse formation in developing interneurons of the adult olfactory bulb. J. Neurosci. 29, 15039–15052.
Petreanu, L., and Avarez-Buylla, A. (2002). Maturation and death of adult-born olfactory bulb granule neurons: role of olfaction. J. Neurosci. 22, 6106–6113.
Price, J. L., and Powell, T. P. (1970). The morphology of the granule cells of the olfactory bulb. J. Cell Sci. 7, 91–123.
Rall, W., Shepherd, G. M., Reese, T. S., and Brightman, M. W. (1966). Dendrodendritic synaptic pathway for inhibition in the olfactory bulb. Exp. Neurol. 14, 44–56.
Rochefort, C., Gheusi, G., Vincent, J. D., and Lledo, P. M. (2002). Enriched odor exposure increases the number of newborn neurons in the adult olfactory bulb and improves odor memory. J. Neurosci. 22, 2679–2689.
Saghatelyan, A., Roux, P., Migliore, M., Rochefort, C., Desmaisons, D., Charneau, P., Shepherd, G. M., and Lledo, P. M. (2005). Activity-dependent adjustments of the inhibitory network in the olfactory bulb following early postnatal deprivation. Neuron 46, 103–116.
Schoppa, N. E., Kinzie, J. M., Sahara, Y., Segerson, T. P., and Westbrook, G. L. (1998). Dendrodendritic inhibition in the olfactory bulb is driven by NMDA receptors. J. Neurosci. 18, 6790–6802.
Sheng, M. (2001). The postsynaptic NMDA-receptor-PSD-95 signaling complex in excitatory synapses of the brain. J. Cell Sci. 114, 1251.
Sholl, D. A. (1953). Dendritic organization in the neurons of the visual and motor cortices of the cat. J. Anat. 87, 387–406.
Sudhof, T. C., and Jahn, R. (1991). Proteins of synaptic vesicles involved in exocytosis and membrane recycling. Neuron 6, 665–677.
Keywords: neurogenesis, morphology, activity, olfaction, naris occlusion, sodium channels
Citation: Dahlen JE, Jimenez DA, Gerkin RC and Urban NN (2011) Morphological analysis of activity-reduced adult-born neurons in the mouse olfactory bulb. Front. Neurosci. 5:66. doi: 10.3389/fnins.2011.00066
Received: 06 December 2010; Paper pending published: 17 January 2011;
Accepted: 27 April 2011; Published online: 09 May 2011.
Edited by:
Jack M. Parent, University of Michigan, USAReviewed by:
Luca Bonfanti, University of Turin, ItalyJürgen Winkler, University Hospital Erlangen, Germany
Copyright: © 2011 Dahlen, Jimenez, Gerkin and Urban. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Nathan N. Urban, Department of Biological Sciences, Carnegie Mellon University, 4400 Fifth Avenue, Pittsburgh, PA 15213, USA. e-mail: nurban@cmu.edu